
Abstract
Utilizing a sequential extraction technique this study provides the first quantitative analysis on the abundance of sedimentary phosphorus and its partitioning between chemically distinguishable phases in sediments of the Bering Sea, the Chukchi Sea and the Mackenzie River Delta in the western Arctic Ocean. Total sedimentary phosphorus (TSP) was fractionated into five operationally defined phases: (1) adsorbed inorganic and exchangeable organic phosphorus, (2) Fe-bound inorganic phosphorus, (3) authigenic carbonate fluorapatite, biogenic apatite and calcium carbonate-bound inorganic and organic phosphorus, (4) detrital apatite, and (5) refractory organic phosphorus. TSP concentrations in surface sediments increased from the Chukchi Sea (18 μmol g−1 of dried sediments) to the Bering Sea (22 μmol g−1) and to the Mackenzie River Delta (29 μmol g−1). Among the five pools, detrital apatite phosphorus of igneous or metamorphic origin represents the largest fraction (~43%) of TSP. The second largest pool is the authigenic carbonate fluorapatite, biogenic apatite as well as CaCO3 associated phosphorus (~24% of TSP), followed by the Fe-bound inorganic phosphorus, representing ~20% of TSP. The refractory organic P accounts for ~10% of TSP and the readily exchangeable adsorbed P accounts for only 3.5% of TSP. Inorganic phosphorus dominates all of phosphorus pools, accounting for an average of 87% of the TSP. Relatively high sedimentary organic carbon and total nitrogen contents and low δ13C values in the Mackenzie River Delta together with the dominance of detrital apatite in the TSP demonstrate the importance of riverine inputs in governing the abundance and speciation of sedimentary phosphorus in the Arctic coastal sediments.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Phosphorus (P) is an essential macronutrient for living organisms on earth. In particular, adenosine tri-phosphate is an universal carrier of chemical energy in all living cells and calcium phosphate is the building blocks for teeth and bonds of vertebrates (Westheimer 1987). However, phosphorus is held largely in earth’s crust as sparingly soluble minerals, such as apatite, fluorapatite and hydroxyapatite (Griffith et al. 1973). Low solubility of P-bearing minerals and strong tendency of dissolved phosphate to absorb onto solid surfaces set an upper limit of dissolved phosphate concentrations in natural waters at micromolar levels (Follmi 1996; Froelich et al. 1982; Kramer 1964). Availability of P often becomes a factor limiting the primary production in many terrestrial and aquatic ecosystems (Krom et al. 1991; Ryther and Dunstan 1971 ; Schindler 1977; Smith 1984; Smith and Atkinson 1984). It is believed that P is the ultimate limiting nutrient on marine productivity which in turn regulates oceanic uptake of atmospheric CO2 on geological time scales (Broecker and Peng 1982).
Continental weathering of P-bearing igneous and sedimentary rocks is the most important pre-industrial source of P to the terrestrial and aquatic ecosystems. Principal P flux to the oceans is carried by river discharges, with suspended particulate matter in river water contributing more than 90% of the riverine P flux (Howarth et al. 1995; Guo et al. 2004a; Meybeck 1982, 1993). A large portion of such riverine suspended particulate matter is aggregated and settled as coastal and shelf sediments (Gibbs 1981; Meade 1982). Particulate P in the suspended matter and sediments has been identified as the dominant reservoir of P in estuarine, coastal and continental shelf ecosystems (e.g., Conley et al. 1995; Lebo 1991; Lebo and Sharp 1992; Zhang et al. 2004). Through desorption, reduction and dissolution processes, sediments can supply dissolved phosphate to the overlying water column. It has been proposed that the sedimentary P functions as a buffer system, regulating dissolved phosphate concentration in the overlying water (Froelich 1988; Zhang and Huang 2007). Quantifying the abundance and chemical forms of different P pools in sediments provides useful information on not only the source of sediments and their depositional environment but also the first-order estimate of the potential bioavailable sedimentary phosphorus in supporting both the benthic and water column productivities.
Although the Arctic Ocean comprises only ~1% of total volume of the global ocean it receives 10% of the global river discharge. Therefore, the Arctic Ocean has the highest input of terrestrial flux of freshwater and associated nutrients in terms of per unit volume of seawater. Among the large Arctic rivers, the Mackenzie River discharges the largest amounts of suspended sediments to the Arctic Ocean (Stein and Macdonald 2004), along with vast fresh water and nutrient fluxes. However, primary productivity in the Arctic estuarine and inner shelf regions was found to be P-limited (Carmack et al. 2004) due to the overall low phosphate concentrations in the Arctic rivers (Cauwet and Sidorov 1996; Guo et al. 2004a). Thus, supply of P from the sediments and suspended particulate matter may play an important role in sustaining the primary production in the Arctic Ocean. Previous studies in the Mackenzie shelf sediments have shown a large fraction of inorganic P and a relative small fraction of organic P (Ruttenberg and Goni 1997). However, specific forms of P associated with this inorganic pool have not been identified. This study provided detailed information on the abundance and chemical forms of P in sediments of the western Arctic Ocean, including the Mackenzie River Delta, the Chukchi Sea, and the Bering Sea, with contrasting environmental setting in terms of riverine inputs, marine primary production, and sediment sources. In conjunction with sedimentation rates, measurements of abundance of sedimentary P can be used to estimate the continental flux of P to the western Arctic Ocean.
2 Methods
2.1 Sample Collection
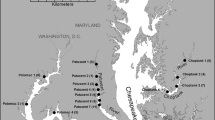
Sediment samples were collected from continental shelf regions of the Bering Sea and the western Arctic Ocean, including the Chukchi Sea and the Beaufort Sea near the Mackenzie River Delta (Fig. 1). The Chukchi Sea is a highly productive marginal sea while the Beaufort Sea receives considerable terrestrial sediments from the Mackenzie River and other continental sources. The Bering Sea lies between these two extreme marine environments. These contrasting depositional environments should provide diverse sediment sources, which might influence the abundance, distribution and geochemical cycling of P in these coastal marine ecosystems.

Locations of sediment sampling in the Bering Sea and western Arctic Ocean. Stations PC and PG are located in the Mackenzie River Delta, station CC in the Chukchi Sea, and stations B1 and B2 in the Bering Sea
Sediment cores were collected from the southeastern Bering Sea in August/September 2001 along a transect at 166°W during MR01-K04 cruise, and from the Chukchi Sea and the Mackenzie River Delta in September 2002 during the MR02-K05 cruise. Both cruises were on board the R/V Mirai and supported by JAMSTEC, Japan (Table 1; Fig. 1).
Three piston cores (PC1, PC2 and PC3) were collected in the Mackenzie River Delta in a transect approaching the river mouth (Fig. 1). In addition, two box cores were collected at a pingo site with different water depth (65 and 38 m, respectively). One box core was collected on Chukchi Sea shelf (CC). Two box cores were collected from the Bering Sea (B1 and B2). The cores were sliced to sections at 1-cm interval and samples were kept frozen until further processing. In a shore-based laboratory, the sediment samples were then freeze-dried to constant weight using a LABCONCO freeze-dryer. The freeze-dried samples were then ground to <125 μm and used for the sequential extraction to quantify the different forms of P in the sediments. The sum of the different forms of P in the sediments was defined as the total sedimentary phosphorus (TSP).
2.2 Measurements of Organic Carbon (OC) and Total Nitrogen (TN) and Their Stable Isotope Composition
Sediments were treated with 2 M HCl solution three times to remove inorganic carbon before OC and stable isotope analyses (Guo et al. 2004b). Contents of OC and TN, and their stable isotopes (δ13C and δ15N) were determined on an elemental analyzer interfaced with isotope ratio mass spectrometry (ISMS) at the Alaska Stable Isotope Facility (Guo et al. 2003). Stable C and N isotope ratios were calculated in terms of δ13C and δ15N, with a formula of (R sample/R standard−1) × 1000, where R is the ratio of 13C:12C, or 15N:14N, in sediment samples or standards (PDB for C and atmospheric N2 for N).
2.3 Sequential Extraction Procedure
Sequential extraction technique is commonly used to separate and quantify different forms of P in solid samples. The extraction procedure used in this study is based upon the scheme developed for marine sediments (Ruttenberg 1992; Zhang et al. 2004). In this study TSP was fractionated into five distinct pools: (1) adsorbed inorganic and exchangeable organic P, (2) Fe-bound inorganic P, (3) authigenic carbonate fluorapatite, biogenic apatite and calcium carbonate-bound inorganic and organic P, (4) detrital apatite of igneous and metamorphic origin, and (5) refractory organic P. The five-step sequential extraction scheme (Fig. 2) allows us to make an operational separation between five different P pools and within some of these into organic and inorganic forms. The major advantage of this method is that it provides a separation of detrital apatite from biogenic carbonate-bound P. It also allows us to quantify the abundance of surface reactive Fe and Si in the Fe-reduction step.

Procedures used for sequential extraction of different forms of sedimentary phosphorus. A 50 ml of extractant was used in all steps. Extracts were determined for dissolved inorganic phosphorus (DIP) and total dissolved phosphorus (TDP), from which dissolved organic phosphorus was calculated as difference
In step 1, loosely adsorbed phosphate, along with some exchangeable organic P, was extracted with MgCl2 solution. The pH of 1 M MgCl2 solution was adjusted to about 8 with dilute NaOH solution. A 0.5 g of dry sediment was placed into a 50 ml of the MgCl2 solution in a 60-ml high-density polypropylene bottle. The mixture was agitated by a bench-top shaking incubator (VMR, USA) at 200 rotations per minute for 2 h at 25 ± 0.1°C. Samples were then filtered through a glass fiber filter (Whatman GF/F) with the aid of a vacuum pump. The filtrate was collected for analysis of dissolved phosphate and total dissolved P from which dissolved organic P was calculated as difference. The filter paper that contains sediment residue was used for subsequent extraction.
In step 2, the sediment residue from the MgCl2 extraction was extracted with bicarbonate dithionite mixed solution (BD solution, 0.11 M NaHCO3 and 0.11 M Na2S2O4 with its final pH adjusted to 7.0). Dithionite is a strong reducing agent that reduces amorphous Fe(OH)3 and Mn(OH)3 to soluble ferrous and Mn2+ ion and therefore releases Fe-bound phosphorus. In previous studies, sodium citrate was added to complex soluble ferric ion released from sediment by reductive dissolution. However, dithionite-citrate reagent was found to be ineffective in separating CaCO3-bound P from Fe-bound P, presumably because citrate is a strong complexing agent for both Ca and Mg (pK of 4.7; Hieltjes and Lijldema 1980). Furthermore, citrate has also been found to interfere with spectrophotometric determination of phosphate. Solvent extraction has been used to circumvent this interference but the resulting analytical precision was poor (Ruttenberg 1992). For these reasons citrate was not used as an extraction reagent for Fe-bound P in recent studies (Jensen et al. 1998; Zhang et al. 2004).
In this step, the sediment residue was extracted in a 50 ml BD solution for 4 h at 25°C and then separated by filtration. In previous studies, the filtrate was usually acidified and the excess dithionite remaining in the samples was decomposed to SO2 under an acidic condition and stripped with nitrogen gas. The acidification also helps to preserve Fe in the samples in case the immediate analysis is not possible. Unfortunately, acidifying samples and bubbling with N2 to remove the SO2 have been found to induce precipitation of elemental sulfur that interferes with subsequent P and Fe analyses by spectrophotometry. To avoid this interference, two aliquots of filtrate were separately collected for phosphate and total Fe analyses. These samples were not acidified. Samples for total Fe analyses were performed immediately after collection, therefore no preservation is necessary. Samples for phosphate analyses were let open to the air for 72 h to allow complete decomposition of excess dithionite to SO2 which was oxidized immediately by dissolved oxygen in sample solution. This procedure prevents the formation of elemental sulfur.
In step 3, the sediment residue from the BD extraction was extracted with an acetate buffer solution (pH = 4) which dissolves authigenic carbonate fluorapatite, biogenic apatite, and calcium carbonate-bound inorganic and organic P, but prevents dissolution of detrital apatite in the sediments. The acetate buffer (HAc/Ac = 1/5.55 for pH 4.0) solution was prepared by dissolving 82 g of NaAc in 317 ml of glacial acetic acid (17.5 M) and then diluting the mixture to a final volume of 1 l with distilled water. Sediment residues were placed in a 50 ml acetate buffer solution and agitated for 6 h at 25°C. Sediment and extractant were then separated by filtration and the filtrates analyzed for dissolved phosphate and total dissolved P. The sample residue was washed with MgCl2 solution to recover any P re-adsorbed on sediment surface during acetate buffer extraction. The MgCl2 wash solutions were then analyzed for both dissolved phosphate and total dissolved P.
In step 4, the sediment residue from step 3 was extracted with 1 M HCl solution to dissolve detrital apatite. The sediment residue was placed in a 50 ml 1 M HCl solution and agitated for 16 h at 25°C. After filtration the filtrates were analyzed for dissolved phosphate.
In step 5, sediment residues from step 4 were ashed to oxidize the refractory organic P to phosphate (Solorzano and Sharp 1980). The sediment residues were placed in 100-ml Pyrex beakers and wetted with a few drops of 1 M Mg(NO3)2 solution and then ashed in a combustion furnace at 550°C for 2 h. After the samples were cooled to room temperature, a 50 ml of 1 M HCl solution was added to each sample. The samples were then agitated at 25°C for 24 h to extract P. Samples were filtered to remove any particulate residuals and the filtrates analyzed for dissolved phosphate.
2.4 Analysis of Dissolved Phosphate
Dissolved phosphate was determined by reacting sample phosphate with ammonium molybdate in the presence of potassium antimony (III) tartrate in an acidic medium to form an antimonyphosphomolybdate complex (Zhang et al. 1999a; Zhang and Chi 2002). This complex is subsequently reduced by ascorbic acid to form a phosphomolybdeum blue. A complete color development takes about 20 min at room temperature. This complex has a typical spectrum characterized by two distinct absorbance maxima at 710 and 890 nm. The absorbance of the latter maximum was measured with a Hewlett Packard 8453 spectrophotometer and a three-point drop technique was used to correct any baseline shift.
In standard phosphate methods used in natural water analysis, the amount of acid in the molybdate reagent is designed to handle samples that contain about 2 mM buffer capacity (mainly bicarbonate ion, Zhang 2000) and can effectively lower the pHT (total proton scale Millero et al. 1993), of mixture (sample plus reagents) to about 1 for optimal color development. This standard method, however, cannot be directly applied to the analysis of phosphate in solutions obtained from the sequential extraction of sediment because the extracts are typically containing high concentrations of acid or base. Although there is little salt effect on the sensitivity of phosphate analysis using the molybdenum blue method, we found that ionic strength does affect the linear dynamic range of the method. The linear dynamic range in 1 M NaCl solution is about one half of that in water (Fig. 3). Therefore the linear dynamic range of phosphate determination in a given matrix needs to be carefully examined for each step of the sequential extraction.

Comparison of the linear dynamic ranges of phosphate determination using molybdenum blue method in two different sample matrices
Because the buffer capacities of extracts are much higher than natural waters, it is essential to neutralize all bases with acid before the addition of the color reagent. The adjustment of individual sample pH was labor intensive and time consuming. The strategy we used was to modify the reagent recipe to accommodate the required acidity in a given extract for optimal color development. This is an efficient procedure for a large number of sediment samples with similar composition. However, if the amount of acid required for a particular extract is determined to be too large for the reagent to accommodate, the only option is then to neutralize the individual sample.
We have verified previous observation that phosphomolybdate blue complex formed in MgCl2 solution was not stable when standard seawater recipe for the molybdate reagent was used (Ruttenberg 1992). Therefore, the molybdate reagent used in the analysis of extract from the step 1 was modified to optimize the stability of molybdenum blue complex in high Mg solution. The optimal molybdate solution was prepared by dissolving 0.71 g ammonium molybdate in 10 ml concentrated sulfuric acid and 50 ml antimony potassium tartrate solution (3 g l−1) and diluting the final volume to 1 l with distilled water. This recipe accommodated 1 M MgCl2 extract of marine sediment and allowed the intensity and stability of the phosphomolybdenum blue complex formed in the MgCl2 solution to be comparable to that in pure water.
In step 2, the molybdate reagent was modified to handle the 0.11 M bicarbonate in the extract. The molybdate reagent was prepared by dissolving 15.38 g ammonium molybdate in 174 ml concentrated sulfuric acid and 50 ml antimony potassium tartrate solution (3 g l−1) and diluting the final volume to 1 l. The high concentration of molybdate used in this reagent is designed to match total H+ concentration, maintaining a ratio of total H+/molybdate at about 70 for optimal color development (Zhang et al. 1999a). The reagents for analysis of phosphate in the NaCl rinse solution in this step are the same as seawater analysis in which the molybdate reagent was prepared by dissolving 2.3 g ammonium molybdate in 26.9 ml concentrated sulfuric acid and 50 ml antimony potassium tartrate solution (3 g l−1) and diluting the final volume to 1 l.
In step 3, an addition of acid to individual samples is required to neutralize carbonate released from the dissolution of CaCO3 in sediments and to diminish the buffer capacity by converting acetate to acetic acid. It was found that 3 ml of 1 M HCl was needed to neutralize the base present in a 3 ml sample extract. Neutralization with sulfuric acid resulted in the formation of a CaSO4 precipitate that interfered with the absorbance measurement. After the pH adjustment, the aforementioned molybdate reagents for seawater analysis could then be used.
In step 4, addition of NaOH to individual samples is required to neutralize 1 M HCl and raise the pH to neutral. A 2.5 ml of 1 M NaOH solution were added to a 3 ml sample extract in drop-wise manner while swirling the solution to avoid any precipitate. After the pH adjustment, the aforementioned molybdate reagent for seawater analysis could then be used.
In step 5, a 3 ml of 1 M NaOH solution was added to a 3 ml sample extract to neutralize the acid in the extract, again permitting the use of the aforementioned molybdate reagent for seawater to analyze dissolved phosphate.
2.5 Determination of Total Dissolved Phosphorus in the Extract
Total dissolved P in the extract was determined by autoclave-assisted boiling temperature persulfate oxidation of the organic P, followed by the standard colorimetric determination of liberated orthorphosphate (Zhang et al. unpublished EPA method). The oxidizing reagent was prepared by dissolving 5.0 g potassium persulfate (K2S2O8) in 100 ml of 0.45 N H2SO4 solution. An aliquot of 10.0 ml of the sample was mixed with 1.0 ml of oxidizing reagent in a 100 ml capped glass tube. Samples were autoclaved for 60 min. at a pressure of 2 atm. and temperature of 120°C. Calibration standards were autoclaved and processed with the same procedures as samples. Phosphate in the digested samples was then determined using the HP 8453 spectrophotometer. Dissolved organic P in the extract was operationally defined as the difference between total dissolved P and dissolved phosphate.
2.6 Determination of Content of Iron Oxides
Reactive Fe oxides in sediments were determined as part of sequential extraction procedure (step 2). Iron oxides were reduced by BD reagent to dissolved Fe. Total dissolved iron, Fe(III) + Fe(II), in the extract solution was reduced with ascorbic acid to Fe(II). The Fe(II) was then spectrophotometrically determined with a ferrozine reagent in a pH 5.5 buffer solution at a maximum absorption wavelength of 562 nm (Zhang et al. 2001).
2.7 Determination of Dissolved Silicate
Dissolved silicate concentration in BD extracts was determined with a autoanalyzer by reacting sample silicate with ammonium molybdate in an acidic medium to form a β-molybdosilicic acid. The β-molybdosilicic acid is then reduced by ascorbic acid to form molybdenum blue. The absorbance of the molybdenum blue is measured at 660 nm. (Zhang et al. 1999b; Amornthammarong and Zhang 2009).
3 Results and Discussion
3.1 Organic Carbon and Total Nitrogen and Their Stable Isotopic Composition
Concentrations of OC and TN along with their stable isotopic composition are given in Table 2. OC and TN contents in sediments increased, in general, from the Bering Sea to the Chukchi Sea to the Beaufort Sea. Molar ratios of organic C/N in sediments also showed a general increase from the Bering Sea and the Chukchi Sea region to the Beaufort Sea, reflecting the influence of terrestrial organic carbon from the Mackenzie River (Stein and Macdonald 2004). The δ13C values ranged from −26.35 to −22.53‰, with a general increase from the Mackenzie River Delta to the Chukchi Sea and to the Bering Sea (Table 2). Similar to stable C isotope composition, the values of δ15N in sediments also increased from the Mackenzie River Delta to the Chukchi and the Bering Sea region, with the δ15N values ranging from 1.8‰ in the Mackenzie River Delta to 5.34‰ in the Bering Sea. Because terrestrial C3 plants have typical δ13C values ranging from −23 to −34‰, and marine plankton are enriched in δ13C relative to C3 terrestrial plants (−18 to −24‰), the δ13C values have been used to distinguish organic matter sources between terrestrial and marine origins (Fry and Sherr 1984; Ruttenberg and Goni 1997). Significant differences in the stable isotope composition (both δ13C and δ15N) further confirm that the terrestrial source dominates the organic matters in Mackenzie River Delta while marine source dominates those in the Bering Sea and the Chukchi Sea.
3.2 Total Sedimentary Phosphorus
Concentrations of TSP ranged from 18.2 to 35.8 μmol g−1 (Table 3) with the highest TSP concentration (35.8 μmol g−1) found at the pingo site near mud volcano on the Beaufort shelf and the lowest TSP concentration (18.2 μmol g−1) in the Chukchi Sea. Similar TSP concentration (36.13 μmol g−1) has been reported for samples taken from the Beaufort Shelf region (Ruttenberg and Goni 1997). Concentrations of TSP found in this study are similar to those reported for the suspended particulate matter (12–30 μmol g−1) in the Yukon River (Guo et al. 2004b). The abundance of TSP in Arctic sediments is generally higher than the sediments from low latitudes locations, such as the Mediterranean Sea (10 μmol g−1, Eijsink et al. 1997), the estuaries of the Gulf of Mexico coast (8–20 μmol g−1, Weng et al. 1994), Florida Bay (1–14 μmol g−1, Zhang et al. 2004), the Yellow Sea (10–20 μmol g−1, Liu et al. 2004) and the East China Sea (13–22 μmol g−1, Fang et al. 2007) in the continental shelf of the western Pacific. Lower TSP have been found in some relatively pristine environments like Davis Reef, Australia (6–11 μmol g−1, Entsch et al. 1983), Indonesia coast (6 μmol g−1, Erftemeijer and Middelburg 1993) and at shallow water carbonate sediments, such as Sharp Bay, western Australia (1 μmol g−1, Atkinson 1987). The only comparable high TSP in low latitude regions is in Tampa Bay (31–38 μmol g−1) along the west coast of central Florida where the largest phosphorite deposits in the United States are located and are currently mined and processed (Weng et al. 1994). High TSP at this location is related to the occurrence of phosphate rock deposits in the area (Cathcart et al. 1984).
3.3 Adsorbed Inorganic and Exchangeable Organic Phosphorus
Total exchangeable P concentrations extracted by MgCl2 solution from these sediments ranged from 0.28 to 1.74 μmol g−1. On average, exchangeable P accounted for 3.5 ± 1.5% of the TSP (Table 4). Further fractionation of the total exchangeable P into organic and inorganic forms indicated that the inorganic P is a dominant form. The percentages of the inorganic P in the total exchangeable P are fairly uniform among the sediments from different locations with an average of 86.2 ± 6.2%.
This exchangeable fraction is generally considered to be bioavailable through desorption process. The average 3.5% of exchangeable P in the TSP in Arctic sediments is lower than the reported values from shallow coastal regions (Koch et al. 2001; Zhang et al. 2004) but closer to oceanic region, such as offshore of Bermuda (Jensen et al. 1998). The dominance of the inorganic P in the total exchangeable P pools found in this study is in contrast to sediments in Florida Bay where organic P accounts for 60% of the total exchangeable P. The difference in the percentage of exchangeable organic P in these sediments might be related to the difference in water depth in these locations. In shallow water (average depth 1 m) of Florida Bay, surface sediments are within the depth of euphotic zone and the benthic productivity is a significant portion of total community production. Benthic algae might contribute significantly to the accumulation of the exchangeable organic P in the surface sediments. In contrast, sediments in deeper waters in Arctic coast (Table 1) might lie below the photic zone, supporting low benthic productivity and thus accumulating little exchangeable organic P.
3.4 Iron oxides and Fe-Bound Phosphorus
Because iron is ubiquitous in aquatic environments, coastal sediments are usually coated with iron hydroxides. Among the various types of natural particles, amorphous iron oxyhydroxide and iron oxides have the strongest sorption capacity for phosphate (Berner 1973; Hingston et al. 1974; Khalid et al. 1977). As a result, the iron oxides content in sediments is an important factor in regulating the P retention capacity of sediments (Zhang and Huang 2007).
In these Arctic sediments, a significant fraction (19.9 ± 5.2%, Table 4) of TSP is bound to the iron oxides. The Fe-bound P ranged from 1.79 to 9.88 μmol g−1. The lowest concentration (1.79 μmol g−1) was found in a deeper sediment layer (≥10 m) of the offshore station (PC1) in the Mackenzie River Delta and is likely due to the mobilization of P as a result of iron oxides reduction as the aged sediment undergone diagenesis. It is noticed that a decrease in surface reactive iron and Fe-bound P with sediment depth was not observed in the near-shore stations PC2 and PC3 (Table 5). These between-station differences are likely a result of a greater sedimentation rate at the near-shore stations PC1 and PC2 than that at the offshore station PC3 (see the station location in Fig. 1 and their water depth in Table 1). It may require deeper sediment core samples at PC1 and PC2 to detect the effect of sediment diagenesis on P transformation in such coastal locations with higher sedimentation rates. Relative low Fe-bound P was found in the Bering Sea and the Chukchi Seas in comparison with those from the Mackenzie River Delta. The highest concentrations of Fe-bound P (9.88 μmol g−1) occurred in sediments from the pingo site with a shallow water depth (38–65 m) in the Mackenzie River Delta.
Surface reactive Fe derived from the reduction of ferric oxides ranged from 14.4 to 137.5 μmol g−1 (Table 5). The lowest concentrations were found in the Bering Sea sediments (14.4 and 17.2 μmol g−1). The concentration increased to 39.5 μmol g−1 in the Chukchi Sea sediments. Much higher concentrations of surface reactive Fe were found in the Mackenzie River Delta sediments (over 100 μmol g−1). The highest Fe concentration of 137.5 μmol g−1 was found at the pingo station. The high abundance of surface reactive Fe in the Mackenzie River Delta sediments demonstrates the importance of riverine runoff as a major pathway that transports the continental Fe, mainly in particulate and colloidal forms, into the Arctic shelf. At locations remote from major riverine influence, such as the stations in the Bering Sea and the Chukchi Sea, the sediments usually contain less Fe. Overall, the surface reactive Fe content in the Arctic sediments is much higher than the other coastal regions. For example, the surface reactive Fe content in the Mackenzie River Delta sediments is approximately 24 times higher than the maximum surface reactive Fe found in the eastern Florida Bay (Zhang et al. 2004). Low Fe content in Florida Bay sediments is a result of combined effects of low Fe content in sedimentary carbonate rocks in Florida Bay watersheds (as compared to igneous rocks in the Arctic regions) and relatively small freshwater discharge via manmade canals in south Florida (as compared to the Mackenzie River in the Arctic).
The Fe to P ratio in this oxide fraction varied over an order of magnitude (from 4.1 to 40.2, Table 5). The lowest ratios were found in the Bering Sea (4.1 and 5.5). The ratio increased to 8.2 in the Chukchi Sea. Most sediment samples from the Mackenzie River Delta have a higher Fe/P ratio ranging between 14 and 25. These ratios are similar to those (8–15) found in clastic surface sediments with similar Fe concentrations (Jensen and Thamdrup 1993). The only exception is the deepest sediment sample from the offshore station PC1 that contains the lowest Fe content and has a Fe/P ratio of up to 40.2, apparently due to the sediment diagenesis.
A significant correlation was observed between the surface reactive Fe and leachable silicate concentrations in the dithionite extract solution (Table 5; Fig. 4) for all sediment samples:
with a correlation coefficient of 0.738. This suggests that reactive Fe was closely associated with silicate on the sediment surface. The Fe/Si molar ratios in this surface iron oxide phase varied from 8.4 to 23.4 with an average of 15.4 ± 4.5. It is known that dissolved silicate in natural waters can be incorporated into iron oxides during the formation of ferric minerals. It has been shown that dissolved silicate can increase the stability of colloidal iron and promote the formation of ferrihydrite over other Fe(III) minerals during the oxidation of Fe(II) (Mayer and Jarrell 1996, 2000). Laboratory experiments demonstrated that dissolved silicate absorbed on iron oxides, and effectively competed with P for surface sites at ambient concentrations (Konhauser et al. 2007). The content of silicon in the iron oxide phases, therefore, has a significant effect on the P sorption capacity of the iron minerals and might influence the Fe/P ratios observed in these sediments.

Correlation of iron with silicate concentration in the dithionite extract solution from all sediment samples
3.5 Authigenic Carbonate Fluorapatite, Biogenic Apatite and CaCO3-Bound Inorganic and Organic Phosphorus
Carbonate minerals are known to strongly adsorb phosphate from ambient seawaters (Millero et al. 2001). The concentration of authigenic carbonate fluorapatite, biogenic apatite and CaCO3 associated P ranged from 2.17 to 10.7 μmol g−1. High CaCO3-bound P (10.7 μmol g−1) was found at the pingo site in the Mackenzie River Delta. Low concentrations (~2 μmol g−1) were found in the Bering Sea and the Chukchi Sea (Table 3). In the Bering Sea and the Mackenzie River Delta, the fractions of CaCO3-bound P are similar to their Fe-bound P (13% for the Bering Sea and 25% for the Mackenzie River Delta, Table 4) but in the Chukchi Sea the CaCO3-bound P (11.9% of TSP) accounts less than a half of Fe-bound P (26.5%, Table 4). Averaged over all samples, CaCO3-bound P represents the second largest pool (23.8% of TSP). Results from further fractionation into inorganic and organic P indicate that the inorganic P is a dominant form in this authigenic fraction, representing 90.2 ± 7.5% of CaCO3-bound P.
This is in contrast to the tropical carbonate sediments such as those in Florida Bay, where CaCO3-bound P represents the largest pool of TSP. In Florida Bay, the percentage of organic P relative to total P in this pool can be as high as 40%. High percentages of organic P in this fraction were found to be associated with higher benthic and water column productivity in the western region of Florida Bay (Zhang et al. 2004).
3.6 Detrital Apatite Phosphorus
Detrital apatite P of igneous or metamorphic origin is the largest fraction (42.5 ± 11.6%) of TSP in these sediments, with concentrations ranging from 6.55 to 13.09 μmol g−1. Similar concentrations of detrital apatite (about 10 μmol g−1) were found in the surface sediments of the Bering Sea, the Chukchi Sea and the Mackenzie River Delta. The high abundance of apatite in the Arctic coastal sediment is similar to that observed in the Yellow Sea (50% of TSP, Liu et al. 2004) and the East China Sea (70% of TSP, Fang et al. 2007) in the continental shelf of the western Pacific. The high percentages of the detrital apatite in the continental shelf of the western Pacific are likely derived from deposition of loess originated from Gobi Desert in China and transported by the Yellow and the Yangtze rivers to the Yellow Sea and the East China Sea, respectively. It has been found that the detrital apatite accounts for 64% of TSP in the Yangtze River sediments (Rao and Berner 1997) and approximately 90% of TSP in loess from China (Liu et al. 2004). However, this is a sharp contrast to the low apatite content in the suspended particles and sediments from the Amazon River where detrital apatite accounts for only about 15% of TSP in the river sediments and decreases to 6% in the estuary sediments (Berner and Rao 1994). Similarly, very low detrital apatite content was measured in Florida Bay sediments, ranging from 0.04 to 1.45 μmol g−1, representing only 4.6 ± 2.2% of TSP (Zhang et al. 2004). Despite the low apatite content in Florida Bay, however, the sharp spatial gradient toward the northwest clearly indicates that the detrital apatite is largely derived from the P mining district in the central Florida and transported by the coast current along the southwest Florida shelf (Zhang et al. 2004).
Similar to the loess origin of the high apatite content in the Yellow Sea and the East China Sea, the abundant apatite in the Arctic sediments suggests a P-rich source terrane in the Mackenzie River watersheds and adjacent regions. However, it might also imply a different extent of biogeochemical cycle of P-bearing minerals in high latitude regions. Cold climate, less extensive vegetation and shorter growing season may result in less uptake of P by living organisms and thus less extensive transformation of inorganic P to organic P in the Arctic environment. Furthermore, suspended particles transported by rivers to estuarine and coastal waters likely undergo less extensive chemical weathering at a low annual temperature in the Arctic region (Ruttenberg and Goni 1997), resulting in higher concentrations of P and Si in particulate phase but lower in dissolved forms (e.g., Guo et al. 2004a).
3.7 Refractory Organic Phosphorus
After HCl extraction of all inorganic P, any P in the remaining sediment residue is defined here as refractory organic P. The refractory organic P content ranged from 1.26 to 5.79 μmol g−1, with lower concentrations (~1 μmol g−1) in the Bering Sea and the Chukchi Sea, moderate concentrations (2–3 μmol g−1) in the Mackenzie River Delta region, and a maximum concentration (5.79 μmol g−1) at the pingo station in the Mackenzie River Delta. Despite contrasting environments, the contents of sedimentary refractory organic P in Arctic coasts are similar to those found in subtropical Florida Bay where it ranged from 0.30 to 3.48 μmol g−1. The positive correlation between the concentration of refractory organic P and the organic carbon content in Florida Bay sediments has been observed and much of the refractory organic P in Florida Bay sediment is derived from in situ biological production, dominated by sea grass and benthic algae (Zhang et al. 2004). The highest refractory organic P and low detrital apatite found at the pingo site suggests a highly productive ecosystem at this station that transformed the detrital apatite to the refractory organic P.
Despite the similar concentrations of refractory organic P concentrations in the coastal Arctic and Florida Bay sediments, its contribution to the TSP in Arctic sediments (10.3 ± 3.0%) is less than a half of that in Florida Bay sediments (24.3 ± 4.3%). Lower percentage of the refractory organic P in the TSP pool in the Arctic sediments is largely due to the high TSP content (mainly as detrital apatite) in the Arctic region.
3.8 Variations in Sedimentary N/P Ratios
As shown in Table 2, the molar ratio of TN/TP ranged from 1.4 to 3.4, with an average of 2.7 ± 0.7. These TN/TP ratios are considerably lower than the Redfield Ratio of 16 (Redfield 1958), indicating the relative high abundance of P in Arctic sediments and non-biogenic in nature. Because of the dominance of inorganic P in TSP pools, the TN/TOP molar ratio become much higher, ranging from 13.4 to 34.3 with an average of 20.8 ± 5.7 (Table 2), and closed to the Redfield Ratio. However, the TN/TOP ratios of surface sediments in the Bering Sea and the Chukchi Sea (14.2–17.5) were lower than those in the surface sediments of the Mackenzie River Delta (17.6–34.3). Higher TN/TOP values in the Mackenzie River Delta are likely resulted from the general P-limitation in the Mackenzie River and its delta regions, consistent with previous reports on P-limitation in the estuarine and coastal region in the Beaufort Sea (e.g., Carmack et al. 2004) and general low dissolved inorganic P in the northern rivers (Guo et al. 2004a).
4 Conclusions
Averaged over all sediment samples collected in the coastal Arctic Ocean in this study, the detrital apatite P of igneous or metamorphic origin is the most abundant among five pools of sedimentary P representing 42.5 ± 11.6% of the TSP (Fig. 5). The authigenic carbonate fluorapatite, and biogenic apatite as well as CaCO3 associated P is the second largest pool (23.8 ± 9.8% of the TSP), followed by the Fe-bound P pool (19.9 ± 5.2% of the TSP). The refractory organic P constitutes 10.3 ± 3.0% of the TSP, and the exchangeable P is the smallest pool with 3.5 ± 1.5% of the TSP. Inorganic P outweighs all P pools, accounting for, on average, 87% of the TSP. Relatively higher sedimentary OC and TN contents and lower δ13C values in the Mackenzie River Delta together with the dominance of detrital apatite in the TSP in the Arctic sediments demonstrate the importance of continental inputs in governing the abundance and speciation of sedimentary P in the Arctic coastal sediments. The relative high refractory organic P in the Mackenzie River Delta, particularly at the pingo site, demonstrates the significance of biological productivity in transforming inorganic P to organic P and its accumulation in the sediments.

a Absolute concentrations and b relative partitioning of five forms of sedimentary phosphorus in the Arctic sediments. The suffixes a, b, c in the sample names denote the subsamples taken at the top, the middle and the bottom layers of a sediment core
References
Amornthammarong N, Zhang J-Z (2009) Liquid—waveguild spectrophotometric measurement of low silicate in natural waters. Talanta 79:621–626
Atkinson MJ (1987) Low phosphorus sediments in a hypersaline marine bay. Estuar Coast Shelf Sci 24:335–347
Berner RA (1973) Phosphate removal from sea water by adsorption on volcanogenic ferric oxides. Earth Planet Sci Lett 18:77–86
Berner RA, Rao JL (1994) Phosphorus in sediments of the Amazon River and estuary: implications for the global flux of phosphorus to the sea. Geochim Cosmochim Acta 58:2333–2339
Broecker WS, Peng TH (1982) Tracers in the sea. Eldigio Press, Lamont Doherty Geological Observatory, New York
Carmack EC, Macdonald RW, Jasper S (2004) Phytoplankton productivity on the Canadian Shelf of the Beaufort Sea. Mar Ecol Prog Ser 277:37–50
Cathcart JB, Sheldon RP, Gulbrandsen RA (1984) Phosphate rock resources of the US. United States Geological Survey, Circular 888
Cauwet G, Sidorov I (1996) The biogeochemistry of Lena River: organic carbon and nutrients distribution. Mar Chem 53:211–227
Conley DJ, Smith WM, Cornwell JC, Fisher TR (1995) Transformation of particle-bound phosphorus at the land–sea interface. Estuar Coast Shelf Sci 40:161–176
Eijsink LM, Krom MD, de Lange GJ (1997) The use of sequential extraction techniques for sedimentary phosphorus in eastern Mediterranean sediments. Mar Geol 139:147–155
Entsch B, Boto KG, Sim RG, Willington JT (1983) Phosphorus and nitrogen in coral reef sediments. Limnol Ocenogr 28:465–476
Erftemeijer PL, Middelburg JJ (1993) Sediment-nutrient interactions in tropical seagrass beds: a comparison between a terrigenous and a carbonate sedimentary environment in South Sulawesi (Indonesia). Mar Ecol Prog Ser 102:187–198
Fang TH, Chen JL, Huh CA (2007) Sedimentary phosphorus species and sedimentation flux in the East China Sea. Cont Shelf Res 27:1465–1476
Follmi KB (1996) The phosphorus cycle, phosphogenesis and marine phosphate-rich deposits. Earth-Sci Rev 40:55–124
Froelich PN (1988) Kinetic control of dissolved phosphate in natural rivers and estuaries: a primer on the phosphate buffer mechanism. Limnol Oceanogr 33:649–668
Froelich PN, Bender ML, Luedtke NA, Heath GB, DeVries T (1982) The marine phosphorus cycle. Am J Sci 282:474–511
Fry B, Sherr EB (1984) δ13C measurements as indicators of carbon flow in marine and freshwater ecosystems. Contrib Mar Sci 27:13–47
Gibbs RJ (1981) Sites of river-derived sedimentation in the ocean. Geology 9:77–80
Griffith EJ, Beeton AM, Spencer JM, Michell DJ (1973) Environmental phosphorus handbook. Wiley, New York
Guo L, Tanaka N, Schell DM, Santschi PH (2003) Nitrogen and carbon isotopic composition of high-molecular-weight dissolved organic matter in marine environments. Mar Ecol Prog Ser 252:51–60
Guo L, Zhang J-Z, Guéguen C (2004a) Speciation and fluxes of nutrients (N, P, Si) from the upper Yukon River. Global Biogeochem Cycles 18(1):GB1038. doi:10.1029/2003GB2152
Guo L, Semiletov I, Gustafsson O, Ingri J, Andersson P, Dudarev O, White D (2004b) Characterization of Siberian Arctic estuarine sediments: implications for terrestrial organic carbon export. Global Biogeochem Cycles 18(1):GB1036. doi:10.1029/2003GB002087
Hieltjes AHM, Lijldema L (1980) Fractionation of inorganic phosphates in calcareous sediments. J Environ Qual 9:405–407
Hingston FJ, Posner AM, Quirk JP (1974) Anion adsorption by goethite and gibbsite. 2. desorption of anions from hydrous oxide surfaces. J Soil Sci 25:16–26
Howarth RW, Jensen HS, Marino R, Postma H (1995) Transport to and processing of P in nearshore and oceanic waters. In: Tiessen H (ed) Phosphorus in the global environment. Wiley, New York, pp 323–328
Jensen HS, Thamdrup B (1993) Iron-bound phosphorus in marine sediments as measured by bicarbonte-dithionite extraction. Hydrobiologia 253:47–59
Jensen HS, McGlathery KJ, Marino R, Howarth RW (1998) Forms and availability of sediment phosphorus in carbonate sand of Bermuda seagrass beds. Limnol Oceanogr 43:799–810
Khalid RA, Patrick WH, Delaune RD (1977) Phosphorus sorption characteristics of flooded soils. Soil Sci Soc Am J 41:305–310
Koch MS, Benz RE, Rudnick DT (2001) Solid-phase phosphorus pools in highly organic carbonate sediments of Northeastern Florida Bay. Estuar Coast Shelf Sci 52:279–291
Konhauser KO, Lalonde SV, Amskold L, Holland HD (2007) Was there really an Archean phosphate crisis? Science 315:1234
Kramer JR (1964) Sea water: saturation with apatites and carbonates. Science 146:637–638
Krom MD, Kress N, Brenner S, Gordon LI (1991) Phosphorus limitation of primary productivity in the eastern Mediterranean Sea. Limnol Oceanogr 36:424–432
Lebo ME (1991) Particle-bound phosphorus along an urbanized coastal plain estuary. Mar Chem 34:225–246
Lebo ME, Sharp JH (1992) Modeling phosphorus cycling in a well mixed coastal plain estuary. Estuar Coast Shelf Sci 35:235–252
Liu SM, Zhang J, Dao JL (2004) Phosphorus cycling in sediments of the Bohai and Yellow Seas. Estuar Coast Shelf Sci 59:209–218
Mayer TD, Jarrell WM (1996) Formation and stability of iron (II) oxidation products under the natural concentrations of dissolved silica. Water Res 30:1208–1214
Mayer TD, Jarrell WM (2000) Phosphorus sorption during iron (II) oxidation in the presence of dissolved silica. Water Res 34:3949–3956
Meade RH (1982) Sources, sinks, and storage of river sediment in the Atlantic drainage of the United States. J Geol 90:235–252
Meybeck M (1982) Carbon, nitrogen and phosphorus transport by world rivers. Am J Sci 282:401–450
Meybeck M (1993) C, N, P and S in rivers: from sources to global inputs. In: Wollast R, Mackenzie FT, Chou L (eds) Interaction of C, N, P and S biogeochemical cycles and global change. Springger-Verlag, Berlin, pp 163–193
Millero FJ, Zhang J-Z, Fiol S, Sotolongo S, Roy RN, Lee K, Mane S (1993) The use of buffers to measure the pH of seawater. Mar Chem 44:143–152
Millero FJ, Huang F, Zhu X, Liu X, Zhang J-Z (2001) Adsorption and desorption of phosphate on the calcite and aragonite in seawater. Aquat Geochem 7(1):33–56
Rao JL, Berner RA (1997) Time variations of phosphorus and sources of sediments beneath the Chang Jiang (Yangtze River). Mar Geol 139:95–108
Redfield AC (1958) The biological control of chemical factors in the environment. Am Sci 46:561–600
Ruttenberg KC (1992) Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnol Oceanogr 37:1460–1482
Ruttenberg KC, Goni MA (1997) Phosphorus distribution, C:N:P ratios, and δ13Coc in arctic, temperate, and tropical coastal sediments: tools for characterizing bulk sedimentary organic matter. Mar Geol 139:123–145
Ryther JH, Dunstan WM (1971) Nitrogen, phosphorus, and eutrophication in the coastal marine environment. Science 171:1008–1013
Schindler DW (1977) Evolution of phosphorus limitation in lakes. Science 195:260–262
Smith SV (1984) Phosphorus versus nitrogen limitation in the marine-environment. Limnol Oceanogr 29:1149–1160
Smith SV, Atkinson MJ (1984) Phosphorus limitation of net production in a confined aquatic ecosystem. Nature 307:626–627
Solorzano C, Sharp J (1980) Determination of total dissolved phosphorus and particulate phosphorus in natural waters. Limnol Oceanogr 25:754–758
Stein R, Macdonald RW (2004) The organic carbon cycle in the Arctic Ocean. New York, Springer
Weng H, Presley BJ, Armstrong D (1994) Distribution of sedimentary phosphorus in Gulf of Mexico Estuaries. Mar Environ Res 37:375–392
Westheimer FH (1987) Why nature chose phosphates. Science 235:1173–1178
Zhang J-Z (2000) The use of pH and buffer intensity to quantify the carbon cycle in the ocean. Mar Chem 70(1–3):121–131
Zhang J-Z, Chi J (2002) Automated analysis of nanomolar concentrations of phosphate in natural waters with liquid waveguide. Environ Sci Technol 36:1048–1053
Zhang J-Z, Huang X-L (2007) Relative importance of solid—phase phosphorus and iron on the sorption behavior of sediments. Environ Sci Technol 41(8):2789–2795. doi:10.1021/es061836q
Zhang J-Z, Fischer C, Ortner PB (1999a) Optimization of performance and minimization of silicate interference in continuous flow phosphate analysis. Talanta 49:293–304
Zhang J-Z, Fischer C, Ortner PB (1999b) Dissolution of silicate from glassware as a contaminant in silicate analysis of natural water samples. Water Res 33:2879–2883
Zhang J-Z, Kelble C, Millero FJ (2001) Gas-segmented continuous flow analysis of iron in water with a long liquid waveguide capillary flow cell. Anal Chim Acta 438:49–57
Zhang J-Z, Fischer CJ, Ortner PB (2004) Potential availability of sedimentary phosphorus to sediment resuspension in Florida Bay. Global Biogeochem Cycles 18(1):GB1038. doi:10.1029/2004GB002255
Acknowledgments
The authors would like to thank Chief Scientists, crew members, Marine Works Japan, and the science party of the R/V Mirai for their assistance in sediment core collection during the MR01-K04 and MR02-K05 cruises to the Bering Sea and the western Arctic Ocean, and Tomoyuki Tanaka and Mindy Juliana for sample processing. This study was supported in part by the JAMSTEC/Japan, the International Arctic Research Center, NSF (#0436179), the University of Southern Mississippi, and NOAA’s Climate Change Program and Coastal Ocean Program. The statements, findings, conclusions, and recommendations are those of the authors and do not necessarily reflect the views of the NOAA or the U.S. Department of Commerce.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhang, JZ., Guo, L. & Fischer, C.J. Abundance and Chemical Speciation of Phosphorus in Sediments of the Mackenzie River Delta, the Chukchi Sea and the Bering Sea: Importance of Detrital Apatite. Aquat Geochem 16, 353–371 (2010). https://doi.org/10.1007/s10498-009-9081-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10498-009-9081-4