
Abstract
Key message
Genetic diversity in elite rye germplasm as well as F 2:3 testcross design enables fast QTL mapping to approach genes controlling grain yield, grain weight, tiller number and heading date in rye hybrids.
Abstract
Winter rye (Secale cereale L.) is a multipurpose cereal crop closely related to wheat, which offers the opportunity for a sustainable production of food and feed and which continues to emerge as a renewable energy source for the production of bioethanol and biomethane. Rye contributes to increase agricultural crop species diversity particularly in Central and Eastern Europe. In contrast to other small grain cereals, knowledge on the genetic architecture of complex inherited, agronomic important traits is yet limited for the outbreeding rye. We have performed a QTL analysis based on a F2:3 design and testcross performance of 258 experimental hybrids in multi-environmental field trials. A genetic linkage map covering 964.9 cM based on SSR, conserved-orthologous set (COS), and mixed-phase dominant DArT markers allowed to describe 22 QTL with significant effects for grain yield, heading date, tiller number, and thousand grain weight across seven environments. Using rye COS markers, orthologous segments for these traits have been identified in the rice genome, which carry cloned and functionally characterized rice genes. The initial genome scan described here together with the existing knowledge on candidate genes provides the basis for subsequent analyses of the genetic and molecular mechanisms underlying agronomic important traits in rye.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rye (Secale cereale L.) belongs to the Triticeae tribe of the grasses and contributes to increase crop species diversity particularly in European agroecosystems. Winter rye offers modest requirements to the germination temperature, low demands on soil and climatic conditions and excellent winter hardiness. This small grain cereal is a multipurpose crop whose grain is traditionally used for bread making and as feed for livestock. In addition, rye continues to emerge as a renewable energy source for the production of bioethanol and biomethane.
Rye is outstanding among the small grain cereals with respect to its outbreeding nature. Cytoplasmic male sterility (CMS) as genetic fertilization control mechanism as well as efficient restorer genes successfully enable hybrid breeding (Geiger and Miedaner 2009). The exploitation of heterosis by a systematic evaluation of the genetic divergent gene pools ‘Petkus’ and ‘Carsten’ (Hepting 1978) resulted in a significant gain in grain yield (Laidig et al. 2017), which is economically the most important trait in rye breeding.
Agronomic important traits like grain yield or grain weight reveal a continuous phenotypic variation and are genetically controlled by a complex network of multiple and interacting loci. Most of these quantitative trait loci (QTL) are sensitive to environmental conditions and have only small effects on the expression of a trait (Mackay et al. 2009). Currently, the phenotypic evolution of rye varieties is achieved by conventional breeding approaches, which select genotypes indirectly through their phenotypic performance. Genetic improvement by artificial selection is known to cause a reduction in genetic diversity (Wright et al. 2005; Yamasaki et al. 2005; Kovach and McCouch 2008), which might be forced in the future by an implementation of the concept of genomic selection (Meuwissen et al. 2001) in hybrid rye breeding programs (Wang et al. 2014; Auinger et al. 2016). Thus, the maintenance of genetic diversity for the long-term success of the genetic improvement of rye is gaining increasing importance. The analysis of functionally characterized genes as components of quantitative genetic variation in agronomic important traits (Hou et al. 2014; Qin et al. 2014; Jiang et al. 2015; Yue et al. 2015) provides an unbiased and precise assessment of crop genetic diversity in space and time. The largest progress among the cultivated cereals in cloning of QTL for grain yield components and other agronomic important traits has been achieved in rice (Yonemaru et al. 2010; Yamamoto et al. 2012), which has a significant impact on the genetic improvement of this important staple food (Xing and Zhang 2010). In species like rye with just emerging genomic tools (Martis et al. 2013; Bauer et al. 2017), knowledge on candidate genes represents an attractive opportunity to elucidate the molecular genetic basis in particular for quantitative inherited traits. The mining of valuable alleles (Kumar et al. 2010), especially those occurring with low frequencies in self incompatible rye germplasm collections, facilitates a targeted identification of new haplotypes, which can, in a first step, be precisely introgressed in self-fertile elite germplasm based on allele-specific markers and subsequently systematically evaluated in terms of their agronomic value. However, knowledge on QTL in the 8 Gb genome of rye (Bartos et al. 2008) is still in its infancy. The initial QTL mapping in rye was based on line per se performance of agronomic traits and RFLP maps of F2-derived mapping populations (Börner et al. 2000; Milczarski and Masojć 2003). QTL governing agronomic traits in genetic resources of rye have been approached by a marker-assisted establishment of introgression line libraries (Falke et al. 2008, 2009). Further QTL mapping experiments were directed towards the dominant dwarfing gene Ddw1 (Börner et al. 1999), in vitro response (Bolibok et al. 2007), α-amylase activity and related traits (Masojć and Milczarski 2005, 2008; Myśków et al. 2011), as well as morphological traits (Myśków et al. 2014) of rye.
The phenotyping of the first comprehensive analysis identifying QTL of yield- and quality related traits was performed on progenies of two elite bi-parental mapping populations within the ‘Petkus’ gene pool (Miedaner et al. 2012). However, knowledge on QTL of agronomic important traits including grain yield and heading date in the ‘Carsten’ gene pool is still lacking. We report on a QTL analysis in elite germplasm of rye hybrids using a phenotyping strategy based on testcrosses with 258 F2:3 lines in multi-environmental field trials. The objectives of our study were to (1) identify QTL for grain yield, thousand grain weight, spikes per square meter, and heading date in the ‘Carsten’ gene pool, and to compare them to companion studies in the ‘Petkus’ gene pool, (2) identify orthologous segments in the rice genome for QTL carrying regions in rye, and (3) identify potential candidate genes based on a comparative genomics approach between rye and rice.
Materials and methods
Plant material
A bi-parental cross between two self-fertile elite inbred lines (HYB201 and HYB202) from the ‘Carsten’ (pollinator) gene pool was established by HYBRO Saatzucht GmbH & Co. KG and advanced to F2:3 lines without selection as recently described (Haffke et al. 2014). In total, 258 out of 272 F2:3 lines as well as both parental genotypes were successfully crossed with a CMS single-cross tester of the ‘Petkus’ (seed parent) gene pool resulting in three-way interpool hybrids. These experimental hybrids together with the six released hybrid varieties Minello, Visello, Palazzo, Brasetto, SU Drive, and SU Stakkato as checks were tested in field trials.
Field experiments and traits
Field experiments were conducted in 2 years (2011 and 2012) in Bornhof, Mecklenburg-Western Pomerania (53°49′N,12°89′W), Groß Lüsewitz, Mecklenburg-Western Pomerania (54°07′N, 12°33′W), Hohenheim, Baden Württemberg (48°72′N, 9°20′W), and Wulfsode, Lower Saxony (53°06′N, 10°24′W). The experiment in Bornhof 2012 failed due to severe pre-flowering drought stress, resulting in a total number of seven environments obtained as location × year combination. Entries were grown on drilled plots of 5–6 m2 size. The experimental design within each trial was a randomized incomplete block design (alpha design, 34 blocks × 8 entries) with two replications except of the parental genotypes, which were repeated four times each.
Plots were harvested with a conventional plot harvester at full ripening (EC 92) for grain yield (GYD). GYD is reported as dt ha−1 at 14% moisture. Additional traits recorded for all plots were heading date (HDT, 1 = very late, 9 = very early), thousand grain weight (TGW, g) and spikes per square meter (SSM), the latter at EC71 and using a counting frame. For statistical analyses, the means of both experiments were calculated as no significant (P > 0.05) difference among experiments was found.
Phenotypic data analyses
Statistical analyses were based on plot data of 258 testcross progenies. Checks were calculated separately. All statistical computations were performed with the PLABSTAT software package in a two-step procedure (Utz 2010). Analyses of variance were first performed for all traits in each environment separately. The adjusted entry means from each location were used in a second step to estimate variance components based on the following linear model:
where G and E denote genotype and environment, respectively. Both factors were treated as random effects. Broad-sense heritability (h 2) on an entry-mean basis was estimated from the variance components as the ratio of genotypic to phenotypic variance (Fehr 1987). Simple correlation coefficients (r) were calculated among all traits based on entry means. Significance of r was tested using tabulated values based on Fischer’s z transformation (Fischer 1921).
Genetic linkage map construction
The F2 population was advanced to F3 lines by selfing each F2 plant and reconstituted by combining leaf tissue from 15 plants each of 272 derived individual F2:3 progenies for DNA extraction. Analysis of SSR and COS markers on genomic DNA was performed in the mapping population as well as in disomic wheat-rye addition lines as previously described (Hackauf et al. 2009, 2012). Detailed information on assay conditions for both marker systems as well as primer sequences of the used COS markers are given in the electronic supplementary material 1 (ESM1). DArT markers have been assessed in the F2 population as well as in the recombinant inbred line population L2039-NxDH (Martis et al. 2013) at Diversity Arrays Ltd., Australia, as described by Bolibok-Bragoszewska et al. (2009). As the majority of genotyped markers in the F2 population followed a dominant inheritance, the linkage map was established by initially splitting the dominant markers into two groups with one group representing the dominant alleles from the female parent and the second group representing the dominant alleles from the male parent (Knapp et al. 1995). The resulting coupling-phase F2 maps were integrated using the codominant SSR and COS markers and the linkage mapping software JoinMap 4.0 (Van Ooijen 2006). The Kosambi function was used to convert recombination values to genetic distances (cM). The assignment of the seven linkage groups to the seven rye chromosomes and the determination of their orientation were performed using previously mapped anchor markers as well as mapping of COS markers to individual rye chromosome arms in disomic wheat-rye addition lines (ESM3).
QTL analyses
QTL analyses were based on the genetic linkage map and adjusted entry means using software PLABMQTL (Utz 2012). Markers with a distance below 1 cM were excluded automatically by the software. Based on testcross performance of F2:3 testcross progenies, main effect QTL for each trait contributing to the additive genetic variation can be detected. A 1-LOD support interval was specified around each QTL. Critical LOD thresholds were analyzed empirically for each trait according to Churchill and Doerge (1994) using 2000 permutation runs. It turned out that critical LOD thresholds corresponding to genome-wide error rates of α ≤ 10% were similar for all agronomic traits. Therefore, the highest LOD threshold (SSM = 3.47) was used for all agronomic traits. The proportion of genetic variance explained by the regression model was calculated as p G = R 2adj /h 2, where R 2adj is the adjusted proportion of phenotypic variance explained by the model. In addition, 1000 cross-validation runs were applied to determine the bias of R 2. For this, the data were resampled independently 200 times at fivefold cross validations. A fivefold cross validation (CV) was performed as follows: the entire data set (DS) was split into five genotypic subsamples and means from four out of five subsamples were used as estimation set (ES) for QTL detection, localization, and estimation of genetic effects. The remaining data group served as test set (TS). Out of this analysis, we give the frequency of recovery, i.e., the percentage of validation runs detecting the respective QTL, and the mean QTL effects in ES and TS for comparison. The identified QTL were designated according to the recommended rules for loci and alleles controlling quantitative characters in wheat and related species (McIntosh et al. 2013).
Comparative QTL mapping
Sequence information on mapped COS markers was used to identify their orthologs in rice as previously described (Hackauf et al. 2009, 2012). The position of individual gene models was obtained from release 6.1 of the rice pseudomolecules (Kawahara et al. 2013). The genomic coordinates of rice QTL were obtained from the Gramene database (Monaco et al. 2014, ESM2). Synteny maps between rye and rice were built using MapChart (Voorrips 2002).
Results
Pronounced genetic variation of experimental hybrids in agronomic traits
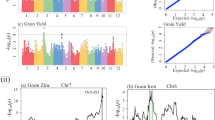
Phenotypic data of both parental genotypes were similar and the segregating progenies represented the mean testcross performance of their parents in most of the assessed traits (Table 1). All traits showed significant (P < 0.01) genotypic and genotype x environment interaction variances. The estimates of broad-sense heritability ranged from h 2 = 0.35 for SSM to h 2 = 0.88 for TGW with h 2 = 0.52 for GYD and h 2 = 0.67 for HDT (Table 1). None of the traits deviates from a normal distribution (Fig. 1). Both parental genotypes performed without significant phenotypic differences in HDT and TGW, while HYB201 performed superior to HYB202 in GYD and SSM (Table 1).

Frequency distribution of the traits spikes per square meter (SSM), heading date (HDT), thousand grain weight (TGW), and grain yield (GYD) in the population HYB201 × HYB202. Dashed lines mark means of parents (red HYB201, blue HYB202) (color figure online)
A genetic linkage map based on SSR, COS, and mixed-phase dominant DArT markers
We have established a genetic linkage map covering 964.9 cM of the rye genome (Table 2, ESM3). A set of 789 DArT markers, which were supplemented by 17 genomic and 40 EST-derived SSR as well as 66 COS markers, provided the framework to establish this map. The map length of the seven rye chromosomes varied between 105.5 cM of chromosome 3R and 165.9 cM of chromosome 5R. Integration of DArT markers in the recently published transcript map of the recombinant inbred line population L2039-NxDH allowed to identify 127 DArT markers (16.1%), which could be mapped in both populations (ESM3). A comparison based on these common markers revealed almost perfect collinearity between both maps.
In total, 26 of the EST-derived SSR (65%) and 60 of the COS markers (91%) correspond to orthologous gene models in rice (ESM4). The collinearity between the rye genetic map of HYB201 × HYB202 and the rice genome (Fig. 2, ESM5-10) identifies 13 conserved blocks, which represent 28.4% of the rye map and 25.2% of the rice genome (Table 3). Each syntenic block has at least two markers in common between individual rye and rice segments. Synteny between this rye map and the rice genome is supported by 157 RFLP and EST-derived SSR markers, which have been mapped in independent populations and which have been integrated in our analysis based on their orthologous rice gene models (ESM4). The chromosomal localization of these 157 markers on individual rye chromosome arms approves the suggested orientation of the 7 rye chromosomes in the genetic map of HYB201 × HYB202 and allows a rough prediction of the centromere position for each chromosome (Fig. 2, ESM5-10). Complementary to the rice gene models with known map position in rye, we have included 156 cloned QTL/genes mapping to the 13 conserved blocks in rice (ESM4). The majority of these genes (62) control flowering time. In addition, we have integrated the identified rice gene models with orthologs of known map position in rye in the recently published virtual linear gene order model (genome zipper) comprising 22,426 rye genes (ESM4, Martis et al. 2013). As already described for the DArT markers, the comparison with the map of the genome zipper, which integrates data from three independent mapping populations, showed a good congruency in the order of the EST-derived SSR and COS markers mapped in the present study (ESM4). Likewise, the rye genome zipper supports the observed syntenic relationship between the genetic map of HYB201 × HYB202 and the rice genome. Furthermore, the rye genome zipper bridges the genetic maps of HYB201 × HYB202 and Lo115 × Lo90 based on 191 SNP markers, which have been mapped in Lo115 × Lo90 (Miedaner et al. 2012) as well as in the genome zipper (ESM4, sheets RyeZipper_1R to RyeZipper_7R). Thus, a comparative QTL mapping between HYB201 × HYB202 and Lo115 × Lo90 based on the collinearity between both maps and the genome zipper becomes feasible as well. This comparison, however, revealed that the orientation of chromosomes 2R, 3R, 4R, 5R, and 7R in Lo115 × Lo90 was published inverted compared to the integrated map of the genome zipper (ESM4).

Comparative QTL mapping between rye and rice. Gene-derived markers are given in bold and allow to integrate a 61.8 cM segment on the long arm of rye chromosome 2R in the physical map of rice chromosomes 4 (R4) and 7 (R7). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of the markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indicate the position of the following quantitative traits: QGyd-2R: grain yield, QHdt-2R.1, QHdt-2R.2: heading date, QTgw-2R: thousand grain weight, qtgwt4: 1000-seed weight, QHd4: heading date, MQGyd4.1: grain yield, sn4.1: spikelet number, qNOS-4-1: spikelet number, qPN-4:panicle number, qtsn4.3: spikelet number, gpp4.1: grain number, pss4.1: seed set percent, dth4.2: day to heading, pss7.1: seed set percent. A description of the genes indicated in the rice physical maps is given in ESM4
Identification of QTL for four agronomic traits
Composite Interval Mapping with a multiple regression approach allowed to detect the positions of 22 QTL with an LOD threshold ≥3.47 (Table 4). Most QTL were found for TGW (10) and HDT (7). The explained genotypic variance of individual QTL ranged from 3 to 40%. The genotypic variance for HDT simultaneously explained by all seven detected QTL reached 85%. For the other traits, this estimate ranged from 44% for SSM to 70% for TGW. Our analysis revealed 11 major QTL explaining more than 10% of the genotypic variance. A major GYD QTL, QGyd-2R (p G = 40%), is located on the long arm of chromosome 2R (Fig. 2). Two major QTL for SSM, QSsm-3R (p G = 23.5%) and QSsm-5R (p G = 20.5%), are located on chromosome 3R and 5R, respectively (Table 4). Significant (P < 0.01) QTL × environment interaction variances could be observed for 10 QTL (45.5%, Table 4).
Comparative QTL mapping between rye and rice
The conserved synteny observed in the present study enables to superimpose QTL identified in rye and rice. For this purpose, we have used the rice genome as surrogate to integrate flanking markers of rye and rice QTL, which enabled to determine consensus among QTL for related traits (Fig. 2, ESM5-10).
Orthologous segments between 0.9 and 8.6 Mb on rice chromosomes R2, R3, R4, and R6 correspond to the six QTL QHdt-2R.1, QHdt-2R.2, QHdt-4R.2, QHdt-5R, QHdt-6R, and QHdt-7R controlling heading date in rye. The syntenic rice segments carry QTL dth4.2, QHd4, hd6, QHd3c, QHd3b, Hd6, dth2.1, and QHd3b, which have an impact on heading date in rice, as well (ESM2). Moreover, 18 cloned genes controlling heading date in rice are located on each of the syntenic fragments (ESM4).
For the four QTL QTgw-2R, QTgw-4R.1, QTgw-4R.3, and QTgw-5R controlling the yield component thousand grain weight in rye, we have identified syntenic segments between 0.8 and 5.2 Mb on rice chromosomes R3, R4, R6, and R11. Four of these syntenic rice segments harbour QTL qtgwt4, tgwt11, gw6, and QKw3a which govern TGW in rice, as well (ESM2). The rice genes, OsFLO, which is involved in the regulation of grain size and starch quality in rice (She et al. 2010) as well as HGW, which regulates grain weight and heading date in rice (Li et al. 2012b), are located in syntenic fragments of QTgw-2R and QTgw-4R.3 (ESM4).
For the QTL QSsm-5R controlling the yield component spikes per square meter in rye, we have identified a syntenic segment on rice chromosome R9. This rice segment carries the QTL qTN-9-1 which governs tiller number in rice (ESM2, ESM8). The gene OsEATB, which is involved in the regulation of tiller number in rice (Qi et al. 2011), is located in this syntenic segment too (ESM4).We have identified two syntenic segments of 0.04 and 4.7 Mb on rice chromosomes R3 and R4 for the 2 QTL QGyd-5R and QGyd-2R controlling GYD in rye. These syntenic rice segments harbour GYD QTL qgy3.1 and the meta QTL MQTL 4.1 for GYD (Swamy et al. 2011), the latter of which is indicated as MQGyld4.1 in Fig. 2. The syntenic segment on rice chromosome R4 is delimited by Xscm188 and Xscm235 and carries additional QTL governing spikelet number (qtsn4.3, qNOS-4.1, and sn4.1), panicle number (qPN-4), grain number (gpp4.1), and seed set percent (pss4.1) (ESM2). Furthermore, this 4.7 Mb rice segment carries the gene OsGA2ox6, a Gibberellin (GA) 2-oxidase (ESM4).
Discussion
Rich genetic diversity for agronomic traits in elite rye germplasm
In this study, we have used an F2:3 design (Austin and Lee 1996; Fisch et al. 1996) and analyzed 258 testcross progenies originating from two elite genotypes of a hybrid rye breeding program to identify QTL for four agronomic traits including grain yield in winter rye hybrids. Although, in this experimental design, only half of the total additive genetic variation can be exploited, this approach considers the limited predictive value of line per se performance for hybrid performance in yield-related traits in rye (Miedaner et al. 2014). A further serious challenge in using testcross performance for mapping in bi-parental populations is dominance. A strong dominant allele of a tester genotype may mask the effect of the allele contributed by the tested genotype. The limitations of strong elite testers have previously been discussed (Hallauer and Miranda 1988; Mihaljevic et al. 2005). However, despite these limitations, we observed a significant genetic variation (P < 0.01) for all analyzed traits. The observation of significant genetic variation between testcross progenies is particularly noteworthy, as in the F2:3 lines, only half of the additive genetic variance was exploited compared to recombinant inbred line populations (Hallauer et al. 2010). Although the resolution of QTL mapping using recombinant inbred lines is higher due to an increased number of recombination events in successive selfing steps, the time consuming development of recombinant inbred lines per se can be biased by inbreeding depression in case of the allogamous rye. Thus, an F2:3 design as described in the present study enables a fast and unbiased approach of QTL mapping in winter rye hybrids.
Notably, the parental mean was identical between both elite genotypes for HDT, which indicates that no major phenology-related gene, for example influencing photoperiod response, is segregating in the F2:3 population. This genetic makeup of both parental elite genotypes minimized masking of QTL by confounding effects of loci governing plant development, a phenomenon that affected QTL analysis in wheat and barley (Fleury et al. 2010). Furthermore, the parental mean did not differ significantly (P < 0.05) from progeny mean in all traits, except for GYD, indicating a predominantly additive inheritance. Although we observed no significant phenotypic differences between the means of the two parents in the testcross performance for HDT and TGW, the analysis of their progeny indicated that both genotypes differ with respect to the genetic constitution at individual QTL for both traits. Our results are in line with research, e.g., in maize which revealed, that parental phenotypic difference in testcross means was only weakly or even unrelated to progeny genetic variances (Melchinger et al. 1998; Hung et al. 2012). Thus, our data represent a further example of the challenge to (1) predict genetic variances in segregating generations from phenotypic means of parental genotypes and (2) appropriately select parents based on their phenotypic performance in plant breeding programs. In contrast, genome-based approaches have a higher power to predict the breeding potential of base populations in rye (Wang et al. 2014; Auinger et al. 2016).
The significance of genotype-by-environment interaction observed for all traits illustrates that multi-environmental phenotyping of the investigated traits is imperative. The number of environments used in the present study fits the suggested prerequisite for a reliable estimation of h 2 (Schön et al. 2004). Entry-mean heritability was high for TGW (h 2 = 0.88) and medium for HDT (h 2 = 0.67), while GYD (h 2 = 0.52) revealed a moderate heritability estimate. Consequently, the chances of QTL detection for these traits with the given sample sizes were assumed to be high. The h 2 estimates for GYD of intrapool testcrosses of two bi-parental populations from the ‘Petkus’ genepool were higher (h 2 = 0.70, Miedaner et al. 2012) compared to the h 2 estimates for GYD in the present study. The moderate heritability for GYD estimated in the present study indicates a reasonable high impact of the different test environments on the performance of the experimental hybrids in our study. Indeed, the year 2011 was one of the five warmest years in Germany since 1881 with 9% less precipitation compared to the long-term average (Deutscher Wetterdienst 2011), while rainfall and temperature were almost close to the average in 2012 throughout Germany (Deutscher Wetterdienst 2012).
A novel genetic linkage map for the analysis of complex inherited traits in rye
The construction of an accurate genetic linkage map with 941 markers provides the pivotal basis for the mapping of QTL and subsequent applications including comparative mapping, positional cloning, as well as the transfer of these results in practical crop improvement programs. We have used dominant inherited DArT markers as an efficient, microarray-based DNA fingerprinting method, which delivered the framework to establish a genetic linkage map in rye. Up to now, DArT markers have successfully been applied in rye to establish genetic linkage maps based on populations of recombinant inbred lines (RIL, Milczarski et al. 2011; Miedaner et al. 2012). By splitting the data set into two complementary subsets, each containing shared codominant and dominant markers in the coupling-phase, and subsequent integrating both data sets into a single map based on the shared codominant markers, we were able to establish a genetic linkage map in a F2 population, whose length (964.6 cM) compares well with data for previously published F2, BC1, and F3:4 mapping populations in rye (Devos et al. 1993; Korzun et al. 2001; Ma et al. 2001; Hackauf et al. 2009; Miedaner et al. 2012). In a previous report, an almost 2.5-fold increase in length has been observed for a genetic linkage map based on DArT and SSR compared to a map based on SNP and SSR markers (Miedaner et al. 2012). The mapping strategy applied in the present study considered that in F2 matings, recombination frequencies and locus orders of markers may be mis-estimated from dominant markers in mixed coupling phases (Knapp et al. 1995; Mester et al. 2003). We demonstrate that genetic map construction in rye can take advantage of an efficient genome-wide DArT fingerprinting without increasing the map length, if genotype data for a reasonable number of codominant markers are available as well. This requirement can be achieved for instance with SSR and COS markers. Likewise, SNP markers from the recently established high-density maps of rye (Martis et al. 2013; Bauer et al. 2017) as well as recently published sequence information on DArT markers (Gawroński et al. 2016) might be used and converted to allele-specific PCR assays for simple and accurate genotyping assays without sophisticated laboratory equipment.
The comprehensive sequence analysis by Gawroński and co-workers (2016) revealed inconsistencies concerning localization and order of DArT markers in a high-density consensus map (Milczarski et al. 2011) compared to the transcript map of rye (Martis et al. 2013). The observed collinearity between the rye map described in the present study and other genetic maps of rye as well as parts of the rice genome indicates a reliable marker order in the established genetic map. In total, 158 DArT markers (20.1%) described here have been mapped in the population Lo115 × Lo117 (Miedaner et al. 2012), as well. Notably, collinearity between both maps could be observed for all chromosomes; however, the orientation of chromosomes 2R, 4R, and 5R in population Lo115 × Lo117 is inverted relative to the map established for the QTL analysis described in the present study (ESM3).
The genetic map described here is characterized by a comparable high marker density. It has recently been reported that high-density maps increase the precision of QTL localization as well as the precision of effect estimates for detected QTL, especially for small- and medium-sized QTL and the power to resolve closely linked QTL (Stange et al. 2013). A further attribute of the established map in terms of a subsequent dissection of individual QTL is the alignment with the recently published transcript map of rye (Martis et al. 2013), which enables a systematic enrichment of markers for individual QTL. Here, we report only on those QTL that surmount the threshold across all seven environments. Although this approach may have contributed to a relatively low number of QTL per trait, these environmentally stable QTL are most promising for breeding purposes.
Large-effect QTL for heading date in conserved segments of the rye genome
Flowering time is a key adaptive trait in wild and crop cereal species which is also known as heading date and warrant plants to switch from vegetative to reproductive growth at a most favourable time for pollination, seed development, and seed distribution. Breeding of varieties with reduced time to flowering has been reported as an effective strategy to increase yield in regions where drought commonly occurs during grain filling (Passioura 1996; Slafer and Whitechurch 2001). In our study, the parents of the F2:3 population HYB201 and HYB202 are both elite winter rye genotypes adapted to the Central European climate. Although, thus, both parental genotypes reveal no phenotypic differences in heading date control, we were able to identify seven QTL explaining 85% of the genotypic variance for HDT in their testcross progenies. This observation indicates the impact of major genes in determining HDT of winter rye. Progress in dissecting the genetic and molecular basis of flowering time control in European elite winter wheat has recently been achieved based on genotypes of contrasting geographic origin, which revealed substantial differences in flowering time behaviour (Langer et al. 2014; Zanke et al. 2014). In both studies, the photoperiod regulator Ppd-D1 was identified as the major factor controlling flowering time in the studied germplasm sets. Interestingly, Zanke and co-workers (loc. cit.) identified a marker locus on wheat chromosome 5BL with homology to the rice photoperiodism gene Hd6, which is associated with the determination of the heading date in wheat. Likewise, QHdt-5R maps to a genomic segment, which is syntenic to a sub-genomic region on rice chromosome R3 including the heading date QTL Hd6. This obvious correspondence between heading date QTL across taxa provides insights in the evolution of the underlying HDT phenotype. The observation suggests that corresponding genes might be involved in the evolution of the relevant phenotypes and identifies QHdt-5R as a rewarding target for a dissection and detailed characterization of a HDT QTL at the molecular level in rye. The further development of molecular tools for a precise genetic improvement of HDT will support the adaptation and performance of winter rye particularly in the light of predicted climate change.
QTL for thousand grain weight are located at different positions in both heterotic groups
In the present study, we have approached TGW as an important component of GYD. In total, 10 QTL were detected explaining 70% of the genotypic variance. Neither of the three major QTL QTgw-1R.1 (p G = 14.68), QTgw-2R (p G = 17.34), and QTgw-4R.3 (p G = 10.41) identified in the present study reached a comparable high p G value as the majority of major TGW QTL identified in testcrosses of the two bi-parental ‘Petkus’ populations (Miedaner et al. 2012).
Based on DArT markers, which have been mapped in the populations Lo115 × Lo117 (‘Petkus’ gene pool) as well as HYB201 × HYB202 (‘Carsten’ gene pool, ESM3), a direct comparison of QTL governing TGW in both bi-parental populations is possible. QTgw-1R.1 maps to an interval which is defined by markers XrPt-399643 mapping at position 147.6 cM and XrPt-401076 mapping at position 175.6 cM on chromosome 1R in Lo115 × Lo117 and can, thus, be classified as not identical to the grain weight QTL #1, which has been mapped distally to the XrPt-399643/XrPt-401076-interval on chromosome 1R (Miedaner et al. 2012). This conclusion can be drawn for QTgw-1R.2 as well, which maps distally to the shared marker XrPt-507636, a marker located at position 270.0 cM in Lo115 × Lo117 (Miedaner et al. 2012). No thousand grain weight QTL have been mapped on chromosomes 2R, 3R, and 4R in population Lo115 × Lo117, thus, turning a comparison for QTgw-2R, QTgw-3R, QTgw-4R.1, QTgw-4R.2, and QTgw-4R.3 not feasible. QTgw-5R is located in an interval marked by XrPt-507948 and XrPt-410783, which have been mapped at positions 41.9 cM and 72.5 cM in Lo115 × Lo117 (Miedaner et al. 2012). Based on the map position of these markers relative to the distally located genetic interval carrying the QTL for TGW on chromosome 5R in Lo115 × Lo117, it can largely be excluded that QTgw-5R corresponds to the thousand grain weight QTL #2 in Lo115xLo117. Rather, QTgw-5R is located in the interval defined by XrPt-400590 and XrPt-507373. These markers are located on the long arm of chromosome 5R in the RIL population L2039-NxDH. In this RIL population, the wheat microsatellite marker Xgwm6-5R maps to this particular interval as well (ESM3). Using its synonymous identifier WMS6, Xgwm6-5R has been reported as being linked with the major gene KW 5 governing TGW in rye (Wricke 2002). This observation renders it likely that QTgw-5R corresponds to KW 5 . However, additional markers linked to Xgwm6-5R in population L2039-NxDH need to be mapped in population HYB201 × HYB202 to validate this hypothesis. The QTL QTgw-6R is located distally from XrPt-411507 and appears to be different from QTL #3, which has been mapped proximal to XrPt-411507 in population Lo115 × Lo117 (Miedaner et al. 2012). Finally, QTgw-7R maps to a segment on the short arm of chromosome 7R, which is located distally from XrPt-508478 at position 17.2 cM in Lo115 × Lo117 (Miedaner et al. 2012), indicating that this QTL does not correspond to the thousand grain weight QTL #4 on the long arm of chromosome 7R in the ‘Petkus’ population. The observed contrasting results in both bi-parental populations with respect to the explained genetic variance and particularly the different map positions of individual QTL indicate that in hybrid rye breeding, the genetic improvement of TGW needs to be independently examined in both heterotic genepools. In conclusion, all QTL detected in this population for TGW are different from those reported earlier in other populations.
A conserved major QTL for grain yield on the long arm of chromosome 2R
The QTL analysis described here allowed to detect a major QTL QGyd-2R, which is characterized by a high frequency of cross validation. In the two recently analyzed mapping populations representing the ‘Petkus’ gene pool, 1 and 7 QTL governing GYD in rye have been reported (Miedaner et al. 2012). Likewise, based on a data set of almost 1000 testcross progenies evaluated in 19 environments, 8 QTL for GYD have been identified in maize at a significance threshold of LOD = 3.21 (Schön et al. 2004). Schön and co-workers (2004) demonstrated by cross validation of experimental data, that increasing sample sizes as well as number of environments increased the number of detected QTL. As a consequence of this so-called Beavis effect (Beavis 1998; Xu 2003), it appears to be evident that the experiment described here was underpowered for QTL analysis of GYD and predisposed us to find large-effect variants, while we could not detect QTL with smaller effects that essentially caused variation in this trait. Indeed, our observation raises the question, if the effect for QGyd-2R is just overestimated or if biological reasons could explain that QTL variants like QGyd-2R with such a substantial influence on a quantitative trait like GYD have arisen under the infinitesimal model, which predicted that heritable quantitative traits would be specified by an innumerable number of minute effects (Fisher 1918)?
The mapping of individual QTL explaining a large proportion of the inherited variability is a common finding from QTL mapping experiments (Farrall 2004; Schön et al. 2004; Phillips 2005). For example, in rice, a major QTL for GYD was identified on chromosome R1 (Vikram et al. 2011), while in wheat, major QTL for GYD have been identified on chromosomes 3A and 6A (Rustgi et al. 2013). The identification of QGyd-2R in rye hybrids is in line with these observations. Interestingly, in the population Lo115 × Lo117, the GYD QTL #3 was mapped to that genetic interval on chromosome 2RL, which harbours QGyd-2R as well and which is defined by the flanking marker loci XrPt-401315 and XrPt-399800, respectively. The effect of GYD QTL #3 was less pronounced compared to QGyd-2R. This might be attributed to the fact that QGyd-2R in our study was identified in testcrosses of the mapping population with a single-cross elite tester from the opposite gene pool, which includes heterotic effects. The congruent map positions of QGyd-2R and GYD QTL #3 in both genetic divergent gene pools suggest that mutations at corresponding genetic loci on chromosome 2RL contribute to determine GYD in rye. Random mutations of large effect were proposed to have a high probability of fixation if they were favourable (Kimura 1983). This proposed modification of the infinitesimal model predicts that mutations of an intermediate size will drive adaptation and balances Fisher’s expectation that most favourable mutations will have small effects with correspondingly small fixation probabilities (Farrall 2004). This process might be enforced by the artificial selection in plant breeding. Likewise, the observed large effect of QGyd-2R fits to another model which suggests that natural selection validates mutations with large effects at the beginning of an adaptation process with a maximum of adaptive space, while later on in the process when the organism has essentially reached its optimum state, the space is narrowed and successful mutations must have smaller effects (Orr 1998). Thus, according to Orr’s model, QGyd-2R might belong to few QTL with (relatively) large effects, which determine the quantitative variation of GYD in rye together with a cumulative number of genes with smaller effects. A comparative genomics approach and the progress achieved in the isolation of rice genes controlling agronomic traits provide a first indication of a candidate gene with such a pronounced effect on grain yield.
Candidate genes exist across the identified QTL regions
In rye, comparative genomic approaches have proven to be successful for the development of molecular markers in different sub-genomic regions carrying major genes on the short arm of chromosome 1R (Mago et al. 2005), the long arm of chromosome 2R (Hackauf and Wehling 2005), the long arm of chromosome 4R (Hackauf et al. 2012), and the short arm of chromosome 7R (Miftahudin and Gustafson 2004; Collins et al. 2008). The observed collinearity between rye and rice in the present study matches the previously established virtual linear gene order model (Martis et al. 2013) almost perfectly and provides the opportunity to investigate overlaps among QTL in the genomes of rye and rice for individual traits.
The syntenic relationships between rye chromosome 3R and rice chromosome R8, 4R and R8, 5R and R6, 6R and R1, as well as 7R and R6 have not yet been described (Martis et al. 2013), and provide further insights in the evolution of the rye genome. The syntenic relationship between 6R and R1 is supported by the map positions of the genomic wheat SSR markers GWM391, GWM247, and GWM340 on the long arm of rye chromosome 6R, which have been described in a previous study (Khlestkina et al. 2004) and could be confirmed here. These markers have been mapped on homeologous group 3 chromosomes in wheat (Röder et al. 1998), which have been shown to correspond to rice chromosome R1 (Munkvold et al. 2004).
Flowering time is a trait that is largely conserved across taxa and the flowering time pathway in temperate grasses is well characterized at the molecular level (Higgins et al. 2010). Thus, the flowering time genes located in corresponding segments of the six QTL QHdt-2R.1, QHdt-2R.2, QHdt-4R.2, QHdt-5R, QHdt-6R, and QHdt-7R provide candidates to further dissect the molecular basis of flowering time control in rye. An interesting candidate gene appears to be the rice HGW gene, which is located 0.02 Mb distally to a 1.7 Mb segment on rice chromosome R6, that is defined by Xtcos1747 and Xtc368556 g and that is syntenic to a segment on rye chromosome 4R including the colocalized QTL QTgw-4R.3 and QHdt-4R.2. HGW encodes an ubiquitin-associated (UBA) domain protein, which delays heading and reduces grain weight in rice (Li et al. 2012a). Likewise, the rice gene OsFLO is involved in the regulation of grain size and starch quality in rice (She et al. 2010) and provides a candidate gene for the QTL QTgw-2R on rye chromosome 2R.
A comprehensive analysis of the official German variety trials, where experimental hybrids are tested for their value for cultivation and use, highlighted that the tremendous genetic gain achieved in grain yield of hybrid rye varieties between 1989 and 2014 is mainly driven by their ability to increase the density of SSM (Laidig et al. 2017). We have identified two QTL, QSsm-3R and QSsm-5R, explaining 44% of the genotypic variance for SSM in our mapping population. This result suggests the impact of major genes in determining SSM of winter rye. The synteny to rice chromosomes R9 discovers the rice gene OsEATB, which encodes an ERF protein associated with tillering and panicle branching (Qi et al. 2011), as an initial target for studying the genetic makeup of QSsm-5R in rye at the sequence level (ESM4).
Interestingly, the meta QTL MQTL 4.1 for GYD in rice is located on the segment on rice chromosome R4 (Swamy et al. 2011) corresponding to the QGyd-2R-interval on rye chromosome 2RL. This observation identifies the Xscm188/Xscm235 interval on rye chromosome 2RL and rice chromosome R4 particularly attractive for further research to improve our understanding concerning the genetic basis of GYD in cereals. Both flanking microsatellite markers Xscm188 and Xscm235 enable a marker-assisted introgression of QGyd-2R in different genetic backgrounds for the validation with the current and alternative CMS single-cross testers in rye. Furthermore, QGyd-2R represents an attractive target to counterbalance linkage drag effects on grain yield that have been observed for efficient restorer-of-fertility genes in rye hybrids (Miedaner et al. 2017).
The co-localization of the grain yield MQTL 4.1 with QTL governing spikelet number (qtsn4.3, qNOS-4.1, sn4.1), panicle number (qPN-4), grain number (gpp4.1), and seed set percent (pss4.1) in the syntenic Xscm188/Xscm235-segment on rice chromosome R4 reveals information on important yield components. In rye, a high amount of heterosis has been reported for seed set percent and grain number (Geiger and Wahle 1978). Thus, these traits provide a possible explanation of the large genetic effect of QGyd-2R on grain yield in the present study compared to the GYD QTL #3 identified in testcrosses within the ‘Petkus’ gene pool (Miedaner et al. 2012) and should be assessed in subsequent experiments on QGyd-2R. Noteworthy, a putative gibberellin (GA) 2-beta-dioxygenase, OsGA2ox6, is located in the syntenic Xscm188/Xscm235-interval on rice chromosome R4 as well. A Gain-of-function GA2ox6-mutant showed increased tillers and adventitious root growth in rice (Lo et al. 2008). Overexpression of GA2ox6 with a mutation at A141E in rice has been reported to result in a significant increase of grain yield by, on average, 24% as compared to the non-transformed control and by, on average, 39% to transgenic rice plants with overexpression of the allele G343A in three tested environments (Lo et al. 2016). Although GA2ox6 is not listed among the candidate genes residing at the MQTL 4.1 region (Swamy et al. 2011), this knowledge of gene function as well the strong correlation between tiller number and grain yield in hybrid rye (Laidig et al. 2017) qualifies GA2ox6 as an interesting target to approach allelic variants of both parental genotypes (HYB201, HYB202) and beyond to elucidate effects on grain yield in rye. The negative heterosis observed for tiller number of rye hybrids compared to the midparent values (Geiger and Wahle 1978) does not exclude GA2ox6 as a candidate gene for QGyd-2R, as light incidence is higher in a plant population of inbred lines, which, in addition, are less vigorous compared to hybrid plants and, thus, have a lower demand for nutrients and water. Altogether, these attributes probably result in a lesser reduction of established tillers in inbred lines at stem elongation compared to hybrids (Geiger and Wahle 1978). However, we observed no co-localization between QGyd-2R and QTL for tiller number, a trait that we have recorded as SSM. This might be explained by the comparable low heritability estimate (h 2 = 0.35) of SSM in our study, as the power of QTL detection is strongly affected by the heritability of a trait (Viana et al. 2017). Likewise, linkage significantly reduced detection power in a simulation study (Li et al. 2012a) and might have compromised to detect a QTL for SSM linked to QGyd-2R. Thus, further research on QGyd-2R including high-resolution mapping is needed to precisely determine the number and linkage relationships of genes with an impact on GYD.
In contrast to the progress achieved in dissecting the molecular basis of complex agronomic traits particularly in rice (Zuo and Li 2014), large knowledge gaps still exists on the genetic and molecular control of biological processes related to grain yield and yield parameters in other small grain cereals (Valluru et al. 2014). Using the rice genome data as a blueprint, comparative genomic approaches enabled to identify genes associated with nitrogen use efficiency (Quraishi et al. 2011a), grain fibre content (Quraishi et al. 2011b), carotinoid content (Dibari et al. 2012), grain weight (Su et al. 2011; Zhang et al. 2012), spikelet number (Zhang et al. 2015), and grain size (Ma et al. 2016) in wheat. These examples demonstrate the potential of comparative genomics approaches to isolate conserved genes controlling QTL in large Triticeae genomes. Hence, the initial genome scan described in the present study can open new perspectives to understand the genetic basis of the analyzed agronomic important traits in rye and to strengthen our understanding of the genetic and molecular mechanisms underlying cereal yield traits.
Conclusions
The QTL analysis of agronomically important traits including grain yield and heading date in the present study revealed a high degree of polymorphism in elite rye genotypes. This rich genetic diversity of the outbreeding rye together with a phenotyping strategy based on a F2:3 design and testcross performance in multi-environmental field trials argues for a stronger utilization of rye in QTL analysis for the identification of genes that genetically control agronomically important traits in small grain cereals.
The large-effect QTL, like those found here for GYD, HDT, SSM, and TGW, could be candidates for successful marker-assisted selection programs in rye practical breeding. In addition, the primary mapping population described here will be advanced to near isogenic lines (NILs) for further fine-mapping and cloning individual QTL. For this purpose, the whole-genome draft sequence of rye (Bauer et al. 2017) will make a crucial contribution to enhance our understanding of complex agronomic traits in this close relative of wheat.
Author contribution statement
The work presented here was carried out in collaboration between all authors. BH, FJF, and TM defined the research theme and supervised the project. BH and FJF conceived the design of this study and coordinated the experiments. FJF developed the plant materials. SH contributed to phenotyping, analyzed and interpreted the phenotypic data statistically, and performed the QTL analysis. BK and SR contributed to the collection of phenotypic data. AK guided the DArT fingerprinting and validated marker scores. DM contributed to the collection of phenotypic data and integrated DArT markers in the L2039-NxDH map. BH guided the genotyping with sequence-specific markers, established the genetic linkage map, performed the comparative mapping, interpreted the results, and drafted the manuscript. TM discussed analyses, interpretation, and presentation. All authors have read and approved the final version of the manuscript.
References
Auinger HJ, Schönleben M, Lehermeier C, Schmidt M, Korzun V, Geiger HH, Piepho HP, Gordillo A, Wilde P, Bauer E, Schön CC (2016) Model training across multiple breeding cycles significantly improves genomic prediction accuracy in rye (Secale cereale L.). Theor Appl Genet 129:2043–2053. doi:10.1007/s00122-016-2756-5
Austin DF, Lee M (1996) Comparative mapping in F2:3 and F6:7 generations of quantitative trait loci for grain yield and yield components in maize. Theor Appl Genet 92:817–826. doi:10.1007/BF00221893
Bartos J, Paux E, Kofler R, Havránková M, Kopecký D, Suchánková P, Safár J, Simková H, Town CD, Lelley T, Feuillet C, Dolezel J (2008) A first survey of the rye (Secale cereale) genome composition through BAC end sequencing of the short arm of chromosome 1R. BMC Plant Biol 8:95. doi:10.1186/1471-2229-8-95
Bauer E, Schmutzer T, Barilar I, Mascher M, Gundlach H, Martis MM, Twardziok SO, Hackauf B, Gordillo A, Wilde P, Schmidt M, Korzun V, Mayer KF, Schmid K, Schön C-C, Scholz U (2017) Towards a whole-genome sequence for rye (Secale cereale L.). Plant J. 89:853–869. doi:10.1111/tpj.13436
Beavis WD (1998) QTL analyses: power, precision and accuracy. In: Paterson AH (ed) Molecular dissection of complex traits. CRC Press, Boca Raton, pp 145–162
Bolibok H, Gruszczyńska A, Hromadajudycka A, Rakoczy-Trojanowska M (2007) The identification of QTL associated with the in vitro response of rye (Secale cereale L.). Cell Mol Biol Lett 12:523–535. doi:10.2478/s11658-007-0023-0
Bolibok-Bragoszewska H, Heller-Uszyńska K, Wenzl P, Uszyński G, Kilian A, Rakoczy-Trojanowska M (2009) DArT markers for the rye genome—genetic diversity and mapping. BMC Genomics 10:578. doi:10.1186/1471-2164-10-578
Börner A, Korzun V, Voylokov AV, Weber WE (1999) Detection of quantitative trait loci on chromosome 5R of rye (Secale cereale L.). Theor Appl Genet 98:1087–1090. doi:10.1007/s11032-011-9627-1
Börner A, Korzun V, Voylokov AV, Worland AJ, Weber WE (2000) Genetic mapping of quantitative trait loci in rye (Secale cereale L.). Euphytica 116:203–209. doi:10.1023/A:1004052505692
Churchill GA, Doerge RW (1994) Empirical threshold values for quantitative trait mapping. Genetics 138:963–971
Collins NC, Shirley NJ, Saeed M, Pallotta M, Gustafson JP (2008) An ALMT1 gene cluster controlling aluminum tolerance at the Alt4 locus of rye (Secale cereale L.). Genetics 179:669–682. doi:10.1534/genetics.107.083451
Deutscher W (2011) Review of the year: The weather in Germany in 2011. www.dwd.de
Deutscher W (2012) Review of the year: The weather in Germany in 2012. www.dwd.de
Devos KM, Atkinson MD, Chinoy CN, Francis HA, Harcourt RL, Koebner RMD, Liu CJ, Masojć P, Xie DX, Gale MD (1993) Chromosomal rearrangements in the rye genome relative to that of wheat. Theor Appl Genet 85:673–680. doi:10.1007/BF00225004
Dibari B, Murat F, Chosson A, Gautier V, Poncet C, Lecomte P, Mercier I, Bergès H, Pont C, Blanco A, Salse J (2012) Deciphering the genomic structure, function and evolution of carotenogenesis related phytoene synthases in grasses. BMC Genomics 6(13):221. doi:10.1186/1471-2164-13-221
Falke KC, Susić Z, Hackauf B, Korzun V, Schondelmaier J, Wilde P, Wehling P, Wortmann H, Mank R, Rouppe van der Voort J, Maurer HP, Miedaner T, Geiger HH (2008) Establishment of introgression libraries in hybrid rye (Secale cereale L.) from an Iranian primitive accession as a new tool for rye breeding and genomics. Theor Appl Genet 117:641–652. doi:10.1007/s00122-008-0808-1
Falke KC, Susić Z, Wilde P, Wortmann H, Möhring J, Piepho HP, Geiger HH, Miedaner T (2009) Testcross performance of rye introgression lines developed by marker-assisted backcrossing using an Iranian accession as donor. Theor Appl Genet 118:1225–1238. doi:10.1007/s00122-009-0976-7
Farrall M (2004) Quantitative genetic variation: a post-modern view. Hum Mol Genet 13 Spec No 1: R1-7. doi: 10.1093/hmg/ddh084
Fehr WR (1987) Heritability. In: Fehr WR (ed) Principles of cultivar development theory and technique. McMillan Pub. Co., New York, pp 95–105
Fisch RD, Ragot M, Gay G (1996) A generalization of the mixture model in the mapping of quantitative trait loci for progeny from a biparental cross of inbred lines. Genetics 143:571–577
Fischer RA (1921) On the “probable error” of a coefficient of correlation deduced from a small sample. Metron 1:1–32
Fisher RA (1918) The correlation between relatives on the supposition of Mendelian inheritance. Trans R Soc Edinb 52:399–433
Fleury D, Jefferies S, Kuchel H, Langridge P (2010) Genetic and genomic tools to improve drought tolerance in wheat. J Exp Bot 61:3211–3222. doi:10.1093/jxb/erq152
Gawroński P, Pawełkowicz M, Tofil K, Uszyński G, Sharifova S, Ahluwalia S, Tyrka M, Wędzony M, Kilian A, Bolibok-Brągoszewska H (2016) DArT markers effectively target gene space in the rye genome. Front Plant Sci. 7:1600. doi:10.3389/fpls.2016.01600
Geiger HH, Miedaner T (2009) Rye breeding. In: Carena MJ (ed), Cereals, pp. 157–181. Handbook of plant breeding, vol. 3. Springer Science + Business Media
Geiger HH, Wahle G (1978) Struktur der Heterosis von Komplexmerkmalen bei Winterrroggen-Einfachhybriden. Z. Pflanzenzüchtg. 80:178–210
Hackauf B, Wehling P (2005) Approaching the self-incompatibility locus Z in rye (Secale cereale L.) via comparative genetics. Theor Appl Genet 110:832–845. doi:10.1007/s00122-004-1869-4
Hackauf B, Rudd S, van der Voort JR, Miedaner T, Wehling P (2009) Comparative mapping of DNA sequences in rye (Secale cereale L.) in relation to the rice genome. Theor Appl Genet 118:371–384. doi:10.1007/s00122-008-0906-0
Hackauf B, Korzun V, Wortmann H, Wilde P, Wehling P (2012) Development of conserved ortholog set markers linked to the restorer gene Rfp1 in rye. Mol. Breed. 30:1507–1518. doi:10.1007/s11032-012-9736-5
Haffke S, Kusterer B, Fromme FJ, Roux S, Hackauf B, Miedaner T (2014) Analysis of covariation of grain yield and dry matter yield for breeding dual use hybrid rye. Bioenergy Res. 7:424–429. doi:10.1007/s12155-013-9383-7
Hallauer AR, Miranda JB (1988) Quantitative genetics in maize breeding. Iowa State University Press, Ames
Hallauer AR, Carena MJ, Miranda Filho JB (2010) Quantitative genetics in maize breeding. Handbook of Plant Breeding vol. 6, 3rd edn, 664 p, Springer, New York
Hepting L (1978) Analysis of a 7 × 7-variety diallel for determination of suitable base materials for hybrid breeding in rye. Z. Pflanzenzüchtg. 80:188–197
Higgins JA, Bailey PC, Laurie DA (2010) Comparative genomics of flowering time pathways using Brachypodium distachyon as a model for the temperate grasses. PLoS ONE 5:e10065. doi:10.1371/journal.pone.0010065
Hou J, Jiang Q, Hao C, Wang Y, Zhang H, Zhang X (2014) Global selection on sucrose synthase haplotypes during a century of wheat breeding. Plant Physiol 164:1918–1929. doi:10.1104/pp.113.232454
Hung HY, Browne C, Guill K, Coles N, Eller M, Garcia A, Lepak N, Melia-Hancock S, Oropeza-Rosas M, Salvo S, Upadyayula N, Buckler ES, Flint-Garcia S, McMullen MD, Rocheford TR, Holland JB (2012) The relationship between parental genetic or phenotypic divergence and progeny variation in the maize nested association mapping population. Heredity 108:490–499. doi:10.1038/hdy.2011.103
Jiang Y, Jiang Q, Hao C, Hou J, Wang L, Zhang H, Zhang S, Chen X, Zhang X (2015) A yield-associated gene TaCWI, in wheat: its function, selection and evolution in global breeding revealed by haplotype analysis. Theor Appl Genet 128:131–143. doi:10.1007/s00122-014-2417-5
Kawahara Y, de la Bastide M, Hamilton JP, Kanamori H, McCombie WR, Ouyang S, Schwartz DC, Tanaka T, Wu J, Zhou S, Childs KL, Davidson RM, Lin H, Quesada-Ocampo L, Vaillancourt B, Sakai H, Lee SS, Kim J, Numa H, Itoh T, Buell CR, Matsumoto T (2013) Improvement of the Oryza sativa Nipponbare reference genome using next generation sequence and optical map data. Rice 6:4. doi:10.1186/1939-8433-6-4
Khlestkina EK, Than MH, Pestsova EG, Röder MS, Malyshev SV, Korzun V, Börner A (2004) Mapping of 99 new microsatellite-derived loci in rye (Secale cereale L.) including 39 expressed sequence tags. Theor Appl Genet 109:725–732. doi:10.1007/s00122-004-1659-z
Kimura M (1983) The neutral theory of molecular evolution. Cambridge University Press, Cambridge
Knapp SJ, Holloway JL, Bridges WC, Liu B-H (1995) Mapping dominant markers using F2 matings. Theor Appl Genet 91:74–81. doi:10.1007/BF00220861
Korzun V, Malyshev S, Voylokov AV, Börner A (2001) A genetic map of rye (Secale cereale L.) combining RFLP, isozyme, protein, microsatellite and gene loci. Theor Appl Genet 102:709–717. doi:10.1007/s001220051701
Kovach MJ, McCouch SR (2008) Leveraging natural diversity: back through the bottleneck. Curr Opin Plant Biol 11:193–200. doi:10.1016/j.pbi.2007.12.006
Kumar GR, Sakthivel K, Sundaramm RM, Neeraja CN, Balachandran SM, Rani NS, Viraktamath BC, Madhav MS (2010) Allele mining in crops: prospects and potentials. Biotechnol Adv 28:451–461. doi:10.1016/j.biotechadv.2010.02.007
Laidig F, Piepho HP, Rentel D, Drobek T, Meyer U, Huesken A (2017) Breeding progress, variation, and correlation of grain and quality traits in winter rye hybrid and population varieties and national on-farm progress in Germany over 26 years. Theor Appl Genet. doi:10.1007/s00122-017-2865-9
Langer SM, Longin CF, Würschum T (2014) Flowering time control in European winter wheat. Front Plant Sci 5:537. doi:10.3389/fpls.2014.00537
Li H, Zhang L, Wang J (2012a) Estimation of statistical power and false discovery rate of QTL mapping methods through computer simulation. Chin Sci Bull 57:2701–2710. doi:10.1007/s11434-012-5239-3
Li J, Chu H, Zhang Y, Mou T, Wu C, Zhang Q, Xu J (2012b) The rice HGW gene encodes a ubiquitin-associated (UBA) domain protein that regulates heading date and grain weight. PLoS ONE 7:e34231. doi:10.1371/journal.pone.0034231
Lo SF, Yang SY, Chen KT, Hsing YI, Zeevaart JA, Chen LJ, Yu SM (2008) A novel class of gibberellin 2-oxidases control semidwarfism, tillering, and root development in rice. Plant Cell 20:2018–2603. doi:10.1105/tpc.108.060913
Lo SF, Ho TD, Liu YL, Jiang MJ, Hsieh KT, Chen KT, Yu LC, Lee MH, Chen CY, Huang TP, Kojima M, Sakakibara H, Chen LJ, Yu SM (2016) Ectopic expression of specific GA2 oxidase mutants promotes yield and stress tolerance in rice. Plant Biotechnol J. doi:10.1111/pbi.12681
Ma XF, Wanous MK, Houchins K, Rodriguez MA, Milla Goicoechea PG, Wang Z, Xie M, Gustafson JP (2001) Molecular linkage mapping in rye (Secale cereale L.). Theor Appl Genet 102:517–523. doi:10.1007/s001220051676
Ma L, Li T, Hao C, Wang Y, Chen X, Zhang X (2016) TaGS5-3A, a grain size gene selected during wheat improvement for larger kernel and yield. Plant Biotechnol J 14:1269–1280. doi:10.1111/pbi.12492
Mackay TFC, Stone EA, Ayroles JF (2009) The genetics of quantitative traits: challenges and prospects. Nat Rev Genet 10:565–577. doi:10.1038/nrg2612
Mago R, Miah H, Lawrence GJ, Wellings CR, Spielmeyer W, Bariana HS, McIntosh RA, Pryor AJ, Ellis JG (2005) High-resolution mapping and mutation analysis separate the rust resistance genes Sr31, Lr26 and Yr9 on the short arm of rye chromosome 1. Theor Appl Genet 112:41–50. doi:10.1007/s00122-005-0098-9
Martis MM, Zhou R, Haseneyer G, Schmutzer T, Vrána J, Kubaláková M, König S, Kugler KG, Scholz U, Hackauf B, Korzun V, Schön CC, Dolezel J, Bauer E, Mayer KF, Stein N (2013) Reticulate evolution of the rye genome. Plant Cell. 25:3685–3698. doi:10.1105/tpc.113.114553
Masojć P, Milczarski P (2005) Mapping QTL for alpha-amylase activity in rye grain. J Appl Genet 46:115–123
Masojć P, Milczarski P (2008) Relationship between QTL for preharvest sprouting and alpha-amylase activity in rye grain. Mol Breed 23:75–84. doi:10.1007/s11032-008-9215-1
McIntosh RA, Yamazaki Y, Dubcovsky J, Rogers J, Morris C, Appels R, Xia XC (2013) Catalogue of gene symbols for wheat. In:12th International Wheat Genetics Symposium 8–13 September 2013, Yokohama, Japan
Melchinger AE, Gumber RK, Leipert RB, Vuylsteke M, Kuiper M (1998) Prediction of testcross means and variances among F3 progenies of F1 crosses from testcross means and genetic distances of their parents in maize. Theor Appl Genet 96:503–512. doi:10.1007/s001220050767
Mester DI, Ronin YI, Hu Y, Peng J, Nevo E, Korol AB (2003) Efficient multipoint mapping: making use of dominant repulsion-phase markers. Theor Appl Genet 107:1102–1112. doi:10.1007/s00122-003-1305-1
Meuwissen THE, Hayes B, Goddard M (2001) Prediction of total genetic value using genome-wide dense marker maps. Genetics 157:1819–1829
Miedaner T, Hübner M, Korzun V, Schmiedchen B, Bauer E, Haseneyer G, Wilde P, Reif JC (2012) Genetic architecture of complex agronomic traits examined in two testcross populations of rye (Secale cereale L.). BMC Genomics 13:706. doi:10.1186/1471-2164-13-706
Miedaner T, Schwegler DD, Wilde P, Reif J (2014) Association between line per se and testcross performance for eight agronomic and quality traits in winter rye. Theor Appl Gen 127:33–41. doi:10.1007/s00122-013-2198-2
Miedaner T, Herter CP, Goßlau H, Wilde P, Hackauf B (2017) Correlated effects of exotic pollen-fertility restorer genes on agronomic and quality traits of hybrid rye. Plant Breed. doi:10.1111/pbr.12456
Miftahudin Scoles GJ, Gustafson JP (2004) Development of PCR-based codominant markers flanking the Alt3 gene in rye. Genome 47:231–238. doi:10.1139/g03-093
Mihaljevic R, Schön CC, Utz HF, Melchinger AE (2005) Correlations and QTL correspondence between line per se and testcross performance for agronomic traits in four populations of European maize. Crop Science 45:114–122
Milczarski P, Masojć P (2003) Interval mapping of genes controlling growth of rye plants. Plant Breed. Seed Sci. 48:135–142
Milczarski P, Bolibok-Brągoszewska H, Myśków B, Stojałowski S, Heller-Uszyńska K, Góralska M, Brągoszewski P, Uszyński G, Kilian A, Rakoczy-Trojanowska M (2011) A high density consensus map of rye (Secale cereale L.) based on DArT markers. PLoS ONE 6:e28495. doi:10.1371/journal.pone.0028495
Monaco MK, Stein J, Naithani S, Wei S, Dharmawardhana P, Kumari S, Amarasinghe V, Youens-Clark K, Thomason J, Preece J, Pasternak S, Olson A, Jiao Y, Lu Z, Bolser D, Kerhornou A, Staines D, Walts B, Wu G, D’Eustachio P, Haw R, Croft D, Kersey PJ, Stein L, Jaiswal P, Ware D (2014) Gramene 2013: comparative plant genomics resources. Nucleic Acids Res 42:D1193–D1199. doi:10.1093/nar/gkt1110
Munkvold JD, Greene RA, Bermudez-Kandianis CE, La Rota CM, Edwards H, Sorrells SF, Dake T, Benscher D, Kantety R, Linkiewicz AM, Dubcovsky J, Akhunov ED, Dvorák J, Miftahudin Gustafson JP, Pathan MS, Nguyen HT, Matthews DE, Chao S, Lazo GR, Hummel DD, Anderson OD, Anderson JA, Gonzalez-Hernandez JL, Peng JH, Lapitan N, Qi LL, Echalier B, Gill BS, Hossain KG, Kalavacharla V, Kianian SF, Sandhu D, Erayman M, Gill KS, McGuire PE, Qualset CO, Sorrells ME (2004) Group 3 chromosome bin maps of wheat and their relationship to rice chromosome 1. Genetics 168:639–650. doi:10.1534/genetics.104.034819
Myśków B, Stojałowski S, Łań A, Bolibok-Bragoszewska H, Rakoczy-Trojanowska M, Kilian A (2011) Detection of the quantitative trait loci for α-amylase activity on a high-density genetic map of rye and comparison of their localization to loci controlling preharvest sprouting and earliness. Mol Breeding 30:367–376. doi:10.1007/s11032-011-9627-1
Myśków B, Hanek M, Banek-Tabor A, Maciorowski R, Stojałowski S (2014) The application of high-density genetic maps of rye for the detection of QTL controlling morphological traits. J Appl Genet 55:15–26. doi:10.1007/s13353-013-0186-5
Orr HA (1998) The population genetics of adaptation: the distribution of factors fixed during adaptive evolution. Evolution 52:935–949. doi:10.2307/2411226
Passioura JB (1996) Drought and drought tolerance. Plant Growth Regul 20:79–83. doi:10.1007/978-94-017-1299-6_1
Phillips PC (2005) Testing hypotheses regarding the genetics of adaptation. Genetica 123:15–24. doi:10.1007/s10709-004-2704-1
Qi W, Sun F, Wang Q, Chen M, Huang Y, Feng YQ, Luo X, Yang J (2011) Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene. Plant Physiol 157:216–228. doi:10.1104/pp.111.179945
Qin L, Hao C, Hou J, Wang Y, Li T, Wang L, Ma Z, Zhang X (2014) Homologous haplotypes, expression, genetic effects and geographic distribution of the wheat yield gene TaGW2. BMC Plant Biol 14:107. doi:10.1186/1471-2229-14-107
Quraishi UM, Abrouk M, Murat F, Pont C, Foucrier S, Desmaizieres G, Confolent C, Rivière N, Charmet G, Paux E, Murigneux A, Guerreiro L, Lafarge S, Le Gouis J, Feuillet C, Salse J (2011a) Cross-genome map based dissection of a nitrogen use efficiency ortho-metaQTL in bread wheat unravels concerted cereal genome evolution. Plant J 65(5):745–756. doi:10.1111/j.1365-313X.2010.04461.x
Quraishi UM, Murat F, Abrouk M, Pont C, Confolent C, Oury FX, Ward J, Boros D, Gebruers K, Delcour JA, Courtin CM, Bedo Z, Saulnier L, Guillon F, Balzergue S, Shewry PR, Feuillet C, Charmet G, Salse J (2011b) Combined meta-genomics analyses unravel candidate genes for the grain dietary fiber content in bread wheat (Triticum aestivum L.). Funct Integr Genomics 11(1):71–83. doi:10.1007/s10142-010-0183-2
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier MH, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 14:2007–2023
Rustgi S, Shafqat MN, Kumar N, Baenziger PS, Ali ML, Dweikat I, Campbell BT, Gill KS (2013) Genetic dissection of yield and its component traits using high-density composite map of wheat chromosome 3A: bridging gaps between QTL and underlying genes. PLoS ONE 8:e70526. doi:10.1371/journal.pone.0070526
Schön CC, Utz HF, Groh S, Truberg B, Openshaw S, Melchinger AE (2004) Quantitative trait locus mapping based on resampling in a vast maize testcross experiment and its relevance to quantitative genetics for complex traits. Genetics 167:485–498. doi:10.1534/genetics.167.1.485
She KC, Kusano H, Koizumi K, Yamakawa H, Hakata M, Imamura T, Fukuda M, Naito N, Tsurumaki Y, Yaeshima M, Tsuge T, Matsumoto K, Kudoh M, Itoh E, Kikuchi S, Kishimoto N, Yazaki J, Ando T, Yano M, Aoyama T, Sasaki T, Satoh H, Shimada H (2010) A novel factor FLOURY ENDOSPERM2 is involved in regulation of rice grain size and starch quality. Plant Cell. 22:3280–32394. doi:10.1105/tpc.109.070821
Slafer GA, Whitechurch EM (2001) Manipulating wheat development to improve adaptation and to search for alternative opportunities to increase yield potential. In: Reynolds MP, Ortiz-Monasterio JI, McNab A (eds) Application of physiology in wheat breeding. CIMMYT, Mexico, pp 160–170
Stange M, Utz HF, Schrag TA, Melchinger AE, Würschum T (2013) High-density genotyping: an overkill for QTL mapping? Lessons learned from a case study in maize and simulations. Theor Appl Genet 126:2563–2574. doi:10.1007/s00122-013-2155-0
Su Z, Hao C, Wang L, Dong Y, Zhang X (2011) Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.). Theor Appl Genet 122:211–223. doi:10.1007/s00122-010-1437-z
Swamy BP, Vikram P, Dixit S, Ahmed HU, Kumar A (2011) Meta-analysis of grain yield QTL identified during agricultural drought in grasses showed consensus. BMC Genomics 12:319. doi:10.1186/1471-2164-12-319
Utz HF (2010) PLABSTAT. Computerprogramm für statistische Analyse für Pflanzenzüchtungsversuche. Institut für Pflanzenzüchtung, Saatgutforschung und Populationsgenetik, Universität Hohenheim, Stuttgart
Utz HF (2012) PlabMQTL—Software for meta-QTL analysis with composite interval mapping. Version 0.5 s. Institute of Plant Breeding, Seed Science, and Population Genetics, University of Hohenheim. PlabMQTL Manual
Valluru R, Reynolds MP, Salse J (2014) Genetic and molecular bases of yield-associated traits: a translational biology approach between rice and wheat. Theor Appl Genet 127:1463–1489. doi:10.1007/s00122-014-2332-9
Van Ooijen JW (2006) JoinMap ® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen, Netherlands
Viana JMS, e Silva FF, Mundim GB, Azevedo CF, Jan HU (2017) Efficiency of low heritability QTL mapping under high SNP density. Euphytica 213:13. doi:10.1007/s10681-016-1800-5
Vikram P, Swamy BP, Dixit S, Ahmed HU, Teresa Sta Cruz M, Singh AK, Kumar A (2011) qDTY1.1, a major QTL for rice grain yield under reproductive-stage drought stress with a consistent effect in multiple elite genetic backgrounds. BMC Genet 12:89. doi:10.1186/1471-2156-12-89
Voorrips RE (2002) MapChart: Software for the graphical presentation of linkage maps and QTL. J Hered 93:77–78
Wang Y, Mette MF, Miedaner T, Gottwald M, Wilde P, Reif JC, Zhao Y (2014) The accuracy of prediction of genomic selection in elite hybrid rye populations surpasses the accuracy of marker-assisted selection and is equally augmented by multiple field evaluation locations and test years. BMC Genom 15:556. doi:10.1186/1471-2156-12-89
Wricke G (2002) Two major genes for kernel weight in rye. Plant Breed 121:26–28. doi:10.1046/j.1439-0523.2002.00666.x
Wright SI, Vroh Bi I, Schroeder SG, Yamasaki M, Doebley JF, McMullen MD, Gaut BS (2005) The effects of artificial selection on the maize genome. Science 308:1310–1314. doi:10.1126/science.1107891
Xing Y, Zhang Q (2010) Genetic and molecular bases of rice yield. Annu Rev Plant Biol 61:421–442. doi:10.1146/annurev-arplant-042809-112209
Xu S (2003) Theoretical basis of the Beavis effect. Genetics 165:2259–2268
Yamamoto E, Yonemaru JI, Yamamoto T, Yano M (2012) OGRO: the overview of functionally characterized genes in rice online database. Rice 5:26. doi:10.1186/1939-8433-5-26
Yamasaki M, Tenaillon MI, Bi IV, Schroeder SG, Sanchez-Villeda H, Doebley JF, Gaut BS, McMullen MD (2005) A large-scale screen for artificial selection in maize identifies candidate agronomic loci for domestication and crop improvement. Plant Cell 17:2859–2872. doi:10.1105/tpc.105.037242
Yonemaru JI, Yamamoto T, Fukuoka S, Uga Y, Hori K, Yano M (2010) Q-TARO: QTL annotation rice online database. Rice 3:194–203. doi:10.1007/s12284-010-9041-z
Yue A, Li A, Mao X, Chang X, Li R, Jing R (2015) Identification and development of a functional marker from 6-SFT-A2 associated with grain weight in wheat. Mol Breed 35:63. doi:10.1007/s11032-015-0266-9
Zanke C, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Hinze M, Beier S, Ganal W, Röder MS (2014) Genetic architecture of main effect QTL for heading date in European winter wheat. Front Plant Sci 5:217. doi:10.3389/fpls.2014.00217
Zhang L, Zhao YL, Gao LF, Zhao GY, Zhou RH, Zhang BS, Jia JZ (2012) TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol 195:184–574. doi:10.1111/j.1469-8137.2012.04194.x
Zhang B, Liu X, Xu W, Chang J, Li A, Mao X, Zhang X, Jing R (2015) Novel function of a putative MOC1 ortholog associated with spikelet number per spike in common wheat. Sci Rep. 5:12211. doi:10.1038/srep12211
Zuo J, Li J (2014) Molecular dissection of complex agronomic traits of rice: a team effort by Chinese scientists in recent years. Natl Sci Rev. 1:253–276. doi:10.1093/nsr/nwt004
Acknowledgements
We highly appreciate the teams at the respective stations of HYBRO Saatzucht GmbH & Co. KG, University of Hohenheim, and Julius Kühn-Institut Groß Lüsewitz for their excellent technical assistance in performing the field trials and data collection. We gratefully acknowledge the excellent technical assistance of Gunda Kölzow in genotyping of the population. This study was financially supported by the Federal Ministry of Education and Research (Grant no. 0315445A, 0315445C, and 0315445D) and the company HYBRO Saatzucht GmbH & Co. KG, Germany. The responsibility of the content of this publication rests with the authors.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Communicated by Aimin Zhang.
Electronic supplementary material
Below is the link to the electronic supplementary material.
122_2017_2926_MOESM5_ESM.jpg
Supplementary material 5 (JPEG 467 kb) Comparative QTL mapping between rye and rice. Gene-derived markers are given in bold and allow to integrate a 98.2 cM segment of rye chromosome 1R into the physical map of rice chromosomes 5 (R5) and 10 (R10). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of he markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indic te the position of the following quantitative traits: QTgw-1R.1, QTgw-1R.2: thousand grain weight, KW10: kernel weight, w5: 1000-seed weight. A description of the genes indicated in the rice physical maps is given in ESM4
122_2017_2926_MOESM6_ESM.jpg
Supplementary material 6 (JPEG 284 kb) Comparative QTL mapping between rye and rice. Gene-derived markers are given in bold andallow to integrate a 6.8 cM segment of rye chromosome 3R into the physical map of rice chromosome 1 (R1). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of the markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indicate the position of the following quantitative traits: QTgw-3R: thousand grain weight, gw1: 1000-seed weight, QSsm-3R: spikes per square meter, QGyd-3R: grain yield. A description of the genes indicated in the rice physical maps is given in ESM4
122_2017_2926_MOESM7_ESM.jpg
Supplementary material 7 (JPEG 451 kb) Comparative QTL mapping between rye and rice. G 1026 ene-derived markers are given in bold and allow to integrate a 44 cM segment of rye chromosome 4R into the physical map of rice chromosomes 6 (R6) and 11 (R11). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of the markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indicate the position of the following quantitative traits: QHdt-4R.1, QHdt-4R.2: heading date, QPh3-4R.1, QPh3-4R.2: plant height, QTgw-4R.1, QTgw-4R.2, QTgw-4R.3: thousand grain weight, hd6: heading date, tgwt11, gw6: 1000-seed weight, ph11: plant height. A description of the genes indicated in the rice physical maps is given in ESM4
122_2017_2926_MOESM8_ESM.jpg
Supplementary material 8 (JPEG 560 kb) Comparative QTL mapping between rye and rice. Gene-derived markers are given in bold and allow to integrate a 33.3 cM segment of rye chromosome 5R into the physical map of rice chromosome 3 (R3). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of the markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indicate the position of the following quantitative traits: QHdt-5R: heading date, QTgw-5R: thousand grain weight, QGyd-5R: grain yield, QSsm-5R: spikes per square meter, Hd3b, Hd3c, Hd6: days to heading, QKw3a: 1000-seed weight, qgy3.1: grain yield. A description of the genes indicated in the rice physical maps is given in ESM4
122_2017_2926_MOESM9_ESM.jpg
Supplementary material 9 (JPEG 334 kb) Comparative QTL mapping between rye and rice. Gene-derived markers are given in bold and allow to integrate a 35.6 cM segment of rye chromosome 6R into the physical map of rice chromosome 2 (R2). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of the markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indicate the position of the following quantitative traits: QHdt-6R: heading date, QTgw-6R: thousand grain weight, dth2.1: days to heading, QKwa2a: 1000-seed weight. A description of the genes indicated in the rice physical maps is given in ESM4
122_2017_2926_MOESM10_ESM.jpg
Supplementary material 10 (JPEG 268 kb) Comparative QTL mapping between rye and rice. Gene-derived markers are given in bold and allow to integrate a 12.4 cM segment of rye chromosome 7R into the physical map of rice chromosome 3 (R3). The gene-derived markers and their orthologs in rice are connected by dotted lines. The positions of the markers in the rye map are given in cM and in the physical map of rice in Mb. The vertical bars and QTL symbols indicate the position of the following quantitative traits: QTgw-7R: thousand grain weight, QHdt-7R: heading date, Qhd3b: days to heading. A description of the genes indicated in the rice physical maps is given ESM4
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Hackauf, B., Haffke, S., Fromme, F.J. et al. QTL mapping and comparative genome analysis of agronomic traits including grain yield in winter rye. Theor Appl Genet 130, 1801–1817 (2017). https://doi.org/10.1007/s00122-017-2926-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-017-2926-0