
Abstract
Context
The investigation of the stability, electronic properties, and catalytic activity of clusters ConMoP holds significant applications and implications in catalyst design, materials science, energy conversion and storage, and environmental protection. The study aims to delve into the unique features of the clusters ConMoP(n = 1 ~ 5), aiming to drive advancements in these related fields. The results obtained from the analysis revealed the stable configurations of the ten clusters, primarily characterized by steric structures. Furthermore, the energy of the clusters was found to increase continuously during growth, as indicated by calculations of atomic fragmentation energy and atomic binding energy. The researchers conducted an analysis of the Natural Population Analysis(NPA) charge, which revealed that Co atoms acted as electron donors, while P and Mo atoms acted as electron acceptors within the clusters. Additionally, an examination of the electrostatic potential indicated that Co and Mo atoms displayed nucleophilic tendencies, while P atoms exhibited electrophilic characteristics. Moreover, the density of states curves, HOMO and LUMO orbitals, and Kooperman's theorem were applied to the clusters ConMoP(n = 1 ~ 5).Through this study, a deeper understanding of the properties and behavior of clusters ConMoP has been achieved, shedding light on their potential as catalysts. The findings contribute to the existing knowledge of these clusters and provide a basis for further research and exploration in this field.
Methods
In this study, we employed the clusters ConMoP(n = 1 ~ 5) to simulate the local structure of the material, enabling us to investigate the stability, electronic properties, and catalytic properties influenced by the metal atoms. By systematically increasing the number of metal atoms and expanding the cluster size, we explored the variations in these properties. Density functional theory (DFT) calculations were performed using the B3LYP hybrid functional implemented in the Gaussian09 software package. The clusters ConMoP(n = 1 ~ 5) underwent optimization calculations and vibrational analysis at the def2-tzvp quantization level, resulting in optimized configurations with different spin multiplet degrees. For data characterization and graphical representation of the stability, electronic properties, and catalytic properties of the optimized configurations, we utilized a range of computational tools. Specifically, the quantum chemistry software GaussView, wave function analysis software Multiwfn were employed. Through the comprehensive utilization of these computational tools, we gained valuable insights into the stability, electronic properties, and catalytic properties of the clusters ConMoP(n = 1 ~ 5) and their dependence on different metal atoms.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Among these amorphous alloys, the CoMoP [1,2,3] system stands out as a promising catalyst with exceptional properties, as evidenced by its impressive performance in experimental studies. The versatility of amorphous alloys extends beyond catalytic applications and encompasses a wide range of fields, including catalytic materials [4], electrode materials [5], magnetic materials [6], and optoelectronic materials. The present study aims to address these gaps in understanding by providing a comprehensive theoretical analysis of the stability, electronic properties, and catalytic performance of clusters ConMoP(n = 1 ~ 5). By employing advanced DFT theory [7], the researchers will explore the intricate details of these clusters and elucidate the impact of varying Co atom numbers on their properties. LU et al. [8] designed and prepared a novel heterostructured electrocatalyst consisting of CoMo alloy particles integrated with CoMoP nanosheets, and the prepared CoMo/CoMoP/NF showed excellent activity. CHANG et al. [9] showed excellent catalytic activity, and this work proposed a simple method to prepare electrocatalysts with fibrous nanostructures. WANG et al. [10] prepared trace amounts of NiP2 coupled CoMoP nanosheets (NCMP) using a one-step hydrothermal method and low-temperature phosphorylation. SHI et al. [11] synthesized a novel carbon-coated cobalt-molybdenum bimetallic phosphide catalyst (Co-Mo-P@C) by chemical vapor deposition and phosphorylation. MAI et al. [12] prepared a CoxMo1P/NiFe-LDH material by a hydrothermal-phosphorylation-electrodeposition process. HUANG et al. [13] synthesized an amorphous bimetallic phosphide (a-CoMoPx/CF), a structure that provides excellent mass transfer capabilities and these fully exposed atoms increase the electrochemically active surface area. HOA V et al. [14] prepared a bifunctional catalyst Co-Mo-P/CoNWs by a simple method and found that the formation of multiple active centers and the increase in the number of active centers due to the double doping of Mo and P synergistically promoted the hydrogen and oxygen precipitation properties. GONG et al. [15] prepared carbon-coated cobalt-molybdenum-phosphorus nanoflakes, Co-Mo-P@C catalysts with high two-dimensional structure, high electrical conductivity and sufficient active centers have good catalytic activity for HER in acidic and basic media. ZHANG et al. [16] prepared an excellent performance CoMoP nanosheet array electrocatalyst (CoMoP Nas@Nf) on nickel foam, and the composite has excellent HER performance, which is expected to replace non-precious metal HER catalysts in practical applications considering its excellent performance and stability. HUANG et al. [17] showed by density functional theory calculations that the interface between CoP and CoMoP can promote H2O dissociation on CoMoP and H+ adsorption on CoP to enhance catalytic activity. LI et al. [18] confirmed the rational design of CoMoP with theoretical calculations down to 0.15 eV DGH, corresponding to (013) and (211) crystal planes. THENUWARA et al. [19] used an amorphous Co-Mo-P model and simulated the energy of its intermediates using DFT. The density flooding study showed that the Co-Mo center acts as a water dissociation center and enhances the basic medium HER.
Transition metal clusters have been extensively studied due to their unique properties and potential applications in various fields. One area of interest is their catalytic activity, as transition metal clusters can serve as efficient catalysts in chemical reactions. For example, studies have shown that transition metal clusters [20] containing Co, Ni, or Cu can exhibit high catalytic activity in the hydrogen evolution reaction (HER) in alkaline media. The interface-engineered crystalline/amorphous Co2P/CoMoPx nanoparticles Chen et al. [21] have been found to be highly efficient electrocatalysts for alkaline HER. Additionally, transition metal clusters supported on amorphous silica surfaces have been investigated to understand the interactions between the metal species and the support material. In the field of nanomaterials, research has focused on nanoalloy clusters, which are composed of two or more different metals. These nanoalloy clusters exhibit unique electronic and catalytic properties, making them promising candidates for various applications. For instance, a theoretical study of (CuS)n clusters [22] has revealed their potential as renewable energy sources in photocatalysis and solar cell applications due to their computed HOMO–LUMO gaps. Furthermore, computational studies have been performed to explore the stability, electronic properties, and catalytic activity of other transition metal clusters, Prabhat et al. [23] employ density functional theory to study the structure, stability, and electronic properties of [AuSin]λ and [Sin+1]λ(λ = 0, + 1;n = 1–12) nanoalloy clusters, investigating their chemical stability, energy gaps, and potential applications in microelectronics and optoelectronics. Moreover, Zeinalipour-Yazdi et al. [24] investigates CO chemisorption on transition metal clusters in supported metal catalysts, revealing the effects of electronic structure on adsorption energy and C-O vibrational frequency, challenging the classical Blyholder model for metal-carbonyl bonds. These works collectively contribute to the understanding of transition metal clusters' behavior and their potential applications in various scientific and technological fields.
In summary, the demand for energy in our society necessitates the search for efficient and environmentally friendly catalysts. Transition metal amorphous alloys, such as the CoMoP system, have emerged as promising candidates due to their accessibility, affordability, and remarkable properties. However, the motivation of this study and the specific objectives of the investigation need clarification. By conducting thorough theoretical analyses using DFT [25], the researchers aim to fill these knowledge gaps and establish correlations between the composition and properties of clusters ConMoP(n = 1 ~ 5). This research will not only contribute to the understanding of CoMoP [26] catalysts but also facilitate future advancements in the field of catalysis.
Theoretical basis
The B3LYP method is widely used for calculating transition metal systems due to its effectiveness and accuracy. However, for elements beyond the fifth period, relativistic effects must be considered, necessitating the use of the def2-TZVP basis set. This basis set combines an all-electron basis set for the first four periods with a small core pseudopotential from Stuttgart for elements beyond the fifth period. It allows for accurate calculations of most elemental systems while reducing the computational workload for transition metal-containing systems. Previous studies by LUO et al. [27,28,29] have validated the reliability of the B3LYP/def2-TZVP quantum chemical level through theoretical simulations of transition metal-containing systems, yielding satisfactory results. In this study, density functional theory (DFT) is employed, and the clusters ConMoP are categorized into various geometries including planar, triangular conical, triangular biconical, quadrilateral, pentagonal, hexagonal, and capped structures. For systems with a small number of atoms, the initial configurations are typically obtained by designing a large number of structures, as demonstrated by Juárez-Sánchez et al. [30, 31] in their calculations of (CuS)N(N = 1–6) and VFn(n = 1–7). Once the initial structures are obtained, the Gaussian09 [32] quantum chemistry software with the B3LYP/def2-TZVP basis set is employed for optimal parameter optimization and frequency corrections. Unstable configurations exhibiting imaginary frequencies or identical shapes are eliminated, resulting in ten optimized steady-state configurations. During the optimization process, certain criteria must be satisfied, including a root mean square value less than 0.00030, a maximum force less than 0.00045, a root mean square displacement less than 0.00120, and a maximum displacement less than 0.00180.
Results and Discussion
Clusters ConMoP(n = 1 ~ 5) stable conformation
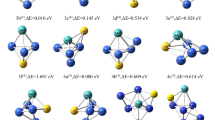
In this study, we performed extensive optimization of configurations for each cluster size. Figure 1 illustrates the most stable geometric configurations of the clusters ConMoP(n = 1 ~ 5) and presents calculated results for their relative energies. To ensure more accurate calculations, we designed a wide range of initial configurations for clusters ConMoP(n = 1 ~ 5) under multiple spin multiplicity conditions. The low-energy isomers are denoted as n-a and n-b, where n represents the number of Co atoms in the ternary clusters ConMoP. Among these clusters, the most stable group is referred to as na. Additionally, we calculated the relative energy of the "nb" configurations by setting the energy of the stable configuration as the reference(0 eV). The configurations were sorted in ascending order of energy, and the numbers in parentheses in the upper corners of the configurations in Fig. 1 indicate the degree of spin multiplicity. Table 1 provides a summary of the corresponding point group symmetry (PG), spin multiplicity (M), electronic state (ES), binding energy per atom (Eb), and relative energy (ΔE) for each configuration.

The optimized configuration of clusters ConMoP(n = 1 ~ 5)
Figure 1 illustrates the optimized geometric configurations of the clusters ConMoP, revealing a predominant three-dimensional arrangement, except for Co1MoP. The calculations indicates that the electronic state of the single multiplet configuration of the clusters ConMoP is 1-A, represents the most stable conformation. The configuration 1a exhibits a planar triangular structure with CS symmetry within the point group, while configuration 1b possesses a slightly higher binding energy (Eb) of 3.237 eV compared to 1a 2.747 eV.
For the cluster Co2MoP, two stable configurations are observed, similar to the n = 1 case. Configurations 2a and 2b adopt a triangular cone shape, with the fundamental planes consisting of Co1, Co2, and P4, and Mo3 acting as the cone's apex. Configuration 2a exhibits a spin multiplicity of two, while configuration 2b has a spin multiplicity of four. Both configurations possess C1 point group symmetry, with electronic states of 2-A and 4-A respectively. Configuration 2a is more stable than 2b by 1.004 eV.
Moving on to the cluster Co3MoP, the stable configuration is 3a, possessing electronic state 1-A and C1 point group symmetry. Interestingly, configuration 3b exhibits the same geometric arrangement as 3a but with electronic state 3-A and an energy 2.72 eV higher than the stable configuration. The triangular biconical geometry of both 3a and 3b involves Co2, Co3, and Mo4 as the primary surfaces, while Co1 and P5 act as the top and bottom of the cone.
In the case of the cluster Co4MoP, all configurations adopt a single-cap triangular biconical shape. The most stable configuration is 4a, with Co1, Co2, and Mo5 forming the reference plane, Co3 and Mo6 serving as the apex and base of the cone, and Co3 acting as the cap. Configuration 4b exhibits an energy 0.082 eV lower than 4a. The spin multiplicity is two for configuration 4a and four for configuration 4b. Their electronic states are 2-A and 4-A, respectively, while the stable configuration retains C1 point group symmetry.
Lastly, for the Co5MoP cluster, the most stable configuration is the double-capped triangular bipyramid 5a, featuring C1 symmetry and corresponding to electronic state 1-A. The 5b configuration, also a double-capped triangular bipyramid with C1 symmetry, follows as the next most stable, albeit with an energy 0.762 eV higher than 5a. Configurations 5a and 5b have Co2, Co5, and Mo6 as the reference planes, with Co1 and Co4 serving as the top and bottom of the cone, while Co3 and P7 form the double caps.
Cluster ConMoP (n = 1 ~ 5) stability analysis
Theoretical investigation of nanoalloy clusters demonstrates the significant influence of binding energies (Eb), fragmentation energies(ΔEf), and second-order difference in energies (Δ2En) on their relative stability. It finds applications in various fields, including high-efficiency electronic devices, catalysts, optical detection elements, and ultraviolet detectors. The calculations was performed using the following equations:
The binding energy per atom (Eb) and Second order difference in energy of the lowest energy structure for the clusters ConMoP(n = 1 ~ 5) were calculated using the following equations:
where E(Con-1MoP) and E(ConMoP) represent the total energy of the clusters Con-1MoP and ConMoP, respectively, and E(Co), E(Mo), and E(P) correspond to the energies of the individual Co, Mo, and P atoms. Table 1 presents the calculated values of the binding energy per atom(Eb) for the clusters ConMoP(n = 1 ~ 5). It is observed that the Eb values vary depending on the number of Co atoms in the cluster. The binding energy values of individual atoms in the clusters ConMoP continuously vary as the number of Co atoms changes, suggesting the clusters' ongoing acquisition of energy from their surroundings to attain structural stability. Additionally, fracture energy serves as a critical parameter for evaluating cluster stability. The variation in binding energy per atom suggests that the cluster requires external energy input to sustain its stability as it undergoes changes with different values of n. The second order difference in energy Δ2E(n) is a valuable tool in chemical computations for assessing the relative stability of different configurations or states. It involves the calculation of energy differences between neighboring structures, providing critical insights into the molecular or cluster stability. Larger Δ2E(n) values typically indicate more stable configurations. In our study, we observed that Co1MoP possesses the largest Δ2E(n), suggesting its superior stability compared to other configurations, as shown in Fig. 2. The fragmentation energy plays a crucial role in determining the stability of the clusters, with higher fragmentation energy corresponding to increased cluster stability. The trend of fragmentation energy variation with the number of Co atoms, as depicted in Fig. 2, exhibits a peak at n = 4. This peak signifies that the cluster Co4MoP possesses higher stability compared to its neighboring clusters.

The change of fragmentation energy of clusters ConMoP(n = 1 ~ 5) with the number of Co atoms and second order difference in energy of the lowest energy structure of clusters ConMoP(n = 1 ~ 5) with the number of Co atoms
Analysis of electronic properties of clusters ConMoP(n = 1 ~ 5)
NPA charge analysis of clusters ConMoP(n = 1 ~ 5)
The investigation of charge distribution is crucial for understanding the electronic properties of clusters ConMoP(n = 1 ~ 5). To achieve precise charge values, we utilized the NBO(Natural Bond Orbital Analysis) method for Natural Population Analysis (NPA) charges, as shown in Table 2. Charge transfer occurs when atoms redistribute their charges, generating distinct potential fields. Positive charge indicates outward electron flow, while negative charge reflects inward electron flow. Table 2 provides valuable insights into the charge distribution within stable clusters ConMoP(n = 1 ~ 5) structures. Co atoms exhibit positive charges, signifying their ability to donate electrons, while Mo and P atoms carry negative charges, indicating their acceptance of electrons. High electron mobility near Co atoms contributes significantly to their exceptional properties.
Electrostatic potential
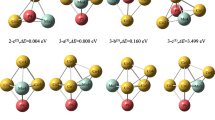
The electrostatic potential is essential for investigating microscopic interactions and predicting the chemical properties of materials. It is a three-dimensional quantity present within the clusters ConMoP(n = 1 ~ 5). Each point in space holds a specific physical meaning: as an infinitely distant positive charge moves, the electrostatic potential varies between that charge and a given point. Due to different forces acting at different points, the distribution of electrostatic potential within the cluster is non-uniform. In this study, we utilized the Multiwfn wave function analysis software and VMD (visualization and analysis program) to generate surface electrostatic potential diagrams for the most stable and sub-stable configurations of clusters ConMoP(n = 1 ~ 5). Figure 3 displays these diagrams.

Surface electrostatic potential diagram of optimized configuration of clusters ConMoP(n = 1 ~ 5)
In the electrostatic potential map, the green region represents areas of positive charge concentration, indicating positive electrostatic potential values. Positive electrostatic potential exhibits strong electrophilic reactivity, attracting electrons towards it. The purple region represents areas of negative charge concentration, indicating negative electrostatic potential values. Negative electrostatic potential demonstrates strong nucleophilic reactivity, attracting holes (electron vacancies) towards it. Overall, the presence of a greater number of green regions around the metal atom Co suggests a predominance of positive electrostatic potential, making it more susceptible to nucleophilic attack and reactions. On the other hand, the presence of more purple regions around the non-metal atom P and the metal atom Mo implies a predominance of negative electrostatic potential, making these atoms more susceptible to electrophilic attack and reactions. Therefore, based on these conclusions and specific experimental requirements, we can design experiments accordingly. For instance, if we want the cluster to exhibit a greater tendency towards nucleophilic reactions, we can increase the proportion of Co atoms in systems where the number of Mo and P atoms is fixed.
Analysis of the catalytic properties of clusters ConMoP(n = 1 ~ 5)
Clusters ConMoP activity energy gap difference analysis
The energy gap difference between the highest occupied molecular orbital(HOMO) and the lowest unoccupied molecular orbital(LUMO) offers valuable insights into the electron activity and spatial distribution within the cluster. Higher energy levels of the HOMO orbitals signify a greater tendency for electron loss, while lower energy levels of the LUMO orbitals suggest a higher likelihood of electron gain. To investigate the catalytic properties of the clusters ConMoP(n = 1 ~ 5), we analyzed the HOMO–LUMO energy difference (EGap = EHOMO-ELUMO) for each cluster configuration. Figure 4 illustrates the HOMO–LUMO energy gap difference. The magnitude of the energy gap difference(EGap) between the HOMO and LUMO orbitals reflects the electron transition capability from HOMO to LUMO, indicating the cluster's electron transfer capability and its potential role in chemical reactions.

HOMO–LUMO energy gap difference of clusters ConMoP(n = 1 ~ 5)
The magnitude of the HOMO–LUMO energy gap(EGap) directly influences the electron transition within the clusters ConMoP(n = 1 ~ 5) and its catalytic activity. A larger EGap indicates a higher barrier for electrons to jump from occupied to empty orbitals, resulting in lower catalytic activity. Conversely, a smaller EGap facilitates electron transitions, leading to higher catalytic activity. The relationship between catalytic activity and EGap in ConMoP(n = 1 ~ 5) is evident. Figure 4 reveals the EGap values for each conformation, with the following size order: 1b > 2a > 3b > 4a > 4b > 2b > 1a > 5b > 5a > 3a. Among the stable configurations within the clusters ConMoP(n = 1 ~ 5), it is observed that configuration 3a exhibits the smallest HOMO–LUMO energy gap. Therefore, compared to other configurations, configuration 3a demonstrates the highest reactivity but the lowest chemical stability. On the other hand, configuration 1b has the largest HOMO–LUMO energy gap, indicating superior chemical stability but lower chemical activity.
HOMO and LUMO track diagrams
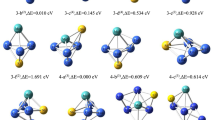
The analysis of HOMO and LUMO orbitals offers insights into the electron-donating and electron-accepting abilities of clusters ConMoP(n = 1 ~ 5) in different conformations. Figure 5 illustrates HOMO and LUMO orbital diagrams for clusters ConMoP(n = 1 ~ 5) in different configurations, visually representing their electronic characteristics and facilitating comprehensive evaluation of their catalytic behavior.

HOMO and LUMO orbital diagrams of various configurations of clusters ConMoP(n = 1 ~ 5)
The size of the region in the HOMO orbital diagram correlates with the energy needed for electron donation in each cluster configuration, whereas the region in the LUMO orbital diagram indicates the intensity of electron acceptance. In both the HOMO and LUMO orbitals, the dark irregular regions near each atom represent off-domain spaces comprising wave functions generated by the orbitals. These regions exhibit a higher likelihood of electron presence and more intense electron flow. The shaded red areas represent the negative phase of the orbital wave function, while the green shading represents the positive phase. The off-domain spaces play a crucial role in determining the catalytic activity of the clusters and are closely associated with their electron dynamics. From Fig. 5, it can be observed that the electrons exhibit delocalization in both the HOMO and LUMO orbitals. Furthermore, the irregular area in the HOMO orbital diagram appears slightly larger than the irregular area in the LUMO orbital diagram. This observation further indicates that the clusters ConMoP(n = 1 ~ 5) possesses certain electron donation and acceptance abilities, with a slightly higher electron-accepting capacity than electron-donating capacity. Taking a closer look at the delocalized region around individual atoms in Fig. 5, within various optimized configurations of the clusters ConMoP(n = 1 ~ 5). The Co atom exhibits the highest contribution percentage, suggesting that it is likely the main potential catalytic active site within the clusters ConMoP(n = 1 ~ 5).
Clusters ConMoP(n = 1 ~ 5) activity Fermi energy level, density of states map analysis
The density of states (DOS) is defined as the number of electronic states within an energy interval of E to E + dE. It characterizes the electron density at a specific energy level, where a higher DOS value indicates a higher probability density of electron occurrence within a given energy range, and vice versa.The Fermi energy level (EFermi) can be considered as the average of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO), serving as an important criterion for evaluating the catalytic activity of a material. The left side of the Fermi energy level is occupied by electrons and can donate electrons, while the unoccupied right side can accept electrons. Figure 6 depicts the density of states around the Fermi energy level, providing a visual representation of the electron density distribution. A higher electron density near EFermi indicates stronger catalytic activity of the cluster, whereas a lower density suggests weaker activity. By examining the changes in electron density within the EFermi-dE range for the HOMO and within the EFermi + dE range for the LUMO, we can assess the cluster's ability to gain or lose electrons and subsequently determine its catalytic reactivity.

Total density of states diagram of each configuration of clusters ConMoP(n = 1 ~ 5)
It can be observed that, among the 10 configurations, both the left and the right side of the Fermi energy level have large or small peaks in the EFermi ± dE range, indicating that the clusters ConMoP(n = 1 ~ 5) can both give and accept electrons in the catalytic reaction and has stronger catalytic activity, but both the peak height and the peak area are slightly larger on the left side than on the right side, indicating that the cluster is more capable of giving electrons. The left side is slightly larger than the right side in both wave height and wave area, indicating that this cluster is more capable of giving electrons. Combined with the analysis of the HOMO and LUMO orbital diagrams of each configuration of the clusters ConMoP (n = 1 ~ 5) in Sect. 2.4.2, it can be seen that the Co atom is most likely the main potential catalytic active site of the clusters ConMoP(n = 1 ~ 5), and it can be guessed that the electron cloud density in the range of EFermi ± dE on the left and right sides of the Fermi energy level is mainly generated by the Co atom, and the Mo and P atoms also contribute to it. also have some contribution, but the contribution is smaller compared to that of P atoms.
The distance between the highest peak on the left side of configuration 4a and configuration 4b from EFermi is greater than the distance between the highest peak on the right side from EFermi, which means that they have a strong electron gaining ability, and the peak on the left side is greater than the peak on the right side, which means that the electron losing ability is greater than the electron gaining ability. The distance between the highest peak on the right side of configuration 3a and EFermi is larger than that on the left side, which means that this configuration has a stronger electron-losing ability, and the peak on the left side of configuration 3a is larger than that on the right side, which means that its electron-losing ability is larger than that of electron gaining ability, indicating that configuration 3a is the easiest to catalyze electrophilic reagent reactants, while the peaks on both sides of configuration 2a are equal and the catalytic ability is weaker. The distance between the left side of conformation 5a and 5b from EFermi and the right side from EFermi are comparable, which means that the two conformations have comparable ability to gain and lose electrons.
Combined with the above analysis of the activity energy gap difference analysis of each configuration of the clusters ConMoP(n = 1 ~ 5) in Sect. 2.4.1, the EGap of configuration 3a is the smallest, and the jump of electrons from the highest occupied orbital to the empty orbital in configuration 3a is the most likely to occur, i.e., the conclusion that configuration 3a has the largest catalytic activity is consistent with the conclusion that configuration 3a is the most likely to catalyze electrophilic reagent reactants.
Clusters ConMoP(n = 1 ~ 5) activity Kooperman's theorem
According to Kooperman's theorem [33], the ionization potential (EI, kJ/mol), electron affinity energy (Eea, kJ/mol), electronegativity (χ, kJ/mol), and electrophilic index (ω, kJ/mol) of each configuration can be determined by combining the energy values of the HOMO and LUMO orbitals of the respective superior configurations of the clusters ConMoP(n = 1 ~ 5). These energy parameters are essential in characterizing the catalytic activity of the different structurally distinct cluster configurations. The calculation equations for these parameters are presented below as Eqs. (1) to (4).
EI represents the energy required for an atom in a cluster molecule to transition into a cation. It is a physical quantity that measures the atom's ability to bind electrons. A lower EI indicates a higher electron loss and greater catalytic activity. Eea represents the energy released by an atom in a cluster after gaining an electron, reflecting the difficulty of electron acquisition. A larger Eea indicates a higher electron-capturing ability and higher catalytic activity. The electronegativity (χ) of an atom reflects its electron adsorption capacity, and as the cluster's χ increases, its catalytic effect strengthens. In chemical reactions within the cluster, the cluster molecule can receive electrons, and an increased electrophilic index (ω) signifies a stronger electron-attracting ability and enhanced catalytic effect. The calculated results of EI, Eea, χ, and ω for clusters of different sizes are presented in Table 3.
Based on the data presented in Table 3, it is evident that configuration 3a has the smallest ionization potential value (4.121 eV) amongthe clusters ConMoP(n = 1 ~ 5), configuration 5b exhibits a higher electron affinity energy compared to other configurations, indicating its advantage over the others. Analyzing the average electron affinity energy, the order of activity is as follows:cluster Co5MoP(2.553eV) > cluster Co1MoP(2.659 eV) > cluster Co4MoP(2.629 eV) > cluster Co3MoP(2.553 eV) > cluster Co2MoP(2.279 eV), indicating the good chemical activity of the cluster Co5MoP Electronegativity analysis reveals the following order of activity:cluster Co1MoP(3.833 eV) > cluster Co5MoP(3.824 eV) > cluster Co4MoP(3.735 eV) > cluster Co3MoP(3.561 eV) > cluster Co2MoP(3.451 eV), highlighting the excellent activity of the Co1MoP cluster. In terms of the electrophilic index, configuration 5b demonstrates a higher value compared to all other sizes, indicating its strongest electron-accepting ability and hence higher activity. According to the average electrophilic index, the order of activity is as follows:cluster Co5MoP(7.705 eV) > cluster Co1MoP(6.514 eV) > cluster Co3MoP(6.455 eV) > cluster Co4MoP(6.305 eV) > cluster Co2MoP(5.104 eV), confirming the superior activity of the Co5MoP cluster compared to other sizes. The electron affinity energy and electronegativity predict excellent activity for the Co5MoP cluster.
Conclusions
The stability, electronic properties and catalytic properties of the clusters ConMoP(n = 1 ~ 5) were studied and analyzed based on the DFT using Gaussian 09 and Multiwfn software.
-
(1)
By optimizing the clusters ConMoP(n = 1 ~ 5), ten stable configurations were obtained, most of which are three-dimensional structures. Co1MoP possesses the largest Δ2E(n), suggesting its superior stability compared to other configurations. Configuration 3a exhibits the highest reactivity but the lowest chemical stability compared to other configurations. Conversely, configuration 1b demonstrates superior chemical stability with the largest HOMO–LUMO energy gap, albeit lower chemical activity.
-
(2)
It can be seen from the NPA charge that Co atoms are electron donors and P and Mo atoms are electron acceptors; through the analysis of electrostatic potential, it is found that Co atoms are easy to be nucleophilic and P、Mo atoms are easy to be electrophilic. The density of states curves, HOMO and LUMO orbitals of ConMoP(n = 1 ~ 5) have been analyzed, and the results show that the clusters have certain electron gaining and losing properties, and the electron losing ability is higher than the electron gaining ability.
The analysis and results presented in this paper demonstrate the wide applicability of ConMoP materials in electrocatalysis, high-efficiency electronic devices, and magnetic materials. The practical significance of this research pave the way for the design and fabrication of earth-abundant nanostructured materials as high-performance HER electrocatalysts, with potential applications in the field of electrocatalysis. These findings serve as a valuable computational reference for future fabrication and utilization of ConMoP materials. In future research, we plan to further reveal the electron transfer pathways in catalytic reactions and gain insight into the interactions and reaction mechanisms between clusters and substrates. The results of these studies will contribute to the development of efficient catalysts, advance the understanding and application of chemical reactions, and meet the needs of the energy and environmental fields.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
Qin Y, Fang ZG, Zhang W, Li LH, Liao W (2020) The study on the catalytic properties of cluster Co3NiB in the hydrogen evolution reaction. J Jiangxi Normal Univ (Nat Sci) 44(01):56–62
Qin Y, Fang ZG, Zhao LL, Liao W, Xu Y (2021) The study on the dynamics and thermodynamicsof isomeric transformation of cluster Co3NiB2 reaction. J Jiangxi Normal Univ (Nat Sci) 45(01):67–74
Fang ZG, Wang ZY, Zheng XX, Qin Y, Mao ZL, Zeng XY, Zhu YW, Wang Q (2022) Study on the dolarizability, dipole moment and density of states of cluster Co3NiB2. J Guizhou Univ (Nat Sci) 39(01):17–24
Zhang YM, Liu Y, Zhao LL, Hou CX, Huang M, Algadi H (2022) Sandwich-like CoMoP2/MoP heterostructures coupling N, P co-doped carbon nanosheets as advanced anodes for high-performance lithium-ion batteries. Adv Compos Hybrid Mater 5(3):2601–2610
Zhang T, Yang TX, Li B, Wai SH, Gao W (2022) Enhancing the electrochemical hydrogen evolution of CoP3/CoMoP nanosheets through the support of black TiO2−x nanotube arrays. J Alloys Compd 905:164165
Zhang TT, Wang YH, Yuan JH, Fang KM, Wang AJ (2022) Heterostructured CoP/CoMoP nanocages as advanced electrocatalysts for efficient hydrogen evolution over a wide pH range. J Colloid Interface Sci 615:465–474
Wu TH, Fang ZG, Wang ZY, Song J, Song JL, Liu LE (2023) The Stable Polarizability of Cluster Co2Mo2P3 Structure. J Jiangxi Normal Univ ( Nat Sci) 47(02):148–153
Lu YK, Zheng XY, Liu Y, Zhu JJ, Li D, Jiang DL (2022) Synergistically Coupled CoMo/CoMoP Electrocatalyst for Highly Efficient and Stable Overall Water Splitting. Inorg Chem 61(21):8328–8338
Chang X, Yan J, Ding XY, Jia YZ, Li SJ, Zhang MY (2022) One-Dimensional CoMoP Nanostructures as Bifunctional Electrodes for Overall Water Splitting. Nanomaterials (Basel) 12(21):3886
Wang YC, Wang YG, Bai J, Lau WM (2021) Trace Amount of NiP2 Cooperative CoMoP Nanosheets Inducing Efficient Hydrogen Evolution. ACS Omega 6(48):33057–33066
Shi JZ, Hou CX, Li L, Xu WC, Fu YB, Huang YZ, Ziyi X, Cheng WJ (2020) Cobalt-Molybdenum Bimetal Phosphides Encapsulated in Carbon as Efficient and Durable Electrocatalyst for Hydrogen Evolution. ChemistrySelect 5(45):14312–14319
Qin W, Wang ZY, Zhao Y, Li FM, Xu L, Wang XM, Jiao H, Chen Y (2020) Self-Supported FeP-CoMoP Hierarchical Nanostructures for Efficient Hydrogen. Evolution 15(10):1590–1597
Huang HW, Cho A, Kim S, Jun H, Lee A, Han JW, Lee J (2020) Structural Design of Amorphous CoMoPx with Abundant Active Sites and Synergistic Catalysis Effect for Effective Water Splitting. Adv Funct Mater 30(43):2003889
Hoa VH, Tran DT, Nguyen DC, Kim DH, Kim NH, Lee JH (2020) Molybdenum and Phosphorous Dual Doping in Cobalt Monolayer Interfacial Assembled Cobalt Nanowires for Efficient Overall Water Splitting. Adv Funct Mater 30(34):2002533
Gong L, Lan K, Wang X, Huang XK, Jiang PB, Wang KZ, Yang M, Ma L, Li R (2020) Carbon-coated Co-Mo-P nanosheets supported on carbon cloth as efficient electrocatalyst for Hydrogen Evolution Reaction. Int J Hydrogen Energy 45(01):544–552
Zhang WG, Liu YH, Zhou HB, Li J, Yao SW, Wang HZ (2019) A high-performance electrocatalyst of CoMoP@NF nanosheet arrays for hydrogen evolution in alkaline solution. J Mater Sci 54(17):11585–11595
Huang XK, Xu XP, Luan XX, Cheng DJ (2020) CoP nanowires coupled with CoMoP nanosheets as a highly efficient cooperative catalyst for hydrogen evolution reaction. Nano Energy 68:104332
Li D, Liu DY, Zhao S, Lu SY, Ma YM, Li MT, Chen GB, Wang YK, Zhou GQ, Xiao CH (2019) Tuning of metallic valence in CoMoP for promoting electrocatalytic hydrogen evolution. Int J Hydrogen Energy 44(59):31072–31081
Thenuwara AC, Dheer L, Attanayake NH, Yan QM, Waghmare UV, Strongin DR (2018) Co-Mo-P Based Electrocatalyst for Superior Reactivity in the Alkaline Hydrogen Evolution Reaction. ChemCatChem 10(21):4832–4837
Alonso JA (2000) Electronic and atomic structure, and magnetism of transition-metal clusters. Chem Rev 100(2):637–678
Chen X, Li Q, Che Q, Chen Y, Xu X (2018) Interface Engineering of Crystalline/Amorphous Co2P/CoMoPx Nanostructure as Efficient Electrocatalysts for Hydrogen Evolution Reaction. ACS Sustain Chem Eng 7(2):2437–2445
Ranjan P, Chakraborty T (2019) Density functional approach: to study copper sulfide nanoalloy clusters. Acta Chim Slov 66(1):173–181
Ranjan P, Chakraborty T (2020) A comparative study of structure, stabilities and electronic properties of neutral and cationic [AuSin]λ and [Sin+1]λ(λ=0,+1;n=1-12) nanoalloy clusters. Mater Today Commun 22:100832
Zeinalipour-Yazdi CD, Cooksy AL, Efstathiou AM (2008) CO adsorption on transition metal clusters: Trends from density functional theory. Surf Sci 602(10):1858–1862
Zl Mao, Fang ZG, Hou QQ, Wang Q, Xu Y, Song JL (2022) The predictive analysis of cluster Co3FeP spectra. J Jiangxi Normal Univ (Nat Sci) 46(1):81–86
Mai WS, Cui Q, Zhang ZQ, Zhang KK, Li GQ, Tian LH, Hu W (2020) CoMoP/NiFe-Layered Double-Hydroxide Hierarchical Nanosheet Arrays Standing on Ni Foam for Efficient Overall Water Splitting. ACS Appl Energy Mater 3(8):8075–8085
Luo SC, Ni D, Li Z, Sun XY, Hu L, Liu XY (2020) Efects of carboxylic acid auxiliary ligands on the magnetic properties of azido-Cu (II) complexes: a density functional theory study. Polyhedron 182:114506
Du JB, Feng ZF, Zhang Q, Han LJ, Tang YL, Li QF (2019) Molecular structure and electronic spectrum of MoS2 under external electric feld. Acta Physica Sinica 68:173101
Kargar H, Behjatmanesh-Ardakani R, Torabi V, Kashani M, Chavoshpour-Natanzi Z, Kazemi Z, Mirkhani V, Sahraei A, TahirMN AM, Munawar KS (2021) Synthesis characterization crystal structures DFT TD-DFT molecular docking and DNA binding studies of novel copper (II) and zinc (II) complexes bearing halogenated bidentate N O-donor Schif base ligands. Polhedron 195:114988
Juárez-Sánchez OJ, Perez-Peralta N, Herrera-Urbina R, Sanchez M, Posada-Amarillas A (2013) Structures and electronic properties of neutral (CuS)N clusters(N=1-6): a DFT approach. ChemPhys Lett 570:132–135
Peng L, Wu SY, Guo JX, Zhong SY, Chen XH (2018) Theoretical investigations on the structural, electronic and spectral properties of VFn(n=1-7) clusters. Zeitschrift für Naturforschung A 73:1091–1104
Haines CRS, Coak MJ, Wildes AR, Lampronti GI, Liu C, Nahai-Williamson P, Saxena SS (2018) Pressure-induced electronic and structural phase evolution in the van der Waals Compound FePS3. Phys Rev Lett 121(26):266801
Ji L, Zeng QX, Wei ML, Jin LW, Zhong QY (2006) J Phys Chem A 110(43):12005–12009
Acknowledgements
Thanks to the National Natural Science Foundation of China and the National Student Innovation and Entrepreneurship Training Program for funding
Funding
This work was supported by National Natural Science Foundation of China Key Project (51634004); National Student Innovation and Entrepreneurship Training Program (202210146008, 202110146027).
Author information
Authors and Affiliations
Contributions
T-H W contributed to writing, data management, methodology. Z-G F contributed to conceptualization, project Management.Z-Y W contributed to article proofreading, data management. L-E L contributed to article proofreading. J-L S contributed to verifcation, methodology. J S contributed to proofreading the article.
Corresponding author
Ethics declarations
Conflict of interest
No potential conflict of interest was reported by the author(s).
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, T., Fang, Z., Wang, Z. et al. Stability, electronic and catalytic properties of ConMoP(n = 1 ~ 5) clusters: A DFT study. J Mol Model 29, 269 (2023). https://doi.org/10.1007/s00894-023-05675-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-023-05675-5