
Abstract
Both intramolecular [4 + 2] and intermolecular [3 + 2] cycloaddition reactions for the synthesis of 1,3-dioxoepoxyisoindole and epoxypyrrolo[3,4-g]indazoles, respectively, experimentally explored by Alizadeh and co-workers, were studied theoretically at the M06-2X/6-311G(d,p) computational level in both gaseous and solution (acetonitrile, benzene and DMSO) phases. In the first stage, it was studied the intramolecular [4 + 2] cycloaddition reaction generating potentially two diastereomeric adducts and it was confirmed the formation of experimentally reported product, 1,3-dioxoepoxyisoindole DEI-1 by the local reactivity indices as well as potential energy surface analysis. In the second stage, the [3 + 2] cycloaddition reaction of DEI-1 with three nitrilimines including NI–H, NI-C and NI–N, which generates potentially four possible regio- and stereoisomeric adducts was studied. The local reactivity indices described satisfactorily the experimentally observed regioselectivities. The results of potential energy surface analysis indicated that among the four possible products, only two exo regioisomeric adducts (namely, epoxypyrrolo[3,4-g]indazoles, CA-x1 and CA-x2) can be formed. In fact, for the reaction of DEI-1 with NI–H, NI-C and NI–H, the ratio of CA-x1:CA-x2 was estimated to be 57:43, 86:14 and 39:61, respectively, which is in satisfactory agreement with the experimental outcomes. The structural variations during the reactions were also studied and the results indicated that the transition states are not completely synchronous.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Natural products such as prostaglandins, terpenoids, and carotenoids are certain chemical compounds produced by living systems. There is a key segment in the natural products known as oxabicyclo template, e.g., cantharidin and norcantharidin. It is found directly in the structure of multiple natural products and, also, is applied in the total synthesis of natural products as an precursor (Chiu and Lautens 1997; Saleem et al. 2007). In recent years, it has been shown many biological and medicinal aspects of oxabicyclo containing compounds. Some oxabicyclo compounds such as cantharidin and norcantharidin and their derivatives can poison the tumour cells through inhibition of a subgroup of human serine/threonine protein phosphatases (Bertini et al. 2009; Hill et al. 2008; McCluskey et al. 2001). In addition, the oxabicyclo compounds extracted from the natural plants show an anti-proliferative effect on breast cancer cells (Huyen et al. 2018).
The oxabicyclo core can undergo ring cleavage reactions to create different ring structures or acyclic chains. The oxabicyclo compounds can be synthesized by the intramolecular Diels–Alder (IMDA) cycloaddition reaction (Schindler and Carreira 2009; Vogel et al. 1999).
Cycloaddition reaction is a pericyclic reaction, in which two unsaturated components interact together to form a cyclic product. The Diels–Alder reaction, also known as [4 + 2] cycloaddition reaction, is one of the most prominent cycloaddition reactions in which a diene and a dienophile react together to form a six-membered cyclic adduct. This reaction is one of the most important procedures in organic synthesis due to its diversity and creation of regio- and stereospecific products. The Diels–Alder reaction can occur as both intramolecular and intermolecular. Intramolecular reactions are faster, cleaner, and more selective than intermolecular ones (Brieger and Bennett 1980; Norman 2017; Smith 2020; Sykes 1986). In addition to the Diels–Alder reactions, the [3 + 2] cycloaddition reactions are another important class of organic transformations, which can be used for the synthesis of different five-membered cyclic compounds (Huisgen 1984). In this reaction, a three-atom-component interacts with an unsaturated system (Emamian 2016).
From the mechanistic point of view, a concerted one-step mechanism was first proposed for the cycloaddition reactions (Woodward and Hoffmann 1965). More studies on these reactions with various reactants have shown that a wide range of cycloaddition reactions proceed via a two-stage one-step mechanism, in which two new bonds are formed between two reactants in one kinetic step but via a two-stage process (Berski et al. 2006). In other word, one of the σ-bonds between two fragments begins to form before the other. For instance, it has been reported that the reaction of nitrilimine with a thione-containing dipolarophile (Bazian et al. 2016), benzonitrile oxide with electron-rich N-vinylpyrrole (Domingo et al. 2016), nitrilimine with alkene (Molteni and Ponti 2017), nitrilimine with allenoate (Soleymani 2018), N‑(p‑methylphenacyl)benzothiazolium ylide with 1‑nitro‑2‑(p‑methoxyphenyl)ethane (Soleymani 2019), 5,5-dimethyl-1-pyrroline N-oxide with 2-cyclopentenone (Soleymani and Chegeni 2019), azoalkenes toward fulvenes (Emamian et al. 2019), para‑quinone methides with nitrile imines (Soleymani and Jahanparvar 2020), C,N-dialkyl nitrones with a series of substituted oxanorbornadienes (Opoku et al. 2020), and 3-(2,2,2-trifluoroethylimino)-1-methylindolin-2-one ylide with cinamaldehyde and the corresponding iminium ion (Soleymani 2020) take place via a two-stage one-step mechanism.
Oxabicyclo[2.2.1]heptenes are a class of the oxabicyclo compounds that came along with improvements in the Diels–Alder reaction of furan. The Diels–Alder reactions of furan as a 4π-diene component have been studied by many chemists such as Diels and Alder (Diels 1929), Brion (Brion 1982) and Vogel et al. (Reymond and Vogel 1990; Vieira and Vogel 1982, 1983). In this regard, Alizadeh et al. reported the synthesis of epoxypyrrolo[3,4-g]indazoles by two sequential [4 + 2] and [3 + 2] cycloaddition reactions (Scheme 1) (Alizadeh et al. 2019). At the first stage, a three-component reaction between an isocyanide 1, a dicarboxylic ester of acetylene 2 and 2-furan carboxylic acid 3 leads to the formation of 1,3-dioxoepoxyisoindole 4. Subsequent [3 + 2] cycloaddition reaction of 4 with hydrazonoyl chloride under basic conditions, produces two regioisomeric adducts 6 and 6′. The amount of 6:6′ ratio is dependent to the nature of the substituents R1, R2 and R3 located on 1, 2 and 5 components. For instance, in the case of R1 = t-Bu, R2 = Me, and R3 = H, both 6 and 6′ adducts are formed in similar yields.

Synthesis of epoxypyrrolo[3,4-g]indazoles by two sequential [4 + 2] and [3 + 2] cycloaddition reactions
Based on the proposed mechanism (Alizadeh et al. 2019), the addition of isocyanide 1 to dicarboxylic ester of acetylene 2 results in an zwitterionic intermediate 7 (Scheme 2). A proton transfer process from 2-furan carboxylic acid 3 into 7 and subsequent Passerini type addition of 2-furan carboxylate to nitrilium ion affords intermediate 8, which undergoes the Mumm rearrangement to form intermediate 9. An IMDA reaction on 9, which is followed by a [3 + 2] cycloaddition reaction with nitrilimine 10 (obtained from dehydrohalogenation of 5 under basic conditions) leads to the formation of two regioisomeric adducts 6 and 6′.

The proposed mechanism for the synthesis of adducts 6 and 6′
Herein, two sequential IMDA and [3 + 2] cycloaddition reactions for the synthesis of 1,3-dioxoepoxyisoindole (DEI-1) and epoxypyrrolo[3,4-g]indazoles (CA-x1 and CA-x2), respectively, experimentally explored by Alizadeh and co-workers (Alizadeh et al. 2019), have been theoretically investigated using density functional theory (DFT) to elucidate regio- and stereochemistry in the reaction (Scheme 3).

Both IMDA and [3 + 2] cycloaddition reactions studied in this work (Alizadeh et al. 2019)
The main aims of the present work are:
-
• Investigation of the global and local reactivity indices at the ground state for the reagents involving in both cycloaddition reactions.
-
• To study the structural variations during the reactions.
-
• To study the regio- and stereochemistry in the reaction.
-
• Analysis of the substituent effects on the [3 + 2] cycloaddition reaction.
-
• Investigation of the reaction from energetic point of view.
Theoretical
All calculations were performed using the Gaussian 09 program based on the density functional theory (DFT) (Frisch 2013). The optimization of the reactants, transition states and products was performed using M062X method, and frequency calculations were carried out on the optimized structures by the same method to verify the stationary points as well as transition states (Zhao and Truhlar 2006). B3LYP/6-31G(d) computational level was used to calculate the global and local DFT reactivity indices (Lee et al. 1988). Solvent effects of acetonitrile, DMSO and benzene were considered using the conductor-like polarizable continuum model (CPCM) (Barone and Cossi 1998). The intrinsic reaction coordinates (IRC) calculations were performed to confirm that each transition state is located between two minima (Gonzalez and Schlegel 1989, 1990).
The global nucleophilicity index N based on the HOMO energies, was obtained by the expression (1):
where EHOMO(Nu) refers to the HOMO energy of the nucleophile and EHOMO(TCE) is the HOMO energy of tetracyanoethylene (TCE) as a reference (Domingo et al. 2013a).
Both chemical potential (µ) and chemical hardness (η) quantities were obtained from the HOMO and LUMO energies according to the Eqs. (2, 3) (Parr and Pearson 1983; Parr and Weitao 1989):
The global electrophilicity index (ω) was calculated according to the Eq. (4) (Parr et al. 1999):
Wiberg bond index (WBI) is a measure of bond strength between atoms A and B and is determined as the sum of the off-diagonal square of the density matrix P between two atoms based on the natural bond order (NBO) according to the Eq. (5) (Wiberg 1968):
Results and discussion
Evaluation of the global reactivity indices for the reactants at the ground state
Global DFT reactivity indices including electronic chemical potential (μ), chemical hardness (η), and global nucleophilicity (N), can be used as a valuable tool to study the reactivity and selectivity in organic reactions (Geerlings et al. 2003). Since we deal with an IMDA reaction in the first stage, the calculation of the global DFT for Int is unnecessary and give no information about the reaction (Scheme 3). On the other hand, in the [3 + 2] cycloaddition reaction of DEI-1 with nitrilimines NI–H, NI-C and NI–N, these parameters were calculated and given in Table 1.
The results given in Table 1 indicate that the electronic chemical potential for DEI-1 (− 4.05 eV) is more negative than that for NI–H (− 3.19 eV) and NI-C (− 3.39 eV). On the other hand, this trend is vice versa for DEI-1 (− 4.05 eV) and NI–N (− 4.19 eV). Therefore, it can be concluded that along the polar [3 + 2] cycloaddition reaction of DEI-1 with either NI–H or NI-C, electron density is transferred from the later ones, as nucleophile, toward the former, as electrophile. Alternatively, it is expected that the electron density fluxes from DEI-1 toward NI–N during the corresponding cycloaddition reaction.
According to a scale introduced by Domingo and coworkers, the electrophilicity as well nucleophilicity power can be evaluated using the values of ω and N (Domingo et al. 2002; Jaramillo et al. 2008). By employing this scale, DEI-1 with the electrophilicity index larger than 1.50 eV is a strong electrophile, and with the nucleophilicity index within the range of 2.00 to 3.00 eV is a moderate nucleophile. On the other hand, NI–H and NI-C are strong electrophiles as well as nucleophiles (their nucleophilicity index is larger than 3.00 eV, the threshold value for the strong nucleophile). NI–N with nucleophilicity and eletrophilicity powers of 3.58 eV and 6.48 eV, respectively, is considered as a strong nucleophile and a very strong electrophile. The increment of the electrophilic power of NI–N compared to NI–H can be attributed to the presence of the nitro group as an electron-withdrawing substituent. Thus, it can be said that we deal with a [3 + 2] cycloaddition reaction between a strong electrophilic/nucleophilic pair (NEI-1 and either NI–H or NI-C), and between a moderate nucleophile (DEI-1) and a very strong electrophile (NI–N), rationalizing why these reactions are performed at room temperature under experimental conditions (Alizadeh et al. 2019).
Analysis of local reactivity indices
In a polar reaction between non-symmetrical reactants, the most favorable interaction can occur between the most nucleophilic center of the nucleophile and the most electrophilic center of the electrophile (Yang and Mortier 1986). There are certain methods to evaluate satisfactorily the local reactivities of the reactants by determination the most electrophilic or nucleophilic centers of the reactants. One of the powerful methods for determination of the reactive sites in the reactants is in terms of Fukui functions, which are calculated based on the Mulliken population analysis of an atom in a molecule (Eqs. 6 and 7) (Chamorro et al. 2013; Domingo et al. 2013b):
The local electrophilicity \({\omega}_{k}\) and nucleophilicity \({N}_{k}\), can be calculated using expressions (8) and (9):
As mentioned in the “Introduction” section, an IMDA reaction on Int results in the corresponding cycloadduct DEI-1, in the first step of the reaction. Since this intramolecular reaction contains only one reactant, the simultaneous calculation of both nucleophilic Nk and electrophilic ωk Fukui functions is impossible. However, we can easily characterise the electrophilic and nucleophilic fragments in Int that participate in the IMDA reaction. In fact, the C1-C2 fragment possessing electron-withdrawing substituents (two CO2Me groups) is electrophilically activated, while the furan moiety is a nucleophilic fragment. Since both fragments have been attached to a C = O group, it can be said that the effect of this additional group is counterbalanced with a good approximation. For determination the local reactivities in Int, it was considered as an electrophile and the local electrophilic Fukui functions ωk for the certain atoms were calculated. Similarly, the local nucleophilic Fukui functions Nk were also calculated by considering Int as a nucleophile. The results are shown in Fig. 1.

B3LYP/6-31G(d) local nucleophilic Nk (in blue) and electrophilic (in red and underlined) Fukui functions ωk for Int obtained by Mulliken population analysis
The analysis of the results presented in Fig. 1 clearly indicates that the C2 carbon atom with the local electrophilic index ωk of 0.211 is the most electrophilic center, and the C3 carbon atom with the local nucleophilicity value of 0.124 is the most nucleophilic one. Thus, it can be concluded that along the IMDA reaction on Int, and in agreement with the experimental outcomes (Alizadeh et al. 2019), the C2 carbon atom tends to interact with the C3 carbon atom to give DEI-1. Of course, it should be here noted that the others alternative interactions (C2⋅⋅⋅C6 and C1⋅⋅⋅C3) are unfavorable geometrically.
For the [3 + 2] cycloaddition reaction of DEI-1 with nitrilimines in the second step of the reaction, the local electrophilic and nucleophilic Fukui functions of DEI-1 (as electrophile toward NI–H and NI-C, and as nucleophile toward NI–N), the local nucleophilic Fukui functions of NI–H and NI-C, and local electrophilic ones of NI–N were calculated. The results are presented in Fig. 2.

B3LYP/6-31G(d) local nucleophilic Nk (in blue) and electrophilic (in red and underlined) Fukui functions ωk for species involving in the studied [3 + 2] cycloaddition reaction, obtained by Mulliken population analysis
Analysis of the local Fukui functions given in Fig. 2 shows that when DEI-1 acts as the electrophile, its C4 carbon atom with an \(\omega_{k}\) value of 0.065, is more electrophilic than C5. On the other hand, the C7 carbon atom of NI–H and NI-C, with the local nucleophilic index of 0.639 and 0.600, respectively, is more nucleophilic than the N9 nitrogen atom with that of 0.535 and 0.497, respectively. When DEI-1 acts as the nucleophile, the C4 carbon atom with a local nucleophilicity of 0.096, is slightly more reactive than the C5 carbon atom with that of 0.069, and tends to interact with the N9 nitrogen atom of NI–N as the most electrophilic center possessing a local electrophilicity of 0.441. These results suggest that the reaction of DEI-1 with either NI–H or NI-C leads mainly to the formation of CA-x1. On the other hand, in the reaction of DEI-1 with NI–N, the CA-x2 adduct should be produced as the major adduct. These predictions are in agreement with the experimental outcomes especially for the reaction of DEI-1 with either NI-C or NI–N, and not satisfactory for the reaction with NI–H (see Scheme 3).
Study of the regio- and stereochemistry in the reactions from energetic point of view
For the IMDA reaction on Int, and in consideration of stereochemistry for the product, two possible cycloadducts, DEI-1 and DEI-2, can be potentially generated via two possible competitive reactive channels (Scheme 4). In fact, due to the trans stereochemistry of two CO2Me groups in Int, and also due to the retention of configuration, two possible diastereomers can be formed, in which the C1 and C2 carbon atoms adopt a trans configuration.

Two possible reactive channels for IMDA reaction on Int
To study the reactive channels from energetic point of view, it was computed the M06-2x/6-311G(d,p) relative Gibbs free energies (ΔG) for transition states, TS-1 and TS-2, and products, DEI-1 and DEI-2, involved in the IMDA reaction on Int in both gaseous and solution phases. In addition, since the IMDA reaction has been performed experimentally in the refluxing acetonitrile, the Gibbs free energies were computed at 85 ℃ (the approximate temperature of the refluxing acetonitrile). The results are summarized in Table 2. To obtain more reliable results, the calculations were also performed by employing 6–311 + G(d, p) basis set in gas phase and acetonitrile.
In addition, it was depicted a diagram for the relative Gibbs free energies in gaseous phase and acetonitrile at 85 ℃ for the studied IMDA reaction in Fig. 3 to elucidate the energetic aspects of two reactive channels.

Free energy diagram for the IMDA cycloaddition reaction on Int in gaseous phase and acetonitrile at 85 ℃
An analysis on the results presented in Table 2 and Fig. 3 indicates that in the presence of acetonitrile TS-1 which is located 73.32 kJ mol−1 above the reactant, Int, is significantly more favorable than TS-2 with an activation barrier of 119.70 kJ mol−1. In addition, it is clear that DEI-I is located 22.31 kJ mol−1 below Int, and in complete agreement with the experimental outcomes (Alizadeh et al. 2019), the corresponding reaction is thermodynamically favorable. On the other hand, the formation of DEI-2 is ruled out, because the corresponding reaction is unfavorable both thermodynamically and kinetically. Finally, the solvent employed in the experimental work (acetonitrile) can slightly increase the rate of the reaction, because it lowers the energy level of the transition state slightly more than that of the reactant and thereby, slightly decreases the activation barriers.
A comparison between the results in three solvents including acetonitrile, DMSO and benzene presented in Table 2 indicates that the polarity of the solvent can slightly affect the activation barriers. In fact, the activation barriers are approximately identical in both polar solvents, acetonitrile and DMSO. On the other hand, the activation barriers are slightly increased in benzene as a non-polar solvent in comparison to the polar solvents. These results indicate that the corresponding transition states are slightly more polar compared to the reactant and a polar solvent can lower the energy surface of transition states more than a non-polar solvent.
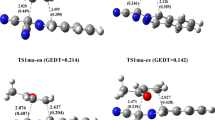
Figure 4 depicts the optimized geometries for the transition states associated with the IMDA reaction on Int along with the interatomic distances for the interacting atoms and the corresponding Wiberg bond indices.

M06-2X/6-311G(d,p) optimized structures for the transition states TS-1 and TS-2 at the presence of acetonitrile along with the interatomic distances (in Å) and the Wiberg bond indices (given in parenthesis) belong to the interacting atoms. The unique imaginary frequencies (in cm−1) are also given in parenthesis in front of each transition state
It is also given the IRC profile associated with the more favorable transition state (TS-1) and the variations in the distances between interacting atoms (C1-C6 and C2-C3) in Fig. 5. The IRC analysis for TS-1 rules out the formation of any stable intermediate during the reaction. In addition, relaxing the end points of the IRC leads to the reactant and products from both sides.

IRC profile (top) and variations in the C1-C6 and C2-C3 lengths (down) associated with the TS-1 for the IMDA reaction on Int
An overview on the results for TS-1 presented in Figs. 4 and 5 shows that, the length of C1-C6 is slightly shorter than C2-C3 and the bond indices are vice versa (Fig. 4). This clearly indicates that the former bond is formed slightly faster than the later one. A similar trend is also observed in the diagram of the bond length during the reaction coordinate (Fig. 5). In fact, in most points, the length of the C1-C6 bond is slightly smaller than that of the C2-C3 one. These results reveal that the studied IMDA reaction is slightly asynchronous. On the other hand, the asynchronicity in TS-2 is more than that in TS-1, because the difference in the bond lengths as well as bond indices for the C1-C6 and C2-C3 bonds is more in TS-2 compared to TS-1.
Finally, it was found that the oxygen atoms are closed together in TS-2 more than TS-1, which leads to electronic repulsions. These repulsive forces are probably responsible of more instability of TS-2 compared to TS-1.
For the [3 + 2] cycloaddition reaction of DEI-1 with nitrilimines, two possible exo and endo stereoselective attacks can be potentially taken place. In fact, in the exo and endo attacks, nitrilimine interacts with DEI-1 from the same and opposite side of the bridged oxygen, respectively. In addition, each of these stereoselective attacks contains two regioselective attacks including C4-C7 and C4-N9 ones, in which the C4 carbon atom of DEI-1 interacts with the C7 and N9 atoms of nitrilimine, respectively. In consideration of four possible stereoselective and regioselective attacks, four possible adducts, CA-x1, CA-x2, CA-n1 and CA-n2, can be formed from the [3 + 2] cycloaddition reaction of DEI-1 and nitrilimine. In addition, the IRC calculations over the transition states associated with the four reactive channels (TS-x1, TS-x2, TS-n1 and TS-n2), show that the reactions take place in one kinetic step and the formation of any stable intermediate is ruled out. Thus, four possible transition states and four possible cycloadducts are proposed for the [3 + 2] cycloaddition reactions of DEI-1 with nitrilimines (Scheme 5). It should be here noted that x and n indicate the exo and endo stereochemistry, respectively.

Four possible reactive channels between DEI-1 and NIs
To investigate the energetic aspects of the reactive channels, it was calculated the relative Gibbs free energies (ΔG) for the transition states and products in both gaseous and solution phases. In addition, since the reaction has been carried out experimentally at room temperature (Alizadeh et al. 2019), the values of the Gibbs free energies were also calculated at this temperature. To obtain more reliable results, the calculations were also performed by employing 6–311 + G(d,p) basis set.
It was also plotted the diagram of the relative Gibbs free energies in gaseous and acetonitrile phases at room temperature for the [3 + 2] cycloaddition reaction of DEI-1 and NI–H in Fig. 6.

Free energy diagram for the [3 + 2] cycloaddition reaction of DEI-1 and NI–H in both gaseous and acetonitrile phases at room temperature obtained by M06-2X/6-311G (d,p) method
The analysis of the results presented in Table 3 and Fig. 6 indicates that:
-
1.
The transition states associated with the exo attack, TS-x1H and TS-x2H, are located about 70.00 kJ mol−1 above the separate reactants. On the other hand, the transition states for the endo attacks are located more than 90.00 kJ mol−1 above the separate reactants. These results clearly reveal that both exo attacks are more favorable kinetically. In addition, since the reaction of DEI-1 with NI–H takes place via the exo stereoselective manner, only the exo attack was investigated for the reaction of DEI-1 with either NI-C or NI–N.
-
2.
It appears that both electronic and steric effects can determine the stereochemistry of the reaction. In fact, when the reactants are closed together from the endo side, some electronic and steric repulsions are created between the NI moiety and the CO2Me group located on the C1 carbon atom of DEI-1. On the other hand, the exo attack is relatively free from both electronic and steric repulsions, and consequently is more favorable kinetically.
-
3.
The Gibbs free energies of activation for formation of adducts CA-x1H and CA-x2H in acetonitrile are 69.54 and 70.21 kJ mol−1, respectively. These values correlate with a CA-x1H:CA-x2H ratio of 57:43, in good agreement with the experimental results, CA-x1C:CA-x2C = 50:50 (Alizadeh et al. 2019).
-
4.
Similarly, the Gibbs free energies of activation for formation of adducts CA-x1C and CA-x2C in acetonitrile were found to be 61.99 and 66.50 kJ mol−1, respectively. These values for activation barriers are equivalent with a CA-x1C:CA-x2C ratio of 86:14, in good agreement with the experimental outcomes (CA-x1C:CA-x2C = 93:7). On the other hand, in the reaction of DEI-1 with NI–N, the cycloadducts CA-x1N and CA-x2N are formed in acetonitrile with the activation Gibbs free energies of 62.59 and 61.49 kJ mol−1, respectively, which are equivalent with a CA-x1N:CA-x2N ratio of 39:61. In consistent with the experimental results, this estimation indicates preference formation of CA-x2N rather than CA-x1N. However, it is not a complete agreement, CA-x1N:CA-x2N = 4:96 (Alizadeh et al. 2019).
-
5.
A comparison between the activation Gibbs free energies in the presence of acetonitrile indicates that the reaction accelerates when both Cl and NO2 groups are introduced in nitrilimine.
-
6.
The activation barriers are more in the solution compared to the gas phase. This can be probably attributed to this fact that acetonitrile as a polar solvent lowers the energies of the reactants more than those of the transition states. As depicted in Fig. 6, in consideration of the solvation effects, the energy surface of the reactants is reduced more than that of the transition states (77.55 against 59.77 or 58.99 kJ mol−1) for the reaction of DEI-1 with NI–H.
-
7.
It is observed a similar trend between the results obtained from the M06-2X/6–311 + G(d, p) method and those obtained from the M06-2X/6-311G(d, p) one.
It was also performed the similar arguments about the geometries and IRC profile associated with the transition states for the second stage, the [3 + 2] cycloaddition reaction of DEI-1 and NIs. The results are given in Figs. 7 and 8.

M06-2X/6-311G(d,p) optimized structures for the transition states at the presence of acetonitrile along with the interatomic distances (in Å) and the Wiberg bond indices (given in parenthesis) belong to the interacting atoms for the [3 + 2] cycloaddition reaction of DEI-1 and NI–H. The unique imaginary frequencies (in cm−1) are also given in parenthesis in front of each transition state

IRC profile (top), variations in the interatomic distances (middle) and Wiberg bond indices (down) associated with the TS-x1H (left) and TS-x2H (right) for the [3 + 2] cycloaddition reaction of DEI-1 and NI–H
The analysis of the results presented in Figs. 7 and 8 suggests the following expressions:
-
1.
A slight asynchronicity is observed in TS-x1H, in which the C4-C7 bond is formed slightly earlier than the C5-N9 one. The variations in bond length (Fig. 8) also confirm this conclusion, because the slope of the decrease in the length of the C4-C7 bond is slightly more than that of the C5-N9 one.
-
2.
The identical values for the C4-N9 and C5-C7 bond indices in TS-x2H (0.215) show that both bonds begin to progress identically and the corresponding reaction proceeds with a high synchronicity. The variations in bond length (Fig. 8) also confirms this conclusion.
-
3.
There is a good agreement between the variations in the interatomic distances and Wiberg bond indices, in which by decreasing the former, the latter is increased, and vice versa.
Conclusions
A theoretical study at the level of M06-2X/6-311G(d,p) was reported on two sequential intramolecular [4 + 2] and intermolecular [3 + 2] cycloaddition reactions. These reactions have been experimentally explored by Alizadeh and co-workers for the synthesis of epoxypyrrolo[3,4-g]indazoles. They have reported the formation of 1,3-dioxoepoxyisoindole DEI-1 from an intramolecular [4 + 2] cycloaddition reaction, and two regioisomeic cycloadducts, CA-x1 and CA-x2, from the intermolecular [3 + 2] one. In the present research, some competitive reactions were considered between the reactants and the results showed a good agreement with the experimental outcomes.
The local reactivity indices based on the Fukui functions and the Gibbs free energy analysis confirmed that the intramolecular [4 + 2] cycloaddition reaction leads to the formation of the experimentally reported product. It was found that the C2 and C3 carbon atoms of Int with maximum values of the local electrophilicity and nucleophilicity, respectively, can interact together to form both DEI-1 and DEI-2 adducts. The Gibbs free energy analysis confirmed that the formation of the experimentally reported product, DEI-1, is favorable both kinetically and thermodynamically.
In the [3 + 2] cycloaddition reaction of DEI-1 with nitrilimines, the results of the local reactivity indices based on the Fukui functions, described satisfactorily the experimentally observed regioselectivities. The results of potential energy surface analysis indicated that among the four possible products, only two regioisomeric exo adducts (CA-x1 and CA-x2) can be formed. It was found that for the reaction of DEI-1 with NI–H, NI-C and NI–H, the ratio of CA-x1:CA-x2 are estimated to be 57:43, 86:14 and 39:61, respectively. These results are in satisfactory agreement with the experimental outcomes.
Finally, the study of the structural variations during both cycloaddition reactions indicated that the reactions are not completely synchronous.
References
Alizadeh A, Asalemi KAA, Halvagar M (2019) Intramolecular Diels-Alder and [3+ 2] Cycloaddition reactions in the one-pot synthesis of epoxypyrrolo [3, 4-g] indazoles. Synthesis 51:2936–2944. https://doi.org/10.1055/s-0037-1612426
Barone V, Cossi M (1998) Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 102:1995–2001. https://doi.org/10.1021/jp9716997
Bazian A, Beyramabadi SA, Davoodnia A, Pordel M, Bozorgmehr MR (2016) Kinetics and mechanism of the 1, 3-dipolar cycloaddition of nitrilimine with thione-containing dipolarophile: a detailed DFT study. Res Chem Intermed 42:6125–6141. https://doi.org/10.1007/s11164-016-2449-3
Berski S, Andrés J, Silvi B, Domingo LR (2006) New findings on the Diels−Alder reactions. An analysis based on the bonding evolution theory. J Phys Chem A 110:13939–13947. https://doi.org/10.1021/jp068071t
Bertini I, Calderone V, Fragai M, Luchinat C, Talluri E (2009) Structural basis of serine/threonine phosphatase inhibition by the archetypal small molecules cantharidin and norcantharidin. J Med Chem 52:4838–4843. https://doi.org/10.1021/jm900610k
Brieger G, Bennett JN (1980) The intramolecular Diels-Alder reaction. Chem Rev 80:63–97. https://doi.org/10.1021/cr60323a004
Brion F (1982) On the lewis acid catalyzed diels-alder reaction of furan. regio-and stereospecific synthesis of substituted cyclohexenols and cyclohexadienols. Tetrahedron Lett 23:5299–5302. https://doi.org/10.1016/S0040-4039(00)85823-2
Chamorro E, Pérez P, Domingo LR (2013) On the nature of Parr functions to predict the most reactive sites along organic polar reactions. Chem Phys Lett 582:141–143. https://doi.org/10.1016/j.cplett.2013.07.020
Chiu P, Lautens M (1997) Using ring-opening reactions of oxabicyclic compounds as a strategy in organic synthesis, Stereoselective Heterocyclic Synthesis II. Springer, Berlin, pp 1–85
Diels O (1929) The Diels-Alder Reaction. Ber Dtsch Chem Ges 62:554–562
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Domingo LR, Pérez P, Ortega DE (2013a) Why do five-membered heterocyclic compounds sometimes not participate in polar diels-alder reactions? J Org Chem 78:2462–2471. https://doi.org/10.1021/jo3027017
Domingo LR, Pérez P, Sáez JA (2013b) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494. https://doi.org/10.1039/C2RA22886F
Domingo LR, Emamian S, Salami M, Ríos-Gutiérrez M (2016) Understanding the molecular mechanism of the [3+ 2] cycloaddition reaction of benzonitrile oxide toward electron-rich N-vinylpyrrole: a DFT study. J Phys Org Chem 29:368–376. https://doi.org/10.1002/poc.3544
Emamian S (2016) How the mechanism of a [3+ 2] cycloaddition reaction involving a stabilized N-lithiated azomethine ylide toward a π-deficient alkene is changed to stepwise by solvent polarity? What is the origin of its regio-and endo stereospecificity? A DFT study using NBO, QTAIM, and NCI analyses. RSC Adv 6:75299–75314. https://doi.org/10.1039/C6RA13913B
Emamian S, Soleymani M, Moosavi SS (2019) Copper (i)-catalyzed asymmetric aza Diels-Alder reactions of azoalkenes toward fulvenes: a molecular electron density theory study. New J Chem 43:4765–4776. https://doi.org/10.1039/C9NJ00269C
Frisch MJTG, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2013) Gaussian 09, Revision E.01. Gaussian Inc., Wallingford CT
Geerlings P, De Proft F, Langenaeker W (2003) Conceptual density functional theory. Chem Rev 103:1793–1874. https://doi.org/10.1021/cr990029p
Gonzalez C, Schlegel HB (1989) An improved algorithm for reaction path following. J Chem Phys 90:2154–2161. https://doi.org/10.1063/1.456010
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527. https://doi.org/10.1021/j100377a021
Hill TA, Stewart SG, Gordon CP, Ackland SP, Gilbert J, Sauer B, Sakoff JA, McCluskey A (2008) Norcantharidin analogues: synthesis, anticancer activity and protein phosphatase 1 and 2A inhibition. ChemMedChem 3:1878–1892. https://doi.org/10.1002/cmdc.200800192
Huisgen R (1984) 1, 3-Dipolar cycloaddition chemistry. Wiley, New York, pp 55–92
Huyen CTT, Luyen BTT, Khan GJ, Oanh HV, Hung TM, Li H-J, Li P (2018) Chemical constituents from Cimicifuga dahurica and their anti-proliferative effects on MCF-7 breast cancer cells. Molecules 23:1083. https://doi.org/10.3390/molecules23051083
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct-THEOCHEM 865:68–72. https://doi.org/10.1016/j.theochem.2008.06.022
Lee C, Yang W, Parr RG (1988) Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37:785. https://doi.org/10.1103/physrevb.37.785
McCluskey A, Walkom C, Bowyer MC, Ackland SP, Gardiner E, Sakoff JA (2001) Cantharimides: a new class of modified cantharidin analogues inhibiting protein phosphatases 1 and 2A. Bioorg Med Chem Lett 11:2941–2946. https://doi.org/10.1016/S0960-894X(01)00594-7
Molteni G, Ponti A (2017) The nitrilimine-alkene cycloaddition regioselectivity rationalized by density functional theory reactivity indices. Molecules 22:202. https://doi.org/10.3390/molecules22020202
Norman RO (1982) Principles of organic synthesis, 2nd edn. Chapman and Hall, London, pp 270–298
Opoku E, Arhin G, Pipim GB, Adams AH, Tia R, Adei E (2020) Site-, enantio-and stereo-selectivities of the 1, 3-dipolar cycloaddition reactions of oxanorbornadiene with C, N-disubstituted nitrones and dimethyl nitrilimines: a DFT mechanistic study. Theor Chem Acc 139:1–15. https://doi.org/10.1007/s00214-019-2529-8
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Parr RG, Weitao Y (1989) Density-functional theory of atoms and molecules. Oxford University Press, Oxford
Parr RG, Lv S, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924. https://doi.org/10.1021/ja983494x
Reymond J-L, Vogel P (1990) New chiral auxiliaries and new optically pure ketene equivalents derived from tartaric acids. Improved synthesis of (−)-7-oxabicyclo [2.2. 1] hept-5-en-2-one. Tetrahedron Asymmetry 1:729–736. https://doi.org/10.1016/S0957-4166(00)82385-X
Saleem M, Ali MS, Hussain S, Jabbar A, Ashraf M, Lee YS (2007) Marine natural products of fungal origin. Nat Prod Rep 24:1142–1152. https://doi.org/10.1039/B607254M
Schindler CS, Carreira EM (2009) Rapid formation of complexity in the total synthesis of natural products enabled by oxabicyclo [2.2. 1] heptene building blocks. Chem Soc Rev 38:3222–3241. https://doi.org/10.1039/B915448P
Smith MB (2020) March's advanced organic chemistry: reactions, mechanisms, and structure. John Wiley & Sons, New Jersey
Soleymani M (2018) DFT study of double 1, 3-dipolar cycloaddition of nitrilimines with allenoates. Monatsh Chem Chem Mon 149:2183–2193. https://doi.org/10.1007/s00706-018-2311-y
Soleymani M (2019) A density functional theory study on the [3+ 2] cycloaddition of N-(p-methylphenacyl) benzothiazolium ylide and 1-nitro-2-(p-methoxyphenyl) ethene: the formation of two diastereomeric adducts via two different mechanisms. Theor Chem Acc 138:87. https://doi.org/10.1007/s00214-019-2477-3
Soleymani M (2020) Regio-, diastereo-and enantioselectivity in the synthesis of CF3-containing spiro [pyrrolidin-3, 2′ oxindole] through the organocatalytic [3+ 2] cycloaddition reaction: A molecular electron density theory study. J Fluorine Chem. https://doi.org/10.1016/j.jfluchem.2020.109566
Soleymani M, Chegeni ZK (2019) A molecular electron density theory study on the [3+ 2] cycloaddition reaction of 5, 5-dimethyl-1-pyrroline N-oxide with 2-cyclopentenone. J Mol Graphics Modell 92:256–266. https://doi.org/10.1016/j.jmgm.2019.08.007
Soleymani M, Jahanparvar S (2020) A computational study on the [3+ 2] cycloaddition of para-quinone methides with nitrile imines: a two-stage one-step mechanism. Monatsh Chem Chem Mon 151:51–61. https://doi.org/10.1007/s00706-019-02531-2
Sykes P (1986) A guidebook to mechanism in organic chemistry. Pearson Education, India
Vieira E, Vogel P (1982) Copper (I)-and Copper (II)-catalyzed Diels-Alder additions of α-substituted acrylonitrile to furan. The synthesis of 7-Oxa-bicyclo [2.2. 1] hept-5-en-2-one. Helv Chim Acta 65:1700–1706. https://doi.org/10.1002/hlca.19820650604
Vieira E, Vogel P (1983) The preparation of optically pure 7-Oxabicyclo [2.2. 1] hept-2-ene derivatives. The CD spectrum of (+)-(1R)-7-Oxabicyclo [2.2. 1] hept-5-en-2-one. Helv Chim Acta 66:1865–1871. https://doi.org/10.1002/hlca.19830660627
Vogel P, Cossy J, Plumet J, Arjona O (1999) Derivatives of 7-oxabicyclo [2.2. 1] heptane in nature and as useful synthetic intermediates. Tetrahedron 55:13521–13642. https://doi.org/10.1016/S0040-4020(99)00845-5
Wiberg KB (1968) Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane. Tetrahedron 24:1083–1096. https://doi.org/10.1016/0040-4020(68)88057-3
Woodward RB, Hoffmann R (1965) Stereochemistry of electrocyclic reactions. J Am Chem Soc 87:395–397. https://doi.org/10.1021/ja01080a054
Yang W, Mortier WJ (1986) The use of global and local molecular parameters for the analysis of the gas-phase basicity of amines. J Am Chem Soc 108:5708–5711. https://doi.org/10.1021/ja00279a008
Zhao Y, Truhlar DG (2006) Comparative DFT study of van der Waals complexes: rare-gas dimers, alkaline-earth dimers, zinc dimer, and zinc-rare-gas dimers. J Phys Chem A 110:5121–5129. https://doi.org/10.1021/jp060231d
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Soleymani, M., Dashti Khavidaki, H. Regio- and stereochemistry in the intramolecular [4 + 2] and intermolecular [3 + 2] cycloaddition reactions in the synthesis of epoxypyrrolo[3,4-g]indazoles: a density functional theory study. Chem. Pap. 75, 951–965 (2021). https://doi.org/10.1007/s11696-020-01359-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11696-020-01359-z