
Abstract
Metabolic disease, including obesity and type 2 diabetes, constitutes a major emerging health crisis in Western nations. Although the symptoms and clinical pathology and physiology of these conditions are well understood, the molecular mechanisms underlying the disease process have largely remained obscure. Sphingolipids, a lipid class with both signaling and structural properties, have recently emerged as key players in most major tissues affected by diabetes and are required components in the molecular etiology of this disease. Indeed, sphingolipids have been shown to mediate loss of insulin sensitivity, to promote the characteristic diabetic proinflammatory state, and to induce cell death and dysfunction in important organs such as the pancreas and heart. Furthermore, plasma sphingolipid levels are emerging as potential biomarkers for the decompensation of insulin resistance to frank type 2 diabetes. Despite these discoveries, the roles of specific sphingolipid species and sphingolipid metabolic pathways remain obscure, and newly developed experimental approaches must be employed to elucidate the detailed molecular mechanisms necessary for rational drug development and other clinical applications.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
Type 2 diabetes is an increasingly important healthcare concern in Western nations, with incidence on the rise in both Europe and the United States (Passa 2002; Wei et al. 2011). This disease, which is diagnosed based on blood plasma hyperglycemia, has been linked to lipid overload and abdominal obesity and may synergize with these conditions to promote negative clinical outcomes (Maheux et al. 1994; Nussey and Whitehead 2001; Pontiroli and Camisasca 2002; Grundy et al. 2004; Banegas et al. 2007; Qiao and Nyamdorj 2010). This connection is significant, as the incidence of obesity is also increasing, with greater than 65 % of individuals classified as overweight or obese in both the United States and the European Union (Martinez et al. 1999; Baskin et al. 2005; Fry and Finley 2005; Samad et al. 2006; Mueller-Riemenschneider et al. 2008). This chapter summarizes current knowledge of the sphingolipid-dependent manifestations of obesity and diabetes and points to areas for future study and potential clinical utility.
2 Pathogenesis of Type 2 Diabetes Mellitus
Diabetes develops not as a single-organ disease but as a syndrome progressively affecting multiple organ systems. Because diabetes is silent early on and often is only discovered after onset of complications, it has been difficult to tease out a complete natural history of this disease. However, careful epidemiological and laboratory studies have revealed a general outline of disease progression.
It is believed that insulin resistance is the primary defect in type 2 diabetes mellitus, preceding frank hyperglycemia (Cavaghan et al. 2000). In many diabetics, the initial cause of insulin resistance may be hyperlipidemia consequent to abdominal obesity (Pouliot et al. 1990). Excess free fatty acids promote insulin resistance in peripheral tissues, including the skeletal muscle and liver; at first, pancreatic β-cells are able to compensate for this by increasing insulin secretion (Bevilacqua et al. 1987; Hirose et al. 1996; Roden et al. 1996). This compensated state, in which hyperinsulinemia effectively controls plasma glucose levels, may persist for years and, in fact, may never progress to overt diabetes mellitus (Leahy 2008). Over time, the ability of the pancreas to produce high levels of insulin may be exhausted due both to aberrant cell signaling and to β-cell loss to apoptosis (Hirose et al. 1996; Pick et al. 1998). This decompensation results in the hyperglycemia observed in type 2 diabetes, which occurs as a result of both continually elevated hepatic glucose output and reduced peripheral glucose uptake and utilization. At this point, the hyperglycemia itself damages pancreatic β-cells, accelerating the course of β-cell insufficiency and disease progression (Maedler et al. 2003).
Type 2 diabetes is not merely a disease of the pancreas. Rather, insulin resistance and dysregulation of plasma glucose and free fatty acid levels can produce severe dysfunction in multiple organs, including the heart, kidneys, liver, and eyes (as discussed in the following sections), as well as the brain and peripheral nervous system. Insidiously, tissue damage can begin before noticeable decompensation of insulin resistance, causing patients to suffer silent tissue damage long prior to diagnosis (Nerpin et al. 2008; Dinh et al. 2010, 2011; Manchanayake et al. 2011). Through this multisystemic onslaught, diabetes slowly saps patients of both longevity and quality of life. Thus, it is critical to understand the mechanisms underlying this disease and to develop rational treatment regimens to halt this cascade of dysfunction.
3 Sphingolipid Metabolism and Regulation
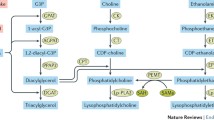
Sphingolipids are a class of structural and signaling molecules incorporating a unique dihydrosphingosine backbone, which may also be present in its desaturated form as sphingosine (Gault et al. 2010). These lipids play important roles in determining membrane biophysical properties, topology, and integrity and in numerous cellular signaling processes, particularly apoptosis and proliferation (Zheng et al. 2006; Hannun and Obeid 2008). Sphingolipid-dependent mechanisms of pathogenesis have been demonstrated in many disease contexts, and sphingolipids have emerged as important players in obesity and diabetes (Shimabukuro et al. 1998; Summers and Nelson 2005; Zheng et al. 2006; Hannun and Obeid 2008). Importantly, sphingolipid metabolism is highly interconnected by a well-characterized network of enzymes, making it possible to study mechanisms of sphingolipid production and signaling with great precision (Gault et al. 2010). This metabolic system, which we recently reviewed (Brice and Cowart 2011), is described in detail in several other chapters.
Given the great complexity of sphingolipid metabolism, it follows that its regulation may also be complex or multifactorial. Indeed, it has been demonstrated that sphingolipid synthesis can be regulated at both the substrate level and through signaling in response to cellular stimuli. In perhaps the most intuitive manner of regulation, which is extremely relevant to the study of type 2 diabetes, fatty acids impact sphingolipid levels through both substrate supply and regulation of the enzymes of sphingolipid metabolism (Shimabukuro et al. 1998; Hu et al. 2009, 2011). Initially, oversupply of saturated fatty acids directly stimulates sphingolipid synthesis via substrate supply (Merrill Jr et al. 1985; Shimabukuro et al. 1998). This excess of fatty acids provides additional acyl-CoA moieties for de novo production of sphingoid bases by serine palmitoyltransferase (SPT) and N-acylation of sphingoid bases by CerS. In a complementary manner, fatty acid supply also regulates expression of the enzymes of sphingolipid metabolism, including SPT, dihydroceramide desaturase 1 (Des1), and sphingosine kinase 1 (SphK1), thus upregulating sphingolipid production in conditions of steatosis (lipid overload) (Hu et al. 2009, 2011). Strikingly, regulation of these enzymes by fatty acids appears to occur in a chain length- and saturation-dependent manner, with unsaturated fatty acids protecting against upregulation of SphK1, SphK2, and Des1 (Hu et al. 2009, 2011).
Sphingolipid metabolism is also regulated in a substrate-independent manner by other components of the diabetic physiologic phenotype. Indeed, sphingolipid metabolism can be activated by several aspects of diabetic physiology, including the presence of inflammatory cytokines, growth factors, and oxidative stress (Goldkorn et al. 1998; Memon et al. 1998; Levy et al. 2006, 2009; Samad et al. 2006; Sultan et al. 2006; Aerts et al. 2007). In vivo, these conditions were all observed in the adipose tissue of genetically obese, diabetic ob/ob mice (Samad et al. 2006). Compared to their lean counterparts, genetically obese mice demonstrated significantly higher expression of enzymes promoting ceramide and sphingosine/sphingosine-1-phosphate accumulation, including acid and neutral sphingomyelinase, alkaline and acid ceramidase, and SPT. In contrast, levels of enzymes that would route ceramide to more complex metabolites, including glucosylceramide synthase and N-acetylneuraminylgalactosylceramide (GM3) synthase, were downregulated in the diabetic mice. This indicated that in the diabetic state, expression of sphingolipid-metabolizing enzymes is recalibrated toward generation of proinflammatory and proapoptotic species. Furthermore, direct exposure of HepG2 hepatocellular carcinoma cells to the inflammatory cytokines TNF-α and IL-1 directly stimulated SPT expression and activity in vitro, providing a direct mechanistic link between these inflammation and sphingolipid synthesis (Memon et al. 1998). Other work has suggested a similar stimulatory effect of inflammatory cytokines on the sphingolipid recycling pathway, in which ceramides are deacylated to form sphingosine and then reacylated (Sultan et al. 2006). Likewise, it has been shown that oxidative stress promotes ceramide accumulation in other disease contexts, particularly through stimulation of neutral sphingomyelinase 2 (NSMase2) and the recycling pathway (Goldkorn et al. 1998; Levy et al. 2006, 2009; Sultan et al. 2006). Thus, the conditions present in diabetes stimulate sphingolipid production and increased expression of the enzymes of sphingolipid metabolism.
4 Adipose Tissue in Obesity and Diabetes
Adipose tissue primarily functions to store free fatty acids (FFA) after food intake and to release FFA in the fasting state (Hajer et al. 2008). However, with the rise in obesity, adipose tissue has emerged as an important tissue in the metabolism of lipids and glucose and possibly the induction of insulin resistance and type 2 diabetes (Samad et al. 2006; Hajer et al. 2008; Bonzon-Kulichenko et al. 2009). Adipose has been demonstrated to act in an endocrine fashion by producing a multitude of factors that include a variety of proinflammatory cytokines and chemokines collectively known as adipokines (Mohamed-Ali et al. 1997; Christiansen et al. 2005; Hotamisligil 2006; Samad et al. 2006, 2011), and the production of these inflammatory mediators is believed to mediate the initiation of insulin resistance in distal tissues such as skeletal muscle and liver.
A proposed mechanism for obesity-induced insulin resistance in distal tissues is by increased intracellular lipid accumulation. Induction of the expression and production of adipokines results in increased lipolysis in adipocytes and elevated release of FFA. Adipocytes at this point lose their ability to store FFA and as a result promote the accumulation of lipids such as liver, skeletal muscle, and pancreas. The inappropriate storage of lipids in these tissues results in the activation of signaling pathways that interfere with insulin signaling and ultimately contribute to insulin resistance in adipose, muscle, and liver. Collectively, these actions have been termed “lipotoxicity.” A key outcome of lipotoxicity is insulin resistance, which precedes type 2 diabetes (Turpin et al. 2009).
Indeed, significant changes in the sphingolipids produced by the adipose tissue have been reported in genetically obese (ob/ob) mice (Samad et al. 2006). Compared to their lean counterparts, ob/ob mice demonstrated significantly higher levels of expression of acid sphingomyelinases (ASMase), NSMase, and SPT. When the levels of sphingolipids within the adipose tissue were measured, it was found that there were decreased levels of total sphingomyelin and ceramide and increased levels of sphingosine. These data suggest that sphingomyelin may be hydrolyzed to ceramide and further converted to sphingosine. Consistent with this, the expression of both alkaline and acid ceramidases was increased in the adipose tissue of ob/ob mice over their lean counterparts. Analysis of plasma sphingolipids from lean and ob/ob mice found that there were increases in sphingomyelin, ceramide, sphingosine, and S1P (Samad et al. 2006). There was no detectable S1P in the adipose tissue of these mice. These data support studies that suggest increased sphingolipid levels in the plasma of obese mice are associated with increased cardiovascular risk in obesity (Auge et al. 2004; Hojjati et al. 2005). Not only have studies evaluated sphingolipid metabolism in genetically obese mice, they have evaluated sphingolipid metabolism in diet-induced obesity models as well. In this study, C57BL/6J mice were maintained on a high-fat (60 % calories from fat) or a low-fat (10 % calories from fat) diet for 16 weeks. In the mice fed with the high-fat diet (HFD), ceramide levels in the adipose and the plasma were increased as evidenced by increases of the enzymes SPT, ASMase, and NSMase (Shah et al. 2008). Studies evaluating changes to sphingolipid metabolism in human adipose tissue remain scarce and have demonstrated that ceramide and triacylglycerides are elevated in the adipose tissue of obese individuals (Kolak et al. 2007). Other studies have demonstrated that inflammation in human adipose tissue affects sphingolipid metabolism in other tissues such as skeletal muscle (Lam et al. 2011).
The data demonstrate that sphingolipid synthesis is disrupted in adipose tissue in response to increased fatty acid influx. As mentioned above, the source of excess FFA delivery from adipose to other tissues such as skeletal muscle leads to loss of insulin sensitivity. In fact, additional roles for sphingolipids in these disease processes are also emerging.
5 Sphingolipids in Skeletal Muscle
The sensitivity of skeletal muscle to insulin is critical for the removal of glucose from the bloodstream as it accounts for approximately 40 % of total body mass and 80 % of whole body insulin-stimulated glucose disposal (Gorski et al. 2002; Bruni and Donati 2008). It is for these reasons that skeletal muscle is believed to contribute most significantly to the glucose intolerance associated with nutrient oversupply and obesity.
In insulin-sensitive tissues, insulin promotes the uptake of glucose (Summers 2006). Insulin signaling is initiated through the activation of the insulin receptor. Insulin receptor substrates (IRS proteins) recruit and activate the phosphatidylinositol 3-kinase (PI3K) (Keller and Lienhard 1994; Summers 2006), which in turn produces phospholipid phosphatidylinositol (3,4,5)-triphosphate (PIP3), a bioactive lipid that mediates recruitment of the cytosolic enzyme Akt/protein kinase B (PKB) to the plasma membrane (Keller and Lienhard 1994). Phosphorylation of the Thr308 in the activation loop and Ser473 in the hydrophobic C-terminal domain by PIP3 is required for full activation of Akt/PKB (Vanhaesebroeck and Alessi 2000; Summers 2006).
Akt/PKB promotes the uptake of glucose in skeletal muscle by stimulating translocation of Glucose Transporter 4 (GluT4) resulting in an increase in the rate of glucose flux into the tissue, creating a state of decreased circulating glucose and stimulating its conversion to energy storage molecules such as glycogen (Hajduch et al. 1998, 2001; Whiteman et al. 2002). Akt/PKB response to insulin can be perturbed by stimuli such as TNF-α, glucocorticoids, and prolonged exposure to long-chain fatty acids such as palmitate, all of which have been implicated in insulin resistance (Chavez et al. 2003; Holland et al. 2007). Intriguingly, a common feature of these stimuli is their ability also to promote the accumulation of sphingolipids (i.e., ceramide) (Schmitz-Peiffer et al. 1999; Teruel et al. 2001; Holland et al. 2007).
Exposure of skeletal muscle to FFA results in the development and accumulation of intramuscular triacylglycerol (IMTG) and other fatty-acid-derived molecules (i.e., diacylglycerol and ceramide) (Itani et al. 2002; Moro et al. 2008; Lipina and Hundal 2011). The accumulation of IMTG in skeletal muscle has been associated with endurance-trained athletes with significant sensitivity to insulin, indicating that the accumulation of IMTG is not the sole mediator of insulin resistance in skeletal muscle (Goodpaster et al. 2001; Bruce et al. 2004); however, it has been demonstrated that exposure of skeletal muscle to exogenous palmitate, the most abundant fatty acid in plasma, impaired insulin signaling by preventing insulin-induced phosphorylation of Akt/PKB (Hajduch et al. 2001; Moro et al. 2008). Several studies have demonstrated that the insulin-desensitizing effects of palmitate can be mimicked through the acute application of ceramide and ceramide analogues (Schmitz-Peiffer et al. 1999; Teruel et al. 2001; Chavez et al. 2003). And, moreover, inhibitors of sphingolipid biosynthesis protect cells from the insulin-desensitizing effects of palmitate (Chavez et al. 2003).
The accumulation of IMTG does not appear to be pathogenic by itself but reflective of lipid oversupply, inducing an increase in free palmitate; however, whether IMTG serve as a source of substrate for sphingolipid synthesis is unknown, but in general, sequestration of cell lipids into TAG is protective (Dimitrios et al. 2010; Amati et al. 2011).
6 Sphingolipids in the Diabetic Heart
Heart disease is a major clinical concern in type 2 diabetes: patients with diabetes are several times more likely to die of heart disease than nondiabetics (Ho et al. 1993; Kucharska-Newton et al. 2010). Significantly, diabetics develop a distinct cardiomyopathy that is independent of traditional risk factors (e.g., hypertension and ischemia/reperfusion) (Guha et al. 2008). This condition, termed diabetic cardiomyopathy, is characterized by systolic and diastolic dysfunction, reduced contractility, fibrosis, hypertrophy, and a profound shift away from utilization of glucose as a substrate (Boudina and Abel 2010). Strikingly, severity of insulin resistance was associated with both the level of myocardial steatosis and the degree of cardiac remodeling (Utz et al. 2011), while the amount of myocardial steatosis was itself an independent predictor of left-ventricular systolic dysfunction in adult humans (Rijzewijk et al. 2008). In fact, induction of myocardial steatosis is used to replicate the phenotype of diabetic cardiomyopathy in mouse models (discussed below). Together, these observations suggested the potential contribution of lipid overload to the development of diabetic cardiomyopathy.
Similarly to what was seen in pancreatic β-cells and other tissues, excess saturated fatty acids were shown to induce both metabolic alterations and apoptosis in cardiomyocytes (de Vries et al. 1997; Hickson-Bick et al. 2000; Sparagna et al. 2000). In isolated neonatal rat cardiac myocytes, it was shown that palmitate induced a dramatic reduction in fatty acid oxidation, as compared to cells treated with oleate (Hickson-Bick et al. 2000). This downregulation of fatty acid oxidation was thought to occur due to reduced AMPK activity and subsequent accumulation of malonyl coenzyme A (CoA), which in turn inhibits fatty acid oxidation through CPT-1. The reduction in fatty acid oxidation shunted fatty acyl-CoAs into non-oxidative pathways: triglyceride and ceramide levels both doubled in response to palmitate, but not oleate, treatment (Hickson-Bick et al. 2000; Sparagna et al. 2000). This increase in ceramide was accompanied by a significant increase in DNA laddering as well as more than twofold increase in caspase 3 activity in palmitate-treated cells (Hickson-Bick et al. 2000). These results were suggestive of a toxic role for ceramides, as had been demonstrated a short time before in pancreatic β-cell lipotoxicity (Shimabukuro et al. 1998) and in TNF-α-mediated cardiomyocyte toxicity (Krown et al. 1996).
Ceramide was first directly implicated in cardiomyocyte apoptosis through the use of a cell-permeable artificial ceramide; this finding was confirmed and extended using a combination of inhibitor studies and ceramide treatments. Initially, it was shown that treatment with C2-ceramide, a cell-permeable synthetic ceramide, induced neonatal cardiomyocyte apoptosis in a caspase 3- and 8-dependent manner (Wang et al. 2000). A subsequent, conceptually important study confirmed that de novo sphingolipid synthesis was required for induction of lipotoxicity in isolated adult rat ventricular cardiomyocytes. In this system, it was shown that inhibition of ceramide synthesis attenuated lipotoxic and glucolipotoxic TUNEL staining and completely ameliorated lipotoxic myofibrillar degeneration (Dyntar et al. 2001). This approach was important for two reasons: first, primary adult cardiomyocytes are postmitotic and, therefore, a better model of the adult heart than neonatal cells, and, second, the use of an inhibitor provided redundancy to the short-chain ceramide analogue-based approach, which has caveats that will be discussed later in this chapter. Therefore, through the use of complementary approaches, these experiments demonstrated that an increase in ceramide levels is both necessary and sufficient to induce lipotoxic cell death and myofibrillar degeneration in cardiomyocytes.
Further studies extended the known role for ceramide in development of the diabetic cardiomyopathy phenotype, including induction of heart failure markers, downregulation of glucose oxidation, and impairment of contractility. Numerous aspects of the response of cardiomyocytes to ceramide were worked out in AC-16 cells, which are an immortalized human ventricular cardiomyocyte-derived cell line (Park et al. 2008). For example, it was shown that C6-ceramide treatment of AC-16 cells induced upregulation of the heart failure markers ANF and BNP (Park et al. 2008). Suggesting a role for ceramide in lipotoxic downregulation of glucose oxidation, C6-ceramide treatment also suppressed expression of the GLUT-4 glucose transporter and potentiated expression of PDK4, which inhibits the pyruvate dehydrogenase complex. Ceramide has also been implicated in the lipotoxic impairment of cardiac contractility. Specifically, it was shown that treatment with either palmitate or C6-ceramide induced β-adrenergic dysfunction in AC-16 cells (Drosatos et al. 2011). Strikingly, the induction of β-adrenergic dysfunction by palmitate was completely ameliorated by co-treatment with the de novo sphingolipid synthesis inhibitor myriocin. This sphingolipid-dependent impairment of β-adrenergic function was thought to occur as a result of activation of PKCα and PKCδ. However, it should be noted that treatment with diacylglycerol was shown to produce the same effects, and it is likely that sphingolipids and diacylglycerol work in concert in this process. Together, these studies showed that, in an immortalized cardiomyocyte cell system, increased cellular ceramide was sufficient to induce expression of heart failure markers, changes in glucose uptake and metabolism, and β-adrenergic dysfunction, and de novo sphingolipid synthesis was required for induction of β-adrenergic dysfunction by palmitate. Critically, results obtained in isolated cell systems have been recapitulated and extended in vivo. One model that has provided a great deal of information is the LpLGPI mouse, which expresses a heart-specific membrane-bound form of lipoprotein lipase and thus develops profound cardiac steatosis (Yagyu et al. 2003). Cardiomyocytes bearing this transgene accumulated ceramides, sphingomyelin, and diacylglycerol (Park et al. 2008). Importantly, while myriocin treatment prevented the increase in cardiac ceramides and sphingomyelin, diacylglycerol levels remained elevated; this allowed the authors to tease out ceramide-specific effects from those of diacylglycerol. The results of this study were striking. It was shown that many of the pathological outcomes in the LpLGPI mouse, including gross cardiac hypertrophy, increased left-ventricular systolic diameter, reduced cardiac efficiency, and increased mortality, were completely prevented by myriocin treatment, which inhibits the first step of de novo sphingolipid synthesis. Glucose oxidation and palmitate oxidation were also restored to normal levels by myriocin treatment, as was expression of molecular markers of heart failure (ANF and BNP), hypertrophy (GSK3β phosphorylation), and insulin signaling (AKT phosphorylation). These sphingolipid-dependent outcome effects were also attenuated upon crossing with a mouse heterozygous null for the serine palmitoyltransferase subunit SPTLC1, which also displays impaired de novo sphingolipid biosynthesis. Thus, the role of sphingolipids in the development of lipotoxic cardiac hypertrophy and heart failure was demonstrated in vivo.
Another transgenic model system provided additional support for the role of sphingolipids in diabetic cardiomyopathy. The ob/ob mouse is a very popular leptin-deficient model of obesity and type 2 diabetes; these mice develop marked cardiac hypertrophy, dysfunction, and steatosis (Barouch et al. 2006; Dobrzyn et al. 2010). Paralleling results in the LpLGPI mouse, these animals dramatically accumulate ceramide and overexpress SPT, the initial enzyme of de novo sphingolipid synthesis (Dobrzyn et al. 2010). However, it was shown that loss of stearoyl-CoA desaturase 1 (SCD1), which catalyzes synthesis of monounsaturated fatty acids, not only protected ob/ob mice from obesity but also reduced SPT expression and ceramide to normal levels and prevented the hypertrophic cardiac phenotype. This study thereby demonstrated that a mutation that indirectly normalized ceramide levels prevented the development of diabetic cardiomyopathy.
While most studies have focused on the role of ceramide in the heart, other work has revealed a protective role for sphingosine-1-phosphate, similar to that demonstrated in other lipotoxic cell models. In isolated primary cardiomyocytes, treatment with sphingosine-1-phosphate protected against palmitate-induced cell death as effectively as myriocin treatment (Holland et al. 2011). Additionally, overexpression of sphingosine kinase 1 protected diabetic mice against the development of cardiomyopathy (Ma et al. 2007). Intriguingly, though, treatment with sphingosine-1-phosphate promoted hypertrophy of neonatal cardiomyocytes, possibly through signaling of the EDG1/S1PR1 receptor (Robert et al. 2001). It remains unknown whether this effect is recapitulated in adult cardiomyocytes. Therefore, although sphingosine-1-phosphate signaling appears to play a protective role in the lipotoxic heart, its function has yet to be completely understood.
The role for sphingolipids other than ceramide and sphingosine-1-phosphate in the diabetic heart remains almost totally uncharacterized. Two studies have indicated that administration of a ganglioside preparation protected atrial function in genetically obese, diabetic mice and prevented reduction in cardiac norepinephrine concentration in diabetes (Prosdocimi et al. 1987; Tessari et al. 1988); potential actions of other sphingolipids in the diabetic heart remain undescribed. Significantly, many studies addressing the roles of sphingolipids in the heart do so by inhibiting de novo sphingolipid synthesis. In addition to reducing ceramide, though, these strategies can reduce levels of downstream metabolites of ceramide (Miyake et al. 1995). Thus, because of the known roles for sphingolipid species other than ceramide and sphingosine-1-phosphate in other diabetic organs, this represents a major gap in knowledge of sphingolipid functions in the diabetic heart and presents a caveat to attribution of cellular outcomes to ceramide per se.
Although numerous studies have addressed the roles of ceramide in diabetic cardiomyopathy, few have addressed the mechanisms by which sphingolipid levels are regulated in diabetes. While general mechanisms parallel those described elsewhere in this chapter, a few cardiac-specific observations are worthy of mention. As discussed above, the prevailing theory of lipotoxicity proposes that accumulation of toxic lipid species, including ceramide, results from the inability of cells to sequester free fatty acids adequately as neutral lipid (Cnop et al. 2001). To test this hypothesis, diacylglycerol acyltransferase 1 (DGAT1), which catalyzes the formation of triacylglycerol from diacylglycerol, was overexpressed in a cardiac-specific manner in mice (Liu et al. 2009). This resulted in accumulation of triacylglycerols and a concomitant reduction in ceramide and diacylglycerol levels. Furthermore, when these animals were crossed with a transgenic strain that developed lipotoxic cardiomyopathy, the resulting mice displayed improved heart function and reduced mortality as well as more normal expression of markers of heart failure, oxidative stress, and apoptosis. Strikingly, statistical analyses revealed a significant correlation between cardiac ceramide levels and the level of cardiac function in a mixed cohort of transgenic and wild-type mice. This study thus provided support for the hypothesis that lipid overload induces steatosis by overwhelming cells’ capacity to sequester fatty acids as neutral lipid.
Other work has addressed a potential role for the transcription factor PPARα in regulation of ceramide levels (Baranowski et al. 2007). In this study, treatment of wild-type rats with a PPARα agonist (WY-14643) synergized with high-fat diet to increased SPT activity in a more dramatic manner than observed with diet alone. Likewise, ceramide levels also increased markedly upon administration of WY-14643 in high-fat diet, and the fatty acyl chain distributions of both ceramide and sphingomyelin were also altered. This latter finding suggests that PPARα could potentially act on other components of the sphingolipid synthetic pathway besides SPT. Strikingly, other studies have implicated PPARα in heart failure [reviewed in Finck (2004) and Madrazo and Kelly (2008)], thus raising the question of whether these effects are mediated by regulation of sphingolipid levels by PPARα. Together, these findings suggest that PPARα-mediated signaling may augment the observed substrate-based effects of lipid oversupply to promote de novo sphingolipid synthesis.
In summary, these findings demonstrate roles for sphingolipids in the pathogenesis of cardiac lipotoxicity and diabetic cardiomyopathy. Ceramide was implicated not only in the induction of cardiomyocyte apoptosis but also in insulin resistance, altered cardiac substrate utilization, hypertrophy, β-adrenergic dysfunction, and impaired cardiac function. In contrast, there are hints that sphingosine-1-phosphate may protect cardiomyocytes from apoptosis while, perhaps, potentiating hypertrophy. As of this writing, no published studies have addressed potential roles for other sphingolipids, including glycosphingolipids, sulfatide, and ceramide-1-phosphate, in the diabetic heart. Furthermore, the contributions of particular enzymes of sphingolipid metabolism and individual N-acyl chain lengths have been utterly neglected. Additionally, it should be noted that, while much of the literature on sphingolipids in the diabetic heart focuses on their roles in diabetic cardiomyopathy, they also play important roles in injury following ischemia and reperfusion (I/R) (Maulik et al. 1993; Bielawska et al. 1997; Theilmeier et al. 2006) and in defects in electrical conductance (Constable et al. 2003) and autonomic nervous function (Tessari et al. 1988; Davis et al. 2006), all of which are major concerns in the diabetic heart. Finally, the role of sphingolipids in diabetic glucotoxicity in the heart has been abjectly neglected, despite its potential importance. Thus, while sphingolipids have been shown to play multiple roles in the pathogenesis of cardiac lipotoxicity, much remains to be learned about their regulation, specific mechanisms of action, and broader significance in the diabetic heart.
7 Sphingolipids in the Diabetic Pancreas
Pancreatic dysfunction is the hallmark disease outcome in diabetes. In insulin resistance, excess plasma free fatty acids initially stimulate insulin secretion by the pancreas, both directly and in response to insulin resistance in peripheral tissues, resulting in a compensatory hyperinsulinemic state (Milburn Jr et al. 1995). However, a gradual decline in insulin release occurs, in part, due to decreased insulin sensitivity in the pancreatic β-cells themselves; this effect has been shown to be mediated by excess uptake of free fatty acids from the plasma (Lee et al. 1994). The resulting loss of compensation results in frank hyperglycemia, which synergizes with the hyperlipidemic state to promote molecular pathology and cell death, as described later in this section.
The process of fatty-acid-mediated β-cell dysfunction and, ultimately, death occurs through lipotoxicity. The ability of lipid oversupply to increase ceramide levels in the diabetic pancreas has long been appreciated (Shimabukuro et al. 1998). Initially, it was observed that both DNA laddering (an indicator of apoptosis) and ceramides increased in the pancreatic islets from obese, diabetic rats, compared to lean controls. Furthermore, treatment of isolated islets with free fatty acids recapitulated this DNA laddering and ceramide production in vitro. Strikingly, it was found that only free fatty acids that promoted ceramide synthesis were able to cause lipotoxicity and insulin resistance in vitro, supporting a link between ceramides and islet lipotoxicity (Maedler et al. 2001, 2003). Substantiating this relationship, treatment with a membrane-permeable synthetic ceramide, C2-ceramide, directly potentiated DNA laddering (Shimabukuro et al. 1998). Intriguingly, this study also noted that islet cells from obese, diabetic rats were more vulnerable to free fatty-acid-mediated lipotoxicity, suggesting that changes in metabolism or protein expression in the diabetic pancreas augmented susceptibility of these cells to lipotoxicity. Together, these findings suggested both that ceramide plays a key role in lipotoxic β-cell apoptosis and that the diabetic pancreas displays an increased vulnerability to toxicity from excess plasma free fatty acids, possibly due to the already-heavy fatty acid loading of these cells.
Glucotoxicity has also been shown to play an important role in pancreatic damage after decompensation of insulin resistance; this process, too, seems to be governed by sphingolipids. Most directly, glucose-stimulated cytotoxicity was shown to be ameliorated by inhibition of ceramide synthase (Maedler et al. 2003). Unfortunately, the effect of this treatment on downstream, complex sphingolipid species was not determined. This point may be significant because, as described later in this section, ceramides may be adorned with carbohydrate headgroups to form complex sphingolipids, which have distinct signaling and membrane roles from those of simple ceramide. Regardless of this point, though, it appears that oversupplies of glucose and free fatty acids work in concert to promote ceramide synthesis in the diabetic pancreas. In fact, neither excess glucose nor a low dose of palmitate alone was able to induce ceramide production profoundly in INS832/13 or INS-1 cells, but the two conditions in combination strongly potentiated this effect (El-Assaad et al. 2010; Veret et al. 2011). In contrast, excess glucose and palmitate each provoked ceramide production in isolated rat islet cells, while the combination of both stimulated an even stronger increase in ceramide levels (Kelpe et al. 2003). This suggests a synergy between excess glucose and excess fatty acids in the induction of sphingolipid-dependent diabetic β-cell toxicity. These findings bring to light several unanswered questions, such as whether glucose and fatty acid oversupply act on distinct metabolic routes of sphingolipid production and clearance, whether these two routes of toxicity promote production of different sphingolipid metabolites, and whether they act on the same or complementary lipotoxic pathways.
Further studies more fully characterized the sphingolipid-dependent molecular features of pancreatic functional deterioration, including apoptosis, endoplasmic reticulum (ER) stress, and insulin resistance. For example, increased intracellular ceramides stimulated several components of the apoptotic cascade, such as cytochrome C release into the cytosol, a reduction in Bcl-2 expression, and loss of mitochondrial membrane potential (Maedler et al. 2003; Veluthakal et al. 2005). Broadly, it was found that ceramide acted to promote apoptosis, in part, by inducing caspase activation (Lupi et al. 2002); studies in the MIN6 insulinoma cell line and in glucolipotoxicity in INS-1 cells specifically implicated caspases 3/7 in sphingolipid-mediated cell death (Boslem et al. 2011; Veret et al. 2011). Ceramide accumulation also induced lipotoxic ER stress, as indicated by increased expression of CHOP, in MIN6 cells (Boslem et al. 2011). Finally, ceramide was directly implicated in insulin resistance at a molecular level, in addition to its nonspecific actions of reducing β-cell proliferation and promoting apoptosis (Maedler et al. 2001). In particular, ceramide accumulation reduced proinsulin mRNA levels via reduced transcription in INS-1 and isolated rat pancreatic islet cells (Kelpe et al. 2003; Guo et al. 2010). This may have been mediated by attenuation of ERK phosphorylation (Guo et al. 2010). Thus, elevated sphingolipid loads in the diabetic pancreas promote insulin resistance and pancreatic failure through a suite of specific molecular outcomes. These findings lead to a larger question of how sphingolipid synthesis is upregulated in the diabetic pancreas, thereby allowing all of these downstream effects to ensue.
As discussed above, lipid oversupply perturbs sphingolipid homeostasis by diverse mechanisms in many organs and cell types, and this is also true in diabetic pancreas. For example, it has been shown that expression of SPT is upregulated in the diabetic pancreas (Shimabukuro et al. 1998). This upregulation may be recapitulated in vitro by oversupply of the fatty acid palmitate as well as by other factors, including exposure to elevated levels of hepatocyte growth factor (HGF), which is overproduced in diabetes (Shimabukuro et al. 1998; Gonzalez-Pertusa et al. 2010). Other routes and enzymes of sphingolipid synthesis have been shown to be regulated by saturated fatty acids in skeletal muscle, as discussed above, and may also play important roles in governance of sphingolipid levels in the diabetic pancreas (Hu et al. 2009). Thus, the diabetic pancreas harbors elevated sphingolipid levels due to both excess substrate supply and upregulation of the enzymes of sphingolipid synthesis. However, while most studies have focused on bulk sphingolipid levels and general routes of metabolism, the roles of individual metabolites and enzymes in this system have remained undetermined. Largely because the distinct functions of individual N-acyl chain lengths of ceramide and specific (dihydro)ceramide synthase (CerS) enzymes have only recently been appreciated, only one study thus far has attempted to implicate an individual CerS isoform in β-cell lipotoxicity: Véret et al. proposed that CerS4 is responsible for glucolipotoxicity in INS-1 cells (Veret et al. 2011). Indeed, overexpression of CerS4 induced caspase 3/7 activity, while siRNA-mediated knockdown of CerS4 partially attenuated caspase activity in response to glucose and fatty acid overload. This work clearly demonstrated a role for CerS4 in glucolipotoxicity; however, additional studies will be required to define or to rule out contributions from other CerS isoforms. There are a few reasons for this. First, although the ceramide chain length profiles in the manuscript are suggestive, they are not sufficiently clear-cut to implicate a specific isoform. Second, the effect of overexpressing or knocking down other CerS isoforms was not tested. Third, and finally, cells were presented with only palmitate in the media. Beta cells encounter a mixed profile of fatty acids in vivo, which are utilized differentially for N-acylation by individual CerS isoforms, and exposure to this characteristic mixture of substrates may reveal roles for other CerS isoforms in a physiologic context. Thus, although CerS4 has been implicated in β-cell lipotoxicity, roles for other CerS isoforms have not been ruled out. Furthermore and importantly, no studies have identified a role for specific ceramide species in β-cell lipoapoptosis. Thus, significant work will be required to elucidate the mechanisms by which diabetes and lipotoxicity upregulate sphingolipid levels in the pancreas, as well as the significance of specific metabolic pathways.
Although most studies discussing sphingolipids in pancreatic lipotoxicity focus on ceramide, ceramide itself is not the only sphingolipid that accumulates during fatty acid overload. Indeed, many downstream metabolites of ceramide are modulated by manipulation of ceramide synthesis, and one must be careful before concluding that it is ceramide, per se, that is responsible for particular molecular outcomes. For example, glucosylceramide was shown to increase in MIN6 insulinoma cells treated with palmitate, and this accumulation was, in fact, much more robust than that of simple ceramide (Boslem et al. 2011). Furthermore, this may have important functional implications, as inhibition of glucosylceramide synthase, which may presumably increase its metabolic precursor, ceramide, partially preserved pancreatic insulin secretion in Zucker diabetic fatty rats (Aerts et al. 2007). This suggests a potential role for glucosylceramide, in particular, and sphingolipids other than ceramide, in general, in pancreatic insulin resistance. An additional example of this principle involves sulfatide (3′-sulfogalactosyl-ceramide), a glycosphingolipid present in the islets of Langerhans in the pancreas (Buschard et al. 2002). Like its metabolic precursor ceramide, this lipid has also been implicated in diabetes and insulin resistance. Mechanistically, sulfatide is involved in proper folding of proinsulin, preservation of insulin crystals, and monomerization of stored insulin hexamers (Osterbye et al. 2001). Furthermore, C16:0 sulfatide, specifically, was shown to be required for stabilization of insulin granules (Blomqvist et al. 2003). C16:0 sulfatide also appears to be required for appropriate management of insulin secretion in normal rat pancreatic islets; it was shown to attenuate insulin secretion by reducing sensitivity of the ATP-dependent potassium channel to inhibition by ATP (Buschard et al. 2002, 2006). Significantly, this particular sulfatide species was deficient in the pancreas of several rodent models of type 2 diabetes (Blomqvist et al. 2003), and administration of sulfatide appeared to ameliorate some aspects of diabetic pathology in vivo. For example, administration of C16:0 sulfatide, specifically, to Zucker fatty rats, which express a defective leptin receptor, reduced fasting insulin and improved the first-phase insulin response and glucose-stimulated insulin response (Blomqvist et al. 2005). More generally, sulfatide was shown to protect isolated islet cells from apoptosis, iNOS expression, and nitric oxide secretion induced by cytokines; this protective effect was not specific to C16:0 sulfatide (Roeske-Nielsen et al. 2010). These findings indicate that sulfatide exerts a protective effect in pancreatic islets via normalization of insulin secretion and protection from inflammation. It is still unknown whether these protective effects are also relevant to the context of fatty acid oversupply, and the mechanism by which C16:0 sulfatide is reduced in the diabetic pancreas remains unknown.
Finally, sphingosine-1-phosphate, the product of sphingosine kinase, has been suggested as an antiapoptotic lipid in inflammation-mediated β-cell death. In particular, supplementation with sphingosine-1-phosphate protected isolated pancreatic islets from IL-1β-induced apoptosis (Rutti et al. 2009), and exposure to both IL-1β and TNF-α upregulated sphingosine kinase activity in INS-1 cells and isolated rat pancreatic islets (Mastrandrea et al. 2005). It was suggested that this increase in activity was primarily mediated by sphingosine kinase 2 (SPHK2), based on in vitro activity assays. These results indicate that, in contrast to ceramide, sphingosine-1-phosphate acts to protect β-cells from the pathological inflammatory state present in diabetes and the metabolic syndrome; indeed, the balance of ceramide and sphingosine-1-phosphate levels has been suggested as a clinically important determinant of β-cell fate (Jessup et al. 2011).
In summary, these findings illustrate an important role for sphingolipids in the pathogenesis of the diabetic pancreas. Fatty acid and glucose oversupply both stimulate ceramide production, which promotes apoptosis and insulin resistance, as well as synthesis of downstream metabolites of ceramide. Like ceramide, glucosylceramide may act in a pro-pathogenic manner by potentiating insulin resistance. In contrast, sulfatide normalizes insulin secretion, and both sulfatide and sphingosine-1-phosphate protect cells from the harmful effects of inflammation. These foundational findings lead to numerous further questions about the detailed mechanisms of sphingolipid action in the diabetic pancreas. For example, it remains unknown which ceramide species play active roles in pathology, which pathways (i.e., salvage or de novo synthesis) generate the bulk of these pathogenic lipids in vivo, and which specific enzyme isoforms are involved. Functions of sphingosine-1-phosphate and other metabolites also remain incompletely characterized. Finally, the roles of sphingolipids in diabetic glucotoxicity are severely understudied and may be distinct but synergistic with those observed in lipotoxicity. Thus, several sphingolipid species play key, but contrasting, roles in β-cells during diabetes, and further studies are needed to clarify them and to understand the underlying molecular mechanisms.
8 Emerging Concepts in the Sphingolipid-Diabetes Axis: Liver, Kidney, and Retina
While many of the changes in sphingolipid synthesis and signaling have been somewhat well defined in tissues such as skeletal muscle, heart, and pancreas, there are several tissues that have yet to be thoroughly examined. One such tissue is the liver. Located between the intestinal tract and the systemic circulation, the liver plays an important role in the metabolism and storage of dietary fats as well as playing a central role in glucose and lipid metabolism (Coleman and Lee 2004; Yosuke et al. 2011). During times of feeding, hepatocytes increase their uptake of glucose and synthesis of glycogen (Yosuke et al. 2011). The liver also acts to synthesize triacylglycerides (TAG) through β-oxidation of fatty acids obtained from overnutrition or adipocyte cell death and/or inflammation (Coleman and Lee 2004; Bijl et al. 2009). In obesity, the combination of increased fatty acid flux from dysfunctional adipose tissue, coupled with induced fatty acid synthesis in the liver, results in the accumulation of TAG-rich lipid droplets in the cytosol (Preiss and Sattar 2008; Bijl et al. 2009; Deevska et al. 2009) and ultimately the development of fatty liver and lipotoxicity (Deevska et al. 2009). The development of these pathologies can lead to loss of insulin sensitivity, which in the liver prevents the suppression of glucose generation, signaling the pancreas to increase insulin production resulting in hyperinsulinemia (Brown and Goldstein 2008; Bijl et al. 2009).
As a result of TAG accumulation, lipid synthesis and metabolism are disrupted and perturbations of these pathways have been demonstrated to contribute to loss of insulin sensitivity and the development of hyperglycemia (Deevska et al. 2009; Yosuke et al. 2011). For example, treatment of hepatocytes with palmitate promoted an increase in TAG. TAG accumulation was attenuated by the inhibition of the activity of acid sphingomyelinase (aSMase), which generates ceramide by hydrolyzing sphingomyelin derived from the recycling/endocytic pathway (Deevska et al. 2009). This was confirmed in mice lacking functional aSMase as well as functional LDL receptors. These animals demonstrated that when maintained on a high-fat diet, there were no increases in TAG content in the cytosol of hepatocytes. They did, however, demonstrate increases in de novo sphingolipid synthesis metabolites sphinganine, dihydroceramide, and ceramide, as well as the activity SPT (Deevska et al. 2009). These animals were also protected against diet-induced hyperglycemia and insulin resistance.
Sphingolipid synthesis in the kidney is also adversely affected by the development of insulin resistance in other distal tissues. Hyperglycemia and insulin resistance induced by fatty acid oversupply to the liver result in dramatic alterations in kidney function, sphingolipid profiles, and the development of diabetic nephropathy. Indeed, complex glycosphingolipids have been indicated in the development of diabetic nephropathy, particularly where changes in glomerular sialic acids and/or sialidase activity correlate with the onset of proteinuria (Cohen-Forterre et al. 1984; Baricos et al. 1986; Cardenas et al. 1991; Mather and Siskind 2011). Ceramide has also been implicated in renal injury as its accumulation is a result of nephropathy induced by the consumption of a high-fat diet (Boini et al. 2010). While this study pointed to ceramide as the causative agent in high-fat diet-induced renal injury and nephropathy, glycosphingolipid levels were not assessed. As ceramide is a necessary for glycosphingolipid synthesis, it is possible that the synthesis of glycosphingolipids is also increased and may play a role (Boini et al. 2010). Indeed, the link between glucose metabolism, insulin resistance, and glycosphingolipid synthesis in the kidney has been established (Zhao et al. 2007), indicating glycosphingolipids in renal injury and the development of nephropathy.
The retina plays a significant role in vision and is adversely affected in type 2 diabetes; however, the effects of changes in sphingolipid metabolism have not been thoroughly evaluated. Studies have indicated that the accumulation of ceramide in response to fatty acid oversupply results in increased apoptosis in retinal pericytes and induction of diabetic retinopathy (Cacicedo et al. 2005; Fox et al. 2006). While these studies provide significant evidence, more has to be done to ascertain the mechanism by which sphingolipid synthesis is perturbed in the retina.
9 Sphingolipids as Biomarkers of Diabetes
In addition to their direct roles in the pathogenesis of diabetes, sphingolipids have also emerged as potential biomarkers of diabetes and the metabolic syndrome. It has long been appreciated that sphingolipid levels are perturbed in the blood plasma of diabetic patients. The first such finding indicated that glycosphingolipids were elevated in the plasma of some groups of diabetic human patients (Kremer et al. 1975). This was later confirmed by a study demonstrating that glucosylceramide was elevated in the plasma of diabetics (Serlie et al. 2007). Similarly, plasma ceramides have emerged as potential biomarkers in diabetes. For example, plasma ceramides were elevated in humans with type 2 diabetes and correlated negatively with the rate of insulin-stimulated glucose disposal, indicating a relationship between plasma ceramide levels and insulin resistance (Haus et al. 2009). Furthermore, the concentration of ceramide in the blood plasma positively correlated with levels of the proinflammatory cytokine IL-6, which has been shown to promote insulin resistance (de Mello et al. 2009). Additionally, it was shown in human patients that sphingomyelin levels in the plasma membranes of erythrocytes positively correlated with circulating insulin levels (Zeghari et al. 2000). These results were echoed in an animal model of diabetes: sphingomyelin, ceramide, sphingosine, and sphingosine-1-phosphate were all elevated in the plasma of ob/ob mice, which are leptin-deficient, diabetic, and obese (Samad et al. 2006). Thus, multiple studies have suggested that sphingolipid levels in the plasma correlate with specific diagnostic criteria for diabetes and the metabolic syndrome in both mice and humans.
Despite these highly suggestive observations, relatively few studies have directly analyzed the predictive value of sphingolipid levels for diabetes. In one such study, sphingomyelin content of erythrocyte plasma membranes was shown to be an independent predictor of fasting insulin levels, degree of insulin resistance, and level of glucose tolerance (Candiloros et al. 1996). Indeed, higher levels of membrane sphingomyelin correlated with increased fasting insulin, insulin resistance, and glucose tolerance in both normal and diabetic human patients.
Another recent study analyzed the potential of uncommon sphingosine bases as biomarkers of type 2 diabetes and the metabolic syndrome (Othman et al. 2011). In this study, plasma sphingolipids were deacylated, and the relative contributions of different sphingoid bases to the total pool were quantified in a small, homogenous population. The authors found that the concentrations of the uncommon sphingoid bases deoxysphinganine and deoxysphingosine, which incorporate the amino acid alanine rather than serine, were increased in patients with metabolic syndrome, with or without type 2 diabetes. Additionally, it was found that the levels of these compounds had a significant positive predictive value for the metabolic syndrome. Furthermore, the concentration of C16-sphingosine, which is derived from myristoyl-CoA rather than palmitoyl-CoA, was decreased in patients with both diabetes and the metabolic syndrome, but not in patients with metabolic syndrome alone. It was also determined that levels of C16-sphingosine had significant predictive value in differentiating patients with type 2 diabetes from prediabetic and control patients. Thus, it was suggested that levels of alanine-derived sphingoid bases are a potential biomarker for metabolic syndrome, with or without type 2 diabetes, while levels of C16-sphingosine could be used to detect the transition from compensated insulin resistance to type 2 diabetes.
Together, these studies demonstrate the potential value of sphingolipids, and particularly uncommon sphingolipids, as biomarkers of diabetes and the metabolic syndrome. However, although strong correlations exist between sphingolipid levels and parameters of these diseases, few studies have performed the statistical analyses necessary to determine their predictive value, including ROC curves, which determine specificity and sensitivity of a potential biomarker. Additionally, further studies will be required to validate the findings of Othman et al. (2011) and Candiloros et al. (1996). These studies would need to incorporate larger and more diverse populations and, ideally, follow a prospective design. Furthermore, studies of ceramides and complex sphingolipids could benefit from analysis of individual N-acyl chain lengths. Thus, while plasma sphingolipids display much potential as biomarkers of diabetes, substantial work remains to be done before these tests reach the clinic.
10 Summary and Future Directions
Diabetes presents as a multifactorial disorder affecting multiple organ systems. Although broader disease outcomes (e.g., liver disease, heart failure, kidney failure) are complex and diverse, the underlying cellular and molecular etiologies flow through some common channels. One major theme seems to be sphingolipid-mediated insulin resistance and cell death, consequent to lipotoxicity. Indeed, a great deal of work has been done to link de novo sphingolipid synthesis and individual sphingolipid metabolites to particular molecular events. Broadly, ceramide has been implicated as a toxic lipid promoting insulin resistance, metabolic derangement, and cell death in skeletal muscle, heart, and pancreas (Shimabukuro et al. 1998; Schmitz-Peiffer et al. 1999; Park et al. 2008). In contrast, sphingosine-1-phosphate has been tentatively shown to exert protective effects in these same organ systems, despite its promotion of inflammation (Rutti et al. 2009; Holland et al. 2011). However, these general findings merely reveal an entire network of questions about the specific molecular mechanisms of disease, especially regarding which pathways, enzyme isoforms, and sphingolipid metabolites are pathogenic or protective. Fortunately, new tools have emerged that will allow researchers to uncover precise aspects of these mechanisms.
Previously, the nature of the available experimental tools limited the resolution to which sphingolipid pathways could be teased apart. For many years, ceramide and its metabolites could only be measured by techniques such as TLC, which revealed changes only in bulk levels of these lipids. However, recent developments now allow researchers to examine detailed changes in sphingolipid N-acyl chain length profiles using quantitative mass spectrometry-based approaches (Bielawski et al. 2010). Results in other disease contexts, particularly cancer, have demonstrated that alterations in levels and proportions of ceramide N-acyl chain lengths are functionally important (Senkal et al. 2010, 2011), and it may now be determined whether the lipotoxic effects of ceramide occur due to increases in bulk ceramide levels or to shifts toward particular chain length profiles. Furthermore, mass spectrometry-based labeling strategies, such as the use of 13C-palmitate, allowed investigators to trace the specific metabolic routes that are invoked in their experimental systems, distinguishing between de novo synthesis and salvage pathways, for instance (Hu et al. 2009). These findings would lead to a better understanding of the underlying molecular mechanisms of ceramide-mediated lipotoxicity.
Critically, determining the nature of any changes in sphingolipid N-acyl profiles would help to reveal which CerS isoforms mediate the lipotoxic effects of ceramide and its downstream metabolites, as each CerS isoform produces a unique N-acyl chain length distribution (Pewzner-Jung et al. 2006; Laviad et al. 2008). Once again, newly developed experimental approaches enable investigators to resolve the cellular functions of particular CerS isoforms. While myriocin, which inhibits SPT, or fumonisin B1, which inhibits all isoforms of CerS, can function as powerful tools to screen for sphingolipid dependence, they reveal very little about involvement of specific sphingolipid metabolic pathways (Wang et al. 1991; Miyake et al. 1995). In contrast, new approaches based on DNA transfection, RNAi, and knockout animals allow precise insight into the roles of specific CerS isoforms in cellular functions and disease, with the caveat that dysregulation of individual isoforms can perturb global CerS expression patterns (Mullen et al. 2011a, b). These approaches may also be applied to other enzymes of SL synthesis that are present in more than one isoform (e.g., ceramidase, sphingosine kinase, sphingomyelinase). Thoughtful employment of these strategies would do much to advance the field beyond a binary sphingolipid-dependent/sphingolipid-independent understanding of diabetic molecular and cellular phenotypes. Successful identification of specific enzymes for therapeutic targeting would also provide rationale for developing inhibitors selective for specific CerS isoforms, a strategy already well underway for sphingosine kinases (ref some Sk1 vs. SK2 inhibitor papers).
Notwithstanding the need for specific inhibitors, due to the highly interconnected nature of sphingolipid metabolism, any approach based on inhibiting or perturbing these pathways should be interpreted with care. In particular, many studies consider sensitivity to myriocin or fumonisin B1 to indicate a dependence on ceramide itself, excluding potential roles for subsequent metabolites. In fact, although such results are suggestive, they can be misleading; inhibition of de novo sphingolipid synthesis can reduce not only levels of ceramide but of its downstream metabolites (Saito et al. 2005; Mullen et al. 2011a, b). Thus, conclusions based on inhibitor studies actually indicate a dependence on either ceramide or its downstream metabolites. Similar caution should be used when employing knockdown or overexpression strategies to manipulate specific enzyme isoforms. Lipid profiles produced by individual isoforms may partition distinctly either at a metabolic level, due to differential acyl chain length preferences of downstream enzymes, or at the organelle level, due to different localization of particular enzymes of sphingolipid metabolism [reviewed in Brice and Cowart (2011)]. Furthermore, knockdown of individual CerS isoforms has been shown to perturb regulation of nontargeted isoforms (Mullen et al. 2011a, b). Thus, results of inhibitor, knockdown, or overexpression experiments should be interpreted cognizant of potential off-target effects on sphingolipid synthesis and metabolism.
Similar caution should be exerted toward experiments using C2- and C6-ceramide, which have served as important tools in determining the roles of sphingolipids in diabetes. This is because these artificial, short-chain ceramides have different biophysical properties than endogenous ceramides and may induce distinct cellular outcomes (Gidwani et al. 2003; Nybond et al. 2005), including lipotoxic outcomes through mechanisms that are not physiologically relevant. For example, treatment with C2-ceramide can provoke mitochondrial depolarization and cytochrome C release in cardiomyocytes (Sparagna et al. 2000; Di Paola et al. 2004; Parra et al. 2008); these results would obviously suggest a role for ceramide. However, these cellular events actually precede ceramide accumulation in the context of palmitate-induced neonatal rat cardiomyocyte lipotoxicity, suggesting that this portion of the lipotoxic cascade is not ceramide-dependent in this system (Sparagna et al. 2000). Similarly, while pyridinium ceramides can provide important clues to intracellular functions of ceramide, it should be kept in mind that these compounds preferentially target to the mitochondria, which could potentially cause their functions to diverge from those of natural ceramides (Dindo et al. 2006). Furthermore, the activity of downstream enzymes, particularly the ceramidases, could be quite different toward endogenous ceramides than toward either short-chain or pyridinium-containing ceramides, which have modified N-acyl chains. Therefore, although treatment with cell-permeable ceramide analogues can activate multiple pathologic pathways and can provide important insights, other modes of experimentation are required to confirm that these mechanisms of pathology are consistent with those induced by endogenous sphingolipids.
In conclusion, it has been shown that sphingolipids play a multiplicity of both harmful and protective roles in the pathogenesis of the metabolic syndrome and diabetes, but these roles are, as yet, only crudely understood. A number of important questions remain unanswered regarding the sphingolipid-diabetes axis, and tools are now available to investigate mechanisms that would have been inscrutable only a decade ago. In particular, it remains to be shown which enzyme isoforms of sphingolipid metabolism are responsible for potentiating specific aspects of the cellular diabetic phenotype. Furthermore, the individual sphingolipid species involved in these processes are almost totally unknown. It is unknown which specific sphingolipid N-acyl chain lengths or classes of chain lengths are important in cellular pathology; just as critically, the roles of complex sphingolipids and, in many tissues, the potently bioactive lipid sphingosine-1-phosphate remain neglected. Furthermore, the vast majority of studies have focused on the roles of sphingolipids in lipotoxicity; this may owe to the intuitive substrate-level connection. However, glucotoxicity is also an important player in the pathogenesis of diabetes, and the literature has provided a few intriguing hints of a synergistic relationship between sphingolipid-dependent glucotoxicity and lipotoxicity that beg for further investigation. Finally, the possibility of using circulating sphingolipids as clinical biomarkers is only now emerging. To develop this area, larger-scale studies with appropriate statistical approaches will need to be conducted. Furthermore, the physiological basis linking levels of particular circulating sphingolipids to diabetes remains to be determined. Thus, while much fundamental research has already implicated sphingolipids in diabetes in a very general way, this field of study blossoms both with questions regarding specific mechanisms of action and with new technologies and strategies to address them.
References
Aerts JM, Ottenhoff R, Powlson AS, Grefhorst A, van Eijk M, Dubbelhuis PF, Aten J, Kuipers F, Serlie MJ, Wennekes T, Sethi JK, O’Rahilly S, Overkleeft HS (2007) Pharmacological inhibition of glucosylceramide synthase enhances insulin sensitivity. Diabetes 56(5):1341
Amati F, Dube J, Alvarez-Carnero E, Edreira M, Chomentowski P, Coen P, Switzer G, Bickel P, Stefanovic-Racic M, Toledo F, Goodpaster B (2011) Skeletal muscle triglycerides, diacylglycerides, and ceramids in insulin resistance: another paradox in endurance-trained athletes? Diabetes 60(10):2588–2597
Auge N, Maupas-Schwalm F, Elbaz M, Thiers J, Waysbort A, Itohara S, Krell H, Salvayre R, Negre-Salvayre A (2004) Role for matrix metalloproteinase-2 in oxidized low-density lipoprotein-induced activation of sphingomyelin/ceramide pathway and smooth muscle cell proliferation. Circulation 110:571–578
Banegas JR, Lopez-Garcia E, Graciani A, Guallar-Castillon P, Gutierrez-Fisac JL, Alonso J, Rodriguez-Artalejo F (2007) Relationship between obesity, hypertension and diabetes, and health-related quality of life among the elderly. Eur J Cardiovasc Prev Rehabil 14(3):456–462
Baranowski M, Blachnio A, Zabielski P, Gorski J (2007) PPARalpha agonist induces the accumulation of ceramide in the heart of rats fed high-fat diet. J Physiol Pharmacol 58(1):57
Baricos W, Cortez-Schwartz S, Shah S (1986) Renal neuraminidase. Characterization in normal rat kidney and measurement in experimentally induced nephrotic syndrome. Biochem J 239(3):705–710
Barouch LA, Gao D, Chen L, Miller KL, Xu W, Phan AC, Kittleson MM, Minhas KM, Berkowitz DE, Wei C, Hare JM (2006) Cardiac myocyte apoptosis is associated with increased DNA damage and decreased survival in murine models of obesity. Circ Res 98(1):119
Baskin M, Ard J, Franklin F, Allison D (2005) Prevalence of obesity in the United States. Obes Rev 6:5–7
Bevilacqua S, Bonadonna R, Buzzigoli G, Boni C, Ciociaro D, Maccari F, Giorico MA, Ferrannini E (1987) Acute elevation of free fatty acid levels leads to hepatic insulin resistance in obese subjects. Metabolism 36(5):502
Bielawska AE, Shapiro JP, Jiang L, Melkonyan HS, Piot C, Wolfe CL, Tomei LD, Hannun YA, Umansky SR (1997) Ceramide is involved in triggering of cardiomyocyte apoptosis induced by ischemia and reperfusion. Am J Pathol 151(5):1257
Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A (2010) Sphingolipid analysis by high performance liquid chromatography-tandem mass spectrometry (HPLC-MS/MS). Adv Exp Med Biol 688:46–59
Bijl N, Sokolovic M, Vrins C, Langeveld M, Moerland P, Ottenhoff R, Roomen Cv, Claessen N, Boot R, Aten J, Groen A, Aerts J, Eijk Mv (2009) Modulation of glycosphingolipid metabolism significantly improves hepatic insulin sensitivity and reverses hepatic steatosis in mice. Hepatology 50(5):1431–1441
Blomqvist M, Osterbye T, Mansson JE, Horn T, Buschard K, Fredman P (2003) Selective lack of the C16:0 fatty acid isoform of sulfatide in pancreas of type II diabetic animal models. Apmis 111(9):867
Blomqvist M, Carrier M, Andrews T, Pettersson K, Mansson JE, Rynmark BM, Fredman P, Buschard K (2005) In vivo administration of the C16:0 fatty acid isoform of sulfatide increases pancreatic sulfatide and enhances glucose-stimulated insulin secretion in Zucker fatty (fa/fa) rats. Diabetes Metab Res Rev 21(2):158
Boini K, Zhang C, Xia M, Poklis J, Li P (2010) Role of sphingolipid mediator ceramide in obesity and renal injury in mice fed a high-fat diet. J Pharmacol Exp Ther 334(3):839–846
Bonzon-Kulichenko E, Schwudke D, Gallardo N, Molto E, Fernandez-Agullo T, Shevchenko A, Andres A (2009) Central leptin regulates total ceramide content and sterol regulatory element binding protein-1C proteolytic maturation in rat white adipose tissue. Endocrinology 150(1):169–178
Boslem E, MacIntosh G, Preston AM, Bartley C, Busch AK, Fuller M, Laybutt DR, Meikle PJ, Biden TJ (2011) A lipidomic screen of palmitate-treated MIN6 beta-cells links sphingolipid metabolites with endoplasmic reticulum (ER) stress and impaired protein trafficking. Biochem J 435(1):267
Boudina S, Abel ED (2010) Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord 11(1):31
Brice SE, Cowart LA (2011) Sphingolipid metabolism and analysis in metabolic disease. Adv Exp Med Biol 721:1–17
Brown M, Goldstein J (2008) Selective versus total insulin resistance: a pathogenic paradox. Cell Metab 7:95–96
Bruce C, Kriketos A, Cooney G, Hawley J (2004) Disassociation of muscle triglyceride content and insulin sensitivity after exercise training in patients with type 2 diabetes. Diabetologia 47:23–30
Bruni P, Donati C (2008) Pleiotropic effects of sphingolipids in skeletal muscle. Cell Mol Life Sci 65:3725–3736
Buschard K, Hoy M, Bokvist K, Olsen HL, Madsbad S, Fredman P, Gromada J (2002) Sulfatide controls insulin secretion by modulation of ATP-sensitive K(+)-channel activity and Ca(2+)-dependent exocytosis in rat pancreatic beta-cells. Diabetes 51(8):2514
Buschard K, Blomqvist M, Mansson JE, Fredman P, Juhl K, Gromada J (2006) C16:0 sulfatide inhibits insulin secretion in rat beta-cells by reducing the sensitivity of KATP channels to ATP inhibition. Diabetes 55(10):2826
Cacicedo J, Benjachareowong S, Chou E, Ruderman N, Ido Y (2005) Palmitate-induced apoptosis in cultured bovine retinal pericytes: role for NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes 54(6):1838–1845
Candiloros H, Zeghari N, Ziegler O, Donner M, Drouin P (1996) Hyperinsulinemia is related to erythrocyte phospholipid composition and membrane fluidity changes in obese nondiabetic women. J Clin Endocrinol Metab 81(8):2912
Cardenas A, Schadeck C, Bernard A, Lauwerys R (1991) Depletion of sialic acid without changes in sialidase activity in glmeruli of uninephrectomized diabetic rats. Biochem Med Metab Biol 46(3):416–421
Cavaghan MK, Ehrmann DA, Polonsky KS (2000) Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J Clin Invest 106(3):329
Chavez JA, Knotts TA, Wang LP, Li G, Dobrowsky RT, Florant GL, Summers SA (2003) A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J Biol Chem 278:10297–10303
Christiansen T, Richelsen B, Bruun J (2005) Monocyte chemoattractant protein-1 is produced in isolated adipocytes, associated with adiposity and reduced after weight loss in morbid obese subjects. Int J Obes 29(1):146–150
Cnop M, Hannaert JC, Hoorens A, Eizirik DL, Pipeleers DG (2001) Inverse relationship between cytotoxicity of free fatty acids in pancreatic islet cells and cellular triglyceride accumulation. Diabetes 50(8):1771
Cohen-Forterre L, Mozere G, Andre J, Sternberg M (1984) Studies on kidney sialidase in normal and diabetic rats. Biochim Biophys Acta 800(1):138–145
Coleman R, Lee D (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res 43:134–176
Constable PD, Smith GW, Rottinghaus GE, Tumbleson ME, Haschek WM (2003) Fumonisin-induced blockade of ceramide synthase in sphingolipid biosynthetic pathway alters aortic input impedance spectrum of pigs. Am J Physiol Heart Circ Physiol 284(6):H2034
Davis S, Deo SH, Barlow M, Yoshishige D, Farias M, Caffrey JL (2006) The monosialosyl ganglioside GM-1 reduces the vagolytic efficacy of delta2-opioid receptor stimulation. Am J Physiol Heart Circ Physiol 291(5):H2318
de Mello VD, Lankinen M, Schwab U, Kolehmainen M, Lehto S, Seppanen-Laakso T, Oresic M, Pulkkinen L, Uusitupa M, Erkkila AT (2009) Link between plasma ceramides, inflammation and insulin resistance: association with serum IL-6 concentration in patients with coronary heart disease. Diabetologia 52(12):2612
de Vries JE, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, van der Vusse GJ, van Bilsen M (1997) Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res 38(7):1384
Deevska G, Rozenova K, Giltiay N, Chambers M, White J, Boyanovsky B, Wei J, Daugherty A, Smart E, Reid M, Merrill JAH, Nikolova-Karakashian M (2009) Acid sphingomyelinase deficiency prevents diet-induced hepatic triacylglycerol accumulation and hyperglycemia. J Biol Chem 284:8359–8368
Di Paola M, Zaccagnino P, Montedoro G, Cocco T, Lorusso M (2004) Ceramide induces release of pro-apoptotic proteins from mitochondria by either a Ca2+ −dependent or a Ca2+ −independent mechanism. J Bioenerg Biomembr 36(2):165
Dimitrios I, Drosatos K, Hiyama Y, Goldberg I, Zannis V (2010) MicroRNA-370 controls the expression of MicroRNA-122 and CPT1a and affects lipid metabolism. J Lipid Res 51(6):1513–1523
Dindo D, Dahm F, Szulc Z, Bielawska A, Obeid LM, Hannun YA, Graf R, Clavien PA (2006) Cationic long-chain ceramide LCL-30 induces cell death by mitochondrial targeting in SW403 cells. Mol Cancer Ther 5(6):1520–1529
Dinh W, Lankisch M, Nickl W, Scheyer D, Scheffold T, Kramer F, Krahn T, Klein RM, Barroso MC, Futh R (2010) Insulin resistance and glycemic abnormalities are associated with deterioration of left ventricular diastolic function: a cross-sectional study. Cardiovasc Diabetol 9:63
Dinh W, Lankisch M, Nickl W, Gies M, Scheyer D, Kramer F, Scheffold T, Krahns T, Sause A, Futh R (2011) Metabolic syndrome with or without diabetes contributes to left ventricular diastolic dysfunction. Acta Cardiol 66(2):167
Dobrzyn P, Dobrzyn A, Miyazaki M, Ntambi JM (2010) Loss of stearoyl-CoA desaturase 1 rescues cardiac function in obese leptin-deficient mice. J Lipid Res 51(8):2202
Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, Agarwal R, Yu S, Jiang H, Steinberg SF, Blaner WS, Koch WJ, Goldberg IJ (2011) Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metab 300(3):E489
Dyntar D, Eppenberger-Eberhardt M, Maedler K, Pruschy M, Eppenberger HM, Spinas GA, Donath MY (2001) Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes 50(9):2105
El-Assaad W, Joly E, Barbeau A, Sladek R, Buteau J, Maestre I, Pepin E, Zhao S, Iglesias J, Roche E, Prentki M (2010) Glucolipotoxicity alters lipid partitioning and causes mitochondrial dysfunction, cholesterol, and ceramide deposition and reactive oxygen species production in INS832/13 ss-cells. Endocrinology 151(7):3061
Finck BN (2004) The role of the peroxisome proliferator-activated receptor alpha pathway in pathological remodeling of the diabetic heart. Curr Opin Clin Nutr Metab Care 7(4):391
Fox T, Han X, Kelly S, Merrill JAH, Martin R, Anderson R, Gardner T, Kester M (2006) Diabetes alters sphingolipid metabolism in the retina: a potential mechanism of cell death in diabetic retinopathy. Diabetes 55(12):3573–3580
Fry J, Finley W (2005) The prevalence and costs of obesity in the EU. Proc Nutr Soc 64(3):359–362
Gault CR, Obeid LM, Hannun YA (2010) An overview of sphingolipid metabolism: from synthesis to breakdown. Adv Exp Med Biol 688:1–23
Gidwani A, Brown HA, Holowka D, Baird B (2003) Disruption of lipid order by short-chain ceramides correlates with inhibition of phospholipase D and downstream signaling by FcepsilonRI. J Cell Sci 116(Pt 15):3177
Goldkorn T, Balaban N, Shannon M, Chea V, Matsukuma K, Gilchrist D, Wang H, Chan C (1998) H2O2 acts on cellular membranes to generate ceramide signaling and initiate apoptosis in tracheobronchial epithelial cells. J Cell Sci 111(Pt 21):3209–3220
Gonzalez-Pertusa JA, Dube J, Valle SR, Rosa TC, Takane KK, Mellado-Gil JM, Perdomo G, Vasavada RC, Garcia-Ocana A (2010) Novel proapoptotic effect of hepatocyte growth factor: synergy with palmitate to cause pancreatic {beta}-cell apoptosis. Endocrinology 151(4):1487
Goodpaster B, He J, Watkins S, Kelley D (2001) Skeletal muscle lipid content and insulin resistance: evidence for a paradox in endurance-trained athletes. J Clin Endocrinol Metab 86:5755–5761
Gorski J, Dobrzyn A, Zendzian-Piotrowska M (2002) The sphingomyelin-signaling pathway in skeletal muscles and its role in regulation of glucose uptake. Ann NY Acad Sci 967:236–248
Grundy SM, Bryan Brewer JH, Cleeman JI, Sidney J, Smith C, Lenfant C (2004) Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation 109:433–438
Guha A, Harmancey R, Taegtmeyer H (2008) Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol 23(3):241
Guo J, Qian Y, Xi X, Hu X, Zhu J, Han X (2010) Blockage of ceramide metabolism exacerbates palmitate inhibition of pro-insulin gene expression in pancreatic beta-cells. Mol Cell Biochem 338(1–2):283
Hajduch E, Alessi D, Hemmings B, Hundal H (1998) Constitutive activation of protein kinase B alpha by membrane targeting promotes glucose and system A amino acid transport, protein synthesis, and inactivation of glycogen synthase kinase 3 in L6 muscle cells. Diabetes 47:1006–1013
Hajduch E, LItherland G, Hundal H (2001) Protein kinase B (PKB/Akt)-a key regulator of glucose transport? FEBS Lett 492:199–203
Hajer GR, Haeften TWv, Visseren FL (2008) Adipose tissue dysfunction in obesity, diabetes, and vascular disease. Eur Heart J 29:2959–2971
Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9(2):139–150
Haus JM, Kashyap SR, Kasumov T, Zhang R, Kelly KR, Defronzo RA, Kirwan JP (2009) Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 58(2):337
Hickson-Bick DL, Buja LM, McMillin JB (2000) Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol 32(3):511
Hirose H, Lee YH, Inman LR, Nagasawa Y, Johnson JH, Unger RH (1996) Defective fatty acid-mediated beta-cell compensation in Zucker diabetic fatty rats. Pathogenic implications for obesity-dependent diabetes. J Biol Chem 271(10):5633
Ho KK, Pinsky JL, Kannel WB, Levy D (1993) The epidemiology of heart failure: the Framingham study. J Am Coll Cardiol 22(4 Suppl A):6A
Hojjati M, Li Z, Zhou H, Tang S, Huan C, Ooi E, Lu S, Jiang X (2005) Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem 280:10284–10289
Holland W, Brozinick J, Wang L, Hawkins E, Sargent S, Liu Y, Narra K, Hoehn K, Knotts T, Siesky A, Nelson D, Karathanasis S, Foentenot G, Birnbaum M, Summers S (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5:167–179
Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE (2011) Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17(1):55
Hotamisligil G (2006) Inflammation and metabolic disorders. Nature 444(7121):860–867
Hu W, Bielawski J, Samad F, Merrill AH Jr, Cowart LA (2009) Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J Lipid Res 50(9):1852
Hu W, Ross J, Geng T, Brice SE, Cowart LA (2011) Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids: implications for insulin resistance. J Biol Chem 286(19):16596
Itani S, Ruderman N, Schmieder F, Boden G (2002) Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protien kinase C, and IkB-alpha. Diabetes 51:2005–2011
Jessup CF, Bonder CS, Pitson SM, Coates PT (2011) The sphingolipid rheostat: a potential target for improving pancreatic islet survival and function. Endocr Metab Immune Disord Drug Targets 11(4):262–272
Keller S, Lienhard G (1994) Insulin signalling the role of insulin receptor substrate 1. Trends Cell Biol 4:115–119
Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V (2003) Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem 278(32):30015
Kolak M, Westerbacka J, Velagapudi V, Wagsater D, Yetukuri L, Makkonen J, Rissanen A, Hakkinen A, Lindell M, Bergholm R, Hamsten A, Eriksson P, Fisher R, Oresic M, Yki-Jarvinen H (2007) Adipose tissue inflammation and increased ceramide content characterize subjects with high liver fat content independent of obesity.". Diabetes 56(8):1960–1968
Kremer GJ, Atzpodien W, Schnellbacher E (1975) Plasma glycosphingolipids in diabetics and normals. Klin Wochenschr 53(13):637
Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, Glembotski CC, Quintana PJ, Sabbadini RA (1996) Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest 98(12):2854
Kucharska-Newton AM, Couper DJ, Pankow JS, Prineas RJ, Rea TD, Sotoodehnia N, Chakravarti A, Folsom AR, Siscovick DS, Rosamond WD (2010) Diabetes and the risk of sudden cardiac death, the Atherosclerosis Risk in Communities study. Acta Diabetol 47(Suppl 1):161
Lam Y, Hatzinikolas G, Weir J, Janovska A, McAinch A, Game P, Meikle P, Wittert G (2011) Insulin-stimulated glucose uptake and pathways regulating energy metabolism in skeletal muscle cells: the effects of subcutaneous and visceral fat, and long-chain saturated, n-3 and n-6 polyunsaturated fatty acids. Biochim Biophys Acta 1811(7–8):468–475
Laviad EL, Albee L, Pankova-Kholmyansky I, Epstein S, Park H, Merrill AH Jr, Futerman AH (2008) Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem 283(9):5677–5684
Leahy JL (2008) Pathogenesis of type 2 diabetes mellitus. In: Feinglos MN, Bethel MA (eds) Type 2 diabetes mellitus. Humana, Amstgerdam, p 17
Lee Y, Hirose H, Ohneda M, Johnson JH, McGarry JD, Unger RH (1994) Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships. Proc Natl Acad Sci USA 91(23):10878
Levy M, Castillo SS, Goldkorn T (2006) nSMase2 activation and trafficking are modulated by oxidative stress to induce apoptosis. Biochem Biophys Res Commun 344(3):900–905
Levy M, Khan E, Careaga M, Goldkorn T (2009) Neutral sphingomyelinase 2 is activated by cigarette smoke to augment ceramide-induced apoptosis in lung cell death. Am J Physiol Lung Cell Mol Physiol 297(1):L125–L133
Lipina C, Hundal H (2011) Sphingolipids: agents provoccateurs in the pathogenesis of insulin resistance. Diabetologia 54:1596–1607
Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ (2009) DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem 284(52):36312
Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, Patane G, Boggi U, Piro S, Anello M, Bergamini E, Mosca F, Di Mario U, Del Prato S, Marchetti P (2002) Prolonged exposure to free fatty acids has cytostatic and pro-apoptotic effects on human pancreatic islets: evidence that beta-cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl-2 regulated. Diabetes 51(5):1437
Ma MM, Chen JL, Wang GG, Wang H, Lu Y, Li JF, Yi J, Yuan YJ, Zhang QW, Mi J, Wang LS, Duan HF, Wu CT (2007) Sphingosine kinase 1 participates in insulin signalling and regulates glucose metabolism and homeostasis in KK/Ay diabetic mice. Diabetologia 50(4):891
Madrazo JA, Kelly DP (2008) The PPAR trio: regulators of myocardial energy metabolism in health and disease. J Mol Cell Cardiol 44(6):968
Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY (2001) Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes 50(1):69
Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY (2003) Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes 52(3):726
Maheux P, Jeppesen J, Sheu WH, Hollenbeck CB, Clinkingbeard C, Greenfield MS, Chen YD, Reaven GM (1994) Additive effects of obesity, hypertension, and type 2 diabetes on insulin resistance. Hypertension 24(6):695–698
Manchanayake J, Chitturi S, Nolan C, Farrell GC (2011) Postprandial hyperinsulinemia is universal in non-diabetic patients with nonalcoholic fatty liver disease. J Gastroenterol Hepatol 26(3):510
Martinez J, Kearney J, Kafatos A, Paquet S, Martinez-Gonzalez M (1999) Variables independently associated with self-reported obesity in the European Union. Public Health Nutr 2(1A):125–133
Mastrandrea LD, Sessanna SM, Laychock SG (2005) Sphingosine kinase activity and sphingosine-1 phosphate production in rat pancreatic islets and INS-1 cells: response to cytokines. Diabetes 54(5):1429
Mather A, Siskind LJ (2011) Glycosphingolipids and kidney disease. Adv Exp Med Biol 721:121–138
Maulik N, Das DK, Gogineni M, Cordis GA, Avrova N, Denisova N (1993) Reduction of myocardial ischemic reperfusion injury by sialylated glycosphingolipids, gangliosides. J Cardiovasc Pharmacol 22(1):74
Memon RA, Holleran WM, Moser AH, Seki T, Uchida Y, Fuller J, Shigenaga JK, Grunfeld C, Feingold KR (1998) Endotoxin and cytokines increase hepatic sphingolipid biosynthesis and produce lipoproteins enriched in ceramides and sphingomyelin. Arterioscler Thromb Vasc Biol 18(8):1257–1265
Merrill AH Jr, Nixon DW, Williams RD (1985) Activities of serine palmitoyltransferase (3-ketosphinganine synthase) in microsomes from different rat tissues. J Lipid Res 26(5):617
Milburn JL Jr, Hirose H, Lee YH, Nagasawa Y, Ogawa A, Ohneda M, BeltrandelRio H, Newgard CB, Johnson JH, Unger RH (1995) Pancreatic beta-cells in obesity. Evidence for induction of functional, morphologic, and metabolic abnormalities by increased long chain fatty acids. J Biol Chem 270(3):1295
Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T (1995) Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem Biophys Res Commun 211(2):396
Mohamed-Ali V, Goodrick S, Rawesh A, Katz D, Miles J, Yudkin J, Klein S, Coppack S (1997) Subcutaneous adipose tissue releases interleukin-6, but not tumor necrosis factor-alpha, in vivo. J Clin Endocrinol Metab 82:4196–4200
Moro C, Bajpeyi S, Smith S (2008) Determinants of intramyocellular triglyceride turnover: implications for insulin sensitivity. Am J Physiol Endocrinol Metab 294:E203–E213
Mueller-Riemenschneider F, Reinhold T, Berghofer A, Willich S (2008) Health-economic burden of obesity in Europe. Eur J Epidemiol 23(8):499–509
Mullen TD, Jenkins RW, Clarke CJ, Bielawski J, Hannun YA, Obeid LM (2011a) Ceramide synthase-dependent ceramide generation and programmed cell death: involvement of salvage pathway in regulating postmitochondrial events. J Biol Chem 286(18):15929–15942
Mullen TD, Spassieva S, Jenkins RW, Kitatani K, Bielawski J, Hannun YA, Obeid LM (2011b) Selective knockdown of ceramide synthases reveals complex interregulation of sphingolipid metabolism. J Lipid Res 52(1):68–77
Nerpin E, Riserus U, Ingelsson E, Sundstrom J, Jobs M, Larsson A, Basu S, Arnlov J (2008) Insulin sensitivity measured with euglycemic clamp is independently associated with glomerular filtration rate in a community-based cohort. Diabetes Care 31(8):1550
Nussey S, Whitehead SA (2001) Endocrinology: an integrated approach. Bios, Oxford, UK
Nybond S, Bjorkqvist YJ, Ramstedt B, Slotte JP (2005) Acyl chain length affects ceramide action on sterol/sphingomyelin-rich domains. Biochim Biophys Acta 1718(1–2):61
Osterbye T, Jorgensen KH, Fredman P, Tranum-Jensen J, Kaas A, Brange J, Whittingham JL, Buschard K (2001) Sulfatide promotes the folding of proinsulin, preserves insulin crystals, and mediates its monomerization. Glycobiology 11(6):473
Othman A, Rutti MF, Ernst D, Saely CH, Rein P, Drexel H, Porretta-Serapiglia C, Lauria G, Bianchi R, von Eckardstein A, Hornemann T (2011) Plasma deoxysphingolipids: a novel class of biomarkers for the metabolic syndrome? Diabetologia 55(2):421–31
Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ (2008) Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49(10):2101
Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Hartel S, Jaimovich E, Zorzano A, Hidalgo C, Lavandero S (2008) Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res 77(2):387
Passa P (2002) Diabetes trends in Europe. Diabetes Metab Res Rev 18(Suppl 3):S3–S8
Pewzner-Jung Y, Ben-Dor S, Futerman AH (2006) When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: insights into the regulation of ceramide synthesis. J Biol Chem 281(35):25001–25005
Pick A, Clark J, Kubstrup C, Levisetti M, Pugh W, Bonner-Weir S, Polonsky KS (1998) Role of apoptosis in failure of beta-cell mass compensation for insulin resistance and beta-cell defects in the male Zucker diabetic fatty rat. Diabetes 47(3):358
Pontiroli AE, Camisasca R (2002) Additive effect of overweight and type 2 diabetes in the appearance of coronary heart disease but not of stroke: a cross-sectional study. Acta Diabetol 39(2):83–90
Pouliot MC, Despres JP, Nadeau A, Tremblay A, Moorjani S, Lupien PJ, Theriault G, Bouchard C (1990) Associations between regional body fat distribution, fasting plasma free fatty acid levels and glucose tolerance in premenopausal women. Int J Obes 14(4):293
Preiss B, Sattar N (2008) Non-alcoholic fatty liver disease: an overview of prevalence, diagnosis, pathogenesis, and treatment considerations. Clin Sci 115:141–150
Prosdocimi M, Paro M, Travagli RA, Tessari F (1987) Alterations of the vegetative nervous system, experimental diabetes and pharmacological use of gangliosides. Funct Neurol 2(4):559
Qiao Q, Nyamdorj R (2010) The optimal cutoff values and their performance of waist circumference and waist-to-hip ratio for diagnosing type II diabetes. Eur J Clin Nutr 64(1):23–29
Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, Romijn JA, de Roos A, Lamb HJ (2008) Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol 52(22):1793
Robert P, Tsui P, Laville MP, Livi GP, Sarau HM, Bril A, Berrebi-Bertrand I (2001) EDG1 receptor stimulation leads to cardiac hypertrophy in rat neonatal myocytes. J Mol Cell Cardiol 33(9):1589
Roden M, Price TB, Perseghin G, Petersen KF, Rothman DL, Cline GW, Shulman GI (1996) Mechanism of free fatty acid-induced insulin resistance in humans. J Clin Invest 97(12):2859
Roeske-Nielsen A, Dalgaard LT, Mansson JE, Buschard K (2010) The glycolipid sulfatide protects insulin-producing cells against cytokine-induced apoptosis, a possible role in diabetes. Diabetes Metab Res Rev 26(8):631
Rutti S, Ehses JA, Sibler RA, Prazak R, Rohrer L, Georgopoulos S, Meier DT, Niclauss N, Berney T, Donath MY, von Eckardstein A (2009) Low- and high-density lipoproteins modulate function, apoptosis, and proliferation of primary human and murine pancreatic beta-cells. Endocrinology 150(10):4521
Saito M, Cooper TB, Vadasz C (2005) Ethanol-induced changes in the content of triglycerides, ceramides, and glucosylceramides in cultured neurons. Alcohol Clin Exp Res 29(8):1374–1383
Samad F, Hester KD, Yang G, Hannun YA, Bielawski J (2006) Altered adipose and plasma sphingolipid metabolism in obesity: a potential mechanism for cardiovascular and metabolic risk. Diabetes 55:2579–2587
Samad F, Badeanlou L, Shah C, Yang G (2011) Adipose tissue and ceramide biosynthesis in the pathogenesis of obesity. Adv Exp Med Biol 721:67–86
Schmitz-Peiffer C, Craig D, Biden T (1999) Ceramide generation is sufficient to account for the inhibition of the insulin-stimulated PKB pathway in C2C12 skeletal muscle cells pretreated with palmitate. J Biol Chem 274:24202–24210
Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B (2010) Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J 24(1):296
Senkal CE, Ponnusamy S, Manevich Y, Meyers-Needham M, Saddoughi SA, Mukhopadyay A, Dent P, Bielawski J, Ogretmen B (2011) Alteration of ceramide synthase 6/C16-ceramide induces activating transcription factor 6-mediated endoplasmic reticulum (ER) stress and apoptosis via perturbation of cellular Ca2+ and ER/Golgi membrane network. J Biol Chem 286(49):42446
Serlie MJ, Meijer AJ, Groener JE, Duran M, Endert E, Fliers E, Aerts JM, Sauerwein HP (2007) Short-term manipulation of plasma free fatty acids does not change skeletal muscle concentrations of ceramide and glucosylceramide in lean and overweight subjects. J Clin Endocrinol Metab 92(4):1524
Shah C, Yang G, Lee I, Bielawski J, Hannun YA, Samad F (2008) Protection from high fat diet-induced increase in ceramide in mice lacking plaminogen activator inhibitor 1. J Biol Chem 283:13538–13548
Shimabukuro M, Zhou Y-T, Levi M, Unger R (1998) Fatty acid-induced beta cell apoptosis: a link between obesity and diabetes. Proc Natl Acad Sci USA 95:2498–2502
Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB (2000) A metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol 279(5):H2124
Sultan I, Senkal CE, Ponnusamy S, Bielawski J, Szulc Z, Bielawska A, Hannun YA, Ogretmen B (2006) Regulation of the sphingosine-recycling pathway for ceramide generation by oxidative stress, and its role in controlling c-Myc/Max function. Biochem J 393(Pt 2):513–521
Summers SA (2006) Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res 45:42–72
Summers S, Nelson D (2005) A role for sphingolipids in producting the common features of type 2 diabetes, metabolic syndrome X, and Cushing’s syndrome. Diabetes 54:591–602
Teruel T, Hernandez R, Lorenzo M (2001) Ceramide mediates insulin resistance by tumor necrosis factor-alpha in brown adipocytes by maintaining Akt in an inactive dephosphorylated state. Diabetes 50:2563–2571
Tessari F, Travagli RA, Zanoni R, Prosdocimi M (1988) Effects of long-term diabetes and treatment with gangliosides on cardiac sympathetic innervation: a biochemical and functional study in mice. J Diabet Complications 2(1):34
Theilmeier G, Schmidt C, Herrmann J, Keul P, Schafers M, Herrgott I, Mersmann J, Larmann J, Hermann S, Stypmann J, Schober O, Hildebrand R, Schulz R, Heusch G, Haude M, von Wnuck Lipinski K, Herzog C, Schmitz M, Erbel R, Chun J, Levkau B (2006) High-density lipoproteins and their constituent, sphingosine-1-phosphate, directly protect the heart against ischemia/reperfusion injury in vivo via the S1P3 lysophospholipid receptor. Circulation 114(13):1403
Turpin SM, Ryall JG, Southgate R, Darby I, Hevener AL, Febbraio MA, Kemp BE, Lynch GS, Watt MJ (2009) Examination of ‘lipotoxicity’ in skeletal muscle of high-fat fed and ob/ob mice. J Physiol 587(7):1598–1605
Utz W, Engeli S, Haufe S, Kast P, Hermsdorf M, Wiesner S, Pofahl M, Traber J, Luft FC, Boschmann M, Schulz-Menger J, Jordan J (2011) Myocardial steatosis, cardiac remodelling and fitness in insulin-sensitive and insulin-resistant obese women. Heart 97(19):1585
Vanhaesebroeck B, Alessi D (2000) The PI3K-PDK1 connection: more than just a road to PKB. Biochem J 346(3):561–576
Veluthakal R, Palanivel R, Zhao Y, McDonald P, Gruber S, Kowluru A (2005) Ceramide induces mitochondrial abnormalities in insulin-secreting INS-1 cells: potential mechanisms underlying ceramide-mediated metabolic dysfunction of the beta cell. Apoptosis 10(4):841
Veret J, Coant N, Berdyshev EV, Skobeleva A, Therville N, Bailbe D, Gorshkova I, Natarajan V, Portha B, Le Stunff H (2011) Ceramide synthase 4 and de novo production of ceramides with specific N-acyl chain lengths are involved in glucolipotoxicity-induced apoptosis of INS-1 beta-cells. Biochem J 438(1):177
Wang E, Norred WP, Bacon CW, Riley RT, Merrill AH Jr (1991) Inhibition of sphingolipid biosynthesis by fumonisins. Implications for diseases associated with Fusarium moniliforme. J Biol Chem 266(22):14486–14490
Wang J, Zhen L, Klug MG, Wood D, Wu X, Mizrahi J (2000) Involvement of caspase 3- and 8-like proteases in ceramide-induced apoptosis of cardiomyocytes. J Card Fail 6(3):243
Wei GS, Coady SA, Goff DC Jr, Brancati FL, Levy D, Selvin E, Vasan RS, Fox CS (2011) Blood pressure and the risk of developing diabetes in african americans and whites: ARIC, CARDIA, and the framingham heart study. Diabetes Care 34(4):873–879
Whiteman E, Cho H, Birnbaum M (2002) Role of Akt/protein kinase B in metabolism. Trends Endocrinol Metab 13:444–451
Yagyu H, Chen G, Yokoyama M, Hirata K, Augustus A, Kako Y, Seo T, Hu Y, Lutz EP, Merkel M, Bensadoun A, Homma S, Goldberg IJ (2003) Lipoprotein lipase (LpL) on the surface of cardiomyocytes increases lipid uptake and produces a cardiomyopathy. J Clin Invest 111(3):419
Yosuke O, Seki E, Kodama Y, Suetsugu A, Miura K, Adachi M, Ito H, Shiratori Y, Banno Y, Olefsky J, Nagaki M, Moriwaki H, Brenner D, Seishima M (2011) Acid sphingomyelinase regulates glucose and lipid metabolism in hepatocytes through Akt activation and AMP-activated protein kinase suppression. FASEB J 25(4):1133–1144
Zeghari N, Younsi M, Meyer L, Donner M, Drouin P, Ziegler O (2000) Adipocyte and erythrocyte plasma membrane phospholipid composition and hyperinsulinemia: a study in nondiabetic and diabetic obese women. Int J Obes Relat Metab Disord 24(12):1600
Zhao H, Przybylska M, Wu I, Siegel C, Komarnitsky S, Yew N, Cheng S (2007) Inhibiting glycosphingolipid synthesis improves glycemic control and insulin sensitivity in animal models of type 2 diabetes. Diabetes 56(5):1210–1218
Zheng W, Kollmeyer J, Symolon H, Momin A, Munter E, Wang E, Kelly S, Allegood JC, Liu Y, Peng Q, Ramaraju H, Sullards MC, Cabot M, Merrill AH Jr (2006) Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta 1758(12):1864–1884
Acknowledgments
This work was supported by Grant Number F30DK092125 from the NIDDK (to S.B.R.), GAANN fellowship in Lipidomics and Systems Biology (to J.S.R), a Merit Award from the Department of Veterans Affairs (to L.A.C.), and the NIH COBRE in Lipidomics and Pathobiology at MUSC (to L.A.C.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Russo, S.B., Ross, J.S., Cowart, L.A. (2013). Sphingolipids in Obesity, Type 2 Diabetes, and Metabolic Disease. In: Gulbins, E., Petrache, I. (eds) Sphingolipids in Disease. Handbook of Experimental Pharmacology, vol 216. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1511-4_19
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1511-4_19
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1510-7
Online ISBN: 978-3-7091-1511-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)