
Abstract
The neutral PrSi n (n = 12–21) species considering various spin configurations were systematically studied using PBE0 and B3LYP schemes in combination with relativistic small-core potentials (ECP28MWB) for Pr atoms and cc-pVTZ basis set for Si atoms. The total energy, growth-pattern, equilibrium geometry, relative stability, hardness, charge transfer, and magnetic moments are calculated and discussed. The results reveal that when n < 20, the ground-state structure of PrSi n evaluated to be prolate clusters. Starting from n = 20, the ground-state structures of PrSi n are evaluated to be endohedral cagelike clusters. Although the relative stabilities based on various binding energies and different functional is different from each other, the consensus is that the PrSi13, PrSi16, PrSi18, and PrSi20 are more stable than the others, especially the PrSi20. Analyses of hardness show that introducing Pr into Si n (n = 12–21) elevates the photochemical sensitivity, especially for PrSi20. Calculated result of magnetic moment and charge transfer shows that the 4f electrons of Pr in the clusters are changed, especially in endohedral structures such as PrSi20, in which one electron transfers from 4f to 5d orbital. That is, the 4f electron of Pr in the clusters participates in bonding. The way to participate in bonding is that a 4f electron transfers to 5d orbital. Although the 4f electron of Pr atom participates in bonding, the total magnetic moment of PrSi n is equal to that of isolated Pr atom. The charge always transfers from Pr atom to Si n cluster for the ground state structures of PrSin (n = 12–19), but charge transfer is reverse for n ≥ 20. The largest charge transfer for endohedral structure reveals that the bonding between Pr and Si n is ionic in nature and very strong. The fullerenelike structure of PrSi20 is the most stable among all of these clusters and can act as the building blocks for novel functional nanotubes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, considerable experimentally and theoretically studies of the metal-doped silicon clusters reported in the literature leave no doubt for their potential important application in nanotechnology and microelectronic industry, which in part is due to their enhanced stabilities and tunable electronic properties by altering composition and shape [1,2,3,4,5,6]. Especially, introducing lanthanide (Ln) atoms into a Si n cluster was regarded as a promising approach to create cluster with new magnetic properties. Since the 4f electron of some of Ln atoms hardly participates in bonding, the atomic magnetic moments of Ln atom in LnSi n clusters (for example, SmSi n and EuSi n ) can be retained. In addition, portions of the 4f electron of Ln atoms are also involved in bonding. The way it can participate in bonding is that a 4f electron of Ln atom is transferred to 5d orbital, and then the 5d electron is involved in bonding. However, the magnetic moment is unchanged (for example PrSi n clusters in this paper), but for late rare earth metal atoms, the total magnetic moment may increase after a 4f electron moved to 5d. In this regard, Ln atoms are different from those of transition metal (TM), the magnetic moment for the latter can be quenched [7,8,9]. In addition, Ln-doped silicon clusters can possess excellently optical and catalytic properties. For instance, a silicon-based optical source can be produced by introducing Er atom into silicon microcrystals [10].
Up to now, many efforts have been focused on Ln-doping Si n clusters. The geometric and electronic structures of LuSi n −, HoSi n −, TbSi n − (6 ≤ n ≤ 20) were experimentally studied by Nakajima and co-workers [4, 5]. The results suggested that Tb atom is encapsulated into the Si n cage at n ≥ 10, and the Ho encapsulation is still incomplete when the size of the Si cage swells to 16 atoms. Bowen et al. [6] probed into the structures and properties of LnSi n − (3 ≤ n ≤ 13; Ln = Ho, Gd, Pr, Sm, Eu, Yb) by means of photoelectron spectroscopy (PES). Based on their appearance, the spectra of Eu and Yb fall into category “A”, the spectra of Gd, Ho, and Pr species fall into category “B”, and the spectra of Sm belong to category “AB”. Our previous studies [24,25,26] show that, however, it is more reasonable for Ln-doped silicon systems to be divided into two groups. Specifically, the group A in which 4f electron of Ln atoms hardly participates in bonding contains LnSi n − (Ln = Eu, Yb, Sm). Group B in which 4f electron of Ln atoms is involved in bonding contains LnSi n − (Ln = Ho, Gd, Pr).
On the theoretical level, many computational investigations on the structures and properties have been performed for LnSi n (Ln = Lu, Yb, Ho, Gd, Eu, Sm, and La, n < 21) by means of density functional theory (DFT), which can provide an interesting example for evaluating the accuracies of various DFT methods [11,12,13,14,15,16,17,18,19,20,21,22]. In addition, the fullerene-like neutral species M@Si20 (M = La, Ac, Sm, Gd, Tm, Ce, Pa, Pu, Th, Np, Pm) and their anions were studied using the ab initio projected augmented wave method. The results showed that significant magnetic moments in the most stable geometries of PaSi20, SmSi20, PuSi20, TmSi20, and GdSi20 − can be retained [23]. Recently, we studied not only the structures and electron affinities of LnSin (Ln = Eu, Yb, Sm, n < 11) and their anions, but also the structures, stabilities, and electronic properties of HoSi n (n = 12–20) using several DFT methods [11, 24,25,26]. The theoretical adiabatic electron affinities evaluated by these schemes can be in excellent agreement with the experimental values.
The objectives of this work are to apply two carefully selected DFT schemes and relativistic small-core potentials (ECP) basis set for Pr atom to the determination of the total energies, growth-pattern, equilibrium geometries, relative stability, hardness, magnetic moments, and charge-transfer of the medium-sized praseodymium-doped silicon clusters PrSi n (n = 12–21) to understand their novel size-dependent electronic properties and the critical size of the Pr encapsulated into the Si frame, which can provide a guide for this type of cluster-assembled material. In consideration of the possible functional dependence of the predicted lowest-energy structures, two different functionals are selected in this work. The reason to use small-core ECP is that the 4f electron can participate in bonding as described above.
Computational details
The two different density functional forms used here are the B3LYP [27, 28] and PBE0 [29] functionals. The cc-pVTZ [30] is employed for Si atoms. The (14s13p10d8f6g)/[10s8p5d4f3g] segmented (SEG) basis sets and relativistic small-core effective potentials (ECP28MWB) [31] are selected for praseodymium (named as SEG/ECP). For clusters of PrSi n (n = 12–21), the stationary point of these geometries are examined by the evaluation of their harmonic vibrational frequencies to insure the optimized structures as local minima. Adopted cc-pVTZ and SEG/ECP basis sets are reasonable because the structural parameters optimized with them are nearly equal to those optimized with aug-cc-pVTZ and aug-SEG/ECP basis sets for LnSi n compounds [25]. All of the calculations were carried out using the Gaussian 09 software package [32].
When determining the most stable structures, a well-known problem is possibly missing the lowest energy isomers. For small sizes, an extensive search such as using a global optimization technique can be adopted to dispose of this problem. However, it is more and more difficult with increasing of the cluster sizes because of the much increased number of low-lying isomers, and the requiring of both an efficient optimization scheme and exact potential functions cannot be done for larger size clusters. Fortunately, based on the previous experience [11, 24,25,26, 33], the ground state exohedral structures of neutral LnSi n clusters can be regarded as a substitution of Ln for a Si in the most stable structure of Si n+1. Therefore, two families of initial geometries are considered in the optimization process. One is exohedral isomers, namely, prolate structures generated by substitution of Pr for a Si in the most stable structure of Si n+1; and another is near-spherical structures generated via constrained search based on the fullerene cage motifs. To search for the ground-state structure of PrSi n clusters the possible ground-state structure of Si n (n = 13–21) reported previously [34,35,36,37,38] has been considered when constructing prolate structures of PrSi n (n = 12–20). Specifically, the tricapped-trigonal-prism (TTP) motif and the six/six (sixfold-puckered hexagonal ring Si6 plus six-atom tetragonal bipyramid Si6, SS) motif [37, 38] of prolate structures are selected. In addition, the spin multiplicities of doublet, quartet, and sextuplet were taken into account, and the results reveal that the most stable structures of PrSi n (n = 12–20) are quartet.
Results and discussion
Lowest-energy structures and isomers
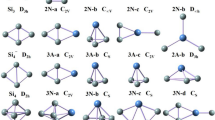
The geometries optimized with the PBE0 and B3LYP methods for PrSi n (n = 12–21) are displayed in Fig. 1. The calculated total energies and relatively energy of the low-lying isomers are summarized in Table 1.

The stable geometries of PrSi n (n = 12–21)
For PrSi12, three low-lying structures are reported. Zhu et al. [34] found that the most stable structure of Si13 is a distorted TTP with an extra rhombus capped on one edge of the prism at the MP2 level. At quantum Monte Carlo level and MP2/aug-cc-pVTZ//B3LYP/6–31+G(d) level, the most stable structure of Si13 is predicted to be a trigonal antiprism with C 3v -symmetry [35] and a C 2v -symmetry geometry [36], respectively. The isomers 12a, 12b, and 12c shown in Fig. 1 can be regarded as a substitution of Pr for a Si of three of these Si13 geometries. The isomers 12d and 12e can be obtained when placing the Pr atom inside the icosahedron and hexagonal prism of Si12 serve as the initio optimized geometries and undergo Jahn-Teller distortion if vibrational analysis yields one or more imaginary frequencies, respectively. The geometries of 12d and 12e reveal that the cage-like structure of PrSi12 is less stable. Energetically, if the B3LYP functional is selected, the 12a is the most stable structure, which is 0.41 eV lower than the 12b, whereas if the PBE0 functional is selected, the 12b is more stable than the 12a by 0.17 eV.
For PrSi13, two isomers are presented. The lowest-energy structure 13a can be viewed as a substitution of Pr atom for a Si atom in the most stable structure of Si14 [34], a faced-capped distorted TTP with an extra rhombus capped on the prism’s edge, by a substitution Pr for a Si atom. The isomer 13b can be generated after a Si atom is added onto the Pr-encapsulated hexagonal prism of Si12. It is higher than that of 13a by 1.72 and 2.60 eV in energy at the B3LYP and PBE0 levels, respectively.
Two isomers are reported for PrSi14. The most stable structure 14a can be regarded as a substitution of Pr for a Si of the ground state structure of Si15 (a TTP with a tricapped trigonal antiprism) [34] with a Pr atom. It is more stable in energy than the cage-like 14b isomer by 1.45 and 3.70 eV at the B3LYP and PBE0 levels, respectively.
Four isomers are presented for PrSi15. Three structures, which contain respectively TTP, SS, and two fused pentagonal prisms, compete with each other for the most stable structure of Si16 [37]. The isomers 15a, 15b, and 15c can be regarded as replacing a Si atom of these isomers with a Pr atom, respectively. The isomer 15d is generated after one silicon atom is removed from the Pr-encapsulated fullerenelike geometry of Si16. Energetically, it is less stable than the 15a by 1.83 and 2.34 eV with the B3LYP and PBE0 schemes, respectively.
For PrSi16, two prolate and one fullerenelike structure are presented. The most stable 16a containing SS motif is generated by replacing a Si atom of the most stable structure of Si17 [37]. The isomer 16b containing TTP motif can be regarded as replacing a Si atom of Si17 [37]. Energetically, the prolate 16a is more stable than the prolate 16b and fullerenelike 16c by 0.14 (PBE0) and 0.38 eV (B3LYP), and 1.50 (PBE0) and 1.85 eV (B3LYP), respectively.
Three isomers are presented for PrSi17. For Si18, both structures containing SS and TTP motif compete the most stable structure with each other [37]. The prolate 17a and 17b are obtained by replacing a Si atom of the two Si18 isomers with a Pr atom, respectively. The isomer 17c is generated after one silicon atom is added to PrSi16 of fullerenelike 16c. Energetically, if the B3LYP functional is selected, the 17a is the stable structure, which is more stable than the 17b by 0.14 eV, whereas if the PBE0 functional is selected, the 17b is more stable than the 17a by 0.15 eV. That is, the predicted lowest-energy structure of PrSi17, analogous to Si18 [37], can be dependent on the functional selected.
For PrSi18, two prolate and one cage-like structure are presented. The most stable 18a is generated by replacing a Si atom of the most stable structure of Si19 containing SS [37]. The isomer 18b can be regarded as supplanting a Si atom of Si19 containing TTP motif [37]. The cage-like 18c is generated after a Si atom is added to PrSi17 of the cake-like 17c. Energetically, the cake-like 18c is less stable than the prolate 18a by 0.56 and 0.54 eV at the PBE0 and B3LYP levels, respectively, but it is more stable than the 18b by about 0.25 eV at the B3LYP level.
For PrSi19, two prolate and one cage-like structure are also reported. The prolate 19a and 19b is regarded as supplanting a Si atom of both Si20 containing SS and TTP motif [37], respectively. The cage-like 19c is generated after a Si atom is removed from the Pr-encapsulated fullerenelike structure of Si20. Energetically, the 19a structure is more stable than those of 19b and 19c by 0.08 and 0.31 eV at the B3LYP level, and by 0.05 and 0.21 eV at the PBE0 level, respectively.
For PrSi20, two isomers are presented. Isomer 20b is regarded as supplanting a Si atom of the most stable structure of Si21 containing SS motif [38]. Energetically, it is less stable than the fullerenelike 20a by 0.42 and 0.57 eV with the PBE0 and B3LYP schemes, respectively.
For PrSi21, two cagelike structures are presented. Isomer 21a which is regarded as adding a Si atom onto the fullerenelike 20a is predicted to be the ground state structure.
From the above described, we can find that the most stable structure of PrSi n , starting from n = 20, are predicted to be endohedral cagelike species. When n < 20, the ground-state structures of PrSi n are prolate clusters, which can be generated by a substitution of Pr for a Si atom of the most stable structures of Si n+1. The ground-state structures of PrSi n are favorable to contain SS motif when n = 16–19, while when n < 16, the ground-state structures of PrSi n are favorable to contain TTP motif.
Binding energies
To probe the inherent stabilities of most stable PrSi n (n = 12–21) clusters, the binding energies per atom (BEPA) (defined as the required energy in the reaction PrSi n → Pr + nSi, namely, BEPA(PrSi n ) = [nE(Si) + E(Pr)-E(PrSi n )]/(n + 1)) of PrSi n are predicted using the PBE0 and B3LYP methods. A plot of the BEPA against the corresponding cluster size shown in Fig. 2 indicates that at the B3LYP level, PrSi13, PrSi16, PrSi18, and PrSi20 are slightly more stable than the others because they correspond to the four “bumps” of curve, respectively. While at the PBE0 level, PrSi13, PrSi16, and PrSi20 are slightly more stable suggested by the smoothly increasing background.

The binding energies per atom (BEPA) for the most stable PrSi n (n = 12–21) clusters calculated with the PBE0 and the B3LYP methods
In additional to the BEPA, the dissociation energies are also illustrated in order to compare the stabilities of various PrSi n species. The dissociation energies are shown in Fig. 3 (defined as the required energies for the disproportionation reaction 2PrSi n → PrSi n+1 + PrSi n-1, namely, DE1(PrSi n ) = [E(PrSi n+1) + E(PrSi n-1)-2E(PrSi n )]). This gives a sensitive measure of relative stability. As can be seen from Fig. 3, three local minimal values with n = 14, 17, and 19 are found, reflecting that PrSi14, PrSi17, and PrSi19 have weaker local stabilities when compared with the others at the B3LYP and PBE0 levels.

DE1 (eV) of PrSin (n = 12–20) versus the number of atoms n
Another measure of the stability is given by the dissociation energies, DE2(PrSi n ) = [E(Si n ) + E(Pr) - E(PrSi n )], DE3(PrSi n ) = [E(PrSi n-1) + E(Si) - E(PrSi n )], and DE4(Si n ) = [E(Si n-1) + E(Si) - E(Si n )], respectively. They are plotted in Figs. 4 and 5. From Fig. 4 we can see that at the B3LYP level, the PrSi n for n = 13, 16, 18, and 20 are more stable than the others because their DE2 are local maximal values. On the other hand, the analysis of DE3 and DE4 reveals that the PrSi n for n = 13, 15, 16, 18, and 20 are more stable than the others in respect that the DE3 is larger than the DE4. In other words, when an extra Si atom is attached to the cluster, it is energetically more favorable to add to PrSi n-1 and to form PrSi n species rather than to add to Si n-1 cluster and to form Si n cluster. From Fig. 5 we can conclude that using the PBE0 scheme, the result of analysis of DE2 is the same as that of analysis of DE3 and DE4. That is, the PrSi n for n = 13, 16, and 20 are more stable than the others in respect that the DE2 are local maximal values.

DE2, DE3, and DE4 (eV) of PrSi n (n = 12–21) versus the number of atoms n calculated with the B3LYP method

DE2, DE3, and DE4 (eV) of PrSi n (n = 12–21) versus the number of atoms n calculated with the PBE0 method
Although the relative stabilities based on various binding energies and different functional is different from each other, the consensus is that the PrSi13, PrSi16, and PrSi20 are more stable than the others, especially the PrSi20, of which various binding energies are obviously larger than those of PrSi13 and PrSi16. It is noticed that PrSi20 structures can act as the building blocks for nanotubes due to their cage-like geometry.
Hardness
In a sense, hardness is not only an important physical property, but also a significant criterion to mirror the chemical reactivity of species, especially for Ln-doped Si clusters which always have good photochemical sensitivity. The hardness (which is defined as the difference between the energy of the HOMO and the LUMO) for the most stable structures of PrSin (n = 12–21) clusters predicted by the two methods are plotted in Fig. 6. The hardness of Si n species for comparison is also sketched in Fig. 6. From Fig. 6 we can conclude that (1) the hardness curves of the B3LYP and the PBE0 are on the whole in parallel, and the hardness of the PBE0 is larger than that of B3LYP. The reason is that the HOMO energies and the LUMO energies predicted in Kohn-Sham (KS) molecular orbital model undergo approximately the same amount of upshift, while Hartree-Fock (HF) hybrids shift the LUMO up to much higher energy levels than the HOMO up [39]. On the other hand, the component of HF hybrid in the PBE0 is larger than that in the B3LYP. So the HOMO-LUMO gap of the PBE0 is larger than that of the B3LYP. The KS HOMO-LUMO gap in molecules approximates the lowest excitation energy much more closely than the HF HOMO-LUMO gap does [39]. So the B3LYP HOMO-LUMO gap may be a better approximation to the optical gap in molecules than the PBE0 HOMO-LUMO gap. (2) The hardness of PrSi n is smaller than that of pure Si n clusters. This shows that introducing Pr atom into Si n clusters elevates the photochemical sensitivity. (3) The smaller the hardness, the easier the PrSin inclines to ignite the photochemical reaction. The hardness of PrSi16, PrSi17, PrSi18, PrSi20, and PrSi21 is smaller than the others, namely, their photochemical sensitivity is better than the others.

The HOMO-LUMO gaps of PrSin and Sin (n = 12–21) calculated with the PBE0 and the B3LYP methods
Magnetic moment and charge transfer
To better understand the interaction between the Pr atom and silicon clusters, natural population analyses (NPA) are carried out with the two schemes. The analyses of charge and NPA valence configurations of Pr atom listed in Table 2 reveal that (1) the valence configurations are 6s0.12–0.314f2.17–2.895d0.90–1.916p0.09–0.33 and 6s0.13–0.384f2.36–2.905d0.82–1.426p0.07–0.29 for Pr in PrSin (n = 12–19) species at the PBE0 and the B3LYP levels, and 6s0.374f 2.03–2.075d 4.52–4.536p 1.53–1.55 and 6s0.33–0.344f 2.05–2.065d4.03–4.166p1.41–1.44 for Pr in PrSi20 and PrSi21, respectively. Evidently, in the clusters the 4f electron of Pr is changed, especially in cagelike structures such as PrSi20 and PrSi20, in which one electron transfers from 4f to 5d orbital (the configuration of isolated Pr atom is [core]6s24f35d06p0). In this regard, Pr atom differs from Eu and Sm. The 4f shell of those in the clusters has almost no change [24, 25]. Apart from the charge transfer from 4f to 5d orbital, charge transfer takes place from 6s to 5d orbital, thus, resulting in hybridization between 6s and 5d orbital. (2) The charge always transfers from Pr atom to Si n cluster for PrSin (n = 12–19), but charge transfer is reversed for PrSi20 and PrSi22, which reveals that Pr atom acts as an electron acceptor in the cage-like ground-state structure. The transferred charges are largest (2.99–3.49e) for PrSi20, which reveals that the bonding between Pr atom and Si n cluster is ionic in nature, and the strong bonding results in the most stable cage-like structure of PrSi20 among all of these species studied in this work. This result is in agreement with that of DE analyses.
Magnetic moments are one of the most interesting properties in physics. The analyses of magnetic moments listed in Table 3 show that the total magnetic moments of PrSi n (n = 12–20) are 3 μB, which is mainly from the 4f state (1.97–2.88 μB) of Pr atom, following this is the 5d state (0.01–0.33 μB), and the remaining states are 3s and 3p (0.06–1.01 μB) of Si atoms (the 6s and 6p states of Pr atom have very little contribution). This is to say that although the 4f electron of Pr atom participates in bonding, the total magnetic moment of PrSi n is idential to that of isolated Pr atom.
Conclusions
The total energies, growth-pattern, equilibrium geometries, relative stability, hardness, magnetic moments, and charge-transfer of PrSi n (n = 12–20) species have been investigated with the PBE0 and the B3LYP functionals in combination with the SEG/ECP basis set for the Pr atoms and cc-PVTZ basis set for the Si atoms. The results reveal that (1) when n < 20, the ground-state structure of PrSi n predicted to be prolate clusters, which can be generated by a substitution of Pr atom for a Si of the ground-state structures of Si n+1. To be more precise, the ground-state structure of PrSi n prefers to contain SS motif when n = 16–19, while it prefers to contain TTP motif when n < 16. Starting from n = 20, the ground-state structures of PrSi n are evaluated to be endohedral cagelike clusters. (2) Although the relative stabilities based on various binding energies and different functional is different from each other, the consensus is that the PrSi13, PrSi16, and PrSi20 are more stable than the others, especially the PrSi20. (3) Analysis of hardness shows that introducing Pr atom to Si n (n = 12–20) clusters elevates the photochemical sensitivity. (4) The natural population analysis (NPA) shows that the 4f electrons of Pr in the clusters are changed, especially in PrSi20 and PrSi21, in which one electron transfers from 4f to 5d orbital. This is to say that the 4f electron of Pr in the clusters participates in bonding. The way to participate in bonding is that a 4f electron transfers to 5d orbital. Although the 4f electron of Pr atom participates in bonding, the total magnetic moment of PrSi n is equal to that of isolated Pr atom. The charge always transfers from Pr atom to Si n cluster for the ground state structures of PrSin (n = 12–19), but charge transfer is reversed for PrSi20 and PrSi21, which reveals that Pr acts as an electron acceptor in the cage-like ground-state structure. The largest charge transfer for PrSi20 reveals that the bonding between Pr atom and Si n cluster is ionic in nature and very strong. As a result, the fullerenelike structure of PrSi20 is the most stable one and can act as the building blocks for novel functional nanotubes.
References
Avaltroni F, Steinmann SN, Corminboeuf C (2012) Phys Chem Chem Phys 14:14842–14849
Tam NM, Tai TB, Ngan VT, Nguyen MT (2013) J Phys Chem A 117:6867–6882
Lin L, Yang JC (2015) J Mol Model 21(155):1–13
Ohara M, Miyajima K, Pramann A, Nakajima A, Kaya K (2002) J Chem Phys A 106:3702–3705
Koyasu K, Atobe J, Furuse S, Nakajima A (2008) J Chem Phys 129(214301):1–7
Grubisic A, Ko YJ, Wang H, Bowen KH (2009) J Am Chem Soc 131:10783–10790
Li JR, Wang GH, Yao CH, Mu YW, Wan JG, Han M (2009) J Chem Phys 130(164514):1–9
Guo LJ, Zhao GF, Gu YZ, Liu X, Zeng Z (2008) Phys Rev B 77(195417):1–8
Wang JG, Zhao JJ, Ma L, Wang BL, Wang GH (2007) Phys Lett A 367:335–344
Kenyon AJ (2005) Semicond Sci Technol 20:R65–R84
Hou LY, Yang JC, Liu YM (2016) J Mol Model 22(193):1–10
Peng Q, Shen J (2008) J. Chem. Phys. 128(084711):1–11
Zhao GF, Sun JM, Gu YZ, Wang YX (2009) J Chem Phys 131:114312-1–114312-7
Li CG, Pan LJ, Shao P, Ding LP, Feng HT, Luo DB, Liu B (2015) Theor Chem Accounts 134(34):1–11
Liu TG, Zhao GF, Wang YX (2011) Phys Lett A 375:1120–1127
Liu TG, Zhang WQ, Li YL (2014) Front Phys 9:210–218
Zhao RN, Ren ZY, Guo P, Bai JT, Zhang CH, Han JG (2006) J Phys Chem A 110:4071–4079
Zhao RN, Han JG, Bai JT, Liu FY, Sheng LS (2010) Chem Phys 372:89–95
Zhao RN, Han JG, Bai JT, Sheng LS (2010) Chem Phys 378:82–87
Cao TT, Zhao LX, Feng XJ, Lei YM, Luo YH (2009) J Mol Struct 895:148–155
Wang HQ, Li HF (2014) RSC Adv 4:29782–29793
Zhao RN, Han JG (2014) RSC Adv 4:64410–64418
Kumar V, Singh AK, Kawazoe Y (2006) Phys Rev B 74(125411):1–5
Yang JC, Wang J, Hao YR (2015) Theor Chem Accounts 134(81):1–11
Xie XH, Hao DS, Liu YM, Yang JC (2015) Comput Theor Chem 1074:1–8
Xie XH, Hao DS, Yang JC (2015) Chem Phys 461:11–19
Becke AD (1993) J Chem Phys 98:5648–5652
Lee C, Yang W, Parr RG (1988) Phys Rev B 37:785–789
Adamo C, Barone V (1999) J Chem Phys 110:6158–6170
Woon DE, Dunning Jr TH (1993) J Chem Phys 98:1358–1371
Cao X, Dolg M (2002) J Mol Struct THEOCHEM 581:139–147
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, et al. (2010) Gaussian 09 revision C.01. Gaussian Inc, Wallingford
Yang JC, Feng YT, Xie XH, Wu HW (2016) Theor Chem Accounts 135(204):1–12
Zhu XL, Zeng XC, Lei YA, Pan B (2004) J Chem Phys 120:8985–8995
Grossman JC, Mitáš L (1995) Phys Rev Lett 74:1323–1326
Nigam S, Majumder C, Kulshreshtha SK (2006) J Chem Phys 125(074303):1–11
Yoo S, Zeng XC (2005) J Chem Phys 123(164303):1–6
Yoo S, Zeng XC (2006) J. Chem. Phys. 124(054304):1–6
Baerends EJ, Gritsenko OV, van Meer R (2013) 15:(39)16408-16425
Acknowledgments
This study was economically supported by the National Natural Science Foundation of China (Grant No. 21263010), by the Inner Mongolia Natural Science Foundation (Grant No. 2015MS0216), and by the Program for Innovative Research Team in University of the Inner Mongolia Autonomous Region (Grant no. NMGIRT-A-1603).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Feng, Y., Yang, J. Stability and electronic properties of praseodymium-doped silicon clusters PrSin (n = 12–21). J Mol Model 23, 180 (2017). https://doi.org/10.1007/s00894-017-3352-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00894-017-3352-6