
Abstract
The equilibrium geometries, electronic structures and electronic properties of PmSi n (n = 3–10) clusters were systematically investigated using the ABCluster global search technique combined with density functional methods. The results revealed that the most stable structure of neutral PmSi n and their anions can be viewed as replacing a Si atom of the ground state structure of Si n+1 with a Pm atom. The adiabatic electron affinities of PmSi n are evaluated, and they differ little from those of SmSi n and EuSi n . Analyses of HOMO–LUMO gaps showed that introducing Pm atom to Si cluster can significantly improve photochemical reactivity of the cluster. And the improved effects are as good as those of the introducing Sm and Eu atom to Si cluster. The NPA calculations indicated that the 4f electrons of Pm atom in PmSi n (n = 3–10) clusters hardly participate in bonding and provide the total magnetic moments. Dissociation energy (DE) of rare earth metal (REM) atom from the lowest energy structure of REMSi n (n = 3–10) and their anions was calculated. The DEs of PmSi n , SmSi n and EuSi n are nearly identical. The DEs of PmSi − n , SmSi − n and EuSi − n are also nearly equal, and they are smaller than those of HoSi − n and PrSi − n .
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Over the past few decades, silicon-based clusters have attracted a great deal of attentions due to their acknowledged importance not only in the modern microelectronics industry, but also as building blocks for the developing new and tunable silicon-based nanomaterials [1,2,3,4,5,6,7,8]. Different from carbon clusters which usually show sp 2 hybridization, pure silicon clusters favor the sp 3 hybridization because Si atom has a larger p-orbital than C atom, and C atom has larger s-orbital than Si atom [9]. As a result, the pure Si clusters are chemically reactive because of the presence of unsaturated dangling bonds and are unsuitable as nanoscale building blocks [10, 11]. It has been found that doping an appropriate foreign atom such as transitional metal (TM) atom and rare earth metal (REM) atom inside silicon clusters can not only solve the deficiency, but also influence impressively the fascinating physical and chemical properties such as narrow HOMO–LUMO gaps, high magnetic moments, bright photoluminescence of these silicon-based clusters [12,13,14,15,16,17,18].
Up to date, there have been some scientific researches on REM-doped silicon clusters. For instance, Nakajima and co-workers firstly studied doping REM atoms into silicon cluster anions REMSi − n (REM = Tb, Ho, Lu, n = 6–20) clusters by using photoelectron spectra (PES) and reported their electronic structures and threshold energies of electron detachment [1, 2]. Subsequently, Grubisic et al. [3, 4] reported the PES of REMSi − n (REM = Ho, Gd, Pr, Sm, Eu, Yb; n = 3–13) and found that the PES can be categorized as three types in light of their motifs. Motivated by these experimental observations, some theoretical simulation and computation have been carried out for REMSi n clusters including PrSi n [7, 19], SmSi n [20], EuSi n [21, 22], GdSi n [23], HoSi n [11, 16, 24], YbSi n [25,26,27] and LuSi n [28] to evaluate their equilibrium configuration and electronic properties such as relative stability, HOMO–LUMO gap, adiabatic electron affinity (AEA), magnetic moment and so forth. Recently, we [29,30,31,32,33,34,35] investigated the most stable structures and properties of PrSi n , SmSi n , EuSi n , GdSi n , HoSi n , YbSi n and LuSi n (n ≤ 10) and their anions and concluded that (1) The REMSi n can be divided into two types according to the situation of 4f electron participation in bonding. One (corresponding to “A” type in Ref. [4]) is that the 4f electron hardly participates in bonding, and another (corresponding to “B” in Ref. [4]) is that the 4f electron prefers to participate in bonding. (2) The double-hybrid mPW2PLYP and/or B2PLYP functional can accurately predict electron affinities of REMSi n including both types of A and B. The pure and single-hybrid density functional theory such as popular PBE, PBE0, TPSSh and B3LYP, especially TPSSh, can only accurately evaluate the electron affinities for REMSi n of type A. Therefore, we applied the mPW2PLYP [36] methods in this paper to the determination of equilibrium configurations and electronic properties (including relative stability, HOMO–LUMO gap, AEA, magnetic moment and charge transfer characteristics) of neutral PmSi n (n = 3–10) and their anions with the target of understanding which type (A, B, or AB) PmSi n clusters belong to and how their properties differ from those of bare Si clusters and other REMSi n species. Our calculations will provide not only specific guidance for the study of medium-size clusters but also strong motivation for further experimental studies of these important PmSi n clusters and their anions.
2 Computational schemes
It is well known that locating the global minimum of the large clusters is usually a rather difficult task. The possibility of missing the ground state structure exists. For small size clusters, this problem can be solved by using a global optimization technique. But with the cluster size increasing, the number of local minimal isomers increases exponentially. The complex distribution of these huge numbers of local minimal isomer makes the potential energy surface locally very rugged, making an “ergodic” sampling on the potential energy surface of the large clusters (especially for heteroatom clusters) by computer simulation nearly impossible [37]. On the other hand, the most stable structure of SmSi n , EuSi n and YbSi n is substitutional structure [30, 31, 34]. Therefore, apart from the ABCluster [37], the “substitutional structure” is also taken into account in order to ensure the lowest energy structure can be selected into initial configuration as much as possible. The detailed description for the ABCluster global search technique combining with the Gaussian 09 codes [38] is as follows. The ABCluster uses the “artificial bee colony” (ABC) algorithm to perform the global optimization. Of course, it can be used as random generator only. The first step is that for n ≤ 7, 100 initial geometries of PmSi n generated by the ABCluster are selected (the ABCluster codes are black-box program, and the first geometry is generated randomly), and 300 are selected for n ≥ 8. Then, the TPSSh functional [39] combining with SMALL basis sets (which contains 6-31G basis set for Si atoms and ECP50MWB basis set [40] for Pm atom) is chosen to optimize the initial geometries of each cluster one by one with doublet electronic state. The second step is that the isomers with their energy differences within 0.8 eV from the lowest energy isomer from the first step for each species are chosen and reoptimized by using the TPSSh functional combined with the LARGE basis sets (which contains Stuttgart-ECP basis set [41, 42] for Pm and the cc-pVTZ [43] for Si atoms). In the third step, the isomers from the second step with their energy value within 0.8 eV from the lowest energy isomer are optimized by using the mPW2PLYP/LARGE method. The substitutional structures for PmSi n are generated by replacing each Si of the ground state structure of Si n+1 with a Pm atom. In fact, our experience is that for pure Si n cluster with n ≤ 11, the ground state structures predicted by using the ABCluster method with 100 initial geometries are the same as the known ones reported previously. For REMSi n clusters, when n ≤ 7, the 100 initial geometries generated from the ABCluster method include all of the substitutional structures. However, from n = 8, even 500 initial geometries generated from the ABCluster method do not include all of the substitutional structures. For instance, the most stable geometries of PmSi8 (and/or PmSi8 −) and PmSi9 − belong to substitutional structure, but cannot be found by ABCluster method. It is to say that the selection of the initial geometries to consider two types is necessary. In addition, only sextuplet electronic state was considered for neutral PmSi n clusters. The reason is that if 4f electrons of Pm atom participate scarcely in bonding (it is to say that electron configurations of Pm is [core]6s 24f 55d 0), the spin multiplicities of neutral PmSi n are sextuplet. If 4f electrons are involved in bonding (the electron configurations of Pm is [core]6s 24f 45d 1), the spin multiplicities of neutral PmSi n are still sextuplet. (Take PmSi10 as an example for this point of view, we have examined the energies of PmSi10 isomers with quartet and octuplet electronic states. The results revealed that the energies of octuplet electronic states are higher than that of sextuplet electronic state. For quartet electronic state, there are serious spin contamination). For their anions, the quintuplet and septet electronic states were considered for n = 3–6. The results revealed that the ground state of PmSi − n anions is septet electronic state. That is, the 4f electrons hardly participate in bonding. Although many structures were gotten, only several selected isomers were reported.
Harmonic frequency analysis was performed only at the TPSSh/LARGE level of theory to assure that the geometries presented in this work are local minima. The mPW2PLYP geometries were applied in single-point calculations with diffuse functions (which contains the aug-cc-pVTZ basis sets [43] for Si atoms and Stuttgart RSC 1997 ECP basis set augmented with 2pdfg diffuse functions [44] for Pm atom, denoted as aug-LARGE). Finally, the mPW2PLYP energies at 0 K are obtained by adding the ZPVE (zero point vibration energy) of the TPSSh. The energies of mPW2PLYP/aug-LARGE//mPW2PLYP/LARGE were used for calculations of properties such as AEA and dissociation energies. All of the calculations were performed using the GAUSSIAN 09 soft package [38]. It is reasonable to adopt the double-hybrid mPW2PLYP scheme. The average absolute deviation of the mPW2PLYP from experiment for 35 calculated AEA of REMSi n (excluded PrSi6) including type of A and B is only 0.05 eV as shown in Table 1. In our previous study, ECP28MWB small-core relativistic potentials and segmented (SEG) valence Gaussian valence basis sets (namely “SEG/ECP”) [45] were employed for the REM atoms [29,30,31,32,33]. At present, they are replaced by the Stuttgart-ECP basis sets. The reason is that (1) SEG/ECP basis set is sometimes excessive mixing of frozen core and valence orbitals; (2) it can save computation time; and (3) the result is agreement with experimental data as described above. The double-hybrid mPW2PYP is susceptible to the initial geometry. A poor initial geometry can result in convergence failure for mPW2PLYP scheme. On the other hand, the cost for frequency calculation of mPW2PLYP is expensive. So the TPSSh method is used for optimization of initial geometries. Our experience is that apart from the TPSSh, the PBE, PBE0 and B3LYP methods can also be recommended for optimization of initial geometries.
3 Results and discussion
3.1 Neutral and anionic geometries
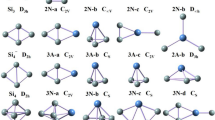
The optimized geometries of neutral PmSi n (n = 3–9) at the mPW2PLYP/LARGE level are shown in Fig. 1. For PmSi3, two isomers are reported. One is a planar rhombus (3a) with 6B2 ground state. And another is a tetrahedron (3b) with 6A1 electronic state. Energetically, it is less stable than that of 3a by 0.74 eV. For PmSi4, three isomers are presented. The 4a structure of 6A″ is predicted to be the ground state. Both 4a and 4c can be regarded as being derived from the ground state trigonal bipyramid structure of Si5 [46, 47] by replacing a Si atom at different positions with a Pm atom. The geometry 4b can be regarded as attaching a Pm atom to the ground state planar rhombus structure of Si4 [46, 47]. For PmSi5, two isomers are reported. Both 5a and 5b belong to substitutional structure. The 5a structure of 6A″ electronic state is predicted to be the ground state structure, analogous to the ground state structure of SmSi5 and EuSi5 [30, 31]. The 5b isomer of 6A′ electronic state is similar to the ground state structure of PrSi5, GdSi5 and HoSi5 [29, 32, 33]. For PmSi6, the 6a structure of 6B1 ground state is a pentagonal bipyramid with Pm atom positioning horizontal axis, analogous to the ground state structure of REMSi6 (REM = Pr, Sm, Eu, Gd, Ho, Yb, Lu) [29,30,31,32,33,34,35]. For PmSi7, four isomers are reported. The structure 7a, which can be viewed as replacing a Si of ground state structure of Si8 [47, 48] with a Pm atom, is predicted to be the ground state structure, analogous to the ground state structure of SmSi7 and EuSi7 [30, 31]. The 7b isomer of 6A″ electronic state can be regarded as attaching a Pm atom to the ground state pentagonal bipyramid structure of Si7 [46, 47]. Both 7c and 7d isomers possess 6A′ electronic state. The 7d geometry is similar to the ground state structures of GdSi7 and HoSi7 [32, 33]. For PmSi8, four isomers are presented. The isomer 8a belongs to substitutional structure and is the most stable structure with 6B2 ground state. The second lowest structure 8b is similar to beetle with 6A′ electronic state. The isomers 8b, 8c and 8d are significantly higher in energy than that of 8a substitutional structure. For PmSi9, five isomers are presented. The 9a, 9b and 9c geometries can be regarded as replacing a Si atom at different position of ground state structure of Si10 [47, 48] with a Pm atom. They compete with each other for the ground state structure of PmSi9 because their energy differences differ little from each other. The 9a structure, analogous to the ground state structure of SmSi9 and EuSi9 [30, 31], is only more stable in energy than the 9b and 9c isomers by 0.09 and 0.10 eV, respectively. The 9d isomer, bicapped tetragonal antiprism, is similar to the ground state structure of PrSi9 [29]. The 9e isomer can be viewed as attaching a Si atom to the ground state structure of PmSi8. Both 9d and 9e structures are less stable in energy than that of 9a. For PmSi10, four isomers are reported. The 10a structure, analogous to the ground state structure of SmSi10 and EuSi10 [30, 31], can be regarded as replacing a Si atom of tricapped tetragonal antiprism of Si11 [48] with a Pm atom. It is predicted to be the most stable structure with 6A′ ground state. The 10b isomer can be viewed as attaching a Pm atom to the ground state tetracapped trigonal prism of Si10 [48]. Energetically, the 10b, 10c and 10d isomers are less stable than that of 10a by 0.21, 0.24 and 0.32 eV, respectively. Take PmSi10 as an example, their energies of quartet and octuplet electronic states are examined. The results revealed that the energies of 10a–10d isomers with octuplet electronic states are higher than those with sextuplet electronic state by 0.95, 0.80, 0.99 and 0.87 eV, respectively. For quartet electronic state, there are serious spin contamination (expectation values of S 2 are about 4.64–5.75).

Neutral geometries optimized with the mPW2PLYP scheme. The Pm–Si bond lengths are in angstroms. The relative energies, ∆, are obtained at the mPW2PLYP/LARGE level and in eV
The optimized geometries of anion PmSi − n (n = 3–9) at the mPW2PLYP level are exhibited in Fig. 2. The Spin, S 2 operator values and relative energy of anion PmSi − n (and neutral PmSi n ) are listed in Table 2. From Table 2, we can see that the energies of quintuplet electronic state for isomers 3a −, 5a − and 6a − are only 0.09, 0.09 and 0.04 eV above those of septet electronic states, respectively. Their S 2 operator analysis reveals that the results of quintuplet electronic state are not trustworthy because of spin contamination. The expectation values (7.01) of S 2 can be expanded for pure states with higher multiplicities. And hence, the septet ground state evaluated for the anions is trustworthy. For 3b −, 4a − and 4b − isomers of quintuplet electronic state, the spin contamination also occurs. Although the spin contamination does not occur for 4c − and 6b − isomers of quintuplet electronic state, of which energies are obviously higher.

Anion geometries optimized with the mPW2PLYP scheme. The Pm–Si bond lengths are in angstroms. The relative energies, ∆, are obtained at the mPW2PLYP/LARGE level and in eV
For negatively charged ion PmSi − n with n = 3–10, their ground state structures are predicted to be 3a −, 4a −, 5a −, 6a −, 7a −, 8a −, 9c − and 10a −, respectively. This result is similar to that of negatively charged ion SmSi − n and EuSi − n [30, 31]. The 6b − geometry, analogous to ground state structure of PrSi6, GdSi6 and HoSi6, is higher in energy than that of 6a − by 0.85 eV. The 7b − isomer competes ground state with 7a − structure because their energy difference falls in 0.10 eV.
Similar to the ground state structures of REMSi n (REM = Sm, Eu) and their anions [30, 31], the ground state structures of PmSi n (n = 3–10) and their anions can be regarded as replacing a Si atom of the most stable structures of Si n+1 clusters with a Pm atom. The most stable geometries of anionic clusters excluded PmSi9 − is similar to their corresponding to neutral ones. The Pm–Si bond distances of the anions are averagely longer than their corresponding neutral ones by 0.146 Å (obtained by using 28 bond distances of 3a, 4a, 5a, 6a, 7a, 8a, 9a, 9c and 10a geometries).
3.2 Stability properties of clusters
It is well known that the dissociation energies (DEs) can be used to examine the relative stability of species. Here, the DEs are defined as the energy required for REM to dissociate from REMSi n or REMSi − n . The DEs calculated at the mPW2PLYP/aug-LARGE//mPW2PLYP/LARGE level for PmSi n and their anions are shown in Figs. 3 and 4, respectively. The DEs of PrSi n , SmSi n , EuSi n , GdSi n , HoSi n and their anions (of which ground state structures are taken from Refs. [29,30,31,32,33]) are also calculated at the same levels and plotted in Figs. 3 and 4, respectively, in order to facilitate comparison. From Figs. 3 and 4, we can see that (1) the DEs of PmSi n are close to those of SmSi n , EuSi n and HoSi n . The DEs of GdSi n are the largest among these data. The reason is that an unpaired d-orbital of Gd atom tends to form ionic polarization. As a result, the covalent bond character of the GdSi n system is enhanced [32]; (2) the consistent change trends of the DE vs n curves occur on all of these species. The larger the DEs, the more stable the clusters. So the REMSi4 and REMSi7 are less stable, and the REMSi5 and REMSi8 are more stable. (3) The DEs of PmSi − n , SmSi − n and EuSi − n are nearly identical. And they are smaller than those of HoSi − n and PrSi − n . The reason can be explained as follows. The PmSi n , SmSi n and EuSi n clusters belong to “A” type, while HoSi n and PrSi n species are “B” type. That is, an 4f electron of Ho and Pr atom transfers to 5d orbital and participates in bonding. This bond is equivalent to the SOMO (singly occupied molecular orbital). When these neutral clusters getting an electron become negatively charged ion, this additional electron going into the SOMO of neutral REMSi n (REM = Ho and Pr) becomes doubly occupied in the anion, which primarily localized on Si n skeleton. However, the electron back donation from the Si n skeleton to REM is induced and makes the bond between Si n and REM strong. This property may be used for the separation of rare earth elements.

Dissociation energy (DE, in eV) of neutral REMSi n (REM = Pm, Sm, Eu, Gd, Ho, Pr, n ≤ 10) calculated at the mPW2PLYP/aug-LARGE//mPW2PLYP/LARGE level of theory

Dissociation energy (DE, in eV) of anionic REMSi n (REM = Pm, Sm, Eu, Gd, Ho, Pr, n ≤ 10) calculated at the mPW2PLYP/aug-LARGE//mPW2PLYP/LARGE level of theory
3.3 Electronic properties of clusters
AEA is not only electronic property but also a key spectroscopic value and vitally important for use in the chemical cycle. The AEA is defined as the energy difference between the neutral ground state structure and the anionic ground state structure. The AEA calculated at the mPW2PLYP level is listed in Table 3. There are no experimental values for comparison. Compared with SmSi n and EuSi n , the AEAs of PmSi n differ little from those of SmSi n and EuSi n . If the numerical size must be distinguished, the AEA of PmSi n is slightly larger than that of SmSi n , but slightly smaller than that of EuSi n as shown in Fig. 5. We hope that our prediction will provide strong motivation for experimental studies of these important Pm-doped Si clusters and their anions.

Adiabatic electron affinity (AEA, in eV) REMSi n (REM = Pm, Sm, Eu, n = 3–10) calculated at the mPW2PLYP/aug-LARGE//mPW2PLYP/LARGE level of theory
HOMO–LUMO gap as an important physical property can also reflect the electronic property. The size dependence of the HOMO–LUMO gaps for the most stable structures of PmSi n (n = 3–10) calculated by the mPW2PLYP method is shown in Fig. 6. To facilitate comparison, the HOMO–LUMO gaps of SmSi n , EuSi n and Si n are also calculated and shown in Fig. 6. From Fig. 6, we can see that the consistent change trends of the HOMO–LUMO gap vs n curves exist in PmSi n , SmSi n and EuSi n , and the HOMO–LUMO gaps of which are nearly identical. The smaller the HOMO–LUMO gap, the more easily the PmSi n tends to set off photochemical reaction. So the photochemical activity of doping Pm atom to Si clusters is very stronger than that of pure Si clusters. This property may be used to produce new functional materials such as environmental catalytic materials.

HOMO-LUMO gaps (eV) of PmSi n , SmSi n , EuSi n and Si n (n = 3–10) species calculated at the mPW2PLYP/aug-LARGE//mPW2PLYP/LARGE level
In addition, the NPA (natural population analysis) charges and valence configurations for the most stable structure are calculated at the mPW2PLYP level of theory to further understand the interaction between Pm and Si clusters. The atomic charges and configurations of Pm are listed in Table 4. Similar to other REM in small REMSi n (n ≤ 10) clusters [29,30,31,32,33], the Pm atom in PmSi n and their anions act as an electron donor. The valence configurations of Pm are 6s 0.13−0.424f 4.97−4.995d 0.25−0.576p 0.06−0.12 and 6 s 0.67−0.964f 4.995d 0.11−0.356p 0.18−0.32 in PmSi n (n = 3–10) and their anions, respectively. It shows that the 4f shell of Pm in the cluster is nearly unchanged, analogous to the situations of Sm and Eu atom in the clusters [30, 31]. The charge transfer takes place largely from 6s to 5d orbitals, leading to hybridization between 6s and 5d orbital. In the case of anion, the majority of the extra electron’s charge is localized on Si n clusters. And the mean charges of 0.63 a.u. go back to Pm from Si n clusters (the mean charges of the anion minus the mean charges of the neutral). This weakened the bonds between Si n clusters and Pm atom. Therefore, the DEs of Pm atom from the lowest energy structures of the anion are less than those of the neutral.
Magnetic moments are the important physical properties for compounds containing REM atoms. The magnetic moments of 6s, 4f, 5d and 6p state for Pm, total magnetic moments of Pm, and total magnetic moments of the lowest energy structures of PmSi n (n = 3–10) and their anions are shown in Table 5. The 4f electrons of Pm atom provide the total magnetic moments for neutral PmSi n . For negatively charged ion, in addition to the 4f electrons, the 6s electrons of Pm also provide little magnetic moments as shown in Table 5.
4 Conclusions
The equilibrium geometries, electronic structures and electronic properties of PmSi n (n = 3–10) clusters have been systematically investigated by using the ABCluster global search technique combined with the TPSSh and mPW2PLYP density functional methods. The results revealed that the most stable structure of neutral PmSi n and their anions belongs to substitutional structure with sextuplet and septet ground state, respectively. The reliable AEAs of PmSi n (n = 3–10) are predicted to 1.44, 1.62, 1.68, 1.55, 1.68, 1.74, 2.05 and 1.95 eV, respectively. The AEAs of PmSi n differ little from those of SmSi n and EuSi n . Analyses of HOMO–LUMO gap showed that introducing Pm atom to Si cluster can significantly improve photochemical reactivity of the cluster. And the improved effects are as good as those of the introducing Sm and Eu atom to Si cluster. The NPA calculations indicated that the 4f electron of Pm atom in PmSi n (n = 3–10) and their anions hardly participates in bonding. That is, PmSi n (n = 3–10) belongs to A type. The total magnetic moments for neutral PmSi n and their anions are mainly provided by the 4f electrons of Pm atom. The DEs of PmSi n , SmSi n and EuSi n are nearly identical. The DEs of PmSi − n , SmSi − n and EuSi − n are also nearly equal, and they are smaller than those of HoSi − n and PrSi − n .
References
Ohara M, Miyajima K, Pramann A, Nakajima A, Kaya K (2002) Geometric and electronic structures of terbium–silicon mixed clusters (TbSi n , 6 ≤ n≤16). J Phys Chem A 106:3702–3705
Koyasu K, Atobe J, Furuse S, Nakajima A (2008) Anion photoelectron spectroscopy of transition metal and lanthanide metal-silicon clusters: MSi − n (n = 6-20). J Chem Phys 129:214301
Grubisic A, Wang HP, Ko YJ, Bowen KH (2008) Photoelectron spectroscopy of europium-silicon clusters anions, EuSi − n (3 ≤ n≤17). J Chem Phys 129:054302
Grubisic A, Ko YJ, Wang HP, Bowen KH (2009) Photoelectron spectroscopy of Lanthanide-silicon cluster anions LnSi − n (3 ≤ n≤13, Ln = Ho, Gd, Pr, Sm, Eu, Yb): prospect for magnetic silicon-based clusters. J Am Chem Soc 131:10783–10790
Han JG, Hagelberg F (2009) Recent progress in the computational study of silicon and germanium clusters with transition metal impurities. J Comput Theor Nanosci 6(2):257–269
Zhao RN, Yuan Y, Han JG, Duan Y (2014) Actinide elements and germanium: a first-principles density functional theory investigation of the electronic and magnetic properties of ApGe (Ap = Ac−Lr) diaotms. RSC Adv 4:59331–59337
Hang TD, Hung HM, Nguyen MT (2016) Structural assignment, and electronic and magnetic properties of lanthanide metal doped silicon heptamers Si7M0/−with M = Pr, Gd and Ho†. Phys Chem Chem Phys 18:31054
Li XJ, Yan ZJ, Li SN (2016) The nature of structure and bonding between transition metal and mixed Si–Ge Tetramers: a 20-electron superatom system. J Comput Chem 37:2316–2323
Pak C, Rienstra-Kiracofe JC, Schaefer HF (2000) Electron affinities of silicon hydrides: SiHn (n = 0–4) and Si2Hn (n = 0–6). J Phys Chem A 104:11232–11242
Li XJ, Claes P, Haertelt M, Lievens P, Janssens E, Fielicke A (2016) Structural determination of niobium-doped silicon clusters by far-infrared spectroscopy and theory. Phys Chem Chem Phys 18:6291
Zhao RN, Han JG (2014) Geometrical stabilities and electronic properties of Si n (n = 12–20) clusters with rare earth holmium impurity: a density functional investigation. RSC Adv 4:64410–64418
Beck SM (1987) Studies of silicon cluster-metal atom compound formation in a supersonic molecular beam. J Chem Phys 87(7):4233–4234
Koyasu K, Akutsu M, Mitsui M, Nakajima A (2005) Selective Formation of MSi16 (M = Sc, Ti, and V). J Am Chem Soc 127:4998–4999
Huang XM, Xu HG, Lu SJ, Su Y, King RB, Zhao JJ, Zheng WJ (2004) Discovery of a silicon-based ferromagnetic wheel structure in VxSi12 −(x = 1–3) clusters: photoelectron spectroscopy and density functional theory investigation†. Nanoscale 6:14617–14621
Li XJ, Han Q, Yang XH, Song RJ, Song LM (2016) Modification of alkali metals on silicon-based nanoclusters: an enhanced nonlinear optical response 659:93–99
Hou LY, Yang JC, Liu Yuming (2016) Reexamination of structures, stabilities, and electronic properties of holmium-doped silicon clusters HoSin (n = 12–20). J Mol Model 22:193
Qi P, Jiang S (2008) Growth behavior of La@Si n (n = 1–21) metal-encapsulated clusters. J Chem Phys 128:084711
Cao TT, Feng XJ, Zhao LX, Liang X, Lei YM, Luo YH (2008) Structure and magnetic properties of La-doped Si n (n = 1–12,24) clusters: a density functional theory investigation. Eur Phys J D 49:343–351
Feng YT, Yang JC (2017) Stability and electronic properties of praseodymium-doped silicon clusters PrSin (n = 12–21). J Mol Model 23:180
Li CG, Pan LJ, Shao P, Ding LP, Feng HT, Luo DB, Liu B (2015) Structures, stabilities, and electronic properties of the neutral and anionic SinSmλ (n = 1–9, λ = 0, − 1) clusters: comparison with pure silicon clusters. Theor Chem Acc 134:34
Zhao GF, Sun JM, Gu YZ, Wang YX (2009) Density-functional study of structural, electronic, and magnetic properties of the EuSi n (n = 1–13) clusters. J Chem Phys 131:114312
Wang J, Liu Y, Li YC (2010) Magnetic silicon fullerene. Phys Chem Chem Phys 12:11428–11431
Liu TG, Zhao GF, Wang YX (2011) Structural, electronic and magnetic properties of GdSi n (n = 1–17) clusters: a density functional study. Phys Lett A 375:1120–1127
Liu TG, Zhang WQ, Li YL (2014) First-principles study on the structure, electronic and magnetic properties of HoSi n (n = 1–12, 20) clusters. Front Phys 9:210–218
Zhao RN, Ren ZY, Guo P, Bai JT, Zhang CH, Han JG (2006) Geometries and electronic properties of the neutral and charged rare earth Yb-doped Si n (n = 1–6) clusters: a relativistic density functional investigation. J Phys Chem A 110:4071–4079
Zhao RN, Han JG, Bai JT, Liu FY, Sheng LS (2010) The medium-sized charged YbSi ± n (n = 7–13) clusters: a relativistic computational investigation. Chem Phys 378:82–87
Zhao RN, Han JG, Bai JT, Liu FY, Sheng LS (2010) A relativistic density functional study of Si n (n = 7–13) clusters with rare earth ytterbium impurity. Chem Phys 372:89–95
Cao TT, Zhao LX, Feng XJ, Lei YM, Luo YH (2009) Structural and electronic properties of LuSi n (n = 1–12) clusters: a density functional theory investigation. J Mol Struct THEOCHEM 895:148–155
Feng YT, Yang JC, Liu YM (2016) Study on the structures and properties of praseodymium-doped silicon clusters PrSin (n = 3–9) and their anions with density functional schemes. Theor Chem Acc 135:258
Xie XH, Hao DS, Liu YM, Yang JC (2015) Samarium doped silicon clusters SmSi n (n = 3–10) and their anions: structures, thermochemistry, electron affinities, and magnetic moments. Comput Theor Chem 1074:1–8
Yang JC, Wang J, Hao YR (2015) Europium-doped silicon clusters EuSi n (n = 3–11) and their anions: structures, thermochemistry, electron affinities, and magnetic moments. Theor Chem Acc 134:81
Yang JC, Feng YT, Xie XH, Wu HW, Liu YM (2016) Gadolinium-doped silicon clusters GdSin (n = 2–9) and their anions: structures, thermochemistry, electron affinities, and magnetic moments. Theor Chem Acc 135:204
Hou LY, Yang JC, Liu YM (2017) Density-functional study of the structures and properties of holmium-doped silicon clusters HoSin (n = 3–9) and their anions. J Mol Model 23:117
Xie XH, Hao DS, Yang JC (2015) Ytterbium doped silicon clusters YbSi n (n = 4-10) and their anions: structures, thermochemistry, and electron affinities. Chem Phys 461:11–19
He S, Yang JC (2017) Study on structure and property of lutetium introduced silicon clusters LuSin (n = 3–10) and their anions with density functional theory. J Clust Sci. doi:10.1007/s10876-017-1225-x
Schwabe T, Grimme S (2006) Towards chemical accuracy for the thermodynamics of large molecules: new hybrid density functionals including non-local correlation effects. Phys Chem Chem Phys 8:4398–4401
Zhang J, Dolg M (2015) ABCluster: the artificial bee colony algorithm for cluster global optimization. Phys Chem Chem Phys 17:24173–24181
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, JA Montgomery Jr, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2010) Gaussian 09 revision C.01. Gaussian Inc, Wallingford
Staroverov VN, Scuseria GE, Tao J (2003) Comparative assessment of a new nonempirical density functional: molecules and hydrogen-bonded complexes. J Chem Phys 119:12129–12137
Dolg M, Stoll H, Savin A, Preuss H (1989) Energy-adjusted pseudopotentials for the rare earth elements. Theor Chim Acta 75:173–194
Feller D (1996) The role of databases in support of computational chemistry calculations. J Comput Chem 17:1571–1586
Schuchardt KL, Didier BT, Elsethagen T (2007) Basis set exchange: a community database for computational sciences. J Chem Inf Model 47(3):1045–1052
Woon DE, Dunning TH (1993) Gaussian basis sets for use in correlated molecular calculations. II. The atoms aluminum through argon. J Chem Phys 98:1358–1371
Buchachenko AA, Chalasiński G, Szeześniak MM (2007) Diffuse basis functions for small-core relativistic pseudopotential basis sets and static dipole polarizabilities of selected lanthanides La, Sm, Eu, Tm and Yb. Struct Chem 18:769–772
Cao X, Dolg M (2002) Segmented contraction scheme for small-core lanthanide pseudopotential basis sets. J Mol Struct THEOCHEM 581:139–147
Raghavachari K (1986) Theoretical study of small silicon clusters: equilibrium geometries and electronic structures of Sin (n = 2–7,10). J Chem Phys 184:5672–5686
Yang JC, Xu WG, Xiao WS (2005) The small silicon clusters Sin, (n = 2–10) and their anions: structures, themochemistry, and electron affinities. J Mol Struct (THEOCHEM) 719:89–102
Zhu X, Zeng XC (2003) Structures and stabilities of small silicon clusters: Ab initio molecular-orbital calculations of Si7–Si11. J Chem Phys 118:3558–3570
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No. 21263010), by Program for Innovative Research Team in Universities of Inner Mongolia Autonomous Region (Gran No. NMGIRT-A1603), by the Inner Mongolia Natural Science Foundation (Grant No. 2015MS0216), and by the Inner Mongolia institutions of higher learning scientific research projects (Grant No. NJZY16419).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
He, S., Yang, J. Promethium-doped silicon clusters PmSi n (n = 3–10) and their anions: structures, thermochemistry, electron affinities and magnetic moments. Theor Chem Acc 136, 93 (2017). https://doi.org/10.1007/s00214-017-2126-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-017-2126-7