
Abstract
Dosage adjustment of anti-epileptic drugs by therapeutic drug monitoring (TDM) is very useful, especially for the first-generation anti-epileptic drugs (AEDs). Microsampling—the collection of small volumes of blood—is increasingly considered a valuable alternative to conventional venous sampling for TDM. Volumetric absorptive microsampling (VAMS) allows accurate and precise collection of a fixed volume of blood, eliminating the volumetric blood hematocrit bias coupled to conventional dried blood spot collection. The aim of this study was to develop and validate an LC-MS/MS method for the determination and quantification of four anti-epileptic drugs (carbamazepine, valproic acid, phenobarbital, and phenytoin) and one active metabolite (carbamazepine-10,11-epoxide) in samples collected by VAMS. The method was fully validated based on international guidelines. Precision (%RSD) was below 10%, while, with a single exception, accuracy (%bias) met the acceptance criteria. Neither carry-over nor unacceptable interferences were observed, the method being able to distinguish between the isomers oxcarbazepine and carbamazepine-10,11-epoxide. All compounds were stable in VAMS samples for at least 1 month when stored at room temperature, 4 °C, and − 20 °C and for at least 1 week when stored at 60 °C. Internal standard-corrected matrix effects were below 10%, with %RSDs below 4%. High (> 85%) recovery values were obtained and the effect of the hematocrit on the recovery was overall limited. Successful application on external quality control materials and on left-over patient samples demonstrated the validity and applicability of the developed procedure.

Graphical representation of the sampling, chemical structures, and the resulting chromatogram for volumetric absorptive microsampling (VAMS)-based therapeutic drug monitoring of first-generation anti-epileptic drugs by liquid chromatography with tandem mass spectrometric detection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Whether a patient with epilepsy is free from seizures and is able to live a normal life depends on the correct administration of appropriate anti-epileptic drugs (AEDs). AEDs can be divided into three subclasses: the “classical” or “first-generation” AEDs, the “second-generation” AEDs, and the “third-generation” AEDs [1]. Carbamazepine, phenobarbital, phenytoin, and valproic acid were introduced prior to 1990 and belong to the first-generation AEDs [1]. Oxcarbazepine, vigabatrin, and topiramate, among others, are examples of the second-generation AEDs, while lacosamide, retigabine, and eslicarbazepine are categorized as third-generation AEDs [1]. Significant interindividual variability in pharmacokinetics (absorption, distribution, metabolism, and excretion) is one of the commonly known properties of first-generation AEDs, making optimization and individualization of the therapy quite challenging [2].
Therapeutic drug monitoring (TDM) serves as an excellent tool in the optimization and individualization of drug therapy. The generally narrow therapeutic indices of first-generation AEDs, causing toxicity to be a common issue, have led to the fact that TDM has become an established application, in general as well as in special populations (e.g., children, elderly, and pregnant women with epilepsy).
TDM is most often performed on venous blood samples (whole blood, plasma, or serum). However, given the invasive nature of the associated sampling and the relatively large amounts of blood that are typically taken, this sampling procedure becomes increasingly less attractive in clinical practice. In addition, as sampling requires a phlebotomist, patients are obliged to visit a hospital or doctor’s office for a venous blood draw. Therefore, there is a growing interest in the use of non- and minimally invasive alternative sampling strategies for TDM [3, 4].
One of the most commonly used alternative sampling strategies is dried blood spot (DBS) sampling. Generally, DBS are prepared by depositing a drop of capillary blood, obtained by a finger or heel prick, on a dedicated filter paper. Over the past years, several methods were published using DBS for the determination of both first- and second-generation AEDs [5,6,7,8,9,10,11,12]. DBS sampling—for TDM and in general—has several advantages over conventional venous blood sampling. As DBS are mostly obtained by a finger prick, the patients themselves can perform sampling at home. Furthermore, as the resulting dried matrix is considered non-contagious, sending DBS via regular mail to the clinical laboratory is allowed [13]. This way, laboratory results may already be available before a patient visits the doctor’s office for follow-up. Besides, sending the samples by airmail can also be advantageous in countries where patients have to cover a long distance to clinical practices. The small sample volume (typically 3–12 μL) associated with DBS sampling is another benefit, particularly for special populations, such as neonates and anemic patients. In addition, the sampling procedure is accompanied by increased analyte stability and by fewer difficulties with respect to sample handling, storage, and transport [13]. Given all these benefits, DBS sampling can serve as an excellent alternative to conventional venous sampling for TDM of AEDs.
On the other hand, DBS sampling is also struggling with some challenges, with the hematocrit (Hct) issue undoubtedly being the most widely discussed one. In essence, because of Hct-dependent spreading of blood on filter paper (blood with higher Hct spreading less), partial punch analysis of a DBS (which is the approach mostly used) will most often yield a bias for DBS generated from blood with divergent (either low or high) Hct. However, several strategies have been developed that allow to cope with the issues coupled to a varying Hct [14,15,16,17,18,19,20,21,22]. One of the proposed approaches is the use of volumetric absorptive microsampling (VAMS) devices [23, 24]. The latter are handheld devices consisting of a hydrophilic polymer tip connected to a plastic handler, which wicks up a fixed volume (approximately 10 or 20 μL) when contacting a blood surface [24]. Using authentic samples with a wide Hct range (0.21–0.50), our lab readily demonstrated that VAMS effectively results in absorption of a fixed volume of blood, irrespective of the hematocrit [23]. Furthermore, VAMS still maintains the benefits associated with DBS sampling and was reported to be preferred over DBS sampling by patients in a home sampling context [25]. The associated cost, as well as current incompatibility with on-line analysis systems, as developed for DBS analysis, may be considered disadvantages. In addition, we—as well as others—found that, while VAMS effectively allows volumetric sampling (thereby not suffering from a Hct effect as observed in DBS), recovery may be impacted by Hct [23, 26].
The aim of this study was to develop, validate, and apply an ultra-performance liquid chromatography–tandem mass spectrometry (UPLC®-MS/MS) method for the determination and quantification of four AEDs and one active metabolite, including carbamazepine (CBZ), valproic acid (VPA), phenytoin (PHT), phenobarbital (PB), and carbamazepine-10,11-epoxide (CBZ-E), making use of VAMS devices. We thereby paid particular attention to the recovery issue associated with analysis of VAMS devices. CBZ, VPA, PHT, and PB were chosen since they belong to the first-generation AEDs class, for which the strongest evidence for TDM exists. Furthermore, this type of AEDs is still frequently used for seizure control in developing countries, where microsampling may offer the largest benefits [27, 28]. CBZ-E, an active metabolite of CBZ, was also incorporated in the multi-analyte method as it is equipotent to CBZ and hence contributes significantly to its therapeutic (or toxic) effects [29]. Furthermore, since in MS/MS the multiple reaction monitoring (MRM) transitions characteristic for CBZ-E are the same as those for its isomer oxcarbazepine (OXC), OXC was also incorporated to assess the capability of the method to distinguish between CBZ-E and OXC, rather than to quantitatively determine OXC.
Materials and methods
Chemicals and stock solutions
LC-MS grade acetonitrile was obtained from Biosolve (Valkenswaard, The Netherlands). A Synergy® Water Purification System (Merck Millipore, Overijse, Belgium) provided ultrapure water. Valproic acid, valproic acid-d6, phenytoin, phenytoin-d5, carbamazepine, carbamazepine-d10, oxcarbazepine, carbamazepine-10,11-epoxide, and ammonium acetate were purchased from Sigma Aldrich (Diegem, Belgium). Phenobarbital and phenobarbital-d5 were derived from LGC standards (Molsheim Cedex, France). Oxcarbazepine-d10 was obtained from Santa Cruz Biotechnology, Inc. (Heidelberg, Germany) and carbamazepine-10,11-epoxide-d10 from J.H. Ritmeester B.V. (Nieuwegein, The Netherlands).
Taking into account the upper and lower limit of the therapeutic range of each compound, methanolic stock solutions were prepared at 40, 50, 10, 10, and 5 mg/mL for VPA, PB, PHT, CBZ, and CBZ-E, respectively. For OXC, a 1.67 mg/mL stock solution was prepared in acetonitrile. For the preparation of the calibrators and quality control samples (QCs), independently prepared stock solutions were used. For the internal standards (IS) of PHT, CBZ, and PB, methanolic stock solutions of 0.1 mg/mL were purchased, while for VPA the concentration of the IS stock solution was 1 mg/mL. IS stock solutions for CBZ-E and OXC (1 mg/mL) were prepared in methanol and acetonitrile, respectively. Working solutions of the standards and the IS were prepared the day of analysis by diluting the stock solutions with water. All solutions—except for the stock solution of CBZ-E (4 °C)—were stored at − 20 °C in 1.5 mL amber glass vials derived from VWR® (Leuven, Belgium).
Sample collection
Venous whole blood from an AED abstinent healthy, female volunteer was collected in EDTA tubes (BD Vacutainer® with BD Hemogard® closure 10 mL) for method development and validation purposes. VAMS devices (Mitra™) were obtained from Neoteryx (Torrance, CA, USA). Samples were prepared by dipping the tip into spiked whole blood in 2 mL Eppendorf tubes. Overfilling of the devices was prevented by not completely immersing the tip into the blood. After completely filling the tips, the devices were dried in the accompanying clamshells for 2 h at ambient temperature. Once dried, the VAMS devices were stored at room temperature in zip-closure plastic bags, containing two 5 g packages of desiccant (Minipax® absorbent packets, Sigma Aldrich) until UPLC®-MS/MS analysis.
Sample preparation
Sample preparation was performed by separating the VAMS tips from the plastic handlers and transferring these into 2 mL Eppendorf cups. Extraction was carried out using a Thermo-shaker TS-100C (BioSan, Riga, Latvia). In order to optimize the extraction conditions, different combinations of water and acetonitrile were evaluated, as well as different extraction solvent volumes (varying from 70 to 140 μL), extraction times, and temperatures. For each of the tested conditions, spiked VAMS devices were analyzed in triplicate and the final sample preparation method was selected based on a comparison of the peak areas obtained for each condition.
Preparation of calibrators and QCs
Calibrators were made at eight concentration levels in blank whole blood. For each compound—except for VPA—the lower limit of the therapeutic range in plasma divided by two was set as the lower limit of quantification (LLOQ) and the upper limit of the therapeutic range times four as the upper limit of quantification (ULOQ). The resulting calibrator concentrations were 1, 1.5, 2, 33.6, 65.2, 96.8, 128.4, and 160 μg/mL for PB; 4, 6, 8, 22.4, 36.8, 51.2, 65.6, and 80 μg/mL for PHT; 2, 3, 4, 12.8, 21.6, 30.4, 39.2, and 48 μg/mL for CBZ; and 0.25, 0.375, 0.5, 5.2, 9.9, 14.6, 19.3, and 24 μg/mL for CBZ-E. For VPA, detector oversaturation occurred with concentrations at four times the upper limit of the therapeutic range, therefore the upper limit of the therapeutic range times 1.5 was used as ULOQ, yielding calibrators at 25, 37.5, 50, 70, 90, 110, 130, and 150 μg/mL. Also when taking into account blood/plasma ratios (see further), these calibration lines cover the anticipated therapeutic ranges in blood. QC solutions (LLOQ, Low, Mid, High, respectively) were prepared in blank whole blood at 25, 55, 100, 112.5 μg/mL for VPA; 1, 3, 40, 120 μg/mL for PB; 4, 8, 20, 60 μg/mL for PHT; 2, 5, 12, 36 μg/mL for CBZ; and 0.25 and 1.50, 6, 18 μg/mL for CBZ-E. Non-matrix solvents were never added in a proportion higher than 5% of the total sample volume.
UPLC®-MS/MS method
A Waters Acquity UPLC® system (Waters, Milford, MA, USA) coupled to a SCIEX API™ 4000 mass spectrometer (SCIEX, Framingham, MA, USA) was used for all analyses. The hardware system was controlled by SCIEX Analyst® 1.6.2 and by the Waters Acquity console software.
The deviating characteristics of the six compounds, combined with the inability of the utilized configuration to switch between positive and negative ionization modes, necessitated development of two different UPLC®-MS/MS methods, one operating in negative ionization mode (method I, monitoring VPA, PB, and PHT) and one in positive ionization mode (method II, monitoring CBZ, CBZ-E, and OXC).
For both methods, a Chromolith® reversed phase (RP)-18 endcapped 100 × 4.60 mm column (Merck Millipore, Overijse, Belgium), equipped with the corresponding guard column, was chosen as it gave the best results in terms of compound separation. The column oven was set at 45 °C. A mobile phase consisting of 5 mM ammonium acetate (A) and 5 mM ammonium acetate in acetonitrile/water (95/5, v/v) (B) at a flow rate of 1.4 mL/min turned out to be the best option. The total run time was 10 min, including a 4-min run for method I and a 6-min run for method II. The mobile phase gradient program for method I started with 20% solvent B, linearly increased to 60% in 1 min, followed by an increase to 98% in 0.5 min, maintained for 1 min, and finally, reversal to starting conditions. For method II, the gradient started with 10% solvent B, followed by a linear increase to 27% in 0.22 min, isocratic conditions for 0.28 min, an increase to 31% in 1.72 min, followed by a rise to 98% in 0.88 min, kept for 0.8 min, and finally, returning to starting conditions.
The API™ 4000 mass spectrometer was equipped with an ESI source (TurboIonSpray®) and used an optimized MRM algorithm for detection. The source temperature was set at 600 °C, the ion spray voltage at − 3000 V for method I and at 2000 V for method II. Nitrogen was used as nebulizer (gas 1), heater (gas 2), curtain (CUR), and collision-activated dissociation (CAD) gas, with following gas pressure settings: 90 psi for gas 1, 10 psi for gas 2, 20 and 40 psi (respectively for method I and II) for CUR and the CAD vacuum was set at 12 for both methods (arbitrary settings).
For PB, PHT, CBZ, CBZ-E, and OXC, two characteristic precursor-to-product ion transitions were monitored, while for the corresponding internal standards one transition was analyzed. Since no stable ion fragments are created for VPA, a pseudo mass transition (143.1/143.1) was monitored. Table S1 in the Electronic Supplementary Material (ESM) shows all MRM transitions, together with the compound-specific MS parameters (optimized following infusion).
Method validation
Method validation was based on U.S. Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for bioanalytical method validation [30, 31] and covered accuracy, precision, carry-over, selectivity, homoscedasticity, calibration model, stability, matrix effect, recovery, and Hct effect. Control blanks (i.e., VAMS samples prepared with blank blood and analyzed without IS in the extraction solvent) and zero samples (i.e., VAMS samples prepared with blank blood and analyzed with the regular extraction solvent) were assessed throughout each sequence.
Accuracy (%bias) and precision (% relative standard deviation, %RSD) were assessed by analyzing QCs (LLOQ, Low, Mid, and High) in duplicate on four different days. The intra- and inter-batch precision were determined using ANOVA, whereas the accuracy was calculated by dividing the difference between the obtained concentration and the nominal value by the nominal value, and multiplying by 100 [32]. The %bias and %RSD should be within ± 15% for the QC samples, except for the LLOQ, where they should be within ± 20% [31].
Carry-over was examined by analysis of two blank samples after measurement of the highest calibrator (ULOQ), on four different days (n = 8). Carry-over for the analytes should not exceed 20% of the peak area found for the LLOQ and 5% for the IS [31]. For selectivity, identical criteria were applied. Selectivity was assessed by analyzing blank VAMS samples prepared with whole blood from six different individuals.
The LLOQ was defined for each of the AEDs as the lower limit of the therapeutic range divided by two. These concentrations gave a signal of at least 10 times the signal of a blank sample. A chromatogram of each compound at the LLOQ level is provided in the ESM.
Homoscedasticity and the calibration model were evaluated by generating eight 8-point calibration curves. Homoscedasticity was tested by performing an F-test (α = 1%) at the lowest and highest calibrator. Furthermore, for the calibration model, both weighted (1/x, 1/x2, 1/√x, 1/y, 1/y2, and 1/√y) and unweighted linear and quadratic regression were performed in order to find the best fitting model. The resulting models were compared by calculating the sum% relative error (%RE) and by plotting the %RE against nominal concentrations. Before accepting a selected model, a back-calculation was performed in which the mean concentrations of the calibrators should be within ± 15% of the nominal value or within ± 20% for the LLOQ [31].
Short- and long-term stability were assessed by analyzing Low and High QCs (n = 3) in duplicate after storage for 4, 7, and 30 days at different temperatures (− 20 °C, 4 °C, room temperature, and 60 °C) in a zip-closure plastic bag containing two 5 g packages of desiccant. As reference, QCs, prepared at the same day of the QCs used for stability testing, were analyzed at time point zero. Autosampler stability (4 °C) was evaluated by storing the extracts of Low and High QCs for 24 h in the autosampler before reinjection. At each day of analysis, an eight-point calibration curve was freshly prepared in order to calculate the concentration of the stored VAMS/extracts. Here again, the mean concentration of the QCs at a particular time point should not deviate more than ± 15% from the nominal concentration [31].
Matrix effects were investigated by comparing the peak areas obtained at two concentration levels (Low or High QC), spiked to blank blood extract (from six different individuals, with a hematocrit ranging from 0.335 to 0.495) (A), with those obtained using a neat aqueous mixture containing the analytes and their IS at corresponding concentrations (B). The ratios of peak areas of (A) to those of (B), multiplied by 100, represent the IS-corrected matrix effect. Overall, the %RSD of the IS-corrected matrix effect should not exceed ± 15% [31].
The impact of the Hct on the recovery was evaluated for Low and High QC’s (n = 6) at four different Hct levels (target values at 0.21, 0.42, 0.52, and 0.62), prepared by centrifuging an aliquot of blood with a hematocrit of 0.40 in 2 mL Eppendorf tubes in an Eppendorf 5804R centrifuge (Hamburg, Germany) for 5 min at 1000×g and by removing or adding plasma. Here, two sets of VAMS samples were compared to one another, i.e., VAMS samples prepared by pipetting 10 μL of spiked blood (C) and VAMS samples prepared by pipetting 10 μL of blank blood and to which the analytes were only spiked post-extraction (D). The absolute recovery values (%) were calculated by multiplying the ratios of peak areas of (C) to those of (D) by 100. To further evaluate the impact of the Hct, VAMS samples (n = 6) were also prepared by dipping them into spiked blood at four different Hct levels (target values at 0.21, 0.42, 0.52, and 0.62). The latter better reflects the reality when compared to pipetting of a fixed volume onto the VAMS devices.
Where relevant, statistical analyses were performed using the Minitab® software.
Application
In order to objectively indicate the validity of the obtained results, three sets of external serum QC materials were used to generate QCs in blood and VAMS samples derived thereof. The QC materials were the ClinCal®-calibrator (Recipe®, Munich, Germany) containing PB 37.5 μg/mL, PHT 18.3 μg/mL, VPA 90.7 μg/mL, CBZ 11.2 μg/mL, and CBZ-E 5.60 μg/mL in serum and the Liquichek™ Therapeutic Drug Monitoring Control (TDM) Levels 2 and 3 (Bio-Rad, California, USA) containing PB 34.7 μg/mL, PHT 15.2 μg/mL, VPA 85.6 μg/mL, and CBZ 9.11 μg/mL in serum (level 2) and PB 65.0 μg/mL, PHT 31.8 μg/mL, VPA 134 μg/mL, and CBZ 13.1 μg/mL in serum (level 3). In order to be comparable with a calibration curve prepared in whole blood, the external QC materials were diluted 1 on 4 with whole blood, by replacing 250 μL of plasma (obtained by centrifugation of 1 mL of whole blood) by 250 μL of the external QC materials. Due to this dilution, some concentrations were no longer within the calibration range and, hence, could not be quantified. The resulting concentrations in blood of the used external QC materials were 33.5 μg/mL for VPA (Liquichek™ Level 3); 8.68, 9.38, and 16.3 μg/mL for PB (Liquichek™ Level 2, ClinCal® and Liquichek™ Level 3, respectively); 4.58 and 7.95 μg/mL for PHT (ClinCal® and Liquichek™ Level 3, respectively); 2.28, 2.80, and 3.28 μg/mL for CBZ (Liquichek™ Level 2, ClinCal® and Liquichek™ Level 3, respectively); and 1.40 μg/mL for CBZ-E (ClinCal®).
Furthermore, as a proof of concept, we analyzed 30 samples, collected at Ghent University Hospital from patients who visited the Hospital for evaluation of a variety of parameters, including follow-up of their AED treatment. VAMS samples were prepared by wicking up EDTA-anticoagulated blood from routine left-over whole blood samples that had been stored at room temperature for maximum 72 h. Approval for this study was provided by the Ethics Committee of Ghent University Hospital (EC2017/0572).
Results and discussion
Sample preparation
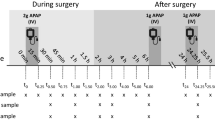
As the IS are in the extraction solvent, these do not compensate for recovery issues [33]. Therefore, optimization of sample extraction was comprehensively implemented. During this optimization, extraction at 22 °C and at 60 °C was compared, using freshly prepared VAMS samples, VAMS samples stored for 3 days at room temperature and VAMS samples stored for 3 days at 60 °C. The VAMS were prepared by using spiked whole blood (Low QC level) with a Hct of 0.41. Also a comparison was made with VAMS samples prepared from blood at a Hct of 0.62, stored for 3 days at 60 °C and extracted at 22 °C and at 60 °C.
As Fig. 1 depicts, extraction of the VAMS at elevated temperature (60 °C) provided overall the best results in terms of absolute recovery. Based on these findings, hundred microliters of an acetonitrile/water (80/20, v/v) mixture, containing 5 mM ammonium acetate and the deuterated internal standards (0.5 μg/mL), was eventually used to extract VAMS devices by shaking for 10 min at 1000 rpm and 60 °C. Following centrifugation at ambient temperature for 10 min at 10,000×g, 70 μL of supernatant was diluted 1 on 1 with water containing 5 mM ammonium acetate. This mixture was transferred to an amber glass vial before injection of 10 μL onto the LC column.

Optimization of sample extraction: comparison of extraction at 22 °C and at 60 °C, using VAMS samples, spiked at Low QC, which were freshly prepared or stored for 3 days at RT (Hct 0.41) or were stored for 3 days at 60 °C (Hct 0.41 and Hct 0.62). Shown are the mean and standard deviation (n = 6). Asterisks denote statistical differences (p < 0.05, two-sided t test) between extraction at 22 and 60 °C
Method validation
With the exception of VPA (18.2%bias at Low QC), the acceptance criteria for accuracy (%bias below 20% at LLOQ and below 15% at the other QC levels) was met. With an intra- and inter-batch precision (%RSD) below 10%, acceptance criteria were met for all compounds (Table 1).
No carry-over was detected when injecting blank samples after the highest calibrator and, regarding selectivity, no unacceptable interferences were observed in VAMS samples prepared from blank blood originating from six different donors. Importantly, a considerable advantage is the possibility to distinguish between CBZ-E and OXC (retention times of 2.64 and 2.88 min, respectively), although they have the same MRM transitions (see chromatograms at LLOQ levels in the ESM). Therefore, the presence of OXC in a patient sample will not interfere with the calculated CBZ-E concentration.
Calibration data for all compounds were found to be heteroscedastic. Only for PHT, weighted regression did not improve the %RE, therefore the simplest model, i.e., unweighted linear regression, was chosen. The selected weighting factors for PB, CBZ, and CBZ-E were 1/x, 1/x2, and 1/x, respectively. Based on %RE values, weighted quadratic regression was chosen for VPA, with a weighting factor 1/x. Using these selected models, mean back-calculated concentrations did not differ more than 7% for all calibrators, which is in line with the acceptance criteria [31].
As displayed in Table 2, all compounds were stable in VAMS samples for at least 1 month when stored at room temperature, 4 °C, and − 20 °C and for at least 1 week when stored at 60 °C. The latter is important when envisaging, e.g., sampling in and/or shipping from countries with high ambient temperatures. Re-analysis of Low and High QCs after storage for 24 h in the cooled autosampler (4 °C) revealed autosampler stability for all compounds and their corresponding IS.
Based on the results provided in Table 3, the values for the non-IS-corrected analyte matrix effects indicated relevant (> 15%) suppression of ionization for PHT, while relevant (> 15%) enhancement of ionization was observed for CBZ and CBZ-E. However, the IS-corrected matrix effects were all within 90–103%, indicating that the IS compensate for the differences in ionization. Importantly, the %RSD of the IS-corrected matrix effects was below 4% in all instances, meeting the pre-set acceptance criterion of 15% [31].
The possibility of a Hct-dependent recovery, when using VAMS, is a well-known issue [23, 26]. Furthermore, as demonstrated by Abu-Rabie et al., a high recovery is important to minimize the risk of being confronted with a significant Hct-based recovery bias [33]. In the first stage, recovery was evaluated by pipetting a fixed volume (10 μL) of blood onto the VAMS. High recoveries were obtained for all compounds, at 85.2 ± 6.1% for VPA, 93.7 ± 4.6% for PB, 85.4 ± 5.9% for PHT, 86.4 ± 5.9% for CBZ, and 91.4 ± 4.6% for CBZ-E, these values corresponding to the averages calculated from all values obtained at all Hct levels and at Low and High QC level. Furthermore, as shown in Fig. 2, overall, apart from VPA (High QC) at high Hct (0.62), the Hct did not significantly affect the recovery. In addition, when normalizing the 0.42 Hct level to 100% (see Fig. S2 in the ESM), all recoveries—except for VPA—were within 15% of the 0.42 Hct reference sample. For VPA, the low QC sample at the extreme Hct of 0.62 Hct differed 18% from the 0.42 Hct sample, which can still be considered acceptable.

IS-compensated recovery (%) at Low and High QC level (n = 6) for VPA, PB, PHT, CBZ, and CBZ-E measured in VAMS samples, prepared by pipetting 10 μL blood at four different Hct levels (target values at 0.21, 0.42, 0.52, and 0.62). Shown are the mean and standard deviation. Asterisk denotes statistical difference (p < 0.05, one-way ANOVA test) from the 0.42 Hct reference sample
To further evaluate the impact of the Hct, VAMS samples were also prepared by dipping them into spiked blood (Low and High QC) at four different Hct levels (target values at 0.21, 0.42, 0.52, and 0.62). As Fig. 3 depicts, all were within 16% of the 0.42 Hct sample, except for PB at 0.62 Hct (Low QC) and 0.52 Hct (High QC). However, one-way ANOVA analyses revealed that the observed differences for PB were not statistically significant (p = 0.303 and 0.082, respectively).

Influence of the hematocrit on the recovery of VPA, PB, PHT, CBZ, and CBZ-E, with the 0.42 Hct sample being normalized to 100%. Here, VAMS samples were prepared by dipping into spiked blood (Low and High QC) at four different Hct levels (target values at 0.21, 0.42, 0.52, and 0.62). The full line indicates the 0.42 Hct sample normalized to 100% and the dotted lines indicate the ± 15% deviation limits
Taking these findings into account, it can be concluded that if there is an effect of the Hct on the recovery, it is overall limited. As in some cases, there is a trend towards somewhat lower recoveries in samples with very high Hct values, it is recommended to be cautious when analyzing patient samples with a Hct above 0.60. Given the overall limited influence at low to normal Hct values and since we aim at applying the developed method on patient samples originating from children living in developing countries, making a high Hct rather exceptional, we do not foresee any Hct-related recovery problems.
Application
The developed method was applied in quadruplicate on three sets of VAMS QCs, generated from blood in which external serum QC materials had been diluted 1 on 4 by replacing 250 μL of plasma with 250 μL of the external QC. As outlined in Fig. 4, 35 out of the 40 measurements deviated less than 20% from the target value and the mean concentrations were within ± 20% in all cases. No trend was evident from the distribution of the means, compared to the target concentrations and, with the exception of PB from set C (owing to one deviating value), the %RSD was below 15% for the quadruplicates. Overall, this objectively indicates the validity of the developed method.

Percentage of the obtained concentration in VAMS samples versus the target concentration present in three sets of external QC materials (n = 4) (A, Liquicheck™ Level 2; B, ClinCal®; C, Liquicheck™ Level 3). The triangles depict the mean concentrations, the full line indicates the target concentration normalized to 100%, and the dotted lines represent a deviation of ± 20%
Next, the method was applied on 30 real-life left-over whole blood samples (Table 4). Analysis was performed within 1 month after collection (storage at − 20 °C). Of the collected samples, 11 contained CBZ (and consequently also CBZ-E), 9 VPA, 6 PB, and 4 PHT. Three of the VPA samples and one of the CBZ samples had a concentration below the used LLOQ and hence were not quantified. As a reference, serum concentrations were obtained using chemiluminescent magnetic microparticle immunoassay technology (CMIA, Architect i2000SR). For VPA, the mean of the VAMS concentrations were 66 ± 9.23% of those measured in serum, for PB 90 ± 12.8%, for PHT 83 ± 16.5%, and for CBZ 114 ± 19.7%. These ratios are in line with published blood/plasma ratios of 0.70 for VPA, 0.90 for PB, 0.71 for PHT, and 1.02 for CBZ [34,35,36]. In theory, blood/plasma ratios could be used to calculate the serum concentrations, based on VAMS concentrations—with as a limitation that other techniques (i.e., immunoassay vs LC-MS/MS) were used for both assessments. However, as is readily clear from our limited dataset, there is a substantial variation in observed blood/plasma ratios between individuals. Also Linder et al. observed substantial variations in blood/plasma ratios for CBZ and VPA, with a role played by Hct and concentration, albeit using spiked samples [37]. More specifically, in our dataset, the %RSD on the observed blood/plasma ratios was between 14 and 20%, yielding a significant level of uncertainty when applying an “average” conversion coefficient. Using the published blood/plasma ratios to calculate serum concentration from VAMS concentrations resulted in a mean bias of − 5.52, 0.42, 17.2, and 12.2%, for VPA, PB, PHT, and CBZ, respectively [34,35,36]. Given the small number of samples, no definitive conclusions can be drawn from this. Therefore, in conclusion, whereas this application on patient samples revealed applicability of the developed method on real-life patient samples, interpretation of the observed concentrations ideally involves the establishment of reference ranges in blood.
Conclusion
In TDM there is a growing interest in the use of non- and minimally invasive alternative sampling strategies, VAMS being one of the recent developments. For anti-epileptic drugs, most evidence for TDM exists for the first-generation anti-epileptic drugs, due to their significant interindividual variability in pharmacokinetics and due to the narrow therapeutic indices related to those drugs.
In this study, an LC-MS/MS method for the determination and quantification of four anti-epileptic drugs and one active metabolite, i.e., CBZ, VPA, PHT, PB, and CBZ-E, making use of VAMS devices, was developed and validated. The final method was extensively validated, including both bioanalytical and VAMS-specific parameters and overall the pre-set acceptance criteria were met. Thorough optimization of the extraction procedure helped enabling a Hct-independent, consistent recovery.
Application of the method on external quality control materials and on real-life patient samples demonstrated the validity and applicability of the developed procedure. We successfully used external serum QCs to replace part of the plasma fraction of control blood, thereby yielding blood (and VAMS) samples with known concentrations. This represents a feasible approach to cope with the lack of external reference materials for dried blood matrices.
To date, the limited availability of clinical validation data still remains one of the constraints preventing the widespread implementation of dried matrix approaches in clinical practice [1]. Furthermore, divergent results have been reported on the ratio between blood and plasma or serum concentrations. Therefore, calculating serum concentrations based on blood concentrations is challenging [9, 37,38,39]. Having at hand reference ranges in blood could allow to cope with this.
As one of the advantages coupled to dried matrices is the extreme usefulness for sampling in remote or resource-limited settings (e.g., ease of collection and storage), in a next step, we aim at applying this newly developed method on patient samples originating from developing countries.
Change history
15 February 2018
We would like to call the reader’s attention to the fact that unfortunately in fig. 2 of the original article the figure headings of both graphs are the same.
References
Milosheska D, Grabnar I, Vovk T. Dried blood spots for monitoring and individualization of antiepileptic drug treatment. Eur J Pharm Sci. 2015;75:25–39.
Krasowski MD, McMillin GA. Advances in anti-epileptic drug testing. Clin Chim Acta. 2014;436:224–36.
Velghe S, Capiau S, Stove CP. Opening the toolbox of alternative sampling strategies in clinical routine: a key-role for (LC-)MS/MS. Trac Trend Anal Chem. 2016;84:61–73.
Capiau S, Alffenaar J-W, Stove CP. Alternative sampling strategies for therapeutic drug monitoring. In: Clarke W, Dasgupta A, editors. Clinical challenges in therapeutic drug monitoring. Amsterdam: Elsevier; 2016. p. 279–336.
Shah NM, Hawwa AF, Millership JS, Collier PS, McElnay JC. A simple bioanalytical method for the quantification of antiepileptic drugs in dried blood spots. J Chromatogr B Analyt Technol Biomed Life Sci. 2013;923-924:65–73.
Popov TV, Maricic LC, Prosen H, Voncina DB. Development and validation of dried blood spots technique for quantitative determination of topiramate using liquid chromatography-tandem mass spectrometry. Biomed Chromatogr. 2013;27(8):1054–61.
Linder C, Andersson M, Wide K, Beck O, Pohanka A. A LC-MS/MS method for therapeutic drug monitoring of carbamazepine, lamotrigine and valproic acid in DBS. Bioanalysis. 2015;7(16):2031–9.
Villanelli F, Giocaliere E, Malvagia S, Rosati A, Forni G, Funghini S, et al. Dried blood spot assay for the quantification of phenytoin using liquid chromatography-mass spectrometry. Clin Chim Acta. 2015;440:31–5.
Shokry E, Villanelli F, Malvagia S, Rosati A, Forni G, Funghini S, et al. Therapeutic drug monitoring of carbamazepine and its metabolite in children from dried blood spots using liquid chromatography and tandem mass spectrometry. J Pharm Biomed Anal. 2015;109:164–70.
AbuRuz S, Al-Ghazawi M, Al-Hiari Y. A simple dried blood spot assay for therapeutic drug monitoring of lamotrigine. Chromatographia. 2010;71(11–12):1093–9.
la Marca G, Malvagia S, Filippi L, Luceri F, Moneti G, Guerrini R. A new rapid micromethod for the assay of phenobarbital from dried blood spots by LC-tandem mass spectrometry. Epilepsia. 2009;50(12):2658–62.
la Marca G, Malvagia S, Filippi L, Fiorini P, Innocenti M, Luceri F, et al. Rapid assay of topiramate in dried blood spots by a new liquid chromatography-tandem mass spectrometric method. J Pharm Biomed Anal. 2008;48(5):1392–6.
Wilhelm AJ, den Burger JC, Swart EL. Therapeutic drug monitoring by dried blood spot: progress to date and future directions. Clin Pharmacokinet. 2014;53(11):961–73.
Fan L, Lee JA. Managing the effect of hematocrit on DBS analysis in a regulated environment. Bioanalysis. 2012;4(4):345–7.
De Kesel PM, Capiau S, Lambert WE, Stove CP. Current strategies for coping with the hematocrit problem in dried blood spot analysis. Bioanalysis. 2014;6(14):1871–4.
Youhnovski N, Bergeron A, Furtado M, Garofolo F. Pre-cut dried blood spot (PCDBS): an alternative to dried blood spot (DBS) technique to overcome hematocrit impact. Rapid Commun Mass Spectrom. 2011;25(19):2951–8.
De Kesel PM, Capiau S, Stove VV, Lambert WE, Stove CP. Potassium-based algorithm allows correction for the hematocrit bias in quantitative analysis of caffeine and its major metabolite in dried blood spots. Anal Bioanal Chem. 2014;406(26):6749–55.
den Burger JC, Wilhelm AJ, Chahbouni AC, Vos RM, Sinjewel A, Swart EL. Haematocrit corrected analysis of creatinine in dried blood spots through potassium measurement. Anal Bioanal Chem. 2015;407(2):621–7.
Li Y, Henion J, Abbott R, Wang P. The use of a membrane filtration device to form dried plasma spots for the quantitative determination of guanfacine in whole blood. Rapid Commun Mass Spectrom. 2012;26(10):1208–12.
Li F, Zulkoski J, Fast D, Michael S. Perforated dried blood spots: a novel format for accurate microsampling. Bioanalysis. 2011;3(20):2321–33.
Meesters RJ, Zhang J, van Huizen NA, Hooff GP, Gruters RA, Luider TM. Dried matrix on paper disks: the next generation DBS microsampling technique for managing the hematocrit effect in DBS analysis. Bioanalysis. 2012;4(16):2027–35.
Leuthold LA, Heudi O, Deglon J, Raccuglia M, Augsburger M, Picard F, et al. New microfluidic-based sampling procedure for overcoming the hematocrit problem associated with dried blood spot analysis. Anal Chem. 2015;87(4):2068–71.
De Kesel PM, Lambert WE, Stove CP. Does volumetric absorptive microsampling eliminate the hematocrit bias for caffeine and paraxanthine in dried blood samples? A comparative study. Anal Chim Acta. 2015;881:65–73.
Denniff P, Spooner N. Volumetric absorptive microsampling: a dried sample collection technique for quantitative bioanalysis. Anal Chem. 2014;86(16):8489–95.
Verougstraete N, Lapauw B, Van Aken S, Delanghe J, Stove C, Stove V. Volumetric absorptive microsampling at home as an alternative tool for the monitoring of HbA1c in diabetes patients. Clin Chem Lab Med. 2017;55(3):462–9.
Kok MGM, Fillet M. Volumetric absorptive microsampling: current advances and applications. J Pharm Biomed Anal. 2018;147:288–96.
Atugonza R, Kakooza-Mwesige A, Lhatoo S, Kaddumukasa M, Mugenyi L, Sajatovic M, et al. Multiple anti-epileptic drug use in children with epilepsy in Mulago hospital, Uganda: a cross sectional study. BMC Pediatr. 2016;16:34.
Gogtay NJ, Kshirsagar NA, Dalvi SS. Therapeutic drug monitoring in a developing country: an overview. Br J Clin Pharmacol. 2001;52(Suppl 1):103S–8S.
Bertilsson L. Clinical pharmacokinetics of carbamazepine. Clin Pharmacokinet. 1978;3(2):128–43.
U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research. Center for Veterinary Medicine. Draft Guidance for Industry. Bioanalytical Method Validation. 2013. https://www.fda.gov/downloads/Drugs/Guidances/ucm368107.pdf. Accessed June 2017.
European Medicines Agency. Guideline on Bioanalytical Method Validation. 2015. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Accessed June 2017.
Wille SMR, Peters FT, Di Fazio V, Samyn N. Practical aspects concerning validation and quality control for forensic and clinical bioanalytical quantitative methods. Accred Qual Assur. 2011;16(6):279–92.
Abu-Rabie P, Denniff P, Spooner N, Chowdhry BZ, Pullen FS. Investigation of different approaches to incorporating internal standard in DBS quantitative bioanalytical workflows and their effect on nullifying hematocrit-based assay bias. Anal Chem. 2015;87(9):4996–5003.
Houts T. Immunochromatography. In: Price CP, editor. Principles and practice of immunoassay. New York: Stockton Press; 1991. p. 563–83.
Launiainen T, Ojanpera I. Drug concentrations in post-mortem femoral blood compared with therapeutic concentrations in plasma. Drug Test Anal. 2014;6(4):308–16.
Morris RG, Schapel GJ. Phenytoin and phenobarbital assayed by the ACCULEVEL method compared with EMIT in an outpatient clinic setting. Ther Drug Monit. 1988;10(4):469–73.
Linder C, Wide K, Walander M, Beck O, Gustafsson LL, Pohanka A. Comparison between dried blood spot and plasma sampling for therapeutic drug monitoring of antiepileptic drugs in children with epilepsy: a step towards home sampling. Clin Biochem. 2017;50(7–8):418–24.
Kong ST, Lim SH, Lee WB, Kumar PK, Wang HY, Ng YL, et al. Clinical validation and implications of dried blood spot sampling of carbamazepine, valproic acid and phenytoin in patients with epilepsy. PLoS One. 2014;9(9):e108190.
Rhoden L, Antunes MV, Hidalgo P, Alvares da Silva C, Linden R. Simple procedure for determination of valproic acid in dried blood spots by gas chromatography-mass spectrometry. J Pharm Biomed Anal. 2014;96:207–12.
Acknowledgements
The authors wish to acknowledge Prof. Veronique Stove, PharmD. Matthijs Oyaert and their team for assistance with blood collection and all volunteers who participated in the study. Furthermore, SV would also like to thank the Special Research Fund (BOF) for granting her a PhD fellowship (application number 01D42414).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Approval for this study was provided by the Ethics Committee of Ghent University Hospital (EC2017/0572). For the use of left-over samples to evaluate an alternative procedure for AED monitoring, the need to obtained individual informed consent was waived by the Ethics Committee.
Conflict of interest
The author declares to not have any financial, commercial, legal, or professional relationship with other organizations, or with the people working with them, that could influence the matter discussed in this manuscript.
Additional information
The original version of this article was revised: the headings of figure 2 of both graphs are the same.
A correction to this article is available online at https://doi.org/10.1007/s00216-018-0951-8.
Electronic supplementary material
ESM 1
(PDF 345 kb)
Rights and permissions
About this article

Cite this article
Velghe, S., Stove, C.P. Volumetric absorptive microsampling as an alternative tool for therapeutic drug monitoring of first-generation anti-epileptic drugs. Anal Bioanal Chem 410, 2331–2341 (2018). https://doi.org/10.1007/s00216-018-0866-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-018-0866-4