
Abstract
To investigate the hydrogen evolution ability of the cluster ConMoS (n = 1–5), this paper based on density functional theory, using the B3LYP generalization and def2-TZVP basis set. In this paper, the cluster was structurally optimized and theoretically analyzed by the Gaussian09 package under multiple spin multiplexes. The optimized conformation of the cluster was mostly in stereo form; in the first step of the hydrogen evolution reaction, the analysis of the cluster HOMO diagram and the water molecule LUMO diagram, the energy level difference, the d-band center, Gibbs free energy and adsorption energy showed that the electrons of configurations 5-a and 4-a were more likely to jump from the HOMO orbital to the water molecule LUMO orbital, and 5-a and 4-a showed excellent activity, they finally formed the ConMoS-Hads model; in the second step of the hydrogen evolution reaction, the Co atom were a potential active site, and analysis of energy level differences and bond level revealed that the configurations 5a-Hads and 4a-Hads showed good desorption capacity compared to the other configurations, and their adsorbed H atoms were more readily shed to complete the reaction with more rapid hydrogen evolution; in summary, configurations 5-a and 4-a were determined to be the favored configurations of the cluster with excellent catalytic activity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In this world, the search for clean energy sources has become crucial for the sustainable development of society, among which hydrogen energy, as a relatively ideal clean energy source, has received a lot of attention from researchers [1, 2]. At the same time, the type of catalyst adopted for efficient and green catalytic production has become a new issue. In recent years, transition metal-based materials have been preferred by researchers due to their abundant sources, low cost and excellent properties [3,4,5,6,7], and the Co-Mo-S system has shown good results as a catalyst for hydrogen evolution reactions in the applications. Li et al. [8] designed an open structure Co-Mo-S which showed high electron transfer efficiency and more active sites in the hydrogen evolution reaction, indicating that Co-Mo-S exhibited excellent hydrogen evolution ability. Zhou et al. [9] produced a synthesis of the Co-Mo-S system by a hydrothermal method and the study showed that the system has high catalytic hydrogen evolution activity and the excellent performance of the catalyst can be attributed to the electron pair interactions and larger surface area. Xu et al. [10] found that the Co-Mo-S material exhibited high catalytic hydrogen evolution activity and stability even after four cycles under light reaction, which indicated that this material has a promising development in the field of green energy. Fan et al. [11] argued that the formation of the Co-Mo-S phase in the composite led to its high catalytic activity in the chemical reaction of hydrogen release. Wang et al. [12] had synthesized a three-dimensional mesh Co-Mo-S material, and they had evaluated the material using electrochemical techniques and found that the material exhibited excellent activity and stability for hydrogen evolution reactions due to its abundant catalytic active sites and fluent electron transport pathway. Lu et al. [13] constructed Co-Mo-S nanomaterials, which were tested to have excellent hydrogen evolution properties derived from the abundance of catalytic active sites, efficient electron transfer and MoS2 edge distribution of the Co promoter. The Co-Mo-S nanomaterials produced by Li et al. [14] demonstrated high activity and persistence in hydrogen evolution experiments due to electronic interactions between Mo and CoS2, abundant active reaction sites and good electrical conductivity, and the study provides a good direction for research in the field of sustainable energy. Wang et al. [15] found that the large CoS2 exchange current density combined with the high MoS2 electrochemical bilayer capacitance combined to enhance the hydrogen evolution activity of Co-Mo-S catalysts. Experiments by Ren et al. [16] showed that Co-Mo-S exhibited significant activity, excellent electrochemical durability and near 100% faraday efficiency when used as a hydrogen evolution electrode. Liu et al. [17] demonstrated that Co-Mo-S can rapidly transfer the trapped electrons to the active site, effectively improving the hydrogen evolution efficiency. Huazhong University of Science and Technology et al. [18] prepared CoMoS nanomaterials by a solvothermal technique, which exhibited good catalytic properties for hydrogen evolution in the alkaline electrolyte, and it was found that the electron transfer from Co to MoS2 and the solvothermal treatment time had significant effects on the catalytic activity. The Co-Mo-S hydrogen evolution catalysts fabricated by China University of Petroleum [19] using low-temperature sulfidation method have good performance and stability. Guo et al. [20] prepared Co-Mo-S nanomaterials with high catalytic activity, low overpotential and good stability in the hydrogen evolution reaction.
The macroscopic experimental studies of Co-Mo-S as a catalyst for the hydrogen resolution reaction of water are abundant, but the microscopic theoretical research is lacking, resulting that it is difficult to fully understand the microscopic mechanism of Co-Mo-S catalysts in the hydrogen evolution reaction. Therefore, in order to gain insight into the micromechanics and properties of the Co-Mo-S system involved in hydrogen evolution reactions, the cluster ConMoS (1–5) was designed based on the literature [14] as the object of research. In this paper, the hydrogen evolution properties of this cluster are investigated at the microscopic level based on density functional theory [21, 22], with a view to explaining its excellent hydrogen evolution ability from a theoretical perspective and providing a theoretical basis for subsequent studies on the influence of more Co atoms on hydrogen evolution activity.
2 Theoretical foundation
2.1 Calculation method
This paper also used the B3LYP function because of its excellent integration performance and the fact that it is still the default function for many researchers in the transition metal system. The subject of this paper was the Co-Mo-S system, which contains elements after the fifth period (Mo), and relativistic effects in theoretical calculations could affect the reliability of the results. The 3-zeta basis set def2-TZVP, which contains a pseudopotential and has a high accuracy, was selected for this paper. This basis set was calculated using the stuttgart small nucleus pseudopotential for the fifth period and beyond, and the all-electron basis set for the first four periods, which was perfectly adequate for the system under study. Luo et al. [23,24,25] used B3LYP/def2-TZVP to investigate the properties of transition metal systems, and their results also confirmed the accuracy and reliability of the density functional and basis set. Meanwhile, in this paper, the theoretical bond length of the Co-Mo-S system (Co-S: 0.227 nm, Mo-S: 0.240 nm, Co-Mo: 0.281 nm) was found to be similar to the experimental value [26] (Co-S: 0.221 nm, Mo-S: 0.241 nm, Co-Mo: 0.285 nm) after using this density functional and basis set, and the theoretical bond length of the Co-Mo system (Co-Mo: 0.248 nm) was also calculated to be in perfect agreement with the experimental value [27] (Co-Mo: 0.248 nm). For the initial configuration of a system with a small number of atoms, it is generally obtained by designing a large number of structures. For example, Juárez-Sánchez et al. [28, 29] calculated (CuS)N (N = 1–6) and VFn (n = 1–7) When the initial structure is obtained, the above method is adopted.
In this paper, we designed as abundant as possible clusters of ConMoS (n = 1–5) with multiple initial configurations of different spin multiplicity and different morphologies at the B3LYP/def2-TZVP quantum chemistry level, based on density functional theory. This paper also adopted the Gaussian09 package and the Multiwfn package [30] for full frequency validation and geometric configuration optimization and data machining. The optimized results were obtained by excluding unstable conformations that contained virtual frequencies and morphologically similar conformations, resulting in 21 optimized stable conformations. The optimization conditions for convergence to be satisfied are: maximum force < 0.00045, root mean square force < 0.00030, maximum displacement < 0.00180 and root mean square displacement < 0.00120. All the above calculations and data machining were done on the computer HP-Z440.
2.2 Mechanism of hydrogen evolution reaction
The research in this paper was performed under neutral condition. In this experiment, the cluster ConMoS (n = 1–5) (hereinafter indicated by M) was used as the theoretical object of the hydrogen evolution reaction, and the mechanism of this cluster when it occurs in catalytic hydrogen evolution is known according to the literature [14]. The reaction was mainly divided into the following two steps, where Hads represent the hydrogen atoms adsorbed to the cluster. The interatomic force analysis of the M-H structure resulting from the Volmer reaction showed that the Chemical Bond interaction between the H atom and the attached metal atom is dominant. Therefore, the theoretical analysis could be mostly satisfied without considering the van der Waals correction.
Step 1
Step 2 There are two reaction pathways in this reaction step, as follows:
3 Results and discussion
3.1 Stable configuration of cluster ConMoS (n = 1–5)
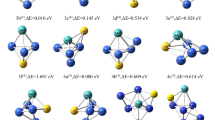
The final 21 stable conformations of the cluster ConMoS (n = 1–5) are obtained, and the geometrical conformational morphology and relative energies are shown in Fig. 1. In Fig. 1, "n-a" show the most stable configurations of the cluster ConMoS for n = 1–5, and the energy of the most stable configuration is set to 0 eV. The energy of “n-a” is used as a reference standard to calculate the relative energy of "n − m" (m = b, c, d, e, f) and rank the cluster configurations according to the lowest to highest energy. The number in brackets at the top of the “n − m” configuration indicate the spin multiplicity.

Optimized stable configuration of cluster ConMoS (n = 1–5)
Figure 1 shows that all the configurations in cluster Co1MoS are planar triangles; cluster Co2MoS contains planar and triangular cone shapes; cluster Co3MoS has a tetragonal cone shape for configuration 3-a, five configurations from 3-b to 3-f are all triangular biconical, and configurations 3-b and 3-c, and configurations 3-d and 3-f have the same geometry, but have different spin multiplicity; the cluster Co4MoS contains five stable configurations, all of which are single-capped triangular bipyramidal, with mirror-symmetric configurations 4-d1 and 4-d2; the cluster Co5MoS contains six double-capped triangular bipyramidal configurations, of which configurations 5-c and 5-d are identical in terms of the relative positions of the atoms at different spin multiplets.
In this paper, the most and second most stable configurations of each size cluster are selected for theoretical analysis of the hydrogen evolution properties.
3.2 Thermodynamic stability of the cluster ConMoS (n = 1–5)
The correction energy, Gibbs free energy, binding energy, and Gibbs free energy variation of the optimized configuration of the cluster ConMoS are listed in Table 1. The smaller the value of the correction energy and the Gibbs free energy variation, the higher the stability, while the reverse is achieved for the binding energy, the larger the value, the more stable the conformation, that is the tighter the bonding between the atoms. The binding energy (EBE) and Gibbs free energy variation (ΔG) were calculated as follows.
From Table 1, the corrected and Gibbs free energies for configurations n-a (n = 1–5) to n-b increase gradually and the binding energy decreases for the same size cluster, indicating that configurations n-a are the most stable configurations in each size cluster.
4 4 Analysis of the hydrogen evolution properties of the cluster ConMoS(n = 1–5)
4.1 Study of hydrogen atom adsorption properties of clusters
4.1.1 Analysis of cluster ConMoS HOMO diagrams and LUMO diagrams of water molecules
From frontline orbital theory it can be seen that the highest energy occupied orbital HOMO and the lowest energy unoccupied orbital LUMO determine the state of a chemical reaction. When the cluster ConMoS reacts with a water molecule, microscopically it is the electrons that leap from the HOMO orbital of the cluster to the LUMO orbital of the water molecule, forming the M-Hads structural model and thus the first step in the hydrogen evolution reaction. Figure 2 shows the HOMO orbital diagrams for the alpha and beta electrons of the cluster ConMoS optimized configuration and the LUMO orbital diagram for the water molecule. Conformation 2-b is a single multiplet state and has only one class of HOMO diagrams due to its closed shell. The graph shows the positive phase of the orbital wave function in lighter shades and the negative phase of the orbital wave function in darker shades, with the area covered by the two representing the region where the cluster is active during hydrogen evolution reactions. The HOMO orbital of the cluster is phased with the LUMO orbital of the water molecule, which makes it easier for the electrons to complete the leap and for the hydrogen evolution reaction to proceed. It is easy to see from Fig. 2 that the LUMO orbital of the water molecule is surrounded by dark shades, indicating that the water molecule is more likely to react with the negative phase of the cluster. The negative phase morphology of the cluster of optimized ConMoS conformations varies in size, indicating that each conformation has a different degree of activity. The dark coverage of the α-HOMO diagrams for configurations 1-a, 1-b, 4-b and 5-a is larger than that of the respective β-HOMO diagrams from Fig. 2, indicating that α-HOMO plays a major role in the reaction of the above configurations with water molecules; configuration 2-a, 3-a, 3-b, 4-a and 5-b are the opposite, with more dark parts of the β-HOMO diagram than the α-HOMO diagram, meaning that when these configurations interact with water molecules, electrons tend to flow more from the β-HOMO to the LUMO of the water molecule. Overall, the α-HOMO of configurations 4-b and 5-a and the β-HOMO of configurations 3-b, 4-a and 5-b are more active in the hydrogen evolution reaction with water molecules.

HOMO diagram of the optimized configuration of cluster ConMoS and LUMO diagram of water molecule
4.1.2 Orbital energy level differences between the cluster ConMoS and water molecules
The energy level difference can be used to further determine the activity of the hydrogen evolution reaction between molecules. According to the frontier orbital theory, the smaller the energy level difference between the reactants and the energy level difference between the HOMO orbital of the reactant and the LUMO orbital of the water molecule is less than 6.004 eV (about 579 kJ/mol) [31], the easier the reaction will be. The results of the energy level difference calculations for the optimized conformations of the cluster ConMoS are shown in Fig. 3. As is seen from Fig. 3, the energy level differences of the optimized conformations of the cluster ConMoS are all less than 6.004 eV, indicating that the reaction between each conformation and the water molecule is possible. The energy level difference of configuration 5-a (4.932 eV) is the smallest of all the optimized configurations, indicating that configuration 5-a is more likely to react with water molecules and adsorb hydrogen atoms than other configurations, completing the electron transfer more quickly and leading to the first step of the hydrogen evolution reaction. The energy level difference gives the following reaction activity for each optimized configuration: 5-a > 3-b > 2-b > 4-b > 4-a > 5-b > 3-a > 1-b > 1-a > 2-a.

Frontier orbital energy levels for the reaction of clustered ConMoS with H2O
4.1.3 Cluster ConMoS d-band center
The d-band center model enables the description and prediction of the extent of the activity capacity of a material surface. The d-band center is defined essentially as the difference between the central position of the fractional density of states corresponding to the d-state of the material surface and the Fermi energy level. The theory bases on the fact that the d-band of a substance mixes with the orbitals of small molecule reactants when adsorption occurs on the surface of the substance with other small molecule reactants, when the bonding and anti-bonding states appear. The higher the d-band energy of a substance relative to the Fermi energy level, the higher the energy of the antibonding state relative to the Fermi energy level, which means that it will be less occupied, and thus the bonding capacity will be weakened, and the stronger the bonding between the substance and the small molecule reactant will be when it adsorbs. The d-band center energy values of the cluster ConMoS are calculated by the wave function software Multiwfn and plotted in Fig. 4. In general, the lower the d-band center value of the cluster, the stronger its ability to adsorb hydrogen atoms. The lowest d-band center value (17.465 eV) for configuration 5-a, indicating that configuration 5-a contains the most superior ability to bind hydrogen atoms compared to the others, which is consistently with the conclusions obtained from the analysis of energy level differences. The relationship between the activity intensity of the clusters when adsorbed with hydrogen atoms is introduced by the d-band center: 5-a > 5-b > 4-a > 3-a > 4-b > 2-a > 3-b > 2-b > 1-b > 1-a.

d-band center of cluster ConMoS optimized configuration
4.1.4 Cluster Gibbs free energy and adsorption energy
To further evaluate the catalytic activity of the adsorbed hydrogen in the Volmer reaction for each configuration, we introduced the Gibbs free energy and the adsorption energy. The Gibbs free energy and adsorption energy of hydrogen adsorbed in conformation 1-b were not discussed because the M-H structure resulting from conformation 1-b does not exist stably. The energy calculation equations were as follows.
ΔGH* represented the Gibbs free energy change of the adsorbed hydrogen and Ead represented the adsorption energy. The nearer the Gibbs free energy (|ΔGH*|) was to 0 eV, the better the catalytic activity of the catalyst [32,33,34].
It is easily seen from Fig. 5a that the Gibbs free energy of configuration 1a was closest to 0, indicating that it had a high catalytic capacity compared to the other configurations. The catalytic ability of configuration 2b was weaker than that of the other configurations. The relationship based on the Gibbs free energy gave the ability to catalyze activity as follows: 1-a > 3-b > 2-a > 3-a > 5-a > 5-b > 4-a > 4-b > 2-b.

Energy variation of adsorbed hydrogen in the optimized configuration of the cluster ConMoS
The change of the adsorption energy of the cluster after the adsorption of hydrogen could be seen in Fig. 5b, where too large and too small values of the adsorption energy are not beneficial for the efficient conduct of the reaction. With too large a value, the H atoms were strongly combined with the clusters and it was more difficult for the H atoms to shed the clusters after the catalytic reaction occurred. The value was too small and again it was difficult to have an active effect. For this cluster, configurations 2-a, 3-a, 4-a, 4-b, 5-a, 5-b were more active than the other configurations.
In the first step, the water molecules first adsorbed onto the clusters, after which the clusters combined with H to form the M-H structure and OH− detachment occurred. The combined HOMO diagram, energy level difference, d-band center, Gibbs free energy and adsorption energy analysis of the cluster ConMoS optimized configuration indicate that the cluster had varying degrees of activity in each optimized configuration in the first step of the hydrogen evolution reaction. Conformation 5-a has better hydrogen evolution activity than the other conformations, meaning that electrons can jump from the HOMO orbital to the LUMO orbital of the water molecule more rapidly. The results of the above analysis are ranked, and the final conclusion was that the strength of the activity of each configuration in the first step of the hydrogen evolution reaction was: 5-a > 3-b > 5-b > 3-a≈4-a > 4-b > 2-a > 1-a≈2-b > 1-b.
4.2 Research of cluster (ConMoS)-Hads desorption process
This paper mainly discussed the Heyrovsky step, the Tafel step will be analyzed during the planned further research. After the reaction of the cluster ConMoS with water to complete the first step of the hydrogen evolution process, the (ConMoS)-Hads (M-Hads) model is formed and the results are shown in Fig. 6. In particular, the M-Hads conformation of conformation 1-b cannot be steadily present after verification by the Gaussian09 package. It is easy to see from the figure that the H atoms are all bound to the Co atoms, indicating that the Co atoms are potential catalytically active sites for this cluster.

(ConMoS)-H configuration diagram of the optimized configuration of cluster ConMoS
4.2.1 Cluster (ConMoS)-Hads and H2O frontline orbital energy level differences
To obtain more accurate information on the catalytic activity of M-Hads in the second step of the hydrogen evolution reaction (desorption reaction), the energy level difference is introduced for determination, and the details are shown in Fig. 7. The energy level difference of each M-Hads configuration of this cluster is less than 6.004 eV, and it can be concluded that each M-Hads structure is capable of reacting with water molecules. In particular, 5a-Hads has the smallest energy difference (5.339 eV) in the M-Hads structure, indicating that 5a-Hads is the most readily available for desorption reactions in which hydrogen atoms can be desorbed, which means that it is the most active. The relationship between the activity capacity of the M-Hads model for the second hydrogen evolution reaction, judging from the energy level difference, is: 5a-Hads > 4a-Hads > 3b-Hads > 3a-Hads > 5b-Hads > 4b-Hads > 2b-Hads > 2a-Hads > 1a-Hads.

The frontier orbital energy level difference of the reaction between M-Hads and H2O
4.2.2 Cluster (ConMoS)-Hads bond level
Figure 8 shows the bond level information for each of the M-Hads models. In this section the bond level between the Co atoms adsorbing H atoms and the H atoms are introduced to further determine the difficulty of the desorption reaction for each M-Hads model. The larger the bond level, the stronger the bond between the two atoms and the less likely to break; the smaller the bond level, the weaker the bond, resulting in the H atom falling off easily and the reaction being completed with the hydrogen evolution. The figure shows that 2a-Hads have the smallest bond level, which means that the adsorbed H atoms are easily shed and hydrogen precipitates more quickly. 1a-Hads has the largest bond level of all the M-Hads models, indicating that Co–H bonds are the most difficult to break and proceed more slowly than the rest of the configurations in the hydrogen evolution reaction. Based on bond levels, the M-Hads model for easy completion of hydrogen evolution reactions is ranked as follows: 2a-Hads > 4a-Hads > 2b-Hads > 4b-Hads > 5a-Hads > 5b-Hads > 3a-Hads > 3b-Hads > 1a-Hads.

Bond level of M-Hads optimized configurations
For the second step of the hydrogen evolution reaction, the analysis of the M-Hads model energy level difference and bonding level ranking judgment, M-Hads model and water molecule reactivity relationship is: 4a-Hads > 5a-Hads > 2a-Hads > 2b-Hads ≈ 4b-Hads > 3a-Hads ≈ 3b-Hads ≈ 5b-Hads > 1a-Hads.
In the first step of the reaction, both the energy level difference and the d-band center analysis showed that configuration 5-a exhibited the best hydrogen evolution activity. The conclusions from the analysis of the HOMO diagram and the adsorption energy also indicated that configuration 5-a also has a better activity. In the second step of the hydrogen evolution reaction, the conformation 5a-Hads contained the best activity capacity in the energy level difference analysis. In both the energy level difference and bond level analyses, the conformation 4a-Hads had the second best activity capacity. Considering the activity of each configuration in the two-step hydrogen evolution reaction, configurations 4-a and 5-a showed an excellent ability to complete the electron transfer and precipitate hydrogen more quickly in both steps.
5 Conclusion
In this paper, the hydrogen evolution properties of the most and least stable conformations of the cluster ConMoS(1–5) were analyzed at the theoretical level based on density functional theory with the support of Gaussian09 and Multiwfn software.
-
(1)
Cluster ConMoS (1–5) contained 21 stable configurations, mostly in stereospecific form.
-
(2)
hydrogen evolution reaction in the first step
This step involved the leap of electrons from the HOMO orbital of the cluster to the LUMO orbital of the water molecule, with the α-HOMO of configurations 1-a, 1-b, 4-b and 5-a being more efficient to interact with the water molecule and the β-HOMO of configurations 2-a, 3-a, 3-b, 4-a and 5-b being more active in the reaction. All configurations had an energy level difference of less than 6.004 eV, meaning that all were able to interact electronically with water normally, with configuration 5-a being the most active. Analysis of the d-band center revealed that configuration 5-a had the lowest value and its ability to adsorb hydrogen atoms was the strongest. Analysis of Gibbs free energy and adsorption energy resulted the activity of configurations 5-a and 4-a can't be neglected. Overall, the conformations 5-a and 4-a showed superior activity in the first step of the reaction compared to the other conformations.
-
(3)
hydrogen evolution reaction in the second step
After the reaction in the previous step, the M-Hads model was formed and the Co atom was found to be a potential active site for the cluster by calculation. From an energy level difference perspective, the M-Hads model still allowed the reaction to proceed smoothly and the conformation 5a-Hads had the smallest energy level difference and the highest activity capacity. The bond level analysis revealed that the conformation 2a-Hads had the smallest bond level and the H atom was easily shed. Overall, the configurations 4a-Hads and 5a-Hads showed good desorption in the second step of the reaction.
The combined analysis of the two steps showed that configurations 5-a and 4-a are the key configurations of the cluster involved in catalytic hydrogen evolution reactions. This conclusion will provide theoretical guidance for the macroscopic experimental study of the clusters as catalytic materials. Based on the present results, it was reasonable to suppose that when Co-M-S contains more Co atoms, its hydrogen precipitation activity may subsequently become excellent. Further research will be required to obtain the exact circumstances.
Data availability
If necessary, the data of this study can be provided to the corresponding author within a reasonable range.
Code availability
This article does not apply the term.
References
Norouzi N (2021) Assessment of technological path of hydrogen energy industry development: a review. Iran (Iranica) J Energy Environ 12:273–284
Falcone PM, Hiete M, Sapio A (2021) Hydrogen economy and sustainable development goals: review and policy insights. Curr Opin Green Sustain Chem 31:100506
Qin Y, Fang ZG, Zhang W, Li LH, Liao W (2020) The study on the catalytic properties of cluster Co3NiB in the hydrogen evolution reaction. J Jiangxi Norm Univ (Nat Sci Ed) 44:56–62
Qin Y, Fang ZG, Zhao LL, Liao W, Xu Y (2021) The study on the dynamics and thermodynamics of isomeric transformation of cluster Co3NiB2. J Jiangxi Norm Univ (Nat Scie Ed) 45:67–74
Zheng XX, Fang ZG, Qin Y, Hou QQ, Wu TH, Mao ZL (2021) Electronic Properties of Cluster Fe3Ni3. J Guizhou Univy (Nat Sci) 38:7–12
Fang ZG, Wang ZY, Zheng XX, Qin Y, Mao ZL, Zeng XY, Zhu YW, Wang Q (2022) Study on the dolarizabilitydipole momentand density of states of cluster Co3NiB. J Guizhou Univ (Nat Sci) 39:17–24
Hou QQ, Fang ZG, Qin Y, Zhu YW (2021) Study on the polarization of Fe4P clusters. J Guangxi Norm Univ (Nat Sci Ed) 39:140–146
Li Y, Hou XW, Gu J, Mikhaylova V, Chen KL, Zhang HM, Han SM (2021) Open and close-ended CoMoS3 nanotubes for hydrogen evolution in acidic and basic conditions. J Energy Chem 57:34–40
Zhou LJ, Han ZX, Li WX, Leng WX, Yu Z, Zhao Z (2020) Hierarchical Co-Mo-S nanoflowers as efficient electrocatalyst for hydrogen evolution reaction in neutral media. J Alloy Compd 844:156108
Xu J, Mao M, Yu H (2020) Functionalization of sheet structure Co-Mo-S with Ni(OH)2 for efficient photocatalytic hydrogen evolution. Res Chem Intermed 46:1823–1840
Fan JP, Ekspong J, Ashok A, Koroidov S, Gracia-Espino E (2020) Solid-state synthesis of few-layer cobalt-doped MoS2 with CoMoS phase on nitrogen-doped graphene driven by microwave irradiation for hydrogen electrocatalysis. RSC Adv 10:34323–34332
Wang S, Ge XB, Xiao JY, Huang LY, Liu J, Wu J, Yue WJ, Yang XH (2020) Amorphous CoMoSx/N-doped carbon hybrid with 3D networks as electrocatalysts for hydrogen evolution. Catal Lett 151:1720–1727
Lu YK, Guo XX, Yang LY, Yang WF, Sun WT, Tuo YX, Zhou Y, Wang ST, Pan Y, Yan WF, Sun D, Liu YQ (2020) Highly efficient CoMoS heterostructure derived from vertically anchored Co5Mo10 polyoxometalate for electrocatalytic overall water splitting. Chem Eng J 394:124849
Li P, Zhuang ZH, Du C, Xiang D, Zheng FQ, Zhang ZW, Fang ZY, Guo JH, Zhu SY, Chen W (2020) Insights into the mo-doping effect on the electrocatalytic performance of hierarchical CoxMoyS nanosheet arrays for hydrogen generation and urea oxidation. ACS Appl Mater Interfaces 12:40194–40203
Wang B, Liu YH, Hao JH, Zhong JB, Yu FR, Zhang KW, Shen H, Mao BD (2018) Synergetic optimization via composition-dependent nanostructuring in Co-Mo-S Electrocatalysts for efficient hydrogen evolution in alkaline solution. Int J Electrochem Sci 13:3501–3515
Ren X, Wu D, Ge RX, Sun X, Ma HM, Yan T, Zhang Y, Du B, Wei Q, Chen L (2018) Self-supported CoMoS4 nanosheet array as an efficient catalyst for hydrogen evolution reaction at neutral pH. Nano Res 11:2024–2033
Liu WJ, Wang XF, Yu HG, Yu JG (2018) Direct photoinduced synthesis of amorphous CoMoSx cocatalyst and its improved photocatalytic H2-evolution activity of CdS. ACS Sustain Chem Eng 6:12436–12445
Wu ZX, Guo JP, Wang J, Liu R, Xiao WP, Xuan CJ, Xia KD, Wang DL (2017) Hierarchically porous electrocatalyst with vertically aligned defect-rich CoMoS nanosheets for the hydrogen evolution reaction in an alkaline medium. ACS Appl Mater Interfaces 9:5288–5294
Liu YR, Shang X, Gao WK, Dong B, Li X, Li XH, Zhao JC, Chai YM, Liu YQ, Liu CG (2017) In situ sulfurized CoMoS/CoMoO4 shell–core nanorods supported on N-doped reduced graphene oxide (NRGO) as efficient electrocatalyst for hydrogen evolution reaction. J Mater Chem A 5:2885–2896
Guo JX, Zhang XQ, Sun YF, Tang L, Zhang X (2017) Self-template synthesis of hierarchical CoMoS3 nanotubes constructed of ultrathin nanosheets for robust water electrolysis. J Mater Chem A 5:11309–11315
Car R, Parrinello M (1985) Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett 55:2471–2474
Purgel M (2022) Rigid, strained, and flexible: a DFT study of a backbone-affected monohydride formation of salen and salan complexes. Theor Chem Acc 141:1–8
Du JB, Feng ZF, Zhang Q, Han LJ, Tang YL, Li QF (2019) Molecular structure and electronic spectrum of MoS2 under external electric field. Acta Physica Sinica 68:173101
Kargar H, Behjatmanesh-Ardakani R, Torabi V, Kashani M, Chavoshpour-Natanzi Z, Kazemi Z, Mirkhani V, Sahraei A, Tahir MN, Ashfaq M, Munawar KS (2021) Synthesis characterization crystal structures DFT TD-DFT molecular docking and DNA binding studies of novel copper (II) and zinc (II) complexes bearing halogenated bidentate N O-donor Schiff base ligands. Polyhedron 195:114988
Luo SC, Nie D, Li Z, Sun XY, Hu L, Liu XY (2020) Effects of carboxylic acid auxiliary ligands on the magnetic properties of azido-Cu (II) complexes: a density functional theory study. Polyhedron 182:114506
Byskov LS, Hammer B, Nørskov JK, Clausen BS, Topsøe H (1997) Sulfur bonding in MoS2 and Co-Mo-S structures. Catal Lett 47:177–182
Liebing S, Martin C, Trepte K, Kortus J (2015) Electronic and magnetic properties of ConMom nanoclusters from density functional calculations (n+m=x and 2≤x≤6 atoms). Phys Rev B 91:155421
Juárez-Sánchez OJ, Perez-Peralta N, Herrera-Urbina R, Sanchez M, Posada-Amarillas A (2013) Structures and electronic properties of neutral (CuS)Nclusters(N = 1–6): a DFT approach. Chem Phys Lett 570:132–135
Peng L, Wu SY, Guo JX, Zhong SY, Chen XH (2018) Theoretical investigations on the structural, electronic and spectral properties of VFn(n=1–7) clusters. Zeitschrift für Naturforschung A 73:1091–1104
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Zhou GD, Duan LY (1995) Fundamentals of Structural Chemistry. Peking University Press
Yang S, Rao D, Ye J, Yang S, Zhang C, Gao C, Zhou X, Yang H, Yan X (2021) Mechanism of transition metal cluster catalysts for hydrogen evolution reaction. Int J Hydrog Energy 46:3484–3492
Zhou X, Yang S, Yang H, Gao S, Yan X (2022) Mechanism of heteroatom-doped Cu5 catalysis for hydrogen evolution reaction. Int J Hydrog Energy 47:7802–7812
Nørskov JK, Bligaard T, Logadottir A, Kitchin JR, Chen JG, Pandelov S, Stimming U (2005) Trends in the exchange current for hydrogen evolution. J Electrochem Soc 152:3
Funding
Project supported by the Key Program of the National Natural Science Foundation of China (Grant No.51634004), National Innovation and Entrepreneurship Training Program for College Students (Grant No.: 202110146027, 202010146009, 202010146016).
Author information
Authors and Affiliations
Contributions
Z-Y W contributed to writing, data management, methodology. Z-G F contributed to conceptualization, project Management. J W contributed to article proofreading. Z-L M contributed to verification, methodology. Q-Q H contributed to article proofreading, software. T-H W contributed to article proofreading, data management. X-X Z and J S contributed to proofreading the article.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wang, ZY., Fang, ZG., Wang, J. et al. Study of the hydrogen evolution properties of cluster ConMoS (n = 1–5) using density functional theory. Theor Chem Acc 142, 2 (2023). https://doi.org/10.1007/s00214-022-02943-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-022-02943-0