
Abstract
Genetic diversity of elite breeding material can be increased by introgression of exotic germplasm to ensure long-term selection response. The objective of our study was to develop and characterize the first two rye introgression libraries generated by marker-assisted backcrossing and demonstrate their potential application for improving the baking quality of rye. Starting from a cross between inbred line L2053-N (recurrent parent) and a heterozygous Iranian primitive population Altevogt 14160 (donor) two backcross (BC) and three selfing generations were performed to establish introgression libraries A and B. Amplified fragment length polymorphisms (AFLP® markers) and simple sequences repeats (SSRs) were employed to select and characterize candidate introgression lines (pre-ILs) from BC1 to BC2S3. The two introgression libraries comprise each 40 BC2S3 pre-ILs. For analyzing the phenotypic effects of the exotic donor chromosome segment (DCS) we evaluated the per se performance for pentosan and starch content in replicated field trials at each of four locations in 2005 and 2006. Introgression library A and B cover 74 and 59% of the total donor genome, respectively. The pre-ILs contained mostly two to four homozygous DCS, with a mean length of 12.9 cM (A) and 10.0 cM (B). We detected eight (A) and nine (B) pre-ILs with a significant (P < 0.05) higher pentosan content and two pre-ILs (B) with a significant (P < 0.05) higher starch content than the elite recurrent parent. Thus, our results indicate that exotic genetic resources in rye carry favorable alleles for baking quality traits, which can be exploited for improving the elite breeding material by marker-assisted selection (MAS). These introgression libraries can substantially foster rye breeding programs and provide a promising opportunity to proceed towards functional genomics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Crop improvement depends highly upon finding and using genetic variation. Decades of cultivation and selection inevitably lead to a reduction in genetic variation in elite germplasm (Hawks 1977; Goodman 1997). To increase genetic diversity and, thus, ensure long-term selection response, the introgression of agriculturally non-adapted germplasm in elite breeding materials was suggested as a promising approach (Tanksley and Nelson 1996). Despite their agronomically inferior phenotypes, exotic germplasm is expected to contain genomic segments that can improve oligo- and polygenically inherited traits, even in highly selected breeding populations (Frey et al. 1981; de Vicente and Tanksley 1993).
Winter rye is an important crop in Germany and Eastern Europe and is grown on about 5.2 million hectares in Europe (http://faostat.fao.org/site/340/default.aspx). The implementation of hybrid breeding in rye (Geiger and Miedaner 1999) resulted in a considerable grain yield increase and, therefore, in Germany about three quarters of the total rye acreage is planted with hybrid varieties. The prerequisite for producing hybrids is self-fertility of the employed inbred lines. The self-fertile gene pools in rye are restricted to two source populations, Petkus and Carsten, thus, they represent only a small part of the worldwide available genetic diversity. To extend the genetic variation of Central European hybrid rye breeding material, Eastern European cultivars, landraces from Asia or South America and primitive populations from the Near East have been employed. These genetic resources are self-incompatible and heterozygous and have been used for extracting monogenically inherited traits such as self-fertility (Ossent 1938) or resistance traits (Rollwitz 1985). The hybridizing mechanism in rye was first enabled by using an Argentinean landrace as source for a cytoplasmic-genic male sterility system (Geiger and Schnell 1970). Moreover, the most effective restorer genes originated from Iranian and South American collections (Miedaner et al. 2000). However, the use of exotic germplasm for improving quantitative traits in the cross-pollinated rye is expensive, time-consuming and complicated due to its (1) low performance level, (2) high mutation load, and (3) unknown affiliation to established heterotic pools.
For broadening the genetic base of elite breeding materials with a minimum of negative side effects Eshed et al. (1992) suggested the use of introgression libraries as a powerful tool. These libraries ideally consist of near-isogenic lines (NILs) which (1) carry single marker-defined, short donor chromosome segments (DCS) introgressed from exotic sources into elite varieties (Zamir 2001) and (2) the introgressed DCS comprise the total donor genome. Thus, by restricting the introgression to defined DCS, the breeder can focus on positive gene effects without risking serious losses due to recombination. The elimination of genetic variation not associated with introgressed DCS is the major advantage of the NILs compared to other segregating populations (Eshed et al. 1996). The establishment of introgression libraries requires a great research effort (Zamir and Eshed 1998), nevertheless, plant breeders express increasing interest in this approach (Mank et al. 2003). So far, introgression libraries have been developed in tomato (Eshed et al. 1992; Eshed and Zamir 1994), rape seed (Howell et al. 1996), cabbage (Ramsay et al. 1996), rice (Lin et al. 1998), barley (Matus et al. 2003, von Korff et al. 2004), soybean (Concibido et al. 2003), lettuce (Jeuken and Lindhout 2004), melon (Eduardo et al. 2005), wheat (Liu et al. 2006), and maize (Ribaut and Ragot 2007; Szalma et al. 2007). However, no experimental results have been published for rye so far.
Introgression libraries provide a useful tool for genetic mapping, identification and localization of quantitative trait loci (QTL), and gene discovery (Zamir 2001; Kearsey 2002). Agriculturally valuable traits of exotic germplasm can be identified in genetic studies and subsequently transferred into commercial varieties by marker-assisted selection (MAS) programs. Moreover, a multitude of genotypes can easily be evaluated in large field experiments due to the homozygosity of ILs. This may enhance the accuracy of phenotyping without increasing the effort of genotyping (Jeuken et al. 2008). The beneficial effects of alleles from exotic germplasm has been illustrated for biotic stress (Von Korff et al. 2005; Finkers et al. 2007; Jeuken et al. 2008) and abiotic stress (Siangliw et al. 2007) as well as for quality traits, like malting, milling, or baking quality (Matus et al. 2003; Pillen et al. 2003; Kunert et al. 2007). Baking quality of rye is determined by a complex group of quantitative traits. Especially pentosan and starch content are two fundamental traits, whose quantity and composition highly affects the baking quality of rye. The potential of exotic germplasm as source for new QTL alleles with favorable effects on baking quality has not been investigated in rye yet.
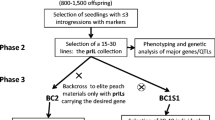
The objectives of our study were to (1) develop the first two rye introgression libraries by marker-assisted backcrossing (2) identify, localize, and characterize DCS of individual candidate introgression lines (pre-ILs), and (3) investigate the per se performance of pre-ILs for baking quality traits to draw conclusion on the potential utility of introgression libraries for improving elite germplasm.
Materials and methods
Plant materials
A homozygous rye inbred line L2053-N and a heterozygous Iranian primitive rye population Altevogt 14160 were crossed by hand emasculation in the greenhouse in 1999 to generate the F1 base population of the first two rye introgression libraries (A and B). As recurrent parent the high performance elite inbred line L2053-N was chosen, which is characterized by a high per se performance, lodging stability, and short plant height and is, therefore, used as parent in several registered hybrid rye varieties (H. Wortmann, personal communication). The donor population Altevogt 14160, provided by the Polish Botanical Garden at Warsaw, represents a primitive rye population found as weed in wheat and barley fields in the Near East. Altevogt 14160 is characterized by self-incompatibility, early heading, non-shattering ears, high susceptibility to lodging, low kernel weight and grain yield. In late 1999, random F1 plants were individually backcrossed to L2053-N in an off-season program. The poor performance of the exotic donor, however, resulted in shortness of seed supply of BC0 plants. For this reason, the progeny of one F1 plant was used to generate introgression library A and the progenies of two further F1 plants were combined to develop introgression library B. BC2 generations were generated in 2001, followed by three selfings to generate the BC2S3 generations.
Marker analyses and linkage map construction
Initially, genetic linkage maps of both introgression libraries were constructed only for amplified fragment length polymorphism (AFLP) markers due to the lack of sufficient simple sequence repeat (SSR) markers in rye at the beginning of this study. These maps were based on 87 randomly chosen BC1 individuals in population A, and 88 random individuals in population B. To identify AFLP primer combinations (PCs) being polymorphic in each population, 48 PCs were screened. Since the donor population was heterozygous, BC0 plant samples were used for the screening. Based on the number and scorability of DNA fragments as well as their distribution over the fingerprint, 13 and 10 PCs, were selected to construct the initial genetic linkage map of population A and B, respectively. The AFLP analyses were assayed according to Vos et al. (1995) and conducted by Keygene N.·V. (Wageningen, The Netherlands).
After the first comprehensive set of rye SSR markers became available (Hackauf and Wehling 2002, 2003), these were stepwise included into the genetic linkage maps. Moreover, SSRs from wheat and barley suitable for cross species amplification were used in both mapping populations. SSR assays were performed as described by Oetting et al. (1995) and assessed by the Federal Research Centre for Cultivated Plants (Julius Kuehn Institute, Quedlinburg, Germany), Lochow-Petkus GmbH (Einbeck, Germany) and Saaten-Union Resistenzlabor GmbH (Leopoldshöhe, Germany). In total, we employed 187 and 143 SSRs as well as 13 and 10 AFLP PCs to genotype 87 BC1 individuals of population A and 88 BC1 individuals of population B, respectively.
Observed genotype frequencies at each marker locus were checked for deviation from Mendelian segregation ratios (1:1) by χ2 tests. Genetic linkage maps were assembled by the software package JoinMap 3.0 (Van Ooijen and Voorrips 2001) using Kosambi's (1944) mapping function. An LOD threshold of 3.0 was employed in two-point analyses. Due to the employment of two F1 plants for the establishment of mapping population B, we first mapped only markers being heterozygous for both F1 gametes. Subsequently the “fixed order” command from JoinMap 3.0 was used for these markers and the remaining markers were added.
Marker-assisted selection
Preliminary genetic maps based on AFLP markers were used to identify and monitor DCS, and further to select the best-suited progenies in generation BC1 and BC2 (Sušić 2005). In generation BC2S1 and BC2S2, identification, monitoring and selection of progenies were conducted by using both SSR and AFLP marker, whereas in BC2S3 only SSRs were employed (Table 1). Monitoring of DCS was carried out by at least two flanking markers. For DCS longer than 10 cM, additional markers were used. During the selection process, the total number of pre-ILs analyzed with molecular markers was increased while the number of markers per pre-IL was reduced.
For selection of progenies as parents for the next generation the following criteria were used: pre-ILs were selected due to (1) the chromosomal localization of DCS to cover most of the donor genome in both introgression libraries, (2) the number of DCS per pre-IL, i.e., a pre-ILs should not carry more than five DCS in BC2 and not more than four in BC2S1, and (3) the proportion of recurrent parent genome (RPG) per pre-IL, i.e., among pre-ILs carrying identical target DCS those with higher proportion of RPG were preferred.
In the final generation BC2S3, the selected 40 progenies per introgression library were analyzed with additional 114 and 77 SSRs to verify DCS and to check the genetic background for further DCS being not discovered in earlier generations. Graphical genotypes of pre-ILs in BC2S3 (Figs. 1, 2) were constructed with software Plabsoft (Maurer et al. 2008). Individual DCS lengths were calculated as the sum of distances between its known markers plus half the distance to the next marker outside the DCS. For determining the number of DCS per pre-IL as well as the characterization of each introgression library (Table 2), we considered only DCS which were separated by >5 cM as two DCS. Moreover, missing marker data were estimated from the flanking markers in the case of tightly linked loci (<5 cM). If the alleles at two adjacent marker loci originated from the same parent missing marker were assumed to have the same genotype as the two flanking markers. If the two flanking markers showed different genotypes, no corrections were made. Heterozygous alleles at AFLP loci in BC2S2 have been assumed as missing data in BC2S3 because no AFLP analyses have been conducted in this population.

Graphical genotypes representing the coverage of the donor genome in the marker-assisted selected set of 40 BC2S3 pre-IL of introgression library A. The respective chromosome and the marker position (vertical bars) are presented above the figure; red coloring indicates homozygous state of the recurrent parent, blue coloring homozygous state of the donor, green coloring heterozygous state, and gray coloring missing data

Graphical genotypes representing the donor genome coverage in the marker-assisted selected set of 40 BC2S3 pre-IL of introgression library B. The respective chromosome and the marker position (vertical bars) are presented above the figure; red coloring indicates homozygous state of the recurrent parent, blue coloring homozygous state of the donor, green coloring heterozygous state, and gray coloring missing data
Agronomic trials
The experimental design at each location was a 10 × 9 α-design (Patterson and Williams 1976) with three replications. Each genotype was grown in plots of 0.83 m width and 1.2 m length, representing about 1 m2. All experiments were machine planted and harvested with a combine. From the harvest a representative sample (500 g) was taken for quality analyses. Agronomic treatments were done as usual, including application of 70–80 kg N ha−1, two sprayings of plant growth regulators (1.5 l ha−1 CCC and 1.5 l ha−1 Terpal C) and one fungicide spraying (1.5 l ha−1 Opus Top). The recurrent parent (L2053-N) was included tenfold to improve accuracy, the donor (Altevogt 14160) as triple entries in each experiment. Because of its high lodging capability, the donor was tied up after flowering by hand to get representative quality data. Data were recorded for pentosan and starch content in grain (%) estimated by near-infrared reflectance spectroscopy. The field trials were conducted in 2005 and 2006 at two sites in the south (Hohenheim, Eckartsweier) and at two in the north (Wulfsode, Bergen) of Germany. For pre-IL 2127 and 2129 in introgression library A as well as 2158, 2159, and 2160 in introgression library B no data could be collected.
Statistical analyses
Ordinary lattice analyses of variance were performed for each experiment and location using software PLABSTAT (Utz 2001). Adjusted entry means were used to compute combined analyses of variance across locations (Cochran and Cox 1957). Variance components were estimated based on adjusted entry means and effective error mean squares from the individual lattice analyses by restricted maximum likelihood (REML), using PROC MIXED of SAS (SAS Institute 2004). To take into account the variation in accuracy of the individual lattices, least square means were weighted with the reciprocal error variance (Piepho 1999). Variance components were estimated combined for both libraries because Akaikes information criterion (AIC) indicated that the increase in estimated parameters, which would result from estimation of separate variance components for the two libraries, would not result in a considerable increase in the fit of the model. The heritability \( \bar H^2 \) was computed with an ad hoc measure for unbalanced test designs (Holland et al. 2003; Piepho and Möhring 2007). A both-sided Dunnett test (Dunnett 1955) for multiple comparisons of least square means was used to determine significant differences for per se performance between the 38 (A) and 37 (B) pre-ILs and the recurrent parent L2053-N as respective control, using a significance level of α = 0.05. The linear model was
where G r (r = 1, …, 75) are the genotypes, L s (s = 1, …, 4) the locations, and J t (t = 1, 2) the years. G r , was considered as fixed factor and the remaining factors were considered as random. The Dunnett test was computed with PROC MIXED of software SAS (SAS Institute 2004).
Results
Significant deviations (P < 0.001) from the expected single-locus genotype frequencies were observed in four cases in the mapping population of introgression library A and in zero cases in the mapping population of introgression library B. The final genetic linkage maps were covered by 131 AFLP and 137 SSR loci in introgression library A and by 182 AFLP and 118 SSR loci in introgression library B. The total map distances spanned 738 cM (A) and 636 cM (B), with an average interval length of 2.8 and 2.2 cM, respectively.
During the marker-assisted introgression process we observed in both introgression libraries an apparent decrease in the (1) number of DCS per pre-IL, (2) length of the individual DCS, and (3) proportion of donor genome per pre-IL in the selected sets of individuals (Table 2). With each backcross and selfing generation the average proportion of the RPG increased and the donor alleles were progressively eliminated.
In introgression library A, the set of nine BC1 plants selected as parents for generation BC2 provided at least a double coverage of the donor genome for most of the chromosome regions. In generation BC2, about 90% of the total donor genome was covered (Table 2), but the coverage declined to 74% in the final BC2S3 generation. Larger gaps occurred on chromosome 1R and in particular on chromosome 2R (Fig. 1). Almost all pre-ILs carried only homozygous DCS. Exceptions were few pre-ILs containing short heterozygous DCS on chromosomes 2R, 3R, 5R, and 6R, which were detected mostly by single markers. Further DCS were discovered only in BC2S3 in particular on chromosomes 5R, 6R, and 7R. Five generations of MAS resulted in 40 BC2S3 pre-ILs carrying on average 4.7 DCS, with a mean length of 12.9 cM. The proportion of RPG ranged from 83.5 to 98.1%, with a mean of 91.8% in generation BC2S3 (Table 2).
In introgression library B, the selected set of nine BC1 plants provided at least a double coverage of the donor genome for most of the chromosome regions. In generation BC2, about 70% of the total donor genome was covered (Table 2). The final set of 40 BC2S3 pre-ILs represented 59% of the donor genome (Table 2). The largest gaps occurred on chromosomes 2R and 7R. The majority of the introgressed DCS was already fixed in homozygous state. Exceptions were some short heterozygous DCS on chromosomes 6R and 7R (Fig. 2). Small new DCS were discovered on chromosomes 1R, 5R, and 6R in BC2S3. In introgression library B, five generations of MAS yielded in 40 BC2S3 pre-ILs containing on average 3.2 DCS with a mean length of 10.0 cM. The finally selected set of BC2S3 pre-ILs contained 88.0–99.9% of the RPG with a mean of 94.9%.
For pentosan content, means of pre-IL progenies ranged from 11.9 to 15.1% and from 12.2 to 15.2% with a mean of 14.0 and 14.1% in introgression libraries A and B, respectively. For starch content, we determined ranges for the means of pre-IL progenies from 50.7 to 55.9% in introgression library A and from 51.2 to 56.8% in introgression library B with a mean of 53.5% (A) and 53.4% (B). REML estimates of the genotypic variance \( \sigma _G^2 \) were significant (P < 0.01) for both traits. Estimates of genotype × year variance \( \sigma _{gj}^2 \) were also significant (P < 0.05) for both traits, while estimates of genotype × location variance \( \sigma _{gl}^2 \) were not significant. Estimates of genotype × year × location variance \( \sigma _{gjl}^2 \) were significant (P < 0.01) for both traits. Heritability \( \bar H^2 \) was relatively high for pentosan (0.78) and starch content (0.87). The Dunnett test was significant (P < 0.05) for 30% out of 76 comparisons between pre-ILs and the recurrent parent in introgression library A (Table 3) and for 31% out of 74 comparisons in introgression library B (Table 4). Out of these, 35% (A) and 48% (B) of the pre-ILs were significantly better than the recurrent parent. We detected eight pre-ILs in introgression library A and nine pre-ILs in introgression library B with significant higher pentosan content than the elite recurrent parent L2053-N. These pre-ILs contained DCS on all chromosomes except for chromosomes 2R in introgression library A and on chromosomes 3R, 4R, 6R and 7R in introgression library B. The Dunnett test was positive significant (P < 0.05) for starch content for two pre-ILs in introgression library B which carried DCS on chromosomes 4R, 5R, and 7R.
Discussion
Breeding plan for establishing introgression libraries
The goal of our study was to develop the first introgression library in rye from an primitive population to illustrate that genetic resources, despite of their poor performance, provide beneficial alleles, which can be used for improving elite breeding material. For the development of our introgression libraries we employed two backcross generations followed by three selfing generations, in all of which only MAS was carried out. This breeding procedure differed from previously published breeding plans, employed in studies to establish introgression libraries for broadening the genetic base of elite germplasm, with respect to (1) the number of backcross and selfing generations and (2) the generations in which selection was carried out. For example, Jeuken and Lindhout (2004) developed an introgression library from a BC5S1 population in lettuce and Chetelat and Meglic (2000) employed a BC1F6 population in tomato. MAS was conducted in the majority of all studies during all backcrossing and selfing generations (Eshed and Zamir 1994; Chetelat and Meglic 2000; Mank et al. 2003; Ribaut and Ragot 2007; Szalma et al. 2007). However, other authors employed random backcrossing followed by marker selection of ILs in the last generations (Matus et al. 2003; Jeuken and Lindhout 2004; von Korff et al. 2004; Eduardo et al. 2005; Liu et al. 2006). First results on the relative advantage of these different breeding plans are available. Jeuken and Lindhout (2004) reported that it is more efficient to apply more backcross and less selfing generations. This result is supported by a simulation study in rye conducted by Sušić (2005), who suggested that BC3S1 introgression libraries are advantageous over BC2S2 libraries, because fewer marker data points were required to establish an introgression library. However, because the employed breeding plan is the major factor determining the costs of an introgression library, a thorough comparison of the alternative breeding plans with computer simulations is a promising topic for further research.
AFLP versus SSR markers
In the BC1 and BC2 generations of the establishment of our introgression libraries, selection of pre-ILs as parents for the next generation was based solely on AFLPs. The reason was the lack of sufficient SSR markers in rye at the beginning of this study in 1999. However, since the number of SSRs was increased during the study (Hackauf and Wehling 2002, 2003; Khlestkina et al. 2004), we were able to employ additional SSRs in the selfing generations such that in the final BC2S3 generation 114 (A) and 77 (B) SSRs were employed in addition to the AFLPs. In general, SSRs are regarded as the superior marker system because they provide a codominant mode of inheritance, are inexpensive and fast (Hackauf and Wehling 2002, 2003), and their application across species is partly possible (Röder et al. 1998; cf. Lübberstedt et al. 1998). We found that in the BC1 and BC2 generation, when a relatively large proportion of the genome was still segregating and only one or a few crossover events have occurred per chromosome, a few AFLPs were sufficient to quickly and efficiently locate DCS across the whole genome. Nevertheless, employing AFLPs with a self-incompatible, heterozygous donor resulted in difficulties to identify whether an AFLP band originated from the donor or the recurrent parent. Therefore, the presence and exact length of some DCS could not be determined unambiguously. This inaccurate monitoring of a few DCS may have additionally reduced the donor genome coverage of our introgression libraries. With the SSR markers in the selfing generations, we were able to verify the DCS and to check the genetic background to find further DCS not being discovered in earlier generations (Table 2). Therefore, we conclude that for the last generations of the establishment of an introgression library SSRs are the marker type of choice to track the DCS.
Assessment of the RPG content and marker map
The average RPG proportions in our introgression libraries A and B increased from 80 to 82% in the selected set of BC1 plants to 92–95% in the final BC2S3 pre-ILs (Table 2). Similar results were observed by Jeuken and Lindhout (2004) in their BC5S1 introgression library in lettuce. Our pre-ILs contained on average three to five DCS (Table 2; Figs. 1, 2). Similar numbers were observed in the introgression libraries of von Korff et al. (2004) in barley and Liu et al. (2006) in wheat. However, there are also reports of introgression libraries where the majority of ILs (Monforte and Tanksley 2000; Jeuken and Lindhout 2004) or even the complete set of ILs (Eshed and Zamir 1994; Eduardo et al. 2005) carried only a single DCS. The number of DCS per pre-IL in our introgression libraries was relatively large partly due to very short donor genome stretches detected by only one single marker. Erroneous marker scorings were at best a marginal factor for these short introgressions, because they were observed in three subsequent selfing generations. However, the probability of frequent double crossovers in very small chromosome regions is extremely low. We therefore assume that incorrect locus orders, caused by the small population sizes (<90) of our mapping populations, resulted in incorrect map positions of the markers for these segments. In the final BC2S3 generation, new DCS were detected when we scanned the whole rye genome with the additional SSRs. This is a strong hint that during the introgression process the limited number of available markers was a constraint. We conclude that a sufficiently saturated map with exactly estimated marker positions is of utmost importance for a assessing the RPG content in establishing intogression libraries.
The mean DCS length in the finally selected set of 40 BC2S3 pre-ILs was 13 cM (introgression library A) and 10 cM (introgression library B, cf. Table 2). Similar results were observed in the introgression library of Liu et al. (2006) in wheat. In contrast, the DCS lengths reported by other authors were considerably longer (25 cM Chetelat and Meglic 2000 in tomato, 47 cM Eshed et al. 1992 in tomato, 33 cM Jeuken and Lindhout 2004 in lettuce, 41 cM Eduardo et al. 2005 in melon, 39 cM Matus et al. 2003 in barley, 35 cM von Korff et al. 2004 in barley). Due to the relatively small fractions of the exotic donor genome in our pre-ILs, the risk of negative effects of this linkage drag on the phenotype is expected to be low. We therefore conclude, that the pre-ILs of our introgression libraries, which have a higher performance than the elite recurrent parent can immediately be used in hybrid rye breeding by crossing with elite breeding material.
Donor genome coverage of the introgression library
In comparison with the introgression libraries of Eshed and Zamir (1994) in tomato, Canady et al. (2005) in tomato, Jeuken and Lindhout (2004) in lettuce, Eduardo et al. (2005) in melon, von Korff et al. (2004) in barley, and Szalma et al. (2007) in maize, which covered almost the complete donor genome (donor genome coverage >89%), our two rye introgression libraries display a lower donor genome coverage (Table 2). Minor gaps appeared across all chromosomes in both rye introgression libraries, with large ones occurring on chromosome 2R in both introgression libraries and on chromosome 4R in introgression library A. A test in generation BC1 detected no distorted segregation ratio favoring the recurrent parent allele. The number and map position of markers could also be excluded as reason for the gaps, because of (1) the additionally employed 114 (A) and 77 (B) SSRs in the final population and (2) a dense and uniform marker coverage over the rye genome with average interval lengths of 2.8 (A) and 2.2 cM (B) in the mapping populations.
The limited recombination due to small progeny sizes per pre-IL during the introgression process is presumably one main reason for the appearance of the gaps in donor genome coverage. Small progeny sizes especially occurred in generation BC2 because of problems in the off-season greenhouse program. Thus, in introgression library B the selection resulted in only three pre-ILs carrying DCS on chromosome 2R (Fig. 2). The second main reason was presumably lethal or sublethal recessive alleles of the donor, because some BC families had to be discarded in the selfing generations due to yellowing and inferior vitality.
Effects of the exotic donor genome on baking quality traits
The estimates of the genotypic variance \( \sigma _G^2 \) in our introgression libraries were significant (P < 0.01) for pentosan and starch content. This demonstrates that new genetic variation for these important baking quality traits was generated from the DCS of the exotic donor Altevogt 14160.
To quantify the effects of the marker-defined DCS on the baking quality traits the evaluation of pre-ILs in large-scale field experiments is required. Therefore, we assessed the traits in eight environments. The phenotypes of most pre-ILs showed the tendency to be more similar to the elite recurrent parent for pentosan content. These results were expected due to the higher pentosan content of the recurrent parent than the donor (Tables 3, 4). However, the Dunnett detected also pre-ILs showing superiority for pentosan content over the elite recurrent parent, indicating their potential for improving baking quality of rye. In contrast, only two pre-ILs in introgression library B could be determined having a significant (P < 0.05) higher starch content than L2053-N, despite of the better performance of Altevogt 14160. We assume that the low association between exotic alleles and starch content can be attributed to (1) the high proportion of the RPG (>92%) and (2) and a complex inheritance of starch content.
The phenotypic superiority of pre-ILs over the elite recurrent parent (Tables 3, 4) can be associated to QTL regions located on single or few DCS (Figs. 1, 2). Consequently, our study revealed that Altevogt 14160 contributed favorable alleles, despite of its general poor per se performance. These favorable alleles for baking quality traits could be identified by using the introgression libraries. We therefore conclude, that exotic germplasm provides a valuable resource for improving quality traits of elite breeding material.
Application of rye ILs in breeding programs
As rye pre-ILs carried only a small fraction of the exotic donor genome, the number of unfavorable or even deleterious genes affecting vigor and fertility is greatly reduced and yield-associated traits harbored in introgressed DCS can be measured with higher accuracy. The pre-ILs performing better than the elite recurrent parent can immediately be used in hybrid rye breeding by recombining elite materials with superior introgression lines (Eshed and Zamir 1995). By crossing ILs to cytoplasmic-male sterile (CMS) testers an “F1 introgression library” can be constructed to map loci contributing to heterosis (Zamir 2001; Peleman and Rouppe van der Voort 2003). A detailed molecular characterization of favorable DCS could pave the way for mapping and verifying candidate genes. A task still to be done is a further backcrossing and marker selection to reduce the number of DCS per pre-IL. The rye introgression libraries can be used as a tool for improvement of other cereals, especially triticale and wheat. Indeed, wheat ILs carrying translocations on the short arm of chromosome 1R possess improved agronomic performance (Moonen and Zeven 1984; Lukaszewski 1990; Carver and Rayburn 1994), particularly when the source of the rye donor chromatin was selected carefully (Kim et al. 2004).
We expect that our rye introgression libraries are a useful tool to make rapidly available a wide array of previously unexplored genetic variation to plant breeders and geneticists. In this respect, our introgression libraries represent a dynamic new resource that could substantially foster future rye breeding programs and provide in addition valuable information for breeding programs in other Triticeae.
References
Canady MA, Meglic V, Chetelat RT (2005) A library of Solanum lycopersicoides introgression lines in cultivated tomato. Genome 48:685–697
Carver BF, Rayburn AL (1994) Comparison of related wheat stocks possessing 1B or 1RS.1BL chromosomes: agronomic performance. Crop Sci 34:1505–1510
Chetelat RT, Meglic V (2000) Molecular mapping of chromosome segments introgressed from Solanum lycopersicoides into cultivated tomato (Lycopersicon esculentum). Theor Appl Genet 100:232–241
Cochran W, Cox GM (1957) Experimental designs, 2nd edn. Wiley, New York
Concibido VC, La Vallee B, Mclaird P, Pineda N, Meyer J, Hummel L, Yang J, Wu K, Delannay X (2003) Introgression of a quantitative trait locus for yield from Glycine soja into commercial soybean cultivars. Theor Appl Genet 106:575–582
De Vicente MC, Tanksley SD (1993) QTL analysis of transgressive segregation in an interspecific tomato cross. Genetics 134:585–596
Dunnett C (1955) A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc 50:1096–1121
Eduardo I, Arus P, Monforte AJ (2005) Development of a genomic library of near isogenic lines (NILs) in melon (Cucumis melo L.) from the exotic accession PI161375. Theor Appl Genet 112:139–148
Eshed Y, Zamir D (1994) A genomic library of Lycopersicon pennellii in L. esculentum: a tool for fine mapping of genes. Euphytica 79:175–179
Eshed Y, Zamir D (1995) An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTL. Genetics 141:1147–1162
Eshed Y, Abu-Abied M, Saranga Y, Zamir D (1992) A genome-wide search for wild-species alleles that increase horticultural yield of processing tomatoes. Theor Appl Genet 93:877–886
Eshed Y, Gera G, Zamir D (1996) Lycopersicon esculentum lines containing small overlapping introgressions from L. pennellii. Theor Appl Genet 83:1027–1034
Finkers R, Van Heusden AW, Meijer-Dekens F, Van Kan JAL, Maris P, Lindhout P (2007) The construction of a Solanum habrochaites LYC4 introgression line population and the identification of QTLs for resistance to Botrytis cinerea. Theor Appl Genet 114:1071–1080
Frey KJ,Cox TS, Rodgers DM, and Bramel-Cox P (1981) Increasing cereal yields with genes from wild and weedy species, pp 51–68. Journal paper No. J-11254 of the Iowa Agric. and Home Econ. Exp. Stn., Ames, Iowa 50011, USA
Geiger HH, Miedaner T (1999) Hybrid rye and heterosis. In: Coors JG, Pandey S (eds) Genetics and exploitation of heterosis in crops. Crop Sci. Soc. America, Madison, pp 439–450
Geiger HH, Schnell FW (1970) Cytoplasmatic male sterility in rye (Secale cereale L.). Crop Sci 35:27–38
Goodman MM (1997) Broadening the genetic diversity in breeding by use of exotic germplasm. In: CIMMYT. Book of abstracts. The genetics and exploitation of heterosis in crops. An intern. symp., pp 58–59, Mexico
Hackauf B, Wehling P (2002) Identification of microsatellite polymorphisms in an expressed portion of the rye genome. Plant Breed 121:17–25
Hackauf B, Wehling P (2003) Development of microsatellite markers in rye: map construction. Plant Breed Seed Sci 48:143–151
Hawks JG (1977) The importance of wild germplasm in plant breeding. Euphytica 26:615–621
Holland J, Nyquist W, Cervantes-Martinez C (2003) Estimating and interpreting heritability for plant breeding: an update. Plant Breed Rev 2:9–112
Howell PM, Marshall DF, Lydiate DJ (1996) Towards developing intervarietal substitution lines in Brassica napus using marker-assisted selection. Genome 39:348–358
SAS Institute (2004) Version 8.2. SAS Institute, Cary
Jeuken MJW, Lindhout P (2004) The development of lettuce backcross inbred lines (BILs) for exploitation of the Lactuca saligna (wild lettuce) germplasm. Theor Appl Genet 109:394–401
Jeuken M, Pelgrom K, Stam P, Lindhout P (2008) Efficient QTL detection for nonhost resistance in wild lettuce: backcross inbred lines versus F2 population. Theor Appl Genet. doi:10.1007/s00122-008-0718-2
Kearsey MJ (2002) QTL analysis: problems and (possible) solutions. In: Kang MS (ed) Quantitative genetics, genomics and plant breeding. CAB International, New York, pp 45–58
Khlestkina EK, Than MHM, Pestsova EG, Röder MS, Malyshev SV, Korzun V, Börner A (2004) Mapping of 99 new microsatellite-derived loci in rye (Secale cereale L.) including 39 expressed sequence tags. Theor Appl Genet 109:725–732
Kim W, Johnson JW, Graybosch RA, Gaines CS (2004) The effect of T1DL.1RS wheat-rye chromosomal translocation on agronomic performance and end-use quality of soft wheat. Cereal Res Comm 31:301–308
Kosambi DD (1944) The estimation of map distances from recombination values. Ann Eugen 12:172–175
Kunert A, Naz AA, Dedeck O, Pillen K, Leon J (2007) AB-QTL analysis in winter wheat: I. Synthetic hexaploid wheat (T. turgidum ssp. dicoccoides + T. tauschii) as a source of favourable alleles for milling and baking quality traits. Theor Appl Genet 115:683–695
Lin SY, Sasaki T, Yano M (1998) Mapping quantitative trait loci controlling seed dormancy and heading date in rice, Oryza sativa L., using backcross inbred lines. Theor Appl Genet 96:997–1003
Liu S, Zhou R, Dong Y, Li P, Jia J (2006) Development, utilization of introgression lines using a synthetic wheat as donor. Theor Appl Genet 112:1360–1373
Lübberstedt T, Dussle C, Melchinger AE (1998) Application of microsatellites from maize to teosinte and other relatives of maize. Plant Breed 117:447–450
Lukaszewski AJ (1990) Frequency of 1RS-1AL and 1RS-1BL translocations in United States wheats. Crop Sci 30:1151–1153
Mank R, Verbakel H, Witsenboer H, and Peleman J (2003) Marker assisted construction of a high resolution introgression line library in tomato using Lycopersicon hirsutum, p 508. In: Plant and animal genomes XI conference, San Diego
Matus I, Corey A, Filichkin T, Hayes PM, Vales MI, Kling J, Riera-Lizarazu O, Sato K, Powell W, Waugh R (2003) Development and characterization of recombinant chromosome substitution lines (RCSLs) using Hordeum vulgare subsp spontaneum as a source of donor alleles in a Hordeum vulgare subsp. vulgare background. Genome 46:1010–1023
Maurer HP, Melchinger AE, Frisch M (2008) Population genetic simulation and data analysis with Plabsoft. Euphytica 161:133–139
Miedaner T, Glass C, Dreyer F, Wilde P, Wortmann H, Geiger HH (2000) Mapping of genes for male-fertility restoration in ‘Pampa’ CMS winter rye (Secale cereale L.). Theor Appl Genet 101:1226–1233
Monforte AJ, Tanksley SD (2000) Development of a set of near isogenic and backcross recombinant inbred lines containing most of the Lycopersicon hirsutum genome in a L. esculentum genetic background: a tool for gene mapping and gene discovery. Genome 43:803–813
Moonen JHE, Zeven AC (1984) SDS-PAGE of the high-molecular-weight subunits of wheat glutenin and characterization of 1R (1B) substitution and 1BL/1RS translocation lines. Euphytica 33:3–8
Oetting WS, Lee HK, Flanders DJ, Wiesner GL, Sellers TA, King RA (1995) Linkage analysis with multiplexed short tandem repeat polymorphisms using infrared fluorescence and M13 tailed primers. Genomics 30:450–458
Ossent HP (1938) Zehn Jahre Roggenzüchtung in Müncheberg. Züchter 10:255–261
Patterson HD, Williams ER (1976) A new class of resolvable incomplete block designs. Biometrica 63:83–92
Peleman JD, Rouppe van der Voort J (2003) Breeding by design. Trends Plant Sci 8:330–334
Piepho H-P (1999) Stability analysis using the SAS system. Agron J 91:154–160
Piepho H-P, Möhring J (2007) Computing heritability and selection response from unbalanced plant breeding trials. Genetics 177:1881–1888
Pillen K, Zacharias A, Léon J (2003) Advanced backcross QTL analysis in barley (Hordeum vulgare L.). Theor Appl Genet 107:340–352
Ramsay LD, Jennings DE, Bohuon EJR, Arthur AE, Lydiate DJ, Kearsey MJ, Marshall DF (1996) The construction of a substitution library of recombinant backcross lines in Brassica oleracea for the precision mapping of quantitative trait loci. Genome 39:558–567
Ribaut J-M, Ragot M (2007) Marker-assisted selection to improve drought adaptation in maize: the backcross approach, perspectives, limitations, and alternatives. J Exp Bot 58:351–360
Röder MS, Korzun V, Wendehake K, Plaschke J, Tixier M-H, Leroy P, Ganal MW (1998) A microsatellite map of wheat. Genetics 149:2007–2023
Rollwitz W (1985) Untersuchungen zur Bewertung von Roggenzuchtmaterial bezüglich Braunrostresistenz und Schaffung von Ausgangsmaterial für die Züchtung. Diss. Rostock, Germany
Siangliw J, Jongdee B, Pantuwan G, Toojinda T (2007) Developing KDML105 backcross introgression lines using marker-assisted selection for QTLs associated with drought tolerance in rice. Sci Asia 33:207–214
Sušić Z (2005) Experimental and simulation studies on introgressing genomic segments from exotic into elite germplasm of rye (Secale cereale L.) by marker-assisted backcrossing. PhD thesis, University of Hohenheim, Stuttgart, Germany
Szalma SJ, Hostert BM, LeDeaux JR, Stuber CW, Holland JB (2007) QTL mapping with near-isogenic lines in maize. Theor Appl Genet 114:1211–1228
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTL from unadapted germplasm into elite breeding lines. Theor Appl Genet 92:191–203
Utz HF (2001) PLABSTAT: a computer program for the statistical analysis of plant breeding experiments. Institute for plant breeding, Seed Science and Population Genetics, University of Hohenheim, Stuttgart
Van Ooijen JW, Voorrips RE (2001) JoinMap version 3.0: software for the calculation of genetic linkage maps. Plant Research International, Wageningen
Von Korff M, Wang H, Léon J, Pillen K (2004) Development of candidate introgression lines using an exotic barley accession (Hordeum vulgare ssp. spontaneum) as donor. Theor Appl Genet 109:1736–1745
Von Korff M, Wang H, Leon J, Pillen K (2005) AB-QTL analysis in spring barley. I. Detection of resistance genes against powdery mildew, leaf rust and scald introgressed from wild barley. Theor Appl Genet 111:583–590
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Zamir D (2001) Improving plant breeding with exotic genetic libraries. Nature Rev Genet 2:983–989
Zamir D, Eshed Y (1998) Case history in germplasm introgression: Tomato genetics and breeding using nearly isogenic introgression lines derived from wild species. In: Paterson AH (ed) Molecular dissection of complex traits. CRC Press, New York, pp 207–217
Acknowledgments
The present study was financially supported by by the German Federal Ministry of Education and Research, Bonn, the private plant breeding companies Hybro GmbH & Co. KG, Schenkendorf, and KWS LOCHOW GmbH, Bergen, in conjunction with the GABI program “Rye Resources” (BMBF Grant #0312289B) and the German Federal Ministry of Economics (BMWi) in the PRO INNO frame work (Aif Grant #KF0141101MD5). The excellent technical work of Mrs. B. Lieberherr, Universitaet Hohenheim, in constructing the two introgression libraries over the years is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by F. Ordon.
K. C. Falke, and Z. Sušić are contributed equally to the manuscript.
Rights and permissions
About this article
Cite this article
Falke, K.C., Sušić, Z., Hackauf, B. et al. Establishment of introgression libraries in hybrid rye (Secale cereale L.) from an Iranian primitive accession as a new tool for rye breeding and genomics. Theor Appl Genet 117, 641–652 (2008). https://doi.org/10.1007/s00122-008-0808-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-008-0808-1