
Abstract
Replacement of red hematopoietic bone marrow with yellow adipocyte-rich marrow is a conserved physiological process among mammals. The extent of this conversion is influenced by a wide array of pathological and non-pathological conditions. Of particular interest is the observation that some marrow adipocyte-inducing factors seem to oppose each other, for instance obesity and caloric restriction. Intriguingly, several important molecular characteristics of bone marrow adipose tissue (BMAT) are distinct from the classical depots of white and brown fat tissue. This depot of fat has recently emerged as an active part of the bone marrow niche that exerts paracrine and endocrine functions thereby controlling osteogenesis and hematopoiesis. While some functions of BMAT may be beneficial for metabolic adaptation and bone homeostasis, respectively, most findings assign bone fat a detrimental role during regenerative processes, such as hematopoiesis and osteogenesis. Thus, an improved understanding of the biological mechanisms leading to formation of BMAT, its molecular characteristics, and its physiological role in the bone marrow niche is warranted. Here we review the current understanding of BMAT biology and its potential implications for health and the development of pathological conditions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
At birth, long bone cavities are filled with active hematopoietic, red marrow which is for the most part composed of immune cells at different maturation stages. By the time healthy humans reach peak bone mass around the age of 25, bone marrow adipose tissue (BMAT) can, depending on the bone compartment, occupy up to 70% of marrow space, generally suggesting a rather non-pathological role for this type of fat [1]. As a rule, bones of the peripheral skeleton accumulate more adipocytes than the axial skeleton, and the distal ends of long bones are infiltrated first [2]. Aging further promotes the increase of marrow fat in the marrow cavities [3]. For instance, vertebral marrow fat of men progressively accumulates with age, whereas a sharp increase between 55 and 65 years of age is observed in women, coinciding with the onset of menopause and leaving them with an approximately 10% higher adipocyte content compared to men [4]. Marrow adipogenesis in rodents follows a comparable, if somewhat delayed, developmental pattern, making them a suitable model for research on BMAT. It should be noted that mice additionally show strong strain-specific variations in marrow fat levels and there exists a loose positive correlation of elevated BMAT presence and increasing mammal size [3].
BMAT localizes to sites of active bone formation and hematopoiesis, which suggests an involvement in skeletal remodeling and blood/immune cell production. Previously, it was suggested that marrow adipocytes are inert under physiological conditions, but may exacerbate pathological effects in states of physiological challenges such as regenerative processes [5]. Studies in humans have revealed an inverse relationship between marrow adiposity and bone volume [1, 6] as well as a negative correlation between marrow adiposity and hematopoiesis [7]. It is not clear, however, whether BMAT is a primary cause or compensatory effect of these processes. For instance, it has also been observed that increased adiposity does not necessarily lead to decreased bone quality in mice and humans [8,9,10]. This is further supported by the observation that, compared to C57BL/6J mice, C3H/HeJ mice have increases in both BMAT and bone mineral density [3]. However, since C3H/HeJ mice also experience increased MAT and reduced trabecular numbers during aging, there may still be a negative correlation between MAT and bone health that remains to be investigated in more detail [11].
Bone and adipose tissues arise from mesenchymal stem cells (MSCs) [12], which acquire their respective cell fates through the activation of specific transcription factors modulating target gene expression [5]. The orchestration of a controlled regulation of cell fate commitment is critical for bone morphology and the functional microenvironment [12]. Osteogenic cells rely on the expression of transcription factors Runt-related protein-2 (Runx2) and Osterix-1 (Osx1) [13], while adipogenic cells require peroxisome proliferator-activated receptor γ (Pparγ) and CCAAT-enhancer-binding protein α (Cebpα) [14]. Upstream, transcription factor Zinc finger protein-521 (Zfp521) controls osteoblast formation, while blocking adipogenesis [15]. Zfp521 simultaneously represses its downstream target Zfp423 by binding and inhibiting the transcriptional activity of pro-adipogenic factor early B cell factor-1 (Ebf1) [16].
Marrow-resident adipocytes display a unilocular morphology, i.e., contain a single large, lipid droplet that is reminiscent of typical white adipocytes [3]. Interestingly, the presence of two distinct types of BMAT has been described, distinguishable by different lipid profiles and histologically by performic acid-Schiff staining (PFAS) [17]. PFAS-positive marrow adipocytes are widely dispersed throughout the hematopoietic tissue and disappear with hematopoietic expansion, while non-stained adipocytes accumulate regionally and remain unaffected by changes in hematopoiesis [17]. A recent study confirmed these findings, describing a regulated BMAT (rBMAT) and a constitutive BMAT (cBMAT) [11]. Postnatally, cBMAT content increases steadily and is mostly inert to external stimuli. In contrast, the inducible rBMAT accrues in skeletal regions with high hematopoiesis, i.e., the proximal limb skeleton, hips, ribs, and the lumbar/thoracic vertebrae. Regulated marrow adipocytes are smaller in diameter (31–33 μm) when compared to cBMAT (38–39 μm) and inguinal white adipocytes (65–69 μm) and seem to undergo a different transcriptional regulation as suggested by lower expression of the adipogenic transcription factors Cebpa and Cebpb. Of note, the saturation degree of fatty acids within the lipid droplets is higher in rBMAT compared to cBMAT, but comparable to white adipose tissue (WAT) in the inguinal depot [11]. Under pathological conditions such as osteoporosis and plasmacytoma, the prevailing lipid species are increasingly saturated, suggesting a switch in marrow fat type [7, 18]. It has been hypothesized that rBMAT forms initially and then matures into cBMAT [19], yet the full developmental relationship between the two BMAT types remains to be elucidated. Interestingly, the resilience of cBMAT against dissolution has led to the suggestion that it might be important for vertebrate development and functions beyond the skeleton [11, 20]. In light of the characteristics of different marrow fat types and the fact that unlike in mice, these two types do not seem to be spatially separated in bones of humans [11], the quality of local BMAT stores might be more important than its overall quantity [21], which could also help explain seemingly opposing observations in recent studies on the pathophysiological effects of BMAT.
Developmental origin and cellular identity of bone marrow adipose tissue
BMAT in all likelihood derives from a mesenchymal origin [22] and genetic lineage tracing in mice has concomitantly revealed a non-endothelial, non-hematopoietic, mesenchymal source for marrow adipocytes [12]. These data support the hypothesis that osteogenic and adipogenic cells derive from a common MSC where distinct genetic and epigenetic factors determine the respective lineage fates [23]. All skeletal multipotent MSC populations are marked by Osx1 in neonatal bone [24], whereas analogous cells express Nestin and Leptin receptor (LepR) in adult bone [25, 26]. The entire marrow adipocyte compartment can be traced to overlapping populations of Prx1-, Osx1-, LepR-, and Adiponectin-expressing cells [12, 26,27,28]. Several marker combinations have been used to prospectively isolate skeletal stem cells [29, 30]. For instance, BMAT progenitors have been described as CD45−Pref1+RANKL+ stromal cells [27]. Independently from this observation, we were recently able to identify defined populations of uniformly committed adipogenic progenitor cells and pre-adipocytes in murine bones [12]. Interestingly, these cell types express the same marker profile as adipogenic progenitor cell populations from classical WAT and brown adipose tissue (BAT) [31,32,33]. In bone they derive from a highly homogeneous population of multipotent MSCs which lack expression of the endothelial and hematopoietic lineage markers, CD31 and CD45, respectively, but express stem cell antigen-1 (Sca1), platelet-derived growth factor α (Pdgfrα), and CD24 [12]. This multipotent population can give rise to two distinct and unilaterally committed populations of osteochondrogenic progenitor cells (surface markers: CD31−CD45−Sca1−Pdgfrα+) or adipogenic progenitor cells (surface markers: CD31−CD45−Sca1+Pdgfrα+CD24−) under in vitro and in vivo conditions. Adipogenic progenitors further mature to a CD31−CD45−Sca1−Zfp423+ pre-adipocyte stage that progresses to a mature adipocyte stage (Fig. 1). In vivo transplantation studies have also shown that these maturation steps are irreversible under normal physiological conditions [12]. Whether such cell populations are also present in human bones remains to be investigated. In humans, bone-derived MSCs may contribute to at least 10% of cells in adipose tissue depots outside the bone when administered by intravenous transplantation routes, but not the other way around [34]. This contribution appears to be doubled in obesity, implying that bone harbors a reserve pool of MSCs for classical WAT. It remains to be verified whether this also occurs in a more physiological context, i.e., not involving application of exogenous MSCs.

Proposed model of the cellular origin of bone marrow adipocytes. A population of stem cell-like multipotent stromal cells gives rise to distinct and unilaterally committed osteochondrogenic and adipogenic lineages. Several markers have been identified to be expressed/not expressed on the individual cellular stages and adipogenic cells of the bone in particular. Genetic lineage tracing by the Cre/loxP system further defines the developmental lineage and restricts BMAT to a mesenchymal, non-hematopoietic (Vav1), non-endothelial (Cdh5) origin that differs from traditional brown adipocytes
Regarding the characteristics of mature marrow adipocytes in comparison to WAT and BAT, lineage tracing to Osx1 is unique to BMAT compared to WAT and BAT [35]. Bone marrow adipocytes are distinct in size, cytokine and adipokine expression, immunomodulatory properties, free fatty acid (FFA) content, and some aspects of stem cell marker expression [3, 11, 36, 37]. Some lack of clarity remains, as adipocytes of long bones trace to a typical white fat-like Prx1+Myf5− origin [12, 22, 38, 39], but are capable of expressing some typical brown adipocyte genes, albeit at much lower expression levels than in BAT [40, 41]. Similarly, our own data suggest that bone-derived progenitors give rise to cells with a very limited brown-adipogenic potential [12]. On the other hand, BMAT phenotypes might be dependent on age and skeletal compartment and clearly warrant further investigation.
Congenital generalized lipodystrophy (CGL) is a rare condition defined by an almost complete loss of adipose tissue [19]. All affected genes (CGL1-4: Loss of Agpat2, Bscl2, Cav1, or Ptrf) are highly enriched in adipogenic cell populations of healthy murine bone [12]. However, only mice and humans with a loss of Agpat2 and Bscl2 (CGL1 and CGL2, respectively) also lack BMAT. Absence of bone fat does not impair patterning of the skeleton, suggesting no role of BMAT in this process. Nonetheless, the complete lack of adipocyte-derived endocrine factors impairs bone integrity [19]. In mice, a CGL4 model specifically reduces rBMAT, but not cBMAT [11], while CGL3 has no effect on any marrow adipose tissue type [11]. These findings further emphasize the existence of variations in genetic determinants of BMAT and WAT/BAT formation and function.
Pathophysiological regulation of bone marrow adipose tissue
Adipocyte development in the bone marrow compartment is regulated by different physiological and pathological processes. With advancing age, depots of BMAT increase in size and number in healthy individuals [42]. Obesity can be a potent driver of bone marrow adipocyte formation in particular in correlation with visceral WAT expansion [43]. Studies in mice show that feeding of a high-fat diet (HFD) rapidly initiates the expansion of BMAT by the activation of adipogenic progenitor cell proliferation [12]. Interestingly, caloric restriction (CR) and conditions such as anorexia nervosa also favor accumulation of marrow adipocytes [19]. Although CR generally counters aging and disease, it is not fully elucidated whether this also applies to bone tissue. For instance, in the growth phase of juvenile mice, CR leads to high marrow adiposity and low bone mass [44]. In patients recovered from anorexia nervosa, marrow adipocytes are depleted suggesting that marrow adipogenesis under these conditions is reversible [45]. While acute starvation does not affect BMAT [46], severe starvation beyond normal CR leads to a loss of bone marrow lipid content [47], altogether indicating that it might be able to serve as energy reservoir in specific situations. Cold exposure selectively reduces rBMAT in mice [11], but effects on bone mass depend on the level of brown adipose tissue (BAT) activation [48]. In contrast, housing at temperatures close to thermoneutrality (32 °C), which inactivates brown adipocytes, seems to be less detrimental to bone mass than room temperature (22 °C), which is a mild cold stimulus in mice [49]. Other physiological changes also regulate BMAT: ovariectomy- and menopause-induced estrogen deficiency favors adipocyte differentiation in the bone and is correlated with osteoporosis and increased fracture risk [3, 50]. Similarly, reduced mechanical stimulation due to extended bed rest induces a persistent bone loss and increased BMAT levels, which are retained even during subsequent exercise programs [51]. In line with the reversible nature of marrow adipogenesis, animal studies have shown that exercise reverses ovariectomy-, HFD-, and rosiglitazone-mediated BMAT accumulation [52,53,54,55]. Rosiglitazone and other thiazolidinediones (TZDs) are Pparγ-agonists with potent insulin-sensitizing effects, thereby improving systemic glucose homeostasis. As a common side effect, TZDs induce BMAT accumulation, which is paralleled by a decrease in BMD [56,57,58]. In mice, the effects of such Pparγ-agonists depend on dosage, age, and genetic background [59, 60]. For instance, the C3H/HeJ strain is highly responsive to rosiglitazone-induced bone loss, while C57BL/6J (B6) mice increase bone marrow adiposity without changes in trabecular bone parameters under TZDs [59]. In summary, the list of BMAT-regulating factors is constantly growing (see also Table 1). Other drivers of red-to-yellow marrow conversion include cancer, chemotherapy, radiation therapy, and hematopoietic malignancies as discussed further below [2, 67, 85, 86].
Endocrine and paracrine regulation of bone marrow adipose tissue
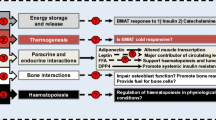
Many hormones involved in metabolic control also regulate marrow adipogenesis (Fig. 2). High systemic levels of growth hormone favor bone over marrow adipocyte formation [71]. Similarly, obese women show an inverse association between vertebral BMAT and circulating insulin-like growth factor-1 (Igf1) levels, which is in congruence with the essential role of Igf1-receptor signaling for bone development [43]. Glucocorticoids, for instance excessive cortisol levels in Cushing’s disease patients, induce marrow adiposity [71, 72]. In the same line, a recent article demonstrated that parathyroid hormone (Pth) stimulates an osteogenic fate. The authors showed that a loss of Pth-receptor activation in mice and humans increases BMAT, inducing the release of osteoclastogenesis-promoting RANKL and thereby promoting bone loss [37]. Increasing BMAT is also observed in response to elevated fibroblast growth factor 21 (Fgf21) levels which can be positively correlated with low bone mass in older men [76, 77]. An increment of osteocyte-derived Sclerostin secretion positively correlates with vertebral BMAT amounts in men [82]. In support of this observation, genetic ablation and pharmacological blockage of Sclerostin prevent BMAT and ameliorate osteoporotic conditions [83, 84]. Moreover, female rats treated with testosterone have less bone marrow fat and high bone mass [49, 75].

The red, predominantly hematopoietic bone marrow is converted into yellow, adipocyte-rich marrow as a normal physiological process, also in response to environmental and pathological cues. BMAT exerts endocrine and paracrine functions, which have been mainly associated with negative effects for local and systemic processes. During caloric restriction, BMAT contributes to significant amounts of circulating Adiponectin, thereby potentially improving metabolic conditions
Adiponectin may act in an anti-osteogenic manner by hampering osteoblast proliferation in short-term conditions, while long-term effects include enhanced bone mass and inhibition of BMAT formation by decreasing sympathetic tone, i.e., pro-adipogenic β3-adrenergic signaling, through central signaling mechanisms of the brain [81]. Intriguingly, BMAT significantly contributes to circulating Adiponectin levels during CR, which may mediate some of the beneficial effects of this dietary intervention [20]. TZD treatment similarly leads to elevated Adiponectin in serum, but it is not clear whether this is due to a general expansion of WAT mass [60]. It also remains to be investigated whether other drivers of BMAT-formation also lead to increased bone-derived Adiponectin secretion or whether these factors induce different types of BMAT. Additionally, BMAT could also be a source of significant amounts of Leptin, an adipokine that regulates fertility, appetite, and energy metabolism [94, 95]. Hypothalamic activity of Leptin increases sympathetic tone and favors marrow adipogenesis over osteogenesis. Locally, however, it may drive osteogenic fates by binding to LepR on osteoblasts [78], whereas other findings imply that activation of LepR signaling in MSCs promotes adipogenesis and impairs fracture healing [79]. In mice lacking Leptin (ob/ob), increased BMAT is observed and this correlates negatively with BMD of the axial skeleton [80]. Further indications of a potential metabolic involvement of bone come from the observation that the pancreatic hormone insulin binds to osteoblasts and thereby contributes to whole-body glucose homeostasis [96]. Specifically, insulin suppresses the expression of Osteoprotegerin (Opg) in osteogenic cells, an inhibitor of the differentiation of bone-resorbing osteoclasts. This stimulates bone degradation leading to a lower site-restricted pH, which in turn leads to decarboxylation of osteoblast-derived Osteocalcin (Ocn). The undercarboxylated form of Ocn promotes insulin secretion and enhances insulin sensitivity in liver, muscle, and WAT [96, 97]. In line with the important metabolic functions of Ocn, known pathologies of reduced bone quality and elevated marrow adiposity correlate with diminishing levels of this skeleton-derived hormone [98, 99]. A recent report shows that osteogenic cell-derived Lipocalin-2 (Lcn2) also controls energy metabolism by stimulating insulin secretion and decreases food intake through binding to melanocortin-4 receptor of neurons in the hypothalamus [100]. This finding is in contrast to two earlier studies that found no effect on appetite in mice lacking Lcn2 systemically [101, 102]. Adipose tissue is a known source of Lcn2, but conditional, adipocyte-specific ablation of Lcn2 driven by the Adipoq gene promoter did not show any effects. Whether BMAT-derived Lcn2 contributes to systemic metabolic effects remains to be determined, since only young mice on a C57BL/6J genetic background known to have negligible amounts of marrow adipocytes were investigated [100]. Lastly, BMAT has been described as a source of Dipeptidyl peptidase-4 (Dpp4), which as a locally secreted negative regulator inhibits the regenerative processes of bone healing and hematopoiesis [12, 103] (Fig. 2).
Implications for bone health and pathology
The potential functions of BMAT include production of adipokines, including several (pro-inflammatory) cytokines, and paracrine effects by direct contact to adjacent cells supporting a pro-adipogenic, bone-resorbing environment which is potentially mediated by lipotoxicity [104]. This is underpinned by the change of bone marrow cytokine profiles with aging [105]. In vitro data show that fatty acids impair osteoblast differentiation [106, 107] and that saturated palmitic acid reduces mineralization by impairing pro-osteogenic Wnt-signaling and related pathways [108]. In accordance with this observation, the fracture risk in postmenopausal women is increased with higher levels of saturated fatty acids in bone marrow fat [21]. Osteoporosis is characterized by an imbalance of bone formation and bone resorption, which is initiated by increased osteoclast activity and a reduced function of osteoblasts due to a clonal switch of MSCs from osteogenesis to adipogenesis during aging [3, 62, 63, 109]. Osteoporosis is particularly prevalent in women after menopause when increased osteoclastogenesis, BMAT accumulation, and fracture risk are evident [109, 110]. In obese, postmenopausal women, a low BMD is associated with high serum levels of Dpp4, a protease that can also be released form adipogenic cells as an adipokine [111]. Importantly, clinically approved Dpp4-inhibitors attenuate bone loss in male diabetic rats [93], decrease fracture risk in diabetic humans [112], and are a promising strategy to improve fracture healing outcomes in healthy, non-diabetic mice [12]. Among the Dpp4-substrates, the incretin hormones glucagon-like peptide-1 (Glp1) and gastric inhibitory polypeptide (Gip) are rapidly inactivated by Dpp4 and play a central role in metabolism [90]. Activation of the Glp1 receptor promotes osteogenesis, which can also be achieved with the synthetic agonist exendin-4, making it a therapeutic target for osteoporosis treatment [91].
Diabetes mellitus increases the risk of osteoporosis and bone fragility in humans [64]. Individuals with diabetes mellitus type 2 present a higher prevalence of fractures and fatty acid saturation in BMAT [21, 66] and experimental data show that type 2 diabetic mice suffer from bone loss and increased bone marrow adiposity [113]. Interestingly, prevention of BMAT accumulation during diabetes mellitus type 1 or after ovariectomy does not prevent bone loss, suggesting additional non-adipocyte-related mechanisms for impairment of bone integrity in diabetics [88, 114]. Under certain circumstances and depending on the BMAT type, bone marrow adipocytes might be supportive, as evidenced by the observation that BMAT-deficiency increases bone loss during unloading [115].
The hematopoietic niche within bones comprises diverse cell types of which osteo-lineage, endothelial, and mesenchymal stromal cells have been assigned hematopoiesis-supportive functions (reviewed in [116]). Obesity, which may induce BMAT, has been shown to negatively affect immune cells [117], but it also promotes white blood cell production through increased circulating Leptin levels [118, 119]. A recent study showed that a 2-week HFD reduces long-term hematopoietic stem cell (HSC) numbers and shifts lymphoid to myeloid differentiation, which leads to a changed bone structure with fewer osteoblasts and more adipocytes [120]. Moreover, blocking BMAT formation by treatment with the Pparγ-agonist bisphenol A diglycidyl ether (BADGE) rescues this phenotype and is potentially mediated by a change in gut microbiota composition [120]. This is in line with the finding that CR impairs lymphoid differentiation after irradiation following hematopoietic reconstitution [121], two mediators of strong BMAT induction. TZD treatment has no effect on the composition of the hematopoietic compartment, leading to the hypothesis that BMAT plays only a minor role during hematopoiesis [122]. In vitro co-culture experiments of bone-derived adipogenic cells and HSCs have yielded supportive as well as inhibitory effects for HSC maintenance [122, 123]. The first in vivo study investigating the role of BMAT for hematopoiesis concluded that marrow adipocytes failed to support hematopoiesis, since HSC number and quiescence were negatively related to adipocyte amount in the bone marrow [87]. Strikingly, genetic ablation of adipocyte development, or application of BADGE, leading to decreased BMAT and also classical adipose depots, rescues impaired hematopoietic recovery after radiation and chemotherapy [27, 85, 87, 89]. Consistent with these observations, our own analyses show that lineage-committed adipogenic progenitor cells co-injected into tibia bones with HSCs following lethal irradiation significantly inhibited the local engraftment of HSCs in competitive reconstitution assays. Conversely, multipotent MSCs isolated simultaneously enhanced hematopoietic reconstitution [12]. Interestingly, a recent study proposed BMAT as an important source of stem cell factor (Scf), a critical HSC niche factor [116], at least during hematopoietic reconstitution [27]. The authors show a reduced hematopoietic recovery in conditional, Adipoq-Cre driven Scf-knockout mice. According to the study, Adiponectin is expressed in all marrow adipocytes and a small subset (ca. 5%) of LepR+ cells, which expands after irradiation [27]. A potential explanation for apparent discrepancies might be that the stromal cells targeted by Adipoq-Cre could be involved in some of the observed effects, as LepR+ cells are highly heterogeneous and contain unilaterally committed osteochondrogenic and adipogenic cells, and also hematopoiesis-supporting multipotent MSCs [12]. Taken together, these data imply a highly context-specific role for bone marrow adipocytes in hematopoiesis and warrant further investigation with stronger emphasis on the definition of potentially distinct BMAT types, closely defined subpopulations of mesenchymal cells, and their distinct micro-anatomical localizations.
Cancer coincides with aging, obesity, and BMAT accumulation [124]. Marrow adipocytes are believed to be involved with the progression of myeloma and support bone metastases of prostate cancer, potentially linking marrow adiposity to an inflammation-induced pathophysiology [68,69,70]. Possible mechanisms include lipid exchange between cells, support of osteoclastogenesis, and contribution to osteolysis through the production of chemokine (C-X-C motif) ligand-1 (CXCL1) and CXCL2 [67, 70, 125]. Interestingly, certain tumor types depend on the local bone marrow status as blood cancers develop in red marrow spaces due to decreased vascularization in BMAT [2]. During stress-related disorders like anemia of chronic disease and childhood leukemia, the hematopoietic marrow becomes hyperplastic, leading to reduced or delayed local adipocyte emergence [2]. Leptin and Adiponectin have potentially opposing roles in cancer progression and thus provide an additional perspective to the endocrine function of BMAT [124]. Together with the observation that CR reduces the incidence of some cancers [126], and despite driving increased marrow adiposity, this underlines the need for a more accurate distinction of a beneficial or detrimental involvement of the prevailing BMAT type in bone-related pathologies.
Future perspectives
Depending on the molecular context, BMAT has pleiotrophic functions and can affect bone and marrow health by exerting beneficial as well as pathological effects (Fig. 2). This clearly emphasizes the need for a more detailed characterization of distinct types of BMAT and the stem/progenitor cells that can give rise to marrow-resident adipocytes. Recent findings highlight its involvement in human health and disease through paracrine and endocrine properties that are worth further examination. Marrow adipocytes that accumulate under physiological or homeostatic conditions may serve as a way to modulate energy-costly hematopoiesis and bone remodeling processes [127]. In contrast, improving osteogenesis by reducing BMAT during pathogenic conditions may not only increase skeletal health, but also other metabolic processes on the systemic level. Supportive clinical evidence along these lines comes from the use of estrogen replacements [74], bisphosphonate [92], and Sclerostin antibodies [84]. Novel candidates are synthetic Glp1-receptor agonists [91] and Dpp4-inhibitors [12] that require further investigation. One major drawback in the field is the lack of BMAT-specific in vivo models. Genetically engineered mice and adipocyte-ablating agents in most cases affect other adipose depots alongside BMAT, making it difficult to deduce the direct contribution of marrow-resident adipocytes. To this end, a detailed analysis of murine BMAT stem/progenitor cells may contribute to novel discoveries. The translation of such findings to the human context will be critical, as is the improvement of non-invasive BMAT analysis and quantification tools for clinical studies. In summary, the research of BMAT holds a significant clinical potential and will contribute to a better understanding of physiological and pathological processes of the musculoskeletal system.
References
Justesen J, Stenderup K, Ebbesen EN, Mosekilde L, Steiniche T, Kassem M (2001) Adipocyte tissue volume in bone marrow is increased with aging and in patients with osteoporosis. Biogerontology 2:165–171
Kricun ME (1985) Red-yellow marrow conversion: its effect on the location of some solitary bone lesions. Skelet Radiol 14:10–19
Fazeli PK, Horowitz MC, MacDougald OA, Scheller EL, Rodeheffer MS, Rosen CJ, Klibanski A (2013) Marrow fat and bone—new perspectives. J Clin Endocrinol Metab 98:935–945
Griffith JF, Yeung DK, Ma HT, Leung JC, Kwok TC, Leung PC (2012) Bone marrow fat content in the elderly: a reversal of sex difference seen in younger subjects. J Magn Reson Imaging 36:225–230
Rosen CJ, Ackert-Bicknell C, Rodriguez JP, Pino AM (2009) Marrow fat and the bone microenvironment: developmental, functional, and pathological implications. Crit Rev Eukaryot Gene Expr 19:109–124
Shen W, Scherzer R, Gantz M, Chen J, Punyanitya M, Lewis CE, Grunfeld C (2012) Relationship between MRI-measured bone marrow adipose tissue and hip and spine bone mineral density in African-American and Caucasian participants: the CARDIA study. J Clin Endocrinol Metab 97:1337–1346
Burkhardt R, Kettner G, Bohm W, Schmidmeier M, Schlag R, Frisch B, Mallmann B, Eisenmenger W, Gilg T (1987) Changes in trabecular bone, hematopoiesis and bone marrow vessels in aplastic anemia, primary osteoporosis, and old age: a comparative histomorphometric study. Bone 8:157–164
Castro JP, Joseph LA, Shin JJ, Arora SK, Nicasio J, Shatzkes J, Raklyar I, Erlikh I, Pantone V, Bahtiyar G et al (2005) Differential effect of obesity on bone mineral density in White, Hispanic and African American women: a cross sectional study. Nutr Metab (Lond) 2:9
Doucette CR, Horowitz MC, Berry R, MacDougald OA, Anunciado-Koza R, Koza RA, Rosen CJ (2015) A high fat diet increases bone marrow adipose tissue (MAT) but does not alter trabecular or cortical bone mass in C57BL/6J mice. J Cell Physiol 230:2032–2037
Douchi T, Yamamoto S, Oki T, Maruta K, Kuwahata R, Yamasaki H, Nagata Y (2000) Difference in the effect of adiposity on bone density between pre- and postmenopausal women. Maturitas 34:261–266
Scheller EL, Doucette CR, Learman BS, Cawthorn WP, Khandaker S, Schell B, Wu B, Ding SY, Bredella MA, Fazeli PK et al (2015) Region-specific variation in the properties of skeletal adipocytes reveals regulated and constitutive marrow adipose tissues. Nat Commun 6:7808
Ambrosi TH, Scialdone A, Graja A, Gohlke S, Jank AM, Bocian C, Woelk L, Fan H, Logan DW, Schurmann A et al (2017) Adipocyte accumulation in the bone marrow during obesity and aging impairs stem cell-based hematopoietic and bone regeneration. Cell Stem Cell 20:771–784
Nakashima K, de Crombrugghe B (2003) Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet 19:458–466
Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM (2000) Transcriptional regulation of adipogenesis. Genes Dev 14:1293–1307
Kang S, Akerblad P, Kiviranta R, Gupta RK, Kajimura S, Griffin MJ, Min J, Baron R, Rosen ED (2012) Regulation of early adipose commitment by Zfp521. PLoS Biol 10:e1001433
Addison WN, Fu MM, Yang HX, Lin Z, Nagano K, Gori F, Baron R (2014) Direct transcriptional repression of Zfp423 by Zfp521 mediates a bone morphogenic protein-dependent osteoblast versus adipocyte lineage commitment switch. Mol Cell Biol 34:3076–3085
Tavassoli M (1976) Marrow adipose cells. Histochemical identification of labile and stable components. Arch Pathol Lab Med 100:16–18
Yeung DK, Griffith JF, Antonio GE, Lee FK, Woo J, Leung PC (2005) Osteoporosis is associated with increased marrow fat content and decreased marrow fat unsaturation: a proton MR spectroscopy study. J Magn Reson Imaging 22:279–285
Scheller EL, Rosen CJ (2014) What’s the matter with MAT? Marrow adipose tissue, metabolism, and skeletal health. Ann N Y Acad Sci 1311:14–30
Cawthorn WP, Scheller EL, Learman BS, Parlee SD, Simon BR, Mori H, Ning X, Bree AJ, Schell B, Broome DT et al (2014) Bone marrow adipose tissue is an endocrine organ that contributes to increased circulating adiponectin during caloric restriction. Cell Metab 20:368–375
Patsch JM, Li X, Baum T, Yap SP, Karampinos DC, Schwartz AV, Link TM (2013) Bone marrow fat composition as a novel imaging biomarker in postmenopausal women with prevalent fragility fractures. J Bone Miner Res 28:1721–1728
Berry R, Rodeheffer MS, Rosen CJ, Horowitz MC (2015) Adipose tissue residing progenitors (adipocyte lineage progenitors and adipose derived stem cells (ADSC)). Curr Molec Biol Rep 1:101–109
Berendsen AD, Olsen BR (2014) Osteoblast-adipocyte lineage plasticity in tissue development, maintenance and pathology. Cell Mol Life Sci 71:493–497
Mizoguchi T, Pinho S, Ahmed J, Kunisaki Y, Hanoun M, Mendelson A, Ono N, Kronenberg HM, Frenette PS (2014) Osterix marks distinct waves of primitive and definitive stromal progenitors during bone marrow development. Dev Cell 29:340–349
Mendez-Ferrer S, Michurina TV, Ferraro F, Mazloom AR, Macarthur BD, Lira SA, Scadden DT, Ma'ayan A, Enikolopov GN, Frenette PS (2010) Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 466:829–834
Zhou BO, Yue R, Murphy MM, Peyer JG, Morrison SJ (2014) Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell 15:154–168
Zhou BO, Yu H, Yue R, Zhao Z, Rios JJ, Naveiras O, Morrison SJ (2017) Bone marrow adipocytes promote the regeneration of stem cells and haematopoiesis by secreting SCF. Nat Cell Biol 19:891–903
Liu Y, Strecker S, Wang L, Kronenberg MS, Wang W, Rowe DW, Maye P (2013) Osterix-cre labeled progenitor cells contribute to the formation and maintenance of the bone marrow stroma. PLoS One 8:e71318
Chan CK, Seo EY, Chen JY, Lo D, McArdle A, Sinha R, Tevlin R, Seita J, Vincent-Tompkins J, Wearda T et al (2015) Identification and specification of the mouse skeletal stem cell. Cell 160:285–298
Worthley DL, Churchill M, Compton JT, Tailor Y, Rao M, Si Y, Levin D, Schwartz MG, Uygur A, Hayakawa Y et al (2015) Gremlin 1 identifies a skeletal stem cell with bone, cartilage, and reticular stromal potential. Cell 160:269–284042
Berry R, Rodeheffer MS (2013) Characterization of the adipocyte cellular lineage in vivo. Nat Cell Biol 15:302–308
Gupta RK, Mepani RJ, Kleiner S, Lo JC, Khandekar MJ, Cohen P, Frontini A, Bhowmick DC, Ye L, Cinti S et al (2012) Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab 15:230–239
Schulz TJ, Huang TL, Tran TT, Zhang H, Townsend KL, Shadrach JL, Cerletti M, McDougall LE, Giorgadze N, Tchkonia T et al (2011) Identification of inducible brown adipocyte progenitors residing in skeletal muscle and white fat. Proc Natl Acad Sci U S A 108:143–148
Arner P, Ryden M (2017) The contribution of bone marrow-derived cells to the human adipocyte pool. Adipocyte 6:1–6
Chen J, Shi Y, Regan J, Karuppaiah K, Ornitz DM, Long F (2014) Osx-Cre targets multiple cell types besides osteoblast lineage in postnatal mice. PLoS One 9:e85161
Tencerova M, Kassem M (2016) The bone marrow-derived stromal cells: commitment and regulation of adipogenesis. Front Endocrinol 7:127
Fan Y, Hanai JI, Le PT, Bi R, Maridas D, DeMambro V, Figueroa CA, Kir S, Zhou X, Mannstadt M et al (2017) Parathyroid hormone directs bone marrow mesenchymal cell fate. Cell Metab 25:661–672
Sanchez-Gurmaches J, Guertin DA (2014) Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat Commun 5:4099
Sanchez-Gurmaches J, Hsiao WY, Guertin DA (2015) Highly selective in vivo labeling of subcutaneous white adipocyte precursors with Prx1-Cre. Stem Cell Rep 4:541–550
Krings A, Rahman S, Huang S, Lu Y, Czernik PJ, Lecka-Czernik B (2012) Bone marrow fat has brown adipose tissue characteristics, which are attenuated with aging and diabetes. Bone 50:546–552
Liu LF, Shen WJ, Ueno M, Patel S, Azhar S, Kraemer FB (2013) Age-related modulation of the effects of obesity on gene expression profiles of mouse bone marrow and epididymal adipocytes. PLoS One 8:e72367
Rozman C, Feliu E, Berga L, Reverter JC, Climent C, Ferran MJ (1989) Age-related variations of fat tissue fraction in normal human bone marrow depend both on size and number of adipocytes: a stereological study. Exp Hematol 17:34–37
Bredella MA, Torriani M, Ghomi RH, Thomas BJ, Brick DJ, Gerweck AV, Rosen CJ, Klibanski A, Miller KK (2011) Vertebral bone marrow fat is positively associated with visceral fat and inversely associated with IGF-1 in obese women. Obesity (Silver Spring) 19:49–53
Devlin MJ, Cloutier AM, Thomas NA, Panus DA, Lotinun S, Pinz I, Baron R, Rosen CJ, Bouxsein ML (2010) Caloric restriction leads to high marrow adiposity and low bone mass in growing mice. J Bone Miner Res 25:2078–2088
Fazeli PK, Bredella MA, Freedman L, Thomas BJ, Breggia A, Meenaghan E, Rosen CJ, Klibanski A (2012) Marrow fat and preadipocyte factor-1 levels decrease with recovery in women with anorexia nervosa. J Bone Miner Res 27:1864–1871
Bathija A, Davis S, Trubowitz S (1979) Bone marrow adipose tissue: response to acute starvation. Am J Hematol 6:191–198
Dietz AA, Steinberg B (1953) Chemistry of bone marrow: VIII. Composition of rabbit bone marrow in inanition. Arch Biochem Biophys 45:10–20
Bredella MA, Fazeli PK, Freedman LM, Calder G, Lee H, Rosen CJ, Klibanski A (2012) Young women with cold-activated brown adipose tissue have higher bone mineral density and lower Pref-1 than women without brown adipose tissue: a study in women with anorexia nervosa, women recovered from anorexia nervosa, and normal-weight women. J Clin Endocrinol Metab 97:E584–E590
Iwaniec UT, Philbrick KA, Wong CP, Gordon JL, Kahler-Quesada AM, Olson DA, Branscum AJ, Sargent JL, DeMambro VE, Rosen CJ et al (2016) Room temperature housing results in premature cancellous bone loss in growing female mice: implications for the mouse as a preclinical model for age-related bone loss. Osteoporos Int 27:3091–3101
Riggs BL, Khosla S, Melton LJ 3rd (2002) Sex steroids and the construction and conservation of the adult skeleton. Endocr Rev 23:279–302
Trudel G, Payne M, Madler B, Ramachandran N, Lecompte M, Wade C, Biolo G, Blanc S, Hughson R, Bear L et al (2009) Bone marrow fat accumulation after 60 days of bed rest persisted 1 year after activities were resumed along with hemopoietic stimulation: the Women International Space Simulation for Exploration study. J Appl Physiol (Bethesda, Md : 1985) 107:540–548
Kurabayashi T, Tomita M, Matsushita H, Honda A, Takakuwa K, Tanaka K (2001) Effects of a beta 3 adrenergic receptor agonist on bone and bone marrow adipocytes in the tibia and lumbar spine of the ovariectomized rat. Calcif Tissue Int 68:248–254
Styner M, Thompson WR, Galior K, Uzer G, Wu X, Kadari S, Case N, Xie Z, Sen B, Romaine A et al (2014) Bone marrow fat accumulation accelerated by high fat diet is suppressed by exercise. Bone 64:39–46
Styner M, Pagnotti GM, Galior K, Wu X, Thompson WR, Uzer G, Sen B, Xie Z, Horowitz MC, Styner MA et al (2015) Exercise regulation of marrow fat in the setting of PPARgamma agonist treatment in female C57BL/6 mice. Endocrinology 156:2753–2761
Chen Y, Wang S, Bu S, Wang Y, Duan Y, Yang S (2011) Treadmill training prevents bone loss by inhibition of PPARgamma expression but not promoting of Runx2 expression in ovariectomized rats. Eur J Appl Physiol 111:1759–1767
Schwartz AV, Sellmeyer DE, Vittinghoff E, Palermo L, Lecka-Czernik B, Feingold KR, Strotmeyer ES, Resnick HE, Carbone L, Beamer BA et al (2006) Thiazolidinedione use and bone loss in older diabetic adults. J Clin Endocrinol Metab 91:3349–3354
Pop LM, Lingvay I, Yuan Q, Li X, Adams-Huet B, Maalouf NM (2017) Impact of pioglitazone on bone mineral density and bone marrow fat content. Osteoporos Int. https://doi.org/10.1007/s00198-017-4164-3
Crossno JT Jr, Majka SM, Grazia T, Gill RG, Klemm DJ (2006) Rosiglitazone promotes development of a novel adipocyte population from bone marrow-derived circulating progenitor cells. J Clin Invest 116:3220–3228
Ackert-Bicknell CL, Shockley KR, Horton LG, Lecka-Czernik B, Churchill GA, Rosen CJ (2009) Strain-specific effects of rosiglitazone on bone mass, body composition, and serum insulin-like growth factor-I. Endocrinology 150:1330–1340
Sulston RJ, Learman BS, Zhang B, Scheller EL, Parlee SD, Simon BR, Mori H, Bree AJ, Wallace RJ, Krishnan V et al (2016) Increased circulating adiponectin in response to thiazolidinediones: investigating the role of bone marrow adipose tissue. Front Endocrinol 7:128
Suchacki KJ, Cawthorn WP, Rosen CJ (2016) Bone marrow adipose tissue: formation, function and regulation. Curr Opin Pharmacol 28:50–56
Verma S, Rajaratnam JH, Denton J, Hoyland JA, Byers RJ (2002) Adipocytic proportion of bone marrow is inversely related to bone formation in osteoporosis. J Clin Pathol 55:693–698
Ralston SH, de Crombrugghe B (2006) Genetic regulation of bone mass and susceptibility to osteoporosis. Genes Dev 20: 2492–2506
Napoli N, Chandran M, Pierroz DD, Abrahamsen B, Schwartz AV, Ferrari SL (2017) Mechanisms of diabetes mellitus-induced bone fragility. Nat Rev Endocrinol 13:208–219
Botolin S, McCabe LR (2007) Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology 148:198–205
Dede AD, Tournis S, Dontas I, Trovas G (2014) Type 2 diabetes mellitus and fracture risk. Metabolism 63:1480–1490
Olechnowicz SW, Edwards CM (2014) Contributions of the host microenvironment to cancer-induced bone disease. Cancer Res 74:1625–1631
Reagan MR, Liaw L, Rosen CJ, Ghobrial IM (2015) Dynamic interplay between bone and multiple myeloma: emerging roles of the osteoblast. Bone 75:161–169
Lwin ST, Olechnowicz SW, Fowler JA, Edwards CM (2015) Diet-induced obesity promotes a myeloma-like condition in vivo. Leukemia 29:507–510
Hardaway AL, Herroon MK, Rajagurubandara E, Podgorski I (2015) Marrow adipocyte-derived CXCL1 and CXCL2 contribute to osteolysis in metastatic prostate cancer. Clin Exp Metastasis 32:353–368
Menagh PJ, Turner RT, Jump DB, Wong CP, Lowry MB, Yakar S, Rosen CJ, Iwaniec UT (2010) Growth hormone regulates the balance between bone formation and bone marrow adiposity. J Bone Miner Res Off J Am Soc Bone Miner Res 25:757–768
Lecka-Czernik B (2012) Marrow fat metabolism is linked to the systemic energy metabolism. Bone 50:534–539
Geer EB, Shen W, Strohmayer E, Post KD, Freda PU (2012) Body composition and cardiovascular risk markers after remission of Cushing’s disease: a prospective study using whole-body MRI. J Clin Endocrinol Metab 97:1702–1711
Syed FA, Oursler MJ, Hefferanm TE, Peterson JM, Riggs BL, Khosla S (2008) Effects of estrogen therapy on bone marrow adipocytes in postmenopausal osteoporotic women. Osteoporos Int 19:1323–1330
Tamura N, Kurabayashi T, Nagata H, Matsushita H, Yahata T, Tanaka K (2005) Effects of testosterone on cancellous bone, marrow adipocytes, and ovarian phenotype in a young female rat model of polycystic ovary syndrome. Fertil Steril 84(Suppl 2):1277–1284
Wei W, Dutchak PA, Wang X, Ding X, Wang X, Bookout AL, Goetz R, Mohammadi M, Gerard RD, Dechow PC et al (2012) Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor gamma. Proc Natl Acad Sci U S A 109:3143–3148
Hanks LJ, Gutierrez OM, Bamman MM, Ashraf A, McCormick KL, Casazza K (2015) Circulating levels of fibroblast growth factor-21 increase with age independently of body composition indices among healthy individuals. J Clin Translat Endocrinol 2:77–82
Upadhyay J, Farr OM, Mantzoros CS (2015) The role of leptin in regulating bone metabolism. Metabolism 64:105–113
Yue R, Zhou BO, Shimada IS, Zhao Z, Morrison SJ (2016) Leptin receptor promotes adipogenesis and reduces osteogenesis by regulating mesenchymal stromal cells in adult bone marrow. Cell Stem Cell 18:1–15
Hamrick MW (2004) Leptin, bone mass, and the thrifty phenotype. J Bone Miner Res 19:1607–1611
Kajimura D, Lee HW, Riley KJ, Arteaga-Solis E, Ferron M, Zhou B, Clarke CJ, Hannun YA, DePinho RA, Guo XE et al (2013) Adiponectin regulates bone mass via opposite central and peripheral mechanisms through FoxO1. Cell Metab 17:901–915
Ma YH, Schwartz AV, Sigurdsson S, Hue TF, Lang TF, Harris TB, Rosen CJ, Vittinghoff E, Eiriksdottir G, Hauksdottir AM et al (2014) Circulating sclerostin associated with vertebral bone marrow fat in older men but not women. J Clin Endocrinol Metab 99:E2584–E2590
Fairfield H, Falank C, Harris E, Demambro V, McDonald M, Pettitt JA, Mohanty ST, Croucher P, Kramer I, Kneissel M et al (2017) The skeletal cell-derived molecule sclerostin drives bone marrow adipogenesis. J Cell Physiol. https://doi.org/10.1002/jcp.25976
Cosman F, Crittenden DB, Adachi JD, Binkley N, Czerwinski E, Ferrari S, Hofbauer LC, Lau E, Lewiecki EM, Miyauchi A et al (2016) Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med 375:1532–1543
Georgiou KR, Hui SK, Xian CJ (2012) Regulatory pathways associated with bone loss and bone marrow adiposity caused by aging, chemotherapy, glucocorticoid therapy and radiotherapy. Am J Stem Cells 1:205–224
Green DE, Adler BJ, Chan ME, Lennon JJ, Acerbo AS, Miller LM, Rubin CT (2013) Altered composition of bone as triggered by irradiation facilitates the rapid erosion of the matrix by both cellular and physicochemical processes. PLoS One 8:e64952
Naveiras O, Nardi V, Wenzel PL, Hauschka PV, Fahey F, Daley GQ (2009) Bone-marrow adipocytes as negative regulators of the haematopoietic microenvironment. Nature 460:259–263
Botolin S, McCabe LR (2006) Inhibition of PPARgamma prevents type I diabetic bone marrow adiposity but not bone loss. J Cell Physiol 209:967–976
Zhu RJ, Wu MQ, Li ZJ, Zhang Y, Liu KY (2013) Hematopoietic recovery following chemotherapy is improved by BADGE-induced inhibition of adipogenesis. Int J Hematol 97:58–72
Rohrborn D, Wronkowitz N, Eckel J (2015) DPP4 in diabetes. Front Immunol 6:386
Meng J, Ma X, Wang N, Jia M, Bi L, Wang Y, Li M, Zhang H, Xue X, Hou Z et al (2016) Activation of GLP-1 receptor promotes bone marrow stromal cell osteogenic differentiation through beta-catenin. Stem Cell Rep 6:633
Duque G, Li W, Adams M, Xu S, Phipps R (2011) Effects of risedronate on bone marrow adipocytes in postmenopausal women. Osteoporos Int 22:1547–1553
Glorie L, Behets GJ, Baerts L, De Meester I, D’Haese PC, Verhulst A (2014) DPP IV inhibitor treatment attenuates bone loss and improves mechanical bone strength in male diabetic rats. Am J Phys Endocrinol Metab 307: E447–E455
Laharrague P, Larrouy D, Fontanilles AM, Truel N, Campfield A, Tenenbaum R, Galitzky J, Corberand JX, Penicaud L, Casteilla L (1998) High expression of leptin by human bone marrow adipocytes in primary culture. FASEB J 12:747–752
Koch L, Wunderlich FT, Seibler J, Konner AC, Hampel B, Irlenbusch S, Brabant G, Kahn CR, Schwenk F, Bruning JC (2008) Central insulin action regulates peripheral glucose and fat metabolism in mice. J Clin Invest 118:2132–2147
Karsenty G, Ferron M (2012) The contribution of bone to whole-organism physiology. Nature 481:314–320
Wei J, Ferron M, Clarke CJ, Hannun YA, Jiang H, Blaner WS, Karsenty G (2014) Bone-specific insulin resistance disrupts whole-body glucose homeostasis via decreased osteocalcin activation. J Clin Invest 124:1–13
Napoli N, Strollo R, Paladini A, Briganti SI, Pozzilli P, Epstein S (2014) The alliance of mesenchymal stem cells, bone, and diabetes. Int J Endocrinol 2014:690783
Singh S, Kumar D, Lal AK (2015) Serum osteocalcin as a diagnostic biomarker for primary osteoporosis in women. J Clin Diagn Res 9:Rc04–Rc07
Mosialou I, Shikhel S, Liu JM, Maurizi A, Luo N, He Z, Huang Y, Zong H, Friedman RA, Barasch J et al (2017) MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543:385–390
Law IK, Xu A, Lam KS, Berger T, Mak TW, Vanhoutte PM, Liu JT, Sweeney G, Zhou M, Yang B et al (2010) Lipocalin-2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes 59:872–882
Guo H, Jin D, Zhang Y, Wright W, Bazuine M, Brockman DA, Bernlohr DA, Chen X (2010) Lipocalin-2 deficiency impairs thermogenesis and potentiates diet-induced insulin resistance in mice. Diabetes 59:1376–1385
Broxmeyer HE, Hoggatt J, O’Leary HA, Mantel C, Chitteti BR, Cooper S, Messina-Graham S, Hangoc G, Farag S, Rohrabaugh SL et al (2012) Dipeptidylpeptidase 4 negatively regulates colony-stimulating factor activity and stress hematopoiesis. Nat Med 18:1786–1796
Kim JE, Ahn MW, Baek SH, Lee IK, Kim YW, Kim JY, Dan JM, Park SY (2008) AMPK activator, AICAR, inhibits palmitate-induced apoptosis in osteoblast. Bone 43:394–404
Gasparrini M, Rivas D, Elbaz A, Duque G (2009) Differential expression of cytokines in subcutaneous and marrow fat of aging C57BL/6J mice. Exp Gerontol 44:613–618
Maurin AC, Chavassieux PM, Frappart L, Delmas PD, Serre CM, Meunier PJ (2000) Influence of mature adipocytes on osteoblast proliferation in human primary cocultures. Bone 26:485–489
Elbaz A, Wu X, Rivas D, Gimble JM, Duque G (2010) Inhibition of fatty acid biosynthesis prevents adipocyte lipotoxicity on human osteoblasts in vitro. J Cell Mol Med 14:982–991
Gunaratnam K, Vidal C, Boadle R, Thekkedam C, Duque G (2013) Mechanisms of palmitate-induced cell death in human osteoblasts. Biol Open 2:1382–1389
Bermeo S, Gunaratnam K, Duque G (2014) Fat and bone interactions. Curr Osteoporos Rep 12:235–242
Wehrli FW, Hopkins JA, Hwang SN, Song HK, Snyder PJ, Haddad JG (2000) Cross-sectional study of osteopenia with quantitative MR imaging and bone densitometry. Radiology 217:527–538
Kim SW, Cho EH (2016) High levels of serum DPP-4 activity are associated with low bone mineral density in obese postmenopausal women. Endocrinol Metab 31:93–99
Dombrowski S, Kostev K, Jacob L (2017) Use of dipeptidyl peptidase-4 inhibitors and risk of bone fracture in patients with type 2 diabetes in Germany—a retrospective analysis of real-world data. Osteoporos Int 28:2421–2428
Devlin MJ, Van Vliet M, Motyl K, Karim L, Brooks DJ, Louis L, Conlon C, Rosen CJ, Bouxsein ML (2014) Early-onset type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology 155:3806–3816
Iwaniec UT, Turner RT (2013) Failure to generate bone marrow adipocytes does not protect mice from ovariectomy-induced osteopenia. Bone 53:145–153
Keune JA, Wong CP, Branscum AJ, Iwaniec UT, Turner RT (2017) Bone marrow adipose tissue deficiency increases disuse-induced bone loss in male mice. Sci Rep 7:46325
Asada N, Takeishi S, Frenette PS (2017) Complexity of bone marrow hematopoietic stem cell niche. Int J Hematol 106:45–54
Adler BJ, Kaushansky K, Rubin CT (2014) Obesity-driven disruption of haematopoiesis and the bone marrow niche. Nat Rev Endocrinol 10:737–748
Trottier MD, Naaz A, Li Y, Fraker PJ (2012) Enhancement of hematopoiesis and lymphopoiesis in diet-induced obese mice. Proc Natl Acad Sci U S A 109:7622–7629
Claycombe K, King LE, Fraker PJ (2008) A role for leptin in sustaining lymphopoiesis and myelopoiesis. Proc Natl Acad Sci U S A 105:2017–2021
Luo Y, Chen GL, Hannemann N, Ipseiz N, Kronke G, Bauerle T, Munos L, Wirtz S, Schett G, Bozec A (2015) Microbiota from obese mice regulate hematopoietic stem cell differentiation by altering the bone niche. Cell Metab 22:886–894
Tang D, Tao S, Chen Z, Koliesnik IO, Calmes PG, Hoerr V, Han B, Gebert N, Zornig M, Loffler B et al (2016) Dietary restriction improves repopulation but impairs lymphoid differentiation capacity of hematopoietic stem cells in early aging. J Exp Med 213:535–553
Spindler TJ, Tseng AW, Zhou X, Adams GB (2014) Adipocytic cells augment the support of primitive hematopoietic cells in vitro but have no effect in the bone marrow niche under homeostatic conditions. Stem Cells Dev 23:434–441
Chitteti BR, Cheng YH, Poteat B, Rodriguez-Rodriguez S, Goebel WS, Carlesso N, Kacena MA, Srour EF (2010) Impact of interactions of cellular components of the bone marrow microenvironment on hematopoietic stem and progenitor cell function. Blood 115:3239–3248
Sulston RJ, Cawthorn WP (2016) Bone marrow adipose tissue as an endocrine organ: close to the bone? Horm Mol Biol Clin Invest 28:21–38
Liu Y (2006) Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis 9:230–234
Meynet O, Ricci JE (2014) Caloric restriction and cancer: molecular mechanisms and clinical implications. Trends Mol Med 20:419–427
Scheller EL, Cawthorn WP, Burr AA, Horowitz MC, MacDougald OA (2016) Marrow adipose tissue: trimming the fat. Trends Endocrinol Metab 27:392–403
Funding
This work was supported by the European Research Council (ERC-StG 311082), the Emmy Noether Program of the German Research Foundation (DFG, grant SCHU 2445/2-1), and a grant from the German Ministry of Education and Research (BMBF) and the State of Brandenburg (DZD grant 82DZD00302).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ambrosi, T.H., Schulz, T.J. The emerging role of bone marrow adipose tissue in bone health and dysfunction. J Mol Med 95, 1291–1301 (2017). https://doi.org/10.1007/s00109-017-1604-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00109-017-1604-7