
Abstract
The 1,3-dipolar cycloaddition reaction of benzaldehyde with diazomethane is investigated, in gas phase and in diverse polar solvents, using the molecular electron density theory through density functional theory calculations at the B3LYP(+D3)/6-31G(d) level. Analysis of the reaction pathway reveals that this reaction takes place along a concerted but asynchronous mechanism. Computations show that the acetophenone product is kinetically and thermodynamically more favored than 2-phenylacetaldehyde product in agreement with experimental outcomes. The favored cyclization mode and the observed regioselectivity of this cycloaddition are rationalized by both activation energy calculations, frontier molecular orbital analysis and reactivity indices. Also, polar solvents effect favors the reaction. Furthermore, we performed electron localization function (ELF) topological analysis. The ELF topological analysis of diazomethane indicates that this reactant presents an allenic pseudoradical electronic structure.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
1,3-Dipolar cycloaddition reactions play an important role in organic chemistry [1]. Indeed, they are routes for the synthesis of a large variety of functionalized five-membered heterocyclic systems, which in turn often lend themselves as ideal synthons of “non heterocyclic” derivatives [2]. The influence of the 1,3-dipolar cycloaddition reactions in the field of heterocyclic synthesis is in many ways comparable to that of Diels–Alder reactions on carbocyclic synthesis [3]. In fact, the availability of various classes of dipoles and dipolarophiles has allowed many perspectives.
The earlier works of Huisgen and coworkers [4,5,6,7,8] revealed that the mechanism of 1,3-dipolar cycloadditions leading to 5-membered rings is governed by the stereoselectivity of dipolarophiles, by the nature of solvent and substituents, by the activation parameters and by orientation phenomena. Since then, 1,3-dipolar cycloadditions represent a key strategy to regio- and stereoselective synthesis of five-membered heterocycles and their ring-opened acyclic derivatives [9]. For explanation, Houk et al. [10] proposed that the frontier molecular orbitals (MOs) allow a specific qualitative treatment of reactivity of individual 1,3 dipoles. Typical dipolarophiles used for that purposes are either an alkene or an alkyne, but can be other π molecular systems. In the case of alkyne, aromatic rings are generally produced [11].
Among the most common dipoles, there are the diazo compounds, despite significant conjugative stabilization. For instance, these compounds are primed for rapid normal-electron-demand during 1,3-dipolar cycloadditions. They have reaction rates with strained alkynes that surpass those of azides. Indeed, Gold et al. [12] have reported that diazoacetamides provide chemoselectivity in the presence of terminal alkynes, a feature unobtainable with an azido moiety. Furthermore, the diazo groups are more nucleophilic than azido groups.
Diazomethane was first synthesized by Pechmann [13, 14] through the reaction of N-nitrosourethane with potassium hydroxide. It is known as 1,3-dipole of the propargyl/allenyl anion types [15]. It has been extensively used for 1,3-dipolar cycloaddition reactions. Diazomethane and its derivatives react with aldehydes and ketones usually under very mild conditions to yield a variety of products. In fact, von Auwers et al. [16, 17] have realized experimentally the first stereospecific 1,3-dipolar cycloaddition by reacting diazomethane with cis/trans-isomeric unsaturated carboxylic acid esters. Theoretically, Branchadell et al. [18] have used both CCSD(T) and B3LYP methods in conjunction with the 6-31G(d) basis set to study the 1,3-dipolar cycloaddition reactions of diazomethane with ethylene and formaldehyde. Their results show that the pyrazoline intermediate is more stable than the reactants and that the Gibbs energy barrier for nitrogen elimination is larger than the barrier corresponding to its formation. In contrast, the reaction of diazomethane with formaldehyde provides the kinetically most favorable cycloadduct (i.e. 1,2,3-oxadiazoline), which is less stable than the reactants and has a lower barrier for nitrogen elimination. Also, Blavins et al. [19] used valence-bond theory to describe the electronic mechanism for the gas phase concerted 1,3-dipolar cycloaddition of diazomethane to ethene through spin-coupled calculations at a sequence of geometries along the intrinsic reaction coordinate (IRC) obtained at the MP2/6-31G(d) level of theory. They showed that the bonding rearrangements during the reaction between diazomethane and ethene follow a heterolytic pattern by moving the well-identifiable orbital pairs that are retained along the entire reaction path from reactants to products. Moreover, Nguyen et al. [20] investigated at the B3LYP/6-311++G(d,p) and CCSD(T)/6-311++G(d,p) levels the regiochemistry of 1,3-dipolar cycloadditions reactions of some simple dipolarophiles with diazomethane. It was established that the presence of two methyl groups in the dipole reinforces the electron-donating capacity of the C-atom in diazomethane as dipole with ethylene as dipolarophile, involving its highest occupied MO (HOMO), whereas it becomes an electrophilic reactant interacting via its lowest unoccupied MO (LUMO) in the reactions with prop-1-yne and fluoroprop-1-yne. Later on, Benchouk and Mekelleche [21] studied the mechanism and regioselectivity of the 1,3-dipolar cycloaddition reaction of diazomethane with methyl acrylate using the B3LYP/6-31G(d) approach. They showed that this cycloaddition reaction follows an asynchronous concerted mechanism via the ortho channel.
In 1907, Schlotterbeck [22] reacted diazomethane with benzaldehyde and obtained acetophenone. The reaction is most often carried out by either allowing the carbonyl compound to react with an ethereal solution of the diazomethane derivative with or without the addition of a catalyst, or by treating a methanol solution of the carbonyl compound with a nitrosoalkylurethane in the presence of a base. The products from aromatic aldehydes depend on the solvent and on the substituents of the ring. For example, benzaldehyde forms acetophenone in 97% yield in the absence of methanol [22,23,24]. A smaller yield (40–50%) is obtained however when methanol is present [24, 25].
The aim of the present work is to investigate in depth the concerted pathway of the cycloaddition reaction between benzaldehyde (1) and diazomethane (2), in gas phase and in solvents, using first principles methods. We would like thus to get insights into the mechanism of this reaction and on its high regioselectivity. Benzaldehyde (the dipolarophile) may approach diazomethane (the dipole) in two parallel plans, where the C of the carbonyl interacts with the nucleophilic nitrogen of compound 2. This interaction leads a priori to 5-phenyl-4,5-dihydro-1,2,3-oxadiazole (P-1), 2-phenyl-2,5-dihydro-1,3,4-oxadiazole (P-2), acetophenone (P-3) and 2-phenylacetaldehyde (P-4) products (Scheme 1). These reactions evolve through the pathways specified in Scheme 1, where four transition states (TS-1, TS-2, TS-3 and TS-4) are involved. Our results include the analysis of the geometrical and electronic structures of the reactants, the transition states and the products, the conceptual density functional theory (CDFT) reactivity indices of the reactants, the electron localization function (ELF) outcomes of the electronic structure and the discussion of the mechanistic study of different paths in gas phase and in polar solvents.

1,3-Dipolar cycloaddition reaction between benzaldehyde (1) and diazomethane (2) producing 5-phenyl-4,5-dihydro-1,2,3-oxadiazole (P-1), 2-phenyl-2,5-dihydro-1,3,4-oxadiazole (P-2), acetophenone (P-3) (+ N2) and 2-phenylacetaldehyde (P-4) (+ N2) products. TS-1, TS-2, TS-3 and TS-4 are transition states along the corresponding reaction pathways
2 Computational details
Geometry optimizations, without constraints, of reactants, transition states and products are carried out with the density functional theory (DFT) [26,27,28,29] by adopting the B3LYP density functional as implemented in GAUSSIAN 09 program [30], where the atoms are described using the 6-31G(d) basis set [31]. This functional has been employed with a great success in reactivity studies, with a good compromise between accuracy and computational cost [27, 28]. However, previous works [29, 32,33,34] showed that one needs to consider an energy correction in order to obtain an accurate determination of the heats of formation and of the reaction barriers. Indeed, the B3LYP triple-hybrid density functional includes a term depending on virtual orbitals but London-dispersion effects are not completely incorporated. An additive dispersion correction should be incorporated ensuring the correct asymptotic behavior of the dispersion energy per construction. This correction can be taken into account using Grimme’s (D3) density-independent DFT [35]. Therefore, we performed computations with and without considering the D3 dispersion correction on top of B3LYP.
For the optimized stationary points, second derivative calculations were carried out to reveal their nature. All minimal structures possess all positive frequencies, whereas first-order saddle points (i.e. transition states) on the respective potential energy surfaces have one imaginary frequency. Solvent effects were taken into account using a self-consistent reaction field (SCRF) [36, 37] based on the polarizable continuum model (PCM) [38,39,40]. Different solvents including mixture of diethylether and methanol, pure diethylether and pure methanol were considered implicitly as implemented in GAUSSIAN 09.
The global reactivity has been analyzed through the condensed Fukui functions by using natural population analysis (NPA) [41]. We also evaluated the electronic chemical potential (\(\mu = \left( {\varepsilon_{\text{HOMO}} + \varepsilon_{\text{LUMO}} } \right)/2\)), chemical hardness (\(\eta = \left( {\varepsilon_{\text{LUMO}} - \varepsilon_{\text{HOMO}} } \right)\)), global electrophilicity (\(\omega = \mu^{2} /2\eta\)) and global nucleophilicity (\(N = (\varepsilon_{{{\text{HOMO}}\left( {\text{nucleophile}} \right)}} - \varepsilon_{{{\text{HOMO}}\left( {\text{TCE}} \right)}}\)), where TCE refers to tetracyanoethylene [27, 42, 43]. N can be considered as a measure of the electrophilic power of a system. It may be noted that a global nucleophilicity index would be superfluous because in comparison, a system with lower electrophilicity will have a more nucleophilic character [44]. We also used Parr functions (P(r)) [45]: \(P^{ - } \left( r \right) = \rho_{\text{s}}^{\text{rc}} \left( r \right)\) for electrophilic attacks; \(P^{ + } \left( r \right) = \rho_{\text{s}}^{\text{ra}} \left( r \right)\) for nucleophilic attacks, where \(\rho_{\text{s}}^{\text{rc}} \left( r \right)\) is the atomic spin density (ASD) at the r atom of the radical cation of a considered molecule and \(\rho_{\text{s}}^{\text{ra}} \left( r \right)\) is the ASD at the r atom of the radical anion. Each ASD gathered at different atoms of the radical cation and the radical anion of a molecule provides the local nucleophilic \(P_{k}^{ - }\) and electrophilic \(P_{k}^{ + }\) Parr functions of the neutral molecule. Moreover, we performed ELF topological analyses, at the B3LYP/6-31G(d) level, of the reactants using the Multiwfn [46] package. The corresponding reaction path was followed by ELF topological analyses along the IRC path. The VMD 1.9.3 and UCSF Chimera 1.14 programs were used to visualize the ELF basin attractor position and the ELF basin domains [47, 48].
3 Results and discussion
Figure 1 gives the optimized structures of the reactants, the transition states and the products as given in Scheme 1. The corresponding main geometrical parameters are listed in Table 1. This figure gives also the outermost MOs of 1 and 2 compounds and their energies. These energies are used later to evaluated the CDFT reactivity indices of the reactants, namely the electronic chemical potential (µ), the chemical hardness (η), the electrophilicity (ω) and the nucleophilicity (N), which are listed in Table 2 [49, 50].

B3LYP-D3/6-31G(d) optimized geometries with the outermost molecular orbitals (HOMO and LUMO) of the reactants and products. We give also the energies (in eV) of these MOs. Egap (in eV) corresponds to the HOMO–LUMO energy gap
3.1 Regioselectivity of the reaction between benzaldehyde and diazomethane
The regioselectivity of the 1,3-dipolar cycloaddition reaction between benzaldehyde and diazomethane was analyzed in light of the CDFT reactivity indices of the reactants. Table 2 shows that the electronic chemical potential [27, 51] of the diazomethane (µ = −3.64 eV) is smaller, in absolute value, than that of benzaldehyde (µ = −4.33 eV), thereby indicating that the global electron density transfer (GEDT) [52] will go from diazomethane toward benzaldehyde [11, 21, 53].
The electrophilicity and nucleophilicity indices of benzaldehyde 1 are evaluated ω = 1.79 eV and N = 2.18 eV. Thus, this compound can be classified as a strong electrophile and a moderate nucleophile [54]. The diazomethane has an electrophilicity ω of 1.41 eV and a nucleophilicity N of 3.11 eV, being classified on the borderline of moderate electrophiles [55] and as a strong nucleophile [56]. Obviously, benzaldehyde 1 will participate in this reaction as electrophile and the diazomethane 2 will participate as nucleophile [57, 58]. Accordingly, the electronic flux will be from diazomethane to benzaldehyde in agreement with the results associated with the electronic chemical potential µ. Also, the values of η confirm the nucleophilic character of diazomethane and electrophilic character of benzaldehyde. Accordingly, the studied reaction has a normal electronic demand (NED).
For further analysis, we have calculated the Parr indices for the centers in interactions associated with the two reactants (Fig. 2). For benzaldehyde, the electrophilic Parr function shows that the C12 carbon, \(P_{k}^{ + }\) = 0.24, is slightly more electrophilically activated than the O13 oxygen, \(P_{k}^{ + }\) = 0.23, these results indicate that the C12 carbon is the most electrophilic center of benzaldehyde 1. On the other hand, the nucleophilic Parr functions of diazomethane 2 indicate that the carbon C15 is the most nucleophilic center of this molecule \(P_{k}^{ - }\) = 0.72. Therefore, the most favorable electrophile–nucleophile interaction along the nucleophilic attack of the diazomethane 2 on the benzaldehyde 1 will take place between the most nucleophilic center of diazomethane 2 (i.e. the carbon C15) and the most electrophilic center of benzaldehyde 1 (i.e. the C12 carbon), leading to P-3 product in agreement with the experimentally observed selectivity [59, 60]. In sum, the regioselectivity of the 1,3-dipolar cycloaddition reaction between benzaldehyde and diazomethane is described correctly using the CDFT reactivity indices and Parr functions in line with the experimental findings [22, 24]. Similar results were obtained by Benchouk and Mekelleche using B3LYP/6-31G(d) level for the 1,3-dipolar cycloaddition reaction of diazomethane with methyl acrylate [21].

3D representations of the ASD of the radical anion of benzaldehyde 1 (\(1^{ \cdot - }\)) and the radical cation of diazomethane 2 (\(2^{ \cdot + }\)) as well as the electrophilic \(P_{k}^{ + }\) Parr functions of 1 and nucleophilic \(P_{k}^{ - }\) Parr functions of 2
3.2 Mechanistic study
The mechanism of the 1,3-dipolar cycloaddition reaction between diazomethane and benzaldehyde was examined in order to reveal the more favorable product by the kinetic and thermodynamic factors. It involves four π electrons from the 1,3-dipole and two electrons from the dipolarophile. Figure 3 displays the energy profile, corresponding to the four modes of cyclization between the two reactants as shown in Scheme 1. These profiles were computed at the B3LYP/6-31G(d) level with and without considering the D3 correction. Those computed in ether, methanol or an ether–methanol mixture solvent are also given. All reactants, transition states and products were optimized at the corresponding level of theory. The total energies and the relative energies of these stationary points are listed in Table S1. This table and Fig. 3 show that the consideration of the D3 correction is needed for better description of non-covalent interactions occurring within the transition states [61, 62]. Indeed, we observe a significant overestimation of the activation energies at the TSs and an underestimation of the products energies in absence of Grimme’s correction. For example, the activation energy associated with TS-1 using DFT/B3LYP and DFT/B3LYP-D3 is 17.67 and 13.63 kcal/mol, respectively. The Er energies are quite different, where B3LYP-D3 values are lower by about 4 kcal/mol than those derived without considering dispersion correction (i.e., B3LYP functional only). In the following, we will refer to the data obtained using B3LYP-D3 except if it is stated specifically.

Energy profiles for the 1,3-dipolar cycloaddition reaction between benzaldehyde 1 and diazomethane 2 at the B3LYP/6-31G(d) and B3LYP-D3/6-31G(d) level of theory in gas phase and in diverse solvents. The energies are given relative to reactants (1 + 2) (in kcal/mol). The corresponding values are listed in Table S1 of the Supplementary Material
The formation reactions of P-1 and P-2 are exothermic by − 7.22 kcal/mol and − 18.19 kcal/mol, respectively. P-2 is thus more stable thermodynamically than P-1, whereas the activation energy associated with the 1 + 2 → P-1 transformation is lower. The first step of the reaction between 1 and 2 should be under kinetic factors control. Afterward, Fig. 3 shows that the P-1 → P-3 and P-1 → P-4 reactions evolve via TS-3 and TS-4 transition states, respectively. TS-3 is associated with a lower activation energy than TS-4, (Table S1) favoring the production of P-3. The formation of P-3 and P-4 is exothermic by − 70.97 kcal/mol and − 61.12 kcal/mol, respectively. In sum, the formation of the cycloadduct P-3 is favored thermodynamically over that of P-4 by ~ 10 kcal/mol, in agreement with the experimental results [22, 24, 63]. Similar results were obtained by Branchadell et al. [18] using B3LYP/6-31G(d) level for the 1,3-dipolar cycloaddition reactions of diazomethane with ethylene and with formaldehyde. For explanation, we present in Fig. 1 the HOMO and LUMO outermost MOs for the reactants and the products and their energies. This figure shows that the HOMO of 1 is located at − 6.94 eV and its LUMO is at − 1.71 eV and the HOMO of 2 is at − 6.01 eV and its LUMO is at − 1.28 eV. The HOMO–LUMO energy gap of 1 is larger than that of 2. Therefore, the best coupling is between the HOMO of 1 and the LUMO of 2. Figure 1 shows also that the energy gaps for products are larger than those of the reactants, which indicates the stability of these products [61, 64, 65].
In laboratory, the reaction of diazomethane with benzaldehyde has been done in different solvents (mixture of ether and methanol, pure ether and pure methanol) [24, 25]. In order to highlight the effect of solvents, we reinvestigated the energy profile of the reaction between 1 and 2 in these solvents (Fig. 3 and Table S1). Computations show that these solvents stabilize all transition states except TS-2, but destabilize the products. Thereby, the inclusion of solvent effects slightly decreases the activation energies and increases the exothermic character of this 1,3-dipolar cycloaddition reaction but does not change the selectivity obtained in gas phase. The formation of the P-3 is more favored when the reaction takes place in a mixture of ether and methanol. Compared to gas phase, the solvent effect decreases the activation energies by about ~ 2 kcal/mol for TS-3; while the formation energy of P-3 increases by 1–2 kcal/mol [64, 66].
3.3 Geometry and vibrational analyses of the transition states
Some selected bond lengths of the all optimized stationary points are given in Table 1. The analysis of the geometric data shows that the N–N and C–N distances of diazomethane as well as the C–O distance of benzaldehyde undergo only small variations from reactants to TSs. For example, the N–N distance is equal to 1.15 Å in diazomethane, 1.14; 1.18 Å at the transition structures (TS-1; TS-2) and ~ 1.23 Å in the cycloadducts (P-1; P-2). Similarly, the C–O distance is equal to 1.22 Å in benzaldehyde, 1.28 Å in the transitions structures TS-1 and TS-2 and 1.45; 1.42 Å in the P-1 and P-2 products. Therefore, we can conclude that we have earlier transition states in these cycloaddition reactions because the TSs look like the reactants rather than the products [67].
The intermolecular geometrical parameters of the newly formed σ bonds (i.e. O13–N19 and C12–C15 bonds in the transition state TS-1, O13–C15 and C12–N19 bonds in the transition state TS-2) differ in TS-1 and in TS-2 (Fig. 4). These distances have also different values in the products. For example, in TS-1, the d(O13–N19) distance is computed 2.20 Å. It reduces by 0.78 to Å in P-1 product. In the other side, the d(C12–C15) distance shrinks from 1.94 Å in TS-1 to 1.55 Å in P-1. Consequently, the C12–C15 bond is more advanced than the O13–N19 bond. Generally, we can conclude that the formation of the new bonds is asynchronous [68, 69]. Furthermore, this is confirmed by the analysis of the B3LYP-D3/6-31G(d) imaginary frequencies of the transition states in gas phase and in solvents (Table S2) and of the corresponding nuclear motions. This analysis reveals that the TS-1-related processes are associated with the heavy atoms’ motions and are also related to the earlier transition states [66, 70], contrariwise for TS-2. Similar findings were obtained by Branchadell et al. [18] for the 1,3-dipolar cycloaddition reactions of diazomethane with ethylene and formaldehyde.

B3LYP-D3/6-31G(d) optimized geometries of TS-1, TS-2, TS-3 and TS-4 transition structures. The distances in the forming bond process are given in Å
3.4 ELF topological analyses of bond changes along the 1,3-dipolar cycloaddition reaction of benzaldehyde with diazomethane
Topological analyses of the electron localization function (ELF) of benzaldehyde and diazomethane were performed in order to characterize the electronic structure of these reactants involved in the 1,3-dipolar cycloaddition reaction. The ELF valence basin populations together with the basin attractor positions and the proposed Lewis-like structures of the reactants are shown in Fig. 5. This ELF quantum chemical analysis provides a straightforward connection between the chemical structure and the distribution of electron density within a compound.

B3LYP/6-31G(d) ELF valence basin populations together with basin attractor positions and proposed Lewis-like structures of benzaldehyde (1) and diazomethane (2)
The ELF topological analysis of benzaldehyde shows the presence of two V(O) and V′(O) monosynaptic basins, integrating for 2.51e and for 2.69e (total 5.20e), and one V(O, C) disynaptic basin with a population of 2.35e (Fig. 5). The reactant 1 has also one V(C, C) disynaptic basin integrating a total electronic density of 2.25e. Moreover, the ELF topological analysis of diazomethane shows the presence of two V(C15) and V′(C15) monosynaptic basins integrating a total population of 1.13e. These ELF valence basin populations allow relating the integrating V(C15) and V′(C15) monosynaptic basins with a sp2 hybridized pseudoradical carbon. Diazomethane also has V(N19) and V′(N19) monosynaptic basins integrating for a total population of 3.89e, one V(C15, N18) disynaptic basin integrating for 3.05e, and two V(N18, N19) and V′(N18, N19) disynaptic basins integrating a total population of 3.72e. These ELF valence basin populations allow relating the V(N19) and V′(N19) monosynaptic basins with the non-bonding electron density of N19, the V(C15, N18) disynaptic basin with an underpopulated C15–N18 double bond and the two V(N18, N19) and V′(N18, N19) disynaptic basins with a N18–N19 double bond as shown in the proposed Lewis-like structure in Fig. 5. The ELF picture of diazomethane is identical to that found for phenyldiazomethane obtained by Domingo and coworkers [71, 72]. It is worth noting that similar results were obtained by Salah et al. [66] using B3LYP/6-31 + g(d,p) level for the 1,3-dipolar cycloaddition reaction between fluorinated alkynes and azides, and by Mohammad-Salim et al. [73] using B3LYP/6-31G(d) level for the [3 + 2] cycloaddition reactions between α,β-unsaturated selenoaldehyde with nitrone and with nitrile oxide.
An ELF topological analysis of the structures along the IRC involved in the bond changes of the studied reaction was performed in order to characterize the occurring bonds formation/breaking. These structures were selected by means of bonding evolution theory (BET) [66, 69, 73,74,75]. The respective bond distances and populations of the most relevant ELF valence basins are listed in Tables S3–S6. The representations of the ELF attractor locations of all transition states are shown in Figs. 6 and 7.

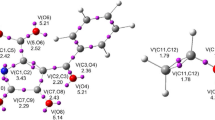
Representations of the ELF attractors of TS-1, TS-2, TS-3 and TS-4 involved in the 1,3-dipolar cycloaddition reaction of benzaldehyde (1) and diazomethane (2)

ELF domains for TS-1, TS-2, TS-3 and TS-4
We have chosen ten significant differentiated points on IRC path associated with P-1 product, which represent the evolution of the formation of the new σ bonds. They are denoted S-1–S-10 (Tables S3–S6). The ELF analysis of these points will be very helpful to locate the evolution of the electronic density, especially between the atomic center C12–C15 and O13–N19 in the two fragments, from reactants, via the transition structures to final products, which helped us to reveal the mechanism of the reaction. Table S3 shows the ELF topological analysis associated with the formation of P-1 along the IRC. The first structure of the IRC, S-1, displays a similar bonding pattern to those of the separated reactants. At TS-1, the N18–N19 bond region has been depopulated by 3.09e and the two V(C15) and V′(C15) monosynaptic basins present in S-1 have merged into one V(C15) monosynaptic basin integrating for 1.41e. As shown in Figs. 6 and 7, the most relevant changes in TS-2 is the presence of two new monosynaptic basins V(N19) and V(N18) integrating for 2.51e and 1.92e, respectively. At S-7, the first most relevant changes take place along the IRC. The V(C15) monosynaptic basin present in TS-1 has disappeared and a new V(C12, C15) disynaptic basin appeared with an initial population of 1.81e. At S-9, the second topological changes along the IRC takes place and a new V(O13, N19) disynaptic basin is formed with a population of 0.81e. This important topological change corresponds to the formation of the second N19–O13 single bond at a N–O distance of 1.44 Å.
Table S4 displays the ELF topological analysis for the formation of P-2 along the IRC. At TS-2, the most relevant changes correspond to the creation of two new monosynaptic basins, V(C12) and V(N18) integrating for 0.11e and 2.12e, respectively. At S-6, the first most relevant topological change along IRC appears and a new V(C15, O13) disynaptic basin is created with an initial population of 0.87e. This significant change indicates that the formation of the first C15–O13 single bond begins at a C–O distance of 1.59 Å. Then, a second major topological change happens at S-7, while the V(C12) and V′(N19) monosynaptic basins present at the previous structure of the IRC merge into a new V(C12, N19) disynaptic basin integrating for 1.71e.
Tables S5 and S6 show the ELF topological analysis associated with the breaking of C15–N18 and N19–O13 single bonds to form P-3 and P-4. For the formation of P-3, the first most relevant topological change occurs at S-2 when a V(N19, O13) disynaptic basin disappears and a V′(N19) monosynaptic basin appears, while the N19–O13 single bond breaks. Afterward, the V(C15, N18) disynaptic basin disappears signature of the breaking of the C15–N18 single bond. Afterward, the V(C15, N18) disynaptic basin disappears signature of the breaking of the C15–N18 single bond. For the formation of P-4, as shown in Table S6, the first most relevant topological change occurs at S-3 when a V(N19, O13) disynaptic basin disappeared, indicating that the N19–O13 single bond is breaking. The second most relevant topological changes take place after disappearing V(C15, N18), highlighting thus the breaking of the C15–N18 single bond. The third change along the IRC occurs at S-7 and the V(C1, C12) disynaptic basin integrating for 1.33e disappears and V(C1, C15) disynaptic basin integrating for 1.68e appears. These significant topological changes document the C1–C12 single bond breaking and the C1–C15 single bond formation.
4 Conclusions
The mechanism and regioselectivity of the 1,3-dipolar cycloaddition reaction between benzaldehyde 1 and diazomethane 2 have been studied using the density functional theory at the B3LYP(+D3)/6-31G(d) levels. The inclusion of the D3 correction has allowed a good estimation of energy associated with each stationary point detected in the energy profile.
The present results of CDFT and mechanistic study allowed explaining the regioselectivity of reaction. For instance, the analysis of the electrophilic \(P_{k}^{ + }\) Parr functions revealed that the C12 carbon atom is the most electrophilic center of benzaldehyde. On the other hand, the nucleophilic \(P_{k}^{ - }\) Parr functions of diazomethane indicate that the C15 carbon is the most nucleophilic center of this molecule, in good agreement with the regioselectivity found in 1,3-dipolar cycloaddition reactions. Moreover, the potential energy surface analysis shows that this reaction follows an asynchronous concerted mechanism, where the formation of the acetophenone cycloadduct P-3 is kinetically and thermodynamically more favored than P-4, in line with the experimental findings. ELF topological analyses of the reactants indicate that diazomethane has an allenic pseudoradical electronic structure. The populations of the most relevant ELF valence basins of the selected structures along the IRC involved in the bond formation/breaking were also determined. They show two stages concerted molecular mechanism for this type of reactions. Finally, the ELF analysis herein allowed a best electronic description of the evolution of the new formed σ bonds, in agreement with the experimentally observed regioselectivity leading to acetophenone.
References
Padwa A (1984) 1,3-dipolar cycloaddition chemistry. Wiley, New York
Carruthers W (2013) Cycloaddition reactions in organic synthesis. Elsevier, Amsterdam
Nair V, Suja TD (2007) Intramolecular 1,3-dipolar cycloaddition reactions in targeted syntheses. Tetrahedron 63:12247–12275
Huisgen R (1963) 1,3-Dipolar cycloadditions. Past and future. Angew Chem Int Ed Engl 2:565–598
Huisgen R (1968) Mechanism of 1,3-dipolar cycloadditions. Reply. J Org Chem 33:2291–2297
Huisgen R (1976) 1,3-Dipolar cycloadditions. 76. Concerted nature of 1,3-dipolar cycloadditions and the question of diradical intermediates. J Org Chem 41:403–419
Huisgen R, Mloston G, Langhals E (1986) The first two-step 1,3-dipolar cycloadditions: interception of intermediate. J Org Chem 51:4085–4087
Mloston G, Langhals E, Huisgen R (1989) 1,3-Cycloadditions of aliphatic thione S-methylides to dimethyl 2,3-dicyanofumarate and 2,3-dicyanomaleate; a test case for steric course and mechanism. Tetrahedron Lett 30:5373–5376
Yazdani H, Kaul E, Bazgir A, Maysinger D, Kakkar A (2020) Telodendrimer-based macromolecular drug design using 1,3-dipolar cycloaddition for applications in biology. Molecules 25:857
Houk KN, Firestone RA, Munchausen LL, Mueller PH, Arison BH, Garcia LA (1985) Stereospecificity of 1,3-dipolar cycloadditions of p-nitrobenzonitrile oxide to cis- and trans-dideuterioethylene. J Am Chem Soc 107:7227–7228
Salah M, Komiha N, Kabbaj OK, Ghailane R, Marakchi K (2017) Computational study of the 1,3-dipolar cycloaddition between methyl 2-trifluorobutynoate and substituted azides in terms of reactivity indices and activation parameters. J Mol Graph Model 73:143–151
Gold B, Aronoff MR, Raines RT (2016) 1,3-Dipolar cycloaddition with diazo groups: noncovalent interactions overwhelm strain. Org Lett 18:4466–4469
Pechmann HV (1894) Ueber Diazomethan. Ber Dtsch Chem Ges 27:1888–1891
Lévai A (1997) Synthesis of pyrazolines by the reactions of α, β-enones with diazomethane and hydrazines (review). Chem Heterocycl Compd 33:647–659
Gothelf KV, Jørgensen KA (1998) Asymmetric 1,3-dipolar cycloaddition reactions. Chem Rev 98:863–910
Auwers KV, Cauer E (1929) Über Δ1- und Δ2-Pyrazoline. Justus Liebigs Annalen Der Chemie 470:284–312
Auwers KV, König F (1932) Über den Aufbau von Pyrazolin-carbonsäureestern. Justus Liebigs Annalen Der Chemie 496:27–51
Branchadell V, Muray E, Oliva A, Ortuño RM, Rodríguez-García C (1998) Theoretical study of the mechanism of the addition of diazomethane to ethylene and formaldehyde. Comparison of conventional ab initio and density functional methods. J Phys Chem A 102:10106–10112
Blavins JJ, Karadakov PB, Cooper DL (2001) Modern valence-bond description of chemical reaction mechanisms: the 1,3-dipolar addition of diazomethane to ethene. J Org Chem 66:4285–4292
Nguyen LT, Proft FD, Dao VL, Nguyen MT, Geerlings P (2003) A theoretical approach to the regioselectivity in 1,3-dipolar cycloadditions of diazoalkanes, hydrazoic acid and nitrous oxide to acetylenes, phosphaalkynes and cyanides. J Phys Org Chem 16:615–625
Benchouk W, Mekelleche SM (2008) Theoretical study of the mechanism and regioselectivity of the 1,3-dipolar cycloaddition of diazomethane with methyl acrylate using theoretical approaches. J Mol Struct (Thoechem) 862:1–6
Schlotterbeck F (1907) Umwandlung von Aldehyden in Ketone durch Diazomethan. Ber Dtsch Chem Ges 40:479–483
Biltz H, Paetzold H (1923) Neue Untersuchungen mit Diazomethan; zugleich ein Beitrag zur Kenntnis des Alloxantins. Justus Liebigs Annalen Der Chemie 433:64–87
Gutsche CD (1954) The reaction of diazomethane and its derivatives with aldehydes and ketones. Org React 8:364–429
Mosettig E, Czadek K (1931) Über die Einwirkung von Diazo-methan auf Piperonal III. Monatsh Chem 57:291–304
Koopmans T (1934) Über die Zuordnung von Wellenfunktionen und Eigenwerten zu den Einzelnen Elektronen Eines Atoms. Physica 1:104–113
Parr RG, Weitao Y (1994) Density-functional theory of atoms and molecules. Oxford University Press, Oxford
Méndez F, Tamariz J, Geerlings P (1998) 1,3-dipolar cycloaddition reactions: a DFT and HSAB principle theoretical model. J Phys Chem A 102:6292–6296
Sheng Y-H, Fang D-C, Wu Y-D, Fu X-Y, Jiang Y (1999) DFT studies on the mechanism of the 1,3–dipolar cycloaddition reaction between methyleneketene and pyrroline-1-oxide. J Mol Struct (Thoechem) 467:31–36
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Ding WF, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ (2009) Gaussian 09 B.01
Hehre WJ, Radom L, von Schleyer PR, Pople J (1986) AB INITIO molecular orbital theory, 1st edn. Wiley-Interscience, New York
Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785–789
Becke AD (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652
Grimme S, Mück-Lichtenfeld C, Würthwein E-U, Ehlers AW, Goumans TPM, Lammertsma K (2006) Consistent theoretical description of 1,3-dipolar cycloaddition reactions. J Phys Chem A 110:2583–2586
Goerigk L (2014) How Do DFT-DCP, DFT-NL, and DFT-D3 compare for the description of london-dispersion effects in conformers and general thermochemistry? J Chem Theory Comput 10:968–980
Tomasi J, Persico M (1994) Molecular interactions in solution: an overview of methods based on continuous distributions of the solvent. Chem Rev 94:2027–2094
Simkin BI (1995) Quantum chemical and statistical theory of solutions: a computational approach/. Ellis Horwood, London
Cossi M, Barone V, Cammi R, Tomasi J (1996) Ab initio study of solvated molecules: a new implementation of the polarizable continuum model. Chem Phys Lett 255:327–335
Cancès E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041
Barone V, Cossi M, Tomasi J (1998) Geometry optimization of molecular structures in solution by the polarizable continuum model. J Comput Chem 19:404–417
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions: a theoretical study. J Org Chem 73:4615–4624
Chattaraj PK, Maiti B, Sarkar U (2003) Philicity: a unified treatment of chemical reactivity and selectivity. J Phys Chem A 107:4973–4975
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494
Lu T, Chen F (2012) Multiwfn: a multifunctional wavefunction analyzer. J Comput Chem 33:580–592
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874
Ess DH, Jones GO, Houk KN (2006) Conceptual, qualitative, and quantitative theories of 1,3-dipolar and diels-alder cycloadditions used in synthesis. Adv Synth Catal 348:2337–2361
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516
Domingo LR (2014) A new C–C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415–32428
Zeroual A, El Idrissi M, El Ajlaoui R, Ourhriss N, Abouricha S, Mazoir N et al (2017) MEDT study of the mechanism and regioselectivity of diazocompounds and alkenes in [3 + 2] cycloaddition reaction. Eur J Mol Biotechnol 5:43–49
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) Applications of the conceptual density functional theory indices to organic chemistry reactivity. Molecules 21:748
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct (Thoechem) 865:68–72
Bakavoli M, Moeinpour F, Sardashti-Birjandi A, Davoodnia A (2013) 1,3-Dipolar cycloaddition of 4-chlorobenzonitrile oxide with some dipolarophiles: theoretical analysis of regioselectivity. J Heterocycl Chem 50:188–193
Marakchi K, Ghailane R, Kabbaj OK, Komiha N (2014) DFT study of the mechanism and stereoselectivity of the 1,3-dipolar cycloaddition between pyrroline-1-oxide and methyl crotonate. J Chem Sci 126:283–292
Zeroual A, Idrissi ME, Zoubir M, Ajlaoui RE, Abouricha S, Hajbi AE (2017) Theoretical study of the mechanism and regioselectivity of prop-2-yn-1-ol with azide in [3 + 2] cycloaddition reactions. Open Access J Transl Med Res 1:1
Ríos-Gutiérrez M, Domingo LR (2019) Unravelling the mysteries of the [3 + 2] cycloaddition reactions. Eur J Org Chem 2019:267–282
Mohammad-Salim HA, Abdallah HH, Maiyelvaganan KR, Prakash M, Hochlaf M (2020) Mechanistic study of the [2 + 2] cycloaddition reaction of cyclohexenone and its derivatives with vinyl acetate. Theor Chem Acc 139:19
Hanachi R, Boughdiri S, Ben Said R, Chambaud G, Hochlaf M (2018) Structural and energetic properties of tautomeric forms of phosphonyl thioamides. Theor Chem Acc 137:55
Hosseini SJ, Emamian S, Domingo LR (2019) Participation of furoxancarbonitrile oxide in [3 + 2] cycloaddition reaction toward C-N triple bond: a Molecular Electron Density Theory study of regioselectivity and mechanistic aspect. Struct Chem 30:317–326
Gouid Z, Said RB, Sanhoury MA, Boughdiri S, Prakash M, Linguerri R et al (2018) Insights into the bonding between tributylphosphine chalcogenides and zinc(II). Theor Chem Acc 137:68
D’Auria M (2012) Regio- and sterochemistry of the [2 + 2]-cycloaddition reaction between enones and alkenes. A DFT study. Tetrahedron 68:8699–8703
Salah M, Zeroual A, Jorio S, El hadki H, Kabbaj OK, Marakchi K et al (2020) Theoretical study of the 1,3-DC reaction between fluorinated alkynes and azides: reactivity indices, transition structures, IGM and ELF analysis. J Mol Graph Model 94:107458
Marakchi K, Abou El Makarim H, Kabbaj OK, Komiha N (2010) Theoretical study of the mechanism of the 1,3-dipolar cycloaddition reaction of methyl-3-fluoro-3-trifluoromethyl prop-2-enoate with pyrroline-1-oxide. Phys Chem News 52:129–137
Kavitha K, Venuvanalingam P (2005) Low-lying stepwise paths for ethylene 1,3-dipolar cycloadditions: a DFT study. Int J Quantum Chem 104:64–78
Marakchi K, Kabbaj OK, Komiha N, Chraı̈bi ML (2001) Etude théorique des réactions de cycloaddition dipolaire-1,3 de la diphénylnitrilimine sur des dipolarophiles hautement fluorés. J Fluor Chem 109:163–171
Bach RD, McDouall JJW, Schlegel HB, Wolber GJ (1989) Electronic factors influencing the activation barrier of the Diels-Alder reaction. An ab initio study. J Org Chem 54:2931–2935
Domingo LR, Ríos-Gutiérrez M, Emamian S (2017) Understanding the domino reaction between 1-diazopropan-2-one and 1,1-dinitroethylene. A molecular electron density theory study of the [3 + 2] cycloaddition reactions of diazoalkanes with electron-deficient ethylenes. RSC Adv 7:15586–15595
Zeroual A, Ríos-Gutiérrez M, Salah M, El Alaoui El Abdallaoui H, Domingo LR (2019) An investigation of the molecular mechanism, chemoselectivity and regioselectivity of cycloaddition reaction between acetonitrile N-Oxide and 2,5-dimethyl-2H-[1,2,3]diazaphosphole: a MEDT study. J Chem Sci 131:75
Mohammad-Salim H, Hassan R, Abdallah HH, Oftadeh M (2020) The theoretical study on the mechanism of [3 + 2] cycloaddition reactions between α, β-unsaturated selenoaldehyde with nitrone and with nitrile oxide. J Mex Chem Soc 64:2594-0317
Krokidis X, Noury S, Silvi B (1997) Characterization of elementary chemical processes by catastrophe theory. J Phys Chem A 101:7277–7282
Zeroual A, Ríos-Gutiérrez M, El Ghozlani M, El Idrissi M, Ouled Aitouna A, Salah M et al (2020) A molecular electron density theory investigation of the molecular mechanism, regioselectivity, stereoselectivity and chemoselectivity of cycloaddition reaction between acetonitrile N-oxide and 2,5-dimethyl-2H-[1,2,3]diazarsole. Theor Chem Acc 139:37
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Supplementary Material:
Tables including: Total energies E (u.a) and relative energies Er (kcal/mol) of the stationary points as computed at the B3LYP(+D3)/6-31G(d) levels of theory in gas phase or in solvents; Imaginary frequencies of all transition states (cm−1) of the 1,3-dipolar cycloaddition reaction as computed at the B3LYP-D3/6-31G(d) level of theory in gas phase and in solvents and Valence basin populations of the most relevant points calculated from the ELF of 1,3-dipolar cycloaddition reaction of diazomethane with benzaldehyde to form P-1, P-2, P-3 and P-4. (DOCX 44 kb)
Rights and permissions
About this article

Cite this article
Abbiche, K., Mohammad-Salim, H., Salah, M. et al. Insights into the mechanism and regiochemistry of the 1,3-dipolar cycloaddition reaction between benzaldehyde and diazomethane. Theor Chem Acc 139, 148 (2020). https://doi.org/10.1007/s00214-020-02662-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-020-02662-4