
Abstract
Restoration of male fertility is a prerequisite for hybrid rye breeding and currently the most straightforward approach to minimize ergot infection in hybrid rye varieties. Molecular markers are important tools for the efficient introgression and management of restorer genes like Rfp1 originating from unadapted genetic resources. Furthermore, closely linked markers flanking Rfp1 are indispensible for identifying and selecting individuals with haplotypes showing recombination between Rfp1 and other gene(s) that reside in close proximity and have a negative influence on yield. We identified orthologous gene sets in rice, Brachypodium, and Sorghum and used these gene models as templates to establish conserved ortholog set (COS) markers for the restorer gene Rfp1 on the long arm of rye chromosome 4R. The novel co-dominant markers delimit Rfp1 within a 0.7-cM interval and allow prediction of Rfp1 genotypes with a precision not feasible before. The COS markers enabled an alignment of the improved genetic map of rye chromosome 4R with wheat and barley maps and allowed identification of regions orthologous to Rfp1 in wheat and barley on the short arms of chromosomes 6D and 6H, respectively. Results obtained in this study revealed that micro-collinearity around the Rfp1 locus in rye is affected by rearrangements relative to other grass genomes. The impact of the novel COS markers for practical hybrid rye breeding is discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Rye (Secale cereale L.) is the only outbreeding species among the small-grain cereals and, thus, has traditionally been bred to population varieties. Based on the so-called ‘Pampa’ (P) cytoplasm as a genetic mechanism of fertilization control for hybrid seed production and due to high levels of heterosis, which can be observed among the cross progeny of parental inbred lines developed from genetically distant genepools, effective hybrid breeding programmes were established during the past four decades. The P cytoplasm was derived from an Argentinian primitive rye population and results in a male-sterile phenotype (Geiger and Schnell 1970). In plants displaying cytoplasmic male sterility (cms), formation of viable pollen grains is disrupted by a mitochondrial mutation (cf. Chase 2007 for review).
Highly productive hybrid varieties keep rye growing competitive in modern agricultural production systems. In Germany, the percentage of released hybrid varieties of rye increased from 37 % in 2002 to 57 % in 2010 (Bundessortenamt 2011). A present drawback of hybrid varieties in rye is their susceptibility to ergot (Claviceps purpurea [Fr.] Tul.), as a consequence of sub-optimal pollen shedding. Particularly under unfavourable, rainy weather conditions during flowering, low abundance of pollen means that, on a stigma, spores of the fungus have an advantage in the competition with germinable pollen grains for sites of entry to the ovary (Miedaner et al. 2005). The elevated risk of ergot incidence as a result of increasing acreages grown with hybrid rye varieties is a major challenge to rye production in Germany. Susceptibility to ergot upon artificial inoculation is a trait assessed for registered rye varieties since 2008 in the descriptive variety list of the Bundessortenamt, an independent German senior federal authority under the supervision of the Federal Ministry of Food, Agriculture and Consumer Protection, which is responsible for granting of Plant Breeders’ Rights, the registration of varieties in the National List and for variety and seed affairs. The compliance of defined thresholds for ergot contamination of the harvest (0.05 % for human consumption, 0.1 % for feeding purposes) is critical for marketing of rye. As a consequence, ergot infection is one of the economically most important diseases in rye.
The restoration of male fertility is a prerequisite for hybrid rye breeding and currently the most straightforward approach to minimize ergot infection in hybrid rye varieties (Miedaner et al. 2000, 2005; Stracke et al. 2003).
Fertility restorer (Rf) genes for the P cytoplasm are designated Rfp (Stracke et al. 2003). First results on the genetics of male-fertility restoration in the P cytoplasm of rye were reported by Scoles and Evans (1979) who suggested the action of three dominant restorer genes with partial and environmentally influenced effects in inbred lines carrying the P cytoplasm. Rfp genes known to date from European rye germplasm lead to incomplete restoration of male fertility in the P cytoplasm (Miedaner et al. 2000, 2005). One of these Rfp genes has been mapped to rye chromosome 1R (Wricke et al. 1993). More effective Rfp genes have been identified in genetic resources collected in Iran and Argentina, respectively. These genes, Rfp1 and Rfp2, were mapped to the long arm of chromosome 4R (Miedaner et al. 2000; Stracke et al. 2003) and contribute to minimize harvest contamination with ergot, as they result in an almost complete restoration of male fertility in hybrid rye varieties. There are, however, other gene(s) associated with these exotic Rfp variants that exert a negative influence on agronomically important traits (Miedaner et al. 2000). To elucidate whether these unfavorable effects are the results of a linkage drag, closely linked molecular markers are needed to identify and select individuals with recombinant haplotypes. In an initial attempt, sequence-tagged site (STS) markers for the restorer genes Rfp1 and Rfp2 were developed from amplified fragment length polymorphism (AFLP) and random amplified polymorphic DNA (RAPD) fragments (Stracke et al. 2003).
The availability of the genome sequences of rice (International Rice Genome Sequencing Project 2005), Sorghum (Paterson et al. 2009), and most recently of Brachypodium (International Brachypodium Initiative 2010) provides the opportunity to identify sets of orthologous genes in species related to these grasses, which have evolved from a common ancestor by speciation. The development of molecular markers representing orthologous genes, so-called COS (conserved ortholog set) markers (Fulton et al. 2002), was successfully applied in rye for different sub-genomic regions on the short arm of chromosome 1R (Mago et al. 2005), the long arm of chromosome 2R (Hackauf and Wehling 2005), and the short arm of chromosome 7R (Miftahudin et al. 2004; Collins et al. 2008). Recently, a draft of the rye/rice genome relationship described conserved orthologous sequences between rye and rice in different sub-genomic regions located on all seven rye chromosomes (Hackauf et al. 2009).
The present study is focussed on the Rfp1 locus located on the long arm of rye chromosome 4R and aims at (1) identifying the Rfp1-orthologous regions in rice, Brachypodium, and Sorghum, (2) using the gene models of these grasses to develop COS markers linked to the Rfp1 locus, and (3) mapping the novel COS markers relative to Rfp1. The potential value of these COS markers for practical hybrid rye breeding is discussed based on knowledge gained from comparative mapping between grass species.
Materials and methods
Assessment of Rfp1 genotypes
F1 plants were established by crossing the male-sterile single-cross tester L2039-P × L145-N from the non-restorer genepool with a BC2 restorer line originating from the restorer genepool of HYBRO GmbH & Co. KG. The restorer line carried a 4RL chromosome-segment with the restorer gene Rfp1, which has its source in the population IRAN IX (Miedaner et al. 2000). Introgression of this donor-chromosomal segment was achieved by marker-assisted backcrossing using the dominant STS marker SCXX04 linked to Rfp1 (Stracke et al. 2003). Two F1 plants were selfed to produce the F2 families JKI-1301 and JKI-1302, respectively. From each of these F2 families segregating for Rfp1, plants were cloned with two clones per individual plant. Male fertility was visually assessed in two environments in 2005 according to Geiger and Morgenstern (1975), with one clonal part of each family being scored by HYBRO and JKI at the experimental field sites of Wulfsode and Groß Lüsewitz, respectively. Two to three main tiller spikes per plant were isolated by bagging. F2:3 progeny testing of all fertile F2 plants was performed in 2006 at Groß Lüsewitz Experimental Station with 16 individuals per F2:3 progeny to identify homozygous and heterozygous Rfp1 genotypes. In addition, three of these F2:3 progenies with 95, 93, and 94 individuals, respectively, were scored in 2007. Additionally, 250 individuals of the F2 population Lo6-P × IRAN IX, which has previously been characterized with respect to the genetic constitution at the Rfp1 locus by KWS LOCHOW GmbH (Stracke et al. 2003), were included for mapping purposes.
Development and mapping of COS markers
The sterility-inducing P cytoplasm was monitored by means of the recently described sequence characterized amplified regions (SCAR) markers to study the mitochondrial genes cox2 and nad6 (Stojałowski et al. 2006). Development, application, and mapping of COS markers on the long arm of chromosome 4R were performed as described previously (Hackauf and Wehling 2005; Hackauf et al. 2009) using GoTaq® DNA polymerase (Promega, Mannheim, Germany) for the amplification of sub-genomic fragments. Sequence information on restriction fragment length polymorphism (RFLP) probes cMWG652 (acc. nos. AJ234404, AJ234405), PSR8 (J02817), HvNAR (X57845), CDO476 (BE439306, BE439373), BCD342 (BE438801), PSR899 (X69817), BCD1821 (BE439006, BE439033), PSR119 (AJ440538, AJ440539) and PSR167 (AJ440600) were downloaded from Genbank (http://www.ncbi.nlm.nih.gov/sites/entrez?db=nucleotide). STS primers derived from wheat and barley unigenes, which were supplemented by primers established from mapped wheat expressed sequence tags (ESTs_ (Akhunov et al. 2010), and PCR conditions are given in Supplemental Table 1.
Amplicons obtained from the non-restorer inbred line L2053-N of the HYBRO breeding programme with the markers TC264604, TC281179, TC176835, and TC300731 were directly sequenced by LGC Genomics (Berlin), using the indicated PCR primers. Information on orthologous gene sets was obtained from the Phytozome project, release v6.0 (http://www.phytozome.net/), as well as from the PLAZA (vers. 2.0) online resource (Proost et al. 2009).
STS markers SCY03 and SCXX04 were assayed as described by Stracke et al. (2003). The waxy locus was addressed using primers described by Mason-Gamer et al. (1998). Multipoint linkage analysis of markers was performed using the software package JoinMap v.4.0 (Van Ooijen 2006) with a LOD score of 7.0. The Kosambi function was applied to convert recombination values to genetic distances (cM). The recombination frequency \( r = \frac{{r_{1} r_{2} }}{{1 - r_{1} - r_{2} + 2r_{1} r_{2} }} \) between Rfp1 and two flanking markers was calculated according to Weber and Wricke (1994). Chromosomal localization of the STS markers was validated using disomic wheat–rye addition lines kindly provided by S.M. Reader (Department of Crop Genetics, John Innes Centre, Norwich, UK). To determine a more precise physical location of the COS markers, sequence information of the analyzed unigenes were aligned to ESTs physically mapped in wheat (Randhawa et al. 2004). Information on mapped ESTs was queried from the GrainGenes-SQL database of Triticeae EST information (http://wheat.pw.usda.gov/cgi-bin/westsql/map_locus.cgi). Rice, Sorghum, and Brachypodium gene models were used to integrate the COS markers mapped in rye into the genome zipper of the short arm of barley chromosome 6H (Mayer et al. 2011). The Gramene QTL database (Youens-Clark et al. 2011) was searched for agronomic traits mapped to chromosomes 2 and 6 in rice and chromosomes 4 and 10 in Sorghum.
Results
Genetic analysis of fertility restoration
Assessment of male fertility revealed a bimodal distribution of pollen-fertility means across locations in the two established F2 populations as well as in the F2:3 families with clear peaks in the male-sterile and fully male-fertile classes (Fig. 1, Table 1). In both F2 populations, ten and nine individuals were scored as partially male fertile (fertility scores 4–6), while in the three F2:3 families all plants appeared either as male sterile or completely male fertile.

Distribution of phenotypic scoring values in populations segregating with male fertile and male sterile phenotypes. Frequency distributions of anther scores (1–3 = male sterile, 4–6 = partially male fertile, 7–9 = male fertile) in the F2 mapping populations JKI-1301 (white bars) with 86 individuals and JKI-1302 with 75 individuals (black bars) as well as F2:3 progenies JKI-200.574 (light gray bars), JKI-200.575 (dotted bars) and JKI-200.638 (dark gray bars) with 95, 93, and 94 individuals, respectively
Four (score 5) and three (score 4) of the 19 plants, which had initially been assessed as partially male fertile in both F2 populations, had no seed set, suggesting false scoring, i.e., that these seven plants indeed should be classified as male sterile. In contrast, seed set for the remaining 12 partially male-fertile individuals ranged from 20 to 60 kernels/ear and enabled analysis of pollen fertility in the F2:3 progenies. In this subset, the observed pollen fertility allowed the definition of three of the parental F2 individuals as male fertile resulting from the expression of a dominant restorer allele. The remaining nine parental F2 plants were deduced to carry the non-restorer allele of the male-sterile tester at the Rfp1 locus, as their progenies were male sterile (data not shown).
Taken together, we were able to determine the genetic constitution at the restorer locus Rfp1 for 161 F2 plants and 282 F2:3 progenies. The observed segregation in the F2 populations as well as in the three selected F2:3 progenies fitted the hypothesis of a monogenic dominant inheritance of male-fertility restoration (Table 1). The 161 F2 plants and 95 progenies of the F2:3 family JKI-200.594, together with a subset of the 250 F2 individuals derived from the cross Lo6-P × IRAN IX previously defined at the Rfp1 locus, were selected for mapping purposes.
Development and mapping of COS markers linked to Rfp1
A comparative-genetic approach of marker development in rye allowed us to establish 22 novel, sequence-specific markers for the long arm of chromosome 4R (Supplemental Table 2). Sequence information of eight RFLP anchor markers located on the long arm of chromosome 4R provided landmarks to identify orthologous regions in other grass genomes by BLASTN analysis, which in turn were used to identify sequence-based markers in the selected sub-genomic region in rye via PCR. Sequence specificity of the PCR was validated by direct sequencing of PCR products followed by BLASTN/BLASTX analysis (data not shown). Two unigene-derived COS markers representing the RFLP probes cMWG652 and CDO476 could not be mapped in rye due to lack of polymorphism in the experimental populations. Seventeen (77 %) of the novel markers revealed a co-dominant inheritance with an insertion/deletion polymorphism in the experimental populations, allowing a simple scoring of the amplification products on agarose gels either directly upon PCR or, in the case of an invariant amplicon, following digestion with restriction enzymes (Supplemental Table 2). Among the remaining dominant markers, four segregated in coupling phase and one in repulsion relative to Rfp1.
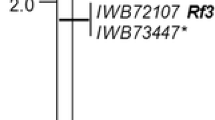
For the 22 markers, the expected 1:2:1 or 3:1 segregation ratios, respectively, were observed. The novel markers, together with five previously developed sequence-tagged sites, allowed the establishment of a genetic linkage map spanning 61 cM (Fig. 2). Localization of the established linkage group on the long arm of chromosome 4R was verified using wheat-rye addition lines and marker loci TC176835 (suppl. Fig. 1), TC120635, and TC112013 (not shown) as anchors. Five of the novel markers are located within the 3.4-cM segment delimited by the STS markers Xp15/55 and Xscxx04, respectively, which have been reported as markers flanking Rfp1 (Stracke et al. 2003). The novel markers allow us to further narrow down the Rfp1 locus to a genetic interval of 0.7 cM, which is defined by the flanking co-dominant markers TC300731 and TC256739 (Fig. 2). The estimated recombination frequencies r Rfp1-TC300731 = 0.4 cM and r Rfp1-TC256739 = 0.3 cM enable calculation of a joint probability for a recombination between markers and Rfp1 of 0.0012 % if both flanking markers are examined concurrently as a single selection criterion.

Comparative mapping of the restorer gene Rfp1 in rye. The integrated linkage map representing part of chromosome 4R is based on 506 Rfp1 genotypes and aligned to physical maps of model grass genomes. Dotted lines indicate orthologous genes in rice, Brachypodium, and Sorghum, which have been used for marker development in rye. COS markers originating from wheat unigenes with EST members of known localization in wheat bins are indicated in bold. Gray boxes indicate segments of the long arm of rye chromosome 4R orthologous to different wheat bins. A wheat bin is defined by two deletion breakpoints and is given as name followed by the arm fraction-length endpoints for which the deletion is diagnostic; e.g., 6DS-6-0.99-1.00 designates the region from a breakpoint at 99 % of the 6DS arm to one at 100 % (Endo and Gill 1996)
Comparison of the Rfp1 locus in rye to orthologous regions in other grass genomes
Fifteen of the unigene sequences used for marker development for the long arm of rye chromosome 4R represent ESTs with known locations in chromosome interval region bins within wheat chromosome (arms) (Fig. 2, Supplemental Table 2). This information enabled us to assign the Rfp1 locus to a rye genome segment orthologous to the major gene-rich region 6S1.0 in wheat (bin 6DS-6(0.99)-1.00; Fig. 2, Supplemental Table 2). Four of the unigene sequences mapped in our study proved to be located in co-linear orders on the short arm of barley chromosome 6H within a 22-cM interval (suppl. Table 2). Comparison of the relative map positions of the unigene sequences in different grass genomes revealed collinearity at the genetic-map level of the distal part of the long arm of rye chromosome 4R with sub-genomic segments of Brachypodium chromosome 3, Sorghum chromosome 4, and rice chromosome 2 (Fig. 2). Three coding sequences, Os02g01440, Os02g01450, and Os02g01480, were reported to be located within a 15.6-kb segment on rice chromosome 2 and allowed us to derive PCR primers for the gene-derived markers TC176835, TC300731, and TC135788. These markers are linked to Rfp1 (Fig. 3).

Close-up of the Rfp1 locus in rye and orthologous sub-genomic regions in Brachypodium, Sorghum, and rice. The genetic map of the Rfp1 locus in rye is illustrated at the top. The physical maps of the orthologous rice, Sorghum, and Brachypodium sub-genomic regions are given below. Orthologous genes in rice, Sorghum, and Brachypodium are given as black boxes. Gray boxes represent genes located at non-collinear positions within rice, Brachypodium, and Sorghum. Empty boxes indicate hypothetical genes which have no orthologs in other species. Orthologous genes are connected at their 3′ ends by dashed lines. Nuclear genes residing in regions orthologous to the Rfp1 locus, which are known to interact with mitochondria, are indicated as bottom-down triangles [pentatricopeptide (PPR) repeat-containing protein], bottom-up triangles [Mitochondrial Rho (Miro) GTPase] and stars [mitochondrial transcription termination factor (mTERF)]
In rice, the 11.2-kb segment defined by Os02g01450 and Os02g01480 harbors two additional genes, namely Os02g01460 and Os02g01470, which are both predicted to encode for transposable elements. For this reason, Os02g01460 and Os02g01470 were not considered for the development of additional markers in the rye target interval. Rather, we included genomic sequence resources from two other grass species, Brachypodium distachyon and Sorghum bicolor. Comparison between rice and Sorghum revealed a conservation of gene order and content (micro-collinearity) between the two species in this sub-genomic segment (Fig. 3). A more complex situation was found for a segment of Brachypodium chromosome 3 containing orthologs of the five TC markers. This segment comprises 10 additional predicted gene-coding sequences in the target region (Fig. 3). Three (Bradi3g00780, Bradi3g00790, Bradi3g00800) of these additional genes are located between Bradi3g00767 and Bradi3g00804, the latter of which is orthologous to Os02g01480 and predicted to encode a pentatricopeptide (PPR) repeat-containing protein. Bradi3g00780 is predicted to encode for a mitochondrial transcription termination factor (mTERF). According to the precomputed PLAZA dataset, Bradi3g00780 belongs to a family of 319 genes in 17 species. The closest related rice gene is Os06g12100 with 54 % identity to Bradi3g00780 at the amino-acid level (E value = 6.8e−104). Os06g12100 is located on rice chromosome 6 within a 29.2-kb segment (position 6,453,328–6,482,604), which contains a cluster of six additional, tandemly duplicated genes (Os06g12040, Os06g12050, Os06g12060, Os06g12070, Os06g12080, Os06g12110) also predicted to encode for mTERF proteins (Fig. 3). BLASTN/BLASTX analysis identified significant sequence similarity between Os06g12100 and the wheat EST BF483174 (63 % amino acid identity, E value = 9e−036), the latter of which maps to wheat bin 6DS6-FL0.99-1.00. Two other rice genes, Os06g12030 (6,444,938–6,439,709) and Os06g12129 (6,490,675–6,488,579), flank the above-mentioned 29.2-kb segment (data not shown). Os06g12030 is orthologous to wheat ESTs BQ161619 and BM134479, and Os06g12129 shows significant sequence similarity to CD453020. Unlike the 29.2-kb segment, these three wheat ESTs are not located on wheat bin 6DS6-FL0.99-1.00; rather, they have been physically mapped to wheat bin 7BS1-0.27-1.00 (BQ161619, BM134479) and chromosome 7D (CD453020). To conclude, use of the Brachypodium gene Bradi3g00780 in combination with the physically mapped wheat EST BF483174 allowed us to identify the 29.2-kb segment on rice chromosome 6 as a specific and promising template for the targeted development of markers for the Rfp1 restorer gene in rye. The orthologous gene set Bradi3g00780/Os06g12100 (cf. PLAZA database, Proost et al. 2009) was used to develop an additional gene-derived marker. Alignment of Os06g12100 with the wheat unigene sequence TC256739 (TBLASTX: E value = 4e−128) enabled us to derive primers which amplify an expected 688-bp fragment from rye-genomic DNA. Restriction digestion of this amplicon revealed a complex fragment pattern which was polymorphic between male-sterile and male-fertile genotypes (Fig. 4). The number and sizes of the observed restriction fragments indicate that the primers amplified several copies of related sequences. Linkage analysis identified the polymorphic TC256739 fragment to be located within the expected genetic interval of TC300731 and TC135788, 0.3 cM proximal of the restorer gene.

Restriction pattern of TC256739. Agarose gel-based restriction analysis of the 688-bp amplicon obtained with primers derived from the wheat EST unigene TC256739 allows a clear visualization of marker genotypes associated with male-sterile and male-fertile individuals. b ms = bulk of 10 male-sterile plants from JKI-1302, b mf = bulk of 10 male fertile plants from JKI-1302, M = size standard
Discussion
Genetic analysis of fertility restoration
The restorer gene Rfp1 in rye was studied in a F2 test-cross progeny with the P-type sterility-inducing cytoplasm. Our data confirms previous findings of Miedaner et al. (2000) and Stracke et al. (2003) who identified Rfp1 as a dominantly acting restorer gene.
Fertility restoration in the P cytoplasm is subject to environmental influence (Scoles and Evans 1979; Geiger et al. 1995). As demonstrated in our study, progeny testing may substantially improve accuracy when assigning Rfp1 genotypes to individual plants. Progeny testing revealed that nine out of 12 F2 individuals which had phenotypically been scored as partially male fertile belong to the class of male-sterile genotypes. This finding is in contrast to the assumption of Stracke et al. (2003) who assigned all partially male-fertile individuals (anther scores 4–6) to the Rfp1. class for segregation analysis. Their classification was based on the genotypes of flanking dominant markers and was not validated by progeny testing.
COS markers for Rfp1
Comparative mapping based on sequenced model genomes such as rice, Sorghum, or Brachypodium is particularly rewarding for a species like rye where species-specific genomic resources lag behind those of other small grain cereals. We have applied this strategy to a region on the distal part of the long arm of rye chromosome 4R to map the Rfp1 gene in a targeted approach.
In the present study, the COS strategy of using ESTs for primer design with a single match to genes in model genomes proved to be successful in the development of closely linked PCR-based markers for Rfp1. In contrast to the previously developed Rfp1 markers, the novel co-dominant markers allow us to unambiguously distinguish heterozygous and homozygous Rfp1 genotypes with a precision not feasible before.
Three Rfp genes have been mapped to the long arm of rye chromosome 4R (Miedaner et al. 2000; Falke et al. 2009). In addition to these Rfp genes, restorer genes for the G (Börner et al. 1998) and C (Stojałowski et al. 2005) cytoplasms, which are both functionally different from P (Geiger et al. 1995) have also been mapped to the long arm of chromosome 4R. Likewise, the rye restorer gene Rfc4, which restores male fertility in hexaploid wheat with timophevii sterility-inducing cytoplasm, has been localized on the long arm of rye chromosome 4R (Curtis and Lukaszewski 1993). The restorer genes Rf6 (Ma et al. 1995) and Rfm1 (Matsui et al. 2001) have been mapped to regions on the short arms of chromosome 6D and 6H in wheat and barley, respectively. These 6DS and 6HS regions, as well as the segment carrying Rfp1 on the long arm of rye chromosome 4R, are orthologous. The COS markers developed for Rfp1 in the present study may thus be useful for comparative mapping to investigate the orthology of the described restorer genes in the Triticeae tribe of the grasses.
Comparative mapping between rye and other grasses
The use of COS markers enabled us to assign the Rfp1 locus to a rye genomic segment orthologous to one of the major gene-rich regions, namely 6S1.0, in wheat (Weng and Lazar 2002; Erayman et al. 2004). The orthologous relationship between the distal segment of the long arm of rye chromosome 4R and the short arms of group 6 chromosomes of the Triticeae was identified by Devos et al. (1993) via comparative mapping with RFLP markers Xpsr167, Xpsr899, XNra1, and XCxp3. In the present study, these RFLP markers have been converted to PCR-based markers. The ~10-Mb sized region 6S1.0 carries 82 % of the genes located on the short arm of chromosome 6D and has the highest recombination rate per chromosome arm in wheat (Weng and Lazar 2002, Erayman et al. 2004). The estimates of the physical to genetic distances for the 6S1.0 bin range from 1.29 Mb/cM (Weng and Lazar 2002) to 164 kb/cM (Erayman et al. 2004) and are, thus, below the whole-genome average of 4.6 Mb/cM in wheat (Weng and Lazar 2002). These ratios found in wheat compare well to estimates reported for the orthologous region on the short arm of barley chromosome 6H (Künzel et al. 2000, Weng and Lazar 2002) and render map-based cloning in this sub-genomic region a feasible task. For the marker interval TC300731–TC256739 in rye, which is orthologous to the above-mentioned wheat and barley sub-genomic regions, the ratios remain to be determined in order to evaluate the prospect of success for a map-based strategy to isolate Rfp1.
The observed macro-collinearity of the distal segment of rye chromosome 4RL, the homoeologous group 6 chromosomes in barley and wheat (International Brachypodium Initiative 2010; Mayer et al. 2011), rice chromosome 2, Brachypodium chromosome 3, and Sorghum chromosome 4 indicates the orthologous relationships of these chromosomal regions. The positions of COS markers determined in the present study on rye chromosome 4RL reveal the same order of common genes as proposed by Mayer et al. (2011) for the “genome zipper” of the short arm of barley chromosome 6H.
The Rfp1-containing target interval of TC300731–TC256739 in rye is orthologous to sub-genomic regions on rice chromosomes 2 and 6. A series of quantitative trait loci (QTL) were reported by several research groups to reside in rice in the vicinity of these genomic regions. These QTL influence agronomically important traits including grain yield and yield components like seed number, spikelet number, panicle length, percent seed set, or 1,000-grain weight (Supplemental Table 3). Several of these QTL were mapped to a relatively small segment of 5 cM on the far-distal part of rice chromosome 2 (cf. Gramene QTL database, Youens-Clark et al. 2011). As shown in our study, this part of rice chromosome 2 and the Rfp1 region on rye chromosome 4RL are orthologous. Another rice QTL, gw6, for thousand-grain weight was mapped to a 6.3-cM interval delimited by the markers R1952 and C226 on rice chromosome 6 (cf. Gramene QTL database, Youens-Clark et al. 2011). The R1951–C226 interval includes a segment which is orthologous to parts of the long arm of rye chromosome 4R including Rfp1, as shown by the COS marker TC256739 in our study. According to rye breeders and consistent with these specific locations of some QTL related to grain yield in rice genomic regions orthologous to parts of 4RL, the Rfp1 gene in rye is associated with donor effects on grain yield after having been introduced into elite germplasm. These effects are negative and thus compromise the use of this restorer gene in hybrid breeding programmes. The unfavorable grain-yield effects can genetically be located somewhere within the STS marker interval of Xp15/55–Xscxx04 (Stracke et al. 2003) and could not to date be separated from Rfp1 via recombination. The present study reveals that five of the novel markers are located within this genomic segment, thereby considerably narrowing down the marker bracket around Rfp1. Hence, the novel markers provide the opportunity to select offspring potentially devoid of the undesirable linkage drag as a result of recombination within the Xp15/55–Xscxx04 segment.
The present study identified collinearity at the macro-level of a sub-genomic region containing Rfp1 on rye chromosome 4RL and a segment of Brachypodium chromosome 3. This allowed us to use the predicted gene sequence of Bradi3g00780 to develop an additional marker, TC256739, which proved to be closely linked to Rfp1. A DNA sequence similar to Bradi3g00780 is absent in the collinear region of rice chromosome 2; however, such a sequence is located on rice chromosome 6. This observation is consistent with the proposal by Bolot et al. (2009) that the orthologous chromosomes rice r2 and wheat w6 as well as rice r6 trace back to an ancestral chromosome A4, which has evolved through a series of duplications, breakages, and fusions.
In the present study (cf. Fig. 3), we have shown that in the genomes of rice and Brachypodium there are sub-genomic regions which are orthologous to the Rfp1-containing interval of TC300731–TC256739 in rye and which contain mTERF- and PPR-encoding genes. Proteins belonging to the mTERF gene family are localized in mitochondria and act as mitochondrial transcription termination factors in humans, sea urchin, and Drosophila (Roberti et al. 2009 for review). Five of eight plant restorer genes isolated so far belong to the family of PPR protein-encoding genes (Chase 2007; Rieseberg and Blackman 2010; Itebashi et al. 2010). The presence of multiple PPR protein-encoding genes linked to these restorer loci (Desloire et al. 2003; Komori et al. 2004) suggests that this class of restorer genes may have evolved through the duplication of PPR protein-encoding genes, followed by functional divergence of the duplicated genes such that the product of just one gene acts on transcripts of a CMS-determining locus (Chase 2007). Recently, phylogenetic analysis of 212 Rf-PPR-like sequences supported the hypothesis that all of these genes have a common evolutionary origin (Fujii et al. 2011). Further experiments are necessary to elucidate whether PPR gene copies are located between the marker loci TC300731 and TC256739 in rye which could serve as candidate genes for Rfp1. Additional markers linked to Rfp1 might be identified using the recently published 5 k SNP chip for rye (Haseneyer et al. 2011).
To summarize, we have identified sub-genomic regions in rice, Brachypodium, and Sorghum which are probably orthologous to a marker interval encompassing the Rfp1 gene in rye. The gene models of these grasses allowed us to establish COS markers for the Rfp1 locus in rye. These novel STS markers may be used for predicting Rfp1 restorer genotypes with a precision not feasible before. Furthermore, they may prove useful in pre-breeding efforts to clean Rfp1 of undesirable linkage drag which currently hampers the use of this valuable restorer gene in hybrid-breeding programmes.
References
Akhunov ED, Akhunova AR, Anderson OD, Anderson JA, Blake N, Clegg MT, Coleman-Derr D, Conley EJ, Crossman CC, Deal KR, Dubcovsky J, Gill BS, Gu YQ, Hadam J, Heo H, Huo N, Lazo GR, Luo MC, Ma YQ, Matthews DE, McGuire PE, Morrell PL, Qualset CO, Renfro J, Tabanao D, Talbert LE, Tian C, Toleno DM, Warburton ML, You FM, Zhang W, Dvorak J (2010) Nucleotide diversity maps reveal variation in diversity among wheat genomes and chromosomes. BMC Genomics 11:702–723
Bolot S, Abrouk M, Masood-Quraishi U, Stein N, Messing J, Feuillet C, Salse J (2009) The ‘inner circle’ of the cereal genomes. Curr Opin Plant Biol 12:119–125
Börner A, Korzun V, Polley A, Malyshev S, Melz G (1998) Genetics and molecular mapping of a male fertility restoration locus (Rfg1) in rye (Secale cereale L.). Theor Appl Genet 97:99–102
Bundessortenamt (2011) Database of descriptive variety lists. http://www.bundessortenamt.de
Chase CD (2007) Cytoplasmic male sterility: a window to the world of plant mitochondrial–nuclear interactions. Trends Genet 23:81–90
Collins NC, Shirley NJ, Saeed M, Pallotta M, Gustafson JP (2008) An ALMT1 gene cluster controlling aluminum tolerance at the Alt4 locus of rye (Secale cereale L.). Genetics 179:669–682
Curtis GA, Lukaszewski AJ (1993) Localization of genes in rye that restore male fertility to hexaploid wheat with timopheevi cytoplasm. Plant Breed 111:106–112
Desloire S, Gherbi H, Laloui W, Marhadour S, Clouet V, Cattolico L, Falentin C, Giancola S, Renard M, Budar F, Small I, Caboche M, Delourme R, Bendahmane A (2003) Identification of the fertility restoration locus, Rfo, in radish, as a member of the pentatricopeptide-repeat protein family. EMBO Rep 4:588–594
Devos KM, Atkinson MD, Chinoy CN, Francis HA, Harcourt RL, Koebner RMD, Liu CJ, Masojć P, Xie DX, Gale MD (1993) Chromosomal rearrangements in the rye genome relative to that of wheat. Theor Appl Genet 85:673–680
Endo TR, Gill BS (1996) The deletion stocks of common wheat. J Hered 87:295–307
Erayman M, Sandhu D, Sidhu D, Dilbirligi M, Baenziger PS, Gill KS (2004) Demarcating the gene-rich regions of the wheat genome. Nucleic Acids Res 32:3546–3565
Falke KC, Wilde P, Miedaner T (2009) Rye introgression lines as source of alleles for pollen-fertility restoration in Pampa CMS. Plant Breed 128:528–531
Fujii S, Bond CS, Small ID (2011) Selection patterns on restorer-like genes reveal a conflict between nuclear and mitochondrial genomes throughout angiosperm evolution. Proc Natl Acad Sci USA 108:1723–1728
Fulton T, van der Hoeven R, Eannetta N, Tanksley S (2002) Identification, analysis and utilization of a conserved ortholog set (COS) markers for comparative genomics in higher plants. Plant Cell 14:1457–1467
Geiger HH, Morgenstern KK (1975) Angewandt-genetische Studien zur cytoplasmatischen Pollensterilität bei Winterroggen. Theor Appl Genet 46:269–276
Geiger HH, Schnell FW (1970) Cytoplasmic male sterility in rye (Secale cereale L.). Crop Sci 10:590–593
Geiger HH, Yuan Y, Miedaner T, Wilde P (1995) Environmental sensitivity of cytoplasmic male sterility (CMS) in Secale cereale L. In: Kück U, Wricke G (eds) Genetic mechanisms for hybrid breeding. Adv Plant Breed 18:7–18, Blackwell Wissenschaftsverlag, Berlin
Hackauf B, Wehling P (2005) Approaching the self-incompatibility locus Z in rye (Secale cereale L.) via comparative genetics. Theor Appl Genet 110:832–845
Hackauf B, Rudd S, van der Voort JR, Miedaner T, Wehling P (2009) Comparative mapping of DNA sequences in rye (Secale cereale L.) in relation to the rice genome. Theor Appl Genet 118:371–384
Haseneyer G, Schmutzer T, Seidel M, Zhou R, Mascher M, Schön CC, Taudien S, Scholz U, Stein N, Mayer KFX, Bauer E (2011) From RNA-seq to large-scale genotyping—genomics resources for rye (Secale cereale L.). BMC Plant Biol 11:131
International Brachypodium Initiative (2010) Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463:763–768
International Rice Genome Sequencing Project (2005) The mapbased sequence of the rice genome. Nature 436:793–800
Itebashi E, Iwata N, Fujii S, Kazama T, Toriyama K (2010) The fertlity restorer gene, Rf2, for lead rice-type cytoplasmic male sterility of rice encodes a mitochondrial gylcine-rich protein. Plant J 65:359–367
Komori T, Ohta S, Murai N, Takakura Y, Kuraya Y, Suzuki S, Hiei Y, Imaseki H, Nitta N (2004) Map-based cloning of a fertility restorer gene, Rf-1, in rice (Oryza sativa L.). Plant J 37:315–325
Künzel G, Korzun L, Meister A (2000) Cytologically integrated physical restriction fragment length polymorphism maps for the barley genome based on translocation breakpoints. Genetics 154:397–412
Ma ZQ, Zhao YH, Sorrells ME (1995) Inheritance and chromosomal locations of male fertility restoring gene transferred from Aegilops umbellulata Zhuk. to Triticum aestivum L. Mol Gen Genet 247:351–357
Mago R, Miah H, Lawrence GJ, Wellings CR, Spielmeyer W, Bariana HS, McIntosh RA, Pryor AJ, Ellis JG (2005) High-resolution mapping and mutation analysis separate the rust resistance genes Sr31, Lr26 and Yr9 on the short arm of rye chromosome 1. Theor Appl Genet 112:41–50
Mason-Gamer RJ, Weil CF, Kellogg EA (1998) Granule-bound starch synthase: structure, function, and phylogenetic utility. Mol Biol Evol 15:1658–1673
Matsui K, Mano Y, Taketa S, Kawada N, Komatsuda T (2001) Molecular mapping of a fertility restoration locus (Rfm1) for cytoplasmic male sterility in barley (Hordeum vulgare L.). Theor Appl Genet 102:477–482
Mayer KF, Martis M, Hedley PE, Simková H, Liu H, Morris JA, Steuernagel B, Taudien S, Roessner S, Gundlach H, Kubaláková M, Suchánková P, Murat F, Felder M, Nussbaumer T, Graner A, Salse J, Endo T, Sakai H, Tanaka T, Itoh T, Sato K, Platzer M, Matsumoto T, Scholz U, Dolezel J, Waugh R, Stein N (2011) Unlocking the barley genome by chromosomal and comparative genomics. Plant Cell 23:1249–1263
Miedaner T, Glass C, Dreyer F, Wilde P, Wortmann H, Geiger HH (2000) Mapping of genes for male-fertility restoration in ‘Pampa’ CMS winter rye (Secale cereale L.). Theor Appl Genet 101:1226–1233
Miedaner T, Wilde P, Wortmann H (2005) Combining ability of non-adapted sources for male-fertility restoration in Pampa CMS of hybrid rye. Plant Breed 124:39–43
Miftahudin, Scoles GJ, Gustafson JP (2004) Development of PCR-based codominant markers flanking the Alt3 gene in rye. Genome 47:231–238
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, McCann MC, Ming R, Peterson DG, Ware D, Mehboob-ur-Rahman, Westhoff P, Mayer KF, Messing J, Rokhsar DS (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457:551–556
Proost S, Van Bel M, Sterck L, Billiau K, Van Parys T, Van de Peer Y, Vandepoele K (2009) PLAZA: a comparative genomics resource to study gene and genome evolution in plants. Plant Cell 21:3718–3731
Randhawa HS, Dilbirligi M, Sidhu D, Erayman M, Sandhu D, Bondareva S, Chao S, Lazo GR, Anderson OD, Miftahudin, Gustafson JP, Echalier B, Qi LL, Gill BS, Akhunov ED, Dvorák J, Linkiewicz AM, Ratnasiri A, Dubcovsky J, Bermudez-Kandianis CE, Greene RA, Sorrells ME, Conley EJ, Anderson JA, Peng JH, Lapitan NL, Hossain KG, Kalavacharla V, Kianian SF, Pathan MS, Nguyen HT, Endo TR, Close TJ, McGuire PE, Qualset CO, Gill KS (2004) Deletion mapping of homoeologous group 6-specific wheat expressed sequence tags. Genetics 168:677–686
Rieseberg LH, Blackman BK (2010) Speciation genes in plants. Ann Bot 106:439–455
Roberti M, Polosa PL, Bruni F, Manzari C, Deceglie S, Gadaleta MN, Cantatore P (2009) The MTERF family proteins: mitochondrial transcription regulators and beyond. Biochim Biophys Acta 1787:303–311
Scoles GJ, Evans LE (1979) The genetics of fertility restoration in cytoplasmic male-sterile rye. Can J Genet Cytol 21:417–422
Stojałowski S, Jaciubek M, Masojć P (2005) Rye SCAR markers for male fertility restoration in the P cytoplasm are also applicable to marker-assisted selection in the C cytoplasm. J Appl Genet 46:371–373
Stojałowski S, Łapinski M, Szklarczyk M (2006) Identification of sterility-inducing cytoplasms in rye using the plasmotype–genotype interaction test and newly developed SCAR markers. Theor Appl Genet 112:627–633
Stracke S, Schilling AG, Förster J, Weiss C, Glass C, Miedaner T, Geiger HH (2003) Development of PCR-based markers linked to dominant genes for male-fertility restoration in Pampa CMS of rye (Secale cereale L.). Theor Appl Genet 106:1184–1190
Van Ooijen JW (2006) JoinMap© 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma B.V, Wageningen
Weber WE, Wricke G (1994) Genetic markers in plant breeding. Adv. Plant Breeding 16, 105 p., Paul Parey, Berlin and Hamburg
Weng Y, Lazar MD (2002) Comparison of homoeologous group-6 short arm physical maps of wheat and barley reveals a similar distribution of recombinogenic and gene-rich regions. Theor Appl Genet 104:1078–1085
Wricke G, Wilde P, Wehling P, Gieselmann C (1993) An isozyme marker for pollen fertility restoration in the Pampa cms system of rye (Secale cereale L.). Plant Breed 111:290–294
Youens-Clark K, Buckler E, Casstevens T, Chen C, Declerck G, Derwent P, Dharmawardhana P, Jaiswal P, Kersey P, Karthikeyan AS, Lu J, McCouch SR, Ren L, Spooner W, Stein JC, Thomason J, Wei S, Ware D (2011) Gramene database in 2010: updates and extensions. Nucleic Acids Res 39(Database issue):D1085–D1094
Acknowledgments
We gratefully acknowledge excellent technical assistance by Marion Hos, Maria T. Goldfisch and Regina Voss.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hackauf, B., Korzun, V., Wortmann, H. et al. Development of conserved ortholog set markers linked to the restorer gene Rfp1 in rye. Mol Breeding 30, 1507–1518 (2012). https://doi.org/10.1007/s11032-012-9736-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-012-9736-5