
Abstract
Ghrelin was first identified as an endogenous ligand of the growth hormone secretagogue receptor (GHSR) in 1999, with the function of stimulating the release of growth hormone (GH), while nesfatin-1 was identified in 2006. Both peptides are secreted by the same kind of endocrine cells, X/A-like cells in the stomach. Compared with ghrelin, nesfatin-1 exerts opposite effects on energy metabolism, glucose metabolism, gastrointestinal functions and regulation of blood pressure, but exerts similar effects on anti-inflammation and neuroprotection. Up to now, nesfatin-1 remains as an orphan ligand because its receptor has not been identified. Several studies have shown the effects of nesfatin-1 are dependent on the receptor of ghrelin. We herein compare the effects of nesfatin-1 and ghrelin in several aspects and explore the possibility of their interactions.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ghrelin and nesfatin-1 are both produced by the same kind of cells, X/A-like cells in the gastric fundus. Ghrelin, an orexigenic peptide, was first discovered to stimulate the release of growth hormone (GH) by Kojima in 1999 [1], while nesfatin-1 was identified as an anorexigenic peptide by Oh-I in 2006 [2]. Although nesfatin-1 and ghrelin exhibit opposite effects on some physiological functions, such as energy metabolism, glucose metabolism, gastrointestinal functions and regulation of blood pressure, they have similar effects in some aspects as for anti-inflammation and neuroprotection. The receptor for nesfatin-1 is still not identified; however, emerging evidences suggest that nesfatin-1 could exert its effects through the ghrelin receptor, growth hormone secretagogue receptor (GHSR) [3, 4]. Co-incubation or pre-incubation with nesfatin-1 or nesfatin-1-like peptide could block the stimulatory effects of ghrelin on GH mRNA and protein expression possibly through the same adenylate cyclase (AC)/protein kinase A (PKA)/cAMP-response element-binding protein (CREB) signaling pathway [3]. Our previous study also showed clearly that GHSR was essential for the effects of nesfatin-1 on regulating glucose metabolism [4]. Therefore, there might be several mechanisms underlying the effects of nesfatin-1, for example, direct effects on GHSR or modulation GHSR function by various pathways, which is of vital importance in the normal physiological functions. We herein compared the effects of nesfatin-1 and ghrelin in several aspects and gave some clues for their possible interactions.
Structure and distribution
Nesfatin-1 is derived from the 396-amino acid precursor, namely nucleobindin 2 (NUCB2) through the post-translational processing [2]. It gains almost all the attentions due to the absence of observed effects of the other two products of NUCB2, that is, nesfatin-2 and nesfatin-3 (Fig. 1). NUCB2/nesfatin-1 is detected in both peripheral tissues and the central nervous system (CNS). In the periphery, nesfatin-1 is mainly secreted by the gastric oxyntic mucosa, which is the main source for the majority of circulating nesfatin-1 [5]. It is also present in the liver, heart, testis, ovaries, uterus, adipose tissue and pancreas [5]. Nesfatin-1 could penetrate the blood–brain barrier (BBB) in a bidirectional pathway without saturation [6]. In the CNS, NUCB2/nesfatin-1 is mainly found in the brain stem, midbrain, hypothalamus, central amygdaloid nucleus, cerebellum, etc. [7]. It is also expressed in the autonomic preganglionic neuronal group in the spinal cord [8].

The production of nesfatin-1 and ghrelin in gastric X/A-like cell. The structure of proghrelin contains both obestatin and ghrelin which is transformed to active peptide after acylation by GOAT. NUCB2 yields three kinds of peptides, nesfatin-1, nesfatin-2, and nesfatin-3. It is known majority of nesfatin-1 and ghrelin co-exist in X/A-like cells of oxyntic gland in stomach, where the PC 1/3 is responsible for the processing of both peptides
Ghrelin is a 28-amino acid brain-gut peptide, derived from its precursor, proghrelin which also yield another peptide, obestatin, with post-translational modification [9]. Ghrelin exists in two forms: unacylated ghrelin and acylated ghrelin whose acylation is catalyzed by ghrelin O-acyltransferase (GOAT) (Fig. 1) [10]. In accordance with nesfatin-1, ghrelin is also mainly secreted by the gastric oxyntic mucosa, which is the main source for the majority of circulating ghrelin. It is also present in other sites of gastrointestinal tract, such as duodenum, jejunum, ileum and colon. Lower expression of ghrelin could be found in the pancreas, adipose tissue, kidneys, testes, placenta and hypophysis. Central source of ghrelin remains questionable and there appears no irrefutable evidence supporting that ghrelin is synthesized in the CNS [11, 12]. Ghrelin signals might be sent to the CNS through the vagal afferent nerve and the blood circulation, but the accessibility of plasma ghrelin to the CNS is limited [13]. Circulating ghrelin might access neurons through fenestrated capillaries of the median eminence (ME) and the area postrema (AP) with incomplete BBB [11, 14]. A recent study reported that circulating ghrelin might cross the blood-cerebrospinal fluid (CSF) barrier mainly through GHSR-dependent pathway [15].
Nesfatin-1 and ghrelin co-exist in X/A-like cells of oxyntic gland in the stomach, where the prohormone convertase (PC) 1/3 is responsible for the processing of both peptides (Fig. 1) [16, 17]. The co-localization was also observed in the pancreatic islets of rats [18] and hypothalamus of goldfish [19]. Besides, the study in pejerrey showed that nesfatin-1 co-localized with ghrelin in enteroendocrine cells, absorptive cells and lamina propria cells of intestine [20]. By comparing with ghrelin, whether the central nesfatin-1 might also at least partly originate from the periphery, is worthy of further investigation.
Receptors and interactions
Up to now, nesfatin-1 remains an orphan ligand because its own receptor has not been identified. The autoradiography showed that the receptors of nesfatin-1 were distributed in the gastrointestinal tract and endocrine tissues, such as the pituitary, pancreas, adrenal gland, testis and adipose tissue, and possibly in some viscera including heart, lung, liver, kidney, and skeletal muscle [21]. In the brain, nesfatin-1 receptors are distributed in the dorsal vagal complex (DVC) of brainstem, hypothalamic paraventricular nucleus (PVN), cerebellum, and cortex (Table 1) [21].
The unknown receptor of nesfatin-1 is presumed to be a kind of G-protein-coupled receptor (GPCR), which is involved in a variety of signaling pathways [22]. Nesfatin-1 could induce the phosphorylation of CREB through the mitogen-activated protein kinase (MAPK) and Ca2+ signaling pathway in neural cell lines [23]. In interleukin (IL)-1β-treated chondrocytes, nesfatin-1 could attenuate inflammation and matrix metalloproteinases expression through the inhibition of the NF-κB and MAPK signaling pathways [24]. Knockdown of hypothalamic nesfatin-1 reduced the phosphorylation of mammalian target of rapamycin (mTOR), as well as signal transducer and activator of transcription 3 (STAT3), and thus increased the expression of phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6pase) to regulate glucose metabolism [25]. In trophoblast cells, nesfatin-1 promoted proliferation, migration, invasion and suppressed oxidative stress by activating phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mTOR and AKT/glycogen synthase kinase 3β (GSK3β) signaling pathways [26]. In type 2 diabetes mellitus (T2DM) mice, nesfatin-1 stimulated free fatty acid utilization through AMP-activated protein kinase (AMPK)-acetyl-CoA carboxylase (ACC) signaling pathway in skeletal muscle cells (Fig. 2A) [27]. Meanwhile, nesfatin-1 promoted anorexia effects through receptors of other hormone, such as corticotropin-releasing 1/2 receptor (CRF 1/2-R) and histamine 1/3 receptors (H1/3-R) [28]. The weight loss effects in obese rats were mediated by elevated nesfatin-1 levels via melanocortin receptors (MC-Rs)-extracellular signal-regulated kinase (ERK) signaling pathway [29]. In adult ventricular myocytes, nesfatin-1 modulated L-type Ca2+ channel through MC4-R resulting in negative inotropic effects [30]. Central administration of oxytocin receptor (Oxy-R) blocker antagonized hypertension and anorexia induced by nesfatin-1 [31, 32], suggesting the involvement of oxytocin system in nesfatin-1’s effects. It was also reported that cholecystokinin receptor (CCK-R) was involved in the effects of nesfatin-1, since CCK receptor selective antagonist reversed the anorectic effect and appetite-related factor expressions induced by nesfatin-1 (Fig. 2B) [33].

The receptor signaling pathways of nesfatin-1 and ghrelin and interactions with other receptors. A The receptor of nesfatin-1 remains unknown and it is supposed to be a G-protein coupled receptor. Nesfatin-1 and ghrelin regulate food intake in the DVC and synthesis of GH through the same signaling pathway with completely opposite effects. B The actions of nesfatin-1 are dependent on CRF1/2-R, H1/3-R, CCK-R, MC-R and Oxy-R, while GHSR heterodimerize with MC-R, Oxy-R, D1/2-R, 5-HT2C-R and SST5. Both peptides are correlated with MC-R and Oxy-R
GHSR, the ghrelin receptor, was found as an orphan receptor in 1996, prior to the discovery of ghrelin. The gene of ghrelin receptor encodes two kinds of receptor, GHSR 1a and 1b, thereinto, GHSR1a is the effective receptor of ghrelin. GHSR1a is a kind of GPCR, which is distributed in both peripheral tissue and the CNS. GHSR1a mRNA exists in peripheral tissue including the pituitary gland, pancreas, adipose tissue, thyroid gland, spleen, kidney, adrenal gland and cardiac muscles [34], and in the CNS including the hindbrain, midbrain, hypothalamus, and hippocampus (Table 1) [14, 35].
Various signaling pathways are involved in the effects of ghrelin, including PKA, Ca2+, MAPK, mTOR, AMPK, and PI3K/AKT [36]. After binding to GHSR1a, ghrelin activated the subsequent G-protein, Gq or Gi/o, then simulated phospholipase C (PLC)-inositol 1,4,5-triphosphate (IP3)-Ca2+-calmodulin-dependent protein kinase II (CaMKII)/AMPK or cAMP-PKA signaling pathway, respectively [37, 38]. It could prevent apoptosis in cortical neuronal cell through the PI3K/AKT-mediated inactivation of GSK-3β pathway [39]. It could inhibit lipogenesis and stimulated fatty acid oxidation via sirtuin1 (Sirt1)/p53/AMPK signaling pathways in the hypothalamus [40]. It promoted cell proliferation and decreased cell apoptosis via MAPK dependent pathway (Fig. 2A) [41]. The seeming paradox that wide distribution of GHSR1a is accompanied by the low expression of ghrelin in the CNS could be explained by the constitutive activity of GHSR and the formation of heterocomplexes. Actually, even in the absence of ghrelin, GHSR1a could activate intracellular signal transduction and display a strikingly strong constitutive activity which accounted for approximately 50% of its maximal capacity [42]. Notably, GHSR1a heterodimerized with a variety of receptors, including dopamine receptor (D-R), MC3-R, 5-serotonin 2c receptor (5-HT2c-R), orexin 1 receptor (OX1-R), Oxy-R, somatostatin receptor 5 (SST5). Bioluminescence resonance energy transfer (BRET) showed the existence of heterodimers formed by D1-R and GHSR1a, which amplified the dopamine (DA) signaling in the presence of ghrelin [12, 43]. The similar effects were observed in the dimerization between D2-R and GHSR1a in hypothalamic neurons [44]. The heteromerization influenced DA-mediated Gi protein activation by modulating the conformation of its α-subunit [45]. The inhibitory effects of D2-R agonist on food intake were dependent on the formation of D2-R and GHSR1a heteromers [46]. The receptors of ghrelin and melanocortin (an anorexigenic peptide) could form MC3-R-GHSR1a dimers identified in the hypothalamic arcuate nucleus (ARC) to enhance melanocortin-induced intracellular cAMP accumulation and impair GHSR signaling of both agonist-independent and ghrelin-induced activity [47]. The formation of 5-HT2C-R-GHSR1a heterodimers inhibited orexigenic signaling whereas 5-HT2c-R antagonist attenuated the formation of dimer and increased GHSR1a-induced food intake [48, 49]. Orexin is another kind of orexigenic peptide, whose receptor OX1-R could form heterodimers with GHSR1a in HEK293 cells. Ghrelin, but not orexin, could activate Gα protein through GHSR1a/OX1-R heterodimers [50]. Oxytocin is a pleiotropic peptide hormone with broad functions, and its receptor, Oxy-R, could also form heterocomplex with GHSR in cultured hypothalamus and hippocampus to impair the downstream Oxy-R signaling pathways [51]. The heteromers of GHSR1a and somatostatin receptor 5 (SST5) could regulate insulin secretion (Fig. 2B) [52].
In summary, nesfatin-1 receptor shares the overlapping distribution and similar intracellular signaling transduction with ghrelin receptor and interacts with analogous receptors (Fig. 2). Several studies simultaneously focused on the interaction of nesfatin-1 and GHSR. Ozturk et al. [53] observed that nesfatin-1 displayed anti-inflammatory and antioxidant effects on colitis via Oxy-Rs and ghrelin receptors. Kerbel et al. [20] demonstrated exogenous nesfatin-1 altered the central expression of ghrelin and GHSR1a in goldfish. Our previous study clearly stated that GHSRs were essential for nesfatin-1 to control glucose metabolism because GHSR antagonist and knockout of ghrelin receptor blocked the effect of peripheral nesfatin-1 on food intake and glucose metabolism [4]. For GH synthesis, nesfatin-1 and ghrelin exerted completely opposite effect via similar signaling pathway, cAMP/PKA/CREB signaling pathway [3]. Even co-incubation or pre-incubation with nesfatin-1 antagonized the stimulatory effects of ghrelin on GH secretion. Therefore, nesfatin-1 might act on GHSRs indirectly, nevertheless, there exits such a possibility that nesfatin-1 could act on GHSRs directly, which deserve further investigations.
The roles of nesfatin-1 and ghrelin in the central functions
Energy metabolism
Plasma nesfatin-1 levels were increased with elevated body mass index (BMI) [54], and a negative correlation between plasma ghrelin concentration and BMI was observed in humans [55]. Nesfatin-1 decreased energy intake and potentiated energy consumption, whereas ghrelin displayed opposite effects. The mechanisms underlying their effects on energy metabolism were discussed hereinafter from lower center to higher center (Fig. 3) [56].

The central mechanisms underlying the effects of nesfatin-1 and ghrelin on energy metabolism. In the hypothalamus, nesfatin-1 inhibits NYP/AgRP neurons and activates Oxy neurons in the PVN which terminates POMC neurons in the DVC. Ghrelin activates NYP/AgRP neurons in the ARC and Ore neurons in the LHA, while inhibits Oxy neurons in the PVN and AMPK activity in the VMH. In the midbrain, nesfatin-1 inhibits DAergic neurons, thereby decreasing DA release in the NAc, whereas ghrelin exerts completely opposite effects. In the LPBN of pons, nesfatin-1 inhibits and excites GE and GI neurons, respectively, while ghrelin reverses these effects. In the hindbrain, nesfatin-1 activates POMC neurons and inhibits mTOR activity in the DVC, while ghrelin activates mTOR activity
In the medulla
In the medulla, the DVC, a central structure involved in the control of food intake, consists of three parts: the nucleus of the tractus solitarius (NTS), which receives and integrates afferent signals from viscera; the dorsal motor nucleus of the vagus (DMV), where the preganglionic vagal motor neurons are located; and the AP, which contains incomplete BBB and permits some circulating substances to contact with.
Peripheral nesfatin-1 might convey signals to NTS through the vagal afferent nerve, since nesfatin-1 could activate vagal afferent neurons by stimulating Ca2+ influx through N-type channels [57]. Nesfatin-1 is expressed in all the three parts of DVC [58] and other appetite suppressing hormones, such as leptin, glucagon-like peptide-1 (GLP-1) and cholecystokinin (CCK)-8 could inhibit food intake via activation of nesfatin-1-positive neurons in the brainstem [59]. Local administration of nesfatin-1 in the DVC inhibited food intake and body weight gain, as shown in our previous study [60]. Peripheral injection of nesfatin-1 midsegment only activated the proopiomelanocortin (POMC, the precursor of alpha-melanocyte-stimulating hormone (α-MSH)) positive neurons in the NTS but not in the ARC, indicating the important role of the NTS in feeding control [61]. In fact, oxytocinergic neurons originating from the PVN, which could be activated by nesfatin-1, enhanced the production of POMC from the NTS [62]. Besides, nesfatin-1 colocalizes with neuropeptide Y (NPY, an orexigenic peptide) and amphetamine-regulated transcript (CART, an anorexigenic peptide) in the NTS [58] and exogenous nesfatin-1 downregulated NPY expression, while CART expression remained unchanged [63].
GHSRs are expressed in all the three parts of the DVC, where c-fos expression increased after intracerebroventricular (i.c.v) injection of ghrelin [64]. Hyperphagia could be induced by injection of ghrelin into either the third ventricle near the ARC or the fourth ventricle near the DVC [65]. However, the essential amount for direct induction of hyperphagia in the DVC [65] is much lower than that in ARC [66]. Despite this, merely selective expression of GHSR in the brainstem was insufficient to reserve peripheral ghrelin-induced feeding [67]. The vagus nerve also plays an important role in ghrelin orexigenic effect, since ghrelin failed to stimulate feeding after vagotomy [68] or impairment of gastric vagal afferent fibers [69]. In addition, antagonist of ghrelin receptor increased the ability of CCK to activate GLP-1 and prolactin-releasing peptide neurons in the hindbrain [70].
Nesfatin-1 and ghrelin shared a common signaling pathway in feeding control by DVC, with decreased food intake through inhibition of mTOR signaling by nesfatin-1 and increased food intake through activation of mTOR signaling by ghrelin [71].
In the pons
The parabrachial nucleus (PBN), a feeding-related center, is located in the pons of the brainstem and transmits gustatory and visceral information to higher centers, which attracts an ever-growing number of concerns [72]. Nesfatin-1 is expressed in the lateral parabrachial nucleus (LPBN) [58] and local injection of nesfatin-1 into the LPBN suppressed food intake and body weight gain, possibly due to the modulation of activities of glucose sensitive neurons, since nesfatin-1 inhibited most glucose-excitatory (GE) neurons and excited most glucose-inhibitory (GI) neurons, respectively [73]. However, ghrelin reversed all above effects [74]. In addition, injection of nesfatin-1 into the LPBN upregulated the expression of uncoupling protein 1 (UCP1) in brown adipose tissue to generate heat with its underlying mechanism related to melanocortin system, as these effects could be blocked by MC3/4-R antagonist [73]. GHSRs are abundantly expressed in the LPBN and functional silencing of GHSR-expressing cells prevented mice from body weight gain and fat accumulation under the condition of a high-fat, high-sugar diet [75]. Most GHSR-positive cells belong to glutamatergic population rather than calcitonin gene-related peptide (CGRP, an anorexigenic peptide) cell [75]. Whether the inhibitory effect of nesfatin-1 on food intake is through CGRP-positive neurons and the relationship between glucose sensitive neurons and CGRP-positive neurons are worthy of further studies.
In the midbrain
In the midbrain, DAnergic rewarding pathway from the ventral tegmental area (VTA) to the nucleus accumbens (NAc) is responsible for hedonic control of feeding. The opposite results are obtained in this pathway between nesfatin-1 and ghrelin.
Our data demonstrated that local administration of nesfatin-1 in the VTA reduced nocturnal food intake through inhibition of DAergic neurons in the VTA and the subsequent DA release in the NAc [76], which has been confirmed by Dore R et al. [77]. Ghrelin reversed all above effects [78, 79], moreover, blockage of D-Rs in the NAc attenuated the effect of ghrelin in the VTA [80]. Central injection of nesfatin-1 abolished the fasting-induced increase in the reward value of sucrose [81], while ghrelin potentiated the rewarding value of high-fat diet [82]. GHSRs are densely expressed in the VTA [35] and GHSR selective antagonist [78] or GHSR knockout blunted the orexigenic effect of ghrelin [83], but reexpression of GHSR1a in DAnergic neurons including those in the VTA partially restored food intake [83]. Ghrelin also interacts with opioid and endocannabinoids system to influence the reward-associated behaviors, since antagonist of opioid receptor or cannabinoid receptor partly blocked the ghrelin-induced food intake and motivated behavior or ghrelin-dependent DA release in the NAc [84], that might inspire to explore the correlation between nesfatin-1 and other transmitters in the midbrain.
In the hypothalamus
The hypothalamus is a classical central area regulating appetite and energy homeostasis. Two types of “first-order” neurons located in the ARC play important roles in the control of energy metabolism. The neurons coexpressing NPY and AgRP are responsible for orexigenic effects, whereas anorexigenic POMC and CART co-exist in the other group of neurons. Bilateral communications between the ARC and other hypothalamic nuclei such as the ventromedial hypothalamus (VMH), PVN and lateral hypothalamic area (LHA) compose the network to regulate energy homeostasis.
Central administration of nesfatin-1 suppressed food intake in rodents or goldfish [85]. The PVN seems to be the key acting site of nesfatin-1. In the PVN, NUCB2/nesfatin-1 expression was significantly decreased under starved conditions [2]. Third ventricle injection of nesfatin-1 upregulated c-fos expression in the PVN [62, 86]. Both oxytocin and melanocortin system are involved in the effects of nesfatin-1 in the PVN, since nesfatin-1 activated PVN oxytocinergic neurons which terminated at the POMC neurons in the NTS and antagonist of Oxy-R [62] or MC3/4-R abolished the anorexigenic effects of central nesfatin-1. In addition, peripheral injection of CCK activated nesfatin-1-positive neurons in the PVN, indicating the interaction between nesfatin-1 and CCK system [87]. The NPY-positive neurons in the ARC seem to be another acting site for nesfatin-1. Application of nesfatin-1 decreased hypothalamic NPY mRNA levels [2] and hyperpolarized NPY-positive neurons [88], but did not influence the POMC/CART mRNA expression [2]. In addition, the CRF2-R-dependent pathway is involved in the inhibitory effect of nesfatin-1 on food intake in forebrain [89].
The ARC serves as the most important central site for the effects of ghrelin on appetite. Ghrelin increased the spontaneous firing rate [90] and miniature frequency of excitatory synaptic current of NPY/AgRP neurons in vivo or in vitro [91]. Gene knockout and antagonist application confirmed the role of NPY/AgRP neurons, since the orexigenic effects of ghrelin could be eliminated in NPY/AgRP ablated mice [92] and antagonists of NPY/Y1 receptor abolished ghrelin-induced feeding [93]. The dense expressions of GHSRs in NPY/AgRP neurons play an important role in orexigenic effect of ghrelin [94] because only deletion of GHSR in AgRP neurons attenuated diet-induced obesity [95]. In contrast to NPY/AgRP neurons, GHSRs are less expressed in POMC/CART neurons [96], suggesting that ghrelin might exert nearly no direct action on POMC/CART neurons. However, due to the reciprocal inhibitory projections between the two groups of first-order neurons, POMC/CART neurons were hyperpolarized after ghrelin application which stimulated the γ-aminobutyric acid (GABA) release from NPY/AgRP to POMC/CART neurons [97]. In addition, central injection of α-MSH inhibited ghrelin-stimulated hyperphagia, suggesting the crosstalk between ghrelin and melanocortin system [98]. The PVN seems to be another important site for ghrelin actions. Administration of ghrelin in the PVN alone is known to stimulate food intake and inhibit fat oxidation [99]. Fasting enhanced the density and strength of ARC nerve fibers projecting to PVN and the activation was impaired in mice lacking GHSR or pretreatment with blockage of GHSR signaling [100]. Inhibition of oxytocinergic neurons in the PVN contributed to orexigenic effects of ghrelin [3]. In the VMH, i.c.v. administration of ghrelin decreased fatty acid synthase mRNA expression via inhibition of AMPK activity [101]. In the LHA, ghrelin depolarized orexin-positive neurons and enhanced the c-fos expression in these neurons [102]. Pretreatment of orexin receptor antagonist partially abolished central ghrelin appetite-stimulating effects [103]. The LHA even serves as a critical downstream site for hippocampus ghrelin-mediated hyperphagia [104]. In the hypothalamus, ghrelin not only promoted food intake but also decreased energy expenditure via inhibition of sympathetic projections to the brown adipose tissue [105]. Even after selective knockout of GHSR in hypothalamic AgRP neurons, mice fed with a high-fat diet exerted elevated energy expenditure and upregulated thermogenesis in both brown and white adipose tissue, suggesting GHSR inhibition in AgRP neurons could increase sympathetic activity [95].
From the above, it is known that nesfatin-1 inhibits NPY-positive neurons in the ARC, different from the activation by ghrelin. Moreover, mTOR signaling is involved in the ghrelin action on NPY/AgRP neurons, since inhibition of mTORC1 abolished ghrelin-induced upregulation of NPY and AgRP [40, 106], while majority mTOR-positive neurons in the ARC are also immunoreactive for nesfatin-1 [107]. One study observed the effects of nesfatin-1 on ghrelin, which showed that i.c.v. administration of nesfatin-1 suppressed preproghrelin, GHSR and NUCB2 mRNA expression in the forebrain of goldfish [51]. In contrast to the effects of nesfatin-1 on promoting activity of oxytocinergic neurons, GHSR forms heterocomplex with Oxy-R and impairs the oxytocinergic signaling [51]. In addition, nesfatin-1 promotes oxytocin release [62], whereas ghrelin inhibits oxytocin expression in the PVN [3]. In neonatal chicks, ghrelin inhibits food intake, opposite to the orexigenic effects of ghrelin in rodents [108, 109]. Coincidentally, both ghrelin and nesfatin-1 inhibit food intake through the CRF-R pathway in chicks [28, 108].
Anxiety, depression and stress
Anxiety
Nesfatin-1 exerts anxiogenic effect in rodents. Central infusion of nesfatin-1 [110] or its middle fragment [111] induced the anxiety-like behaviors in several maze tests, or in unfamiliar environment, while central blockage of endogenous nesfatin-1 with antibody attenuated anxiety-like behavior in male rats [112]. Peripheral continuous administration of nesfatin-1 also exhibited anxiogenic effect by downregulating expression of phosphorylated-ERK1/2 and the brain-derived neurotropic factor (BDNF) in the prefrontal cortex and hippocampus [113]. Human studies showed that alteration of nesfatin-1 levels in anxiety was inconsistent between genders. In male patients suffering from anxiety disorder, plasma nesfatin-1 levels were decreased [114], whereas in female obese patients, NUCB2/nesfatin-1 levels were upregulated [115]. However, improvement of anxiety did not alter NUCB2/nesfatin-1 levels [116].
Ghrelin seems to exert dual effects on anxiety. I.c.v. [117], amygdala, hippocampus [118] and hypothalamus [119] injections of ghrelin induced anxiety-like behaviors in the open field test, plus-maze test, or elevated plus maze, while antisense DNA for ghrelin exerted an anxiolytic effects in the elevated plus maze test, black and white test, or conditioned fear tests [120]. Conversely, Jensen et al. [121] reported overexpression of GHSRs in the amygdala or peripheral injection of ghrelin caused anxiolytic effect, which could be abolished in GHSR knockout mice. Acute energy restrictions stimulated endogenous ghrelin secretion and thus exerted anti-anxiety effects and the anxiolytic effects could be abolished by GHSR1a antagonist [122] and GHSR knockout [123], confirming the GHSR-dependent pattern.
Depression
Nesfatin-1 levels were profoundly increased in subjects suffering from depression [124] and in depressed patients with subclinical hypothyroidism [125]. NUCB2 mRNA contents in Edinger–Westphal (EW) nucleus were significantly higher in male depressed suicide victims than those in the control, but not in female ones [126]. The rats exhibited more despair after chronic nesfatin-1 application in forced swimming test and open field test, accompanied by hypothalamic–pituitary–adrenal (HPA) axis activation [127]. The higher nesfatin-1 levels in depressive disorder were correlated with elevated corticosterone, IL-6, and C-reactive protein (CRP) levels [128, 129]. Pretreatment with a traditional Chinese antidepressant medicine, Xiaoyaosan, downregulated nesfatin-1 levels in the PVN [130]. In contrast to above studies, Korucu et al. reported serum nesfatin-1 levels were decreased obviously in major depressive disorder with suicidal ideation [131]. In adolescent depression, nesfatin-1 levels were significantly lower than in the control group [132]. Hence, we performed a meta-analysis on 567 depressive patients and 447 control subjects and the result showed higher plasma levels of nesfatin-1 were associated with an increased risk of depression, indicating that nesfatin-1 might act as a potential novel biomarker for the diagnosis of depressive disorder [133].
Similar to the controversial effects of ghrelin on anxiety, the links between ghrelin and depression are also inconsistent. In humans [134,135,136] and rodents [137, 138], ghrelin levels were elevated in depressive status. Exogenous ghrelin generated antidepressant-like effect in tail-suspended mice and in the bilateral olfactory bulbectomy mice [139]. In humans, ghrelin exerted antidepressant effects in patients with major depression [140]. However, in contrast to above results obtained from adult rodents, Jackson et al. [141] reported that acute i.c.v. administration of ghrelin displayed depressive-like effects in male juvenile rats by prolonging immobility time in forced swimming test. Similarly, Guo et al. [142] observed that GHSR knockout mice did not exert apparent depression after chronic social, but the control mice did, possibly due to the differential expressions of brain BDNF and IL-6.
One study simultaneously focusing on nesfatin-1 and ghrelin in depression showed that plasma levels of both peptides were increased in depressive patients and even associated with the severity of depression [129].
Stress
Restraint stress mobilized nesfatin-1-positive neurons in the hypothalamus, NTS [143], locus coeruleus (LC), rostral raphe pallidus (rRPa), and the non-preganglionic Edinger–Westphal nucleus (npEW) [144], most of which could also be activated by abdominal surgery, a kind of visceral stressor [145]. Only acute stress, instead of chronic stress, increased the plasma and hypothalamic nesfatin-1 levels in rats [146]. The elevated nesfatin-1 levels attributed to the activation all the three parts of HPA axis during stress. In the hypothalamus, nesfatin-1 activated the CRH-positive neurons by increasing cytosolic Ca2+ concentration [147]. Central administration of nesfatin-1 elevated ACTH and corticosterone levels in pituitary and adrenal gland, respectively [148]. In mouse corticotrophs, nesfatin-1 stimulated the synthesis of POMC which serves as the precursor of ACTH [149]. Glucocorticoid receptors were detected in nesfatin-1-positive neurons in the PVN [150], indicating a possible feedback control through adrenal signaling. Actually, nesfatin-1 expression is negatively regulated by adrenal steroids, as bilateral adrenalectomy triggered an increase in nesfatin-1/NUCB2 mRNA expression in the HPA axis [148]. Similar results were obtained in goldfish. NUCB2/nesfatin-1 was detected in all the three parts of the stress axis in goldfish [151] and restraint stress upregulated NUCB2/nesfatin-1 mRNA levels in hypothalamus and pituitary; moreover, nesfatin-1 application stimulated the release of cortisol and ACTH in vivo and in vitro [151].
When stress occurs, the changes in ghrelin levels seem to be controversial. Under acute stress, ghrelin levels were decreased by orderly activation of CRF1-R, 5-HT(1B)/(2C)-R and MC4-R in mice [152]. However, Kristenssson et al. [153] reported that acute psychological stress led to elevated ghrelin levels. Chronic social defeat stress increased ghrelin levels in rats [137], while ghrelin mRNA levels of hypothalamus were significantly decreased by chronic restraint stress [154]. Nahata et al. [155] even found that plasma ghrelin concentration was not altered by restraint stress. In humans, one meta-analysis based on the data of 348 patients showed a short-term increase of ghrelin level following acute stress [156]. The altered ghrelin levels seem to influence the HPA axis, while the results also remain controversial. In rodents [157] and humans [158], ghrelin increased CRH, ACTH or corticosterone levels. However, Jensen M. et al. [121] reported no effect of ghrelin on plasma ACTH levels in mice. Alterations in HPA axis induced by stress also affect ghrelin secretion. Cortisol decreased serum ghrelin levels and its mRNA expression in the stomach in tilapia [159]. In patients with Cushing’s syndrome, hypercortisol reduced plasma ghrelin levels, which were restored by surgical treatment [160].
Learning and memory
There are only a few studies that focus on the effects of nesfatin-1 on learning and memory and the results remain conflicting, while ghrelin exerts dual effects on learning and memory.
As demonstrated by Zhu Q et al. [161], nesfatin-1 did not affect learning and memory after infusion into the lateral amygdala or area CA1 of the dorsal hippocampus. Similarly, Ge et al. [113] verified that continuous intraperitoneal (i.p) injection of nesfatin-1 did not alter the performance of rats in the Morris water maze. In rat model with non-alcoholic fatty liver disease, increased plasma nesfatin-1 levels were found to be at least partly associated with impaired learning and memory [162]. Only one study showed that nesfatin-1 improved memory impairment induced by transient global cerebral ischemia/reperfusion (I/R), and the underlying mechanism might be related to its inhibitory effects on activation of microglial cells and caspase-3 [163].
GHSR mRNA is profoundly expressed in the CA1, CA2, and CA3 regions of hippocampus and the dentate gyrus in rat brain [35]. Memory could be improved in a dose-dependent manner through administration of ghrelin in the cerebroventricles, hippocampus, amygdala and dorsal raphe nucleus (DRN) [117, 118]. In ghrelin knockout mice, the exploration time of novel object decreased, which could be rescued by ghrelin re-supplement [164]. Blocking GHSR1a worsened memory encoding [165], while ghrelin receptor agonists ameliorated object recognition memory [166]. The mechanisms underlying memory enhancement of ghrelin partly lie in synaptic plasticity and hippocampal neurogenesis, since ghrelin increased the spine synapse density in CA1 region [164, 167] and promoted proliferation and differentiation of adult hippocampal progenitors [168]. In depressed mice, lower ghrelin levels caused decreased hippocampal BDNF levels and resulted in cognitive decline [169]. NMDA receptor is another site for ghrelin action. Ghrelin attenuated memory impairment induced by antagonist of NMDA receptor [170] and upregulated NMDA receptor and MAPK1 expressions to improve memory [171]. In contrast, some studies documented the negative effects of ghrelin on memory. Spitznagel et al. demonstrated that cognitive function was negatively associated with ghrelin levels in non-demented elder adults [172]. In GHSR1a knockout mice, spatial memory was improved [173]. In addition, injection of ghrelin into lateral amygdala prevented acquisition of conditioned taste aversion [174]. A study done by Li N et al. might explain these contradictory results. They found that a selective increase of GHSR1a expression in hippocampus CA1 excitatory pyramidal neurons impaired hippocampus-dependent memory, whereas its upregulation in CA1 inhibitory interneurons improved hippocampus-dependent memory. [175].
Neuroprotection
Nesfatin-1 reduced the gene expression of NF-κB, levels of tumor necrosis factor alpha (TNF-α), IL-1β, IL-6 and activity of caspase-3 in traumatic brain of rat, which implies its anti-inflammatory and anti-apoptotic effects in the CNS [176]. In patients with traumatic brain injury (TBI), nesfatin-1 reflected severity of trauma and even served as a prognostic biomarker following TBI [177]. Nesfatin-1 protected PC12 cells against high glucose-induced cell injury by inhibition of apoptosis, autophagy and ROS production [178]. In chronic high-fat diet mice, obesity was associated with upregulated pro-inflammatory factors and NF-κB signaling components in hypothalamus, which was accompanied by decreased NUCB2/nesfatin-1 levels and diet and/or exercise interventions reversed above alterations [179].
Ghrelin ameliorated secondary brain injury induced by intracerebral hemorrhage through inhibition of nucleotide-binding oligomerization domain-like receptor pyrin domain-containing 3 (NLRP3) inflammasome and activation of nuclear factor-E2-related factor 2 (Nrf2)/anti-oxidative response element (ARE) signaling pathway [180]. Ghrelin restored cerebral microvascular integrity, reduced vascular leakage [181] and improved neural survival [182] by inhibition of apoptosis, inflammation or oxidative stress. In amyloid β induced AD mouse models, ghrelin ameliorated cognition, synaptic plasticity deficiency and neuroinflammation [183].
Our previous data have elucidated the neuroprotective effect of both nesfatin-1 and ghrelin on Parkinson's disease (PD). Nesfatin-1 protected nigral DAergic neurons against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced neurotoxicity via C-Raf-ERK 1/2 pathway in vivo and in vitro [184]. Moreover, nesfatin-1 prevented the neurotoxicity of rotenone in DAergic cells [185]. Reduced nesfatin-1 in the brain might induce degeneration of nigrostriatal DAnergic system, which was mediated via mitochondrial dysfunction-related apoptosis [186]. Paralleled with nesfatin-1, similar results were obtained with application of ghrelin which antagonized the impairment of 1-methyl-4-phenyl-pyridinium (MPP+) [187], except that ghrelin increased [188] and nesfatin-1 decreased the firing rate of nigral DAergic neurons [189]. Stutz et al. found that the protective effect on DAergic neurons mediated by GHSR was independent of their electric activity, which could explain the similar protective effects and opposite actions on the firing of DAergic neuron between nesfatin-1 and ghrelin [190]. In PD models, ghrelin blocked the activation of microglia, inducible nitric oxide synthase, reduced the expression of TNF-α and IL-1β [191], improved motor symptoms and partly restored tyrosine hydroxylase in substantia nigra via improving autophagic flux dysfunction and recovery of lysosome functions [192]. In PD patients, total and active plasma ghrelin levels were decreased [193]. Deep brain stimulation of subthalamic nucleus, as a type of treatment of PD [191], increased active ghrelin levels and body weight gain [194].
The roles of nesfatin-1 and ghrelin in the peripheral functions
Gastrointestinal functions
Nesfatin-1-positive neurons in the NTS were activated by gastric distension (GD) and exogenous nesfatin-1 modulated mechanosensitivity of gastric vagal afferent fibers [195]. Nesfatin-1 affects not only gastrointestinal sensory input but also gastrointestinal motility. Central injections of nesfatin-1, such as lateral ventricle [196], ARC [197], PVN [198], VMH [199] and amygdala [200], inhibited gastric motility and the underlying mechanisms were linked to oxytocin and melanocortin systems. Besides, nesfatin-1 affects gastric secretion. Central infusion of nesfatin-1 reduced gastric acid secretion dependent upon the inhibition of H+/K+-ATPase expression in the gastric mucosa [201]. The elevated nesfatin-1 levels also contributed to the protection of gastric mucosa impaired by stress, partly through the inhibition of gastric acid secretion [202]. In addition, nesfatin-1 affected the secretion of other gastrointestinal hormones, including CCK and peptide YY in mice intestine [203].
Ghrelin exerts different effects from nesfatin-1 on gastrointestinal functions. Activities of GD sensitive neurons in the DVC [204], hippocampus [205], and lateral septum [206] were modulated by micro-injection of ghrelin. Both ghrelin and GHSRs were detected in vagus nerve and ghrelin inhibited the mechanosensitivity of vagal afferent [207], while ghrelin receptor inverse agonist increased the firing rate of vagal afferent fibers [208]. Ghrelin regulates gastrointestinal motility during both digestive and interdigestive period. In rodents and humans, administration of ghrelin promoted gastric emptying [98, 209, 210], which might be mediated by GABAergic neurons located in the AP [211]. During interdigestive period, ghrelin promoted migrating motility complex (MMC) phase III-like movement [212] and the promotion extended to the small intestine [213]. Ghrelin stimulated gastric acid secretion through vagus nerve and the underlying mechanism involved in the histamine and gastrin [214].
Despite the same origin in gastrointestinal tract, nesfatin-1 mainly exerts inhibitory effects on gastrointestinal functions, opposite to the major stimulatory effect of ghrelin. The studies focusing on nesfatin-1 and ghrelin showed that fasting generated opposite impacts on both peptides in the stomach, as ghrelin expression was upregulated and NUCB2/nesfatin-1 production was suppressed [215]. Histone deacetylases5 (HDAC5)-mTORC1 signaling served as common pathway leading to reciprocal changes in ghrelin and NUCB2/nesfatin-1 secretions [215]. In dogs, NUCB2 mRNA was expressed in all digestive organs including digestive tract and digestive glands [216], while ghrelin gene was only detected in the stomach [217]. A research in dogs showed that plasma nesfatin-1 concentrations might mark the inversed kinetics for ghrelin during MMC, since plasma ghrelin hit its nadir just when plasma nesfatin-1 levels peaked in late phase I [218]. Peripheral administration of nesfatin-1 inhibited gastric MMC in the fasted dogs, but did not affect gastrointestinal motility in the fed ones [218].
Blood glucose and diabetes mellitus
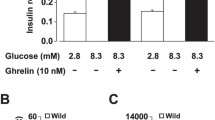
Nesfatin-1 is co-distributed with insulin in pancreatic β-cells and promotes the secretion of insulin, indicating the correlation between nesfatin-1 and glucose metabolism. In vitro study showed that nesfatin-1 potentiated glucose-induced insulin production by activation of L-type Ca2+ channels [219] and inhibition of voltage-gated K+ channels [220] in isolated β-cells of pancreatic islet. In pancreatic β-cell-specific NUCB2 knockout mice, blood glucose levels were elevated and insulin secretion was inhibited [221]. Notably, nesfatin-1 exhibited direct hypoglycemic effect in peripheral tissue, since it enhanced phosphorylation of AKT and glucose transporter 4 (GLUT4) expressions in skeletal muscle and adipose tissue [222], decreased the glucose intake in the gut and increased glucose uptake in liver and muscle [223]. Nesfatin-1 exerts hypoglycemic effects in physiological state, which makes nesfatin-1 a possible candidate for hypoglycemic hormones besides insulin. In T2DM rats, nesfatin-1 levels decreased in the blood, which could be reversed by hypoglycemic agents [224]. In T2DM mice, central injection of nesfatin-1 decreased plasma levels of free fatty acid and promoted fatty acid oxidation in skeletal muscle [225]. In T2DM patients, higher plasma nesfatin-1 concentrations in the early stages might compensate for hyperglycemia and nesfatin-1 concentrations were reduced in patients receiving antidiabetic therapy [226]. In addition, NUCB2 gene polymorphism might be associated with T2DM, as a consequence of lower CG and GG genotype of NUCB2 gene in T2DM patients [227].
In contrast to the hypoglycemic effects of nesfatin-1, ghrelin exerts hyperglycemic actions. In rodents and humans, exogenous ghrelin suppressed glucose-induced insulin secretion [228,229,230] and increased plasma glucose levels [231]. In pancreatic β-cells, ghrelin reduced insulin secretion by activating voltage-dependent K+ channels and by weakening Ca2+ signaling [232]. In addition, ghrelin stimulated somatostatin secretion from pancreatic δ-cells, which in turn suppressed insulin secretion [233]. The formation of GHSR1a-SST5 heterocomplex also contributed to the inhibition of insulin secretion [52]. Moreover, ghrelin stimulated glucagon secretion from α-cells, which elevated plasma glucose levels [234]. In T2DM patient, plasma ghrelin levels were decreased and were associated with insulin resistance [235]. Due to hyperglycemic effects of ghrelin, GHSR1a receptor antagonist, PF-05190457, was explored to treat T2DM [208].
In short, nesfatin-1 stimulates insulin secretion and decreases plasma glucose levels, while ghrelin exerts opposite effects. In T2DM, plasma nesfatin-1 levels are elevated and ghrelin levels are decreased. Bertucci et al. observed the alteration of both peptides after exposure to glucose in goldfish, concluding that preproghrelin was upregulated and NUCB2/nesfatin-1 was downregulated in cultured intestine sections [236]. GHSRs are involved in insulin secretion, since GHSR knockout mice showed significantly lower blood glucose levels under caloric restriction [237] and the glucose-stimulated insulin secretion was inhibited both in vivo and in vitro after specific GHSR knockout in β cells [238]. The direct effect of ghrelin on insulin secretion is inhibitory, but after deleting its receptors on β cells, insulin secretion was still inhibited, indicating the possible involvement of other insulinostatic hormones. It could not be ruled out that nesfatin-1 might be one of the candidates. Our results demonstrated that the improvement of nesfatin-1 on glucose tolerance could be abolished by pretreatment of GHSR antagonist or GHSR knockout, which implies the dependence of nesfatin-1 on GHSR in the regulation of glucose metabolism [4]. Whether nesfatin-1 could regulate pancreatic insulin release through GHSRs is worthy of further investigation.
Reproduction
The reproduction is mainly regulated by the hypothalamic-pituitary–gonadal (HPG) axis, which originates from hypothalamus, also serving as a feeding control center. In fact, reproductive functions are closely correlated with energy metabolism. It is vital for reproduction that the feeding related hormones are maintained in appropriate range. The effects of nesfatin-1 and ghrelin on the three respective levels of the HPG axis will be discussed, with regards to different species.
Protein or mRNA of NUCB2/nesfatin-1 was detected in all the three parts of the HPG axis indicating the link between nesfatin-1 and reproduction. The actions of nesfatin-1 on reproduction have been observed in different species. In adult male rats, i.c.v. injection of nesfatin-1 suppressed gene expression of gonadotropin-releasing hormone (GnRH) in hypothalamus, follicle-stimulating hormone β (FSHβ) and luteinizing hormone β (LHβ) in the pituitary gland, as well as genes for testosterone synthesis in the testis [239]. Most results obtained in male pubertal rats were similar to those of the adults except for the elevated genes for testosterone synthesis [239]. In female pubertal rats, elevated NUCB2/nesfatin-1 mRNA levels in hypothalamus promoted pubertal maturation, since the central infusion of nesfatin-1 created a slight increase in circulating gonadotropins and continuous infusion of antisense morpholino oligonucleotides of NUCB2 prevented vaginal opening and reduced ovarian weights and plasma LH concentration [240]. In both male and female goldfish, peripheral injection of nesfatin-1-like peptide suppressed hypothalamic GnRH mRNA expression, as well as pituitary LHβ and FSHβ. In gonads, nesfatin-1 downregulated LH receptor, FSH receptor and enzymes for sex hormone synthesis [241]. Accordingly, the circulating testosterone and estradiol were decreased [241]. Nesfatin-1-like peptide inhibited oocyte maturation in zebrafish [241]. In general, nesfatin-1 has an inhibitory effect on reproduction in vivo in adult rodents and fishes. However, some results obtained in vitro seem to be different. Synthetic nesfatin-1 increased GnRH and LHβ expressions in hypothalamic and pituitary cell lines [242] and nesfatin-1 enhanced the testicular testosterone secretion in Leydig cells [239]. In addition to fishes and rodents, there were several studies focusing on porcine. I.c.v. administration of nesfatin-1 stimulated LH secretion in prepubertal gilts [243]. Nesfatin-1 significantly promoted the meiotic maturation [244] and stimulated cell proliferation and progesterone production in granulosa cells from large follicles [245].
The effects of ghrelin on reproduction have been studied in different species, with different results. In rodents, in vivo results showed that ghrelin inhibited hypothalamic GnRH secretion [246, 247], pituitary LH or FSH expressions or secretion [248,249,250], ovarian and testicular steroid hormone secretion [250, 251]. In vitro results showed that ghrelin stimulated GnRH release in hypothalamic explant of the prepubertal [246], and enhanced basal LH and FSH secretions in pituitary tissue [247], but decreased testosterone secretion in testicular slices [252]. In female rats, the main effect of chronic subcutaneous injection of ghrelin on the ovaries was inhibitory, including the number of corpora lutea, the mean diameter of follicle, corpora lutea, luteal cell, theca layer, oocyte and zona pellucida, as well as the whole ovarian volume [253]. Ghrelin inhibited male puberty onset, as balano-preputial separation was delayed. However, in prepubertal female rats, daily subcutaneous administration of ghrelin failed to influence puberty onset and the sexual hormone secretions [254]. In humans, ghrelin decreased both LH and FSH levels in females [255], but only LH in males [256]. Ghrelin attenuated basal and hCG-stimulated progesterone release in human luteal cells [257]. A human study showed that high serum ghrelin levels were correlated with constitutional delay of growth and puberty in adolescence [258]. In goldfish, ghrelin stimulated LH release in vivo and in vitro [259], while inhibited germinal vesicle breakdown of zebrafish oocyte maturation in vitro [260]. The effects of ghrelin on porcine reproduction were mainly concentrated in experiments in vitro and the results were not completely consistent. Some studies showed that ghrelin promoted reproductive functions: ghrelin reinforced estradiol and progesterone secretions, increased ovarian cell proliferation and enhanced blastocyst formation [261,262,263]. However, ghrelin inhibited LH and FSH induced follicular steroid secretion [264], as well as the organization of microtubules and microfilaments, potentially being a contributing factor in the lowered maturation rate [265].
In general, the regulation of nesfatin-1 and ghrelin in reproductive axis are complicated, varying with different species, genders and stages of development. The main roles of nesfatin-1 and ghrelin in rodents are similar, with inhibitory effects in vivo and excitatory effects in vitro. Nesfatin-1 inhibits reproduction in fish and promotes reproduction in porcine, while the effects of ghrelin on reproduction are inconsistent in above two species.
Cardiovascular functions
The distribution of ghrelin and nesfatin-1 in the cardiovascular centers and in myocardium suggests their role in controlling cardiovascular functions. The effects of the two peptides on the heart and blood vessels will be discussed, respectively.
In myocardial I/R injury models, nesfatin-1 exerted cardioprotective effects through anti-inflammation, anti-apoptosis, anti-oxidative stress and anti-autophagy [266]. Nesfatin-1 decreased cardiac troponin-T and pro-inflammatory cytokines, increased the expressions of p-AKT/AKT and p-GSK-3β/GSK-3βa [267] and suppressed necroptosis via modulation of receptor interacting kinase 1 (RIPK1)-RIPK3-the mixed lineage kinase domain-like protein (MLKL) axis and RhoA/Rho-associated coiled-coil-containing protein kinase (ROCK)/RIP3 signaling pathway [268] as well as attenuation of endoplasmic reticulum stress via AKT/ERK pathway after I/R injury [269]. In addition, high levels of nesfatin-1 were probably associated with left ventricle myocardial remodeling [270]. In blood vessels, nesfatin-1 could alleviate endothelial inflammation induced by free fatty acids via the growth factor independent-1 transcriptional repressor (GFI1)/NF-κB signaling pathway [271]. However, nesfatin-1 promoted vascular smooth muscle cells proliferation [272], migration and neointimal hyperplasia by increasing matrix metalloproteinase 2 (MMP2)/MMP-9 levels and decreasing peroxisome proliferator-activated receptor γ (PPARγ) gene expression [273], indicating the role of nesfatin-1 in the formation of atherosclerotic plaque. Compared with the control group, higher plasma levels of nesfatin-1 were found in patients with coronary artery stenosis [274]. In the regulation of blood pressure, central nesfatin-1 significantly increased mean arterial pressure (MAP) by acting on MC3/4-Rs in the NTS and sympathetic nervous system, since the vasopressor effect could be abolished by pretreatment with the MC3/4-R antagonist or the alpha-adrenergic antagonist. In addition, after intravenous infusion of nesfatin-1, the arterial vessels contracted, resulting in increased peripheral resistance in rats [275] and chronic infusion of nesfatin-1 induced severe hypertension [272].
Ghrelin plays a protective role in myocardial I/R injury. Ghrelin reversed the alteration of infarct size and the levels of creatine kinase (CK), lactate dehydrogenase (LDH), TNF‑α, IL‑6, malondialdehyde (MDA), caspase-3, caspase-9, iNOS, superoxide dismutase (SOD), and glutathione (GSH)‑peroxidase (PX) after myocardial I/R injury via the high mobility group box 1 (HMGB1)/Toll-like receptor 4 (TLR4)/NF-κB pathway [276]. In addition to the anti-inflammatory and antioxidant effects, ghrelin resisted myocardial I/R injury by enhancing autophagy. After delivering ghrelin to the infarcted myocardium with an adeno-associated virus 9 vector, the ratio of LC3-II/LC3-I, formation of autophagosomes and genes expression involved in the autophagic pathways were elevated [277]. Chronic application of ghrelin improved ultrastructural changes after myocardial infarction, including increased numbers of intracellular organelles in endoplasmic reticulum, and decreased numbers of atrophic nuclei, phagocytes, irregular nuclear membrane and chromatin condensation [278]. Myocardial hypertrophy and myocardial fibrosis are important pathological features of myocardial remodeling. Karcz-Socha I et al. reported that circulating ghrelin levels were associated with left ventricular mass index [279]. After transverse aortic constriction, ghrelin knockout mice exhibited severer cardiac hypertrophy and inhibition of parasympathetic activities, while re-application of ghrelin attenuated cardiac hypertrophy by activating the cholinergic anti-inflammatory pathway [280]. Both in vivo and in vitro experiments confirmed that ghrelin inhibited angiotensin-induced myocardial fibrosis via PPAR-γ/transforming growth factor-β1 (TGF-β1) [281] or Nrf2/NADPH/ROS pathway [282]. Ghrelin protects not only the myocardium but also the vascular endothelium. Ghrelin ameliorated endothelial inflammation via AMPK/NF-κB signaling pathway [283], and inhibited atherosclerosis by preventing endoplasmic reticulum stress [284]. Ghrelin reduced intraplaque angiogenesis and lowered the thickness ratio of the intima to media by downregulating vascular endothelial growth factor (VEGF) and VEGF receptor 2 levels in an atherosclerotic rabbit model [285]. In the regulation of blood pressure, low circulating ghrelin levels are correlated with elevated blood pressure [286]. Exogenous administration of ghrelin decreased MAP in rabbits, rats and even humans [287]. The underlying mechanisms included vasorelaxation, inhibition of sympathetic activity, kidney diuresis, alleviation of oxidative stress and regulation of renin–angiotensin system [287,288,289,290].
Both nesfatin-1 and ghrelin exert similar protective effects on the cardiovascular system, but they have hypertensive and hypotensive effects, respectively, in the regulation of MAP.
Anti-inflammatory and anti-oxidative effects
Despite their previously stated opposite effects, nesfatin-1 and ghrelin exhibit similar anti-inflammation and antioxidation effects in various organs and tissues. (The cardiovascular system is not covered in this section, as discussed above.)
Serum nesfatin-1 levels were elevated in patients with Crohn's disease and ulcerative colitis [291], which indicates the anti-inflammatory effects of nesfatin-1. Actually, exogenous nesfatin-1 ameliorated necrotizing enterocolitis [292] and acetic acid-induced colitis [53]. Nesfatin-1 improved gastric ulcer by downregulating myeloperoxidase (MPO), MDA, chemiluminescence, IL-6, TNF-α and by upregulating GSH levels [293], in a COX-dependent manner [294]. Nesfatin-1 attenuated inflammation and oxidative stress in alveolar epithelial cell and in animal models of acute lung injury by inhibiting HMGB1, p38MAPK and NF-κB signaling pathways [295], while in NUCB2/nesfatin-1 knockout mice, acute lung injury caused by lipopolysaccharide was exacerbated [296]. In testicular torsion rat models, nesfatin-1 regulated pro-inflammatory/anti-inflammatory cytokine balance and reduced AKT/CREB expressions [297]. Nesfatin-1 alleviated osteoarthritis by suppressing the activation of NF-κB, MAPK, and the Bax/Bcl-2 signaling pathway [24].
Elevated plasma ghrelin levels have been observed in various inflammatory diseases, such as colitis [298], Crohn’s disease [299], sepsis [300] and pancreatitis [301]. Ghrelin inhibited the expression of pro-inflammatory cytokines such as IL-1β, IL-6 and TNF-α in human T lymphocytes and monocytes [302]. In sepsis-induced inflammation, ghrelin downregulated the pro-inflammatory cytokines mediated by the MAPK phosphatase-1 [303] and elevated the levels of antioxidants such as GSH, catalase (CAT) and SOD against the oxidative stress [304]. Ghrelin improved acute pancreatitis via inhibiting NF-κB signaling pathway, thus reducing the release of inflammatory cytokines [305].
Both nesfatin-1 and ghrelin possess anti-inflammatory and anti-oxidative effects. One group simultaneously observed the protective effects of both peptides on gastric ulcer. The results showed that a much lower dose of nesfatin-1 (0.02 nmol/Kg) appeared to be slightly less effective than ghrelin (4 nmol/Kg) according to the loss of gastric surface epithelium, glands erosion, cell inflammation and bleeding scores [306]. In colitis [53] and acute pancreatitis models [307], nesfatin-1 could alleviate inflammations via ghrelin receptor, since GHSR antagonist attenuated this protective effect, suggesting the interaction between nesfatin-1 and GHSR.
Conclusion and perspectives
Generally, nesfatin-1 displays opposite effects compared to ghrelin in the regulation of energy metabolism, blood glucose levels, gastrointestinal functions, blood pressure, but exhibits similar effects in cardiovascular protection, anti-inflammation, antioxidation and neuroprotection via distinct or similar mechanisms (Fig. 4). The early identifications of ghrelin and GHSR have led to numerous related studies. Therefore, clinical applications of ghrelin could enlighten those of nesfatin-1. For example, it should help with exploring the roles of nesfatin-1 in the treatment of obesity, neurodegenerative diseases, diabetes, myocardial I/R injury, inflammation, oxidative stress-related diseases, etc. [308, 309]. The antagonist of nesfatin-1 receptor might be applied in the treatment of cachexia, anorexia, GH deficiency and gastroparesis [310]. Therefore, the discovery of nesfatin-1 receptor could potentially contribute to the treatment of certain diseases and further clarification of the mechanisms of nesfatin-1 actions. The overlapping receptor distribution and the interaction between nesfatin-1 and ghrelin indicate the potential crosstalk. The dependence of nesfatin-1 on GHSR provides some strategies for searching the receptor of this orphan ligand. It could not be ruled out that nesfatin-1 might serve as an endogenous ligand for other known receptors, that the unknown nesfatin-1 receptor might form heterocomplex with other known receptors, or that nesfatin-1 even serves as an allosteric modulator for other GPCRs. Experimental techniques such as Biacore, proximity ligation assay (PLA), co-immunoprecipitation and BRET, may be conducive to exploring the interactions between nesfatin-1 and other receptors.

The comparison of nesfatin-1 and ghrelin in central and peripheral functions. In the CNS, nesfatin-1 inhibits food intake and induces anxiety. Stress activates nesfatin-1-positive neurons. Ghrelin stimulates food intake and exerts dual effects on learning and memory. Both nesfatin-1 and ghrelin exhibit neuroprotective effects. In peripheral system, nesfatin-1 inhibits gastrointestinal motility and secretion, stimulates insulin secretion, decreases blood glucose levels, and increases arterial blood pressure, while ghrelin reverses all above effects. Both nesfatin-1 and ghrelin attenuates myocardial I/R injury and exerts anti-inflammation and antioxidation effects
Data availability
Enquiries about data availability should be directed to the authors.
References
Kojima M, Hosoda H, Date Y, Nakazato M, Matsuo H, Kangawa K (1999) Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature 402(6762):656–660
Oh IS, Shimizu H, Satoh T, Okada S, Adachi S, Inoue K, Eguchi H, Yamamoto M, Imaki T, Hashimoto K et al (2006) Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature 443(7112):709–712
Velez EJ, Unniappan S (2020) Nesfatin-1 and nesfatin-1-like peptide suppress growth hormone synthesis via the AC/PKA/CREB pathway in mammalian somatotrophs. Sci Rep 10(1):16686
Fan XT, Tian Z, Li SZ, Zhai T, Liu JL, Wang R, Zhang CS, Wang LX, Yuan JH, Zhou Y et al (2018) Ghrelin receptor is required for the effect of nesfatin-1 on glucose metabolism. Front Endocrinol (Lausanne) 9:633
Feijoo-Bandin S, Rodriguez-Penas D, Garcia-Rua V, Mosquera-Leal A, Gonzalez-Juanatey JR, Lago F (2016) Nesfatin-1: a new energy-regulating peptide with pleiotropic functions. Implic Cardiovasc Lev Endocr 52(1):11–29
Pan W, Hsuchou H, Kastin AJ (2007) Nesfatin-1 crosses the blood-brain barrier without saturation. Peptides 28(11):2223–2228
Stengel A (2015) Nesfatin-1—more than a food intake regulatory peptide. Peptides 72:175–183
Goebel M, Stengel A, Wang L, Lambrecht NW, Tache Y (2009) Nesfatin-1 immunoreactivity in rat brain and spinal cord autonomic nuclei. Neurosci Lett 452(3):241–246
Gualillo O, Lago F, Casanueva FF, Dieguez C (2006) One ancestor, several peptides post-translational modifications of preproghrelin generate several peptides with antithetical effects. Mol Cell Endocrinol 256(1–2):1–8
Davis TR, Pierce MR, Novak SX, Hougland JL (2021) Ghrelin octanoylation by ghrelin O-acyltransferase: protein acylation impacting metabolic and neuroendocrine signalling. Open Biol 11(7):210080
Cabral A, Lopez Soto EJ, Epelbaum J, Perello M (2017) Is ghrelin synthesized in the central nervous system? Int J Mol Sci 18(3):638
Wellman M, Abizaid A (2015) Growth hormone secretagogue receptor dimers: a new pharmacological target. eNeuro 2(2):1–16
Perello M, Cabral A, Cornejo MP, De Francesco PN, Fernandez G, Uriarte M (2019) Brain accessibility delineates the central effects of circulating ghrelin. J Neuroendocrinol 31(7):e12677
Yanagi S, Sato T, Kangawa K, Nakazato M (2018) The homeostatic force of ghrelin. Cell Metab 27(4):786–804
Uriarte M, De Francesco PN, Fernandez G, Castrogiovanni D, D’Arcangelo M, Imbernon M, Cantel S, Denoyelle S, Fehrentz JA, Praetorius J et al (2021) Circulating ghrelin crosses the blood-cerebrospinal fluid barrier via growth hormone secretagogue receptor dependent and independent mechanisms. Mol Cell Endocrinol 538:111449
Yang J, Brown MS, Liang G, Grishin NV, Goldstein JL (2008) Identification of the acyltransferase that octanoylates ghrelin, an appetite-stimulating peptide hormone. Cell 132(3):387–396
Shimizu H, Oh IS, Okada S, Mori M (2009) Nesfatin-1: an overview and future clinical application. Endocr J 56(4):537–543
Mohan H, Gasner M, Ramesh N, Unniappan S (2016) Ghrelin, ghrelin-O-acyl transferase, nucleobindin-2/nesfatin-1 and prohormone convertases in the pancreatic islets of Sprague Dawley rats during development. J Mol Histol 47(3):325–336
Kerbel B, Unniappan S (2012) Nesfatin-1 suppresses energy intake, co-localises ghrelin in the brain and gut, and alters ghrelin, cholecystokinin and orexin mRNA expression in goldfish. J Neuroendocrinol 24(2):366–377
Bertucci JI, Blanco AM, Sanchez-Bretano A, Unniappan S, Canosa LF (2019) Ghrelin and NUCB2/nesfatin-1 co-localization with digestive enzymes in the intestine of pejerrey (Odontesthes bonariensis). Anat Rec (Hoboken) 302(6):973–982
Prinz P, Goebel-Stengel M, Teuffel P, Rose M, Klapp BF, Stengel A (2016) Peripheral and central localization of the nesfatin-1 receptor using autoradiography in rats. Biochem Biophys Res Commun 470(3):521–527
Rupp SK, Wolk E, Stengel A (2021) Nesfatin-1 receptor: distribution, signaling and increasing evidence for a G protein-coupled receptor—a systematic review. Front Endocrinol (Lausanne) 12:740174
Ishida E, Hashimoto K, Shimizu H, Okada S, Satoh T, Kato I, Yamada M, Mori M (2012) Nesfatin-1 induces the phosphorylation levels of cAMP response element-binding protein for intracellular signaling in a neural cell line. PLoS ONE 7(12):e50918
Jiang L, Xu K, Li J, Zhou X, Xu L, Wu Z, Ma C, Ran J, Hu P, Bao J et al (2020) Nesfatin-1 suppresses interleukin-1beta-induced inflammation, apoptosis, and cartilage matrix destruction in chondrocytes and ameliorates osteoarthritis in rats. Aging (Albany NY) 12(2):1760–1777
Wu D, Yang M, Chen Y, Jia Y, Ma ZA, Boden G, Li L, Yang G (2014) Hypothalamic nesfatin-1/NUCB2 knockdown augments hepatic gluconeogenesis that is correlated with inhibition of mTOR-STAT3 signaling pathway in rats. Diabetes 63(4):1234–1247
Li T, Wei S, Fan C, Tang D, Luo D (2021) Nesfatin-1 promotes proliferation, migration and invasion of HTR-8/SVneo trophoblast cells and inhibits oxidative stress via activation of PI3K/AKT/mTOR and AKT/GSK3beta pathway. Reprod Sci 28(2):550–561
Dong J, Xu H, Xu H, Wang PF, Cai GJ, Song HF, Wang CC, Dong ZT, Ju YJ, Jiang ZY (2013) Nesfatin-1 stimulates fatty-acid oxidation by activating AMP-activated protein kinase in STZ-induced type 2 diabetic mice. PLoS ONE 8(12):e83397
Heidarzadeh H, Zendehdel M, Babapour V, Gilanpour H (2018) The effect of Nesfatin-1 on food intake in neonatal chicks: role of CRF1/CRF2 and H1/H3 receptors. Vet Res Commun 42(1):39–47
Zhang T, Wang M, Liu L, He B, Hu J, Wang Y (2019) Hypothalamic nesfatin-1 mediates feeding behavior via MC3/4R-ERK signaling pathway after weight loss in obese Sprague–Dawley rats. Peptides 119:170080
Ying J, Zhang Y, Gong S, Chang Z, Zhou X, Li H, Tao J, Zhang G (2015) Nesfatin-1 suppresses cardiac L-type Ca(2)(+) channels through melanocortin type 4 receptor and the novel protein kinase C theta isoform pathway. Cell Physiol Biochem 36(2):555–568
Yosten GL, Samson WK (2010) The anorexigenic and hypertensive effects of nesfatin-1 are reversed by pretreatment with an oxytocin receptor antagonist. Am J Physiol Regul Integr Comp Physiol 298(6):R1642-1647
Florent G, Guenievre R, Bernadette F, Jean-Denis T, Michel D, Anne A (2019) Interaction between nesfatin-1 and oxytocin in the modulation of the swallowing reflex. Brain Res 1711:173–182
Zhang X, Qi J, Tang N, Wang S, Wu Y, Chen H, Tian Z, Wang B, Chen D, Li Z (2018) Intraperitoneal injection of nesfatin-1 primarily through the CCK-CCK1R signal pathway affects expression of appetite factors to inhibit the food intake of Siberian sturgeon (Acipenser baerii). Peptides 109:14–22
Howick K, Griffin BT, Cryan JF, Schellekens H (2017) From belly to brain: targeting the ghrelin receptor in appetite and food intake regulation. Int J Mol Sci 18(2):273
Zigman JM, Jones JE, Lee CE, Saper CB, Elmquist JK (2006) Expression of ghrelin receptor mRNA in the rat and the mouse brain. J Comp Neurol 494(3):528–548
Yin Y, Li Y, Zhang W (2014) The growth hormone secretagogue receptor: its intracellular signaling and regulation. Int J Mol Sci 15(3):4837–4855
Xiao X, Bi M, Jiao Q, Chen X, Du X, Jiang H (2020) A new understanding of GHSR1a–independent of ghrelin activation. Ageing Res Rev 64:101187
Blanco AM, Bertucci JI, Sanchez-Bretano A, Delgado MJ, Valenciano AI, Unniappan S (2017) Ghrelin modulates gene and protein expression of digestive enzymes in the intestine and hepatopancreas of goldfish (Carassius auratus) via the GHS-R1a: possible roles of PLC/PKC and AC/PKA intracellular signaling pathways. Mol Cell Endocrinol 442:165–181
Chung H, Seo S, Moon M, Park S (2008) Phosphatidylinositol-3-kinase/Akt/glycogen synthase kinase-3 beta and ERK1/2 pathways mediate protective effects of acylated and unacylated ghrelin against oxygen-glucose deprivation-induced apoptosis in primary rat cortical neuronal cells. J Endocrinol 198(3):511–521
Martins L, Fernandez-Mallo D, Novelle MG, Vazquez MJ, Tena-Sempere M, Nogueiras R, Lopez M, Dieguez C (2012) Hypothalamic mTOR signaling mediates the orexigenic action of ghrelin. PLoS ONE 7(10):e46923
Mazzocchi G, Neri G, Rucinski M, Rebuffat P, Spinazzi R, Malendowicz LK, Nussdorfer GG (2004) Ghrelin enhances the growth of cultured human adrenal zona glomerulosa cells by exerting MAPK-mediated proliferogenic and antiapoptotic effects. Peptides 25(8):1269–1277
Damian M, Marie J, Leyris JP, Fehrentz JA, Verdie P, Martinez J, Baneres JL, Mary S (2012) High constitutive activity is an intrinsic feature of ghrelin receptor protein: a study with a functional monomeric GHS-R1a receptor reconstituted in lipid discs. J Biol Chem 287(6):3630–3641
Jiang H, Betancourt L, Smith RG (2006) Ghrelin amplifies dopamine signaling by cross talk involving formation of growth hormone secretagogue receptor/dopamine receptor subtype 1 heterodimers. Mol Endocrinol 20(8):1772–1785
Kern A, Albarran-Zeckler R, Walsh HE, Smith RG (2012) Apo-ghrelin receptor forms heteromers with DRD2 in hypothalamic neurons and is essential for anorexigenic effects of DRD2 agonism. Neuron 73(2):317–332
Damian M, Pons V, Renault P, M’Kadmi C, Delort B, Hartmann L, Kaya AI, Louet M, Gagne D, Salah BH, K., et al (2018) GHSR-D2R heteromerization modulates dopamine signaling through an effect on G protein conformation. Proc Natl Acad Sci USA 115(17):4501–4506
Kern A, Grande C, Smith RG (2014) Apo-ghrelin receptor (apo-GHSR1a) regulates dopamine signaling in the brain. Front Endocrinol (Lausanne) 5:129
Rediger A, Piechowski CL, Yi CX, Tarnow P, Strotmann R, Gruters A, Krude H, Schoneberg T, Tschop MH, Kleinau G et al (2011) Mutually opposite signal modulation by hypothalamic heterodimerization of ghrelin and melanocortin-3 receptors. J Biol Chem 286(45):39623–39631
Schellekens H, De Francesco PN, Kandil D, Theeuwes WF, McCarthy T, van Oeffelen WE, Perello M, Giblin L, Dinan TG, Cryan JF (2015) Ghrelin’s orexigenic effect is modulated via a serotonin 2C receptor interaction. ACS Chem Neurosci 6(7):1186–1197
Huang XF, Weston-Green K, Yu Y (2018) Decreased 5-HT2cR and GHSR1a interaction in antipsychotic drug-induced obesity. Obes Rev 19(3):396–405
Xue Q, Bai B, Ji B, Chen X, Wang C, Wang P, Yang C, Zhang R, Jiang Y, Pan Y et al (2018) Ghrelin through GHSR1a and OX1R reveals a heterodimers galphas-cAMP-cAMP response element binding protein signaling pathway in vitro. Front Mol Neurosci 11:245
Wallace Fitzsimons SE, Chruscicka B, Druelle C, Stamou P, Nally K, Dinan TG, Cryan JF, Schellekens H (2019) A ghrelin receptor and oxytocin receptor heterocomplex impairs oxytocin mediated signalling. Neuropharmacology 152:90–101
Park S, Jiang H, Zhang H, Smith RG (2012) Modification of ghrelin receptor signaling by somatostatin receptor-5 regulates insulin release. Proc Natl Acad Sci U S A 109(46):19003–19008
Ozturk CC, Oktay S, Yuksel M, Akakin D, Yarat A, Kasimay Cakir O (2015) Anti-inflammatory effects of nesfatin-1 in rats with acetic acid—induced colitis and underlying mechanisms. J Physiol Pharmacol 66(5):741–750
Ramanjaneya M, Chen J, Brown JE, Tripathi G, Hallschmid M, Patel S, Kern W, Hillhouse EW, Lehnert H, Tan BK et al (2010) Identification of nesfatin-1 in human and murine adipose tissue: a novel depot-specific adipokine with increased levels in obesity. Endocrinology 151(7):3169–3180
Shiiya T, Nakazato M, Mizuta M, Date Y, Mondal MS, Tanaka M, Nozoe S, Hosoda H, Kangawa K, Matsukura S (2002) Plasma ghrelin levels in lean and obese humans and the effect of glucose on ghrelin secretion. J Clin Endocrinol Metab 87(1):240–244
Schalla MA, Stengel A (2018) Current understanding of the role of nesfatin-1. J Endocr Soc 2(10):1188–1206
Iwasaki Y, Nakabayashi H, Kakei M, Shimizu H, Mori M, Yada T (2009) Nesfatin-1 evokes Ca2+ signaling in isolated vagal afferent neurons via Ca2+ influx through N-type channels. Biochem Biophys Res Commun 390(3):958–962
Psilopanagioti A, Makrygianni M, Nikou S, Logotheti S, Papadaki H (2020) Nucleobindin 2/nesfatin-1 expression and colocalisation with neuropeptide Y and cocaine- and amphetamine-regulated transcript in the human brainstem. J Neuroendocrinol 32(9):e12899
Saito R, So M, Motojima Y, Matsuura T, Yoshimura M, Hashimoto H, Yamamoto Y, Kusuhara K, Ueta Y (2016) Activation of nesfatin-1-containing neurones in the hypothalamus and brainstem by peripheral administration of anorectic hormones and suppression of feeding via central nesfatin-1 in rats. J Neuroendocrinol 28(9)
Dong J, Guan HZ, Jiang ZY, Chen X (2014) Nesfatin-1 influences the excitability of glucosensing neurons in the dorsal vagal complex and inhibits food intake. PLoS ONE 9(6):e98967
Shimizu H, Oh IS, Hashimoto K, Nakata M, Yamamoto S, Yoshida N, Eguchi H, Kato I, Inoue K, Satoh T et al (2009) Peripheral administration of nesfatin-1 reduces food intake in mice: the leptin-independent mechanism. Endocrinology 150(2):662–671
Maejima Y, Sedbazar U, Suyama S, Kohno D, Onaka T, Takano E, Yoshida N, Koike M, Uchiyama Y, Fujiwara K et al (2009) Nesfatin-1-regulated oxytocinergic signaling in the paraventricular nucleus causes anorexia through a leptin-independent melanocortin pathway. Cell Metab 10(5):355–365
Wernecke K, Lamprecht I, Johren O, Lehnert H, Schulz C (2014) Nesfatin-1 increases energy expenditure and reduces food intake in rats. Obesity (Silver Spring) 22(7):1662–1668
Date Y, Shimbara T, Koda S, Toshinai K, Ida T, Murakami N, Miyazato M, Kokame K, Ishizuka Y, Ishida Y et al (2006) Peripheral ghrelin transmits orexigenic signals through the noradrenergic pathway from the hindbrain to the hypothalamus. Cell Metab 4(4):323–331
Faulconbridge LF, Cummings DE, Kaplan JM, Grill HJ (2003) Hyperphagic effects of brainstem ghrelin administration. Diabetes 52(9):2260–2265
Wren AM, Small CJ, Abbott CR, Dhillo WS, Seal LJ, Cohen MA, Batterham RL, Taheri S, Stanley SA, Ghatei MA et al (2001) Ghrelin causes hyperphagia and obesity in rats. Diabetes 50(11):2540–2547
Scott MM, Perello M, Chuang JC, Sakata I, Gautron L, Lee CE, Lauzon D, Elmquist JK, Zigman JM (2012) Hindbrain ghrelin receptor signaling is sufficient to maintain fasting glucose. PLoS ONE 7(8):e44089
Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Ueno N, Makino S, Fujimiya M, Niijima A, Fujino MA et al (2001) Ghrelin is an appetite-stimulatory signal from stomach with structural resemblance to motilin. Gastroenterology 120(2):337–345
Date Y, Murakami N, Toshinai K, Matsukura S, Niijima A, Matsuo H, Kangawa K, Nakazato M (2002) The role of the gastric afferent vagal nerve in ghrelin-induced feeding and growth hormone secretion in rats. Gastroenterology 123(4):1120–1128
Maniscalco JW, Edwards CM, Rinaman L (2020) Ghrelin signaling contributes to fasting-induced attenuation of hindbrain neural activation and hypophagic responses to systemic cholecystokinin in rats. Am J Physiol Regul Integr Comp Physiol 318(5):R1014–R1023
Zhang W, Zhang C, Fritze D, Chai B, Li J, Mulholland MW (2013) Modulation of food intake by mTOR signalling in the dorsal motor nucleus of the vagus in male rats: focus on ghrelin and nesfatin-1. Exp Physiol 98(12):1696–1704
Bake T, Le May MV, Edvardsson CE, Vogel H, Bergstrom U, Albers MN, Skibicka KP, Farkas I, Liposits Z, Dickson SL (2020) Ghrelin receptor stimulation of the lateral parabrachial nucleus in rats increases food intake but not food motivation. Obesity (Silver Spring) 28(8):1503–1511
Yuan JH, Chen X, Dong J, Zhang D, Song K, Zhang Y, Wu GB, Hu XH, Jiang ZY, Chen P (2017) Nesfatin-1 in the lateral parabrachial nucleus inhibits food intake, modulates excitability of glucosensing neurons, and enhances UCP1 expression in brown adipose tissue. Front Physiol 8:235
Zhang C, Yuan J, Lin Q, Li M, Wang L, Wang R, Chen X, Jiang Z, Zhu K, Chang X et al (2020) Ghrelin in the lateral parabrachial nucleus influences the excitability of glucosensing neurons, increases food intake and body weight. Endocr Connect 9(12):1168–1177
Le May MV, Peris-Sampedro F, Stoltenborg I, Schele E, Bake T, Adan RAH, Dickson SL (2021) Functional and neurochemical identification of ghrelin receptor (GHSR)-expressing cells of the lateral parabrachial nucleus in mice. Front Neurosci 15:633018
Chen X, Shu X, Cong ZK, Jiang ZY, Jiang H (2015) Nesfatin-1 acts on the dopaminergic reward pathway to inhibit food intake. Neuropeptides 53:45–50
Dore R, Krotenko R, Reising JP, Murru L, Sundaram SM, Di Spiezio A, Muller-Fielitz H, Schwaninger M, Johren O, Mittag J et al (2020) Nesfatin-1 decreases the motivational and rewarding value of food. Neuropsychopharmacology 45:1645–1655
Abizaid A, Liu ZW, Andrews ZB, Shanabrough M, Borok E, Elsworth JD, Roth RH, Sleeman MW, Picciotto MR, Tschop MH et al (2006) Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J Clin Invest 116(12):3229–3239
Pierre A, Van Schuerbeek A, Allaoui W, Van Laere S, Singewald N, Van Eeckhaut A, Smolders I, De Bundel D (2020) Effects of ghrelin receptor activation on forebrain dopamine release, conditioned fear and fear extinction in C57BL/6J mice. J Neurochem 154(4):389–403
Skibicka KP, Shirazi RH, Rabasa-Papio C, Alvarez-Crespo M, Neuber C, Vogel H, Dickson SL (2013) Divergent circuitry underlying food reward and intake effects of ghrelin: dopaminergic VTA-accumbens projection mediates ghrelin’s effect on food reward but not food intake. Neuropharmacology 73:274–283
Dore R, Krotenko R, Reising JP, Murru L, Sundaram SM, Di Spiezio A, Muller-Fielitz H, Schwaninger M, Johren O, Mittag J et al (2020) Nesfatin-1 decreases the motivational and rewarding value of food. Neuropsychopharmacology 45(10):1645–1655
Perello M, Sakata I, Birnbaum S, Chuang JC, Osborne-Lawrence S, Rovinsky SA, Woloszyn J, Yanagisawa M, Lutter M, Zigman JM (2010) Ghrelin increases the rewarding value of high-fat diet in an orexin-dependent manner. Biol Psychiatry 67(9):880–886
Chuang JC, Perello M, Sakata I, Osborne-Lawrence S, Savitt JM, Lutter M, Zigman JM (2011) Ghrelin mediates stress-induced food-reward behavior in mice. J Clin Invest 121(7):2684–2692
Al Massadi O, Nogueiras R, Dieguez C, Girault JA (2019) Ghrelin and food reward. Neuropharmacology 148:131–138
Stengel A, Mori M, Tache Y (2013) The role of nesfatin-1 in the regulation of food intake and body weight: recent developments and future endeavors. Obes Rev 14(11):859–870
Gotoh K, Masaki T, Chiba S, Ando H, Shimasaki T, Mitsutomi K, Fujiwara K, Katsuragi I, Kakuma T, Sakata T et al (2013) Nesfatin-1, corticotropin-releasing hormone, thyrotropin-releasing hormone, and neuronal histamine interact in the hypothalamus to regulate feeding behavior. J Neurochem 124(1):90–99
Noetzel S, Stengel A, Inhoff T, Goebel M, Wisser AS, Bannert N, Wiedenmann B, Klapp BF, Tache Y, Monnikes H et al (2009) CCK-8S activates c-Fos in a dose-dependent manner in nesfatin-1 immunoreactive neurons in the paraventricular nucleus of the hypothalamus and in the nucleus of the solitary tract of the brainstem. Regul Pept 157(1–3):84–91
Price CJ, Samson WK, Ferguson AV (2008) Nesfatin-1 inhibits NPY neurons in the arcuate nucleus. Brain Res 1230:99–106
Stengel A, Goebel M, Wang L, Rivier J, Kobelt P, Monnikes H, Lambrecht NW, Tache Y (2009) Central nesfatin-1 reduces dark-phase food intake and gastric emptying in rats: differential role of corticotropin-releasing factor2 receptor. Endocrinology 150(11):4911–4919
Cowley MA, Smith RG, Diano S, Tschop M, Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M, Heiman ML et al (2003) The distribution and mechanism of action of ghrelin in the CNS demonstrates a novel hypothalamic circuit regulating energy homeostasis. Neuron 37(4):649–661
Yang Y, Atasoy D, Su HH, Sternson SM (2011) Hunger states switch a flip-flop memory circuit via a synaptic AMPK-dependent positive feedback loop. Cell 146(6):992–1003
Cornejo MP, Denis RGP, Garcia Romero G, Fernandez G, Reynaldo M, Luquet S, Perello M (2021) Ghrelin treatment induces rapid and delayed increments of food intake: a heuristic model to explain ghrelin’s orexigenic effects. Cell Mol Life Sci 78(19–20):6689–6708
Shintani M, Ogawa Y, Ebihara K, Aizawa-Abe M, Miyanaga F, Takaya K, Hayashi T, Inoue G, Hosoda K, Kojima M et al (2001) Ghrelin, an endogenous growth hormone secretagogue, is a novel orexigenic peptide that antagonizes leptin action through the activation of hypothalamic neuropeptide Y/Y1 receptor pathway. Diabetes 50(2):227–232
Seoane LM, Lopez M, Tovar S, Casanueva FF, Senaris R, Dieguez C (2003) Agouti-related peptide, neuropeptide Y, and somatostatin-producing neurons are targets for ghrelin actions in the rat hypothalamus. Endocrinology 144(2):544–551
Wu CS, Bongmba OYN, Yue J, Lee JH, Lin L, Saito K, Pradhan G, Li DP, Pan HL, Xu A et al (2017) Suppression of GHS-R in AgRP Neurons Mitigates Diet-Induced Obesity by Activating Thermogenesis. Int J Mol Sci 18(4):832
Willesen MG, Kristensen P, Romer J (1999) Co-localization of growth hormone secretagogue receptor and NPY mRNA in the arcuate nucleus of the rat. Neuroendocrinology 70(5):306–316
Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB (2008) Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11(9):998–1000
Huang HH, Chen LY, Doong ML, Chang SC, Chen CY (2017) alpha-melanocyte stimulating hormone modulates the central acyl ghrelin-induced stimulation of feeding, gastrointestinal motility, and colonic secretion. Drug Des Devel Ther 11:2377–2386
Dos-Santos RC, Reis LC, Perello M, Ferguson AV, Mecawi AS (2019) The actions of ghrelin in the paraventricular nucleus: energy balance and neuroendocrine implications. Ann N Y Acad Sci 1455:81–97
Cabral A, Fernandez G, Tolosa MJ, Rey Moggia A, Calfa G, De Francesco PN, Perello M (2020) Fasting induces remodeling of the orexigenic projections from the arcuate nucleus to the hypothalamic paraventricular nucleus, in a growth hormone secretagogue receptor-dependent manner. Mol Metab 32:69–84
Lopez M, Lage R, Saha AK, Perez-Tilve D, Vazquez MJ, Varela L, Sangiao-Alvarellos S, Tovar S, Raghay K, Rodriguez-Cuenca S et al (2008) Hypothalamic fatty acid metabolism mediates the orexigenic action of ghrelin. Cell Metab 7(5):389–399
Yamanaka A, Beuckmann CT, Willie JT, Hara J, Tsujino N, Mieda M, Tominaga M, Yagami K, Sugiyama F, Goto K et al (2003) Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38(5):701–713
So M, Hashimoto H, Saito R, Yamamoto Y, Motojima Y, Ueno H, Sonoda S, Yoshimura M, Maruyama T, Kusuhara K et al (2018) Inhibition of ghrelin-induced feeding in rats by pretreatment with a novel dual orexin receptor antagonist. J Physiol Sci 68(2):129–136
Hsu TM, Hahn JD, Konanur VR, Noble EE, Suarez AN, Thai J, Nakamoto EM, Kanoski SE (2015) Hippocampus ghrelin signaling mediates appetite through lateral hypothalamic orexin pathways. Elife 4:e11190
Mano-Otagiri A, Ohata H, Iwasaki-Sekino A, Nemoto T, Shibasaki T (2009) Ghrelin suppresses noradrenaline release in the brown adipose tissue of rats. J Endocrinol 201(3):341–349
Stevanovic D, Trajkovic V, Muller-Luhlhoff S, Brandt E, Abplanalp W, Bumke-Vogt C, Liehl B, Wiedmer P, Janjetovic K, Starcevic V et al (2013) Ghrelin-induced food intake and adiposity depend on central mTORC1/S6K1 signaling. Mol Cell Endocrinol 381(1–2):280–290
Stengel A, Goebel M, Tache Y (2011) Nesfatin-1: a novel inhibitory regulator of food intake and body weight. Obes Rev 12(4):261–271
Saito ES, Kaiya H, Tachibana T, Tomonaga S, Denbow DM, Kangawa K, Furuse M (2005) Inhibitory effect of ghrelin on food intake is mediated by the corticotropin-releasing factor system in neonatal chicks. Regul Pept 125(1–3):201–208
Jonaidi H, Abbassi L, Yaghoobi MM, Kaiya H, Denbow DM, Kamali Y, Shojaei B (2012) The role of GABAergic system on the inhibitory effect of ghrelin on food intake in neonatal chicks. Neurosci Lett 520(1):82–86
Merali Z, Cayer C, Kent P, Anisman H (2008) Nesfatin-1 increases anxiety- and fear-related behaviors in the rat. Psychopharmacology 201(1):115–123
Kuhne SG, Schalla MA, Friedrich T, Kobelt P, Goebel-Stengel M, Long M, Rivalan M, Winter Y, Rose M, Stengel A (2018) Nesfatin-130–59 injected intracerebroventricularly increases anxiety, depression-like behavior, and anhedonia in normal weight rats. Nutrients 10(12):1889
Schalla MA, Kuhne SG, Friedrich T, Kobelt P, Goebel-Stengel M, Long M, Rivalan M, Winter Y, Mori M, Rose M et al (2020) Central blockage of nesfatin-1 has anxiolytic effects but does not prevent corticotropin-releasing factor-induced anxiety in male rats. Biochem Biophys Res Commun 529(3):773–777
Ge JF, Xu YY, Qin G, Pan XY, Cheng JQ, Chen FH (2015) Nesfatin-1, a potent anorexic agent, decreases exploration and induces anxiety-like behavior in rats without altering learning or memory. Brain Res 1629:171–181
Gunay H, Tutuncu R, Aydin S, Dag E, Abasli D (2012) Decreased plasma nesfatin-1 levels in patients with generalized anxiety disorder. Psychoneuroendocrinology 37(12):1949–1953
Hofmann T, Stengel A, Ahnis A, Busse P, Elbelt U, Klapp BF (2013) NUCB2/nesfatin-1 is associated with elevated scores of anxiety in female obese patients. Psychoneuroendocrinology 38(11):2502–2510
Hofmann T, Weibert E, Ahnis A, Obbarius A, Elbelt U, Rose M, Klapp BF, Stengel A (2017) Alterations of circulating NUCB2/nesfatin-1 during short term therapeutic improvement of anxiety in obese inpatients. Psychoneuroendocrinology 79:107–115
Carlini VP, Monzon ME, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR (2002) Ghrelin increases anxiety-like behavior and memory retention in rats. Biochem Biophys Res Commun 299(5):739–743
Carlini VP, Varas MM, Cragnolini AB, Schioth HB, Scimonelli TN, de Barioglio SR (2004) Differential role of the hippocampus, amygdala, and dorsal raphe nucleus in regulating feeding, memory, and anxiety-like behavioral responses to ghrelin. Biochem Biophys Res Commun 313(3):635–641
Currie PJ, Khelemsky R, Rigsbee EM, Dono LM, Coiro CD, Chapman CD, Hinchcliff K (2012) Ghrelin is an orexigenic peptide and elicits anxiety-like behaviors following administration into discrete regions of the hypothalamus. Behav Brain Res 226(1):96–105
Kanehisa M, Akiyoshi J, Kitaichi T, Matsushita H, Tanaka E, Kodama K, Hanada H, Isogawa K (2006) Administration of antisense DNA for ghrelin causes an antidepressant and anxiolytic response in rats. Prog Neuropsychopharmacol Biol Psychiatry 30(8):1403–1407
Jensen M, Ratner C, Rudenko O, Christiansen SH, Skov LJ, Hundahl C, Woldbye DP, Holst B (2016) Anxiolytic-like effects of increased ghrelin receptor signaling in the amygdala. Int J Neuropsychopharmacol 19(5):123
Lu Y, Niu M, Qiu X, Cao H, Xing B, Sun Y, Zhou Z, Zhou Y (2019) Acute but not chronic calorie restriction defends against stress-related anxiety and despair in a GHS-R1a-dependent manner. Neuroscience 412:94–104
Zhang F, Xu F, Mi X, Dong L, Xiao Y, Jiang S, Li GD, Zhou Y (2020) Ghrelin/GHS-R1a signaling plays different roles in anxiety-related behaviors after acute and chronic caloric restriction. Biochem Biophys Res Commun 529(4):1131–1136
Ari M, Ozturk OH, Bez Y, Oktar S, Erduran D (2011) High plasma nesfatin-1 level in patients with major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 35(2):497–500
Xu YY, Liang J, Cao Y, Shan F, Liu Y, Xia QR (2017) High levels of nesfatin-1 in relation to the dysfunction of the hypothalamic-pituitary-adrenal and hypothalamus-pituitary-thyroid axes in depressed patients with subclinical hypothyroidism. Neuropsychiatr Dis Treat 13:1647–1653
Bloem B, Xu L, Morava E, Faludi G, Palkovits M, Roubos EW, Kozicz T (2012) Sex-specific differences in the dynamics of cocaine- and amphetamine-regulated transcript and nesfatin-1 expressions in the midbrain of depressed suicide victims vs. controls. Neuropharmacology 62(1):297–303
Ge JF, Xu YY, Qin G, Peng YN, Zhang CF, Liu XR, Liang LC, Wang ZZ, Chen FH (2015) Depression-like behavior induced by nesfatin-1 in rats: involvement of increased immune activation and imbalance of synaptic vesicle proteins. Front Neurosci 9:429
Xia QR, Liang J, Cao Y, Shan F, Liu Y, Xu YY (2018) Increased plasma nesfatin-1 levels may be associated with corticosterone, IL-6, and CRP levels in patients with major depressive disorder. Clin Chim Acta 480:107–111
Algul S, Ozcelik O (2018) Evaluating the levels of nesfatin-1 and ghrelin hormones in patients with moderate and severe major depressive disorders. Psychiatry Investig 15(2):214–218
Ma Q, Li X, Yan Z, Jiao H, Wang T, Hou Y, Jiang Y, Liu Y, Chen J (2019) Xiaoyaosan ameliorates chronic immobilization stress-induced depression-like behaviors and anorexia in rats: the role of the nesfatin-1-oxytocin-proopiomelanocortin neural pathway in the hypothalamus. Front Psychiatry 10:910
Korucu CC, Atay IM, Zayif SS, Gultekin F (2018) May nesfatin-1 be a state marker in major depressive disorder with suicidal ideation? Psychiatry Res 267:272–276
Karadeniz S, Yaman H, Bilginer C, Hizarci Bulut S, Yaman SO (2020) Serum nesfatin-1, ghrelin, and lipid levels in adolescents with first episode drug naive unipolar depression. Nord J Psychiatry 74(8):613–619
Liu M, Shen X, Du X, Jiang H (2020) Plasma levels of nesfatin-1 as a new biomarker in depression in Asians: evidence from meta-analysis. Biomarkers 25(3):228–234
Wittekind DA, Kluge M (2015) Ghrelin in psychiatric disorders—a review. Psychoneuroendocrinology 52:176–194
Agawa S, Futagami S, Yamawaki H, Ikeda G, Noda H, Kirita K, Higuchi K, Murakami M, Kodaka Y, Ueki N et al (2019) Acylated ghrelin levels were associated with depressive status, physical quality of life, endoscopic findings based on Kyoto classification in Japan. J Clin Biochem Nutr 65(1):65–70
Naufel MF, Pedroso AP, Oyama LM, Telles MM, Hachul H, Ribeiro EB (2021) Preliminary evidence of acylated ghrelin association with depression severity in postmenopausal women. Sci Rep 11(1):5319
Lutter M, Sakata I, Osborne-Lawrence S, Rovinsky SA, Anderson JG, Jung S, Birnbaum S, Yanagisawa M, Elmquist JK, Nestler EJ et al (2008) The orexigenic hormone ghrelin defends against depressive symptoms of chronic stress. Nat Neurosci 11(7):752–753
Kumar J, Chuang JC, Na ES, Kuperman A, Gillman AG, Mukherjee S, Zigman JM, McClung CA, Lutter M (2013) Differential effects of chronic social stress and fluoxetine on meal patterns in mice. Appetite 64:81–88
Carlini VP, Machado DG, Buteler F, Ghersi M, Ponzio MF, Martini AC, Schioth HB, de Cuneo MF, Rodrigues AL, de Barioglio SR (2012) Acute ghrelin administration reverses depressive-like behavior induced by bilateral olfactory bulbectomy in mice. Peptides 35(2):160–165
Kluge M, Schussler P, Dresler M, Schmidt D, Yassouridis A, Uhr M, Steiger A (2011) Effects of ghrelin on psychopathology, sleep and secretion of cortisol and growth hormone in patients with major depression. J Psychiatr Res 45(3):421–426
Jackson TM, Ostrowski TD, Middlemas DS (2019) Intracerebroventricular ghrelin administration increases depressive-like behavior in male juvenile rats. Front Behav Neurosci 13:77
Guo L, Niu M, Yang J, Li L, Liu S, Sun Y, Zhou Z, Zhou Y (2019) GHS-R1a deficiency alleviates depression-related behaviors after chronic social defeat stress. Front Neurosci 13:364
Goebel M, Stengel A, Wang L, Tache Y (2009) Restraint stress activates nesfatin-1-immunoreactive brain nuclei in rats. Brain Res 1300:114–124
Okere B, Xu L, Roubos EW, Sonetti D, Kozicz T (2010) Restraint stress alters the secretory activity of neurons co-expressing urocortin-1, cocaine- and amphetamine-regulated transcript peptide and nesfatin-1 in the mouse Edinger–Westphal nucleus. Brain Res 1317:92–99
Stengel A, Goebel M, Wang L, Tache Y (2010) Abdominal surgery activates nesfatin-1 immunoreactive brain nuclei in rats. Peptides 31(2):263–270
Xu YY, Ge JF, Qin G, Peng YN, Zhang CF, Liu XR, Liang LC, Wang ZZ, Chen FH, Li J (2015) Acute, but not chronic, stress increased the plasma concentration and hypothalamic mRNA expression of NUCB2/nesfatin-1 in rats. Neuropeptides 54:47–53
Yoshida N, Maejima Y, Sedbazar U, Ando A, Kurita H, Damdindorj B, Takano E, Gantulga D, Iwasaki Y, Kurashina T et al (2010) Stressor-responsive central nesfatin-1 activates corticotropin-releasing hormone, noradrenaline and serotonin neurons and evokes hypothalamic-pituitary-adrenal axis. Aging (Albany NY) 2(11):775–784
Konczol K, Bodnar I, Zelena D, Pinter O, Papp RS, Palkovits M, Nagy GM, Toth ZE (2010) Nesfatin-1/NUCB2 may participate in the activation of the hypothalamic-pituitary-adrenal axis in rats. Neurochem Int 57(3):189–197
Nasri A, Unniappan S (2021) Nucleobindin-derived nesfatin-1 and nesfatin-1-like peptide stimulate pro-opiomelanocortin synthesis in murine AtT-20 corticotrophs through the cAMP/PKA/CREB signaling pathway. Mol Cell Endocrinol 536:111401
Ekizceli G, Halk KZ, Minbay Z, Eyigor O (2021) Nesfatin-1 and neuronostatin neurons are co-expressed with glucocorticoid receptors in the hypothalamus. Biotech Histochem 96(7):555–561
Pham V, Pemberton JG, Chang JP, Blanco AM, Nasri A, Unniappan S (2021) Nesfatin-1 stimulates the hypothalamus-pituitary-interrenal axis hormones in goldfish. Am J Physiol Regul Integr Comp Physiol 321(4):R603–R613
Saegusa Y, Takeda H, Muto S, Nakagawa K, Ohnishi S, Sadakane C, Nahata M, Hattori T, Asaka M (2011) Decreased plasma ghrelin contributes to anorexia following novelty stress. Am J Physiol Endocrinol Metab 301(4):E685-696
Kristenssson E, Sundqvist M, Astin M, Kjerling M, Mattsson H, Dornonville de la Cour C, Hakanson R, Lindstrom E (2006) Acute psychological stress raises plasma ghrelin in the rat. Regul Pept 134(2–3):114–117
Jeong JY, Lee DH, Kang SS (2013) Effects of chronic restraint stress on body weight, food intake, and hypothalamic gene expressions in mice. Endocrinol Metab (Seoul) 28(4):288–296
Nahata M, Saegusa Y, Sadakane C, Yamada C, Nakagawa K, Okubo N, Ohnishi S, Hattori T, Sakamoto N, Takeda H (2014) Administration of exogenous acylated ghrelin or rikkunshito, an endogenous ghrelin enhancer, improves the decrease in postprandial gastric motility in an acute restraint stress mouse model. Neurogastroenterol Motil 26(6):821–831
Bouillon-Minois JB, Trousselard M, Thivel D, Gordon BA, Schmidt J, Moustafa F, Oris C, Dutheil F (2021) Ghrelin as a biomarker of stress: a systematic review and meta-analysis. Nutrients 13(3):784
Asakawa A, Inui A, Kaga T, Yuzuriha H, Nagata T, Fujimiya M, Katsuura G, Makino S, Fujino MA, Kasuga M (2001) A role of ghrelin in neuroendocrine and behavioral responses to stress in mice. Neuroendocrinology 74(3):143–147
Takaya K, Ariyasu H, Kanamoto N, Iwakura H, Yoshimoto A, Harada M, Mori K, Komatsu Y, Usui T, Shimatsu A et al (2000) Ghrelin strongly stimulates growth hormone release in humans. J Clin Endocrinol Metab 85(12):4908–4911
Janzen WJ, Duncan CA, Riley LG (2012) Cortisol treatment reduces ghrelin signaling and food intake in tilapia. Oreochromis mossambicus Domest Anim Endocrinol 43(3):251–259
Otto B, Tschop M, Heldwein W, Pfeiffer AF, Diederich S (2004) Endogenous and exogenous glucocorticoids decrease plasma ghrelin in humans. Eur J Endocrinol 151(1):113–117
Zhu Q, Xiao K, Yu M, Niu M, Li C, Gao Y, Li GD, Zhou Y (2013) Ghrelin but not nesfatin-1 affects certain forms of learning and memory in both rats and mice. Brain Res 1541:42–51
Chen Z, Xu YY, Wu R, Han YX, Yu Y, Ge JF, Chen FH (2017) Impaired learning and memory in rats induced by a highfat diet: involvement with the imbalance of nesfatin-1 abundance and copine 6 expression. J Neuroendocrinol 29(4)
Erfani S, Moghimi A, Aboutaleb N, Khaksari M (2018) Nesfatin-1 improve spatial memory impairment following transient global cerebral ischemia/reperfusion via inhibiting microglial and caspase-3 activation. J Mol Neurosci 65(3):377–384
Diano S, Farr SA, Benoit SC, McNay EC, da Silva I, Horvath B, Gaskin FS, Nonaka N, Jaeger LB, Banks WA et al (2006) Ghrelin controls hippocampal spine synapse density and memory performance. Nat Neurosci 9(3):381–388
Beheshti S, Shahrokhi S (2015) Blocking the ghrelin receptor type 1a in the rat brain impairs memory encoding. Neuropeptides 52:97–102
Atcha Z, Chen WS, Ong AB, Wong FK, Neo A, Browne ER, Witherington J, Pemberton DJ (2009) Cognitive enhancing effects of ghrelin receptor agonists. Psychopharmacology 206(3):415–427
Berrout L, Isokawa M (2012) Ghrelin promotes reorganization of dendritic spines in cultured rat hippocampal slices. Neurosci Lett 516(2):280–284
Moon M, Kim S, Hwang L, Park S (2009) Ghrelin regulates hippocampal neurogenesis in adult mice. Endocr J 56(3):525–531
Li YH, Qing-Xiu L, Wang JS, Xiang H, Zhang RF, Huang CQ (2021) Ghrelin improves cognition via activation of the cAMP–CREB signalling pathway in depressed male C57BL/6J mice. Int J Neurosci :1–13
Zahiri, H., Rostampour, M., Khakpour, B., and Rohampour, K. (2021). The effect of ghrelin injection in the CA1 region of hippocampus on the MK801- induced memory impairment in wistar rats. Behav Brain Res 405(113209.
Bianconi S, Poretti MB, Rodriguez P, Maestri G, Rodriguez PE, de Barioglio SR, Schioth HB, Carlini VP (2021) Ghrelin restores memory impairment following olfactory bulbectomy in mice by activating hippocampal NMDA1 and MAPK1 gene expression. Behav Brain Res 410:113341
Spitznagel MB, Benitez A, Updegraff J, Potter V, Alexander T, Glickman E, Gunstad J (2010) Serum ghrelin is inversely associated with cognitive function in a sample of non-demented elderly. Psychiatry Clin Neurosci 64(6):608–611
Albarran-Zeckler RG, Brantley AF, Smith RG (2012) Growth hormone secretagogue receptor (GHS-R1a) knockout mice exhibit improved spatial memory and deficits in contextual memory. Behav Brain Res 232(1):13–19
Song L, Zhu Q, Liu T, Yu M, Xiao K, Kong Q, Zhao R, Li GD, Zhou Y (2013) Ghrelin modulates lateral amygdala neuronal firing and blocks acquisition for conditioned taste aversion. PLoS ONE 8(6):e65422
Li N, Li N, Xu F, Yu M, Qiao Z, Zhou Y (2021) Selectively increasing GHS-R1a expression in dCA1 excitatory/inhibitory neurons have opposite effects on memory encoding. Mol Brain 14(1):157
Tang CH, Fu XJ, Xu XL, Wei XJ, Pan HS (2012) The anti-inflammatory and anti-apoptotic effects of nesfatin-1 in the traumatic rat brain. Peptides 36(1):39–45
Wu GQ, Chou XM, Ji WJ, Yang XG, Lan LX, Sheng YJ, Shen YF, Li JR, Huang GZ, Yu WH et al (2016) The prognostic value of plasma nesfatin-1 concentrations in patients with traumatic brain injury. Clin Chim Acta 458:124–128
Nazarnezhad S, Rahmati M, Shayannia A, Abbasi Z, Salehi M, Khaksari M (2019) Nesfatin-1 protects PC12 cells against high glucose-induced cytotoxicity via inhibiting oxidative stress, autophagy and apoptosis. Neurotoxicology 74:196–202
Chen D, Cao S, Chang B, Ma T, Gao H, Tong Y, Li T, Han J, Yi X (2019) Increasing hypothalamic nucleobindin 2 levels and decreasing hypothalamic inflammation in obese male mice via diet and exercise alleviate obesity-associated hypogonadism. Neuropeptides 74:34–43
Cheng Y, Chen B, Xie W, Chen Z, Yang G, Cai Y, Shang H, Zhao W (2020) Corrigendum to “Ghrelin attenuates secondary brain injury following intracerebral hemorrhage by inhibiting NLRP3 inflammasome activation and promoting Nrf2/ARE signaling pathway in mice” [Int Immunopharmacol 79 (2020) 106180]. Int Immunopharmacol 84:106461
Wu CR, Yang QY, Chen QW, Li CQ, He WY, Zhao YP, Wang L (2021) Ghrelin attenuate cerebral microvascular leakage by regulating inflammation and apoptosis potentially via a p38 MAPK-JNK dependent pathway. Biochem Biophys Res Commun 552:37–43
Jiao Q, Du X, Li Y, Gong B, Shi L, Tang T, Jiang H (2017) The neurological effects of ghrelin in brain diseases: beyond metabolic functions. Neurosci Biobehav Rev 73:98–111
Santos VV, Stark R, Rial D, Silva HB, Bayliss JA, Lemus MB, Davies JS, Cunha RA, Prediger RD, Andrews ZB (2017) Acyl ghrelin improves cognition, synaptic plasticity deficits and neuroinflammation following amyloid beta (Abeta1–40) administration in mice. J Neuroendocrinol 29(5)
Shen XL, Song N, Du XX, Li Y, Xie JX, Jiang H (2017) Nesfatin-1 protects dopaminergic neurons against MPP(+)/MPTP-induced neurotoxicity through the C-Raf-ERK1/2-dependent anti-apoptotic pathway. Sci Rep 7:40961
Tan Z, Xu H, Shen X, Jiang H (2015) Nesfatin-1 antagonized rotenone-induced neurotoxicity in MES23.5 dopaminergic cells. Peptides 69:109–114
Chen H, Li X, Ma H, Zheng W, Shen X (2021) Reduction in nesfatin-1 levels in the cerebrospinal fluid and increased nigrostriatal degeneration following ventricular administration of anti-nesfatin-1 antibody in mice. Front Neurosci 15:621173
Dong J, Song N, Xie J, Jiang H (2009) Ghrelin antagonized 1-methyl-4-phenylpyridinium (MPP(+))-induced apoptosis in MES23.5 cells. J Mol Neurosci 37(2):182–189
Shi L, Bian X, Qu Z, Ma Z, Zhou Y, Wang K, Jiang H, Xie J (2013) Peptide hormone ghrelin enhances neuronal excitability by inhibition of Kv7/KCNQ channels. Nat Commun 4:1435
Li C, Zhang F, Shi L, Zhang H, Tian Z, Xie J, Jiang H (2014) Nesfatin-1 decreases excitability of dopaminergic neurons in the substantia nigra. J Mol Neurosci 52(3):419–424
Stutz B, Nasrallah C, Nigro M, Curry D, Liu ZW, Gao XB, Elsworth JD, Mintz L, Horvath TL (2019) Dopamine neuronal protection in the mouse Substantia nigra by GHSR is independent of electric activity. Mol Metab 24:120–138
Moon M, Kim HG, Hwang L, Seo JH, Kim S, Hwang S, Kim S, Lee D, Chung H, Oh MS et al (2009) Neuroprotective effect of ghrelin in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease by blocking microglial activation. Neurotox Res 15(4):332–347
He X, Yuan W, Liu F, Feng J, Guo Y (2020) Acylated ghrelin is protective against 6-OHDA-induced neurotoxicity by regulating autophagic flux. Front Pharmacol 11:586302
Song N, Wang W, Jia F, Du X, Xie A, He Q, Shen X, Zhang J, Rogers JT, Xie J et al (2017) Assessments of plasma ghrelin levels in the early stages of parkinson’s disease. Mov Disord 32(10):1487–1491
Pietraszko W, Furgala A, Gorecka-Mazur A, Kwinta B, Kaszuba-Zwoinska J, Polak J, Fiszer U, Gil K, Krygowska-Wajs A (2020) Assessments of plasma acyl-ghrelin levels in Parkinson’s disease patients treated with deep brain stimulation. Peptides 128:170299
Kentish SJ, Li H, Frisby CL, Page AJ (2017) Nesfatin-1 modulates murine gastric vagal afferent mechanosensitivity in a nutritional state dependent manner. Peptides 89:35–41
Atsuchi K, Asakawa A, Ushikai M, Ataka K, Tsai M, Koyama K, Sato Y, Kato I, Fujimiya M, Inui A (2010) Centrally administered nesfatin-1 inhibits feeding behaviour and gastroduodenal motility in mice. NeuroReport 21(15):1008–1011
Li ZL, Xu L, Sun XR, Guo FF, Gong YL, Gao SL (2013) Central nesfatin-1 influences the excitability of ghrelin-responsive gastric distension neurons in the arcuate nucleus and reduces gastric motility in rats. Eur J Neurosci 38(11):3636–3643
Guo FF, Xu L, Gao SL, Sun XR, Li ZL, Gong YL (2015) The effects of nesfatin-1 in the paraventricular nucleus on gastric motility and its potential regulation by the lateral hypothalamic area in rats. J Neurochem 132(3):266–275
Gao S, Guo F, Sun X, Zhang N, Gong Y, Xu L (2016) The inhibitory effects of nesfatin-1 in ventromedial hypothalamus on gastric function and its regulation by nucleus accumbens. Front Physiol 7:634
Wang Q, Guo F, Sun X, Gao S, Li Z, Gong Y, Xu L (2014) Effects of exogenous nesfatin-1 on gastric distention-sensitive neurons in the central nucleus of the amygdala and gastric motility in rats. Neurosci Lett 582:65–70
Yang GT, Zhao HY, Kong Y, Sun NN, Dong AQ (2017) Study of the effects of nesfatin-1 on gastric function in obese rats. World J Gastroenterol 23(16):2940–2947
Szlachcic A, Sliwowski Z, Krzysiek-Maczka G, Majka J, Surmiak M, Pajdo R, Drozdowicz D, Konturek SJ, Brzozowski T (2013) New satiety hormone nesfatin-1 protects gastric mucosa against stress-induced injury: mechanistic roles of prostaglandins, nitric oxide, sensory nerves and vanilloid receptors. Peptides 49:9–20
Ramesh N, Mortazavi S, Unniappan S (2016) Nesfatin-1 stimulates cholecystokinin and suppresses peptide YY expression and secretion in mice. Biochem Biophys Res Commun 472(1):201–208
Wang WG, Chen X, Jiang H, Jiang ZY (2008) Effects of ghrelin on glucose-sensing and gastric distension sensitive neurons in rat dorsal vagal complex. Regul Pept 146(1–3):169–175
Xu L, Gong Y, Wang H, Sun X, Guo F, Gao S, Gu F (2014) The stimulating effect of ghrelin on gastric motility and firing activity of gastric-distension-sensitive hippocampal neurons and its underlying regulation by the hypothalamus. Exp Physiol 99(1):123–135
Gong Y, Xu L, Guo F, Pang M, Shi Z, Gao S, Sun X (2014) Effects of ghrelin on gastric distension sensitive neurons and gastric motility in the lateral septum and arcuate nucleus regulation. J Gastroenterol 49(2):219–230
Page AJ, Slattery JA, Milte C, Laker R, O’Donnell T, Dorian C, Brierley SM, Blackshaw LA (2007) Ghrelin selectively reduces mechanosensitivity of upper gastrointestinal vagal afferents. Am J Physiol Gastrointest Liver Physiol 292(5):G1376-1384
Kong J, Chuddy J, Stock IA, Loria PM, Straub SV, Vage C, Cameron KO, Bhattacharya SK, Lapham K, McClure KF et al (2016) Pharmacological characterization of the first in class clinical candidate PF-05190457: a selective ghrelin receptor competitive antagonist with inverse agonism that increases vagal afferent firing and glucose-dependent insulin secretion ex vivo. Br J Pharmacol 173(9):1452–1464
Cao SG, Wu H, Cai ZZ (2016) Dose-dependent effect of ghrelin on gastric emptying in rats and the related mechanism of action. Kaohsiung J Med Sci 32(3):113–117
Falken Y, Webb DL, Abraham-Nordling M, Kressner U, Hellstrom PM, Naslund E (2013) Intravenous ghrelin accelerates postoperative gastric emptying and time to first bowel movement in humans. Neurogastroenterol Motil 25(6):474–480
Cabral A, Cornejo MP, Fernandez G, De Francesco PN, Garcia-Romero G, Uriarte M, Zigman JM, Portiansky E, Reynaldo M, Perello M (2017) Circulating ghrelin acts on GABA neurons of the area postrema and mediates gastric emptying in male mice. Endocrinology 158(5):1436–1449
Ariga H, Tsukamoto K, Chen C, Mantyh C, Pappas TN, Takahashi T (2007) Endogenous acyl ghrelin is involved in mediating spontaneous phase III-like contractions of the rat stomach. Neurogastroenterol Motil 19(8):675–680
Fujino K, Inui A, Asakawa A, Kihara N, Fujimura M, Fujimiya M (2003) Ghrelin induces fasted motor activity of the gastrointestinal tract in conscious fed rats. J Physiol 550(Pt 1):227–240
Yakabi K, Kawashima J, Kato S (2008) Ghrelin and gastric acid secretion. World J Gastroenterol 14(41):6334–6338
Ma L, Tang H, Yin Y, Yu R, Zhao J, Li Y, Mulholland MW, Zhang W (2015) HDAC5-mTORC1 interaction in differential regulation of ghrelin and nucleobindin 2 (NUCB2)/nesfatin-1. Mol Endocrinol 29(11):1571–1580
Jiang S, Zhou W, Zhang X, Wang D, Zhu H, Hong M, Gong Y, Ye J, Fang F (2016) Developmental expression and distribution of nesfatin-1/NUCB2 in the canine digestive system. Acta Histochem 118(2):90–96
Tomasetto C, Wendling C, Rio MC, Poitras P (2001) Identification of cDNA encoding motilin related peptide/ghrelin precursor from dog fundus. Peptides 22(12):2055–2059
Watanabe A, Mochiki E, Kimura A, Kogure N, Yanai M, Ogawa A, Toyomasu Y, Ogata K, Ohno T, Suzuki H et al (2015) Nesfatin-1 suppresses gastric contractions and inhibits interdigestive migrating contractions in conscious dogs. Dig Dis Sci 60(6):1595–1602
Nakata M, Manaka K, Yamamoto S, Mori M, Yada T (2011) Nesfatin-1 enhances glucose-induced insulin secretion by promoting Ca(2+) influx through L-type channels in mouse islet beta-cells. Endocr J 58(4):305–313
Maejima Y, Horita S, Kobayashi D, Aoki M, O’Hashi R, Imai R, Sakamoto K, Mori M, Takasu K, Ogawa K et al (2017) Nesfatin-1 inhibits voltage gated K(+) channels in pancreatic beta cells. Peptides 95:10–15
Yang Y, Zhang B, Nakata M, Nakae J, Mori M, Yada T (2019) Islet beta-cell-produced NUCB2/nesfatin-1 maintains insulin secretion and glycemia along with suppressing UCP-2 in beta-cells. J Physiol Sci 69(5):733–739
Li Z, Gao L, Tang H, Yin Y, Xiang X, Li Y, Zhao J, Mulholland M, Zhang W (2013) Peripheral effects of nesfatin-1 on glucose homeostasis. PLoS ONE 8(8):e71513
Bertucci JI, Blanco AM, Unniappan S (2019) Nesfatin-1 regulates glucoregulatory genes in rainbow trout (Oncorhynchus mykiss). Comp Biochem Physiol A Mol Integr Physiol 235:121–130
Algul S, Ozcelik O, Oto G, Sarikaya M, Goceroglu RT, Embiyaoglu NM, Caf F, Oner S, Akcan AG (2021) Effects of curcumin administration on Nesfatin-1 levels in blood, brain and fat tissues of diabetic rats. Eur Rev Med Pharmacol Sci 25(3):1616–1621
Liu Y, Chen X, Qu Y, Song L, Lin Q, Li M, Su K, Li Y, Dong J (2020) Central nesfatin-1 activates lipid mobilization in adipose tissue and fatty acid oxidation in muscle via the sympathetic nervous system. BioFactors 46(3):454–464
Zhai T, Li SZ, Fan XT, Tian Z, Lu XQ, Dong J (2017) Circulating nesfatin-1 levels and type 2 diabetes: a systematic review and meta-analysis. J Diabetes Res 2017:7687098
Wang C, Wang Y, Hu W (2017) Association of the polymorphism in NUCB2 gene and the risk of type 2 diabetes. Diabetol Metab Syndr 9:39
Reimer MK, Pacini G, Ahren B (2003) Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology 144(3):916–921
Egido EM, Rodriguez-Gallardo J, Silvestre RA, Marco J (2002) Inhibitory effect of ghrelin on insulin and pancreatic somatostatin secretion. Eur J Endocrinol 146(2):241–244
Lindqvist A, Shcherbina L, Prasad RB, Miskelly MG, Abels M, Martinez-Lopez JA, Fred RG, Nergard BJ, Hedenbro J, Groop L et al (2020) Ghrelin suppresses insulin secretion in human islets and type 2 diabetes patients have diminished islet ghrelin cell number and lower plasma ghrelin levels. Mol Cell Endocrinol 511:110835
Dezaki K, Hosoda H, Kakei M, Hashiguchi S, Watanabe M, Kangawa K, Yada T (2004) Endogenous ghrelin in pancreatic islets restricts insulin release by attenuating Ca2+ signaling in beta-cells: implication in the glycemic control in rodents. Diabetes 53(12):3142–3151
Dezaki K, Kakei M, Yada T (2007) Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes 56(9):2319–2327
DiGruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing-Zitron C, van der Meulen T, Huising MO (2016) Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab 5(7):449–458
Chuang JC, Sakata I, Kohno D, Perello M, Osborne-Lawrence S, Repa JJ, Zigman JM (2011) Ghrelin directly stimulates glucagon secretion from pancreatic alpha-cells. Mol Endocrinol 25(9):1600–1611
Ukkola O (2011) Ghrelin in Type 2 diabetes mellitus and metabolic syndrome. Mol Cell Endocrinol 340(1):26–28
Bertucci JI, Blanco AM, Canosa LF, Unniappan S (2017) Glucose, amino acids and fatty acids directly regulate ghrelin and NUCB2/nesfatin-1 in the intestine and hepatopancreas of goldfish (Carassius auratus) in vitro. Comp Biochem Physiol A Mol Integr Physiol 206:24–35
Sun Y, Butte NF, Garcia JM, Smith RG (2008) Characterization of adult ghrelin and ghrelin receptor knockout mice under positive and negative energy balance. Endocrinology 149(2):843–850
Pradhan G, Wu CS, Villarreal D, Lee JH, Han HW, Gaharwar A, Tian Y, Fu W, Guo S, Smith RG et al (2021) Beta Cell GHS-R regulates insulin secretion and sensitivity. Int J Mol Sci 22(8):3950
Gao X, Zhang K, Song M, Li X, Luo L, Tian Y, Zhang Y, Li Y, Zhang X, Ling Y et al (2016) Role of nesfatin-1 in the reproductive axis of male rat. Sci Rep 6:32877
Garcia-Galiano D, Navarro VM, Roa J, Ruiz-Pino F, Sanchez-Garrido MA, Pineda R, Castellano JM, Romero M, Aguilar E, Gaytan F et al (2010) The anorexigenic neuropeptide, nesfatin-1, is indispensable for normal puberty onset in the female rat. J Neurosci 30(23):7783–7792
Rajeswari JJ, Hatef A, Unniappan S (2020) Nesfatin-1-like peptide suppresses hypothalamo-pituitary-gonadal mRNAs, gonadal steroidogenesis, and oocyte maturation in fishdagger. Biol Reprod 103(4):802–816
Hatef A, Unniappan S (2017) Gonadotropin-releasing hormone, kisspeptin, and gonadal steroids directly modulate nucleobindin-2/nesfatin-1 in murine hypothalamic gonadotropin-releasing hormone neurons and gonadotropes. Biol Reprod 96(3):635–651
Lents CA, Barb CR, Hausman GJ, Nonneman D, Heidorn NL, Cisse RS, Azain MJ (2013) Effects of nesfatin-1 on food intake and LH secretion in prepubertal gilts and genomic association of the porcine NUCB2 gene with growth traits. Domest Anim Endocrinol 45(2):89–97
Cao Z, Luo L, Yang J, Zhang L, Gao D, Xu T, Tong X, Zhang D, Wang Y, Li Y et al (2019) Stimulatory effects of NESFATIN-1 on meiotic and developmental competence of porcine oocytes. J Cell Physiol 234(10):17767–17774
Ciccimarra R, Bussolati S, Grasselli F, Grolli S, Paolucci M, Basini G (2020) Potential physiological involvement of nesfatin-1 in regulating swine granulosa cell functions. Reprod Fertil Dev 32(3):274–283
Lebrethon MC, Aganina A, Fournier M, Gerard A, Parent AS, Bourguignon JP (2007) Effects of in vivo and in vitro administration of ghrelin, leptin and neuropeptide mediators on pulsatile gonadotrophin-releasing hormone secretion from male rat hypothalamus before and after puberty. J Neuroendocrinol 19(3):181–188
Fernandez-Fernandez R, Tena-Sempere M, Navarro VM, Barreiro ML, Castellano JM, Aguilar E, Pinilla L (2005) Effects of ghrelin upon gonadotropin-releasing hormone and gonadotropin secretion in adult female rats: in vivo and in vitro studies. Neuroendocrinology 82(5–6):245–255
Babaei-Balderlou F, Khazali H (2016) Effects of ghrelin on sexual behavior and luteinizing hormone beta-subunit gene expression in male rats. J Reprod Infertil 17(2):88–96
Martini AC, Fernandez-Fernandez R, Tovar S, Navarro VM, Vigo E, Vazquez MJ, Davies JS, Thompson NM, Aguilar E, Pinilla L et al (2006) Comparative analysis of the effects of ghrelin and unacylated ghrelin on luteinizing hormone secretion in male rats. Endocrinology 147(5):2374–2382
Pan D, Wang K, Cao G, Fan K, Liu H, Li P, Li H, Chenguang D (2020) Inhibitory effect of central ghrelin on steroid synthesis affecting reproductive health in female mice. J Steroid Biochem Mol Biol 204:105750
Wang L, Fang F, Li Y, Zhang Y, Pu Y, Zhang X (2011) Role of ghrelin on testosterone secretion and the mRNA expression of androgen receptors in adult rat testis. Syst Biol Reprod Med 57(3):119–123
Afsar T, Jahan S, Razak S, Almajwal A, Abulmeaty M, Wazir H, Majeed A (2017) Obestatin modulates ghrelin’s effects on the basal and stimulated testosterone secretion by the testis of rat: an in vitro study. Physiol Res 66(1):93–98
Kheradmand A, Roshangar L, Taati M, Sirotkin AV (2009) Morphometrical and intracellular changes in rat ovaries following chronic administration of ghrelin. Tissue Cell 41(5):311–317
Fernandez-Fernandez R, Navarro VM, Barreiro ML, Vigo EM, Tovar S, Sirotkin AV, Casanueva FF, Aguilar E, Dieguez C, Pinilla L et al (2005) Effects of chronic hyperghrelinemia on puberty onset and pregnancy outcome in the rat. Endocrinology 146(7):3018–3025
Kluge M, Schussler P, Schmidt D, Uhr M, Steiger A (2012) Ghrelin suppresses secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) in women. J Clin Endocrinol Metab 97(3):E448-451
Lanfranco F, Bonelli L, Baldi M, Me E, Broglio F, Ghigo E (2008) Acylated ghrelin inhibits spontaneous luteinizing hormone pulsatility and responsiveness to naloxone but not that to gonadotropin-releasing hormone in young men: evidence for a central inhibitory action of ghrelin on the gonadal axis. J Clin Endocrinol Metab 93(9):3633–3639
Tropea A, Tiberi F, Minici F, Orlando M, Gangale MF, Romani F, Miceli F, Catino S, Mancuso S, Sanguinetti M et al (2007) Ghrelin affects the release of luteolytic and luteotropic factors in human luteal cells. J Clin Endocrinol Metab 92(8):3239–3245
El-Eshmawy MM, Abdel Aal IA, El Hawary AK (2010) Association of ghrelin and leptin with reproductive hormones in constitutional delay of growth and puberty. Reprod Biol Endocrinol 8:153
Unniappan S, Peter RE (2004) In vitro and in vivo effects of ghrelin on luteinizing hormone and growth hormone release in goldfish. Am J Physiol Regul Integr Comp Physiol 286(6):R1093-1101
Shepperd E, Peng C, Unniappan S (2012) Ghrelinergic system in fish ovaries and ghrelin inhibition of germinal vesicle breakdown in zebrafish oocytes. Gen Comp Endocrinol 176(3):426–431
Rak A, Gregoraszczuk EL (2008) Modulatory effect of ghrelin in prepubertal porcine ovarian follicles. J Physiol Pharmacol 59(4):781–793
Sirotkin AV, Meszarosova M (2010) Comparison of effects of leptin and ghrelin on porcine ovarian granulosa cells. Domest Anim Endocrinol 39(1):1–9
Zhang K, Wei HX, Zhang YH, Wang SH, Li Y, Dai YP, Li N (2007) Effects of ghrelin on in vitro development of porcine in vitro fertilized and parthenogenetic embryos. J Reprod Dev 53(3):647–653
Rak-Mardyla A, Wrobel A, Gregoraszczuk EL (2015) Ghrelin negatively affects the function of ovarian follicles in mature pigs by direct action on basal and gonadotropin-stimulated steroidogenesis. Reprod Sci 22(4):469–475
Suzuki H, Sasaki Y, Shimizu M, Matsuzaki M, Hashizume T, Kuwayama H (2010) Ghrelin and leptin did not improve meiotic maturation of porcine oocytes cultured in vitro. Reprod Domest Anim 45(5):927–930
Naseroleslami M, Sharifi M, Rakhshan K, Mokhtari B, Aboutaleb N (2020) Nesfatin-1 attenuates injury in a rat model of myocardial infarction by targeting autophagy, inflammation, and apoptosis. Arch Physiol Biochem :1–9
Tasatargil A, Kuscu N, Dalaklioglu S, Adiguzel D, Celik-Ozenci C, Ozdem S, Barutcigil A, Ozdem S (2017) Cardioprotective effect of nesfatin-1 against isoproterenol-induced myocardial infarction in rats: role of the Akt/GSK-3beta pathway. Peptides 95:1–9
Sharifi M, Nazarinia D, Ramezani F, Azizi Y, Naderi N, Aboutaleb N (2021) Necroptosis and RhoA/ROCK pathways: molecular targets of Nesfatin-1 in cardioprotection against myocardial ischemia/reperfusion injury in a rat model. Mol Biol Rep 48(3):2507–2518
Su RY, Geng XY, Yang Y, Yin HS (2021) Nesfatin-1 inhibits myocardial ischaemia/reperfusion injury through activating Akt/ERK pathway-dependent attenuation of endoplasmic reticulum stress. J Cell Mol Med 25(11):5050–5059
Shaparenko OV, Kravchun PG, Kravchun PP, Kadykova OI (2018) Nesfatin-1 role in remodeling of the left ventricle myocardium in patients with arterial hypertension and obesity. Wiad Lek 71(5):1006–1009
Meng Q, Lu Q, Zhang Z, Liu J, Lou Y, Wang Y, Liu J (2021) Nesfatin-1 inhibits free fatty acids (FFAs)-induced endothelial inflammation via Gfi1/NF-kappaB signaling. Biosci Biotechnol Biochem 86(1):47–55
Lu QB, Wang HP, Tang ZH, Cheng H, Du Q, Wang YB, Feng WB, Li KX, Cai WW, Qiu LY et al (2018) Nesfatin-1 functions as a switch for phenotype transformation and proliferation of VSMCs in hypertensive vascular remodeling. Biochim Biophys Acta Mol Basis Dis 1864(6 Pt A):2154–2168
Zhang JR, Lu QB, Feng WB, Wang HP, Tang ZH, Cheng H, Du Q, Wang YB, Li KX, Sun HJ (2018) Nesfatin-1 promotes VSMC migration and neointimal hyperplasia by upregulating matrix metalloproteinases and downregulating PPARgamma. Biomed Pharmacother 102:711–717
Ibe S, Kishimoto Y, Niki H, Saita E, Umei T, Miura K, Ikegami Y, Ohmori R, Kondo K, Momiyama Y (2019) Associations between plasma nesfatin-1 levels and the presence and severity of coronary artery disease. Heart Vessels 34(6):965–970
Yamawaki H, Takahashi M, Mukohda M, Morita T, Okada M, Hara Y (2012) A novel adipocytokine, nesfatin-1 modulates peripheral arterial contractility and blood pressure in rats. Biochem Biophys Res Commun 418(4):676–681
Sun N, Wang H, Wang L (2016) Protective effects of ghrelin against oxidative stress, inducible nitric oxide synthase and inflammation in a mouse model of myocardial ischemia/reperfusion injury via the HMGB1 and TLR4/NF-kappaB pathway. Mol Med Rep 14(3):2764–2770
Ruozi G, Bortolotti F, Falcione A, Dal Ferro M, Ukovich L, Macedo A, Zentilin L, Filigheddu N, Gortan Cappellari G, Baldini G et al (2015) AAV-mediated in vivo functional selection of tissue-protective factors against ischaemia. Nat Commun 6:7388
Eid RA, Zaki MSA, Al-Shraim M, Eleawa SM, El-Kott AF, Al-Hashem FH, Eldeen MA, Ibrahim H, Aldera H, Alkhateeb MA (2018) Subacute ghrelin administration inhibits apoptosis and improves ultrastructural abnormalities in remote myocardium post-myocardial infarction. Biomed Pharmacother 101:920–928
Karcz-Socha I, Zwirska-Korczala K, Zembala M, Borgiel-Marek H, Karcz K (2011) Ghrelin PYY 3–36 serum changes in left ventricular hypertrophic, insulin-resistant, hypertensive obese patients. Obes Facts 4(5):386–392
Mao Y, Tokudome T, Kishimoto I, Otani K, Nishimura H, Yamaguchi O, Otsu K, Miyazato M, Kangawa K (2015) Endogenous ghrelin attenuates pressure overload-induced cardiac hypertrophy via a cholinergic anti-inflammatory pathway. Hypertension 65(6):1238–1244
Wang Q, Sui X, Chen R, Ma PY, Teng YL, Ding T, Sui DJ, Yang P (2018) Ghrelin ameliorates angiotensin II-induced myocardial fibrosis by upregulating peroxisome proliferator-activated receptor gamma in young male rats. Biomed Res Int 2018:1–14
Wang Q, Liu AD, Li TS, Tang Q, Wang XC, Chen XB (2021) Ghrelin ameliorates cardiac fibrosis after myocardial infarction by regulating the Nrf2/NADPH/ROS pathway. Peptides 144:170613
Zhang M, Wang S, Pan Z, Ou T, Ma J, Liu H, Li R, Yang P, Han W, Guan S et al (2018) AMPK/NF-kappaB signaling pathway regulated by ghrelin participates in the regulation of HUVEC and THP1 Inflammation. Mol Cell Biochem 437(1–2):45–53
Ai W, Wu M, Chen L, Jiang B, Mu M, Liu L, Yuan Z (2017) Ghrelin ameliorates atherosclerosis by inhibiting endoplasmic reticulum stress. Fundam Clin Pharmacol 31(2):147–154
Wang L, Chen Q, Ke D, Li G (2017) Ghrelin inhibits atherosclerotic plaque angiogenesis and promotes plaque stability in a rabbit atherosclerotic model. Peptides 90:17–26
Poykko SM, Kellokoski E, Horkko S, Kauma H, Kesaniemi YA, Ukkola O (2003) Low plasma ghrelin is associated with insulin resistance, hypertension, and the prevalence of type 2 diabetes. Diabetes 52(10):2546–2553
Mao Y, Tokudome T, Kishimoto I (2016) Ghrelin and blood pressure regulation. Curr Hypertens Rep 18(2):15
Matsumura K, Tsuchihashi T, Fujii K, Abe I, Iida M (2002) Central ghrelin modulates sympathetic activity in conscious rabbits. Hypertension 40(5):694–699
Schwenke DO, Tokudome T, Kishimoto I, Horio T, Shirai M, Cragg PA, Kangawa K (2008) Early ghrelin treatment after myocardial infarction prevents an increase in cardiac sympathetic tone and reduces mortality. Endocrinology 149(10):5172–5176
Boshra V, Abbas AM (2017) Effects of peripherally and centrally applied ghrelin on the oxidative stress induced by renin angiotensin system in a rat model of renovascular hypertension. J Basic Clin Physiol Pharmacol 28(4):347–354
Beyaz S, Akbal E (2021) Increased serum nesfatin-1 levels in patients with inflammatory bowel diseases. Postgrad Med J 2020–139227
Karadeniz Cerit K, Koyuncuoglu T, Yagmur D, Peker Eyuboglu I, Sirvanci S, Akkiprik M, Aksu B, Dagli ET, Yegen BC (2020) Nesfatin-1 ameliorates oxidative bowel injury in rats with necrotizing enterocolitis: The role of the microbiota composition and claudin-3 expression. J Pediatr Surg 55(12):2797–2810
Kolgazi M, Cantali-Ozturk C, Deniz R, Ozdemir-Kumral ZN, Yuksel M, Sirvanci S, Yegen BC (2015) Nesfatin-1 alleviates gastric damage via direct antioxidant mechanisms. J Surg Res 193(1):111–118
Kolgazi M, Ozdemir-Kumral ZN, Cantali-Ozturk C, Demirci EK, Yuksel M, Sirvanci S, Yegen BC (2017) Anti-inflammatory effects of nesfatin-1 on acetic acid-induced gastric ulcer in rats: involvement of cyclo-oxygenase pathway. J Physiol Pharmacol 68(5):765–777
Wang ZZ, Chen SC, Zou XB, Tian LL, Sui SH, Liu NZ (2020) Nesfatin-1 alleviates acute lung injury through reducing inflammation and oxidative stress via the regulation of HMGB1. Eur Rev Med Pharmacol Sci 24(9):5071–5081
Hui J, Aulakh GK, Unniappan S, Singh B (2021) Loss of Nucleobindin-2/Nesfatin-1 increases lipopolysaccharide-induced murine acute lung inflammation. Cell Tissue Res 385(1):87–103
Arabaci Tamer S, Yildirim A, Koroglu MK, Cevik O, Ercan F, Yegen BC (2018) Nesfatin-1 ameliorates testicular injury and supports gonadal function in rats induced with testis torsion. Peptides 107:1–9
Jung JY, Jeong JB, Kim JW, Kim SH, Koh SJ, Kim BG, Lee KL (2015) Circulating ghrelin levels and obestatin/ghrelin ratio as a marker of activity in ulcerative colitis. Intest Res 13(1):68–73
Ates Y, Degertekin B, Erdil A, Yaman H, Dagalp K (2008) Serum ghrelin levels in inflammatory bowel disease with relation to disease activity and nutritional status. Dig Dis Sci 53(8):2215–2221
Maruna P, Gurlich R, Frasko R, Rosicka M (2005) Ghrelin and leptin elevation in postoperative intra-abdominal sepsis. Eur Surg Res 37(6):354–359
Kerem M, Bedirli A, Pasaoglu H, Unsal C, Yilmaz TU, Ofluoglu E, Sahin TT (2007) Role of ghrelin and leptin in predicting the severity of acute pancreatitis. Dig Dis Sci 52(4):950–955
Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, Lillard JW Jr, Taub DD (2004) Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest 114(1):57–66
Jacob A, Rajan D, Pathickal B, Balouch I, Hartman A, Wu R, Zhou M, Wang P (2010) The inhibitory effect of ghrelin on sepsis-induced inflammation is mediated by the MAPK phosphatase-1. Int J Mol Med 25(1):159–164
Koyuturk M, Sacan O, Karabulut S, Turk N, Bolkent S, Yanardag R, Bolkent S (2015) The role of ghrelin on apoptosis, cell proliferation and oxidant-antioxidant system in the liver of neonatal diabetic rats. Cell Biol Int 39(7):834–841
Zhou X, Xue C (2009) Ghrelin inhibits the development of acute pancreatitis and nuclear factor kappaB activation in pancreas and liver. Pancreas 38(7):752–757
Kalayci M, Kocdor MA, Kuloglu T, Sahin I, Sarac M, Aksoy A, Yardim M, Dalkilic S, Gursu O, Aydin S et al (2017) Comparison of the therapeutic effects of sildenafil citrate, heparin and neuropeptides in a rat model of acetic acid-induced gastric ulcer. Life Sci 186:102–110
Buzcu H, Ozbeyli D, Yuksel M, Cilingir Kaya OT, Kasimay Cakir O (2019) Nesfatin-1 protects from acute pancreatitis: role of melanocortin receptors. J Physiol Pharmacol 70(6)
Reich N, Holscher C (2020) Acylated ghrelin as a multi-targeted therapy for Alzheimer’s and Parkinson’s disease. Front Neurosci 14:614828
Liang Y, Yin W, Yin Y, Zhang W (2021) Ghrelin based therapy of metabolic diseases. Curr Med Chem 28(13):2565–2576
Strasser F (2012) Clinical application of ghrelin. Curr Pharm Des 18(31):4800–4812
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31771110, 82071429, 32171131), Shandong Province Natural Science Foundation (ZR2019ZD31, ZR2020MC072, ZR2020QH125) and Innovative Research Team of High-Level Local Universities in Shanghai. The authors would like to thank Prof. Linxiang Guo and Honglin Zhu for their kind assistance in polishing this manuscript.
Author information
Authors and Affiliations
Contributions
XC wrote the manuscript; XC and QJ created and edited the figures; MB and XD outlined the review and supervised the text; JD and HJ conceived and edited the final work. All the authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
We have no conflicts of interest to declare.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chen, X., Dong, J., Jiao, Q. et al. “Sibling” battle or harmony: crosstalk between nesfatin-1 and ghrelin. Cell. Mol. Life Sci. 79, 169 (2022). https://doi.org/10.1007/s00018-022-04193-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-022-04193-6