
Abstract
In the field of bioanalysis, dried matrix spot sampling is increasingly receiving interest, as this alternative sampling strategy offers many potential benefits over traditional sampling, including matrix volume-sparing properties. By using a microsampling strategy, e.g., volumetric absorptive microsampling (VAMS), the number of samples that can be collected from a patient can be increased, as a result of the limited sample volume that is required per sample. To date, no VAMS-based methods have been developed for the quantification of analytes in cerebrospinal fluid (CSF). The objective of this study was to develop and validate two LC-MS/MS methods for the quantification of paracetamol in dried blood and dried CSF, with both matrices sampled using VAMS. Both methods were fully validated based on internationally accepted guidelines. Paracetamol was chromatographically separated from its glucuronide and sulfate metabolites and no carry-over or unacceptable interferences were detected. The total precision (%RSD) was below 15% for all QC levels and accuracy (%bias) was below 7% (17% for the LLOQ of aqueous VAMS). The influence of the hematocrit on the recovery of blood VAMS samples appeared to be limited within the hematocrit range of 0.21 to 0.62. The blood VAMS samples were stable for 1 week if stored at 50 °C, and for at least 8 months when stored between − 80 °C and room temperature. The aqueous VAMS samples were stable for at least 9 months when stored between − 80 and 4 °C, and for 1 month when stored at room temperature. Application of the methods on external quality control material and analysis of patient samples demonstrated the validity and utility of the methods and provided a proof of concept for the analysis of CSF microsamples obtained via VAMS devices.

ᅟ
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dried matrix spot (DMS) sampling is an alternative sampling strategy, typically using sample volumes less than 50 μL, which has many potential benefits over traditional sampling [1,2,3,4,5]. Therefore, this sampling strategy is receiving increasing interest from the bioanalytical field. Most methods using micro volumes of matrix have been developed and applied for the analysis of small molecules in blood- or plasma-based samples. The most commonly used dried microsampling technique still remains dried blood spots (DBS), but over the years, many innovative alternatives have been developed. One of these alternatives is an approach coined volumetric absorptive microsampling (VAMS). This device has been commercially available for several years (brand name Mitra™), and many research groups have developed methods for different types of clinical and toxicological determinations using this device [6]. The device (see Supplementary Electronic Material (ESM) Fig. S1) consists of a plastic handle and a polymeric tip, which absorbs a fixed volume of a given biofluid. The main advantage of the VAMS devices, when used for blood sampling, is that the hematocrit (Hct) of a sample does not influence the amount of blood that is absorbed [7, 8]. Thereby, the VAMS devices partially overcome the most discussed issue in DBS sampling, which is the Hct-based area bias [6, 9]. Besides its use for blood and plasma sampling, microsampling, including VAMS, can also be used for the analysis of other biological matrices, such as urine, oral fluid, and cerebrospinal fluid (CSF).
Specifically for CSF, the number of methods and applications making use of DMS is limited [10, 11]. In certain specific situations, dried CSF microsampling offers important advantages over traditional sampling, e.g., pharmacokinetic (PK) and toxicokinetic (TK) studies of drugs in CSF. By using a microsampling strategy, e.g., VAMS, the number of samples that can be collected from a patient can be increased, as a result of the limited sample volume that is required per sample. This low sample volume is even more beneficial when performing PK or TK studies in pediatric patient groups. Moreover, VAMS overcomes the need to handle micropipettes and accurate pipetting of small volumes in a clinical setting and facilitates sample storage and transport, which is especially interesting for analytes suffering from stability issues. As automated VAMS sample extraction and consecutive LC-MS/MS analysis are most likely to be available in the near future, this would allow a uniform workflow for blood and CSF VAMS samples. To date, no VAMS-based methods have been developed for sampling and subsequent quantification of analytes in CSF.
To establish a proof of concept for the use of VAMS for CSF microsampling, we chose paracetamol as a model compound. Paracetamol is one of the most widely used analgesics, for both children and adults. Many different research groups have studied the mechanism of action of paracetamol and recent studies have shown involvement of the central nervous system [12, 13]. Despite this insight, and the many studies that are being performed on the effect and the use of paracetamol in different patient groups, knowledge about its CSF disposition is still limited. The lack of PK studies can, at least partly, be explained by the high patient burden related to sampling and the relatively small CSF volume available for sampling.
The objective of this study was to develop and fully validate methods for the quantification of paracetamol in dried blood and dried CSF utilizing VAMS. To the best of our knowledge, this is the first method in which analytes were quantified in dried CSF samples obtained via VAMS devices.
Materials and methods
Chemicals and materials
Paracetamol (pharmaceutical secondary standard), paracetamol sulfate (potassium salt), paracetamol β-d-glucuronide, formic acid (FA), the internal standard (IS) paracetamol-D4, calcium chloride, sodium phosphate dibasic (anhydrous), sodium phosphate monobasic (monohydrate), and bovine serum albumin were purchased from Sigma-Aldrich (Diegem, Belgium). LC-MS grade methanol was obtained from Biosolve (Valskenswaard, The Netherlands). Ultrapure water (18.2-MΩ resistivity) was generated in-house by a Synergy® Water Purification System (Merck Millipore, Overijse, Belgium). Potassium chloride and magnesium chloride (hexahydrate) were obtained from Merck (Darmstadt, Germany) and sodium chloride from VWR International (Radnor, PA, USA). Glucose was obtained from Acros Organics (Geel, Belgium). Ten microliter VAMS devices (brand name Mitra™) were purchased from Neoteryx (Torrance, CA, USA).
Standard solutions, calibrators, and quality control samples
Stock solutions (10 mg/mL) were prepared by dissolving 10 mg of the paracetamol standard in 1 mL of methanol-water (50:50, v/v). Working solutions were prepared on the day of analysis by diluting the stock solution in water. Independently prepared solutions were used for the working solutions to prepare calibrators and quality control samples (QCs). For the IS, a stock solution (5 mg/mL) was prepared in a similar way and further diluted to 10 μg/mL with methanol. All stock solutions were stored at − 20 °C.
Blood samples
Paracetamol-free whole blood was collected from a healthy volunteer, using K2EDTA as anticoagulant. The blood was fortified with paracetamol by adding working solutions (non-matrix spiked volume was 5% v/v). Calibrators were prepared at eight concentration levels in blank whole blood with concentrations of 0.10, 0.20, 0.50, 1.0, 5.0, 10.0, 50.0, and 100 μg/mL. QCs were prepared at four concentration levels: 0.10 μg/mL (lower limit of quantification, LLOQ); 0.25 μg/mL (low QC); 7.5 μg/mL (Mid QC); and 80.0 μg/mL (high QC). VAMS was performed on the same day as the preparation of calibrators and QCs. To avoid overfilling of the device, only the lower part of the VAMS device was dipped in the blood samples. After the tip had turned completely red, an additional 2 s of sampling time was added. After sampling, the VAMS device was air-dried for at least 2 h prior to sample extraction. When samples were not analyzed on the day of preparation, they were stored in airtight bags containing desiccant. Blood with varying Hct levels was prepared by centrifuging an aliquot of the blood (5 min, 5000g) and by removing or adding plasma. The Hct level of the donor blood was measured using a Sysmex XE-5000 hematology analyzer (Sysmex, Kobe, Japan).
CSF samples
No blank CSF was available for the preparation of calibrators and QCs. Therefore, water was evaluated (see the “Method validation” section) as an alternative matrix replacing blank CSF. Calibrators and QCs were prepared by spiking water with the paracetamol working solutions. Calibrators were prepared at eight concentration levels: 0.10, 0.20, 0.50, 1.0, 5.0, 10.0, 20.0, and 40.0 μg/mL. QCs were prepared at the same concentration levels as the blood QC samples, except for the high QC (35.0 μg/mL). For the absorption of blanks, calibrators, and QCs onto the VAMS device tip, the lower part of the tip was dipped in the fluid for 6 s, as recommended by the manufacturer. After sampling, the VAMS device was air-dried for at least 2 h prior to sample extraction. When samples were not analyzed on the day of preparation, they were stored in airtight bags containing desiccant.
Sample preparation
VAMS sampler tips were removed from the handle and put into a 2-mL cup and extracted by adding 300 μL of extraction solvent (methanol-water-FA, 80:20:0.01, v/v/v), containing the IS (0.04 μg/mL). The extraction temperature was optimized by the evaluation of peak area ratios after extraction at 22 °C, 37 °C, and 60 °C. For this evaluation, blood VAMS samples, with medium (0.44) and high (target value 0.60) Hct levels and spiked with paracetamol (1.0 μg/mL), were analyzed for each condition (n = 3). The tips were extracted for 15 min by shaking in a Thermomixer comfort (Eppendorf, Hamburg, Germany) (1400 rpm), followed by centrifugation at ambient temperature for 10 min at 10,000g. Next, 250 μL of the supernatants was transferred to a vial and diluted with 750 μL of water (0.01% FA). Injection volumes onto the UPLC® column were 3 μL for blood samples and 4 μL for CSF samples.
Liquid chromatographic and mass spectrometric conditions
A Waters Acquity UPLC® system (Waters, Milford, MA, USA) coupled to a SCIEX API 4000™ triple quadrupole mass spectrometer (SCIEX, Framingham, MA, USA) equipped with a TurboIonSpray® source was used for all experiments. The liquid chromatography tandem mass spectrometry (LC-MS/MS) system was controlled by SCIEX Analyst (version 1.6.2) and Waters Acquity console software.
For the chromatographic separation of paracetamol and its metabolites, a Waters Acquity UPLC® HSS-T3 column (100 mm × 2.1 mm, 1.8-μm particle size) was used, equipped with a corresponding Waters VanGuard pre-column (5 mm × 2.1 mm; 1.8-μm particle size), held at 45 °C and using a gradient mobile phase consisting of water-methanol-FA (95:5:0.01, v/v/v) (A) and methanol-water-FA (95:5:0.01, v/v/v) (B) with a flow rate of 0.55 mL/min. Mobile phase was held at 20% B for 0.7 min, increased linearly to 66% B over 1.3 min, further increasing linearly to 95% B over 0.2 min, held at 95% B for 1 min, followed by an immediate return to 20% B and re-equilibrating at 20% B for 1 min, yielding a total runtime of 4.2 min.
The mass spectrometer was operated in positive electrospray ionization (ESI) mode and multiple reaction monitoring (MRM). The mass transitions were m/z 152.04 to m/z 110.20 and to m/z 93.00 respectively for the quantification and qualification ion pairs of paracetamol. All MRM transitions, together with compound-specific parameters, are listed in the ESM Table S1, and source and gas parameters are tabulated in ESM Table S2.
Method validation
The methods and acceptance criteria used for method validation were based on the US Food and Drug Administration (FDA) and European Medicines Agency (EMA) guidelines for bioanalytical method validation [14, 15]. The validation encompassed an evaluation of the following characteristics: selectivity, carry-over, calibration model, precision, accuracy, matrix effect (ME), recovery, Hct effect, stability, and dilution integrity. In each analytical run, a control blank (i.e., VAMS sample prepared from blank matrix and processed with extraction solvent that does not contain the IS), a zero sample (i.e., VAMS sample prepared from blank matrix and extracted with extraction solvent including the IS), and QC samples were included.
Since no analyte-free CSF was available, we investigated whether water was suitable to be used as a surrogate matrix for CSF for the preparation of blanks, calibrators, and QCs. For this purpose, patient CSF samples (n = 6), containing paracetamol, were used. First, VAMS samples were made from CSF samples and from the same CSF samples diluted 1/10 in water, the latter resulting in overall aqueous samples. As a next step, both the original and diluted VAMS samples were analyzed against a calibration curve obtained by VAMS samples prepared from spiked water. For the evaluation, the %bias (diluted vs. original; taking into account the dilution factor) and the relative standard deviation (%RSD) were calculated. Acceptance criteria were ± 10% for the mean %bias and 15% for the %RSD [14, 15].
Selectivity was assessed by analysis of analyte-free blood samples from six individual donors for blood samples. For CSF samples, blank artificial CSF, prepared in-house, was used (see ESM Table S3 for composition). In addition, transition ratios were compared between calibrators and QCs (n = 16) on the one hand, and patient CSF VAMS samples (n = 55) on the other hand. The absence of interfering compounds was accepted if the transition ratio of the sample was ± 20% of the average transition ratio of the calibrators and QCs. Furthermore, separate standard solutions were prepared from the metabolites paracetamol glucuronide and paracetamol sulfate (50 ng/mL for all compounds) to investigate whether these metabolites interfered with the paracetamol assay by monitoring of the paracetamol channel. These metabolites were not determined qualitatively nor quantitatively in this method. The absence of interfering components was accepted if the response at the retention time of paracetamol was less than 20% of the LLOQ for paracetamol and 5% for the IS [14, 15].
Carry-over was assessed by injection of blank solvent (water) after the highest calibrator (n = 4) using the same acceptance criteria as for selectivity [14, 15].
Calibration lines were constructed by plotting the ratio of the analyte area and IS area versus the nominal concentration. To determine the best fitting calibration model, six eight-point calibration curves were constructed over a total of four different days. An F test was performed to investigate homoscedasticity (α = 1%) at the highest and lowest calibrator level. Selection of the calibration model was based on the calculation of the sum % residual error (%RE) and a plot of the %RE against the nominal concentrations. A linear and a quadratic regression model were evaluated with or without weighing factors (1/x, 1/x2, 1/y, 1/y2, 1/√x, and 1/√y). The calibration model with the lowest %RE and that showed no trend in the %RE plot was chosen. To test the selected models for goodness of fit, the %bias of the back-calculated concentrations of the calibrators from their nominal value was determined. The %bias should be less than 15% (and 20% for the LLOQ) for 75% of all calibrators to accept the chosen model [14, 15].
The accuracy of the methods was assessed by calculating the percent difference from the nominal concentration (%bias). The intra-day and total assay precision (%RSD) was calculated using single-factor analysis of variance (ANOVA), according to the CLSI EP05-A3 guideline [16]. When the intra-day %RSD exceeded the inter-day %RSD, the inter-day %RSD was set to zero for the calculation of the total precision. All QC levels were prepared and analyzed in duplicate on three different days (n = 3 × 2). Accuracy and precision were considered acceptable if %RSD and %bias were less than 15% and ± 20% for the LLOQ [14, 15].
The determination of ME and recovery was based on the approach suggested by Matuszewski et al. [17]. For the evaluation of ME in blood VAMS samples, analyte-free blood from six individual donors was used (Hct values ranging from 0.43 to 0.47). Extracts (i.e., the 250-μL supernatants mentioned in the “Sample preparation” section) prepared from blank blood were spiked at concentrations corresponding to low QC and high QC level in blood (B). Neat solvent (methanol-water-FA, 20:80:0.01, v/v/v) was spiked at the same concentration levels (A). Absolute ME was calculated by dividing the peak area of (B) by the peak area obtained from (A), multiplied with 100% (n = 6). Internal standard-compensated ME was calculated similarly by taking into account the IS peak area for (A) and (B). The relative ME was determined by calculation of the %RSD and should not exceed 15%. For the evaluation of recovery, water and blood with a medium (0.40) Hct level were spiked at low QC and high QC level before (C) and after (B) extraction. Recovery was determined by dividing the peak area ratio of (C) by the peak area ratio of (B) multiplied with 100% (n = 6). While no acceptance criteria were set for the recovery, %RSD must be less than 15% [14]. To investigate the effect of the Hct level of a sample on the recovery, blood VAMS samples with low (0.21), intermediate (0.30 and 0.50), and high (0.62) Hct levels were also prepared (n = 6). The mean recovery of the VAMS samples with a Hct of 0.40 was normalized to 100%, and a relative recovery was calculated for all Hct levels by dividing the mean calculated concentration by the mean calculated concentration at Hct 0.40. For the CSF VAMS samples, ME was assessed via an evaluation of the IS peak area in 55 patient samples, compared to the IS peak area of calibrators and QCs (n = 16).
Short- and long-term stability of VAMS was assessed by analyzing VAMS samples spiked at low QC and high QC levels prepared in blood and water (for CSF) at time point zero (t0) and after 1 week at 50 °C, 9 months at 4 °C, 8 and 9 months at room temperature (RT) for blood and aqueous VAMS samples respectively, 9 months at − 20 °C, and 9 months at − 80 °C (n = 3; duplicate measurements). VAMS samples were analyzed against a freshly prepared calibration curve. The mean concentration of the QC samples at a given storage condition should be within ± 15% from the concentration at t0 [14, 15]. Furthermore, the stability of processed samples in the autosampler (held at 4 °C) for 1 week, after two freeze-thaw cycles and after 1 week at − 20 °C was determined (n = 3; duplicate measurements).
The calibration range encompasses the therapeutic range in blood, but higher blood concentrations can be found in patients with a paracetamol intoxication [18]. We therefore investigated whether final extracts of samples above the upper limit of quantification (ULOQ) can be safely diluted. Blank whole blood and water were spiked with paracetamol at concentrations above the ULOQ (150 μg/mL and 300 μg/mL for blood and 60 μg/mL and 125 μg/mL for water); VAMS samples were made and processed as described above. The final extracts were diluted 2-fold and 5-fold with water (0.01% FA) (n = 6). With these dilution factors, the peak area of the IS was still high enough to be integrated accurately. The peak area of the IS was multiplied with the dilution factor and used to determine the ratio analyte area/IS area. This way, the concentration of the diluted extracts can be calculated via the calibration curve. Finally, the concentrations found were multiplied with the dilution factor. The maximum acceptable %bias and %RSD were 15%.
Statistical analyses were performed with GraphPad Prism version 7.02 and Microsoft Excel 2016, where necessary.
Application
For an objective evaluation of our method, we applied it on QCs based upon the external quality control (EQC) material. The available EQC material, Liquichek™ Therapeutic Drug Monitoring (TDM) Control (Bio-Rad, CA, USA), was serum-based. Hence, in order to somewhat mimic the matrices of interest as closely as possible that are to be analyzed with our method, blood QCs were prepared by replacing 250 μL of plasma (from a total blood volume of 1 mL) with 250 μL of the EQC material. For the generation of CSF QCs, the EQC material was diluted 1/10 with water. This procedure was carried out for all three available levels of EQC material, resulting in nominal concentrations of 4.0, 10.1, and 32.8 μg/mL and 1.6, 4.0, and 13.1 μg/mL for blood QCs and CSF QCs respectively. VAMS samples were made from these QCs and processed as described above.
As a proof of concept, blood and CSF VAMS samples from a patient admitted to the pediatric intensive care unit receiving intravenous paracetamol were analyzed. Five blood VAMS samples and five CSF VAMS samples were collected at consecutive time points. At the pediatric intensive care unit, a small amount of arterial blood and CSF (via an external ventricular drain) was collected, after which VAMS samples were immediately prepared on-site as described in the “Blood samples” and “CSF samples” sections. These samples were stored at room temperature for a maximum of 2 weeks, and then kept at − 80 °C until analysis. This study was approved by the Ethics Committee of Ghent University Hospital (B670201629325). Informed consent was provided by their parents or their legal representative.
Results and discussion
Sample preparation
As it is known that extraction of VAMS may be subject to Hct-dependent recovery issues [6, 9, 19], we evaluated three different extraction temperatures (22 °C, 37 °C, and 60 °C) by comparing peak area ratios of spiked VAMS samples (1.0 μg/mL) with intermediate (0.44) and high (target value 0.60) Hct (n = 3) levels. As depicted in Fig. 1, there were no major differences in recovery over the different conditions. Furthermore, the only statistically significant difference detected was between extraction at 22 and 60 °C for the intermediate Hct VAMS samples. Although statistically significant, this difference was limited (0.362 vs. 0.384), and therefore we opted for 22 °C as the extraction temperature.

Effect of the extraction temperature on the recovery of blood VAMS samples with intermediate (0.44) and high (target value 0.60) Hct levels. Extraction temperatures of 22 °C, 37 °C, and 60 °C were evaluated by comparison of peak area ratios. The mean peak area ratio and SD are shown (n = 3). Asterisk depicts significant statistical difference from extraction at 22 °C (p < 0.05, two-sided t test)
The choice of the extraction solvent (methanol-water-FA, 80:20:0.01, v/v/v) was based on a previously published method and was not further optimized since the desired sensitivity was reached with this procedure [9]. Furthermore, the %RSD in the experiment described above was below 8% for all conditions tested, demonstrating the reproducibility of the extraction. The same extraction conditions for blood VAMS samples were also applied for the analysis of CSF VAMS samples.
Method validation
The first step in order to validate the developed method was the evaluation of water as a surrogate matrix for blank CSF, since it was not possible to obtain the latter in sufficient quantities. To this end, we compared the results obtained from neat (undiluted) CSF from paracetamol-treated patients with those from CSF that had been diluted 10-fold in water, both against a calibration line set-up in water. All the diluted and undiluted samples fell within the calibration range. This approach is based on the publication of Matuszewski about the measure of relative ME via standard line slopes [20]. After correction for the dilution, the mean difference in concentrations found for diluted and undiluted samples was 3.33%, with a %RSD of 7.71%. These results were well within the predefined acceptance criteria (± 10% bias and < 15% RSD), which were based on the EMA criteria for the assessment of ME [14]. Although this approach does not give absolute values for ME and recovery in both matrices (i.e., CSF VAMS samples and aqueous VAMS samples), the equality of the process efficiency (a combination of matrix effect and recovery) for aqueous and CSF VAMS samples analyzed with this method can be demonstrated based upon an experiment with real-life patient samples. As the values obtained for neat and diluted samples (after taking into account the 10-fold dilution factor) were within 13% and as no consistent bias was observed after the dilution of CSF in water, it can be concluded that VAMS devices absorb the same volume of water and CSF. Therefore, we can safely use water as an alternative matrix for the preparation of blanks, calibrators, and QCs for the bioanalytical validation of the method and for the analysis of patient CSF samples.
No carry-over or unacceptable interferences were observed for both blood and artificial CSF VAMS samples. Representative chromatograms of blank artificial CSF and blood VAMS samples and QC samples spiked at LLOQ level are shown in Fig. 2, together with a patient CSF VAMS sample and patient blood VAMS sample. The transition ratios of the patient samples (n = 55) were all between 97.9 and 103.4% of the average transition ratio of the calibrators and QCs (n = 16), indicating no interferences are present in these patient samples.

Chromatograms obtained with the analysis of a blank blood VAMS sample (a), blank artificial CSF VAMS sample (b), spiked blood VAMS sample at LLOQ level (0.10 μg/mL) (c), spiked aqueous VAMS sample at LLOQ level (0.10 μg/mL) (d), patient blood VAMS sample (e), and patient CSF VAMS sample (f). Two transitions for paracetamol (APAP 1, blue line and APAP 2, red line) and two transitions for the IS (APAP-D4 1, green line and APAP-D4 2, gray line) are shown in each chromatogram
Paracetamol and its sulfate and glucuronide metabolites are chromatographically separated (respective retention times 0.93 min, 0.70 min, and 0.60 min; ESM Fig. S2), which is necessary to eliminate interferences from these metabolites by in-source formation. The stability of paracetamol sulfate and paracetamol glucuronide in dried VAMS samples made from spiked rat blood has been evaluated previously by Denniff et al., and no response at the paracetamol retention time was observed after 72 h of drying [21]. Furthermore, Li et al. determined paracetamol, paracetamol sulfate, and paracetamol glucuronide in DBS generated from spiked human blood [22]. Also there, no issues were seen with the stability of the sulfate and glucuronide metabolite after a longer storage period (27 days; RT). The same stability can be assumed for these compounds in human blood VAMS samples and CSF VAMS samples and was therefore not further investigated in this validation process.
The calibration data for blood and CSF VAMS samples were heteroscedastic. For the quantification of paracetamol in VAMS devices made from blood samples, a quadratic regression model with 1/x2 weighing gave the lowest %RE. Linear regression with 1/x2 weighing was the second best option; however, in the %RE plot, a trend was seen. Therefore, the quadratic regression model was preferred. For the CSF VAMS samples, a linear model with a weighing factor 1/x2 was the best fitting model based upon back-calculation of the concentrations of the calibrators. The selected regression models met the pre-set acceptance criteria [14, 15]. The calibration range chosen for both the CSF VAMS method and the blood VAMS method includes sub-therapeutic, therapeutic, and supra-therapeutic concentrations (in blood). Hence, these methods cannot only be used for the analysis of samples from patients receiving therapeutic concentrations of paracetamol, but also for overdose cases and for PK or TK studies.
The accuracy, intra-day precision, and total precision for QCs, analyzed in duplicate on three different days, fulfilled the predefined criteria (%bias and %RSD < 15%, 20% for LLOQ), as shown in Table 1.
The absolute ME and IS-corrected ME for blood VAMS samples approximated 100% for both the low QC and high QC level, indicating no significant enhancement or suppression of ionization. The relative ME fulfilled the predetermined acceptance criteria, with a maximum value of 3.8% (Table 2).
The experiment in which water was investigated as surrogate matrix for analyte-free CSF showed that the process efficiency of aqueous VAMS samples and CSF VAMS samples is equivalent. A matrix effect experiment with spiked water (as was done for blood) was not performed, since this does not provide any additional relevant information for the validation of this method. The evaluation of IS areas between patient samples and aqueous calibrators and QCs resulted in an overall %RSD of 7.3%, demonstrating that no significant ion suppression or ion enhancement was present in these patient samples. In order to conclude this, we assumed that the effects of interfering compounds on the ionization are the same for the analyte and the IS, which can be assumed as the internal standard is a stably isotope-labeled (D4) analogue of the analyte.
The absolute recovery has been determined for blood VAMS samples derived from blood with intermediate Hct (0.40) and water spiked at low QC and high QC level. Values (mean ± SD) obtained were 75 ± 9% for the low QC and 78 ± 4% for high QC blood VAMS samples and 70 ± 6% and 78 ± 3% for low and high QC aqueous VAMS samples respectively. For these calculations, it was not taken into account that only 250 μL out of 300 μL extraction solvent is used to proceed to the next step of the sample preparation. Therefore, the recovery value provides the relative amount of analyte recovered from the sample after the complete sample preparation process, and not solely the relative amount of analyte recovered from the VAMS sample during the extraction step. Hence, the maximum obtainable recovery is 83.3%. Statistical analysis via one-way ANOVA did not reveal any significant difference in recovery between the groups.
Hct-dependent recovery is a well-known issue when using VAMS as a sampling device [6, 9, 19]. Furthermore, it is known that a high recovery minimizes the chance of a Hct-dependent recovery. In the experiments discussed by Abu-Rabie et al., paracetamol was even used as a case example of a compound that suffers from Hct-dependent recovery when using DBS as a sampling technique [23]. Moreover, as the IS is added in the extraction solvent, it cannot compensate for variations in recovery. We therefore evaluated the impact of the Hct on the recovery in more detail. Recoveries for VAMS samples derived from spiked blood with Hct values ranging from 0.21 to 0.62 were determined and compared to the recovery of blood samples with Hct 0.40. All samples were analyzed against a calibration curve with VAMS samples derived from spiked blood with Hct 0.40.
Even though we noticed a trend towards decreasing recovery with increasing Hct values, only the recovery of the low QC level VAMS samples with Hct 0.62 differed more than 15% (15.8%) from the recovery at Hct 0.40 (Fig. 3). The difference is statistically significant (p = 0.0053) but the result is only just outside of the acceptance limit. These values are comparable with the results from Denniff et al. for the analysis of paracetamol in rat blood VAMS samples [21]. Therefore, we conclude that there is an impact of the Hct value on the recovery, but overall this impact is limited and will not significantly affect the analytical result. If possible, adjustment of the Hct value of the blood used to prepare calibrators to the median expected Hct of the patient population is recommended. This way, the influence of the Hct on recovery will be reduced as much as possible.

Impact of the Hct on recovery. VAMS samples (low QC and high QC level) were prepared from spiked blood with five different Hct values (target values 0.21, 0.30, 0.40, 0.50, and 0.62). The mean IS-corrected recovery of samples with Hct 0.40 is normalized to 100%. Mean (%) ± SD is shown. The dashed lines represent the ± 15% acceptance limits. Asterisks depict statistically significant difference from Hct 0.40 (p < 0.05, two-sided t test)
The stability in blood VAMS samples was found to be within the acceptance criteria after storage at 50 °C for 1 week; − 80 °C, − 20 °C, and 4 °C for 9 months; and RT for at least 8 months. The stability in aqueous VAMS samples was within ± 15% limits after storage at 4 °C, − 20 °C, and − 80 °C for 9 months. The stability at RT was within the acceptance criteria for aqueous VAMS samples stored up to 1 month. The results are summarized in Table 3. Overall, for blood VAMS, the data do not indicate any issue with stability, which is in line with earlier reports [21, 22]. In contrast, readily after 4 days, concentrations determined in aqueous VAMS samples stored at 50 °C had considerably decreased (30% and 32% of the concentrations measured at t0 for the low QC and high QC respectively) and these values decreased even further after storage for 7 days (Table 3). Furthermore, also the aqueous VAMS samples stored for 9 months at RT showed a notable decrease in measured concentrations (56% and 57% of the concentrations measured at t0 for the low QC and high QC respectively). As there is no reason to believe that there would be an issue with paracetamol stability as such, as demonstrated by the results from the blood VAMS samples, this apparent instability is most likely attributable to a recovery issue. A similar phenomenon was also described by Xie et al., where optimization of the extraction procedure led to an increased recovery and thereby elimination of an extractability-mediated recovery bias [24]. Our results demonstrate that it is indeed highly important to carefully evaluate the recovery, after different (long-term) storage conditions. Furthermore, these results indicate that an extractability-mediated recovery bias can be matrix-dependent. The underlying cause for this unexpected matrix-dependent finding is unknown, but might be related to an “aging-phenomenon” of VAMS. Indeed, it is known that, upon aging of unused VAMS, the speed at which a biofluid is absorbed decreases. The process which takes place upon storage of the aqueous matrix-filled VAMS might be similar to changes taking place in unused devices. However, further research is required to reveal the exact cause of this unexpected decreased extractability. To the best of our knowledge, we are the first to report on a matrix-dependent “aging-phenomenon” in filled VAMS. Although these observations do demand caution, the extreme storage conditions evaluated here are not relevant for our applications, in which patient CSF VAMS samples are collected in a hospital setting where they are stored at room temperature (for a maximum of 2 weeks), after which they are transported to the lab for further storage at − 80 °C. Three sets of processed sample extracts from blood and aqueous VAMS samples (n = 3; duplicate measurements), stored in the autosampler (4 °C) for 1 week, stored at − 20 °C for 1 week, and after 2 freeze-thaw cycles were re-analyzed. None of the conditions gave a decrease in concentration of more than 8%, with a maximum %RSD of less than 4% (see ESM Fig. S3).
Finally, a dilution integrity experiment showed that VAMS blood and aqueous samples can be diluted 2-fold as well as 5-fold without compromising the accuracy and precision, as is shown in Table 4. A maximum %bias of 7.8% and maximum %RSD of 5.9% was found. The highest concentration of samples that can be quantified is thereby increased to 500 μg/mL for blood VAMS samples and 200 μg/mL for CSF VAMS samples, which is sufficient for the purpose of this method.
Application
Since neither blood- nor CSF-based EQC material was available, serum-based EQC material (Liquichek™ Therapeutic Drug Monitoring (TDM) Control level 1, level 2, and level 3) was used to prepare blood and aqueous QC material and VAMS samples derived thereof. The calculated concentrations were compared to the nominal value and relative values are depicted in Fig. 4.

Analysis of QC samples derived from external quality control material. The obtained concentrations are depicted as percentage of the nominal value. Individual values (open squares and open circles) and mean values (black squares) are presented for each level of blood VAMS samples and aqueous VAMS samples (n = 6). The solid line represents the 100% target value. The dashed lines represent the ± 20% acceptance criteria
For the blood VAMS samples, all means and individual sample values were within the ± 20% acceptance interval. Although for the aqueous VAMS samples 4/18 individual values had a %bias outside of the acceptance interval (with a maximum %bias of 26%), all means were within the ± 20% acceptance criterion. The concentration range of these QCs (4.0–32.8 μg/mL in blood and 1.6–13.1 μg/mL in water) covers therapeutically relevant concentrations. Therefore, this experiment, using lab-prepared QCs, starting from EQCs, supports the validity of our method.
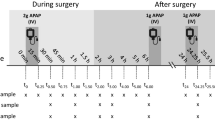
To have a proof of concept that the analysis of real-life patient samples does not pose any unexpected issues, five blood VAMS samples and five CSF VAMS samples were analyzed. The samples were collected from a patient receiving treatment with intravenous paracetamol. The results, displayed in Fig. 5, show that the concentration range of the method was suitable and all samples could be quantified.

Analysis of patient CSF VAMS samples (n = 5; dashed black line) and blood VAMS samples (n = 5; solid gray line) with the measured paracetamol concentrations shown over time. The shaded area represents the period during which paracetamol was intravenously administered to the patient
One point of attention is that a visual evaluation of VAMS sample quality (e.g., under- and overfilling) is difficult for a non-colored matrix like CSF. Therefore, an appropriate training of the staff responsible for the collection of study samples is highly recommended.
Conclusion
The use of microsampling strategies for the collection of patient samples is increasingly receiving interest, as these offer many advantages over traditional sampling. To date, the use of these microsampling strategies for the collection of CSF has remained limited. However, this approach can facilitate the implementation of drug PK and TK studies in CSF.
In this study, LC-MS/MS methods were developed and validated for the quantitative analysis of paracetamol in blood and CSF VAMS samples. The methods fulfilled the pre-set validation acceptance criteria and their application on EQC material and patient samples demonstrated their validity. For the analysis of blood VAMS samples, special attention was paid to the effect of the Hct on the recovery, which appeared to be limited. Furthermore, our study revealed that an extractability-mediated recovery bias can be matrix dependent and that long-term storage and/or storage at elevated temperature should be carefully evaluated when developing methods utilizing VAMS for sampling.
The most important advantage of using VAMS for the collection of CSF is the increased number of samples that can be collected from a single patient without increased patient burden. As a next step, these methods will be used to measure paracetamol concentrations in blood and CSF VAMS samples from a larger number of pediatric patients.
Abbreviations
- ANOVA:
-
Analysis of variance
- CSF:
-
Cerebrospinal fluid
- DMS:
-
Dried matrix spot
- EMA:
-
European Medicines Agency
- EQC:
-
External quality control
- ESI:
-
Electrospray ionization
- FA:
-
Formic acid
- FDA:
-
US Food and Drug Administration
- Hct:
-
Hematocrit
- IS:
-
Internal standard
- LC:
-
Liquid chromatography
- LLOQ:
-
Lower limit of quantification
- ME:
-
Matrix effect
- MRM:
-
Multiple reaction monitoring
- MS/MS:
-
Tandem mass spectrometry
- QC:
-
Quality control
- RSD:
-
Relative standard deviation
- ULOQ:
-
Upper limit of quantification
- VAMS:
-
Volumetric absorptive microsampling
References
Edelbroek PM, van der Heijden J, Stolk LML. Dried blood spot methods in therapeutic drug monitoring: methods, assays, and pitfalls. Ther Drug Monit. 2009;31(3):327–36.
Stove CP, Ingels AS, De Kesel PM, Lambert WE. Dried blood spots in toxicology: from the cradle to the grave? Crit Rev Toxicol. 2012;42(3):230–43.
De Kesel PM, Sadones N, Capiau S, Lambert WE, Stove CP. Hemato-critical issues in quantitative analysis of dried blood spots: challenges and solutions. Bioanalysis. 2013;5(16):2023–41.
Wilhelm AJ, den Burger JC, Swart EL. Therapeutic drug monitoring by dried blood spot: progress to date and future directions. Clin Pharmacokinet. 2014;53(11):961–73.
Velghe S, Capiau S, Stove CP. Opening the toolbox of alternative sampling strategies in clinical routine: a key-role for (LC-)MS/MS. TrAC Trends Anal Chem. 2016;84:61–73.
Kok MGM, Fillet M. Volumetric absorptive microsampling: current advances and applications. J Pharm Biomed Anal. 2018;147:288–96.
Denniff P, Spooner N. Volumetric absorptive microsampling: a dried sample collection technique for quantitative bioanalysis. Anal Chem. 2014;86(16):8489–95.
Spooner N, Denniff P, Michielsen L, De Vries R, Ji QC, Arnold ME, et al. A device for dried blood microsampling in quantitative bioanalysis: overcoming the issues associated blood hematocrit. Bioanalysis. 2015;7(6):653–9.
De Kesel PM, Lambert WE, Stove CP. Does volumetric absorptive microsampling eliminate the hematocrit bias for caffeine and paraxanthine in dried blood samples? A comparative study. Anal Chim Acta. 2015;881:65–73.
Zimmer JSD, Christianson CD, Johnson CJL, Needham SR. Recent advances in the bioanalytical applications of dried matrix spotting for the analysis of drugs and their metabolites. Bioanalysis. 2013;5(20):2581–8.
Hirtz C, Lehmann S. What is the potential of dried matrix spot sampling for cerebrospinal fluid analysis? Bioanalysis. 2015;7(22):2849–51.
Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, Leone S. Paracetamol: new vistas of an old drug. Cns Drug Rev. 2006;12(3–4):250–75.
Sharma CV, Long JH, Shah S, Rahman J, Perrett D, Ayoub SS, et al. First evidence of the conversion of paracetamol to AM404 in human cerebrospinal fluid. J Pain Res. 2017;10:2703–9.
European Medicines Agency. Guideline on bioanalytical method validation 2011 [Available from: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf.
U.S. Department of Health and Human Services Food and Drug Administration. Bioanalytical method validation. Guidance for industry 2018 [Available from: https://www.fda.gov/downloads/drugs/guidances/ucm070107.Pdf.
Clinical and Laboratory Standards Institute. CLSI document EP05-A3: evaluation of precision of quantitative measurement procedures; approved guideline—Third Edition 2014.
Matuszewski BK, Constanzer ML, Chavez-Eng CM. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal Chem. 2003;75(13):3019–30.
Winek CL, Wahba WW, Winek CLJ, Balzer TW. Drug and chemical blood-level data 2001. Forensic Sci Int. 2001;122(2–3):107–23.
Velghe S, Stove CP. Volumetric absorptive microsampling as an alternative tool for therapeutic drug monitoring of first-generation anti-epileptic drugs. Anal Bioanal Chem. 2018;410(9):2331–41.
Matuszewski BK. Standard line slopes as a measure of a relative matrix effect in quantitative HPLC-MS bioanalysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2006;830(2):293–300.
Denniff P, Parry S, Dopson W, Spooner N. Quantitative bioanalysis of paracetamol in rats using volumetric absorptive microsampling (VAMS). J Pharm Biomed Anal. 2015;108:61–9.
Li W, Doherty JP, Kulmatycki K, Smith HT, Tse FL. Simultaneous LC-MS/MS quantitation of acetaminophen and its glucuronide and sulfate metabolites in human dried blood spot samples collected by subjects in a pilot clinical study. Bioanalysis. 2012;4(12):1429–43.
Abu-Rabie P, Denniff P, Spooner N, Chowdhry BZ, Pullen FS. Investigation of different approaches to incorporating internal standard in DBS quantitative bioanalytical workflows and their effect on nullifying hematocrit-based assay bias. Anal Chem. 2015;87(9):4996–5003.
Xie I, Xu Y, Anderson M, Wang M, Xue L, Breidinger S, et al. Extractability-mediated stability bias and hematocrit impact: high extraction recovery is critical to feasibility of volumetric adsorptive microsampling (VAMS) in regulated bioanalysis. J Pharm Biomed Anal. 2018;156:58–66.
Acknowledgements
The authors wish to acknowledge Prof. Veronique Stove and her team for assistance with blood collection and hematocrit measurements and all volunteers who participated in the study.
Funding
This study was supported by the Research Foundation – Flanders (G0E010916N).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Approval for the clinical proof-of-concept part of this study was provided by the Ethics Committee of Ghent University Hospital (B670201629325). Informed consent has been obtained from the participants involved or their legal representatives.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(pdf 671 kb)
Rights and permissions
About this article

Cite this article
Delahaye, L., Dhont, E., De Cock, P. et al. Volumetric absorptive microsampling as an alternative sampling strategy for the determination of paracetamol in blood and cerebrospinal fluid. Anal Bioanal Chem 411, 181–191 (2019). https://doi.org/10.1007/s00216-018-1427-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00216-018-1427-6