

The selectivity and the molecular mechanism of the intramolecular [3+2] cycloaddition reaction of a nitrone–vinylphosphonate adduct was computationally studied within the molecular electron density theory using density functional theory method at the B3LYP/6-31G(d,p) level of theory. Conceptual density functional theory indices show that the nitrone–vinylphosphonate adduct has dual strong electrophilic and nucleophilic character. Local Parr functions reactivity indices reveal that this reaction favors the formation of the fused regioisomers in accordance with the experimental data. Analysis of different energetic profiles indicates that the fused-endo competitive pathway is favored kinetically, whereby this intramolecular reaction is characterized by exothermic and exergonic character. The geometry of transition states structures shows that the mechanism of this cycloaddition reaction is synchronous. Electron localization function topological analysis of the changes in electron density during the most favored reaction pathway shows that the mechanism is synchronous non-concerted.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Heterocyclic compounds and their derivatives are of great interest in pharmaceutical research because they have diverse useful biological activities. Since many medicinal and pharmaceutical commercial substances contain a heterocyclic moiety in their structure, numerous experimental and theoretical studies have been performed to develop new economic and efficient synthetic methods for the preparation of these structures.1
One of the most exploited strategies for the preparation of various heterocycles, is the reactions of cycloaddition,2 including the [3+2] cycloaddition (32CA) reaction.3 The inter- and intramolecular 32CA reaction between nitrone (e.g., compound 1) and ethylene (2) or its derivatives (alkenes) has found a wide use as an efficient method of synthesizing isoxazolidine heterocycles like compound 3 (Scheme 1).3,4 Isoxazolidines have been used as starting materials or important intermediates for the synthesis of natural products and biologically active molecules.5 These heterocycles have been shown to possess antimicrobial,6 antifungal,7 anti-inflammatory,8 and antioxidant activity.9

32CA reaction between nitrone 1 and ethylene (2) for the synthesis of isoxazolidine heterocycle 3
Phosphonates are analogs of natural phosphates and phosphonic acid and are characterized by the presence of one or more C−P bonds. An interest related to these compounds has increased tremendously because of their potential as drugs (also prodrugs).10 Phosphonates were observed to have antibacterial, anticancer, and antiparasitic activity.10,11 These compounds can be also regarded as analogs of carboxylic acids, amino acids, and peptides.12
In recent years, the intramolecular 32CA reactions have been well studied experimentally and theoretically in order to understand the mechanism and the origin of the selectivity of this type of cycloaddition.11,13,14,15,16,17,18,19 A major challenge in organic synthesis is to control the stereochemistry of the addition step. The stereochemistry of these reactions can be controlled either by choosing the appropriate reactants or by governing the reaction with a molecular complex acting as catalyst.4 In some cases, cycloaddition reactions proceed through a stepwise mechanism.17,20,21,22,23,24 Recent molecular electron density theory (MEDT)25 studies of 32CA reactions involving the simplest three-atom components (TACs) have established a relationship between their electronic structure and reactivity.26
The intramolecular 32CA reactions of nitrones, leading to the formation of heterocycles via the bridged and/or the fused modes, constitute a versatile and efficient approach to the stereo- and regioselective construction of complex heterocyclic architectures having both nitrogen and oxygen atoms in a five-membered ring.27,28 Recently Huang et al.29 reported the intramolecular 32CA reaction of bifunctional nitrone–vinylphosphonate 6 prepared in situ from O-vinylphosphonylated salicylaldehyde 4 and N-methylhydroxylamine hydrochloride (5). The authors found that this 32CA reaction is characterized by complete fused-endo regio- and stereoselectivity in obtaining isoxazole derivative 7 (Scheme 2). The regioisomeric C3–C4 and O1–C5 reaction channel corresponds to the fused mode, while the C3–C5 and O1–C4 interaction corresponds to the bridged mode (Fig. 1).

Synthesis of polycyclic phosphonated isoxazolidine 7 via an intramolecular 32CA reaction of nitrone–vinylphosphonate adduct 6

The fused and bridged regioisomeric modes of the intramolecular 32CA reaction of nitrone–vinylphosphonate adduct 6, together with the numbering of the atoms involved in the reaction.
The main goal of this work is to undertake a theoretical study of intramolecular 32CA reaction of nitrone–alkene 6, in which we investigate the experimentally observed selectivity, i.e., the regio- and stereoselectivity, in order to shed the light on the molecular mechanism of this reaction.
The present MEDT study has been divided into three parts; starting with the analysis of conceptual density functional theory (CDFT) reactivity indices, then energies and geometries, followed by the study of the nature of the molecular mechanism based on electron localization function (ELF) topological analysis. In the following investigation, the E-configuration of the nitrone double bond of adduct 6 will be used.
CDFT reactivity indices analysis
Global indices
The values of chemical potential (μ), chemical hardness (η), electrophilicity index (ω), and nucleophilicity index (N), as well as energies of frontier molecular orbital (FMO), i.e., the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) of nitrone–vinylphosphonate 6 have been calculated as shown below (see Computation details) and collected in Table 1.
From Table 1, we can notice that nitrone–vinylphosphonate 6 has a high electrophilicity index ω of 1.44 eV, being classified as the borderline between moderate and strong electrophile according to the electrophilicity scale.30 On the other hand, compound 6 has also high nucleophilicity index and, therefore, may be classified as a strong nucleophile.31 The polar 32CA reactions require participation of good electrophiles and good nucleophiles. Therefore, the intramolecular 32CA of nitrone–vinylphosphonate 6 will occur with a polar character demanding low activation energy.32
Local indices
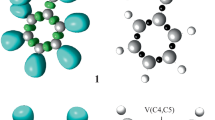
In polar cycloaddition reactions involving the participation of non-symmetric reagents, the most favorable reaction channel is that involving the initial two-center interaction between the most electrophilic center of the electrophile and the most nucleophilic center of the nucleophile. In this context, Domingo et al.33 proposed the electrophilic \({P}_{\mathrm{k}}^{+}\) and nucleophilic \({P}_{\mathrm{k}}^{-}\) Parr functions, derived from the changes of spin electron density via the global electron density transfer34 process from the nucleophile to the electrophile, as powerful tools in the study of the local reactivity in polar processes. Accordingly, the electrophilic \({P}_{\mathrm{k}}^{+}\) and the nucleophilic \({P}_{\mathrm{k}}^{-}\) Parr functions of nitrone–vinylphosphonate 6 were analyzed in order to characterize the most electrophilic and nucleophilic centers in this molecule involved in the intramolecular 32CA reaction and, thus, to explain the fused regioselectivity experimentally observed. The 3D representations of the atomic spin density (ASD) of the radical anion and the radical cation of nitrone–vinylphosphonate 6 including the values of electrophilic and nucleophilic Parr functions \({P}_{\mathrm{k}}^{+}\) and \({P}_{\mathrm{k}}^{-}\) are given in Figure 2.

Maps of ASD representation of radical cation 6·+ and radical anion 6·– of nitrone–vinylphosphonate 6 with local Parr function values on the reactive atoms.
As shown in Figure 2, the electrophilic Parr functions \({P}_{\mathrm{k}}^{+}\) of nitrone–vinylphosphonate 6 are mainly concentrated on the C4 carbon atom (\({P}_{\mathrm{k}}^{+}\) 0.17) (see Figure 1 for atom numbering). On the other hand, the nucleophilic Parr functions \({P}_{\mathrm{k}}^{-}\) of nitrone–vinylphosphonate 6 are mainly concentrated on the O1 oxygen atom (\({P}_{\mathrm{k}}^{-}\) 0.57). Consequently, the most favored regioisomeric interaction will occur between the O1 oxygen atom and C4 carbon atom, giving the fused cycloadducts, as observed experimentally.29
Energies and geometries
The intramolecular reactions are characterized by a single reagent which reacts with itself to form a new product. Therefore, in the intramolecular 32CA reactions, the reagent contains two reactive parts, the TAC35 part and the ethylene part. Thus, two reaction regioisomeric channels, namely the fused mode and the bridged mode, are possible in this intramolecular 32CA reaction. In addition, in each regioisomeric channel, there are two stereoisomeric approaches, the endo and exo. Thereby, in this intramolecular 32CA reaction, there are four possible transition states (TSs), fused-endo (TSfn), fused-exo (TSfx), bridged-endo (TSbn), and bridged-exo (TSbx), leading to the formation of four possible cycloadducts, 7fn, 7fx, 7bn, and 7bx, respectively (Scheme 3).

The possible reaction pathways for the intramolecular 32CA reaction of nitrone–vinylphosphonate 6
Table 2 shows the values of the relative electronic energies (in kcal·mol–1) in gas phase and in solution of DCM, while total energies are given in Table S1 in Supplementary information file. These relative energies are calculated with respect to energy of nitrone–vinylphosphonate 6 that was taken as reference. From the analysis of the activation energies associated with the different reaction pathways we can notice that the fused-endo (TSfn) approach is the more kinetically favored (ΔE 8.41 kcal·mol–1) one, the second favored approach is TSbn (ΔE 18.50 kcal·mol–1, indicating for total regio- and stereoselectivity.
On the other hand, we can notice from the relative energies of the cycloadducts, that the fused and bridged-endo approaches are only under kinetic control, because of their exothermic character, while that of the bridged-exo (ΔE 38.08 kcal·mol–1) is at the same time under kinetic and thermodynamic control due to its endothermic character. The instability of the bridged-exo cycloadduct 7bx may be attributed to the high ring tension caused by the sevenmembered ring. Therefore, the fused and bridged-endo pathways are irreversible processes and the bridged-exo pathway is reversible, and this intramolecular 32CA reaction thus leads to the formation of the kinetically favored cycloadduct 7fn generated by the fused-endo pathway as a single regio- and stereoisomer, as observed experimentally,29 and in accordance with the local indices analysis based on Parr functions.
DCM was used experimentally as a reaction solvent, which can have a certain influence on the energy values in the case of the better solvation of the reagents. Table 2 clearly shows an increase in the activation energies of the fused modes in comparison with those obtained in the gas phase. These increases in fused mode energies are due to stabilization of the nitrone–vinylphosphonate 6 reagent in the DCM.36 Note that for the non-favored bridged pathway (bridged-exo), we noticed a slight decrease of activation energy, but it is always the least favored pathway. The main change in activation energy is related to the more favored approach fused-endo, which amounts to 1.59 kcal·mol–1. We can also observe that the solvent effects decrease the exothermic character of this intramolecular 32CA reaction by a values of 2.60, 2.77, 1.60, and 2.36 kcal·mol–1 for cyclization products 7fn, 7fx, 7bn, and 7bx, respectively. However, the solvent effects have no influence on the obtained gas phase regio- and stereoselectivities.
In order to take in consideration all the used experimental conditions, further calculations were performed which included the DCM solvent, temperature of 298K and 1 atmosphere of pressure. Thus, the values of relative enthalpies, entropies, and free energies for the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 are collected in Table 3, while total energy values are given in Table S2 in Supplementary information file.
A comparison between the activation enthalpies indicates that the fused-endo mode (TSfn) leading to isoxazolidine 7fn is still the most favorable approach by 4.75 kcal·mol–1 over the second most favored, i.e., bridgedendo mode (TSbn). In addition, the fused pathways and bridged-endo one are exothermic, while the bridged-exo is endothermic. Thus, the inclusion of thermal corrections in calculation does not change the selectivity and the thermal character obtained in the gas phase calculations.
On the other hand, we can notice that the values of relative activation entropies are low, which may be due to the unimolecular character of this 32CA reaction. The addition of entropic contribution to the enthalpies slightly increases the activation free energies by values in the range of 2–3 kcal·mol–1, and always the fused-endo reaction pathway is the more favored one, TSfn having by about 4 kcal·mol–1 lower ΔG≠ value than the second lowest value (bridged-endo path). Also, we can remark that the fused and the bridged-endo paths are exergonic, because they have negative relative free energies of the associated cycloadducts, while the bridged-exo path has an endergonic character, and therefore, it is the less favored path. Consequently, this intramolecular 32CA reaction of nitrone–vinylphosphonate 6 in DCM is characterized by an exothermic and exergonic characters favoring the formation of the fused-endo cycloadduct, in good agreement with experiment.
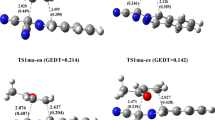
Figure 3 illustrates the optimized geometries of the TSs associated with the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 together with the length of the new forming bonds. The Cartesian coordinates of the stationary points involved in the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 together with the imaginary frequency of transition states are given in Supplementary information file.

Optimized structures of the TSs associated with the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 together with the lengths of the newly forming bonds (Å).
From Figure 3, the lengths of the new O1–C5 and C3–C4 bonds in the fused mode are 1.921 and 2.321 Å for TSfn and 1.751 and 2.391 Å for TSfx, while for the bridged mode, the lengths of the newly formed bonds O1–C4 and C3–C5 are 2.199 and 2.077 Å for TSbn and 2.704 and 2.129 Å for TSbx. We clearly notice that the length of the C−O bond is shorter than that of the C–C bond, and if we take in consideration that a fully formed C–O bond is shorter than a C–C bond, we can conclude that the mechanism of formation of the new bonds is synchronous in the fused and bridged-endo pathways, and asynchronous in the unfavorable bridged-exo pathway.
ELF topological analysis of the cleavage and formation of bonds
An attractive procedure that gives us a direct link between the distribution of electron density and the chemical structure is the quantum-chemical topological analysis of ELF established by Becke and Edgecombe.37 In several studies we applied ELF topological analysis to different chemical systems in order to study the reaction mechanism and the reactivity of molecular systems.21,23,38,39,40
ELF localization domains and their attractor positions, together with the most representative valence basin populations of some pertinent points (MC, TSfn, P1–3, 7fn) selected from the intrinsic reaction coordinate (IRC) curve (Fig. 4) are shown in Figure 5. ELF valence basin populations of these selected points corresponding to the formation of the O1–C5 and C3–C4 single bonds along the most favorable path (fused-endo) of the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 are gathered in Table 4.

Relative energy curve of the fused-endo path of the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 together with selected IRC points.

ELF basins attractor positions of relevant points of the IRC curve together with the populations of the relevant valence basins along the most favorable fused-endo path of the intramolecular 32CA reaction of nitrone–vinylphosphonate 6.
At the stage of molecular complex (MC), we can notice that the O1···N2···C3 and C4···C5 reactive regions of this system are characterized by two monosynaptic basins V(O1) and V'(O1) with a summary population of 5.90 e, accounting for the presence of three-electron non-pair region at the atomic center O1. The O1···N2 region is characterized by a valence basin with 1.34 e population associated to the O1−N2 single bond. In addition, we can notice the presence of V(C3,N2) disynaptic basin with population 3.99 e, which accounts for the C3=N2 double bond. On the other hand, the C4···C5 reactive region is characterized by the presence of two disynaptic basins V(C4,C5) and V'(C4,C5) with 1.70 and 2.09 e, respectively, accounting for C4=C5 double bond character.
At TSfn, the most noticeable change is the apparition of two V(C3) and V(C4) monosynaptic basins with 0.31 and 0.46 e of electron density, indicating the creation beginning of two reactive pseudoradical centers.41 Also, a V(N2) monosynaptic basin appears at this point integrating 1.36 e which is associated with N2 non-bonding electron density region. This new electron population region is created from the decrease of electron density from the disynaptic basins V(C4,C5), V'(C4,C5), and V(C3,N2) that become populated 2.79 and 2.64 e, respectively. Note that the C4···C5 reactive region develops to contain only one disynaptic basin associated with the forming C4−C5 single bond.
At the IRC point P1, which is the first selected point after TSfn, the main change is presented by the increase of electron density of the monosynaptic basins V(C3) and V(C4) to achieve 0.53 and 0.74 e, respectively. In the same time, the V(C4,C5) and V(C3,N2) disynaptic basins continue their depopulation to become 2.25 and 2.26 e, respectively.
At the IRC point P2, the remarkable change is the formation of two disynaptic basins V(O1,C5) and V(C3,C4) with a population of 0.99 and 1.55 e, respectively. These new basins are associated with the new forming C3–C4 and O1–C5 bonds. Thus, the formation of these disynaptic basins in the same point account for that the formation of the new bonds is slightly synchronous, in which the C3–C4 new bond is formed through pseudodiradical mechanism, while the O1–C5 one is formed via a dative mechanism through the donation of a part of the non-bonding electron density of the oxygen O1 atom to the C5 atomic center. In addition to the previous remarks, the electronic population of disynaptic basins V(C4,C5) and V(C3,N2) decreases slightly to 2.01 and 1.99 e, respectively.
At the IRC point P3 which is the last point before the full formation of cycloadduct 7fn, the electron density of the new V(O1,C5) and V(C3,C4) disynaptic basins increases to become 1.14 and 1.71 e, respectively. On the other hand, the electronic population of the V(C4,C5) and V(C3,N2) disynaptic basins slightly decreases further to arrive at 1.94 and 1.88 e, respectively.
Upon the arrival at cycloadduct 7fn, which is the last point on the IRC curve, the population of the reactive basins changes slightly, whereby the values associated with the newly forming bond C3–C4 and O1–C5 increase to become 1.82 and 1.21 e, respectively, and those of the reactive region C3···N2 (V(C3,N2)) and C4···C5 (V(C4,C5)) decrease to 1.82 and 1.89 e, respectively.
In conclusion of this ELF topological analysis, the intramolecular 32CA reaction of the studied nitrone–vinylphosphonate adduct begins with decreasing bond order of the C4=C5 and C3=N2 double bonds forming pseudodiradical centers at C3 and C4 atoms. These pseudodiradical centers are necessary for the formation of the new C3–C4 bond. The second O1–C5 single bond is formed through the donation of part of the non-bonding electron density of the O1 oxygen on the C5 carbon. The two new bonds are formed simultaneously, which accounts for a synchronous non-concerted mechanism.
In this work, we have performed a MEDT study at the DFT B3LYP/6-31G(d,p) computational level of theory of the intramolecular 32CA reaction of a nitrone–vinylphosphonate adduct, in order to investigate the experimentally observed selectivity and to shed light on the nature of molecular mechanism of this type of 32CA reaction. The main conclusions of this study are as follows:
1) CDFT reactivity indices analysis shows that the nitrone–vinylphosphonate adduct is a strong nucleophile and moderate electrophile which may undergo cyclization through a polar 32CA reaction with low activation energy.
2) Analysis of local reactivity indices based on Parr functions indicates that the main interaction is that between the most nucleophilic O1 atom center and the most electrophilic C5 atom center, leading to the formation of the fused regioisomers, as observed experimentally.
3) Analysis of energetic profiles, shows that the fusedendo reaction pathway is kinetically favored leading to the formation of the corresponding cycloadduct as a single product.
4) The presence of solvent DCM has no qualitative influence on the regio- and diastereoselectivity of the reaction, but it increases slightly the activation energies and decreases the exothermic character of this intramolecular 32CA reaction.
5) Analysis of thermodynamic parameters associated with this intramolecular 32CA reaction, indicates that it is exothermic and exergonic, characterized by low entropy values due to its monomolecular nature.
6) Analysis of TSs geometries indicates that this intramolecular 32CA reaction of the nitrone–vinylphosphonate adduct proceeds via a synchronous mechanism for the more favored pathways.
7) ELF topological analysis of the intramolecular 32CA reaction of cyclization shows that it proceeds through a non-concerted synchronous mechanism.
Computational details
In this theoretical study, for the geometry optimization of all stationary points, we have used the Gaussian 09 program.42 The theoretical method used for this study is the DFT with the B3LYP hybrid functional in conjunction with the basis set 6-31G(d,p).43,44 This level is shown to be suitable for geometry optimization and electronic property analysis of 32CA reactions and [4+2] Diels–Alder cycloaddition reactions.23,38,40,45,46 The structures of the TSs have been confirmed by the existence of a single imaginary frequency and correspond to the new forming bonds. Several studies devoted to 32CA reactions have reported that the inclusion of solvent effects in the optimization of the geometry causes slight changes in the geometries of the gas phase.13,47 Thus, the effects of DCM solvent were also taken into consideration through single point calculation from the gas phase optimized geometries using the polarizable continuum model48 within the self-coherent reaction field.49,50 Values of thermodynamic properties such as enthalpies, entropies, and Gibbs free energies in DCM were calculated using standard statistical thermodynamics at 298K and 1 atm through the optimized gas phase structures.51 The natural bond orbitals method52,53 was used for the analysis of electronic structures in TSs. The IRC54 paths were traced in order to verify the energy profiles connecting each TS to the two associated minima of the proposed mechanism using the second order González–Schlegel integration method.55
The CDFT global reactivity descriptors,56,57 i.e., the electronic potential (μ), the chemical hardness (η),58 the electrophilicity index (ω),59 and the empirical nucleophilicity index (N)60 have been calculated from the energies of the FMO (HOMO and LUMO), according to the following equations: μ = (εHOMO + εLUMO)/2, η = εLUMO – εHOMO, ω = μ2/2η, N = εHOMO(Nu) – εHOMO(TCE), where, TCE is the abbreviation of tetracyanoethylene, which is taken as reference, because it has the lowest HOMO energy among most widely used ethylenic molecules.
The electrophilic (Pk+) and nucleophilic (Pk–) Parr functions33 which enable characterization of the electrophilic and nucleophilic centers of a molecule were obtained by analysis of the Mulliken atomic spin density of the radical anion and the radical cation, respectively, of the studied molecule. Electronic structures of the selected structures were studied by ELF37 over the B3LYP/6-31G(d,p) single determinant wave functions using the Multiwfn program.61
Supplementary information file containing values of total energies in gas phase and in dichloromethane, total enthalpies, entropies, and free energies, as well as the Cartesian coordinates of the stationary points involved in the intramolecular 32CA reaction of nitrone–vinylphosphonate 6 is available at the journal website http://springerlink.bibliotecabuap.elogim.com/journal/10593.
References
Kaur, N. J. Heterocycl. Chem. 2015, 52, 953.
Carruthers, W. Cycloaddition Reactions in Organic Synthesis; Pergamon Press: London, 1990. eBook; Elsevier, 2013.
Huisgen, R. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; Wiley: New York, 1984, Vol. 1, p. 55.
Gothelf, K. V.; Jørgensen, K. A. Chem. Rev. 1998, 98, 863.
Berthet, M.; Cheviet, T.; Dujardin, G.; Parrot, I.; Martinez, J. Chem. Rev. 2016, 116, 15235.
Sadashiva, M. P.; Mallesha, H.; Murthy, K. K.; Rangappa, K. S. Bioorg. Med. Chem. Lett. 2005, 15, 1811.
Żelechowski, K.; Gołębiewski, W. M.; Krawczyk, M. Monatsh. Chem. 2015, 146, 1895.
Mullen, G. B.; Swift, P. A.; Georgiev, V. S. J. Pharm. Sci. 1987, 76, 930.
Kaur, M.; Singh, B.; Singh, B.; Arjuna, A. J. Heterocycl. Chem. 2017, 54, 1348.
Groaz, E.; De Jonghe, S. Front. Chem. 2020, 8, 616863.
Sennikova, V. V; Zalaltdinova, A. V; Sadykova, Y. M.; Khamatgalimov, A. R.; Gazizov, A. S.; Voloshina, A. D.; Lyubina, A. P.; Amerhanova, S. K.; Voronina, J. K.; Chugunova, E. A.; Appazov, N. O.; Burilov, A. R.; Pudovik, M. A. Int. J. Mol. Sci. 2022, 23, 14348.
Turhanen, P. A. J. Biomed. Res. Environ. Sci. 2022, 3, 195.
Duret, G.; Quinlan, R.; Yin, B.; Martin, R. E.; Bisseret, P.; Neuburger, M.; Gandon, V.; Blanchard, N. J. Org. Chem. 2017, 82, 1726.
Chafaa, F.; Hellel, D.; Khorief, A.; Djerourou, A. A. Tetrahedron Lett. 2016, 57, 67.
Chafaa, F.; Nacereddine, A. K.; Djerourou, A. Lett. Org. Chem. 2020, 17, 260.
Domingo, L. R.; Ríos-Gutiérrez, M.; Adjieufack, A. I.; Ndassa, I. M.; Nouhou, C. N.; Mbadcam, J. K. ChemistrySelect 2018, 3, 5412.
Jasiński, R.; Dresler, E. Organics 2020, 1, 49.
Jasiński, R. Tetrahedron 2013, 69, 927.
Sobhi, C.; Khorief Nacereddine, A.; Djerourou, A.; Ríos-Gutiérrez, M.; Domingo, L. R. J. Phys. Org. Chem. 2017, 30, e3637.
Żmigrodzka, M.; Sadowski, M.; Kras, J.; Dresler, E.; Demchuk, O. M.; Kula, K. Sci. Rad. 2022, 1, 26.
Barama, L.; Bayoud, B.; Chafaa, F.; Nacereddine, A. K.; Djerourou, A. Struct. Chem. 2018, 29, 1709.
Lamri, S.; Heddam, A.; Kara, M.; Yahia, W.; Khorief Nacereddine, A. Organics 2021, 2, 57.
Hamdaoui, H.; Khorief Nacereddine, A.; Djerourou, A. J. Phys. Org. Chem. 2022, 36, e4462.
Sadi, S.; Khorief Nacereddine, A.; Djerourou, A. J. Phys. Org. Chem. 2022, 35, e4311.
Domingo, L. R. Molecules 2016, 21, 1319.
Ríos-Gutiérrez, M.; Domingo, L. R. Eur. J. Org. Chem. 2019, 267.
Koumbis, A. E.; Gallos, J. K. Curr. Org. Chem. 2003, 7, 585.
Tufariello, J. J. In 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; Wiley: New York, 1984, vol. 2, p. 83.
Huang, T.; Wang, Q.; Kong, D.; Wu, M. Tetrahedron Lett. 2019, 60, 150913.
Domingo, L. R.; Aurell, M. J.; Pérez, P.; Contreras, R. Tetrahedron 2002, 58, 4417.
Jaramillo, P.; Domingo, L. R.; Chamorro, E.; Pérez, P. A J. Mol. Struct.: THEOCHEM 2008, 865, 68.
Domingo, L. R.; Sáez, J. A. Org. Biomol. Chem. 2009, 7, 3576.
Domingo, L. R.; Pérez, P.; Sáez, J. A. RSC Adv. 2013, 3, 1486.
Domingo, L. R. RSC Adv. 2014, 4, 32415.
Emamian, S.; Lu, T.; Domingo, L. R.; Heidarpoor Saremi, L.; Ríos-Gutiérrez, M. Chem. Phys. 2018, 501, 128.
Benchouk, W.; Mekelleche, S. M.; Silvi, B.; Aurell, M. J.; Domingo, L. R. J. Phys. Org. Chem. 2011, 24, 611.
Becke, A. D.; Edgecombe, K. E. J. Chem. Phys. 1990, 92, 5397.
Nacereddine, A. K. J. Mol. Model. 2020, 26, 328.
Yahia, W.; Nacereddine, A. K.; Djerourou, A. Intl. J. Quantum Chem. 2017, 118, e25540.
Chafaa, F.; Nacereddine, A. K.; Djerourou, A. Theor. Chem. Acc. 2019, 123.
Domingo, L. R.; Sáez, J. A. J. Org. Chem. 2011, 76, 373.
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, Ma.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Li, X.; Caricato, M.; Marenich, A.; Bloino, J.; Janesko, B. G.; Gomperts, R.; Mennucci, B.; Hratchian, H. P.; Ortiz, J. V.; Izmaylov, A. F.; Sonnenberg, J. L.; Williams-Young, D.; Ding, F.; Lipparini, F.; Egidi, F.; Goings, J.; Peng, B.; Petrone, A.; Henderson, T.; Ranasinghe, D.; Zakrzewski, V. G.; Gao, J.; Rega, N.; Zheng, G.; Liang, W.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Throssell, K.; Montgomery, J. A., Jr.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Keith, T.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Millam, J. M.; Klene, M.; Adamo, C.; Cammi, R.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Farkas, O.; Foresman, J. B.; Fox, D. J. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, 2016.
Lee, C.; Yang, W.; Parr, R. G. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785.
Becke, A. D. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098.
Ríos-Gutiérrez, M.; Domingo, L. R.; Jasiński, R. RSC Adv. 2021, 11, 9698.
Mitka, K.; Fela, K.; Olszewska, A.; Jasiński, R. Molecules 2021, 26, 7147.
Layeb, H.; Nacereddine, A. K.; Djerourou, A.; Ríos-Gutiérrez, M.; Domingo, L. R. J. Mol. Model. 2015, 21, 104.
Tomasi, J.; Persico, M. Chem. Rev. 1994, 94, 2027.
Cancès, E.; Mennucci, B.; Tomasi, J. J. Chem. Phys. 1997, 107, 3032.
Cossi, M.; Barone, V.; Cammi, R.; Tomasi, J. Chem. Phys. Lett. 1996, 255, 327.
Becke, A. D. J. Chem. Phys. 1993, 98(7), 5648.
Reed, A. E.; Weinstock, R. B.; Weinhold, F. J. Chem. Phys. 1985, 83, 735.
Reed, A. E.; Curtiss, L. A.; Weinhold, F. Chem. Rev. 1988, 88, 899.
Fukui, K. J. Phys. Chem. 1970, 74, 4161.
Gonzalez, C.; Schlegel, H. B. J. Chem. Phys. 1991, 95, 5853.
Geerlings, P.; De Proft, F.; Langenaeker, W. Chem. Rev. 2003, 103, 1793.
Domingo, L. R.; Ríos-Gutiérrez, M.; Pérez, P. Molecules 2016, 21, 748.
Parr, R. G.; Pearson, R. G. J. Am. Chem. Soc. 1983, 105, 7512.
Parr, R. G.; Szentpály, L. v.; Liu, S. J. Am. Chem. Soc. 1999, 12, 1922.
Domingo, L. R.; Chamorro, E.; Pérez, P. J. Org. Chem. 2008, 73, 4615.
Lu, T.; Chen, F. J. Comput. Chem. 2012, 33, 580.
This work was supported by the Ministry of Higher Education and Scientific Research of the Algerian Government (project PRFU Code: B00L01EN210120220001).
Author information
Authors and Affiliations
Corresponding author
Additional information
Published in Khimiya Geterotsiklicheskikh Soedinenii, 2023, 59(3), 171–178
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Chafaa, F., Nacereddine, A.K. A molecular electron density theory study of mechanism and selectivity of the intramolecular [3+2] cycloaddition reaction of a nitrone–vinylphosphonate adduct. Chem Heterocycl Comp 59, 171–178 (2023). https://doi.org/10.1007/s10593-023-03179-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10593-023-03179-x