
Abstract
Dihydrosphingolipids refer to sphingolipids early in the biosynthetic pathway that do not contain a C4-trans-double bond in the sphingoid backbone: 3-ketosphinganine (3-ketoSph), dihydrosphingosine (dhSph), dihydrosphingosine-1-phosphate (dhS1P) and dihydroceramide (dhCer). Recent advances in research related to sphingolipid biochemistry have shed light on the importance of sphingolipids in terms of cellular signalling in health and disease. However, dihydrosphingolipids have received less attention and research is lacking especially in terms of their molecular mechanisms of action. This is despite studies implicating them in the pathophysiology of disease, for example dhCer in predicting type 2 diabetes in obese individuals, dhS1P in cardiovascular diseases and dhSph in hepato-renal toxicity. This review gives a comprehensive summary of research in the last 10–15 years on the dihydrosphingolipids, 3-ketoSph, dhSph, dhS1P and dhCer, and their relevant roles in different diseases. It also highlights gaps in research that could be of future interest.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Since their discovery in the 1800s, sphingolipids have been shown to play key roles in physiological and pathological states by functioning as mediators or effectors of cellular signals. They are integral components of all eukaryotic cell membranes. It is now known that sphingolipids play a role in cell apoptosis, autophagy, oxidative stress and inflammation [1, 2, 3] and in disease states such as cancer, multiple sclerosis and diabetes [4, 5, 6]. These cellular events are effected through activation and interaction of the sphingosine 1 phosphate receptors (S1PR1–5), enzymes such as sphingosine kinases (SK 1 and 2), ceramide synthases (CerS1–6) or sphingolipids such as sphingosine 1 phosphate (S1P), and ceramides (Cers) [7]. Accordingly, there is significant interest in targeting the enzyme of sphingolipid metabolism and S1PRs in the discovery of new therapies. The term sphingolipids extends to a lot of other lipids and enzymes within the sphingolipid de novo biosynthesis pathway (Fig. 1). These include 3-ketoSph, dhSph, dhS1P and dhCer, as well as enzymes such as serine palmitoyltransferase (SPT), dihydroceramide desaturases (Des 1 and 2), and ceramidases (CDases). Here we, attempt to give a comprehensive review of literature focusing on the evidence for the role of the aforementioned dihydropshingolipids in relevant disease states and the associative effects they may have or the possible roles they may play. The information presented in this review was derived through data searches in Ovid, Medline and Embase using the MeSH terms (dihydrosphingosine 1-phsophate, sphinganine 1 phosphate, 3-ketosphinganine, dihydroceramide, dihydrosphinganine and dihydrosphingosine) and keyword searches of the same. The articles derived from the search were limited to human and animal studies and the English language. It is hoped that this review will also shed light on much needed areas of research on the relevance of dihydrosphingolipids and their roles in diseases.

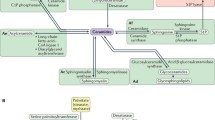
De novo sphingolipid biosynthesis pathway. In the de novo pathway, the condensation of palmitoyl-CoA and serine by the enzyme SPT forms 3-ketoSph. This is then reduced by 3-KR to dhSph. The acylation and phosphorylation of dhSph by CerS1-6 and SK 1 and 2 leads to the formation of dhCer and dhS1P, respectively. Des-1 and -2 then catalyze the desaturation of dhCer to Cer, which is a non-reversible reaction. The metabolization of Cer by CDase produces Sph. The production of S1P from Sph is exclusively phosphorylated by SK 1 and 2. S1P is then degraded to ethanolamine phosphate (EAP) and trans-2-hexadecenal by S1P lyase (SPL). DhS1P and S1P can be converted back to dhSph and Sph by S1P phosphatase (S1PP) and dhSph and Sph to dhCer and Cer, respectively, by cDase
De novo synthesis of sphingolipids
Briefly, apart from the de novo synthesis pathway (Fig. 1), sphingolipids are also synthesized through the salvage pathway and the sphingomyelin pathway. Regulation of plasma levels of sphingolipids generally occurs through the de novo synthesis pathway [8]. The backbone of the sphingolipids are Sph and dhSph, which are composed of an amino alcohol, from which all the other sphingolipids are derived by the enzymatic activity of a number of enzymes along the pathway. Most of the enzymatic activities along the de novo synthesis pathway are reversible except a few, including the conversion of dhCer to Cer. Thus, the enzymes responsible for this, Des-1 and -2, have now been described as gatekeepers [9].
The segment of the pathway that begins at Des-1 and -2 to S1P which includes Cer, Sph and S1P has been studied the most and their relevance in disease is well documented by other reviewers [10,11,12,13]. Therefore, in this review, the focus is on highlighting dhCer, dhS1P, dhSph and 3-ketoSph; dihydrosphingolipids; and the possible regulatory and contributory effects of these dihydrosphingolipids in diseases.
DhCer in disease
Overview and structure
DhCers lack the C4-double bond observed in Cers (Fig. 2); however, they also serve as precursors of complex sphingolipids such as dihydrosphingomyelins and dihydrogangliosides. For years, dhCers were thought to be biologically inactive due to them being less abundant, compared to Cers. This perception changed with the development of fenretinide [(N-(4-hydroxyphenyl)retinamide]-(4-HPR), which was found to inhibit Des-1 by the Merill Group [14]. Des-1 is found in all tissues, whereas Des-2 has been found in skin, intestine and kidney [15]. A later study further showed that the ablation of Des-1 and 2 shifts sphingolipid synthesis pathway toward the sphingolipid lacking the double bond introduced by Des-1 and -2, such as dhS1P, dhSph, dhsphingomyelin (dhSM) and especially dhCer [16]. Together, these discoveries led to new functional discoveries for dhCers in apoptosis, autophagy, hypoxia and cell proliferation, as reviewed by Siddique et al. [17].

Comparison of C2-ceramide with C2-dihydroceramide, without the double bond
In the conventional sphingolipid synthesis pathway, dhCers are produced as a result of the addition of fatty acyl-CoAs of differing chain lengths to dhSph by the enzyme CerS. The six isoforms of CerS expressed in mammalians are encoded by different chromosomes and exhibit preference for a defined chain length of fatty acyl-CoA [18], therefore portraying different functional, structural and biochemical attributes [19]. The dhCer chain lengths that are mentioned in this review are summarized in Table 1, except for the studies in cancer cells. It should be noted that most of the studies referenced in the table also had alterations in the Cer levels; however, they have not been mentioned due to the focus of the review in highlighting dhCers and the other dihydrosphingolipids. The majority of the studies in which dhCer has been mentioned, from 1990s to 2009, used cell penetrant dhCer bearing short acyl chains as negative controls in experiments tailored toward elucidating the effects of Cers in biological systems or disease conditions [20,21,22]. Due to the way in which these were used, most reported no effects and thus will not be included here. However, evidence contained in more recent studies paint a different picture of longer chain dhCers in terms of diseases.
DhCer in brain diseases
Research on sphingolipids in the brain has focussed on the glycolipids which include the cerebrosides, gangliosides and ceramide oligosaccharides as well as on Cers. Though Sun et al. [60] give a comprehensive review of the role of sphingolipids in stroke, the review does not highlight dihydrosphingolipids, which may be due to most of the studies focussing on other sphingolipids. Here, we highlight studies that have mentioned dhCer levels in brain-related diseases.
A study investigating the effects of hypoxia on sphingolipid metabolism in human cerebral endothelial cells found that dhCers (long chains) were increased together with other sphingolipids [23]. In addition, increased dhCer levels were also seen after subarachnoid haemorrhage [24]. Both of these studies allude to the involvement of dhCer in the mechanisms of disease in oxygen deprivation states such as stroke. Not only this, but dhCer levels have also been noted to be altered in studies related to certain neuronal diseases such as luekodystrophia [26], Alzheimer’s [61], Huntington’s disease (HD) [62] and in episodic migraineurs [25]. Though the cause of migraines is not so clear, genetic anomalies in the enzymes could have played a part in the reduced levels of dhCer seen in the migraine study, as shown by Matesanz et al. [63]. This study hypothesized that the splice variant of the acyl-coenzyme A synthase 5 (ACSL5) gene which lacked exon 20 (ACSL5-Δ20), could have led to the decrease in CerS, and thus dhCer levels. On the other hand, genetic mutations in other enzymes such as ACER3, which is an alkaline ceramidase (CDase), have been linked to elevated dhCer (C18:1 and C20:1) levels observed in the plasma of childhood leukodystrophic twin patients with a genetic mutation at p.E33G, responsible for the catalytic activity of ACER3. In Alzheimer’s disease, the inhibition of the gatekeeper enzyme, Des-1 byXM461 and XM462, increased dhCer levels in Alzheimer’s transgenic mice, which led to the induction of autophagy and reduced amyloid secretion by neuronal cells through loss of ribosomal protein S6 kinase (S6K) activity due to reduced mammalian target of rapamycin complex 1 (MTORC1) activity [64].The autophagy effect exerted by increased dhCer observed in this study is corroborated by studies in cancer cells that have shown similar effects [65, 66]. However, clinically, others have shown that increased plasma dihydroshingomyelin/dhCer and sphingomyelin/Cer ratios are predictive of slower progression among Alzheimer’s disease patients [61]. In addition, reduced dhCer (C18:0) including dhSph and dhS1P levels and mRNA expression of the enzymes CerS1 and serine palmotyltransferase long chain base 1 (SPLTC1) have been observed in transgenic mice brains manifesting HD. These reductions may be a result of the reduced level of SPLTC1, which impacts the entire de novo sphingolipid synthesis pathway. These studies show an association of dhCer with the progression of degenerative brain diseases as well as in other brain-related diseases, which makes it a potential target as a biomarker. There are also conceivable genetic associations of the enzymes in the sphingolipid pathway with neurodegenerative diseases. However, whether or not dhCer has a causal effect is an area that warrants further research.
DhCer in diabetes
It is now known that dyslipidaemia commonly occurs in diabetes [67], which is a major risk factor for developing cardiovascular diseases (CVDs) [68]. The main characteristic of dyslipidemia in diabetes is high triglyceride levels, reduced high-density lipids (HDL) and slightly elevated low-density lipids (LDL)-cholesterol, with a dominance of the atherogenic small dense LDL [69]. Studies have shown that the sphingolipid, S1P, is bound to HDL in plasma and its distribution is shifted to other non-HDL carriers in the plasma, when HDL levels are low [70]. A number of studies also support the role of Cer and Cer16:0 in particular, in insulin resistance and glucose intolerance [71,72,73,74]. These evidences show that sphingolipid metabolism and transport, including dhCer, can be altered in diabetes affecting insulin resistance and mitochondrial and adipose tissue homeostasis.
Insulin resistance
Insulin resistance is an important factor in type 2 diabetes and pre-diabetes [75], while chronic exposure to free fatty acids (FFA), such as palmitate, causes insulin resistance. In cellular models (C2C12 myotubes and isolated β-islet cells) of insulin resistance induced by palmitate, increased dhCer (C16:0), Cer (C16:0) and dhSph have been noted [27, 29]. The study in C2C12 myotubes also indicated the inhibition of the Akt/PKB pathway in promoting the insulin resistance [27], which is similar to how Cer has been shown to antagonize insulin signalling [72]. Others have shown that palmitate causes an increase in specific dhCer (C16:0, C18:0, C22:0, C24:1) and Cer chain lengths, resulting in glucolipotoxicity in beta cells [28]. These studies denote the changes in dhCer as associative effects, rather than a causal effect. There are recent studies which imply that dhCer and the de novo sphingolipids could have an additive effect to that of Cers. For example, Reali et al. [71] showed in their model of ob/ob mice macrophage that increases in the enzymatic activity of CerS6 led to increased Cer C16:0 and that the impairment of insulin signalling in these model occurred at 16 weeks when the levels of all the sphingolipids were upregulated. This increase in all sphingolipids provides a link to the clinical [76] and animal [36] studies that have shown increases in both dhCer and Cer levels. This is further supported by findings that the enzymes SPT, CerS and Des-1 are not specific to one type of sphingolipid in their sensitivity but quite diffuse [71], implying that they contribute towards balancing the regulation of sphingolipids. Perhaps, this is one of the reasons for the insignificant changes in Cer levels seen in the same cohort of patients with significant increase in dhCer levels [76]. Moreover, the type of abundant saturated fats available in the system could also determine the type of dhCer species produced. For example, when SPT is induced by high saturated fats, it has been shown to switch substrate specificity (palmitate to myristate), producing different dhCer C16:0 species [49]. In terms of therapy, increasing the expression of adiponectin receptors in single muscles of rats fed a high fat diet did increase the insulin sensitivity and also reduced the level of dhCer and Cer [77], which may be occurring through the adiponectin/AMP-activated protein kinase (AMPK) pathway. Activation of the adiponectin–AMPK pathway leads to inhibition of manoyl-CoA resulting in the increase of cartinine palmitoyltransferase 1 (CPT1), the rate-limiting step in fatty acid oxidation [78]. Furthermore, two other studies have also noted increase in the levels of dhCer and Cer in primary human trophoblasts (PHT) [35], and in cows transitioning from gestation to lactation [38], implicating these sphingolipids in gestational diabetes.
These studies show that the changes in dhCer levels in lipid-driven insulin signalling are directly related to it being a precursor to Cers and that Cer is involved in insulin resistance. However, it should be noted that these studies were aimed at Cer; therefore, the question of the effect that dhCers has on insulin signalling still remains unanswered.
Mitochondrial homeostasis
The clinical complications associated with type 2 diabetes such as dyslipidaemia, hyperglycaemia and insulin resistance are linked to mitochondrial defragmentation [79]. Mitochondrial homeostasis is maintained through a balance of fusion and fission, mitochondrial biogenesis and degradation. Increased longer chain dhCer due to ablation of Des-1−/− in mouse embryonic fibroblasts and Des-1 inhibition in C2C12 myotubes reduced mitochondrial respiration and complex IV (cytochrome c oxidase) expression in the presence of lipopolysaccharides (LPS) [31, 80]. Complex IV catalyses the final step in the mitochondrial electron transfer chain and is thought to be a major regulation site for oxidative phosphorylation [30]. A reduction in complex IV would impair ATP synthesis. Introduction of LPS to the C2C12 myotubes caused an increase in Cers and had opposite effect to dhCers. LPS also increased oxidative stress and mitochondrial fission through dynamin-related protein 1 (DRP1) which was inhibited when SPT was inhibited by myriocin. Increase in DRP1 and oxidative stress leads to increased mitochondrial defragmentation and insulin resistance [81]. Another study has shown that dhCer (C2, 95% and C16, 51%) can inhibit Cer channel formation in mitochondria [32], inhibiting apoptosis. The study in mouse embryonic fibroblasts also found the Des-1−/− cells to be resistant to apoptosis through the Akt/PkB pathway, but had increased autophagy through AMPK activation as a result of the impaired ATP synthesis. These studies show that dhCer can disrupt the processes of mitochondrial biogenesis and degradation, and contribute towards improving mitochondrial function by increasing autophagy and decreasing apoptosis, inhibiting mitochondrial respiration and possibly inhibiting DRP1 and oxidative stress.
Apidose tissue homeostasis
A number of researchers have shown how the selective manipulation of Des-1 and its substrates may be a pathophysiologically advantageous strategy to improve adipose tissue homeostasis and ameliorate the burden of obesity-associated metabolic complications. For example, Barbarroja et al. [33] showed that an ablation in expression of Des-1 or the pharmacological inhibition of Des-1 in 3T3-L cells led to an increase in dhCer/Cer ratio with concurrent increases in oxidative stress, cell death and inhibition of cell differentiation. Their results also showed an increase in the protein expression of GLUT4, which facilitates the uptake of glucose from the plasma. Moreover, 5- to 16-fold increases in dhCer with activation of p38-MAPK, protein phosphorylated eukaryotic translation initiation factor 2α (PeIF2α) and autophagy markers (Beclin1 and LC3B II) have been observed in mature adipocytes treated with 4-HPR-fenretinide [34]. PeIF2α is involved in the nutrient stress response pathway, which has been shown to contribute to the pathogenesis of diabetes. In this study, 4-HPR-fenretinide was shown to utilize both retinoic acid receptor (RAR)-dependent and -independent pathways to regulate adipogenesis and prevent obesity in mice fed a high fat diet. The RA-dependent pathway results in increased Cer despite the presence of 4-HPR-fenretinide, an example of which is given by Bikman et al. [37]. 4-HPR-fenretinide is a structural derivative of retinoic acid, and research in cancer cells has also shown that this compound and dhCer are associated with the activation of cellular stress responses and induction of autophagy [65, 82]. In fact, a recent study in kidney cells has shown that 4-HRP-fenretinide induced polyubiquitination of Des-1, which exhibited “gain of function” and activated pro-survival pathways, p38 MAPK, JNK and X-Box Protein-1s [83]. In addition, dhCers directly suppressed the transcriptional activity of peroxisome proliferator-activated receptor gamma (PPARγ) similar to that seen in Degs1 (Des-1 regulatory gene) ablation, which also suppressed cyclins (D1, D3 and E) and cyclin-dependent kinase 2 (cdk2), thus impairing adipocyte programming in pre-adipocytes [33]. PPARγ plays a central role in adipogenesis and lipid metabolism [84]. We would like to note that the inhibition or ablation of Des-1 led to feedback inhibition and downregulation of SPLTC1 and CerS6, a systemic counter balancing mechanism which could be triggered by the increased dhCer levels.
These studies showed that dhCer could be involved in the disruption of adipogenesis and cause cell death either as a direct result of Des-1 inhibition or by itself. Since the inhibition of Des-1 certainly leads to dhCer accumulation, it is possible that it disrupted adipogenesis early on through inhibition of PPARγ transcription, which is necessary for the terminal differentiation of the adipocytes, and the increased oxidative stress and cell death through autophagy can be attributed to dhCer. However, whether it functions as a ligand or has lipid–protein interactions or lipid–enzyme interactions is elusive since these studies focussed on Des-1.
Epidemiological findings
Epidemiological studies aimed at decoding the associations between sphingolipids and known risk factors [42, 43, 85] or markers for diabetes [39, 40] show increases in dhCer to be precedent of increases in Cer, with concomitant reductions seen when diet, exercise and anti-diabetics are introduced [45, 46]. While others found it to have no longitudinal or cross-sectional association with pre-diabetes or diabetes, Cer (C18:0, C22:1) did [39]. This can be attributed to the progression of the de novo synthesis pathway towards Cer. However, there are at least two studies which show dhCer levels to be opposite to that of Cers. One study found dhCer to be genetically correlated with waist circumference [41], while Cer was not, even after adjusting for confounders such as age and sex, and accounting for genetic differences by using polygenic models. The other study found dhCer to be elevated in the abdominal adipose tissue of obese and non-obese diabetics when compared to lean non-diabetics [39, 41, 86], with negative correlation between homeostasis model of insulin resistance score (HOMA-IR) and Cer. It is possible that sampling differences (plasma vs. adipose tissue from abdominal area) could account for the differences; however, the latter study did not adjust for patients taking the anti-diabetic metformin, which could have had an effect on the HOMA-IR scores. Despite these, there is evidence for dhCer to be used as a predictor for developing type 2 diabetes. A study showed dhCer C18:0 to be the single best predictor for progression to diabetes, with those progressing from non-diabetic to diabetic within 10 years having higher dhCer C18:0 at baseline [87]. Furthermore, researchers in the USA have recommended that the lipidomic risk score (LRS) assessment criteria—dhCer (C18:0) included in the criteria—be used in conjunction with metformin supplementation for individuals with high risk of developing type 2 diabetes [88]. The LRS score predicted future type 2 diabetes independently of prediabetes with an accuracy of 76%. Therefore, dhCer lipid profiling in obese patients could be a tool for predicting the onset of pre-diabetes and diabetes in this population.
In summary, apart from the epidemiological evidence showing its value as a predictor for developing type 2 diabetes, the in vitro and in vivo studies show a possible therapeutic potential in targeting the Des-1 enzyme and elevating dhCer, which could increase autophagy, reduce adipogenesis and lipid accumulation, leading to increased insulin sensitivity and glucose uptake as summarized in Fig. 3.

Possible effects of dhCer on adipocytes. The ablation or inhibition of Des-1 by drugs such as Fen (4-HRP-fenretinide) in adipocytes leads to increased dhCer, (1) reducing adipogenesis and (2) increasing autophagy and resulting in increased insulin sensitivity and glucose uptake. (1) Increased dhCer reduces adipogenesis by (a) causing endoplasmic reticulum (ER) stress or nutrient stress which then phosphorylates eIF2alpha downstream of PERK (Protein Kinase R-like Endoplasmic Reticulum Kinase), resulting in cell cycle arrest at G1, and (b) the increased dhCer also inhibits ligand activation of PPARγ. Both of these lead to reduced differentiation of adipocytes due to reduced expression of cyclins D1, D3 and E and cdk2. (2) DhCer also increases autophagy by reducing mitochondrial respiration and complex IV, which results in reduced ATP synthesis. The impaired ATP synthesis leads to increased AMPK, activating the phosphorylation of ULK1 (unc-51 like Autophagy Activating Kinase 1), Beclin 1 and LC3B II, which are involved in the initiation and formation of autophagomsomes. This leads to increased expression of autophagy genes such as atg7 and E1-like, thus increasing autophagy. An increase in AMPK also increases GLUT4 translocation to the cell membrane, leading to increased glucose uptake. The hypothesis of dhCer acting as a ligand to activate RARα thus inhibiting PPARγ remains to be deciphered (light blue dotted line)
DhCer in aging and disease
As age increases, lipid dysregulation increases also and gives rise to the risk of developing CVDs. A current epidemiological report released by the American Heart Association (AHA) highlighted that 48.6% of adults aged ≥ 40 years in the USA are eligible for statin “lipid-lowering” therapy [89]. Chronological aging has a tremendous effect on cardiorespiratory fitness (CRF) and low levels are representative of risk factors for CVDs, dyslipidaemia and hypertension [90,91,92]. CRF refers to the ability of the cardiac and respiratory systems to supply oxygen to skeletal muscles during sustained physical activity. Increased C20:0 dhCer was found to be strongly associated with lower CRF in both men and women aged 54–96 years [47], while C24:0 dhCer was not. This connection of dhCer to hypoxia is supported by evidence in mice hypoxia models, which showed elevated levels of dhCer C16:0 in the right ventricles [48] and in the heart [93] from week 4 to week 8, with a concomitant decrease in Cer and expression of Des-1. The latter study identified that the Des-1 promoter harbours overlapping sites for HAND2 and nuclear factor of activated T cell (NFATC) transcription factors, which have been shown to be important in the development of cardiac systems. Both of these factors were required for upregulation of Des-1, while the re-activation of HAND2 in failing hearts due to co-operation between NFATC and miRNA-125 has been shown to aid cardiac dysfunction [94]. Whether the hypoxia-induced dhCer is a protective mechanism even in reduced CRF through autophagic flux remains to be answered. Furthermore, increased local dhCer levels were shown to be associated with reductions in thymocyte apoptosis and age-associated thymic involution in aged mice, when growth hormones were introduced [95]. This most likely fostered autophagy in thymic epithelial cells, which shapes the T cell repertoire and tolerance. These contrasting effects of hypoxia and autophagy point to tissue-specific associations of dhCer. However, this remains inconclusive due to the lack of evidence with regard to dhCer in aging. Therefore, including dhCer and dihydrosphingolipids in future lipidomic profiling studies in the elderly should be encouraged.
DhCer in cardiovascular disease
Though cholesterol is vital for healthy bodily functions, excess amounts in the blood due to increased dietary intake of saturated fats can lead to buildup of atherosclerotic plaque and coronary artery disease (CAD), increasing the risk for heart attacks. Cers are known to be associated with cholesterol in terms of lipid rafts formation [96], which serve as the basis for signal transduction during inflammatory responses. In human atherosclerotic plaques [50] and rat models of hypercholesterolaemia [97], dhCers were found to be increased. Both dhCer and Cer correlated with the release of the inflammatory cytokine interleukin 6 (IL-6), but only dhCer correlated with macrophage inflammatory protein 1β (MIP-1β) release [50]. Elevated IL-6 levels in atherosclerosis results in effects on endothelial cells (activation), platelets (prothrombotic effect), muscle cells (proliferation) and macrophages (lipid accumulation) that are involved in lipid processing and plaque formation [98], while increased MIP-1β (also known as chemokine CC motif ligand 4—CCL4) in patients was linked to atherosclerosis and plaque instability [99]. What role this increase in dhCer plays in plaque stability is still debatable, since the extracellular addition of dhCer to human aortic smooth muscle cells did not cause apoptosis, whereas Cer did [50]. Apoptosis of cells in the vessel walls increases plaque instability. Apart from these CAD-related studies, dhCer levels have also been found to be elevated in patients with rheumatoid arthritis [100], patients with “HeartWare” left ventricular assist devices [101], hypertensive rats [102] and in doxorubicin-induced cardiac toxicity [103]. These studies point to the possible role of dhCer as a marker for cardiac pathology. The correlation between MIP-1β and dhCer should also be investigated further, since MIP-1β is also implicated in type 2 diabetes. However, there is a lack of mechanistic studies that are directed at determining whether dhCer has an associative or causal effect in CVDs.
DhCer in lung disease
Studies in lung diseases investigating dhCer were outnumbered by studies investigating Cer, S1P and Sph. As can be seen below, the few studies that did mention dhCer compared its role in hypoxia as opposed to Cer. 4-HPR-fenretinide treatment of Sprague–Dawley (SD) rat lungs with emphysema showed that there was increase in the dhCer levels with a decrease in hypoxia-inducible factor1-α (HIF1-α) and vascular endothelial growth factor (VEGF) protein expression [54], which was rescued with concurrent S1P treatment. Additionally, it is now known that dhCer does accumulate in states of hypoxia through the induction of autophagy and inhibits proliferation of primary rat lung-transformed cells [55]. These researchers proposed that the dhCer desaturation step acts as an oxygen sensor, based on the amplitude and kinetics of increased dhCer at physiological alterations of oxygen concentration. This can be explained by the requirement for oxygen by Des-1 and -2 to convert dhCer to Cer in the reaction involving nicotinamide adenine dinucleotide phosphate (NADPH) and nicotinamide adenine dinucleotide (NADH) [15]. In immortalized lung epithelial cells (IB3, A549 and C38) with defective expression of the cystic fibrosis transmembrane conductance regulator (CFTR) gene, the levels of C16:0 dhCer, Sph, SM and Cer (C22, C24 and C26) were increased [52]. The use of 4-HPR-fenretinide and fumonisin (FB1) reduced the level of these sphingolipids (individual species measurement not given) without affecting the level of CFTR, showing that CFTR could function in a feedback loop manner, sequestering sphingolipids and or altering the membrane structure. The increase in dhCer in mice with defective CFTR gene expression is comparable to the increase seen in those with emphysema, since both pathologies have an underlying hypoxic condition. However, in states of infection, the response differs, as shown by the increased airway sensitivity caused by reduced levels of de novo sphingolipids including dhCer (due to deletion of SPLTC2) in mice lung infected with rhinovirus [104], showing that sphingolipids may be protective in lung hypersensitivity reactions. These studies show regulating dhCer levels by targeting the enzymes involved in its modulation could be potential therapeutic targets for hypoxia-related disorders in the lung. However, whether the increased dhCer contributes to the disease or occurs as a coping mechanism is yet to be deciphered.
DhCer in liver disease
The excessive accumulation of lipids within hepatocytes is one of the factors listed in the pathogenesis of fatty liver or non-alcoholic fatty liver disease (NAFLD), which can progress to hepatic fibrosis and cancer if not managed well. Raised dhCer levels together with Cer have been observed in both NAFLD and hepatocellular carcinoma patients when compared to hepatitis C infection and cirrhosis patients, respectively [58, 59, 105]. However in diabetic patients with NAFLD, up to 12% of increase in dhCer has been noted, with negative correlations with insulin resistance [106]. However, cell and animal studies show some conflicting results. For example, reductions in the de novo sphingolipid pathway (knockout of SPTLC1) led to the occurrence of fatty liver, insulin resistance and elevated fasting glucose in mice [107], while knockdown of Des-1 in Huh7 hepatocyte cells led to increased dhCer, FFAs and diacylglrcerol [57]. This study also showed that silencing SPLTC1–3 showed positive effects such as increased nutrient uptake and reduction in lipid synthesis, whereas Des-1 silencing led to prominent changes in amino acid, sugar, and nucleotide metabolism and vesicle trafficking between organelles in Huh7 hepatocyte cells. These contrasting effects may be reconciled if SPLTC2 and 3 are considered to be still functional in the former study. This also implies that the ablation of Des-1 in hepatocytes may be detrimental, since increasing levels of FFA and diacylglycerol can cause lipotoxicity which activates a chain of events that eventually leads to hepatocyte death.
In human hepatocarcinoma (HepG2) cells, interleukin 1 (IL-1)-mediated sterile inflammation downregulated oroscomucoid like protein 3 (ORMDL3), a key regulator of SPT, leading to increased dhCers, dhSph and Cers [56]. Also, an integrated lipidomics and transcriptomics study in balb/c mice showed that the anti-inflammatory and immunosuppressive drug triptolide caused reductions in dhCer C18:0, C18:1, C20:0, C22:0 and C24:0 in the liver, and C22:0, C24:0, and C24:1 in plasma [108]. These studies suggest that inflammatory processes can also affect alterations in the level of individual species of dhCer in the liver and contribute to liver pathologies.
In the liver, dhCer together with sphingolipids seems to be part of the lipid pool that accumulates in disease states. However, due to the limited amount of studies specifically targeting dhCer in the liver, whether it has any effect remains to be answered.
DhCer in cancer and cancer therapy
As the investigation on Cer increased in cancer cells for combination therapy with various cancer treatments, due to its apoptotic property [109] it became apparent that dhCer could be bioactive. Most studies have regarded dhCer as a precursor to Cer [110,111,112]. However, there are studies that have demonstrated dhCer’s potential role in cancer cell autophagy [14, 66, 113], in cancer induced bone pain [114] and cell cytotoxicity [115]. The changes in the levels of dhCer and Cer in cancer cells also seem to differ according to the site of origin of the cancer. For example, in melanoma cells, dhCers (d18:0/16:0) and Cers were significantly lowered compared to non-malignant melanocytes [116], while in cancerous tissue of human endometrial cells the level of dhCer was increased 3- to 4.6-fold, and Cer and S1P were increased 1.6- to 1.9-fold [117]. The most effective way to understand the effects of dhCer on a biological system is through the inhibition of the gatekeeper enzyme, Des-1, which is now a target for cancer therapy.
DhCer induced autophagy as a result of Des-1 inhibition
The Des-1 inhibitor, 4-HPR-fenretinide, is currently under clinical trial for use in breast cancer therapy [118]. The anti-cancer effects of 4-HPR-fenretinide are thought to occur through the modulation of endogenous sphingolipids. A study by Rahmaniyan et al. [119] showed that 4-HPR-fenretinide does directly inhibit Des-1 with an IC50 of 2.32 µM in SMS-KCNR neuroblastoma cells. Others have shown that inhibiting SK sensitizes cells to 4-HPR-fenretinide’s cytotoxic effects due to increased dhCers [120]. These show that there is possible interaction between 4-HPR-fenretinide inhibition of Des-1 and SK activity, which has also been noted by others [113, 121,122,123]. Apart from these pharmacological agents, oxidative stress can also inhibit Des-1 in cancer cell lines such as HEK293, MCF 7, 549 and SMS-KCNR cells, leading to increased dhCers [124]. The raised exogenous dhCer levels seems to be capable of inducing autophagy; as shown inT98G, U87MG glioblastoma cells [66] and DU145 cells [14] and also reduce the proliferation of castration-resistant prostate cancer cells [125]. In the prostate cancer cells, reduction in proliferation occurred without inducing apoptosis and autophagy, perhaps through effects on the cell cycle. Additional support for dhCers autophagic effects in cancer cells is found in a study on human gastric cancer cell line, HGC-27, where the inhibition of Des-1 by XM462 and resveratrol led to the accumulation of dhCer at 16 h with induction of autophagy, whereas Cer was increased only slightly [113]. In addition, another study on U937 cells showed that dhCer did not induce apoptosis through DNA fragmentation, compared to Cers and tumour necrosis factor-α (TNF-α) [126]. The autophagy effect of dhCer seems to occur only when the de novo sphingolipid biosynthesis pathway is altered. This is because in studies where dhCer levels increased together with Cers, apoptosis occurred rather than autophagy. For instance, the anti-tumour effect of TNF-α in MCF-7 cells occurred through increased activity of CerS, which then drove the de novo sphingolipid synthesis forward, leading to accumulation of dhCers (C16:0, C18:0, C20:0, C22:0, C24:0, C24:1) and Cers [127] and thus regulating focal adhesion kinase (FAK) and apoptosis. Since the role of autophagy in tumours is highly context driven and can lead to either regression or advancement of tumours [128], this could also apply to targeting Des-1 inhibition as an anti-cancer therapy. This is evident in a recent study in leukaemia cells which found that dhCer accumulation and ROS generation were distinct and non-essential events in 4-HPR-fenretinide-induced cell death [129]. This is further confounded considering that 4-HPR-fenretinide can have both retinoic acid (RA)-dependent and -independent effects [34], and that it induces polyubiquitination of the enzyme [83]. Apart from these, Des-1 inhibition is also promising in terms of restraining metastasis. Studies have linked Des-1 to promotion of metastasis in prostate cancer cells [130], and oesophageal carcinoma [131]. It is worth mentioning that this promotional effect was regulated by RA without affecting the proliferative potential of the cell [130], maybe because Des-1 also increases cyclin D1 expression as a result of NF-кB activation [131].
In an effort to beat resistance to Foscan photodynamic therapy (PDT), some have studied its combination with 4-HPR-fenretinide. Their findings showed that the apoptotic effect was greater when combined, compared to either alone in SCC19 cell by increasing dhCer C16:0 and not Cer [132]. This combination also enhanced mitochondrial depolarization. PDT alone has been shown to induce accumulation of dhCer in SCC cells [133, 134] and was thought to effect the resistance by inhibiting the formation of ceramide channels in the mitochondria [133]. The reason for the enhanced effect when combined may be due to enhanced CerS activity and mitochondrial dysfunction [132]. This is supported by two different studies by Separovic et al. [135], which showed that SCC cells with silenced CerS1 or knockout of CerS6 genes treated with PDT had reduced levels of global Cers, dhCers (C18:0, C18:1 and C20:0) and decreased apoptosis. These findings also imply ROS as a mediator between Des-1 and CerS, since PDT induces cell cytotoxicity through ROS generation.
These studies contribute to the evidence that raised dhCer levels could potentially mean increased autophagic flux. Collectively, increasing dhCer levels to increase autophagy and inhibiting metastasis through Des-1 inhibition are promising targets for cancer therapy.
DhCer induced ER stress
Vitamin E, γ-tocotrienol (γ-TE), has been demonstrated to confer its anti-cancer effects through modulation of dhCer. A study by Jiang et al. [136] showed that γ-TE induced autophagy, necrosis and apoptosis in prostate cancer cells by increasing intracellular dhCer and dhSph, suppressing Akt phosphorylation. Suppression of the PI3K/Akt signalling pathway which leads to inhibition of NF-кB is a known target for γ-TEs anti- breast cancer effects [137]. In fact, in RAW264.7 macrophages, the shorter chain dhCer, C8:0, was linked to the anti-NF-кB effects of γ-TE, by enhancing ER stress and attenuating TNF-α-triggered increase in NF-кB [138]. What is interesting to note in this study is that dhCer C8:0 mimicked the effects of γ-TE by increasing the expression of the zinc finger protein A20, which is a negative feedback regulator of NF-кB. This also led to increased phosphorylation of eIF2α, cJun N-terminal kinase (JNK) and NF-кB inhibitor α (IкBα). Phosphorylation of the ER stress marker, eIF2α, has also been noted in adipocytes treated with 4-HPR-fenretinide [34]. In contrast, increased A20 in adipocytes has been shown to enhance adipogenesis by supressing NF-кB even in the presence of TNF-α [139]. These differences may be due to different NF-кB pathways being activated: canonical (involves TNF-α) vs. non canonical, apart from cellular differences. It is also possible that γ-TE may be inhibiting Des-1 or even the expression of certain CerS. Interference of the expression of CerS2, 5 and 6 has been shown to increase dhCer C16:0 and hexosyl-ceramide which also promoted ER stress [140]. This study also noted that the observation of the expression levels of individual CerS in MCF-7 cells leads to counter regulation of non-targeted CerS species with no significant differences in total sphingolipids.
DhCer in other diseases
In the kidney, Cer triggers the mitogen-activated protein kinase (MAPK/ ERK) cascade in glomerular mesangial cells [141] and the stress-activated protein kinase (SAPK/JNK) cascade in the endothelial cell; however, dhCer was not able to trigger the SAPK/JNK cascade [142] and whether it triggers the MAPK/ERK is yet to be deciphered. Dermatological studies have indicated dhCer’s possible role in heterogeneity of the stratum corneum layer [143]. In addition, others have found altered expressions of the enzymes CerS, cDase and SPT in the skin disease, “hidradenitis suppurativa” [144]. However, lack of measurement of the different sphingolipids was a limitation in this study. In the eye, increased dhCer (C18:1, C16:0) has been indicated as a possible contributor to cataracts in 64–70 year old [145].
Collectively, dhCers’ association with hypoxia possibly triggering autophagy is a recurrent finding in the brain, diabetes, aging, lung and cancer. The relevance of this effect depends on the pathophysiology of the disease, therefore indicating its potential applications as a biomarker or therapeutic target. The mechanistic aspects of this link between dhCer and hypoxia remain to be elucidated. Figure 4 gives a summary of the possible effects of increased dhCer as highlighted in this review.

DhCer in diseases. A summary of the potential effects of increased dhCer as highlighted in this review
DhS1P in disease
Overview and structure
DhS1P is derived from the phosphorylation of dhSph by SK1 and 2, and it is known to accumulate when CerS is inhibited [146]. It differs from S1P in that its backbone structure is composed of dhSph instead of Sph. The role of its chemical analog, S1P, as a signalling molecule in the regulation of cellular processes such as cell proliferation [147, 148] and neuroprotection [149] are now known and are being targeted for therapy. As in the case of the other dihydropshingolipids, research into the relevance of dhS1P in the cellular mechanisms of disease is fairly new and quite limited.
DhS1P in cerebrovascular disease
DhS1P has been shown to activate S1PRs [150] in neuronal progenitor cells, and the orphaned receptor GPR63 in the thalamus and nuclear-caudatus of the brain [151]. Recent studies have demonstrated reduced dhS1P levels in the brains of rat models of Alzheimer’s disease [152] and HD [62]. The reduced availability of dhSph due to a perturbation in the de novo synthesis pathway may led to reduced dhS1P levels, since it occurred in conjunction with reduced levels of dhSph and dhCer and the enzymes SPTLC1 and CerS1. Raised DhS1P may have a protective role in HD, since the accumulation of nuclear dhS1P has been shown to inhibit histone deacetylases (HDAC) [146], which is being targeted for HD therapy [153, 154]. The inhibition of HDAC results in increased gene expression that leads to increased cell proliferation, migration and decreased cell apoptosis. In addition, studies in neuronal cells also found that dhS1P increases Smad phosphorylation compared to S1P [150]. Smads are involved in neuronal precursor proliferation and differentiation. However, in nerve cells (PC12), dhS1P did not protect the cells from apoptosis, whereas S1P did [155]. The different cellular microenvironments could be the reason for this difference. This is exemplified by the inhibition of TGFβ-induced Smad 2/3 phosphorylation by dhS1P in dermal fibroblasts [156], which is opposite to the effects seen in neuronal cells. Other studies have also shown that the pharmacological inhibition of Des-1 in cerebellar neuron cells [157], hypoxia in cerebellar endothelial cells [23], and CerS inhibition by FB1 in neuronal progenitor cells [158] can raise the dhS1P levels. In addition, dhS1P has been identified as a potential marker in FB1–neural tube defect risk assessment [158]. These studies show that the inhibition of Des-1 or CerS reverses the sphingolipid metabolism reaction towards the dihydrosphingolipids and that of dhS1P, possibly by interfering with the activity levels of SPT and S1P lyase. It is obvious that DhS1P does have some form of influence on neuronal cells proliferation and differentiation, and could be a potential therapeutic target for neurodegenerative diseases such as HD.
DhS1P in cardiovascular disease
Similar to S1P, plasma erythrocyte and platelet levels of dhS1P differ in physiological states. In states of physical strain such as exercise, the dhS1P levels differ according to the type of activity, duration and training [159,160,161]. For example, in untrained man, the erythrocyte levels of dhS1P at 60 min of pedalling were elevated and remained markedly elevated post-exercise [159]. Thus, these differences are also most likely to be present in pathophysiological states.
Both in animal models of cardiomyopathies and patients with cardiomyopathies, altered sphingolipid levels have been noted. Having a major cardiac event such as a myocardial infarct (MI) has been shown to alter the levels of dhS1P in plasma (reduced at 1–6 h), erythrocytes (increased at 6 and 24 h), and platelets (reduced) in rats [162]. Reduced dhS1P and S1P have also been observed in left ventricular tissue of Wistar rats subjected to tachycardia [163]. Similar trends in plasma (reduced early on) and erythrocytes (increased early on) have been observed in patients with acute ST-segment elevation myocardial infarct (STEMI) [164], and MI [165]. It has been suggested that reduced plasma S1P enables erythrocytes to increase S1P production by increasing SK1 protein expression and activity [166]. Hypothetically, this may also be the case for dhS1P, since both were incidentally increased or decreased. Samples from patients in the Copenhagen City Heart Study (CCHS) showed that there was an inverse relationship between reduced dhS1P, S1P and Cer C24:1, and the occurrence of ischaemic heart disease (IHD) in the plasma fraction containing HDL [167]. This may be due to the decreased availability of HDL, implying that dhS1P may be bound to HDL just as S1P [168]. S1P is known to be positively and negatively correlated to CAD depending on the plasma HDL or non-HDL fraction it is bound to [70], while S1P released from activated platelets preferentially binds to the non-HDL fraction—Albumin [169]. Studies that have investigated dhS1P together with S1P have shown that dhS1P is found in non-activated platelets [170, 171], and it was increased in and released by activated platelets [170, 172]. Whether albumin-bound dhS1P and S1P influenced the outcomes observed in the CCHS study was not investigated.
Moreover, a shift in the balance between dhS1P, S1P and Cer within the platelets rather than erythrocytes may be aiding the cross talk in CAD, as observed in patients with multi-vessel CAD [171]. Together with the findings of reduced dhS1P contributing to reduced endothelial barrier [173], its positive correlation with increased miRNA-122 and 126 in improved endothelial barrier function [174], and dhS1P as a potent inducer of S1PR1-dependent endothelial barrier function and endothelial cell migration [167], it can be inferred that dhS1P may promote plaque stability. Even in human umbilical vein endothelial cells (HUVEC), dhS1P has been shown to inhibit chemotaxis and Rac activation stimulated by platelet-derived growth factor (PDGF) [175], which is known to promote atherosclerosis. Furthermore, dhS1P has been shown to induce matrix metalloproteinase 1 (MMP1) in dermal and scleroderma fibroblasts [176, 177], which is involved in plaque stability [178] and linked to reduced risk of coronary heart disease [179]. The downregulation of MMP1 is also a known marker for cardiac fibrosis. The study on scleroderma fibroblasts showed that dhS1P not only normalized MMP1 expression through the upregulation of phosphatase and tensin homolog (PTEN), but also inhibited factors known to promote fibrosis such as phosphorylated Smad3 (pSmad3), and collagen [177]. In the dermal fibroblasts, dhS1P induced the ERK 1/2-Etsl pathway, leading to increased MMP1 through one of its pertussis toxin-dependent receptors. In the setting of atherosclerosis, this pathway facilitates and promotes vascular smooth muscle cell proliferation, thus promoting fibrous cap stability, while S1P led to the induction of the inflammatory factor, cyclooxygenase 2 (COX-2) in the same study. However, MMP1 activation or reduction by dhS1P in endothelial cells is not known, It is possible to hypothesize from these studies that dhS1P may also play a role in vascular fibrosis. Considering the common factors involved in fibrosis in the cardiac and circulatory system such as the renin–angiotensin–aldosterone system (RAAS), the effects of dhS1P in the cardiac system needs to be investigated. Furthermore, raised dhS1P levels were demonstrated to have a strong relationship with survival from cardiac arrest in SK1-knockout mice, while S1P did not [180]. The increase in dhS1P can be attributed to the increased activity of the enzyme SK2, which is localized in the nucleus [181]. Taking into account the inhibitory effects of dhS1P on HDAC in the nucleus [146], the potential for it to impact on survival through increased proliferation is highly likely. Another area that warrants further research is studies detailing what impact commonly prescribed cardiac medications may have on dhS1P’s role in CVDs, since dhS1P levels were shown to be reduced in plasma of healthy subjects taking a 300 mg loading dose of aspirin [182].
These animal and clinical studies clearly show that dhS1P may be involved in the pathophysiology of CVDs and that platelets and erythrocyte levels of dhS1P influence the plasma levels of dhS1P and S1P for that matter. Applying this to CAD, hypothetically, there could be increased albumin-bound dhS1P. However, how this may influence the outcome of the disease is unknown, especially since the studies also show that dhS1P may promote plaque stability through improved endothelial barrier function. Another area that warrants further research is dhS1P’s role in cardiac fibrosis.
DhS1P in lung disease
In terms of lung diseases, sphingolipids and sphingolipid metabolism have been suggested as potential contributors to the pathogenesis of asthma [183], especially in relation to the interactions between ORMDL3 and SPT. A recent study has shown that the inhibition of ORMDL3 increased SPTLC1 and S1P, which then increased smooth muscle contraction rather than inflammation, causing airway hypersensitivity (AHR) [184]. Increases in both S1P and dhS1P have been noted in relation to dust mite allergy, increasing AHR and the asthmatic phenotype [185]. However, it is likely that the prominent increase in S1P led to the effects. The immunomodulatory molecule FTY720, which is known to reduce ORMLD3 leading to reduced AHR and inflammation [186], was able to inhibit CerS4 and increase SK1, leading to decreased S1P and increased dhS1P levels in human lung endothelial cells [53]. This suggests that therapeutic agents such as FTY720 could be more useful than those that inhibit ORMDL3 alone, assuming dhS1P potentially has a different effect than S1P. Furthermore, Berdyshev et al. [187] have shown in their study that the increase in SK1 derails the metabolic pathway of sphingolipids towards that of dhS1P generation, rather than S1P in respiratory syncytial virus (RSV) infection of human bronchial epithelial cells (HBEpC) and HPAEC. They also suggested that SK1 forms a substrate membrane enzymatic complex that impacts on this derailment. Additionally, dhS1P has been shown to compete for cystic fibrosis transmembrane receptor uptake with S1P in C127 cells [188], while in the setting of radiation-induced pulmonary fibrosis both S1P and dhS1P, and the expression of SK1 were increased [189]. Considering the contrasting findings in dermal cells and neuronal cells in terms of dhS1P in activating or inhibiting certain fibrotic factors, and those of S1P in cardiac fibrosis, the role of dhS1P in pulmonary fibrosis needs to be investigated. What is apparent in these latter studies is the regulation of dhS1P and S1P by SK1 increase may be stimulus, cell type, and complex dependent as hinted by Berdyshev et al. [187].
DhS1P in liver and kidney disease
Studies have demonstrated the protective effects of dhS1P against ischaemic–reperfusion injury (IRI) in mice hepatic and renal tissues [190, 191]. DhS1P was able to confer protection against IRI by activating S1PR1, which led to phosphorylation of MAPK/ERK, Akt, and heat shock protein 27 (HSP27) [190]. Exogenous treatment of the mice subjected to hepatic IRI with low doses of dhS1P led to reduced hepatic and renal necrosis and apoptosis, neutrophil infiltration, preserved endothelial cell integrity and reduced pro-inflammatory mRNA [191]. It should be noted that there were no changes observed in S1P levels and S1P conferred protection through S1PR3. DhS1P has also been recommended as a marker for FB1 toxicity [192]. This is supported by studies in cells [193], ducks [194] and human [195] serum or tissue, which showed an increase in dhS1P after exposure to FB1. Apart from it being a marker for toxicity, it may also contribute to cell proliferation. The accumulation of dhS1P due to FB1 toxicity in renal cells led to transient activation of PKCα within 5 min of exposure, compared to dhSph, Sph, S1P and Cer [193]. PKCα mediates the mitogenic effect of PDGF in renal mesangial cells (RMC) [196]. PDGF has been shown to induce increased expression of SK1 mRNA [197], which diverts dhSph towards phosphorylation to give dhS1P instead of Cer, promoting cell survival [198]. In addition, both S1P and dhS1P were able to stimulate similar gene expression waves as PDGF in RMC [199]. In dhS1P-stimulated cells, the angiotensin II receptor type 2 (AT2R) expression was lower than in S1P-stimulated cells, implying that dhS1P has a higher mitogenic effect. In fact, this study also showed that dhS1P had a greater degree of intracellular calcium mobilization than S1P, which explains the transient activation of PKCα seen in FB1 toxicity [193]. The calcium/ PKC pathway is one of the signal transduction pathway for growth factors such as PDGF. Both dhS1P and S1P also induced growth factors such as heparin-binding EGF-like growth factor (HB-EGF) and connective tissue growth factor (CTGF), a fibrotic protein, which was not induced upon stimulation with PDGF [199].
It can be summarized from these studies that dhS1P is able to activate proliferation either on its own through the calcium/ PKC pathway or by interacting with other signalling molecules, including ERK, MAPK and Akt, and HSP27 in the kidney and liver at lower doses while conferring toxic effects at higher doses. How this may impact upon the long-term systemic effects such as fibrosis and in the setting of different pathologies needs to be evaluated.
DhS1P in cancer and cancer therapy
In terms of cancer therapy, dhS1P may help promote survival of neuronal cells [200], inhibit migration, invasion of melanomas through S1PR2 activation [201] and could even be harnessed as a therapeutic tool for tumours [202]. In C6 glioma cells, dhS1P was able to activate the ERK/early growth factor response 1 (EGR-1)/fibroblast growth factor 2 (FGF-2) pathway through S1PR1 [200]. FGF-2 is a neurotrophic factor involved in neuronal differentiation and survival. DhS1P also activated phospholipase D (PLD), a mitogenic factor, through S1PR2 but at lower levels than S1P. The activation of S1PR2 by dhS1P and S1P in B16 melanoma cells led to inhibition of cell migration through regulation of RhoA and Rac which are involved in cell motility [201]. One of the limitations to cancer treatment has been the systemic immune suppression caused by tumour-associated inflammation effected through myeloid lineage cells. Barth and colleagues showed that a recent therapeutic tool targeted at this phenomenon, termed “Photo-ImmunoNanoTherapy”, improved the outcome in mice models as a result of dhS1P (S1P to a lesser degree) abrogating myeloid lineage cells and allowing the expansion of anti-tumour lymphocytes [202]. The increase in dhS1P was attributed to increase in SK2, which is known to have epigenetic effects [203], rather than SK1. They also injected tumour-bearing mice with dhS1P and found it to have anti-tumour effect, while S1P promoted tumour growth. Incubation of T cells stimulated with the immune-suppressive drugs anti-CD3 and anti-CD28 with dhS1P induced the release of interleukin 2 (IL-2) and interferon-γ (INF-γ), respectively [204]. Thus, dhS1P inhibits T cell proliferation which could suppress tumour growth and survival. However, this may not be true for all types of cancers, since patients with hepatocellular carcinoma were found to have raised serum dhS1P levels. Despite this, it can be surmised that dhS1P is a potential anti-cancer biomolecule that needs to be further investigated (Fig. 5).
DhSph in disease
Overview and structure
Sphinganine or dihydrosphingosine (dhSph) forms the backbone of dihydrosphingolipids. It has a molecular weight of 301.5 g/mol and is produced mostly in the endoplasmic reticulum. DhSph serves as a precursor to dhS1P synthesis by SK1 and 2 and dhCer by ceramide synthases. In biological systems, early studies in the 1990s seem to have used dhSph as a protein kinase C (PKC) inhibitor with regard to cell proliferation and vasoconstriction studies [205,206,207]. Here, we look at its role in different diseases.
DhSph in hepatic and renal diseases
Much relevance has been given to the enzymes involved in dhSph metabolism, thus overlooking its role in pathophysiology. Only a few studies have considered dhSph, especially in terms of FB1 toxicity which increases the dhSph and dhSph/Sph ratio. The extent of FB1 toxicity in humans has been reviewed by Voss et al. [208]. A number of studies have found raised dhSph levels due to FB1 exposure in the liver and kidney [209], the brain of calves [210], gastrointestinal tract (GIT) of chickens [211], pregnant mice and fish (with no fetal toxicity) [212, 213], and in urine samples from humans [214]. Apart from FB1 toxicity, dhSph was also increased in the plasma in other instances, such as in hepatotoxicity due to Guynuria Segetum, Fabry’s disease, endemic nephropathy, hepatitis C infection, type 2 diabetes-induced NAFLD, disease models of glucocorticoid-induced osteoporotic rats and dyslipidaemia, remote ischaemic preconditioning (RIPC) strategy for IRI, and genetic ablation of CerS2 in the liver [106, 215,216,217,218,219,220,221,222]. The key factor in all of these increases is the inhibition of the CerS enzyme which catalyses the acylation of dhSph to dhCer. FB1 competitively inhibits CerS due to it being structurally similar to dhSph and differing only in the free amino group at C1 [223]. This inhibition not only raises dhSph levels, but also the levels of dhS1P which is known to have autocrine–paracrine functions on the S1PRs, further complicating the mechanistic pathways of FB1 toxicity. Whether or not the increased dhS1P is also able to inhibit CerS2 by directly interacting with the S1P receptor-like motif on CerS2 is unknown [224]. FB1 toxicity is accompanied by an increase in TNF-α expression causing increased cell apoptosis and induction of cytokines such as IL-12 p40 and IFNγ [225, 226]. However, He et al. [227] stated that this is not directly related to the increase in dhSph or Sph as shown by the continuous expression of TNF-α despite the inhibition of SPT in the presence of FB1 in kidney cells. However, earlier studies by Sharma et al. [228] in TNF-α receptor knockout mice showed that there was some increase in dhSph in the liver and kidney, but these were lower than in the wild types. These studies imply that there may be partial interactions between TNF-α and dhSph or the de novo pathway.
Reduced dhSph levels have been noted in the seminal plasma of infertile male patients with Kidney–Yang syndrome [229], adenine-induced chronic renal failure in rats [230], and in type 2 diabetes-induced diabetic nephropathy [231]. These studies were metabolomics and metabonomics studies aimed at discovering biomarkers for these disease conditions. Their findings showed the sphingolipid metabolism may be perturbed, the mechanisms of which remain unknown. Regardless, it is likely that the beginning of the de novo pathway is perturbed in these disease conditions, causing the reduced levels seen. DhSph has also been mentioned as a possible biomarker for kidney cancer [232]. It is worth considering the causal increase in dhS1P levels in these studies which could influence the outcomes observed. Therefore, to explore the effect of dhSph, research that takes into account this aspect would be valuable.
DhSph in cardiovascular disease
Elevated levels of dhSph have been noted in the hearts of rats exercising to exhaustion in 30 min [233], or pacing for 60 min [234], both of which show that increased cardiac workload not only affects SLs levels, but dhSLs as well.
Cardiomyopathies
In terms of cardiomyopathies, various other researchers have shown altered dhSph levels. For example, raised levels of dhSph were shown in plasma and tissues from rat MI models [162, 235,236,237], in the right ventricle after 60 min of tachycardia [163] and in cardiac muscle of male Wistar rats with drug-induced hyperthyroidism [238]. DhSph and phytosphingosine were identified as biomarkers in relation to the efficacy of traditional Chinese medicine (TCM) therapies in two of the MI studies [236, 237]. Phytosphingosine is derived from dhSph (as characterized in yeast) and causes apoptosis of cancer cells by caspase 8 activation and Bcl-2- associated X protein (Bax) translocation [239]. However, another study employing similar analytical methods and experimental conditions for MI indicated phytosphingosine as a biomarker and not dhSph [240]. The reason for this may lie in the rate of metabolism of dhSph in the tissue and plasma. The latter study was carried out on heart tissue. Reducing solid tissue de novo synthesis of sphingolipids were also shown to affect the level of dhSph in plasma (decreased) and platelets (increased) [241]. The disruption of the sphingolipid metabolic pathway showing increases in dhSph in cardiomyopathies has also been shown in the plasma of young (STEMI) patients [164, 242]. This study showed that dhSph had high specificity and sensitivity to the prognosis related to major adverse cardiovascular events after patients were discharged [242]. Prior to this study, 25–27% reductions in plasma dhSph were reported in chronic systolic heart failure patients, independent of the underlying cause of heart failure [243], with no changes observed in the plasma level of S1P and dhS1P, perhaps due to metabolic clearance as noted in another study where urine levels of dhSph and phytosphingosine were increased in HF patients [244]. Disease onset and duration could have also influenced these findings. For example, the STEMI study reported elevated levels upon admission, which were reduced at 1, 5, and 30 days after admission, while others have shown no changes in plasma dhSph levels in MI patients at the time of admission and 5 days after [165].
Coronary artery disease
Raised dhSph levels have been indicated in the progression of atherosclerotic dyslipidaemia [245], in spontaneously hypertensive rats [246], and has also been investigated as a biomarker for atherosclerosis in a rabbit model [247]. In patients with multi-vessel CAD, the level of dhSph and Sph in platelets has been shown to be higher than in the controls, whereas their levels in plasma and erythrocytes were stable or similar [171]. In addition, a study in patients with temporary coronary occlusion found that 1 min after PCI in the coronary sinus, dhSph levels were raised to 614%, and 272% in peripheral blood, but dropped below baseline at 12 h [248]. The inhibition of SPT by myriocin in apolipoprotein E (ApoE)-deficient mice led to significant reductions in dhSph and other sphingolipids levels, with a stable plaque formation and reductions in cholesterol and LDL [249], but in ApoE null mice fed with a high fat diet, dhSph levels were raised which positively correlated with total cholesterol and LDL-C [245]. Thus, inhibition of the sphingolipid de novo synthesis pathway may be beneficial to lowering atherogenic plasma lipids and encourage stable plaque formation. However, studies that could inform the mechanisms of this interaction between cholesterol and dhSph or sphingolipids are lacking. Therefore, these findings are speculative at this time.
DhSph in other diseases
The intracellular increase in dhSph is either as a result of overall increase in the de novo sphingolipid synthesis leading to effects similar to that of Cer, or due to inhibitions at the CerS enzymes, the effects of which are still elusive. The extracellular addition of dhSph also leads to Cer-type effects such as apoptosis in cancer cells [250]. De novo sphingolipid synthesis can be perturbed by inhibiting or overexpressing the enzyme SPT. The yeast orthologues of ORMDLs have been shown to inhibit SPT by forming a conserved complex with SPT reducing sphingolipids such as dhSph [251, 252]. Lowering the level of the enzymes at both ends of the de novo pathway such as that seen HD rat models [62]: SPLTC1 and CerS1, results in reductions in dhSph, dhCer and dhS1P. However, dhSph could be a promising target for therapy in dermatological diseases such as atopic dermatitis, where Sph and dhSph ratios were found to influence barrier abnormalities observed in human stratum corneum (SC) [253]. For example, dhSph was found to play a role in contributing to the formation of more rigid lattice of lipids in the SC [254]. It has also been suggested as a biomarker in neurodegenerative disease and diabetes. The altering of dhSph levels in diabetic disease states and models by the inhibition of the sphingolipid pathway or anti-diabetics that regulate lipid and cholesterol also supports dhSph being a possible biomarker for diabetes and diabetes therapy [255,256,257,258]. Such applications could allow for early detection of insulin resistance and patient response to therapy, because it is a necessary step in the de novo pathway that leads to Cers. Overall, studies in which dhSph is implicated are sporadic, which makes them difficult to discuss; therefore we have collated them in Table 2.
3-Ketosphinganine in disease
3-KetoSph is the product of the condensation of palmitoyl-CoA and serine catalysed by the enzyme SPT in the ER, which is the rate-limiting enzyme in the de novo sphingolipid metabolism pathway. The inhibition of the enzyme SPT in relation to disease seems to be studied more than the effects of the product 3-ketoSph, due to it being metabolized rapidly. In fact, an increasing number of studies are reporting links of mutations in the gene that encodes SPT, SPLTC1 and 2, to hereditary peripheral neuropathies [266,267,268]. There are also reports of new novel SPT inhibitors for cancer that have shown to reduce 3-ketoSph in human lung adenocarcinoma cells [269]. Mutations or missense in the enzyme that reduces 3-ketoSph, 3-ketodihydrosphingosine reductase (KDSR), have been linked to recessive progressive symmetric erythrokeratoderma [270], keratinization disorders associated with thrombocytopaenia [271] and bovine spinal muscular atrophy [272]. Long-term exposure of cancer cells (HGC27, T98G and U87MG) to 3 ketoSph has been shown to induce autophagy and overexpression of Des-1 [273].

DhS1P in disease. A summary of the potential effects of increased dhS1P as highlighted in this review
The evidence for 3 ketoSph in disease is quite scarce owing to its rapid metabolism in the de novo sphingolipid synthesis pathway; however, the enzymes involved in its synthesis and metabolism are targets for further studies.
Conclusion and perspectives
Collectively, the evidence for dihydrosphingolipids in disease is spatial across the board and thus requires a lot more research in terms of their roles in disease, especially the mechanistic pathways through which they could contribute to disease. There are a number of areas that have been examined in this review that should be the focus of further research. These include: (1) the value of dhCers in predicting type 2 diabetes in relation to obesity, (2) the possible role of dhCer in reducing adipogenesis and increasing autophagy in adipocytes, (3) the reoccurring theme of dhCer in association with hypoxia, (4) the role of dhS1P and dhSph in plaque stability, (5) the anti-tumour effects of dhS1P conferred through suppression of T cell proliferation, (6) the binding of dhS1P to albumin and the effects of this in terms of IHD, (7) the possible therapeutic effect of dhS1P in terms of HD, and (8) the stimulus, cell type and complex-dependent regulation of dhS1P by SK1. There are also a number of studies in terms of CVDs showing alterations in sphingolipid levels; however, what is lacking are mechanistic studies to show if these alterations can contribute to the pathophysiology of the disease. The role of dhSph as a biomarker in cardiomyopathies, drug-induced toxicities, as well as liver and kidney toxicity due to FB1 is imperative, especially in determining if the de novo sphingolipid synthesis pathway is perturbed. Future studies applying current lipidomics tools should be encouraged, together with studies that take into consideration both the metabolites and the enzymatic interactions of the de novo pathway. The use of more potent and selective Des-1 inhibitors should be encouraged for investigating the effects of dhCer or Des-1 inhibition in light of the recent polyubiquitination findings for 4-HRP-fenretinide. Finally, the altering role of dihydrosphingolipids in the different organs seems to depend not only upon the initial insults and the disease processes, but also the key players along the de novo sphingolipid pathway.
Abbreviations
- 3-KR:
-
3-Ketosphinganine Reductase
- 4-HPR:
-
N-(4-Hydroxyphenyl) retinamideFenretinide
- γ-TE:
-
γ-Tocotrienol
- ACER3:
-
Alkaline ceramidase 3
- ACSL5:
-
Acyl-coenzyme A synthase
- ACSL5:
-
Δ20 acyl-coenzyme A synthase lacking exon 20
- ADH:
-
Adiponectin hormone
- AHA:
-
American Heart Association
- Akt:
-
Protein kinase B
- AMPK:
-
AMP activated protein kinase
- BMI:
-
Body mass index
- CAD:
-
Coronary artery disease
- cAMP:
-
Cyclic adenosine 3ʹ,5ʹ-monophosphate
- cDase:
-
Ceramidase
- cdk2:
-
Cyclin dependent kinase 2
- Cer:
-
Ceramide
- CERKL:
-
Ceramide like kinase
- CERK:
-
Ceramide kinase
- CerS:
-
Ceramide synthase
- CFTR:
-
Cystic fibrosis transmembrane conductance regulator
- COX-2:
-
Cyclooxygenase 2
- CRF:
-
Cardiorespiratory fitness
- CTGF:
-
Connective tissue growth factor
- CVD:
-
Cardiovascular disease
- Des1:
-
Dihydroceramide desaturase 1
- Des2:
-
Dihydroceramide desaturase 2
- DhCer:
-
Dihydroceramide
- DhSph:
-
Dihydrosphingosine/Dihydrosphinganine
- DhS1P:
-
Dihydrosphingosine 1 phosphate/dihydrosphinganine 1 phosphate
- EAP:
-
Ethanolamine phosphate
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal regulated kinases
- FAK:
-
Focal adhesion kinase
- FB1:
-
Fumonisin B 1
- FFA:
-
Free fatty acid
- HDAC2:
-
Histone deacetylase 2
- HDL:
-
High density lipid
- HepG2:
-
Human hepato-carcinoma cell
- HIF1-α:
-
Hypoxia inducible factor 1-α
- HOMA-IR:
-
Homestasis model of insulin resistance
- HSP27:
-
Heat shock protein 27
- HUVEC:
-
Human umblical endothelial cell
- FTY720:
-
Fingolimod
- IL-1:
-
Interleukin 1
- IL-6:
-
Interleukin 6
- JNK:
-
c-Jun N terminal kinase
- LDL:
-
Low density lipid
- LPS:
-
Lipopolysaccharide
- LRS:
-
Lipidomic risk score
- MAPK:
-
Mitogen activated protein kinases
- MI:
-
Myocardial infarct
- MnTBAP:
-
Manganese(III) tetrakis (4-benzoic acid) porphyrin
- MTORC1:
-
Mammalian target of rapamycin complex 1
- NADH:
-
Nicotinamide adenine nucleotide
- NADPH:
-
Nicotinamide adenine nucleotide phosphate
- NAFLD:
-
Non-alcoholic fatty liver disease
- NFATC:
-
Nuclear factor of activated T cells
- NK-kβ:
-
Nuclear factor kappa light chain enhancer of B cell
- Nrf2:
-
Nuclear factor erythroid related factor 2
- PDGF:
-
Platelet derived growth factor
- PDT:
-
Photodynamic therapy
- PeIF2α:
-
Phosphorylated eukaryotic translation initiation factors 2α
- PERK:
-
PKR like endoplasmic reticulum kinase
- PKCα:
-
Protein kinase Cα
- PLD:
-
Phospholipase D
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- RAR:
-
Retinoic acid receptor
- RMC:
-
Renal mesengial cell
- ROS:
-
Reactive oxygen species
- S6K:
-
Ribosomal protein S6 kinase
- SAFHS:
-
San Antonio Family Heart Study
- SEK-1:
-
Dual specificity mitogen activated protein kinase kinase 1
- SD:
-
Sprague Dawley
- SK 1 and 2:
-
Sphingosine kinase 1 and 2
- S1P:
-
Sphingosine 1 phosphate
- S1PP:
-
Sphingosine 1 phosphate phophatase
- S1PR1–5:
-
Sphingosine 1 phosphate receptor 1–5
- SPL:
-
Sphingosine 1 phosphate lyase
- SPT:
-
Serine palmitoyltransferase
- SPLTC1:
-
Serine palmitoyltransferase long chain base 1
- SPTLC3:
-
Serine palmitoyltransferase long chain base 3
- STEMI:
-
ST-segment elevation myocardial: infarct
- SCC19:
-
Squamous cell carcinoma cell
- T2DM:
-
Type 2 diabetes mellitus
- TNF-α:
-
Tumour necrosis factor α
- VEGF:
-
Vascular endothelial growth factor
- WC:
-
Waist circumference
References
Young MM, Kester M, Wang H-G (2013) Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res 54(1):5–19. https://doi.org/10.1194/jlr.R031278
Cutler RG, Kelly J, Storie K, Pedersen WA, Tammara A, Hatanpaa K, Troncoso JC, Mattson MP (2004) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci USA 101(7):2070–2075. https://doi.org/10.1073/pnas.0305799101
Kitatani K, Iwabuchi K, Snider A, Riboni L (2016) Sphingolipids in inflammation: from bench to bedside. Mediators Inflamm 2016:7602526. https://doi.org/10.1155/2016/7602526
Ogretmen B (2017) Sphingolipid metabolism in cancer signalling and therapy. Nat Rev Cancer 18:33. https://doi.org/10.1038/nrc.2017.96
Halmer R, Walter S, Fassbender K (2014) Sphingolipids: important players in multiple sclerosis. Cell Physiol Biochem 34(1):111–118. https://doi.org/10.1159/000362988
Russo SB, Ross JS, Cowart LA (2013) Sphingolipids in obesity, Type 2 diabetes, and metabolic disease. Handb Exp Pharmacol 216:373–401. https://doi.org/10.1007/978-3-7091-1511-4_19
Gulbins E, Petrache I (2013) Sphingolipids in disease. In: Gulbins E, Petrache I (eds) Hand book of experimental pharmacology, vol 216. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1511-4
Giles C, Takechi R, Mellett NA, Meikle PJ, Dhaliwal S, Mamo JC (2017) Differential regulation of sphingolipid metabolism in plasma, hippocampus, and cerebral cortex of mice administered sphingolipid modulating agents. J Neurochem 141(3):413–422. https://doi.org/10.1111/jnc.13964
Rodriguez-Cuenca S, Barbarroja N, Vidal-Puig A (2015) Dihydroceramide desaturase 1, the gatekeeper of ceramide induced lipotoxicity. Biochim Biophys Acta Mol Cell Biol Lipids 1851(1):40–50. https://doi.org/10.1016/j.bbalip.2014.09.021
Levkau B (2013) Cardiovascular effects of sphingosine-1-phosphate (S1P). Handb Exp Pharmacol 216:147–170. https://doi.org/10.1007/978-3-7091-1511-4_8
Colacios C, Sabourdy F, Andrieu-Abadie N, Ségui B, Levade T (2015) Basics of Sphingolipid Metabolism and Signalling. In: Hannun YA, Luberto C, Mao C, Obeid LM (eds) Bioactive sphingolipids in cancer biology and therapy. Springer, Cham, pp 1–20. https://doi.org/10.1007/978-3-319-20750-6_1
Stiban J, Tidhar R, Futerman AH (2010) Ceramide synthases: roles in cell physiology and signaling. Adv Exp Med Biol 688:60–71
Petrache I, Berdyshev EV (2016) Ceramide signaling and metabolism in pathophysiological states of the lung. Annu Rev Physiol 78:463–480. https://doi.org/10.1146/annurev-physiol-021115-105221
Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, Ramaraju H, Sullards MC, Cabot M, Merrill AH (2006) Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta Biomembr 1758(12):1864–1884. https://doi.org/10.1016/j.bbamem.2006.08.009
Casasampere M, Ordonez YF, Pou A, Casas J (2016) Inhibitors of dihydroceramide desaturase 1: therapeutic agents and pharmacological tools to decipher the role of dihydroceramides in cell biology. Chem Phys Lipids 197:33–44. https://doi.org/10.1016/j.chemphyslip.2015.07.025
Siddique MM, Bikman BT, Wang L, Ying L, Reinhardt E, Shui G, Wenk MR, Summers SA (2012) Ablation of dihydroceramide desaturase confers resistance to etoposide-induced apoptosis in vitro. PLoS One 7(9):e44042. https://doi.org/10.1371/journal.pone.0044042
Siddique MM, Li Y, Chaurasia B, Kaddai VA, Summers SA (2015) Dihydroceramides: from bit players to lead actors. J Biol Chem 290(25):15371–15379. https://doi.org/10.1074/jbc.R115.653204
Wegner MS, Schiffmann S, Parnham MJ, Geisslinger G, Grosch S (2016) The enigma of ceramide synthase regulation in mammalian cells. Prog Lipid Res 63:93–119. https://doi.org/10.1016/j.plipres.2016.03.006
Cingolani F, Futerman AH, Casas J (2016) Ceramide synthases in biomedical research. Chem Phys Lipids 197:25–32. https://doi.org/10.1016/j.chemphyslip.2015.07.026
Samadi A (2007) Ceramide-induced cell death in lens epithelial cells. Mol Vis 13:1618–1626
Chien CC, Shen SC, Yang LY, Wu CY, Liau JS, Chen YC (2009) Activation of telomerase and cyclooxygenase-2 in PDGF and FGF inhibition of C2-ceramide-induced apoptosis. J Cell Physiol 218(2):405–415. https://doi.org/10.1002/jcp.21613
Wong K, Li X-B, Hunchuk N (1995) N-Acetylsphingosine (C-ceramide) inhibited neutrophil superoxide formation and calcium influx. J Biol Chem 270(7):3056–3062. https://doi.org/10.1074/jbc.270.7.3056
Testai FD, Kilkus JP, Berdyshev E, Gorshkova I, Natarajan V, Dawson G (2014) Multiple sphingolipid abnormalities following cerebral microendothelial hypoxia. J Neurochem 131(4):530–540. https://doi.org/10.1111/jnc.12836
Testai FD, Xu HL, Kilkus J, Suryadevara V, Gorshkova I, Berdyshev E, Pelligrino DA, Dawson G (2015) Changes in the metabolism of sphingolipids after subarachnoid hemorrhage. J Neurosci Res 93(5):796–805. https://doi.org/10.1002/jnr.23542
Peterlin BL, Mielke MM, Dickens AM, Chatterjee S, Dash P, Alexander G, Vieira RV, Bandaru VV, Dorskind JM, Tietjen GE, Haughey NH (2015) Interictal, circulating sphingolipids in women with episodic migraine: a case–control study. Neurology 85(14):1214–1223. https://doi.org/10.1212/WNL.0000000000002004
Edvardson S, Yi JK, Jalas C, Xu R, Webb BD, Snider J, Fedick A, Kleinman E, Treff NR, Mao C, Elpeleg O (2016) Deficiency of the alkaline ceramidase ACER3 manifests in early childhood by progressive leukodystrophy. J Med Genet 53(6):389
Park M, Kaddai V, Ching J, Fridianto KT, Sieli RJ, Sugii S, Summers SA (2016) A role for ceramides, but not sphingomyelins, as antagonists of insulin signaling and mitochondrial metabolism in C2C12 myotubes. J Biol Chem 291(46):23978–23988
Veret J, Coant N, Berdyshev EV, Skobeleva A, Therville N, Bailbe D, Gorshkova I, Natarajan V, Portha B, Le Stunff H (2011) Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 beta-cells. Biochem J 438(1):177–189. https://doi.org/10.1042/BJ20101386
Roomp K, Kristinsson H, Schvartz D, Ubhayasekera K, Sargsyan E, Manukyan L, Chowdhury A, Manell H, Satagopam V, Groebe K, Schneider R, Bergquist J, Sanchez J-C, Bergsten P (2017) Combined lipidomic and proteomic analysis of isolated human islets exposed to palmitate reveals time-dependent changes in insulin secretion and lipid metabolism. PLoS One 12(4):e0176391. https://doi.org/10.1371/journal.pone.0176391
Li Y, Park J-S, Deng J-H, Bai Y (2006) Cytochrome c oxidase Subunit IV is essential for assembly and respiratory function of the enzyme complex. J Bioenerg Biomembr 38(5–6):283–291. https://doi.org/10.1007/s10863-006-9052-z
Hansen ME, Simmons KJ, Tippetts TS, Thatcher MO, Saito RR, Hubbard ST, Trumbull AM, Parker BA, Taylor OJ, Bikman BT (2015) Lipopolysaccharide disrupts mitochondrial physiology in skeletal muscle via disparate effects on sphingolipid metabolism. Shock 44(6):585–592. https://doi.org/10.1097/SHK.0000000000000468
Stiban J, Fistere D, Colombini M (2006) Dihydroceramide hinders ceramide channel formation: implications on apoptosis. Apoptosis 11(5):773–780. https://doi.org/10.1007/s10495-006-5882-8
Barbarroja N, Rodriguez-Cuenca S, Nygren H, Camargo A, Pirraco A, Relat J, Cuadrado I, Pellegrinelli V, Medina-Gomez G, Lopez-Pedrera C, Tinahones FJ, Symons JD, Summers SA, Oresic M, Vidal-Puig A (2015) Increased dihydroceramide/ceramide ratio mediated by defective expression of degs1 impairs adipocyte differentiation and function. Diabetes 64(4):1180–1192. https://doi.org/10.2337/db14-0359
McIlroy GD, Tammireddy SR, Maskrey BH, Grant L, Doherty MK, Watson DG, Delibegovic M, Whitfield PD, Mody N (2016) Fenretinide mediated retinoic acid receptor signalling and inhibition of ceramide biosynthesis regulates adipogenesis, lipid accumulation, mitochondrial function and nutrient stress signalling in adipocytes and adipose tissue. Biochem Pharmacol 100:86–97. https://doi.org/10.1016/j.bcp.2015.11.017
Aye ILMH, Gao X, Weintraub ST, Jansson T, Powell TL (2014) Adiponectin inhibits insulin function in primary trophoblasts by PPARα-mediated ceramide synthesis. Mol Endocrinol 28(4):512–524. https://doi.org/10.1210/me.2013-1401
Brozinick JT, Hawkins E, Hoang Bui H, Kuo MS, Tan B, Kievit P, Grove K (2013) Plasma sphingolipids are biomarkers of metabolic syndrome in non-human primates maintained on a Western-style diet. Int J Obes 37(8):1064–1070. https://doi.org/10.1038/ijo.2012.191
Bikman BT, Guan Y, Shui G, Siddique MM, Holland WL, Kim JY, Fabriàs G, Wenk MR, Summers SA (2012) Fenretinide prevents lipid-induced insulin resistance by blocking ceramide biosynthesis. J Biol Chem 287(21):17426–17437. https://doi.org/10.1074/jbc.M112.359950
Rico JE, Saed Samii S, Mathews AT, Lovett J, Haughey NJ, McFadden JW (2017) Temporal changes in sphingolipids and systemic insulin sensitivity during the transition from gestation to lactation. PLoS One 12(5):e0176787. https://doi.org/10.1371/journal.pone.0176787
Mielke MM, Bandaru VV, Han D, An Y, Resnick SM, Ferrucci L, Haughey NJ (2015) Demographic and clinical variables affecting mid- to late-life trajectories of plasma ceramide and dihydroceramide species. Aging Cell 14(6):1014–1023. https://doi.org/10.1111/acel.12369
Weir JM, Wong G, Barlow CK, Greeve MA, Kowalczyk A, Almasy L, Comuzzie AG, Mahaney MC, Jowett JBM, Shaw J, Curran JE, Blangero J, Meikle PJ (2013) Plasma lipid profiling in a large population-based cohort. J Lipid Res 54(10):2898–2908. https://doi.org/10.1194/jlr.P035808
Mamtani M, Meikle PJ, Kulkarni H, Weir JM, Barlow CK, Jowett JB, Bellis C, Dyer TD, Almasy L, Mahaney MC, Duggirala R, Comuzzie AG, Blangero J, Curran JE (2014) Plasma dihydroceramide species associate with waist circumference in mexican american families. Obesity (Silver Spring, MD) 22(3):950–956. https://doi.org/10.1002/oby.20598
Meikle PJ, Wong G, Barlow CK, Weir JM, Greeve MA, MacIntosh GL, Almasy L, Comuzzie AG, Mahaney MC, Kowalczyk A, Haviv I, Grantham N, Magliano DJ, Jowett JBM, Zimmet P, Curran JE, Blangero J, Shaw J (2013) Plasma lipid profiling shows similar associations with prediabetes and Type 2 diabetes. PLoS One 8(9):e74341. https://doi.org/10.1371/journal.pone.0074341
Lopez X, Goldfine AB, Holland WL, Gordillo R, Scherer PE (2013) Plasma ceramides are elevated in female children and adolescents with type 2 diabetes. J Pediatr Endocrinol Metab 26(9–10):995–998. https://doi.org/10.1515/jpem-2012-0407
Racette SB, Evans EM, Weiss EP, Hagberg JM, Holloszy JO (2006) Abdominal adiposity is a stronger predictor of insulin resistance than fitness among 50–95 year olds. Diabetes Care 29(3):673–678
Dubé JJ, Amati F, Toledo FGS, Stefanovic-Racic M, Rossi A, Coen P, Goodpaster BH (2011) Effects of weight loss and exercise on insulin resistance, and intramyocellular triacylglycerol, diacylglycerol and ceramide. Diabetologia 54(5):1147–1156. https://doi.org/10.1007/s00125-011-2065-0
Warshauer JT, Lopez X, Gordillo R, Hicks J, Holland WL, Anuwe E, Blankfard MB, Scherer PE, Lingvay I (2015) Effect of pioglitazone on plasma ceramides in adults with metabolic syndrome. Diabetes Metab Res Rev 31(7):734–744. https://doi.org/10.1002/dmrr.2662
Fabbri E, Yang A, Simonsick EM, Chia CW, Zoli M, Haughey NJ, Mielke MM, Ferrucci L, Coen PM (2016) Circulating ceramides are inversely associated with cardiorespiratory fitness in participants aged 54–96 years from the Baltimore Longitudinal Study of Aging. Aging Cell 15(5):825–831. https://doi.org/10.1111/acel.12491
Noureddine L, Azzam R, Nemer G, Bielawski J, Nasser M, Bitar F, Dbaibo GS (2008) Modulation of total ceramide and constituent ceramide species in the acutely and chronically hypoxic mouse heart at different ages. Prostaglandins Other Lipid Mediat 86(1–4):49–55. https://doi.org/10.1016/j.prostaglandins.2008.02.003
Russo SB, Tidhar R, Futerman AH, Cowart LA (2013) Myristate-derived d16:0 sphingolipids constitute a cardiac sphingolipid pool with distinct synthetic routes and functional properties. J Biol Chem 288(19):13397–13409. https://doi.org/10.1074/jbc.M112.428185
Edsfeldt A, Dunér P, Ståhlman M, Mollet IG, Asciutto G, Grufman H, Nitulescu M, Persson AF, Fisher RM, Melander O, Orho-Melander M, Borén J, Nilsson J, Gonçalves I (2016) Sphingolipids contribute to human atherosclerotic plaque inflammation. Arterioscler Thromb Vasc Biol 36(6):1132
Ellims AH, Wong G, Weir JM, Lew P, Meikle PJ, Taylor AJ (2014) Plasma lipidomic analysis predicts non-calcified coronary artery plaque in asymptomatic patients at intermediate risk of coronary artery disease. Eur Heart J Cardiovasc Imaging 15(8):908–916. https://doi.org/10.1093/ehjci/jeu033
Hamai H, Keyserman F, Quittell LM, Worgall TS (2009) Defective CFTR increases synthesis and mass of sphingolipids that modulate membrane composition and lipid signaling. J Lipid Res 50(6):1101–1108. https://doi.org/10.1194/jlr.M800427-JLR200
Berdyshev EV, Gorshkova I, Skobeleva A, Bittman R, Lu X, Dudek SM, Mirzapoiazova T, Garcia JGN, Natarajan V (2009) FTY720 inhibits ceramide synthases and up-regulates dihydrosphingosine 1-phosphate formation in human lung endothelial cells. J Biol Chem 284(9):5467–5477. https://doi.org/10.1074/jbc.M805186200
Yasuo M, Mizuno S, Allegood J, Kraskauskas D, Bogaard HJ, Spiegel S, Voelkel NF (2013) Fenretinide causes emphysema, which is prevented by sphingosine 1-phoshate. PLoS One 8(1):e53927. https://doi.org/10.1371/journal.pone.0053927
Devlin CM, Lahm T, Hubbard WC, Van Demark M, Wang KC, Wu X, Bielawska A, Obeid LM, Ivan M, Petrache I (2011) Dihydroceramide-based response to hypoxia. J Biol Chem 286(44):38069–38078. https://doi.org/10.1074/jbc.M111.297994
Cai L, Oyeniran C, Biswas DD, Allegood J, Milstien S, Kordula T, Maceyka M, Spiegel S (2016) ORMDL proteins regulate ceramide levels during sterile inflammation. J Lipid Res 57(8):1412–1422. https://doi.org/10.1194/jlr.M065920
Ruangsiriluk W, Grosskurth SE, Ziemek D, Kuhn M, des Etages SG, Francone OL (2012) Silencing of enzymes involved in ceramide biosynthesis causes distinct global alterations of lipid homeostasis and gene expression. J Lipid Res 53(8):1459–1471. https://doi.org/10.1194/jlr.M020941
Grammatikos G, Schoell N, Ferreirós N, Bon D, Herrmann E, Farnik H, Köberle V, Piiper A, Zeuzem S, Kronenberger B, Waidmann O, Pfeilschifter J (2016) Serum sphingolipidomic analyses reveal an upregulation of C16-ceramide and sphingosine-1-phosphate in hepatocellular carcinoma. Oncotarget 7(14):18095–18105. https://doi.org/10.18632/oncotarget.7741
Grammatikos G, Mühle C, Ferreiros N, Schroeter S, Bogdanou D, Schwalm S, Hintereder G, Kornhuber J, Zeuzem S, Sarrazin C, Pfeilschifter J (2014) Serum acid sphingomyelinase is upregulated in chronic hepatitis C infection and non alcoholic fatty liver disease. Biochim Biophys Acta Mol Cell Biol Lipids 1841(7):1012–1020. https://doi.org/10.1016/j.bbalip.2014.04.007
Sun N, Keep RF, Hua Y, Xi G (2016) Critical role of the sphingolipid pathway in stroke: a review of current utility and potential therapeutic targets. Transl Stroke Res 7(5):420–438. https://doi.org/10.1007/s12975-016-0477-3
Mielke MM, Haughey NJ, Bandaru VV, Weinberg DD, Darby E, Zaidi N, Pavlik V, Doody RS, Lyketsos CG (2011) Plasma sphingomyelins are associated with cognitive progression in Alzheimer’s disease. J Alzheimers Dis 27(2):259–269. https://doi.org/10.3233/JAD-2011-110405
Di Pardo A, Basit A, Armirotti A, Amico E, Castaldo S, Pepe G, Marracino F, Buttari F, Digilio AF, Maglione V (2017) De novo synthesis of sphingolipids is defective in experimental models of Huntington’s disease. Front Neurosci 11:698. https://doi.org/10.3389/fnins.2017.00698
Matesanz F, Fedetz M, Barrionuevo C, Karaky M, Catalá-Rabasa A, Potenciano V, Bello-Morales R, López-Guerrero J-A, Alcina A (2016) A splice variant in the ACSL5 gene relates migraine with fatty acid activation in mitochondria. Eur J Hum Genet 24(11):1572–1577. https://doi.org/10.1038/ejhg.2016.54
Ordóñez-Gutiérrez L, Benito-Cuesta I, Abad JL, Casas J, Fábrias G, Wandosell F (2018) Dihydroceramide desaturase 1 inhibitors reduce amyloid-β levels in primary neurons from an Alzheimer’s disease transgenic model. Pharm Res 35(3):49. https://doi.org/10.1007/s11095-017-2312-2
Gagliostro V, Casas J, Caretti A, Abad JL, Tagliavacca L, Ghidoni R, Fabrias G, Signorelli P (2012) Dihydroceramide delays cell cycle G1/S transition via activation of ER stress and induction of autophagy. Int J Biochem Cell Biol 44(12):2135–2143. https://doi.org/10.1016/j.biocel.2012.08.025
Casasampere M, Ordóñez YF, Casas J, Fabrias G (2017) Dihydroceramide desaturase inhibitors induce autophagy via dihydroceramide-dependent and independent mechanisms. Biochim Biophys Acta Gen Subj 1861(2):264–275. https://doi.org/10.1016/j.bbagen.2016.11.033
Schofield JD, Liu Y, Rao-Balakrishna P, Malik RA, Soran H (2016) Diabetes dyslipidemia. Diabetes Ther 7(2):203–219. https://doi.org/10.1007/s13300-016-0167-x
Mooradian AD (2009) Dyslipidemia in type 2 diabetes mellitus. Nat Clin Pract Endocrinol Metab 5:150. https://doi.org/10.1038/ncpendmet1066
Mark L, Dani G (2016) Diabetic dyslipidaemia and the atherosclerosis. Orv Hetil 157(19):746–752. https://doi.org/10.1556/650.2016.30441
Sattler KJ, Elbasan S, Keul P, Elter-Schulz M, Bode C, Graler MH, Brocker-Preuss M, Budde T, Erbel R, Heusch G, Levkau B (2010) Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res Cardiol 105(6):821–832. https://doi.org/10.1007/s00395-010-0112-5
Reali F, Morine MJ, Kahramanoğulları O, Raichur S, Schneider H-C, Crowther D, Priami C (2017) Mechanistic interplay between ceramide and insulin resistance. Sci Rep 7:41231. https://doi.org/10.1038/srep41231. https://www.nature.com/articles/srep41231#supplementary-information
Stratford S, Hoehn KL, Liu F, Summers SA (2004) Regulation of insulin action by ceramide: dual mechanisms linking ceramide accumulation to the inhibition of AKT/protein kinase B. J Biol Chem 279(35):36608–36615. https://doi.org/10.1074/jbc.M406499200
Turpin SM, Nicholls HT, Willmes DM, Mourier A, Brodesser S, Wunderlich CM, Mauer J, Xu E, Hammerschmidt P, Bronneke HS, Trifunovic A, LoSasso G, Wunderlich FT, Kornfeld JW, Bluher M, Kronke M, Bruning JC (2014) Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab 20(4):678–686. https://doi.org/10.1016/j.cmet.2014.08.002
Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Ohman MK, Takeda K, Sugii S, Pewzner-Jung Y, Futerman AH, Summers SA (2014) CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 20(4):687–695. https://doi.org/10.1016/j.cmet.2014.09.015
Taylor R (2012) Insulin resistance and type 2 diabetes. Diabetes 61(4):778
Szpigel A, Hainault I, Carlier A, Venteclef N, Batto A-F, Hajduch E, Bernard C, Ktorza A, Gautier J-F, Ferré P, Bourron O, Foufelle F (2018) Lipid environment induces ER stress, TXNIP expression and inflammation in immune cells of individuals with type 2 diabetes. Diabetologia 61(2):399–412. https://doi.org/10.1007/s00125-017-4462-5
Patel SA, Hoehn KL, Lawrence RT, Sawbridge L, Talbot NA, Tomsig JL, Turner N, Cooney GJ, Whitehead JP, Kraegen EW, Cleasby ME (2012) Overexpression of the adiponectin receptor AdipoR1 in rat skeletal muscle amplifies local insulin sensitivity. Endocrinology 153(11):5231–5246. https://doi.org/10.1210/en.2012-1368
Qu Q, Zeng F, Liu X, Wang QJ, Deng F (2016) Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis 7:e2226. https://doi.org/10.1038/cddis.2016.132
Williams M, Caino MC (2018) Mitochondrial dynamics in Type 2 diabetes and cancer. Front Endocrinol 9:211. https://doi.org/10.3389/fendo.2018.00211
Siddique MM, Li Y, Wang L, Ching J, Mal M, Ilkayeva O, Wu YJ, Bay BH, Summers SA (2013) Ablation of dihydroceramide desaturase 1, a therapeutic target for the treatment of metabolic diseases, simultaneously stimulates anabolic and catabolic signaling. Mol Cell Biol 33(11):2353–2369. https://doi.org/10.1128/MCB.00226-13
Jheng H-F, Tsai P-J, Guo S-M, Kuo L-H, Chang C-S, Su I-J, Chang C-R, Tsai Y-S (2012) Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle. Mol Cell Biol 32(2):309
Lai WL, Wong NS (2008) The PERK/eIF2 alpha signaling pathway of unfolded protein response is essential for N-(4-hydroxyphenyl)retinamide (4HPR)-induced cytotoxicity in cancer cells. Exp Cell Res 314(8):1667–1682. https://doi.org/10.1016/j.yexcr.2008.02.002
Alsanafi M, Kelly SL, Jubair K, McNaughton M, Tate RJ, Merrill AH, Pyne S, Pyne NJ (2018) Native and polyubiquitinated forms of dihydroceramide desaturase are differentially linked to human embryonic kidney cell survival. Mol Cell Biol 38:e00222
Grygiel-Górniak B (2014) Peroxisome proliferator-activated receptors and their ligands: nutritional and clinical implications—a review. Nutr J 13(1):17. https://doi.org/10.1186/1475-2891-13-17
Bergman BC, Brozinick JT, Strauss A, Bacon S, Kerege A, Bui HH, Sanders P, Siddall P, Wei T, Thomas MK, Kuo MS, Perreault L (2016) Muscle sphingolipids during rest and exercise: a C18:0 signature for insulin resistance in humans. Diabetologia 59(4):785–798. https://doi.org/10.1007/s00125-015-3850-y
Blachnio-Zabielska AU, Pulka M, Baranowski M, Nikolajuk A, Zabielski P, Gorska M, Gorski J (2012) Ceramide metabolism is affected by obesity and diabetes in human adipose tissue. J Cell Physiol 227(2):550–557. https://doi.org/10.1002/jcp.22745
Curran JE, Weir, Jacquelyn M., Bellis, Claire., Carless, Melanie A., Jowett, Jeremy B., Mahaney, Michael C., Dyer, Thomas D., Goring, Harald H., Comuzzie, Anthony G., Almasy, Laura., Meikle, Peter J., Blangero, John. (2011) Genetic analysis of lipidomic profiles influencing diabetes risk in Mexican Americans. Paper presented at the American Diabetes Association, 71st Scientific Sessions
Mamtani M, Kulkarni H, Wong G, Weir JM, Barlow CK, Dyer TD, Almasy L, Mahaney MC, Comuzzie AG, Glahn DC, Magliano DJ, Zimmet P, Shaw J, Williams-Blangero S, Duggirala R, Blangero J, Meikle PJ, Curran JE (2016) Lipidomic risk score independently and cost-effectively predicts risk of future type 2 diabetes: results from diverse cohorts. Lipids Health Dis 15(1):67. https://doi.org/10.1186/s12944-016-0234-3
Benjamin EJ, Virani SS, Callaway CW, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jiménez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O’Flaherty M, Palaniappan LP, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JHY, Alger HM, Wong SS, Muntner P (2018) Heart disease and stroke statistics—2018 update: a report from the American Heart Association. Circulation 137(12):e67–e492
Fagard RH (1999) Physical activity in the prevention and treatment of hypertension in the obese. Med Sci Sports Exerc 31(11 Suppl):S624–S630
Paterson DH, Govindasamy D, Vidmar M, Cunningham DA, Koval JJ (2004) Longitudinal study of determinants of dependence in an elderly population. J Am Geriatr Soc 52(10):1632–1638. https://doi.org/10.1111/j.1532-5415.2004.52454.x
Laaksonen DE, Lakka HM, Salonen JT, Niskanen LK, Rauramaa R, Lakka TA (2002) Low levels of leisure-time physical activity and cardiorespiratory fitness predict development of the metabolic syndrome. Diabetes Care 25(9):1612–1618
Azzam R, Hariri F, El-Hachem N, Kamar A, Dbaibo G, Nemer G, Bitar F (2013) Regulation of de novo ceramide synthesis: the role of dihydroceramide desaturase and transcriptional factors NFATC and Hand2 in the hypoxic mouse heart. DNA Cell Biol 32(6):310–319. https://doi.org/10.1089/dna.2013.1993
Dirkx E, Gladka MM, Philippen LE, Armand A-S, Kinet V, Leptidis S, el Azzouzi H, Salic K, Bourajjaj M, da Silva GJJ, Olieslagers S, van der Nagel R, de Weger R, Bitsch N, Kisters N, Seyen S, Morikawa Y, Chanoine C, Heymans S, Volders PGA, Thum T, Dimmeler S, Cserjesi P, Eschenhagen T, da Costa Martins PA, De Windt LJ (2013) Nfat and miR-25 cooperate to reactivate the transcription factor Hand2 in heart failure. Nat Cell Biol 15:1282. https://doi.org/10.1038/ncb2866. https://www.nature.com/articles/ncb2866#supplementary-information
de Mello-Coelho V, Cutler RG, Bunbury A, Tammara A, Mattson MP, Taub DD (2017) Age-associated alterations in the levels of cytotoxic lipid molecular species and oxidative stress in the murine thymus are reduced by growth hormone treatment. Mech Ageing Dev 167:46–55. https://doi.org/10.1016/j.mad.2017.08.015
Brown DA, London E (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 275(23):17221–17224. https://doi.org/10.1074/jbc.R000005200
González-Peña D, Checa A, de Ancos B, Wheelock CE, Sánchez-Moreno C (2017) New insights into the effects of onion consumption on lipid mediators using a diet-induced model of hypercholesterolemia. Redox Biol 11:205–212. https://doi.org/10.1016/j.redox.2016.12.002
Reiss AB, Siegart NM, De Leon J (2017) Interleukin-6 in atherosclerosis: atherogenic or atheroprotective? Clin Lipidol 12(1):14–23. https://doi.org/10.1080/17584299.2017.1319787
Chang T-T, Chen J-W (2016) Emerging role of chemokine CC motif ligand 4 related mechanisms in diabetes mellitus and cardiovascular disease: friends or foes? Cardiovasc Diabetol 15(1):117. https://doi.org/10.1186/s12933-016-0439-9
Fang L, Mundra PA, Fan F, Galvin A, Weir JM, Wong G, Chin-Dusting J, Cicuttini F, Meikle P, Dart AM (2016) Plasma lipidomic profiling in patients with rheumatoid arthritis. Metabolomics 12(8):136. https://doi.org/10.1007/s11306-016-1086-6
Fine B, Marx A, Topkara V, Gomez EA, Vunjak-Novakovic G, Colombo P (2017) (223)—An integrated analysis of metabolomics after left ventricular assist device implantation. J Heart Lung Transplant 36(4, Supplement)):S93. https://doi.org/10.1016/j.healun.2017.01.235
Liu A, Chu Y-J, Wang X, Yu R, Jiang H, Li Y, Zhou H, Gong L-L, Yang W-Q, Ju J (2018) Serum metabolomics study based on LC-MS and antihypertensive effect of uncaria on spontaneously hypertensive rats. Evid Based Complement Altern Med 2018:11. https://doi.org/10.1155/2018/9281946
Ji R, Chang JY, Liao X, Zhang X, Kennel P, Castillero E, Brunjes D, Akashi H, Homma S, Goldberg I, Schulze PC (2015) Abstract 17320: inhibition of ceramide synthesis preserves cardiac function and increases survival in doxorubicin-induced cardiomyopathy. Circulation 132(Suppl 3):A17320
Sharma A, Sung B, Veerappan A, Silver RB, Kim B, Worgall TS, Worgall S (2017) Decreased Sphingolipid Synthesis Enhances Rhinovirus-Triggered Airway Hyperreactivity. In: A36. Host defense against viral infection. American Thoracic Society International Conference Abstracts, American Thoracic Society, pp A1381–A1381. https://doi.org/10.1164/ajrccm-conference.2017.195.1_meetingabstracts.a1381
Mucke VT, Gerharz J, Jakobi K, Thomas D, Ferreiros Bouzas N, Mucke MM, Trotschler S, Weiler N, Welker MW, Zeuzem S, Pfeilschifter J, Grammatikos G (2018) Low serum levels of (dihydro-)ceramides reflect liver graft dysfunction in a real-world cohort of patients post liver transplantation. Int J Mol Sci. https://doi.org/10.3390/ijms19040991
Apostolopoulou M, Gordillo R, Koliaki C, Gancheva S, Jelenik T, De Filippo E, Herder C, Markgraf D, Jankowiak F, Esposito I, Schlensak M, Scherer PE, Roden M (2018) Specific hepatic sphingolipids relate to insulin resistance, oxidative stress, and inflammation in nonalcoholic steatohepatitis. Diabetes Care. https://doi.org/10.2337/dc17-1318
Alexaki A, Clarke BA, Gavrilova O, Ma Y, Zhu H, Ma X, Xu L, Tuymetova G, Larman BC, Allende ML, Dunn TM, Proia RL (2017) De novo sphingolipid biosynthesis is required for adipocyte survival and metabolic homeostasis. J Biol Chem 292(9):3929–3939. https://doi.org/10.1074/jbc.M116.756460
Qu L, Qu F, Jia Z, Wang C, Wu C, Zhang J (2015) Integrated targeted sphingolipidomics and transcriptomics reveal abnormal sphingolipid metabolism as a novel mechanism of the hepatotoxicity and nephrotoxicity of triptolide. J Ethnopharmacol 170:28–38. https://doi.org/10.1016/j.jep.2015.05.010
Li F, Zhang N (2015) Ceramide: therapeutic potential in combination therapy for cancer treatment. Curr Drug Metab 17(1):37–51
Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Häussinger D, Reinehr R, Larner A, Spiegel S, Fisher PB, Voelkel-Johnson C, Ogretmen B, Grant S, Dent P (2010) Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca2+-de novo ceramide-PP2A-ROS dependent signaling pathway. Can Res 70(15):6313–6324. https://doi.org/10.1158/0008-5472.CAN-10-0999
Gencer EB, Ural AU, Avcu F, Baran Y (2011) A novel mechanism of dasatinib-induced apoptosis in chronic myeloid leukemia; ceramide synthase and ceramide clearance genes. Ann Hematol 90(11):1265–1275. https://doi.org/10.1007/s00277-011-1212-5
Maeng HJ, Song J-H, Kim G-T, Song Y-J, Lee K, Kim J-Y, Park T-S (2017) Celecoxib-mediated activation of endoplasmic reticulum stress induces de novo ceramide biosynthesis and apoptosis in hepatoma HepG2 cells. BMB Rep 50(3):144–149. https://doi.org/10.5483/bmbrep.2017.50.3.197
Signorelli P, Munoz-Olaya JM, Gagliostro V, Casas J, Ghidoni R, Fabriàs G (2009) Dihydroceramide intracellular increase in response to resveratrol treatment mediates autophagy in gastric cancer cells. Cancer Lett 282(2):238–243. https://doi.org/10.1016/j.canlet.2009.03.020
Grenald SA, Doyle TM, Zhang H, Slosky LM, Chen Z, Largent-Milnes TM, Spiegel S, Vanderah TW, Salvemini D (2017) Targeting the S1P/S1PR1 axis mitigates cancer-induced bone pain and neuroinflammation. Pain 158(9):1733–1742. https://doi.org/10.1097/j.pain.0000000000000965
Holliday MW Jr, Cox SB, Kang MH, Maurer BJ (2013) C22:0- and C24:0-dihydroceramides confer mixed cytotoxicity in T-Cell acute lymphoblastic leukemia cell lines. PLoS One 8(9):e74768. https://doi.org/10.1371/journal.pone.0074768
Realini N, Palese F, Pizzirani D, Pontis S, Basit A, Bach A, Ganesan A, Piomelli D (2016) Acid ceramidase in melanoma: expression, localization, and effects of pharmacological inhibition. J Biol Chem 291(5):2422–2434. https://doi.org/10.1074/jbc.M115.666909
Knapp P, Baranowski M, Knapp M, Zabielski P, Błachnio-Zabielska AU, Górski J (2010) Altered sphingolipid metabolism in human endometrial cancer. Prostaglandins Other Lipid Mediat 92(1):62–66. https://doi.org/10.1016/j.prostaglandins.2010.03.002
Cazzaniga M, Varricchio C, Montefrancesco C, Feroce I, Guerrieri-Gonzaga A (2012) Fenretinide (4-HPR): a preventive chance for women at genetic and familial risk? J Biomed Biotechnol 2012:172897. https://doi.org/10.1155/2012/172897
Rahmaniyan M, Curley RW, Obeid LM, Hannun YA, Kraveka JM (2011) Identification of dihydroceramide desaturase as a direct in vitro target for fenretinide. J Biol Chem 286(28):24754–24764. https://doi.org/10.1074/jbc.M111.250779
Illuzzi G, Bernacchioni C, Aureli M, Prioni S, Frera G, Donati C, Valsecchi M, Chigorno V, Bruni P, Sonnino S, Prinetti A (2010) Sphingosine kinase mediates resistance to the synthetic retinoid N-(4-hydroxyphenyl)retinamide in human ovarian cancer cells. J Biol Chem 285(24):18594–18602. https://doi.org/10.1074/jbc.M109.072801
Noack J, Choi J, Richter K, Kopp-Schneider A, Regnier-Vigouroux A (2014) A sphingosine kinase inhibitor combined with temozolomide induces glioblastoma cell death through accumulation of dihydrosphingosine and dihydroceramide, endoplasmic reticulum stress and autophagy. Cell Death Dis 5:e1425. https://doi.org/10.1038/cddis.2014.384
Valsecchi M, Aureli M, Mauri L, Illuzzi G, Chigorno V, Prinetti A, Sonnino S (2010) Sphingolipidomics of A2780 human ovarian carcinoma cells treated with synthetic retinoids. J Lipid Res 51(7):1832–1840. https://doi.org/10.1194/jlr.M004010
Wang H, Maurer BJ, Liu YY, Wang E, Allegood JC, Kelly S, Symolon H, Liu Y, Merrill AH Jr, Gouaze-Andersson V, Yu JY, Giuliano AE, Cabot MC (2008) N-(4-Hydroxyphenyl)retinamide increases dihydroceramide and synergizes with dimethylsphingosine to enhance cancer cell killing. Mol Cancer Ther 7(9):2967–2976. https://doi.org/10.1158/1535-7163.MCT-08-0549
Idkowiak-Baldys J, Apraiz A, Li L, Rahmaniyan M, Clarke CJ, Kraveka JM, Asumendi A, Hannun YA (2010) Dihydroceramide desaturase activity is modulated by oxidative stress. Biochem J 427(2):265–274. https://doi.org/10.1042/BJ20091589
Venant H, Rahmaniyan M, Jones EE, Lu P, Lilly MB, Garrett-Mayer E, Drake RR, Kraveka JM, Smith CD, Voelkel-Johnson C (2015) The sphingosine kinase 2 inhibitor ABC294640 reduces the growth of prostate cancer cells and results in accumulation of dihydroceramides in vitro and in vivo. Mol Cancer Ther 14(12):2744–2752. https://doi.org/10.1158/1535-7163.MCT-15-0279
Obeid LM, Linardic CM, Karolak LA, Hannun YA (1993) Programmed cell death induced by ceramide. Science 259(5102):1769
Hernandez-Corbacho MJ, Canals D, Adada MM, Liu M, Senkal CE, Yi JK, Mao C, Luberto C, Hannun YA, Obeid LM (2015) Tumor necrosis factor-alpha (TNFalpha)-induced ceramide generation via ceramide synthases regulates loss of focal adhesion kinase (FAK) and programmed cell death. J Biol Chem 290(42):25356–25373. https://doi.org/10.1074/jbc.M115.658658
Singh SS, Vats S, Chia AY, Tan TZ, Deng S, Ong MS, Arfuso F, Yap CT, Goh BC, Sethi G, Huang RY, Shen HM, Manjithaya R, Kumar AP (2018) Dual role of autophagy in hallmarks of cancer. Oncogene 37(9):1142–1158. https://doi.org/10.1038/s41388-017-0046-6
Apraiz A, Idkowiak-Baldys J, Nieto-Rementería N, Boyano MD, Hannun YA, Asumendi A (2012) Dihydroceramide accumulation and reactive oxygen species are distinct and nonessential events in 4-HPR-mediated leukemia cell death. Biochem Cell Biol 90(2):209–223. https://doi.org/10.1139/o2012-001
McNair C, Urbanucci A, Comstock CES, Augello MA, Goodwin JF, Launchbury R, Zhao SG, Schiewer MJ, Ertel A, Karnes J, Davicioni E, Wang L, Wang Q, Mills IG, Feng FY, Li W, Carroll JS, Knudsen KE (2017) Cell cycle-coupled expansion of AR activity promotes cancer progression. Oncogene 36(12):1655–1668. https://doi.org/10.1038/onc.2016.334
Zhou W, Ye X-L, Sun Z-J, Ji X-D, Chen H-X, Xie D (2009) Overexpression of degenerative spermatocyte homolog 1 up-regulates the expression of cyclin D1 and enhances metastatic efficiency in esophageal carcinoma Eca109 cells. Mol Carcinog 48(10):886–894. https://doi.org/10.1002/mc.20533
Boppana NB, DeLor JS, Van Buren E, Bielawska A, Bielawski J, Pierce JS, Korbelik M, Separovic D (2016) Enhanced apoptotic cancer cell killing after Foscan photodynamic therapy combined with fenretinide via de novo sphingolipid biosynthesis pathway. J Photochem Photobiol B Biol 159(Supplement C):191–195. https://doi.org/10.1016/j.jphotobiol.2016.02.040
Breen P, Joseph N, Thompson K, Kraveka JM, Gudz TI, Li LI, Rahmaniyan M, Bielawski J, Pierce JS, Van Buren E, Bhatti G, Separovic D (2013) Dihydroceramide desaturase knockdown impacts sphingolipids and apoptosis after photodamage in human head and neck squamous carcinoma cells. Anticancer Res 33(1):77–84
Separovic D, Bielawski J, Pierce JS, Merchant S, Tarca AL, Ogretmen B, Korbelik M (2009) Increased tumour dihydroceramide production after Photofrin-PDT alone and improved tumour response after the combination with the ceramide analogue LCL29. Evidence from mouse squamous cell carcinomas. Br J Cancer 100(4):626–632. https://doi.org/10.1038/sj.bjc.6604896
Separovic D, Breen P, Joseph N, Bielawski J, Pierce JS, Van Buren E, Gudz TI (2012) siRNA-mediated down-regulation of ceramide synthase 1 leads to apoptotic resistance in human head and neck squamous carcinoma cells after photodynamic therapy. Anticancer Res 32(7):2479–2485
Jiang Q, Rao X, Kim CY, Freiser H, Zhang Q, Jiang Z, Li G (2012) Gamma-tocotrienol induces apoptosis and autophagy in prostate cancer cells by increasing intracellular dihydrosphingosine and dihydroceramide. Int J Cancer 130(3):685–693. https://doi.org/10.1002/ijc.26054
Sylvester PW, Ayoub NM (2013) Tocotrienols target PI3K/Akt signaling in anti-breast cancer therapy. Anticancer Agents Med Chem 13(7):1039–1047
Wang Y, Park NY, Jang Y, Ma A, Jiang Q (2015) Vitamin E gamma-tocotrienol inhibits cytokine-stimulated NF-kappaB activation by induction of anti-inflammatory A20 via stress adaptive response due to modulation of sphingolipids. J Immunol 195(1):126–133. https://doi.org/10.4049/jimmunol.1403149
Dorronsoro A, Lang V, Jakobsson E, Ferrin I, Salcedo JM, Fernández-Rueda J, Fechter K, Rodriguez MS, Trigueros C (2013) Identification of the NF-κB inhibitor A20 as a key regulator for human adipogenesis. Cell Death Dis 4:e972. https://doi.org/10.1038/cddis.2013.494
Mullen TD, Spassieva S, Jenkins RW, Kitatani K, Bielawski J, Hannun YA, Obeid LM (2011) Selective knockdown of ceramide synthases reveals complex interregulation of sphingolipid metabolism. J Lipid Res 52(1):68–77. https://doi.org/10.1194/jlr.M009142
Huwiler A, Brunner J, Hummel R, Vervoordeldonk M, Stabel S, van den Bosch H, Pfeilschifter J (1996) Ceramide-binding and activation defines protein kinase c-Raf as a ceramide-activated protein kinase. Proc Natl Acad Sci USA 93(14):6959–6963
Huwiler A, Xin C, Brust AK, Briner VA, Pfeilschifter J (2004) Differential binding of ceramide to MEKK1 in glomerular endothelial and mesangial cells. Biochim Biophys Acta 1636(2–3):159–168. https://doi.org/10.1016/j.bbalip.2003.08.010
Skolova B, Jandovska K, Pullmannova P, Tesar O, Roh J, Hrabalek A, Vavrova K (2014) The role of the trans double bond in skin barrier sphingolipids: permeability and infrared spectroscopic study of model ceramide and dihydroceramide membranes. Langmuir 30(19):5527–5535. https://doi.org/10.1021/la500622f
Dany M, Elston D (2017) Gene expression of sphingolipid metabolism pathways is altered in hidradenitis suppurativa. J Am Acad Dermatol 77(2):268.e266–273.e266. https://doi.org/10.1016/j.jaad.2017.03.016
Deeley JM, Hankin JA, Friedrich MG, Murphy RC, Truscott RJW, Mitchell TW, Blanksby SJ (2010) Sphingolipid distribution changes with age in the human lens. J Lipid Res 51(9):2753–2760. https://doi.org/10.1194/jlr.M007716
Gardner NM, Riley RT, Showker JL, Voss KA, Sachs AJ, Maddox JR, Gelineau-van Waes JB (2016) Elevated nuclear sphingoid base-1-phosphates and decreased histone deacetylase activity after fumonisin B1 treatment in mouse embryonic fibroblasts. Toxicol Appl Pharmacol 298:56–65. https://doi.org/10.1016/j.taap.2016.02.018
Zhang H, Desai NN, Olivera A, Seki T, Brooker G, Spiegel S (1991) Sphingosine-1-phosphate, a novel lipid, involved in cellular proliferation. J Cell Biol 114(1):155–167
Olivera A, Spiegel S (1993) Sphingosine-1-phosphate as second messenger in cell proliferation induced by PDGF and FCS mitogens. Nature 365(6446):557–560. https://doi.org/10.1038/365557a0
Couttas TA, Kain N, Tran C, Chatterton Z, Kwok JB, Don AS (2018) Age-dependent changes to sphingolipid balance in the human hippocampus are gender-specific and may sensitize to neurodegeneration. J Alzheimers Dis 63(2):503–514. https://doi.org/10.3233/JAD-171054
Callihan P, Zitomer NC, Stoeling MV, Kennedy PC, Lynch KR, Riley RT, Hooks SB (2012) Distinct generation, pharmacology, and distribution of sphingosine 1-phosphate and dihydrosphingosine 1-phosphate in human neural progenitor cells. Neuropharmacology 62(2):988–996. https://doi.org/10.1016/j.neuropharm.2011.10.005
Niedernberg A, Tunaru S, Blaukat A, Ardati A, Kostenis E (2003) Sphingosine 1-phosphate and dioleoylphosphatidic acid are low affinity agonists for the orphan receptor GPR63. Cell Signal 15(4):435–446
Lin W, Zhang J, Liu Y, Wu R, Yang H, Hu X, Ling X (2017) Studies on diagnostic biomarkers and therapeutic mechanism of Alzheimer’s disease through metabolomics and hippocampal proteomics. Eur J Pharm Sci 105:119–126. https://doi.org/10.1016/j.ejps.2017.05.003
Gray SG (2011) Targeting Huntington’s disease through histone deacetylases. Clin Epigenet 2(2):257–277. https://doi.org/10.1007/s13148-011-0025-7
Sharma S, Taliyan R (2015) Transcriptional dysregulation in Huntington’s disease: the role of histone deacetylases. Pharmacol Res 100:157–169. https://doi.org/10.1016/j.phrs.2015.08.002
Edsall LC, Pirianov GG, Spiegel S (1997) Involvement of sphingosine 1-phosphate in nerve growth factor-mediated neuronal survival and differentiation. J Neurosci 17(18):6952–6960
Bu S, Kapanadze B, Hsu T, Trojanowska M (2008) Opposite effects of dihydrosphingosine 1-phosphate and sphingosine 1-phosphate on transforming growth factor-beta/Smad signaling are mediated through the PTEN/PPM1A-dependent pathway. J Biol Chem 283(28):19593–19602. https://doi.org/10.1074/jbc.M802417200
Triola G, Fabrias G, Dragusin M, Niederhausen L, Broere R, Llebaria A, van Echten-Deckert G (2004) Specificity of the dihydroceramide desaturase inhibitor N-[(1R,2S)-2-hydroxy-1-hydroxymethyl-2-(2-tridecyl-1-cyclopropenyl)ethyl]octanami de (GT11) in primary cultured cerebellar neurons. Mol Pharmacol 66(6):1671–1678. https://doi.org/10.1124/mol.104.003681
Gelineau-van Waes J, Rainey MA, Maddox JR, Voss KA, Sachs AJ, Gardner NM, Wilberding JD, Riley RT (2012) Increased sphingoid base-1-phosphates and failure of neural tube closure after exposure to fumonisin or FTY720. Birth Defects Res A Clin Mol Teratol 94(10):790–803. https://doi.org/10.1002/bdra.23074
Baranowski M, Charmas M, Dlugolecka B, Gorski J (2011) Exercise increases plasma levels of sphingoid base-1 phosphates in humans. Acta Physiol (Oxf) 203(3):373–380. https://doi.org/10.1111/j.1748-1716.2011.02322.x
Baranowski M, Gorski J, Klapcinska B, Waskiewicz Z, Sadowska-Krepa E (2014) Ultramarathon run markedly reduces plasma sphingosine-1-phosphate concentration. Int J Sport Nutr Exerc Metab 24(2):148–156. https://doi.org/10.1123/ijsnem.2013-0093
Baranowski M, Błachnio-Zabielska AU, Charmas M, Helge JW, Dela F, Książek M, Długołęcka B, Klusiewicz A, Chabowski A, Górski J (2015) Exercise increases sphingoid base-1-phosphate levels in human blood and skeletal muscle in a time- and intensity-dependent manner. Eur J Appl Physiol 115(5):993–1003. https://doi.org/10.1007/s00421-014-3080-x
Knapp M, Zendzian-Piotrowska M, Blachnio-Zabielska A, Zabielski P, Kurek K, Gorski J (2012) Myocardial infarction differentially alters sphingolipid levels in plasma, erythrocytes and platelets of the rat. Basic Res Cardiol 107(6):294. https://doi.org/10.1007/s00395-012-0294-0
Górski J, Baranowski M, Wójcik B, Chabowski A (2015) Effect of atrial pacing on the level of bioactive sphingolipids in the heart ventricles of the rat. Atherosclerosis 241(1):e122–e123. https://doi.org/10.1016/j.atherosclerosis.2015.04.425
Knapp M, Lisowska A, Zabielski P, Musiał W, Baranowski M (2013) Sustained decrease in plasma sphingosine-1-phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediat 106:53–61. https://doi.org/10.1016/j.prostaglandins.2013.10.001
Knapp M, Baranowski M, Czarnowski D, Lisowska A, Zabielski P, Gorski J, Musial W (2009) Plasma sphingosine-1-phosphate concentration is reduced in patients with myocardial infarction. Med Sci Monit 15(9):CR490–CR493
Książek M, Chacińska M, Chabowski A, Baranowski M (2015) Sources, metabolism, and regulation of circulating sphingosine-1-phosphate. J Lipid Res 56(7):1271–1281. https://doi.org/10.1194/jlr.R059543
Argraves KM, Sethi AA, Gazzolo PJ, Wilkerson BA, Remaley AT, Tybjaerg-Hansen A, Nordestgaard BG, Yeatts SD, Nicholas KS, Barth JL, Argraves WS (2011) S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease. Lipids Health Dis 10:70. https://doi.org/10.1186/1476-511X-10-70
Levkau B (2015) HDL-S1P: cardiovascular functions, disease-associated alterations, and therapeutic applications. Front Pharmacol 6:243. https://doi.org/10.3389/fphar.2015.00243
Aoki S, Yatomi Y, Ohta M, Osada M, Kazama F, Satoh K, Nakahara K, Ozaki Y (2005) Sphingosine 1-phosphate-related metabolism in the blood vessel. J Biochem 138(1):47–55. https://doi.org/10.1093/jb/mvi100
Dahm F, Nocito A, Bielawska A, Lang KS, Georgiev P, Asmis LM, Bielawski J, Madon J, Hannun YA, Clavien PA (2006) Distribution and dynamic changes of sphingolipids in blood in response to platelet activation. J Thromb Haemost 4(12):2704–2709. https://doi.org/10.1111/j.1538-7836.2006.02241.x
Knapp M, Lisowska A (2016) Blood bioactive sphingolipids and activity of acid sphingomyelinase in patients with multivessel coronary artery disease. J Clin Exp Cardiol 7:12. https://doi.org/10.4172/2155-9880.1000482
Ono Y, Kurano M, Ohkawa R, Yokota H, Igarashi K, Aoki J, Tozuka M, Yatomi Y (2013) Sphingosine 1-phosphate release from platelets during clot formation: close correlation between platelet count and serum sphingosine 1-phosphate concentration. Lipids Health Dis 12:20. https://doi.org/10.1186/1476-511X-12-20
Dobierzewska A, Soman S, Illanes SE, Morris AJ (2017) Plasma cross-gestational sphingolipidomic analyses reveal potential first trimester biomarkers of preeclampsia. PLoS One 12(4):e0175118. https://doi.org/10.1371/journal.pone.0175118
Sugiura T, Dohi Y, Yamashita S, Ohte N, Ito S, Sanagawa A, Iwaki S, Ohkawa R, Yatomi Y, Fujii S (2013) Circulating microRNA-126 as a potential biomarker for recovery from smoking-related vascular endothelial damage. Eur Heart J 34(suppl_1):P2417. https://doi.org/10.1093/eurheartj/eht308.p2417
Ryu Y, Takuwa N, Sugimoto N, Sakurada S, Usui S, Okamoto H, Matsui O, Takuwa Y (2002) Sphingosine-1-phosphate, a platelet-derived lysophospholipid mediator, negatively regulates cellular rac activity and cell migration in vascular smooth muscle cells. Circ Res 90(3):325
Bu S, Yamanaka M, Pei H, Bielawska A, Bielawski J, Hannun YA, Obeid L, Trojanowska M (2006) Dihydrosphingosine 1-phosphate stimulates MMP1 gene expression via activation of ERK1/2-Ets1 pathway in human fibroblasts. FASEB J 20(1):184–186. https://doi.org/10.1096/fj.05-4646fje
Bu S, Asano Y, Bujor A, Highland K, Hant F, Trojanowska M (2010) Dihydrosphingosine-1 phosphate has a potent anti-fibrotic effect in Scleroderma fibroblasts via normalization of PTEN levels. Arthritis Rheum 62(7):2117–2126. https://doi.org/10.1002/art.27463
Ruddy JM, Ikonomidis JS, Jones JA (2016) Multidimensional contribution of matrix metalloproteinases to atherosclerotic plaque vulnerability: multiple mechanisms of inhibition to promote stability. J Vasc Res 53(1–2):1–16. https://doi.org/10.1159/000446703
Ye S, Gale CR, Martyn CN (2003) Variation in the matrix metalloproteinase-1 gene and risk of coronary heart disease. Eur Heart J 24(18):1668–1671
Gorshkova IA, Wang H, Orbelyan GA, Goya J, Natarajan V, Beiser DG, Vanden Hoek TL, Berdyshev EV (2013) Inhibition of sphingosine-1-phosphate lyase rescues sphingosine kinase-1-knockout phenotype following murine cardiac arrest. Life Sci 93(9):359–366. https://doi.org/10.1016/j.lfs.2013.07.017
Igarashi N, Okada T, Hayashi S, Fujita T, Jahangeer S, S-i Nakamura (2003) Sphingosine kinase 2 is a nuclear protein and inhibits DNA synthesis. J Biol Chem 278(47):46832–46839
Knapp M, Lisowska A, Knapp P, Baranowski M (2013) Dose-dependent effect of aspirin on the level of sphingolipids in human blood. Adv Med Sci 58(2):274–281. https://doi.org/10.2478/ams-2013-0021
Ono JG, Worgall TS, Worgall S (2015) Airway reactivity and sphingolipids—implications for childhood asthma. Mol Cell Pediatr 2:13. https://doi.org/10.1186/s40348-015-0025-3
Miller M, Tam AB, Mueller JL, Rosenthal P, Beppu A, Gordillo R, McGeough MD, Vuong C, Doherty TA, Hoffman HM, Niwa M, Broide DH (2017) Cutting edge: targeting epithelial ORMDL3 increases, rather than reduces, airway responsiveness and is associated with increased sphingosine-1-phosphate. J Immunol
Kowal K, Zebrowska E, Chabowski A (2018) Plasma concentration of selected sphingolipids correlates with lung function parameters in house dust mite allergic patients. J Allergy Clin Immunol 141(2, Supplement):AB113. https://doi.org/10.1016/j.jaci.2017.12.360
Oyeniran C, Sturgill JL, Hait NC, Huang WC, Avni D, Maceyka M, Newton J, Allegood JC, Montpetit A, Conrad DH, Milstien S, Spiegel S (2015) Aberrant ORM (yeast)-like protein isoform 3 (ORMDL3) expression dysregulates ceramide homeostasis in cells and ceramide exacerbates allergic asthma in mice. J Allergy Clin Immunol 136(4):1035 e1036–1046 e1036. https://doi.org/10.1016/j.jaci.2015.02.031
Berdyshev EV, Gorshkova IA, Usatyuk P, Zhao Y, Saatian B, Hubbard W, Natarajan V (2006) De novo biosynthesis of dihydrosphingosine-1-phosphate by sphingosine kinase 1 in mammalian cells. Cell Signal 18(10):1779–1792. https://doi.org/10.1016/j.cellsig.2006.01.018
Boujaoude LC, Bradshaw-Wilder C, Mao C, Cohn J, Ogretmen B, Hannun YA, Obeid LM (2001) Cystic fibrosis transmembrane regulator regulates uptake of sphingoid base phosphates and lysophosphatidic acid: modulation of cellular activity of sphingosine 1-phosphate. J Biol Chem 276(38):35258–35264. https://doi.org/10.1074/jbc.M105442200
Gorshkova I, Zhou T, Mathew B, Jacobson JR, Takekoshi D, Bhattacharya P, Smith B, Aydogan B, Weichselbaum RR, Natarajan V, Garcia JG, Berdyshev EV (2012) Inhibition of serine palmitoyltransferase delays the onset of radiation-induced pulmonary fibrosis through the negative regulation of sphingosine kinase-1 expression. J Lipid Res 53(8):1553–1568. https://doi.org/10.1194/jlr.M026039
Park SW, Kim M, Chen SW, Brown KM, D’Agati VD, Lee HT (2010) Sphinganine-1-phosphate protects kidney and liver after hepatic ischemia and reperfusion in mice through S1P1 receptor activation. Lab Invest 90(8):1209–1224. https://doi.org/10.1038/labinvest.2010.102
Park SW, Kim M, Chen SW, D’Agati VD, Lee HT (2010) Sphinganine-1-phosphate attenuates both hepatic and renal injury induced by hepatic ischemia and reperfusion in mice. Shock 33(1):31–42. https://doi.org/10.1097/SHK.0b013e3181c02c1f
Kim DH, Yoo HS, Lee YM, Kie JH, Jang S, Oh S (2006) Elevation of sphinganine 1-phosphate as a predictive biomarker for fumonisin exposure and toxicity in mice. J Toxicol Environ Health A 69(23):2071–2082. https://doi.org/10.1080/15287390600746215
Gopee NV, Sharma RP (2003) Sphingoid bases and their phosphates: transient activation and delayed repression of protein kinase C isoforms and their possible involvement in fumonisin B1 cytotoxicity. Toxicology 187(2):239–250. https://doi.org/10.1016/S0300-483X(03)00048-9
Tardieu D, Tran ST, Auvergne A, Babile R, Benard G, Bailly JD, Guerre P (2006) Effects of fumonisins on liver and kidney sphinganine and the sphinganine to sphingosine ratio during chronic exposure in ducks. Chem Biol Interact 160(1):51–60. https://doi.org/10.1016/j.cbi.2005.11.004
Riley RT, Torres O, Matute J, Gregory SG, Ashley-Koch AE, Showker JL, Mitchell T, Voss KA, Maddox JR, Gelineau-van Waes JB (2015) Evidence for fumonisin inhibition of ceramide synthase in humans consuming maize-based foods and living in high exposure communities in Guatemala. Mol Nutr Food Res 59(11):2209–2224. https://doi.org/10.1002/mnfr.201500499
Choudhury GG, Biswas P, Grandaliano G, Abboud HE (1993) Involvement of PKC-alpha in PDGF-mediated mitogenic signaling in human mesangial cells. Am J Physiol 265(5 Pt 2):F634–F642. https://doi.org/10.1152/ajprenal.1993.265.5.F634
Katsuma S, Hada Y, Ueda T, Shiojima S, Hirasawa A, Tanoue A, Takagaki K, Ohgi T, Yano J, Tsujimoto G (2002) Signalling mechanisms in sphingosine 1-phosphate-promoted mesangial cell proliferation. Genes Cells 7(12):1217–1230
Veret J, Coant N, Gorshkova IA, Giussani P, Fradet M, Riccitelli E, Skobeleva A, Goya J, Kassis N, Natarajan V, Portha B, Berdyshev EV, Le Stunff H (2013) Role of palmitate-induced sphingoid base-1-phosphate biosynthesis in INS-1 beta-cell survival. Biochim Biophys Acta 1831(2):251–262. https://doi.org/10.1016/j.bbalip.2012.10.003
Katsuma S, Hada Y, Shiojima S, Hirasawa A, Tanoue A, Takagaki K, Ohgi T, Yano J, Tsujimoto G (2003) Transcriptional profiling of gene expression patterns during sphingosine 1-phosphate-induced mesangial cell proliferation. Biochem Biophys Res Commun 300(2):577–584. https://doi.org/10.1016/S0006-291X(02)02850-4
Sato K, Ui M, Okajima F (2000) Differential roles of Edg-1 and Edg-5, sphingosine 1-phosphate receptors, in the signaling pathways in C6 glioma cells. Brain Res Mol Brain Res 85(1–2):151–160
Arikawa K, Takuwa N, Yamaguchi H, Sugimoto N, Kitayama J, Nagawa H, Takehara K, Takuwa Y (2003) Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J Biol Chem 278(35):32841–32851. https://doi.org/10.1074/jbc.m305024200
Barth BM, Shanmugavelandy SS, Kaiser JM, McGovern C, Altinoglu EI, Haakenson JK, Hengst JA, Gilius EL, Knupp SA, Fox TE, Smith JP, Ritty TM, Adair JH, Kester M (2013) PhotoImmunoNanoTherapy reveals an anticancer role for sphingosine kinase 2 and dihydrosphingosine-1-phosphate. ACS Nano 7(3):2132–2144. https://doi.org/10.1021/nn304862b
Hait NC, Allegood J, Maceyka M, Strub GM, Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S, Spiegel S (2009) Regulation of histone acetylation in the nucleus by sphingosine-1-phosphate. Science 325(5945):1254–1257. https://doi.org/10.1126/science.1176709
Jin Y, Knudsen E, Wang L, Bryceson Y, Damaj B, Gessani S, Maghazachi AA (2003) Sphingosine 1-phosphate is a novel inhibitor of T-cell proliferation. Blood 101(12):4909–4915. https://doi.org/10.1182/blood-2002-09-2962
Xu Y, Stenmark KR, Das M, Walchak SJ, Ruff LJ, Dempsey EC (1997) Pulmonary artery smooth muscle cells from chronically hypoxic neonatal calves retain fetal-like and acquire new growth properties. Am J Physiol 273(1 Pt 1):L234–L245. https://doi.org/10.1152/ajplung.1997.273.1.L234
Das M, Stenmark KR, Dempsey EC (1995) Enhanced growth of fetal and neonatal pulmonary artery adventitial fibroblasts is dependent on protein kinase C. Am J Physiol 269(5 Pt 1):L660–L667. https://doi.org/10.1152/ajplung.1995.269.5.L660
Mann J, Farrukh IS, Michael JR (1991) Mechanisms by which endothelin 1 induces pulmonary vasoconstriction in the rabbit. J Appl Physiol (1985) 71(2):410–416. https://doi.org/10.1152/jappl.1991.71.2.410
Voss KA, Riley RT (2013) Fumonisin toxicity and mechanism of action: overview and current perspectives. Food Saf 1(1):2013006. https://doi.org/10.14252/foodsafetyfscj.2013006
Riley RT, Voss KA (2006) Differential sensitivity of rat kidney and liver to fumonisin toxicity: organ-specific differences in toxin accumulation and sphingoid base metabolism. Toxicol Sci 92(1):335–345. https://doi.org/10.1093/toxsci/kfj198
Mathur S, Constable PD, Eppley RM, Waggoner AL, Tumbleson ME, Haschek WM (2001) Fumonisin B(1) is hepatotoxic and nephrotoxic in milk-fed calves. Toxicol Sci 60(2):385–396
Grenier B, Schwartz-Zimmermann HE, Caha S, Moll WD, Schatzmayr G, Applegate TJ (2015) Dose-dependent effects on sphingoid bases and cytokines in chickens fed diets prepared with fusarium verticillioides culture material containing fumonisins. Toxins (Basel) 7(4):1253–1272. https://doi.org/10.3390/toxins7041253
Collins TF, Sprando RL, Black TN, Shackelford ME, Laborde JB, Hansen DK, Eppley RM, Trucksess MW, Howard PC, Bryant MA, Ruggles DI, Olejnik N, Rorie JI (1998) Effects of fumonisin B1 in pregnant rats. Part 2. Food Chem Toxicol 36(8):673–685. https://doi.org/10.1016/s0278-6915(98)00036-2
LaBorde JB, Terry KK, Howard PC, Chen JJ, Collins TFX, Shackelford ME, Hansen DK (1997) Lack of embryotoxicity of fumonisin B1in New Zealand White Rabbits. Fundam Appl Toxicol 40(1):120–128. https://doi.org/10.1006/faat.1997.2380
Domijan AM, Peraica M, Markov K, Fuchs R (2009) Urine ochratoxin a and sphinganine/sphingosine ratio in residents of the endemic nephropathy area in Croatia. Arh Hig Rada Toksikol 60(4):387–393. https://doi.org/10.2478/10004-1254-60-2009-1938
Grammatikos G, Dietz J, Ferreiros N, Koch A, Dultz G, Bon D, Karakasiliotis I, Lutz T, Knecht G, Gute P, Herrmann E, Zeuzem S, Mavromara P, Sarrazin C, Pfeilschifter J (2016) Persistence of HCV in acutely-infected patients depletes C24-ceramide and upregulates sphingosine and sphinganine serum levels. Int J Mol Sci 17(6):922. https://doi.org/10.3390/ijms17060922
Qiu S, Zhang H, Fei Q, Zhu F, Wang J, Jia X, Chen B (2018) Urine and plasma metabolomics study on potential hepatoxic biomarkers identification in rats induced by Gynura segetum. J Ethnopharmacol 216:37–46. https://doi.org/10.1016/j.jep.2018.01.017
Touboul D, Roy S, Germain DP, Baillet A, Brion F, Prognon P, Chaminade P, Laprevote O (2005) Fast fingerprinting by MALDI-TOF mass spectrometry of urinary sediment glycosphingolipids in Fabry disease. Anal Bioanal Chem 382(5):1209–1216. https://doi.org/10.1007/s00216-005-3239-8
Ribar S, Mesarić M, Bauman M (2001) High-performance liquid chromatographic determination of sphinganine and sphingosine in serum and urine of subjects from an endemic nephropathy area in Croatia. J Chromatogr B Biomed Sci Appl 754(2):511–519. https://doi.org/10.1016/S0378-4347(01)00041-X
Cho K, S-i Min, Ahn S, Min S-K, Ahn C, Yu K-S, Jang I-J, Cho J-Y, Ha J (2017) Integrative analysis of renal ischemia/reperfusion injury and remote ischemic preconditioning in mice. J Proteome Res 16(8):2877–2886. https://doi.org/10.1021/acs.jproteome.7b00167
Dekker MJ, Baker C, Naples M, Samsoondar J, Zhang R, Qiu W, Sacco J, Adeli K (2013) Inhibition of sphingolipid synthesis improves dyslipidemia in the diet-induced hamster model of insulin resistance: evidence for the role of sphingosine and sphinganine in hepatic VLDL-apoB100 overproduction. Atherosclerosis 228(1):98–109. https://doi.org/10.1016/j.atherosclerosis.2013.01.041
Huang Y, Liu X, Zhao L, Li F, Xiong Z (2014) Kidney tissue targeted metabolic profiling of glucocorticoid-induced osteoporosis and the proposed therapeutic effects of Rhizoma Drynariae studied using UHPLC/MS/MS. Biomed Chromatogr 28(6):878–884. https://doi.org/10.1002/bmc.3194
Pewzner-Jung Y, Park H, Laviad EL, Silva LC, Lahiri S, Stiban J, Erez-Roman R, Brügger B, Sachsenheimer T, Wieland F, Prieto M, Merrill AH, Futerman AH (2010) A critical role for ceramide synthase 2 in liver homeostasis: I. Alterations in lipid metabolic pathways. J Biol Chem 285(14):10902–10910. https://doi.org/10.1074/jbc.m109.077594
Merrill AH Jr, Sullards MC, Wang E, Voss KA, Riley RT (2001) Sphingolipid metabolism: roles in signal transduction and disruption by fumonisins. Environ Health Perspect 109(Suppl 2):283–289. https://doi.org/10.1289/ehp.01109s2283
Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH Jr, Futerman AH (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem 283(9):5677–5684. https://doi.org/10.1074/jbc.M707386200
Dugyala RR, Sharma RP, Tsunoda M, Riley RT (1998) Tumor necrosis factor-α as a contributor in Fumonisin B1 toxicity. J Pharmacol Exp Ther 285(1):317
Bhandari N, He Q, Sharma PR (2001) Gender-related differences in subacute fumonisin B1 hepatotoxicity in BALB/c mice, vol 165. https://doi.org/10.1016/s0300-483x(01)00449-8
He Q, Riley RT, Sharma RP (2001) Fumonisin-induced tumor necrosis factor-alpha expression in a porcine kidney cell line is independent of sphingoid base accumulation induced by ceramide synthase inhibition. Toxicol Appl Pharmacol 174(1):69–77. https://doi.org/10.1006/taap.2001.9189
Sharma RP, Bhandari N, Riley RT, Voss KA, Meredith FI (2000) Tolerance to fumonisin toxicity in a mouse strain lacking the P75 tumor necrosis factor receptor. Toxicology 143(2):183–194. https://doi.org/10.1016/S0300-483X(99)00168-7
Chen X, Hu C, Dai J, Chen L (2015) Metabolomics analysis of seminal plasma in infertile males with kidney-yang deficiency: a preliminary study. Evid Based Complement Alternat Med 2015:892930. https://doi.org/10.1155/2015/892930
Zhao YY, Cheng XL, Wei F, Xiao XY, Sun WJ, Zhang Y, Lin RC (2012) Serum metabonomics study of adenine-induced chronic renal failure in rats by ultra performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. Biomarkers 17(1):48–55. https://doi.org/10.3109/1354750X.2011.637180
Zhang J, Yan L, Chen W, Lin L, Song X, Yan X, Hang W, Huang B (2009) Metabonomics research of diabetic nephropathy and type 2 diabetes mellitus based on UPLC—oaTOF-MS system. Anal Chim Acta 650(1):16–22. https://doi.org/10.1016/j.aca.2009.02.027
Lin L, Huang Z, Gao Y, Chen Y, Hang W, Xing J, Yan X (2012) LC-MS-based serum metabolic profiling for genitourinary cancer classification and cancer type-specific biomarker discovery. Proteomics 12(14):2238–2246. https://doi.org/10.1002/pmic.201200016
Baranowski M, Zabielski P, Blachnio A, Gorski J (2008) Effect of exercise duration on ceramide metabolism in the rat heart. Acta Physiol (Oxf) 192(4):519–529. https://doi.org/10.1111/j.1748-1716.2007.01755.x
Wojcik B, Baranowski M, Chabowski A, Gorski J (2015) Effect of atrial pacing on the level of bioactive sphingolipids in the heart ventricles of the rat. J Physiol Pharmacol 66(3):385–389
Sun L, Liu J, Sun M, Lin L, Miao L, Ge Z, Yang B (2017) Comprehensive metabonomic analysis of heart tissue from isoproterenol-induced myocardial infarction rat based on reversed-phase and hydrophilic interaction chromatography coupled to mass spectrometry. J Sep Sci 40(10):2198–2206. https://doi.org/10.1002/jssc.201601013
Qi Y, Gu H, Song Y, Dong X, Liu A, Lou Z, Fan G, Chai Y (2013) Metabolomics study of resina draconis on myocardial ischemia rats using ultraperformance liquid chromatography/quadrupole time-of-flight mass spectrometry combined with pattern recognition methods and metabolic pathway analysis. Evidence Based Complement Altern Med 2013:10. https://doi.org/10.1155/2013/438680
Y-t Liu, H-m Jia, Chang X, W-h Cheng, Zhao X, Ding G, H-w Zhang, D-y Cai, Zou Z-M (2014) Metabolic pathways involved in Xin-Ke-Shu protecting against myocardial infarction in rats using ultra high-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. J Pharm Biomed Anal 90:35–44. https://doi.org/10.1016/j.jpba.2013.11.008
Miklosz A, Lukaszuk B, Chabowski A, Rogowski F, Kurek K, Zendzian-Piotrowska M (2015) Hyperthyroidism evokes myocardial ceramide accumulation. Cell Physiol Biochem 35(2):755–766. https://doi.org/10.1159/000369735
Park MT, Kang JA, Choi JA, Kang CM, Kim TH, Bae S, Kang S, Kim S, Choi WI, Cho CK, Chung HY, Lee YS, Lee SJ (2003) Phytosphingosine induces apoptotic cell death via caspase 8 activation and Bax translocation in human cancer cells. Clin Cancer Res 9(2):878–885
Yang Y, Jia H, Yu M, Zhou C, Sun L, Zhao Y, Zhang H, Zou Z (2018) Chinese patent medicine Xin-Ke-Shu inhibits Ca2+ overload and dysfunction of fatty acid β-oxidation in rats with myocardial infarction induced by LAD ligation. J Chromatogr B 1079:85–94. https://doi.org/10.1016/j.jchromb.2018.01.038
Błachnio-Zabielska A, Baranowski M, Wójcik B, Górski J (2016) Reduction of ceramide de novo synthesis in solid tissues changes sphingolipid levels in rat plasma, erythrocytes and platelets. Adv Med Sci 61(1):72–77. https://doi.org/10.1016/j.advms.2015.09.006
Huang L, Li T, Liu YW, Zhang L, Dong ZH, Liu SY, Gao YT (2016) Plasma metabolic profile determination in young ST-segment elevation myocardial infarction patients with ischemia and reperfusion: ultra-performance liquid chromatography and mass spectrometry for pathway analysis. Chin Med J (Engl) 129(9):1078–1086. https://doi.org/10.4103/0366-6999.180527
Knapp M, Baranowski M, Lisowska A, Musiał W (2012) Decreased free sphingoid base concentration in the plasma of patients with chronic systolic heart failure. Adv Med Sci 57(1):100–105. https://doi.org/10.2478/v10039-011-0057-4
Sun M, Miao Y, Wang P, Miao L, Liu L, Liu J (2014) Urinary metabonomics study of heart failure patients with HILIC and RPLC separation coupled to TOF–MS. Chromatographia 77(3):249–255. https://doi.org/10.1007/s10337-013-2585-5
Chen Y, Wen S, Jiang M, Zhu Y, Ding L, Shi H, Dong P, Yang J, Yang Y (2017) Atherosclerotic dyslipidemia revealed by plasma lipidomics on ApoE(−/−) mice fed a high-fat diet. Atherosclerosis 262:78–86. https://doi.org/10.1016/j.atherosclerosis.2017.05.010
Jiang H, Shen Z, Chu Y, Li Y, Li J, Wang X, Yang W, Zhang X, Ju J, Xu J, Yang C (2015) Serum metabolomics research of the anti-hypertensive effects of Tengfu Jiangya tablet on spontaneously hypertensive rats. J Chromatogr B Analyt Technol Biomed Life Sci 1002:210–217. https://doi.org/10.1016/j.jchromb.2015.08.010
Liu Y-T, Peng J-B, Jia H-M, Cai D-Y, Zhang H-W, Yu C-Y, Zou Z-M (2014) UPLC-Q/TOF MS standardized Chinese formula Xin-Ke-Shu for the treatment of atherosclerosis in a rabbit model. Phytomedicine 21(11):1364–1372. https://doi.org/10.1016/j.phymed.2014.05.009
Egom EE, Mamas MA, Chacko S, Stringer SE, Charlton-Menys V, El-Omar M, Chirico D, Clarke B, Neyses L, Cruickshank JK, Lei M, Fath-Ordoubadi F (2013) Serum sphingolipids level as a novel potential marker for early detection of human myocardial ischaemic injury. Front Physiol 4:130. https://doi.org/10.3389/fphys.2013.00130
Park TS, Rosebury W, Kindt EK, Kowala MC, Panek RL (2008) Serine palmitoyltransferase inhibitor myriocin induces the regression of atherosclerotic plaques in hyperlipidemic ApoE-deficient mice. Pharmacol Res 58(1):45–51. https://doi.org/10.1016/j.phrs.2008.06.005
Dolgachev V, Nagy B, Taffe B, Hanada K, Separovic D (2003) Reactive oxygen species generation is independent of de novo sphingolipids in apoptotic photosensitized cells. Exp Cell Res 288(2):425–436. https://doi.org/10.1016/S0014-4827(03)00235-0
Miller M, Rosenthal P, Beppu A, Gordillo R, Broide DH (2017) Oroscomucoid like protein 3 (ORMDL3) transgenic mice have reduced levels of sphingolipids including sphingosine-1-phosphate and ceramide. J Allergy Clin Immunol 139(4):1373.e1374–1376.e1374. https://doi.org/10.1016/j.jaci.2016.08.053
Breslow DK, Collins SR, Bodenmiller B, Aebersold R, Simons K, Shevchenko A, Ejsing CS, Weissman JS (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463(7284):1048–1053. https://doi.org/10.1038/nature08787
Loiseau N, Obata Y, Moradian S, Sano H, Yoshino S, Aburai K, Takayama K, Sakamoto K, Holleran WM, Elias PM, Uchida Y (2013) Altered sphingoid base profiles predict compromised membrane structure and permeability in atopic dermatitis. J Dermatol Sci 72(3):296–303. https://doi.org/10.1016/j.jdermsci.2013.08.003
Obata Y, Sano H, Ohta N, Moriwaki T, Ishida K, Uchida Y, Takayama K (2017) Characterization of simple intercellular lipid model of atopic dermatitis stratum corneum containing sphingosine and sphinganine. J Dermatol Sci 86(2):e43–e44. https://doi.org/10.1016/j.jdermsci.2017.02.128
Kurek K, Miklosz A, Lukaszuk B, Chabowski A, Gorski J, Zendzian-Piotrowska M (2015) Inhibition of ceramide de novo synthesis ameliorates diet induced skeletal muscles insulin resistance. J Diabetes Res 2015:154762. https://doi.org/10.1155/2015/154762
Dong Y, Chen YT, Yang YX, Zhou XJ, Dai SJ, Tong JF, Shou D, Li C (2016) Metabolomics study of type 2 diabetes mellitus and the antidiabetic effect of berberine in Zucker diabetic fatty rats using Uplc-ESI-Hdms. Phytother Res 30(5):823–828. https://doi.org/10.1002/ptr.5587
Jiang W, Gao L, Li P, Kan H, Qu J, Men L, Liu Z, Liu Z (2017) Metabonomics study of the therapeutic mechanism of fenugreek galactomannan on diabetic hyperglycemia in rats, by ultra-performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. J Chromatogr B 1044–1045:8–16. https://doi.org/10.1016/j.jchromb.2016.12.039
Jung S, Kim M, Ryu HJ, Chae JS, Lee S-H, Lee JH (2015) Age-related increase in LDL-cholesterol is associated with enhanced oxidative stress and disturbed sphingolipid metabolism. Metabolomics 11(1):40–49. https://doi.org/10.1007/s11306-014-0669-3
Oertel S, Scholich K, Weigert A, Thomas D, Schmetzer J, Trautmann S, Wegner M-S, Radeke HH, Filmann N, Brüne B, Geisslinger G, Tegeder I, Grösch S (2017) Ceramide synthase 2 deficiency aggravates AOM-DSS-induced colitis in mice: role of colon barrier integrity. Cell Mol Life Sci 74(16):3039–3055. https://doi.org/10.1007/s00018-017-2518-9
Choi S, Kim JA, Kim TH, Li HY, Shin KO, Lee YM, Oh S, Pewzner-Jung Y, Futerman AH, Suh SH (2015) Altering sphingolipid composition with aging induces contractile dysfunction of gastric smooth muscle via K(Ca) 1.1 upregulation. Aging Cell 14(6):982–994. https://doi.org/10.1111/acel.12388
Xu H, Zhang L, Kang H, Zhang J, Liu J, Liu S (2016) Serum metabonomics of mild acute pancreatitis. J Clin Lab Anal 30(6):990–998. https://doi.org/10.1002/jcla.21969
Barbas-Bernardos C, Armitage EG, García A, Mérida S, Navea A, Bosch-Morell F, Barbas C (2016) Looking into aqueous humor through metabolomics spectacles—exploring its metabolic characteristics in relation to myopia. J Pharm Biomed Anal 127:18–25. https://doi.org/10.1016/j.jpba.2016.03.032
Sui Z, Li Q, Zhu L, Wang Z, Lv C, Liu R, Xu H, He B, Li Z, Bi K (2017) An integrative investigation of the toxicity of Aconiti kusnezoffii radix and the attenuation effect of its processed drug using a UHPLC-Q-TOF based rat serum and urine metabolomics strategy. J Pharm Biomed Anal 145:240–247. https://doi.org/10.1016/j.jpba.2017.06.049
Charkiewicz K, Goscik J, Blachnio-Zabielska A, Raba G, Sakowicz A, Kalinka J, Chabowski A, Laudanski P (2017) Sphingolipids as a new factor in the pathomechanism of preeclampsia—mass spectrometry analysis. PLoS One 12(5):e0177601. https://doi.org/10.1371/journal.pone.0177601
Zmyslowska A, Ciborowski M, Borowiec M, Fendler W, Pietrowska K, Parfieniuk E, Antosik K, Pyziak A, Waszczykowska A, Kretowski A, Mlynarski W (2017) Serum metabolic fingerprinting identified putatively annotated sphinganine isomer as a biomarker of Wolfram syndrome. J Proteome Res 16(11):4000–4008. https://doi.org/10.1021/acs.jproteome.7b00401
Auer-Grumbach M, Bode H, Pieber TR, Schabhüttl M, Fischer D, Seidl R, Graf E, Wieland T, Schuh R, Vacariu G, Grill F, Timmerman V, Strom TM, Hornemann T (2013) Mutations at Ser331 in the HSN type I gene SPTLC1 are associated with a distinct syndromic phenotype. Eur J Med Genet 56(5):266–269. https://doi.org/10.1016/j.ejmg.2013.02.002
Hsiao CT, Chao HC, Liao YC, Lin KP, Soong BW, Lee YC (2017) Investigation for SPTLC1 mutations in a Taiwanese cohort with hereditary neuropathies. J Neurol Sci 381:463–464. https://doi.org/10.1016/j.jns.2017.08.3516
Murphy SM, Ernst D, Wei Y, Laura M, Liu YT, Polke J, Blake J, Winer J, Houlden H, Hornemann T, Reilly MM (2013) Hereditary sensory and autonomic neuropathy type 1 (HSANI) caused by a novel mutation in SPTLC2. Neurology 80(23):2106–2111. https://doi.org/10.1212/WNL.0b013e318295d789
Kojima T, Asano Y, Kurasawa O, Hirata Y, Iwamura N, Wong T-T, Saito B, Tanaka Y, Arai R, Yonemori K, Miyamoto Y, Sagiya Y, Yaguchi M, Shibata S, Mizutani A, Sano O, Adachi R, Satomi Y, Hirayama M, Aoyama K, Hiura Y, Kiba A, Kitamura S, Imamura S (2018) Discovery of novel serine palmitoyltransferase inhibitors as cancer therapeutic agents. Bioorg Med Chem 26(9):2452–2465. https://doi.org/10.1016/j.bmc.2018.04.008
Boyden LM, Vincent NG, Zhou J, Hu R, Craiglow BG, Bayliss SJ, Rosman IS, Lucky AW, Diaz LA, Goldsmith LA, Paller AS, Lifton RP, Baserga SJ, Choate KA (2017) Mutations in KDSR cause recessive progressive symmetric erythrokeratoderma. Am J Hum Genet 100(6):978–984. https://doi.org/10.1016/j.ajhg.2017.05.003
Takeichi T, Torrelo A, Lee JYW, Ohno Y, Lozano ML, Kihara A, Liu L, Yasuda Y, Ishikawa J, Murase T, Rodrigo AB, Fernández-Crehuet P, Toi Y, Mellerio J, Rivera J, Vicente V, Kelsell DP, Nishimura Y, Okuno Y, Kojima D, Ogawa Y, Sugiura K, Simpson MA, McLean WHI, Akiyama M, McGrath JA (2017) Biallelic mutations in KDSR disrupt ceramide synthesis and result in a spectrum of keratinization disorders associated with thrombocytopenia. J Invest Dermatol 137(11):2344–2353. https://doi.org/10.1016/j.jid.2017.06.028
Krebs S, Medugorac I, Röther S, Strässer K, Förster M (2007) A missense mutation in the 3-ketodihydrosphingosine reductase FVT1 as candidate causal mutation for bovine spinal muscular atrophy. Proc Natl Acad Sci USA 104(16):6746–6751. https://doi.org/10.1073/pnas.0607721104
Ordonez YF, Gonzalez J, Bedia C, Casas J, Abad JL, Delgado A, Fabrias G (2016) 3-Ketosphinganine provokes the accumulation of dihydroshingolipids and induces autophagy in cancer cells. Mol BioSyst 12(4):1166–1173. https://doi.org/10.1039/C5MB00852B
Funding
This research was supported by National Health and Medical Research Council of Australia Program Grants (1092642) (BHW, DL, CR and DJK) and Project Grant (1087355) (BHW). RM and FS are sponsored by a Monash Graduate Scholarship and Monash International Postgraduate Research Scholarship for their doctoral studies.
Author information
Authors and Affiliations
Contributions
RM and BW conceived and designed review question, conducted preliminary data search, and sorting the papers. FS and YH conducted literature search and screening. DK, CR, BF and DL assisted drafting and edited of the paper and had responsibility for its final content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Magaye, R.R., Savira, F., Hua, Y. et al. The role of dihydrosphingolipids in disease. Cell. Mol. Life Sci. 76, 1107–1134 (2019). https://doi.org/10.1007/s00018-018-2984-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-018-2984-8
Keywords
- Adipocyte
- Aging
- Airway hypersensitivity
- Apoptosis
- Autophagy
- Cancer
- Cardiomyopathy
- Ceramide
- Ceramide synthase
- Dihydroceramide desaturase 1-Des-1
- Diabetes
- Dihydrosphinganine
- FB1 toxicity
- Hypoxia
- Neurodegenerative
- Sphingosine kinase
- Serine palmitoyl transferase
- Sphingosine-1-phosphate—S1P
- Sphingosine-1-phosphate receptors
- 4-HRP fenretinide