
Abstract
The traceability of meat origin has become a necessity to safeguard consumer's confidence in commercial meat products. Our study recommends a meat species detection using qualitative approach based on PCR–RFLP and species-specific primers PCR. Here, we targeted a 359 bp fragment of the mitochondrial cytochrome b gene amplified by PCR using universal primers followed by three enzymatic digestions. Seven animal species including dromedary, rabbit, goat, turkey, rat, donkey and pork, have been efficiently detected in pure and mixed samples. The combination of PCR–RFLP and triplex PCR assays offers, in addition, the identification of chicken, dog and cat species in meat. In conclusion, by the mean of PCR-based techniques using universal primers followed by enzymatic digestion and multiplex primer-specific approach, we developed an extensible protocol by which we identified 10 animal species that could be integrated in meat analysis daily routine.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Meat adulteration, with other meat of inexpensive or objectionable species, has become a frequent practice in both developing and developed countries [1,2,3]. Beyond posing a health risk to people including metabolic disorders, especially for dyslipidemia patients [4] or allergies, which mainly affected atopic persons [5], meat impurities may lead to religious apprehensions and financial gain [6,7,8]. For instance, in 2013, the horse meat scandal in Europe has aroused global attention of meat adulteration [9]. In fact, adulterated horse meat [10, 11] and halal beef burgers adulterated with pork were discovered in some European countries. Similarly, in the same year, 37% of horse DNA and 85% of pork DNA were found by Irish authorities in beef burgers, ground beef products and sausages [12]. Comparable phenomena have been widely reported in many places in China, such as wholesale markets, village fairs and even some supermarkets [13]. Recently, in 2017 and 2019, based on social media and electronic newspapers, undeclared donkey and cat meat were used as a raw material for making cooked sausage ‘Sausage and "Chawarma" in Tunisia (supplementary Material S4).
The drastic consequences on the meat industry highlighted the urgent necessity to control the products quality and to point out the complexity of both supply and processing circuits. Due to the expansion of this problem, the authentic testing meat and meat products, is deemed crucial to avoid unfair market competition and to protect consumers from fraudulent practices of meat adulteration [14, 15]. The adoption of authentication methods by the food meat-control laboratories is becoming a priority issue. However, in some developing countries, the number of food tests is still insignificant although a variety of processed and tradition meat products are widely consumed (e.g. “Sausage”, “merguez”, “kebab”, “kofta” and “hamburgers”, etc.). Little attention has been paid to provide an easy, fast, reproducible and low-cost molecular test, which could be conducted in basic laboratory. In the meat industry, there is a need to implement such protocol for food authentication to encourage analysis of processed meat in daily routines [16]. In this line, technologies for identifying meat contamination have been investigated and key approaches for accurately detecting have been developed. Several investigated techniques, primarily relying on protein and DNA analysis, have been proposed to identify meat species in composite mixtures so far [4]. Based on DNA and protein, numerous assays have been widely used for the identification of meat species [17, 18]. Compared to protein, DNA is a remarkably stable molecule, which can be extracted from many different types of samples [19]. Indeed, techniques using DNA as a template are cutting-edge tools to check the corruption and the authenticity of the products [20]. Mitochondrial DNA (mtDNA) is often favored as a genetic marker over nuclear DNA for species identification. In fact, the recognition of mtDNA is easier in highly processed and degraded tissue as its stability is higher than the genomic DNA [21]. Moreover, the mtDNA is distributed in all the tissues with multiple copies per cell. Thus, its sequences are preferential for DNA barcoding in goat, sheep, deer, buffalo, cattle, yak, pig, and camel species identification [22].
On the other hand, multiple methods based on polymerase chain reaction (PCR) have been proposed to identify the different species composing the meat products ranging from DNA hybridization to detect chicken, turkey, pork, horse, sheep and beef [23], conventional polymerase chain reaction (PCR) to reveal the presence of pork, horse, beef, chicken, turkey and goat [24], species-specific PCR, multiplex PCR to identify beef and sheep [25, 26], Droplet digital PCR to detect the contaminant pork, horse and chicken species [27], PCR–RFLP to detect cattle, sheep, pork, chicken, donkey and horse [28], PCR-SSCP to identify pork [29, 30] to PCR sequencing [29,30,31].
In recent years, universal primer and species-specific primers PCRs have been used and further developed for animal species identification. This choice is due to their high specificity and sensitivity, as well as rapid processing time and low cost [16, 32].
Based on the last assumptions, this study aimed to set up a primary test for the identification of ten animal species based on mitochondrial D-loop [33] and cytochrome b gene [34]. This can be achieved through the use of a PCR–RFLP and a species-specific PCR as potential screening test methods to promote their adoption as a routine procedure.
Materials and methods
Biological samples collection
Fresh blood samples
Fifty-six animals' peripheral blood was collected from dog, cat, chicken, goat, dromedary, rat, pig, rabbit, donkey, and turkey species from 2013 till 2015. Characteristics of all included DNA samples are detailed in supplementary materials (Table S1).
Fresh and processed meat samples
Samples from six commercialized turkey ‘Sausage brands were purchased from local suppliers Sfax-Tunisia (N: 34.4426°, E: 10.4537°) and stored at − 20 °C. The manufacture and expiry dates as well as the batch numbers were recorded for each sample. Authentic fresh meat samples from chickens and turkey were collected as mentioned in Table S1. To obtain a homogeneous paste, 50 mg of raw and processed meat samples were blended using a vibromill MM 400 (Retsch, France).
DNA extraction
DNA was extracted from blood collected in EDTA-containing vacutainer (BD, USA) and from raw and processed meat using, respectively, a modified phenol–chloroform protocol as described by Kawazaki [35] and the phenol–chloroform protocol adopted by Gargouri and HadjKacem [16].
PCR optimization for animal species DNA amplification
Universal primer and species-specific primers specificity
In silico study
mtDNA sequences of 45 species were selected from the RefSeq database of the NCBI Website (Table S2) and for the multiple sequence alignment (MSA), Clustal W (http:/www.ebi.ac.uk/clustalw/) was used.
In vitro study
For PCR amplification, three species-specific and one universal pairs of primers designed by Abdulmawjood et al. [36], Mane et al. [33] and Kocher et al. [34] for the mitochondrial cytochrome b and D-loop (Table 1) were used. Species-specific primers were tested for inter and intraspecific discrimination.
PCR amplifications were carried out using the same protocol and the same reagents. All reactions were conducted using DNA extracted from blood in a final volume of 50 µl containing 200 ng of genomic DNA, 2 mmol/l MgCl2, 0.2 mmol/l dNTP, 0.10 µmol/l of each primer, 1 × reaction buffer (Thermoscientific, Waltham, USA), and 1 U of Taq DNA polymerase (Thermoscientific, Waltham, Massachusetts, USA) [16]. PCR was conducted for 38 cycles following the conditions of 94 °C for 30 s, 60 °C for 30 s and 72 °C extension for 30 s, with an initial denaturation step at 94 °C for 4 min. Whether using universal or species-specific primers, the first operation was a PCR amplification handled with an individual DNA sample [16]. The second step was carrying out a PCR amplification using different mixtures of animal DNA samples in the same proportion (Table S3).
Multiplex PCR amplification using species-specific primers
DNA amplifications were performed in a final volume of 50 µl containing 200 ng of DNA, 4 mmol/l MgCl2, 0.4 mmol/l dNTP, 0.2 µmol/l of each primer, 4 U Gotaq polymerase (Promega), reaction buffer 10X and 1 µl DMSO. The PCRs amplifications were conducted with the following cycling conditions: an initial denaturation step at 95 °C for 5 min, 40 cycles of denaturation at 95 °C for 45 s, annealing at 58 °C for 30 s and extension at 72 °C for 45 s. A final extension step at 72 °C for 5 min was added.
PCR products purification and sequencing
PCR products were purified with enzymatic treatment using 10 U of Exonuclease I (Thermoscientific, EU) and then sequenced by automated DNA sequencing analysis with fluorescence-labeledideoxy terminators (Big Dye Terminator V3.1 Cycle Sequencing Kits, Applied Biosystems, Foster City, CA, USA) according to the manufacturer’s instructions (ABI 3100-4 Genetic Analyser, Applied Biosystems). The electropherograms were analyzed by the BioEdit software (Ibis Biosciences, Carlsbad, CA, USA).
Restriction fragment length polymorphism (RFLP) analysis
PCR products of the 359 bp cytochrome b gene from different animal DNA were sequenced. Amplicons and RefSeq sequences restriction maps were established and compared using NEBcutter web server (data not shown). The relevant restriction profiles were selected according to the following conditions; 1-minimize the number of enzymes and digestion reactions, 2- avoid the sites of heteroplasmy and 3-identification of a specific profile for each species considering all selected enzymes. Under the above conditions, three restriction enzymes were selected— SspI, TaaI and BsmAI (Fermentas, EU) (Table 2). The PCR–RFLP was first performed with an individual DNA sample and then using different mixtures of animal DNA samples at the same proportion (Table S3). Fragments amplified from 10 animal species were digested with the three selected restriction enzymes as recommended by the supplier for optimal reaction conditions. Digestion reactions were incubated overnight at 37 °C. The digestion products were then loaded on 3% Agarose gel with a suitable DNA ladder (Catalog Number: 15,628,019; ThermoFisher Scientific).
Synthesis mixture preparation
Experimental synthesis mixtures were prepared using the same DNA mixture proportion. Species’ compositions of each mixture are mentioned in Table S3. M1, M2 and M3 were amplified by PCR using universal primers and digested with BsmAI, SspI, and TaaI, respectively. M1, M2, M3 and M4 were also prepared and amplified by species-specific primers PCR using D-loop, CaSSR and DoSSR pair of specific primers. The amount of DNA extracted from blood and used in both PCR–RFLP and in triplex PCR was 30 ng of each DNA sample.
Experimental procedure assessment on standard commercial samples
To identify the animal species in processed meat (Fig. 1), DNA was extracted first from turkey Sausage using the phenol–chloroform protocol, as described by Gargouri and HadjKacem [16]. Then, the extracted DNA was amplified using a universal and species-specific set of primers. The digestion of the 359 bp PCR fragment was done with a single digest enzyme according to the manufacturer’s instructions. PCR triplex was also carried out using the three species-specific primers. PCR products were purified, and both cytochrome b and D-loop fragments were directly sequenced as previously mentioned.

Multiple Alignment of 24 mitochondrial genomes from 24 animals’ species. Analysis indicates a similar forward and reverse sequence between all animals’ species
Results
Universal and species-specific primers specificity
Multiple sequence alignments were investigated to confirm the primer pairs’ specificities. A universal primer and three sets of chicken, cat, and dog species-specific primers were selected from the literature (Table 1) and tested in silico and in vitro using DNA extracted from fresh blood to check its specificity/sensitivity for the detection of animal species.
In silico study
CYT b1 and CYT b2 pair of primers, amplifying the 359 bp region of the mitochondrial cytochrome b gene, were screened for their specificity. The results of the MSA of the CYT b target regions using 24 mitochondrial genomes showed the presence of highly conserved sequences flanking the discrimination variable region (Fig. 1 and supplementary data Table S2). Our findings were coherent with several previous reports defining CYT b1 and CYT b2 sequences as a PCR universal primer [37,38,39,40,41].
The specificity of the CaSSR, DoSSR and D-loop pairs of primers corresponding to the three species of interest (chicken, cat, and dog) was investigated. These pairs of primers cover the 442 bp D-loop, the 672 bp and the 808 bp cytochrome b regions. The MSA of the mentioned regions using 45 different animal species sequences was exclusively matched with the chicken, the cat and the dog, respectively.
In vitro study
To assess the specificities of the CaSSR, DoSSR and D-loop species-specific primers, individual PCRs were performed using mitochondrial DNAs of Tunisian animal including dog, cat, goat, chicken, turkey, rabbit, rat, camel, donkey, and pork. As shown in Fig. 3, the expected specific amplicons are obtained only with cat, dog and chicken DNA template. Additionally, no cross-reactivities have been observed with all remaining animal DNA. Afterwards, the generated PCR amplicons were purified and sequenced. Blast analysis indicated that the sizes and sequences of all the PCR products correspond exactly to the expected amplicons (Fig. 3).
A specific 672 bp amplicon was generated and sequenced using the DNA of a Tunisian cat as a template (Fig. 3a). The sequence alignment showed 100% of identity with sequences of cats from different origins; Portugal (NCBI/Nucleotide accession number: EF689045), Mexico (NCBI/Nucleotide accession number: FJ160761) and the USA (NCBI/Nucleotide accession number: KP202275)).
DoSSR, the second used pair of primers, was reported to be the dog mitochondrial genome-specific amplifying 808 bp fragment (Fig. 3b). The sequence alignment revealed a 100% identity between the UK dog (NCBI/Nucleotide accession number: KU291088, KU291086), the Tunisian dog, and the Spain wolf (NCBI/Nucleotide accession number: KU696399).
The D-loop set of primers, previously designed by Mane et al. [33], was also tested to detect chicken meat. As expected, a fragment of 442 bp from D-loop region was generated using Tunisian chicken DNA.
DNA amplification and PCR–RFLP analysis
The PCR–RFLP technique has been widely applied as a valuable DNA fingerprint analysis for the identification of animal species, especially in food products [42,43,44]. This method is based on the digestion of the Cyt b region on specific recognition sites using specific restriction endonucleases (RE).
Universal primers were used to amplify the cytochrome gene region of 359 bp of the ten investigated species. Then, three different restriction enzymes (BsmAI, TaaI, and SspI) were selected from the restriction map to distinguish the different species.
The in silico digestion results of the amplicons (Table 2) and the agarose gel electrophoresis profile (Fig. 2) demonstrated that all PCR products were digested at least by one restriction enzyme and, thereby, gave rise to a unique pattern for all animal species, except the chicken amplicon. Thus, PCR–RFLP analysis was not suitable to identify chicken DNA, due to the absence of a restriction site of the applied RE. In addition, the presence of intact fragments (359 bp) in the restriction products using cat and dog mtDNA, suggested a mitochondrial heteroplasmy confirmed by direct sequencing.

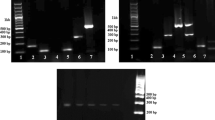
Agarose gel electrophoresis of amplified cytochrome b gene following digestion with TaaI (a), BsmAI (b), SspI (c). a Lane 1: turkey, Lane 2: dromedary, L: 100 bp Dna ladder (Catalog Number: 15,628,019; thermoFisher Scientific). b Lane 1: goat, Lane 2: donkey, Lane 3: rat, L: 100 bp Dna ladder (Catalog Number: 15,628,019; thermoFisher Scientific). c Lane 1: rabbit, Lane 2: rat, Lane 3: pork, L: 100 bp Dna ladder (Catalog Number: 15,628,019; thermoFisher Scientific)
Triplex PCR amplification analysis using species-specific primers
As previously mentioned, the PCR–RFLP was not a suitable technique for the identification of DNA extracted from meat products, due to the possibility of the presence of more than one DNA species in the sample. The PCR–RFLP method was combined with a triplex PCR using species-specific primers. Two targeted mtDNA regions are simultaneously amplified— D-loop and cytochrome b. Figure 3f showed that three specific fragments, corresponding to the expected sizes of 442 bp, 674 bp, and 808 bp, were generated. As a consequence, it was concluded that Triplex PCR might allow the identification and differentiation of three animal species.

PCR amplification results for chicken, cat and dog DNA using a specific primer. a Determination of the specificity of the chicken specific primer Lane 2: Chicken DNA, Lanes 3–12 represent goat, rabbit, dog, cat, donkey, rat, turkey, pork, and dromedary DNA. b Determination of the specificity of the cat specific primer Lane 2: cat DNA, Lanes 3–12 represent goat, rabbit, dog, chicken, donkey, rat, turkey, pork, and dromedary DNA. c Determination of the specificity of the dog specific primer Lane 2: dog DNA, Lanes 3–12 represent goat, rabbit, cat, chicken, donkey, rat, turkey, pork, and dromedary DNA. d Analysis of the chicken DNA race. Lane 2–5: arbor acres + meat, Novogen White and Ross Coqs, arbor acros + blood d. Analysis of the cat DNA race. Lane 7–11: different DNA cat samples. e. Analysis of the dog DNA race. Lane 2- 10: American Pitt Bull Terrier, German Shepherd dog, Rottweiler, Braque, Poodle Dog, Labrador Retriever, Bichon Frise, and Stafford shire Bull Terrier. f Triplex PCR amplification of the fragments corresponding to chicken, cat and dog species. Lane 2, 4, 6: Separate PCR with specific primer Lane 2: Chicken, Lane 4: Cat, Lane 6: Dog. Lane 8–9: Triplex PCR by combining the 3 specific primers. Lane 1 and 12: negative control Lane L: 100 bp Dna ladder (Catalog Number: 15,628,019; thermoFisher Scientific)
DNA mixtures analysis
The combination of PCR–RFLP and triplex PCR assays was sufficient to distinguish separately all included species in our investigation. These assays were further tested to screen all ten targeted animal species in different DNA mixtures (Supplementary data Table S3).
The PCR–RFLP was coupled with a single digestion using each of the three restriction endonucleases (BsmAI, TaaI and SspI) separately. PCR–RFLP analysis was applied to mixed ratios of the seven animal species as listed in Table S3 (Supplementary data). Thus, 30 ng of DNA was sufficient for the identification of the different animal species applying PCR amplification and restriction enzyme digestion on mixed samples.
The electrophoretic DNA pattern, presented in Fig. 4, revealed the presence of different amplicon fragments for every single species with the absence of any cross-reaction. Furthermore, the multiplex PCR assay was successful in detecting the amplicons of the cat, dog, and chicken in the reaction. Hence, this assay enabled us to differentiate between the three mentioned animal species and the other ones using four different reaction mixtures, as mentioned in Table S3 (Supplementary data).

Agarose gel electrophoresis. a of the amplified fragment of the cytochrome b gene in mixture M1 composed of DNA of dog, donkey, rat and goat after digestion with BsmAI (L1) in mixture M2 composed of DNA of rabbit, dog and cat, donkey, rat, pig after digestion with SSPI (L2) in mixture M3 composed of DNA of dromedary, turkey and cat after digestion with TaaI (L3) L: 100 bp DNA Ladder (Invitrogen 100 bp DNA ladder, ref: 15,628,019; thermoFisher Scientific). b of the product amplified by PCR of the mixture M1 composed by the DNA of chicken, cat and dog using the specific primers of cat, dog and chicken. L1: negative control, L2: the DNA mixture M2 composed by dog, donkey, rat, goat, L3: the DNA mixture M3 composed by rabbit, dog, cat, donkey, rat, pig, L4: the DNA mixture M4 composed by dromedary, turkey, cat, L: 100 bp DNA Ladder (catalog number: GM343/GM344; BioBasicInc). c Tunisian turkey Sausage amplified by cytochrome b pairs of primers after digestion with TaaI (L1), with SSPI (L2), with BsmAI (L3), negative control (L4), Tunisian turkey Sausage amplified with specific primers of chicken, cat and dog (L5), L: 100 bp DNA Ladder (catalog number: GM343/GM344; BioBasicInc)
Processed meat analysis
The effectiveness and the sensibility of the DNA identification technique developed in the present study have been evaluated for the identification of animal species in turkey Sausage products. To obtain the characteristic of restriction profiles, the 359 pb Cyt b region was digested by restriction endonucleases. As shown in Fig. 4c, the quantity of DNA isolated from turkey sausage was sufficient for the amplification by PCR reaction. In addition, the PCR–RFLP results demonstrated two fragments of length 158 and 201 bp (Fig. 4c, lane 1), which resulted from TaaI digestion of the PCR products of Cyt b regions. However, no BsmAI nor SspI restriction sites were found in the DNA extracted from turkey Sausage (Fig. 4c, lane 2 and 3). Furthermore, the results from multiplex PCR analysis showed the presence of a specific chicken 442 bp band Fig. 4c, lane 5. To confirm this finding, all the electrophoretic bands were isolated and sequenced. Sequencing results confirmed that the turkey Sausage contained both turkey and undeclared chicken meat.
Discussion
Species identification of animals in meat products is an outstanding topic claimed for economic, religious, and public health purposes. It is an important issue to prevent and detect frauds. Most typical approaches used to identify meat have relied upon the identification of species DNA. Therefore, highly sensitive and modern methods based on DNA sequencing were described in the literature, particularly the Next-Generation Sequencing (NGS) technique. NGS is a non-target technique applied for meat species identification [45, 46]. It is a fast and high-throughput technology and could operate multiple sequencing samples in a single run.
However, due to its relatively high costs, PCR and its derived methods have been commonly applied for meat authentication [16, 32]. Among these methods, PCR–RFLP is recognized as a highly discriminatory, reliable, and reproducible method. PCR–RFLP of a mitochondrial cytochrome b segment has been reported by Maede et al. [47]. Indeed, PCR–RFLP amplification techniques targeting Cyt b gene (359 pb) could be applied as a quick recognizing approach for suspect raw and processed meat.
The discrimination pattern of the 359 bp Cyt b gene has been reported in the literature. Bellagamba et al. [44] stated the use of the PCR–RFLP products of Cyt b for the identification of species in meat and animal feed stuffs. Bravi et al. [48] amplified a fragment of Cyt b to identify meat of cattle, horse, donkey, pig, sheep, dog, cat, rabbit, chicken, and human using universal primers and three restriction enzymes (AluI, HaeIII, and HinfI). Additionally, the amplification of Cyt b gene (359 bp) followed by its digestion with Taa I was carried out to differentiate between cattle and buffalo meats [49]. Abdel-Rahman et al. [39, 50] and Doosti et al. [28] reported the ability to discriminate between donkey and horse species meat using a single restriction enzyme (AluI). Ahmad et al. [51] reported in their study the identification of buffalo, cow, sheep, goat and chicken species with two restriction enzymes (Tas1 and Hinf1). In the present study, the PCR–RFLP amplification of the 359 bp Cyt b and its digestion using BsmAI, Taal, and SspI, restriction enzymes were developed for simultaneous identification of seven species including donkey, pork, rat, turkey, dromedary, rabbit, and goat, reducing so the running cost of the experiment and simplifying digestion protocols using restriction enzymes working at the same conditions. The selected enzymes have no restriction site on the generated fragment using chicken DNA. In addition, intact copies have been obtained using cat and dog DNA with TaaI/SspI and BsmAI/SspI digestions, respectively (Table 2). Therefore, to cover all included species in our work and to reach the identification of 10 different species, a species-specific primer mixture targeting cytochrome b (CaSSR and DoSSR primers) and D-loop regions of mitochondrial DNA was used for the identification of the cat, dog, and chicken DNAs in meat products. The sensitivity of PCR reaction using D-loop primers was tested previously on turkey Sausage DNA mixed with 1, 2, 5 or 10% of chicken DNA [16]. Direct sequencing confirmed that these sets of primers presented no cross-reaction with other species.
This assay was also successfully applied for turkey Sausage authentication [52, 53]. Contrary to their labeling, triplex PCR-assay data revealed the presence of chicken DNA in turkey Sausage samples. This finding clearly indicates the presence of fraudulent substitution of turkey meat by chicken products.
Regarding the overall results of species detection, several inconsistencies with labeling can be pointed out, namely the presence of undeclared chicken meats. The triplex PCR results confirmed also the absence of dog and cat meat.
Consequently, the combination of PCR–RFLP and triplex-PCR assays was successfully developed as a suitable, quick and reliable strategy for detection of adulteration and identification of 10 animal species in daily routine.
Conclusion
The PCR-based methods with specific and predefined primers/probes showed during the last 30 years acceptable efficiencies to characterize animal and plant species in food. However, they displayed limited power with unknown food composition. Nevertheless, using universal fragments, PCR amplification followed by indirect (RFLP) or direct sequencing (Sanger) could reach higher efficiencies and the reduction of the running time and cost with the possibility of identification of fraudulently added species. The validation of the authentication power of species of several small universal genomic regions could be the rapid way to the extensive use of the parallel DNA sequencing (amplicon-based NGS) in food control.
Data availability
All data analyzed during this study are included in this published article (and its supplementary information files).
Abbreviations
- DNA:
-
Deoxyribonucleic acid
- mtDNA:
-
Mitochondrial deoxyribonucleic acid
- MSA:
-
Multiple sequence alignment
- PCR:
-
Polymerase chain reaction
- PCR–RFLP:
-
Restriction fragment length polymorphism polymerase chain reaction
- PCR-SSCP:
-
Single-strand conformation polymorphism polymerase chain reaction
- EDTA:
-
Ethylenediaminetetraacetic acid
- D-loop:
-
Displacement loop
- MgCl2 :
-
Magnesium chloride
- dNTP:
-
Deoxynucleotide
- DMSO:
-
Dimethyl sulfoxide
- Cyt b:
-
Cytochrome b
- CaSSR:
-
Cat species-specific region
- DoSSR:
-
Dog species-specific region
- NEBcutter:
-
NEB—New England Biolabs cutter
References
Wu H, Qian C, Wang R, Wu C, Wang Z, Wang L, Zhang M, Ye Z, Zhang F, He J-S, Wu J (2020) Identification of pork in raw meat or cooked meatballs within 20 min using rapid PCR coupled with visual detection. Food Control 109:106905
Kang T-S (2019) Basic principles for developing real-time PCR methods used in food analysis: a review. Trends Food Sci Technol 91:574–585
Song Q, Chen Y, Zhao L, Ouyang H, Song J (2019) Monitoring of sausage products sold in Sichuan Province, China: a first comprehensive report on meat species’ authenticity determination. Sci Rep 9:1–9
Drupad KT, Katherine AH, Nicholas JWR, Holli W, Dakshat KT, Joseph G, David IE, Royston G (2016) Meat, the metabolites: an integrated metabolite profiling and lipidomics approach for the detection of the adulteration of beef with pork. Analyst 141:2155
Ahmed N, Sangale D, Tiknaik A, Prakash B, Hange R, Sanil R, Khan S, Khedkar G (2018) Authentication of origin of meat species processed under various Indian culinary procedures using DNA barcoding. Food Control 90:259–265
Hossain MM, Uddin SMK, Sultana S, Wahab YA, Sagadevan S, Johan MR, Ali ME (2020) Authentication of Halal and Kosher meat and meat products: Analytical approaches, current progresses and future prospects. Crit Rev Food Sci Nutr 1–26
Li Y-C, Liu S-Y, Meng F-B, Liu D-Y, Zhang Y, Wang W, Zhang J-M (2020) Comparative review and the recent progress in detection technologies of meat product adulteration. CRFSFS 19:2256–2296
Ross A, Brunius C, Chevallier O, Dervilly G, Elliott C, Guitton Y, Prenni JE, Savolainen O, Hemeryck L, Vidkjaer NH, Scollan N, Stead SL, Zhang R, Scollan N (2020) Making complex measurements of meat composition fast: Application of rapid evaporative ionisation mass spectrometry to measuring meat quality and fraud. Meat Sci 108333
Abbots E-J, Coles B (2013) Horsemeat-gate: the discursive production of a neoliberal food scandal. Food Cult Soc 16:535–550
Nau J-Y (2013) Horse meat: first lessons of a scandal. Rev Med Suisse 9:532
Hsieh Y-H-P, Ofori J-A (2014) Detection of horse meat contamination in raw and heat-processed meat products. J Agric Food Chem 62:12536–12544
Kane D (2015) Identification of species in ground meat products sold on the U.S. commercial market using DNA-based methods. Master's thesis, Chapman University
Qiuchi S, Yiwu C, Liming Z, Hongsheng O, Jun S (2019) Monitoring of sausage products sold in Sichuan Province, China: a first comprehensive report on meat species’ authenticity determination. Sci Rep 9:1
Abbas O, Zadravec M, Baeten V, Mikuš T, Lešić T, Vulić A, Prpić J, Jemeršić L, Pleadin J (2018) Analytical methods used for the authentication of food of animal origin. Food Chem 246:6–17
Cavin C, Cotteneta G, Kevin M-C, Pascal Z (2018) Meat vulnerabilities to economic food adulteration require new analytical solutions. Food AnAlysis MeAt And MeAt Prod 72:697–703
Gargouri H, HadjKacem H (2018) Evaluation of alternative DNA extraction protocols for the species determination in turkey salami authentication tests. Int J Food Prop 21:733
Alikord M, Momtaz H, Keramat J, Kadivar M, Homayouni RA (2017) Species identification and animal authentication in meat products: a review. Food Measure 12:145
Kaltenbrunner M, Hochegger R, Cichna-Markl M (2018) Development and validation of a fallow deer (Dama dama)- specific TaqMan real-time PCR assay for the detection of food adulteration. Food Chem 243:82–90
Liu W, Tao J, Xue M, JiJ ZY, Zhang L, Sun W (2019) A multiplex PCR method mediated by universal primers for the identification of eight meat ingredients in food products. Eur Food Res Tech 245:2385–2392
Lo Y-T, Shaw P-C (2018) DNA-based techniques for authentication of processed food and food supplements. Food Chem 240:767–774
Hang J, Sejeong K, Jeeyeon L, Soomin L, Heeyoung L, Yukyung C, Hyemin O, Yohan Y (2017) Identification of pork adulteration in processed meat products using the developed mitochondrial DNA-based primers. Korean J Food Sci Anim Resour 37:464–468
Guan F, Jin Y, Zhao J, XU A, Luo Y (2018) A PCR method that can be further developed into PCR-RFLP assay for eight animal species identification. J Anal Methods Chem 5890140
Jonker KM, Tilburg JJHC, Hägele GH, de Boer E (2008) Species identification in meat products using real-time PCR. Food Addit Contam Part A 25:527–533
Thienes Ha Y, Agapov CP, Laznicka AA, Han AV, Nadala S, Samadpour MC (2018) Comparison of ELISA and DNA lateral flow assays for detection of pork, horse, beef, chicken, turkey, and goat contamination in meat products. J AOAC Int 102:189–195
Li XN, Guan YF (2019) Specific identification of the adulterated components in beef or mutton meats using multiplex PCR. J AOAC Int 102:1181–1185
Liu WW, Tao J, Xue M, Ji JG, Zhang YH, Zhang LJ, Sun WP (2019) A multiplex PCR method mediated by universal primers for the identification of eight meat ingredients in food products. Eur Food Res Tech 245:2385–2392
Naaum A, Shehata H, Chen S, Li J, Tabujara N, Awmack D, Hanner R (2018) Complementary molecular methods detect undeclared species in sausage products at retail markets in Canada. Food Control 84:339–344
Doosti A, Dehkordi PG, Rahim E (2014) Molecular assay to fraud identification of meat products. J Food Sci Technol 51:148–152
Kumar A, Rajiv R-K, Brahm D-S, Palanisamy G, Sanjod K-M, Deepak S (2015) Identification of species origin of meat and meat products on the DNA basis: a review. Crit Rev Food Sci Nutr 55:1340–1351
Shabani H, Mehdizadeh M, Mousavi S-M, Dezfouli E-A, Solgi T, Khodaverdi M, Rabiei M, Rastegar H, Alebouyeh M (2015) Halal authenticity of gelatin using species-specific PCR. Food Chem 184:203–206
Amorim A, Fernandes T, Taveira N (2019) Mitochondrial DNA in human identification: a review. PeerJ 7:7314
Floren C, Wiedemann I, Brening B, Schutz E, Beck J (2015) Species identification and quantification in meat and meat products using droplet digital PCR (ddPCR). Food Chem 173:1054–1058
Mane B-G, Mendiratta S-K, Tiwari A-K (2009) Polymerase chain reaction assay for identification of chicken in meat and meat products. Food Chem 116:806–810
Kocher T-D, Thomas W-K, Meyer A, Edwards S-V, Paabo S, Villablanca F-X (1989) Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc Natl Acad Sci USA 86:6196–6200
Kawazaki E (1990) Sample preparation from blood, cells and other fluids. In: Innis M, Gelffand D, Snisky G, White T (eds) PCR protocols. A guide to methods and application. Academic Press, San Diego, pp 146–152
Abdulmawjood A, Schönenbrücher H, Bülte M (2003) Development of a polymerase chain reaction system for the detection of dog and cat meat in meat mixtures and animal feed. J Food Sci 68:1757
Steven M-C, Marshall W-D (1991) Detection of intraspecific DNA sequence variation in the mitochondrial cytochrome b gene of Atlantic Cod (cadusMorhua) by the polymerase chain reaction. Can J Fish Aquat Sci 48:48–52
Parson W, Karin P, Niederstätter H, Föger M, Martin S (2000) Species identification by means of cytochrome b gene. Int J Legal Med 114:23–28
Abdel-Rahman S-M, El-Saadani M-A, Ashry K-M, Haggag A-S (2009) Detection of adulteration and identification of Cat’s, Dog’s, Donkey’s and Horse’s meat using species-specific PCR and PCR-RFLP techniques. Aust J Basic Appl Sci 3:1716–1719
Minarovič T, Trakovická A, Rafayová A, Lieskovská Z (2010) Animal Species Identification by PCR – RFLP of Cytochrome b. J Anim Sci Biotechnol 43:296
D’Amato M-E, Alechine E, Cloete K-W, Davison S, Corach D (2013) Where is the game? Wild meat products authentication in South Africa: a case study. Investig Genet 4:1
Meyer R, Hoefelein C, Luethy J, Candrian U (1995) Polymerase chain reaction restriction fragment length polymorphism analysis : a simple method for species identification in food. J AOAC Int 78:1542–1551
Partis L, Croan D, Guo Z, Clark R, Coldham T, Murby J (2000) Evaluation of a DNA fingerprinting method for determining the species origin of meats. Meat Sci 54:369–376
Bellagamba F, Moretti V-M, Comincini S, Valfrè F (2001) Identification of species in animal feed-stuffs by polymorphism chain reaction-restriction fragment length polymorphism analysis of mitochondrial DNA. J Agric Food Chem 49:3775–3781
Xing R-R, Wang N, Hu R-R, Zhang J-K, Han J-X, Chan Y (2019) Application of next generation sequencing for species identification in meat and poultry products: a DNA metabarcoding approach. Agris 101:173–179
Cottenet G, Carine B, Poh Fong C, Christophe C (2020) Evaluation and application of a next generation sequencing approach for meat species identification. Food Control 110:107003
Maede D (2006) A strategy for molecular species detection in meat and meat products by PCR-RFLP and DNA sequencing using mitochondrial and chromosomal genetic sequences. Eur Food Res Tech 224:209–217
Bravi C-M, Liron J-P, Mirol P-M, Ripoli M-V, Garcia P-P, Giovambattista G (2004) A simple method for domestic animal identification in Argentina using PCR-RFLP analysis of cytochrome b gene. J Legal Med 6:246–251
Ahmed M-M-M, Abdel-Rahman S-M, El-Hanafy A-A (2007) Application of species-specific polymerase chain reaction (PCR) for different meat species authentication. Biotechnol 6:426–430
Abdel-Rahman SM (2017) Detection of adulteration and identification of meat and milk species using molecular genetic techniques. Agrotechnology
Ahmad K, Ashfaq CM (2018) Water buffalo production in Turkey part 1: global trend and geographical distribution. Livestock 23:1
Cho A-R, Dong H-J, Cho S (2014) Meat species identification using loop-mediated isothermal amplification assay targeting species-specific mitochondrial DNA. Korean J Food Sci Anim Resour 34:799–807
Ha J, Kim S, Lee J, Lee S, Lee H, Choi Y, Yoon Y (2017) Identification of pork adulteration in processed meat products using the developed mitochondrial DNA-based primers. Korean J Food Sci Anim Resour 37:464–468
Acknowledgements
The authors are thankful to Dr. Fatma KARRAY, Riadh BEN MARZOUG and Mohamed Arbi BEN YOUNES for providing necessary help. We also thank Prof. Saber MASMOUDI for pertinent discussions. The authors are also grateful to Kamel MAALOUL, translator and English professor for having proofread the manuscript.
Funding
The research was funded by the Ministry of Higher Education and Scientific Research of Tunisia.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This research does not include any studies with human subjects or animal experiments.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Gargouri, H., Moalla, N. & Kacem, H.H. PCR–RFLP and species-specific PCR efficiency for the identification of adulteries in meat and meat products. Eur Food Res Technol 247, 2183–2192 (2021). https://doi.org/10.1007/s00217-021-03778-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-021-03778-y