
Abstract
Maltosine (6-(3-Hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-l-norleucine) is formed in the late stage of the Maillard reaction. Quantitative data on the occurrence in food of this metal-chelating 3-hydroxy-4(1H)pyridinone are not available. In the present study, the formation of the compound was studied first in model systems simulating bread crust and crumb. Maltosine was formed predominantly in the presence of di- and oligosaccharides and glycosylated isomaltol derivatives under conditions resembling crust formation. Disaccharide derivatives with β-glycosidic bonds were less potent precursors than derivatives with α-glycosidic bonds. Moreover, a method was developed for the quantification of maltosine in food. The compound was released from proteins by enzymatic hydrolysis due to artefact formation during acid hydrolysis. [13C6,15N2] labelled maltosine was synthesized and utilized as an internal standard, and the analysis of 62 commercial food samples was performed by HPLC–MS/MS in the multiple reaction monitoring mode. Maltosine was present predominantly in bread samples (0.1–4.2 mg/kg). The highest concentrations of the compound were found in the crust of wheat bread (up to 19.3 mg/kg), representing a maximal lysine modification of 0.4 %. The concentrations of maltosine in most food samples reached approximately 10 % of the concentrations of the advanced glycation end product pyrraline. From these data, an intake of maltosine of 1–2 mg per day can be expected from the diet.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
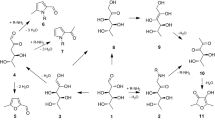
During the Maillard reaction (synonyms: non-enzymatic browning or glycation), reducing carbohydrates react with free amino groups of amino acids, amines, peptides and proteins, and particularly with the side-chains of protein-bound lysine and arginine residues to form a complex structural network that ultimately causes the brown colour of heated foods [1, 2]. Reactions of the disaccharide degradation products maltol 1 and isomaltol 2a with primary amines, leading to N-substituted 3-hydroxy-4(1H)pyridinones (3,4-HP), had already been described in the 1970s [3]. The corresponding lysine derivative maltosine 3 (6-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-l-norleucine) was first isolated from model glycation mixtures of N-α-acetyl lysine with maltose 4b and lactose 4c [4]. The disaccharides 4b, c are first degraded to the 1-deoxy-2,3-diulose derivatives 6 via decomposition of intermediate Amadori products 5 [5] (Fig. 1). A dehydration reaction of the respective ring structure leads to β-pyranones 7b, c as comparatively stable intermediates, of which 7c has already been detected in milk [6]. The β-pyranones 7b, c, which only result from disaccharides, are important intermediates in maltosine formation. Already after short heating times, they represent the principal reaction products of lactose or maltose in the presence of primary and secondary amines [1, 7]. Further dehydration and loss of the glycosyl moiety lead to 1 [8]. In parallel, the isomaltol derivatives 2b, c are formed after acetal cleavage in 7b, c, ring formation in the enol ethers 8 and further dehydration [7–9]. Loss of the glycosyl moiety from 2b, c also gives rise to 2a. While 3 can then be formed directly from 1 and 2, respectively [4], a pyridinium betaine 9 is formed as an intermediate from the derivatives 2b, c [7].

Proposed pathways for the formation of 6-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-l-norleucine (maltosine, 3) during the Maillard reaction (Literature see text)
At present, maltosine 3 is the only known glycated amino acid with a 3,4-HP structure. Compounds bearing this structural element are capable of chelating iron, and therefore, new individual compounds are still developed in pharmaceutical research [10, 11]. The 3,4-HP deferiprone 10 (Fig. 2) is currently used as a drug in chelation therapy for treatment of iron overload resulting from haemochromatosis or β-thalassaemia (secondary iron overload) and serves as a substitute of deferoxamine [12]. The non-proteinogenic natural 3,4-HP amino acid mimosine 11 can account for 4 % of the dry weight of Leucaena leucocephala, which is used as a forage plant. Most probably due to metal chelation, 11 provokes toxic effects such as alopecia and growth depression and acts as a goitrogen, which restricts the use of Leucaena in animal feeding [13].

Chemical structures of compounds structurally similar to maltosine 3. 3-Hydroxy-1,2-dimethyl-4(1H)pyridinone (deferiprone, 10), 3-(3-hydroxy-4-oxo-4(1H)-pyridin-1-yl)-l-alanine (mimosine, 11) and 6-(3-hydroxy-4-oxo-5-methyl-4(1H)-pyridin-1-yl)-l-norleucine (pyridosine 12)
Excellent iron-binding properties have also been measured for 3 [14, 15]. When applied orally as the free amino acid, 3 led to an inhibition of iron absorption and to a fourfold to fivefold increase in renal iron excretion as compared to the non-treated controls [16]. MRP-rich diets may also influence the bioavailability of iron in humans [17]; however, it is not known up to now to what extent 3 might be responsible for such an effect. In a study using the human cell line Caco-2, we were able to show that 3 cannot pass the intestinal barrier as the free amino acid. In contrast, when bound in dipeptides, it is effectively transported into intestinal cells by the peptide transporter PEPT1 and can thus be taken up into the systemic circulation [18, 19].
Despite its possible physiological properties as an iron chelator in vivo, 3 has only once been analysed in food up to now. Approximately 100 mg maltosine/kg of protein was quantified in heated whey powder by amino acid analysis [20]. Moreover, the significance of carbohydrates to act as a precursor of 3 in foods and the way of formation are not known. It was the aim of this study to identify the relevant precursors of 3 in food model systems and to get an insight into its occurrence in food.
Materials and methods
Chemicals
N-ε-Carboxybenzyllysine (H-Lys(Z)-OH) was purchased from Bachem (Bubendorf, Switzerland). Palladium on activated charcoal, sodium heptanesulfonate monohydrate, maltotriose, lactulose, maltulose, pepsin (10 FIP-U/mg protein), leucine aminopeptidase (19 U/mg protein; 70.4 U/mL) and prolidase (553 U/mg protein; 100 U/mL) were purchased from Sigma-Aldrich (Steinheim, Germany) and glucose and galactose from Alfa Aesar (Karlsruhe, Germany). Pronase E (4000 PU/mg protein), trisodium citrate dihydrate and maltose monohydrate from Merck (Darmstadt, Germany) were used. N-α-butyloxycarbonyllysine (N-α-Boc-lysine) was purchased from Fluka (Steinheim, Germany). Lactose monohydrate and fructose were obtained from Roth (Karlsruhe, Germany) and [13C6,15N2]lysine from Cambridge Laboratories (Andover, MA, USA). HPLC gradient-grade acetonitrile and methanol were from VWR Prolabo (Leuven, Belgium). The water used for the preparation of buffers and solutions was prepared using a Purelab plus purification system (USFilter, Ransbach-Baumbach, Germany). The water for HPLC–MS/MS eluents was double distilled (first over potassium permanganate). Benzylmaltol [18], isomaltol [21], glucosyl isomaltol [22, 23], galactosyl isomaltol [24], pyrraline [25] and formyline [26] were synthesized according to the literature stated.
Food samples
A total of 53 food samples were obtained from local retail stores, 5 malt samples were from a local brewery, and 4 samples of milk and whey powders were from industrial suppliers. Samples of milk and milk products were directly used for enzymatic hydrolysis. Bakery and pasta products and breakfast cereals were ground using a laboratory mill (Grindomix GM 100, Retsch, Haan, Germany). Fat-rich samples such as chocolate and cream candies were defatted as follows: approximately 3 g of the food sample was extracted with 25 mL of dichloromethane in a tube. The suspension was centrifuged (10,000 rpm, 20 °C, 10 min), and the supernatant was discarded. The pellet was extracted once more in the same way. After complete drying under air in a hood, the residue was utilized for enzymatic hydrolysis. Candy samples were dissolved in water at a concentration of 15 g/100 mL. Beer samples (50 mL) were dialysed against water for 4 days (MWCO 3.5 kDa, ZelluTrans, Carl Roth, Karlsruhe, Germany), and the retentate was lyophilized [27]. If necessary, protein contents were determined by the Kjeldahl method using the factor 6.25 for cereal products.
Protein hydrolysis
Portions of food samples representing 3–4 mg of protein were subjected to enzymatic hydrolysis as described previously [27]. The incubations were performed at 37 °C. The samples were first suspended in 1.0 mL of 0.02 M HCl, and 50 µL of a solution of pepsin (1 FIP-U per sample) in 0.02 M HCl was added. After 24 h, 250 µL 2 M TRIS buffer (pH 8.2) and 50 µL of a solution of pronase E (400 PU per sample) in 2 M TRIS buffer were added. After 24 h, 5.68 µL of aminopeptidase solution (0.4 U per sample) and 10 µL of prolidase solution (1 U per sample) were added. After 24 h, the samples were lyophilized. The lyophilizate was taken up in 950 µL of 5 mM NFPA, and 50 µL of a solution (c = 2.31 mg/L) of the internal standard [13C6,15N2]maltosine was added. After centrifugation (5000 rpm, 10 min) and membrane filtration (0.45 µm), the samples were subjected to HPLC–MS/MS analysis.
Boc-lysine/sugar incubations
Incubations of Boc-lysine with different potential precursors (glucose, fructose, galactose, maltose, lactose, maltulose, lactulose, cellobiose, maltotriose, 2b, c) were performed as in our previous study [26]; 50 µL of a 25 mM Boc-lysine solution in 0.2 M sodium acetate buffer, pH 5.5, containing 2 % (w/v) of suspended cellulose was pipetted into a sealable glass tube, and 50 µL of a 100 mM precursor solution was added. The suspensions were freeze-dried overnight. Samples for the investigation of crumb conditions were then stored in a desiccator for 48 h. The desiccator had been equilibrated for 3 days with a saturated solution of potassium sulphate (aW = 0.97, [28]). Samples for the investigation of crust conditions were stored analogously in the presence of a saturated solution of sodium chloride (aW = 0.75, [28]). The sealed tubes were then incubated in a sand bath in a drying chamber either at 100 °C for 60 min (crumb conditions) or at 130 °C for 30 min (crust conditions). After addition of 1.5 mL of 10 % acetic acid, the Boc protecting group was hydrolysed by heating the samples in a water bath (70 °C, 4 h). An aliquot of the sample was then centrifuged (10,000 U/min, 10 min, 20 °C), and the supernatant was directly used for HPLC–UV, or HPLC–MS/MS, respectively. All incubations were performed in triplicate.
High-pressure liquid chromatography with UV detection
HPLC was performed on an Äkta Basic high-pressure gradient system from Amersham Pharmacia Biotech (Uppsala, Sweden), consisting of a pump P-900 with an online degasser (Knauer, Berlin, Germany), an autosampler A-900 and a UV detector UV-900. A polymer-based RP-18 column (PLRP-S, 250 mm × 4.6 mm, 8 µm, 100 Å) with a guard column (5 × 4 mm) of the same material (Polymer Laboratories, Darmstadt, Germany) was used for the quantification of maltosine in model mixtures [18]. The column temperature was maintained at 30 °C in a column oven. As eluent A, a solution of 5 mM sodium heptanesulfonate in water was used (the pH was adjusted to 2.0 with 6 M H2SO4), and eluent B was a mixture of eluent A and acetonitrile (1/1, v/v). The injection volume was 50 µL. A linear gradient from 4 % B to 70 % B in 20 min was applied at a flow rate of 1 mL/min. The absorbance was simultaneously read at 280, 293 and 297 nm. External calibration was performed with the synthesized standards of maltosine, pyrraline and formyline. The limits of detection (LOD) and quantification (LOQ), respectively, were determined as the concentrations of the analytes that are necessary to show peaks with signal-to-noise ratios of 3 and 10, respectively.
High-pressure liquid chromatography with mass-spectrometric detection
The analysis of maltosine in food samples and selected samples from model experiments was performed on the high-pressure gradient system 1200 Series (Agilent Technologies, Böblingen, Germany), consisting of a binary pump, an online degasser, a column oven, an autosampler and a diode array detector. The HPLC was coupled to the mass spectrometer 6410 Triple Quad (Agilent Technologies). HPLC–MS/MS conditions are compiled in Tables 1 and 2. Data were acquired and evaluated with the software Mass Hunter B.02.00 (Agilent).
Semi-preparative high-pressure liquid chromatography
Semi-preparative HPLC was performed on a Wellchrom high-pressure gradient pump system consisting of two HPLC pumps K-1001 with an online degasser, a UV detector K-2501 and a fraction collector K-16 (all from Knauer, Berlin, Germany). All separations were performed at room temperature using an RP-18 column (Eurospher 100, 300 mm × 8 mm, 5 µm, Knauer, Berlin, Germany) with a guard column (30 mm × 8 mm) filled with the same material. UV detection was performed at 280 nm. The raw synthesis solutions were membrane filtered (0.45 mm) before injection.
Characterization of synthesis products
Proton NMR spectra were recorded on a Bruker DRX 500 instrument (Rheinstetten, Germany) at 500 MHz using D2O as the solvent. Chemical shifts are given in parts per million (ppm), relative to the internal HOD signal (4.70 ppm). HPLC–UV–ESI–MS/MS was used to determine the chromatographic purity, the UV maxima, the molecular mass and the fragmentation behaviour of the compounds. The same device and column as above and the chromatographic conditions described in Table 1 were used. The absorbance was read at 280 nm, and UV spectra were recorded between 190 and 400 nm (step size, 2 nm). For the acquisition of product ion spectra, the fragmentor voltage was set at 105 V, and a collision energy of 10 eV was applied. Elemental analysis was used to calculate the product content of substances. Data were obtained on a Euro EA 3000 elemental analyser (Eurovector, Milano, Italy). The measured percentage of nitrogen was divided by the theoretical percentage of nitrogen in the target substance and the content expressed in per cent by weight.
Synthesis of [13C6,15N2]6-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-l-norleucine 13 ([13C6,15N2]maltosine)
257.2 mg (1.13 mmol) of [13C6,15N2]lysine dihydrochloride was dissolved in 50 mL of 0.2 M sodium borate buffer, pH 10.0, and 294.3 mg (1.4 mmol) of benzyl maltol in a small volume of ethanol was added. The pH was adjusted to 10.0 with sodium hydroxide solution. The mixture was incubated at 80 °C for 24 h in a drying chamber. After cooling, the pH was set to 7.0, and the solution was extracted with diethyl ether (3 × 20 mL). The aqueous phase was evaporated to dryness and reconstituted in 50 mL of a mixture of methanol and water (1/9, v/v). Fractionation was performed by semi-preparative HPLC using 0.075 % acetic acid as eluent A and a mixture of 20 % eluent A and 80 % of methanol as eluent B, and a linear gradient from 10 % B to 70 % B in 30 min was applied at a flow rate of 1.4 mL/min. The injection volume was 2 mL. The absorbance was recorded at 280 nm, and the eluates of the prominent peaks were collected and evaporated to dryness. The residues were dissolved in 40 mL of a mixture of methanol and water (1/1, v/v) and incubated in a hydrogen atmosphere overnight after addition of 50 mg of palladium on charcoal. After hydrogenation, 100 µL of the mixtures was added to 900 µL 5 mM heptanesulfonate, pH 2.0, and analysed by HPLC–UV as described above. The analysis revealed that the fraction eluting between 33 and 38 min on semi-preparative HPLC contained a compound yielding maltosine after hydrogenation. As the compound was chromatographically pure, the hydrogenated solution was filtered, and the filtrate was evaporated to dryness and lyophilized to yield [13C6,15N2]maltosine as a white powder, which was stored at −20 °C.
Analytical data: ESI–MS, positive mode, [M + H]+ m/z 263.2; HPLC–MS/MS (System 1): tR, 15.1 min; λ max, 278 nm; fragmentation (105 V, 10 eV) of [M + H]+ (m/z 263), 263 (100), 127 (66), 90 (49), 137 (42), 216 (10). Content = 87.0 %, based on HPLC–UV, calibration with the unlabelled standard. Yield: 148.7 mg (43.5 %).
Synthesis of 2-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-6-amino-l-hexanoic acid 14
3.09 g (11.0 mmol) of H-Lys(Z)-OH and 2.62 g (12.0 mmol) of benzyl maltol were dissolved in 50 mL of a mixture of ethanol and water (1/1, v/v), and the pH was set to 12.8 by addition of concentrated sodium hydroxide. The mixture was heated under reflux for 7 h. The reddish-brown solution was evaporated in vacuo. The residue was suspended in 50 mL of water, and the pH was set to 1.0 with 6 M HCl. The solution was extracted with ethyl acetate (2 × 100 mL). The organic phases were combined and evaporated to dryness, and the residue was taken up in 50 mL of 0.1 M NaOH. The pH of this solution was set to 7.0, and the solution was extracted with diethyl ether (3 × 100 mL). A voluminous white solid that formed at the phase interface was discarded together with the organic phases. The pH of the aqueous phase was then adjusted to 1.0 with 6 M HCl, the solution was extracted with ethyl acetate (3 × 100 mL), then the pH was adjusted to 2.1 and a further extraction with ethyl acetate (3 × 100 mL) was performed. The combined organic extracts were evaporated to dryness using a rotary evaporator. The crude reaction product (1.33 g) was dissolved in 50 mL methanol. For hydrogenation, 100 mg of palladium on activated charcoal was added to 37.6 mL of this solution, and the suspension was stirred under a hydrogen atmosphere at room temperature and atmospheric pressure for 18 h. The catalyst was removed by filtration, and ethanol was evaporated in vacuo.
The residue was dissolved in 30 mL of 0.1 N sodium citrate buffer, pH 3.0, and the pH was adjusted to 3.0. The solution was applied to a column (1.5 cm × 50 cm) of 80 mL strongly acidic cation exchange resin DOWEX 50 WX-8 (200–400 mesh; Acros, Geel, Belgium) that had previously been equilibrated with 250 mL of 6 M HCl, 250 mL of water, 250 mL of 1 M sodium hydroxide, 250 mL of water and 250 mL of 0.1 N sodium citrate buffer, pH 3.0. The product was eluted with 0.3 N sodium citrate buffer, pH 5.25, and fractions of 10 mL were collected using a fraction collector (RediFrac, Pharmacia Biotech, Uppsala, Sweden). Of all fractions, 1 µL was spotted on a TLC plate which was then sprayed with a 0.1 % solution of ninhydrin in ethanol containing 3 % (v/v) acetic acid. Heating of the plate (50 °C) revealed ninhydrin-positive fractions to elute between 160 and 210 mL. These fractions were combined, and the pH was adjusted to 2.0 with 6 M HCl. The solution was loaded on a column (2.5 cm × 15 cm) of DOWEX 50 WX-8 (200–400 mesh) previously equilibrated with 250 mL of 6 M HCl and 250 mL of water. After two elution steps with water (250 mL) and 1 M HCl (250 mL), the target product was eluted with 250 mL of 4 M HCl. The eluate was repeatedly evaporated in vacuo and the residue was dissolved in a small volume of water; the solution was lyophilized to give a light grey solid, which was stored at −20 °C.
Analytical data: ESI–MS, positive mode, [M + H]+ m/z 255.2; 1H-NMR (500 MHz, D2O), δ [ppm]: 1.14 (m, 1H, Lys-H4A); 1.36 (m, 1H, Lys-H4B); 1.59 (m, 2H, Lys-H5); 2.09 (m, 1H, Lys-H3A); 2.33 (m, 1H, Lys-H3B); 2.47 (s, 3H, Mal-CH3); 2.83 (t, 2H, J = 7.7 Hz, Lys-H6); 5.23 (dd, 1H, J = 5.7 Hz, 9.5 Hz, Lys-H2); 7.10 (d, 1H, J = 7.2 Hz, Mal-H4); 8.02 (d, 1H, J = 7.2 Hz, Mal-H5). HPLC–MS/MS (System 1): tR, 10.4 min; λ max, 276 nm; fragmentation (105 V, 10 eV) of [M + H] + (m/z 255), 126 (100), 255 (41), 84 (36), 86 (35), 130 (10), 95 (3), 237 (1). Elemental analysis: C12H18N2O4 (MW = 254.28) requires C 56.68 %, H 7.13 %, N 11.02 %, C/N = 5.14; found, C 39.72 %, H 6.44 %, N 7.87 %, C/N = 5.05; content = 71.4 %, based on nitrogen. Yield: 291.6 mg (9.9 %).
Statistical analysis
Comparison of means was performed by one-way ANOVA and Scheffé’s post hoc test using PASW Statistics 18.0. Differences were considered significant at a significance value P < 0.05.
Results and discussion
Formation of maltosine in a model system
Maltosine 3 was first isolated from heated mixtures of lysine and disaccharides [4]. Since degradation of starch during the baking process leads to the formation of maltose and maltooligosaccharides [29], a model system based on the baking process was chosen. Maltose and lactose along with the corresponding ketoses and constituting sugars as well as maltotriose were taken as model carbohydrates. Maltol 1 and isomaltol 2a could not be introduced into the model system due to their volatility during lyophilization. However, glucosyl isomaltol 2b and galactosyl isomaltol 2c, two derivatives of 2a described as degradation products of maltose and lactose in model systems and food [23, 30, 31], were synthesized and investigated. Model systems representing the crust and crumb of bakery products, which are characterized by strong differences in the conditions of the Maillard reaction, were used. According to our previous study [26], Boc-lysine was incubated in the presence of a fourfold excess of potential carbohydrate precursors on cellulose as a solid support. During baking, the dough surface temperature initially rises quickly to 100–110 °C and then slowly increases further depending on the oven temperature [32]. The crust of wheat bread shows a water activity of 0.65–0.77 directly after baking [33]. We therefore chose an incubation temperature of 130 °C for 30 min for the samples preconditioned over a saturated solution of sodium chloride at a water activity of 0.75 [28] to simulate crust conditions. On the contrary, the crumb temperature does not exceed 100 °C due to continued water mobilization, and the water activity in the crumb is between 0.97 and 0.995 [34]. An incubation temperature of 100 °C for 60 min for samples stored over a saturated solution of potassium chloride at a water activity of 0.97 [28] was thus chosen to simulate crumb conditions.
After incubation, the Boc protecting group was hydrolysed in the presence of 10 % acetic acid. When Boc-lysine and maltose solutions were mixed and subjected to the Boc cleavage procedure, no formation of 3 was detected by HPLC–UV. When Boc-lysine and maltose solutions were mixed, lyophilized and subjected to the Boc cleavage procedure without incubation at higher temperatures, 3 was not detected as well. Furthermore, compound 3 was not formed when Boc-lysine was incubated in the presence of cellulose under “crust conditions” without added sugars. Detection of 3 must therefore result from the reaction of the precursors with the ε-amino group of lysine during heating. The performance parameters of our HPLC–UV method are compiled in Table 3. When samples are worked up as described here, the LOD is equivalent to a conversion of 0.2 % lysine to maltosine and the LOQ to a conversion of 0.7 %. The results from the precursor study were later corroborated by measurement of a subset of the samples with the HPLC–MS/MS method.
Generally, less 3 was produced under crumb than under crust conditions (Table 4). No significant conversion of lysine to 3 was noticed when monosaccharides were used as potential precursors (P > 0.05). However, the compound was formed from all disaccharides, maltotriose and both isomaltol derivatives. During incubation under crust conditions, 3 turned out to be the most important lysine derivative absorbing at 280 nm (Fig. 3). In most cases, the contents of 3 in the incubated mixtures exceeded those of pyrraline. In the presence of the ketoses maltulose and lactulose, less lysine is converted to 3 than in the presence of the corresponding aldoses, possibly because Amadori products as intermediates are formed rapidly from the aldoses and during degradation give rise to 1-deoxyosones of the type 6a–c [5]. The direct formation of these osones by 2,3-enolization during ketose degradation obviously only plays a minor role in the presence of amines.

RP-HPLC with UV detection of (a) a mixture of maltose and Boc-lysine incubated under crust conditions, (b) a mixture of maltose and Boc-lysine incubated under crumb conditions and (c) a mixture of lactose and Boc-lysine incubated under crust conditions
Comparatively strong differences were observed in the yields of 3 between di- and trisaccharide derivatives with an α-glycosidic bond (maltose, maltulose, maltotriose, 2b) and those with a β-glycosidic bond (lactose, lactulose, cellobiose, 2c). It has been reported in the literature that the glucopyranosyl substituent in maltose rather gives rise to maltol 1, while the galactopyranosyl substituent in lactose makes 2c prevail during thermal treatment [1, 8, 24]. In our model system, the maltol precursor can alternatively react with the ε-amino group of lysine and generate 3 instead of forming the intramolecular maltol pyranone ring. Since cellobiose behaves like lactose, we suppose that these differences should not primarily result from the type of sugar attached, but from the conformation of the glycosidic bond. An increased formation of 1 from the α-substituted di- and trisaccharides compared to β-substituted derivatives could be shown qualitatively in this study (Fig. 3). Moreover, the conformational differences might also influence the stability of the intermediates 6b, c, 8b, c and even 9b, c during the course of the reaction as shown in Fig. 1. Information on the chemistry of these derivatives should be the key for a deeper understanding on the pathway of formation of 3. Further studies are needed to elucidate the mechanisms behind these findings on a molecular level.
Quantification of maltosine in food samples
Before the concentration of 3 in food samples could be measured, suitable protein hydrolysis and analysis methods had to be optimized. It is known that Amadori products such as fructoselysine and lactuloselysine are converted during common acid hydrolysis (6 M HCl, 110 °C, 23 h) to furosine, lysine, CML and pyridosine 12 [35, 36]. The structural similarity of 3 and 12 posed a strong challenge to analysis, which could not be solved by changes of HPLC columns, eluents and gradient systems. We then decided to avoid the formation of 12 by using enzymatic hydrolysis as in previous works [20, 27]. Unfortunately, the gradient system used above for the quantification of 3 in model mixtures could not be applied for enzymatic hydrolysates of food samples because UV detection was not sufficiently sensitive. We therefore decided to develop an assay based on HPLC with MS/MS detection. Porous graphite as a stationary HPLC phase had already been used in the literature for the separation of 3,4-HPs [37]. In the present study, we used a similar material and an eluent system with NFPA as an ion-pairing agent. A stable isotope labelled derivative of 3 was synthesized for use as an internal standard according to our established method [18], but starting from unprotected [13C6,15N2]lysine. Before cleavage of the benzyl protecting group, the separation from by-products was accomplished by semi-preparative HPLC. From the most prominent peak, [13C6,15N2]maltosine 13 was obtained in comparatively high molar yield of 43.5 %. However, when 3 is synthesized from unprotected lysine, there is also the possibility of modification at the α-amino group of lysine. In order to verify the purity of 13, the respective isomer 2-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-6-amino-l-hexanoic acid 14 was synthesized and characterized. HPLC–MS/MS showed that the lysine derivative 14 eluted before maltosine and that the internal standard 13 only showed one peak coeluting with unlabelled 3 (Fig. 4). Interferences during analysis caused by the presence of 14 in samples could thus be excluded. Hydrogenation and analysis of the eluates of the other peaks from semi-preparative HPLC revealed that 14 was formed only as a minor by-product. These results show that the synthesis of 3 by our established protocol [18] can generally be performed starting from unlabelled lysine without significant loss in molar yield.

HPLC with UV detection (a) and product ion spectra of maltosine 3 (b), [13C6,15N2]maltosine 13 (c) and 2-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-6-amino-l-hexanoic acid 14 (d); isotopically labelled atoms are marked by asterisks. Proposed fragmentation reaction of maltosine 3 and [13C6,15N2]maltosine 13 (e). Mass shifts between the fragments of 3 and 13 are given in brackets. HPLC was performed with the conditions in Table 1. Product ion spectra were recorded at a fragmentor voltage of 105 V and a collision energy of 10 eV
Unlabelled 3 and the internal standard 13 were then subjected to HPLC–MS/MS analysis in order to optimize the working conditions. The MS/MS spectra showed only few fragment peaks at different collision energies (Fig. 4). The product ion spectrum of 3 results from the loss of the pyridinone ring (m/z = 126, [M + H]+). The ions resulting from the decomposition of the lysine residue (m/z = 84; 130) are known from several Maillard reaction products of lysine [38, 39]. The mechanism proposed in Fig. 4e is substantiated by comparison of the mass shifts of the respective ions with the product ion spectrum of 13 (Fig. 4c). The most selective transitions, m/z 255 → 126 for 3 and 263 → 127 for 13, were chosen for the quantification of the substances in the multiple reaction monitoring (MRM) mode (Table 2). The LOD and LOQ for 3 with the HPLC–MS/MS method were considerably lower than with the HPLC–UV method (Table 3). When aliquots of 4 mg of protein per sample were subjected to enzymatic hydrolysis, the LOD and LOQ, respectively, were 0.2 mg/kg protein and 0.5 mg/kg protein, respectively. With the enzyme preparations and conditions of enzymatic hydrolysis applied in this study, it was possible to release at least 70–85 % of the aliphatic and aromatic amino acids isoleucine, leucine, tyrosine and phenylalanine, as was determined by comparison of the amino acid concentrations in acid and enzymatic hydrolysates. This was also in agreement with literature data [40]. The quantitative data presented in Table 5 should thus be slightly underestimated; however, as discussed above, acid hydrolysis would not have been suitable.
Maltosine 3 was then qualitatively analysed in enzymatic hydrolysates of rye bread. A comparison of the product ion spectrum of the ion m/z = 255 between a standard solution (Fig. 4) and this food item (Fig. 5b) showed excellent similarity (Figs. 4, 5) for the peak eluting at 13.5 min. In many samples, two further peaks with different product ion spectra eluted after 3 (Fig. 5). By comparison of the spectra with literature data [27], the second peak could be assigned to pyrraline. The third peak represents a product originating from the reaction of lysine and maltulose (M. Hellwig, T. Henle, unpublished results).

HPLC with MS/MS detection of an enzymatic hydrolysate of a rye bread sample (black) and a maltosine standard (grey) showing the chromatograms of the transitions used for qualification and quantification of maltosine 3 (a). The product ion spectra of the three prominent peaks b–d were recorded at a fragmentor voltage of 105 V and a collision energy of 10 eV
The measurement of 3 in a broader spectrum of food items (Table 5) revealed that milk products only contain small amounts of the substance, which is in accordance with the low maltosine formation potential of lactose found in the precursor study. However, the formation of 3 was strongly inducible by dry heating of milk powder, resulting in contents of up to 200 mg/kg protein after 4 h of heating at 100 °C. A comparably high concentration of 3 has already been reported in the literature [20]. The highest amounts of 3 were quantified in bakery products, predominantly in crust samples, which is also in agreement with the precursor study. The concentration in wheat bread crust of up to 19.3 mg/kg represents a maximal lysine modification of approximately 0.4 %. When milk is used as an ingredient, less 3 is formed than in the presence only of flour as a carbohydrate source. In the presence of high concentrations of lactose, the precursor of 3 should have to compete against the precursors of other lysine derivatives, especially of pyrraline. A similar effect was also observed in our previous study, where comparatively low contents of formyline were measured in milk bread samples [27]. When compared to literature data, the concentrations of formyline were approximately 20 % higher than the concentrations of 3, but the pyrraline concentrations exceed those of 3 about tenfold in most of the samples [27]. In the precursor study, however, 3 was always more abundant than formyline (Table 4). This indicates an instability of 3 in food proteins possibly due to reaction with food components that were not examined in the model system. Moreover, 3 might be introduced into the melanoidin network.
Based on a model diet consisting of coffee, milk and bakery products as sources of Maillard reaction products [41], the data in Table 5 allow an estimation of the daily intake of 3 of about 1–2 mg. Maltosine can be transported across the intestinal epithelium in the form of dipeptides [18, 19]. Regarding the fact that 3 is a good iron chelator [14, 15] that can increase iron elimination in vivo, it will now be necessary to obtain further knowledge on the physiological effects of the compound. Interestingly, the 3,4-HP 11 (Fig. 2) can induce apoptosis [42] and shows antiviral [43] and antiproliferative [44] activity in vitro and antitumour activity in vivo [45]. It could be possible by adaptation of the baking technology to produce products with a high content of 3 that could be utilized as a means to reduce the in vivo concentrations of iron and other heavy metals such as copper or aluminium, because 3,4-HPs are vastly discussed in the literature as sequestering agents for metal overload [11].
Abbreviations
- 3,4-HP:
-
3-Hydroxy-4(1H)-pyridinone
- AGE:
-
Advanced glycation end product
- aW :
-
Water activity
- HPLC:
-
High-pressure liquid chromatography
- LOD:
-
Limit of detection
- LOQ:
-
Limit of quantification
- MRM:
-
Multiple reaction monitoring
- MWCO:
-
Molecular weight cut-off
- NFPA:
-
Nonafluoropentanoic acid
- NMR:
-
Nuclear magnetic resonance
References
Ledl F, Schleicher E (1990) Angew Chem Int Ed Engl 29:565–594
Hellwig M, Henle T (2014) Angew Chem Int Ed Engl 53:10316–10329
Severin T, Loidl A (1976) Z Lebensm Unters Forsch 161:119–124
Ledl F, Osiander H, Pachmayr O, Severin T (1989) Z Lebensm Unters Forsch 188:207–211
Beck J, Ledl F, Severin T (1989) Z Lebensm Unters Forsch 188:118–121
Pellegrino L, Cattaneo S (2001) Nahrung/Food 45:195–200
Kramhöller B, Pischetsrieder M, Severin T (1993) J Agric Food Chem 41:347–351
Ledl F, Ellrich G, Klostermeyer H (1986) Z Lebensm Unters Forsch 182:19–24
Estendorfer S, Ledl F, Severin T (1990) Tetrahedron 46:5617–5620
Liu ZD, Hider RC (2002) Med Res Rev 22:26–64
Santos MA, Chaves S (2015) Fut Med Chem 7:383–410
Galanello R, Campus S (2009) Acta Haematol 122:155–164
Hegarty MP, Schinckel PG, Court RD (1964) Aust J Agric Res 15:153–167
Seifert S (2008) PhD Thesis, TU Dresden
Katoh A, Hikita Y, Harata M, Ohkanda J, Tsubomura T, Higuchi A, Saito R, Harada K (2001) Heterocycl 55:2171–2187
Rehner G, Walter T (1991) Z Ernährungswiss 30:50–55
Mesías García M, Seiquer I, Delgado-Andrade C, Galdo G, Navarro MP (2009) Mol Nutr Food Res 53:1551–1560
Geissler S, Hellwig M, Markwardt F, Henle T, Brandsch M (2011) Eur J Pharm Biopharm 78:75–82
Hellwig M, Geissler S, Matthes R, Peto A, Silow C, Brandsch M, Henle T (2011) ChemBioChem 12:1270–1279
Henle T, Walter AW, Klostermeyer H (1994) In: Labuza TP (ed) Maillard reactions in food chemistry, and health. The Royal Society of Chemistry, Cambridge, pp 195–200
Fox RC, Taylor PD (1999) Synth Commun 29:989–1001
Goodwin JC (1983) Carbohydr Res 115:281–287
Guerra-Hernández E, Ramirez-Jiménez A, García-Villanova B (2002) J Agric Food Chem 50:7282–7287
Hodge JE, Nelson EC (1961) Cereal Chem 38:207–221
Hellwig M, Geissler S, Peto A, Brandsch M, Henle T (2009) J Agric Food Chem 57:6474–6480
Hellwig M, Henle T (2010) Eur Food Res Technol 230:903–914
Hellwig M, Henle T (2012) Eur Food Res Technol 235:99–106
Rockland LB (1960) Anal Chem 32:1375–1376
Król B, Grzelak K (2006) Eur Food Res Technol 223:755–758
Morales FJ, Arnoldi A (1999) Food Chem 67:185–191
Rufián-Henares JA, Delgado-Andrade C, Morales FJ (2006) J Cereal Sci 43:63–69
Purlis E, Salvadori VO (2009) Food Res Int 42:865–870
Primo-Martín C, van de Pijpekamp A, van Vliet T, de Jongh HHJ, Plijter JJ, Hamer RJ (2006) J Cereal Sci 43:342–352
Puhr DP, D’Appolonia BL (1992) Cereal Chem 69:582–586
Krause R, Knoll K, Henle T (2003) Eur Food Res Technol 216:277–283
Finot PA, Mauron J (1969) Helv Chim Acta 52:1488–1495
Epemolu RO, Singh S, Hider RC, Damani LA (1990) J Chromatogr 519:171–178
Yaylayan V, Sporns P (1989) Food Chem 33:81–91
Hegele J, Buetler T, Delatour T (2008) Anal Chim Acta 617:85–96
Wellner A, Huettl C, Henle T (2011) J Agric Food Chem 59:7992–7998
Henle T (2003) Kidney Int 63:S145–S147
Hallak M, Vazana L, Shpilberg O, Levy I, Mazar J, Nathan I (2008) Apoptosis 13:147–155
Upadhyay A, Chompoo J, Taira N, Fukuta M, Gima S, Tawata S (2011) J Agric Food Chem 59:12858–12863
Ju H, Hao J, Zhao S, Dixon IMC (1998) Biochim Biophys Acta 1448:51–60
DeWys W, Hall TC (1973) Eur J Canc 9:281–283
Acknowledgments
We are grateful to Karla Schlosser, Institute of Food Chemistry, for performing the amino acid analyses. We thank the members of the Institute of Organic Chemistry, namely Dr. Margit Gruner and Anett Rudolph, for recording the NMR spectra and Anke Peritz for performing the elemental analyses.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Rights and permissions
About this article

Cite this article
Hellwig, M., Kiessling, M., Rother, S. et al. Quantification of the glycation compound 6-(3-hydroxy-4-oxo-2-methyl-4(1H)-pyridin-1-yl)-l-norleucine (maltosine) in model systems and food samples. Eur Food Res Technol 242, 547–557 (2016). https://doi.org/10.1007/s00217-015-2565-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00217-015-2565-0