
Abstract
Nonsense-mediated mRNA decay (NMD) is a translation-dependent, multistep process that degrades irregular or faulty messenger RNAs (mRNAs). NMD mainly targets mRNAs with a truncated open reading frame (ORF) due to premature termination codons (PTCs). In addition, NMD also regulates the expression of different types of endogenous mRNA substrates. A multitude of factors are involved in the tight regulation of the NMD mechanism. In this review, we focus on the molecular mechanism of mammalian NMD. Based on the published data, we discuss the involvement of translation termination in NMD initiation. Furthermore, we provide a detailed overview of the core NMD machinery, as well as several peripheral NMD factors, and discuss their function. Finally, we present an overview of diseases associated with NMD factor mutations and summarize the current state of treatment for genetic disorders caused by nonsense mutations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gene expression in eukaryotes is a compartmentalized process consisting of several different, yet connected steps. The importance of gene expression for living cells and organisms is exemplified by the existence of diverse molecular mechanisms that detect errors and thereby ensure the accuracy of gene expression. A well-known quality control process, referred to as nonsense-mediated mRNA decay (NMD) or alternatively mRNA surveillance, limits the expression of mRNAs with premature termination codons (PTCs) and other aberrant termination events. NMD prevents the biosynthesis of C-terminally truncated proteins from PTC-containing mRNAs, which may exert dominant-negative activities and interfere with the normal function of full-length proteins present in the cell (Fig. 1). NMD recognizes not only PTC-containing transcripts, which could result for example from frameshift or nonsense mutations, but also regulates the expression of many physiological mRNAs, the so-called endogenous NMD substrates. Hence, NMD represents a molecular quality control mechanism, but in addition also plays an important role during post-transcriptional gene expression.

Function of nonsense-mediated mRNA decay. Top A normal transcript consists of a 5′ cap, followed by a 5′ UTR, the ORF, a 3′ UTR, and a poly(A) tail. The ORF of a transcript with a normal termination codon is translated into a full length protein. Bottom A transcript carrying a premature termination codon (PTC) will host a shortened ORF as well as an elongated 3′ UTR. Instead of continuously translating the transcript into a truncated protein with possible deleterious function in the cell, the PTC-containing transcript will be degraded via NMD
In this review, we aim to provide a comprehensive overview about the factors involved in NMD and their characteristics and molecular functions. We also describe NMD-activating features of different types of NMD substrates. Furthermore, we present a model of the NMD mechanism, which explains how the interplay of different factors helps to recognize different classes of mRNAs. Finally, we discuss the function of NMD factors during normal embryonic development and how NMD influences the phenotypes of human disorders. This review focuses on mammalian NMD but we provide information about essential aspects of NMD in different organisms.
Terminating the message: steps in normal translation termination
The expression of protein coding genes is essential for cellular functionality, with the final step being cytoplasmic translation of mature mRNA transcripts into proteins by the ribosome. Translation termination plays a pivotal role in the lifespan of an mRNA, because the position of the termination codon and efficiency of the termination process determines mRNA stability [1–5]. The termination process begins when a stop codon (UAA, UAG, or UGA) enters the A-site of a translating ribosome, causing it to stall. Two important factors involved in the ensuing steps of translation termination are the class I release factor 1 (in humans eRF1) and the class II release factor 3 (in humans eRF3). Using RNA interference, it was shown that human eRF3 is directly involved in translation termination, albeit in a cell-type specific manner and is required for eRF1 stability in the cell [6, 7]. Of the two human eRF3 isoforms (eRF3a and eRF3b), eRF3a has been shown in HEK 293 cells to act as the main factor in translation termination [7–9]. A termination codon within the ribosomal A-site is decoded by eRF1, which is known to bind the ribosomal A-site and mimic a tRNA [10, 11]. Instead of promoting peptidyl transfer like a regular tRNA, eRF1 mediates via its GGQ motif the hydrolysis of the synthesized polypeptide chain, which is still attached to the ribosomal P-site [12, 13]. The role of the GTPase eRF3 is to enhance the activity of its interaction partner eRF1 [14]. Indeed, it has been shown in yeast that GTP hydrolysis by eRF3 stimulates the release of the polypeptide chain via eRF1 [15]. Although eRF3 is able to bind GTP, its weak inherent GTPase activity is stimulated by a quaternary complex consisting of eRF1, eRF3, GTP, and the ribosome [16, 17].
Upon peptide release mediated by eRF1, the mRNA-bound ribosome needs to be removed to free up space for any approaching, upstream ribosomes and any subsequent termination events. This dissociation of the 80S complex is executed by the ATP-binding cassette subunit family E member 1 (ABCE1) ATPase and involves ATP hydrolysis. The exact mechanism by which ABCE1 mediates ribosome recycling is not yet fully understood; however, several steps of this process have already been illuminated. It is known, for example, that eRF1 and ABCE1 interact with each other and that this interaction is required for efficient ribosome splitting [18]. Furthermore, ABCE1 is known to interact with the ribosome and this interaction is mediated by a FeS cluster domain of ABCE1 [19, 20]. This multistep process of translation termination ensures efficient ribosome recycling with continuous rounds of translation and increases mRNA survivability in general.
Several other factors besides the aforementioned ones are involved in translation termination. One such prominent factor in human cells is cytoplasmic poly(A)-binding protein 1 (PABPC1), which binds to the 3′ poly(A) tail of an mRNA. PABPC1 consists of four RNA recognition motifs (RRMs) in the N-terminal region of the protein [21, 22]. The first two RRMs are responsible for PABPC1-binding to the poly(A) tail [23]. The C-terminal region of PABPC1 contains the so-called MLLE (pronounced Mademoiselle) domain, which mediates the interaction with eRF3a [24, 25]. Two PAM2 motifs in the N terminus of eRF3a interact directly with the MLLE domain of PABPC1 and this interaction is known to stimulate polypeptide release as well as the consequent ribosome recycling [26–28]. PABPC1 interacts via its second RRM with eukaryotic initiation factor 4G (eIF4G), which is another important binding partner [29, 30]. eIF4G is part of a larger, multisubunit complex termed the eIF4F complex consisting of eIF4E, eIF4G, and eIF4A. This eIF4F complex is bound to the cap of mRNAs in the cytoplasm via its eIF4E subunit [31]. The interaction between PABPC1 and eIF4G is believed to give rise to a looped mRNA termed the closed loop. The closed loop model predicts a close proximity between a 3′ terminating ribosome and the 5′ end of the mRNA, which is believed to play a role in facilitating ribosome recycling as well as translation initiation [32–34].
PTC carrying transcripts activate NMD
NMD as a quality control mechanism targets aberrant mRNAs harboring a PTC at approximately 50 to 55 nucleotides upstream of the final exon–exon junction of a transcript. PTCs downstream of this border fail to initiate efficient degradation of a transcript via NMD [35–38]. It is of interest to note that not all mRNAs fully adhere to this 50 to 55 nucleotide rule. For example the T-cell receptor β (TCR-β) transcript represents one well known exception [39]. These findings suggest that intron position and nuclear splicing are important determinants for whether NMD is activated or not. This is emphasized by the fact that PTC-containing, but intronless transcripts are immune to degradation via NMD [40–42]. It was later found that a multisubunit protein complex termed exon junction complex (EJC) is deposited 20 to 24 nucleotides upstream of an exon–exon junction during splicing and remains bound to the mRNA until it is displaced during translation in the cytoplasm [43–46]. Although ribosomal transit is likely sufficient to remove EJCs located within the ORF, the protein PYM acts as an additional specific EJC disassembly factor [47]. It has been shown that PYM interacts with the heterodimer MAGOH/Y14, part of the EJC core, via its N-terminal region, thereby causing the disassembly of the EJC from the mRNA [47]. The EJC core complex is composed of four proteins: the heterodimer MAGOH/Y14, Barentzs (BTZ, MLN51 or CASC3), and the DEAD box protein eukaryotic initiation factor 4A-3 (eIF4A3). This core is deposited onto the RNA in the nucleus and stays attached during export to the cytoplasm, as well as in the cytoplasm by inhibition of the ATPase activity of eIF4A3 [48, 49]. The crystal structure of the EJC core complex revealed that the heterodimer Y14/MAGOH stabilizes the closed conformation of eIF4A3, effectively locking the EJC to the mRNA [50]. BTZ has been shown to interact with eIF4A3 by wrapping around the protein as well as interacting with MAGOH [49]. The EJC serves as a binding platform for NMD factors, thereby providing a direct molecular link between splicing and NMD [51, 52]. The role of the EJC in NMD has further been elucidated by tethering and RNA interference assays. The mRNA levels of a β-globin reporter construct were reduced when artificially tethering Y14 to the reporter [53]. At the same time, the significance of the EJC for NMD was shown by the knockdown of Y14, which impaired the degradation of a PTC-containing β-globin reporter construct [53].
NMD substrates lacking an EJC
Despite the fact that EJCs have been shown to activate NMD, further evidence also suggests that NMD can be activated without the need for an EJC. A set of experiments examined the effect of EJC-dependent and EJC-independent NMD. A reporter with an intron downstream of a PTC was susceptible to NMD even after knocking down the EJC core factor eIF4A3, albeit to a lesser extent compared to non-knockdown conditions [54]. Degradation of a second reporter with no intron downstream of a PTC was unaffected by the eIF4A3 knockdown [54]. This suggests that an EJC downstream of a PTC certainly has an NMD-enhancing effect, but an EJC is not mandatory for NMD activation.
Several long 3′ UTR containing mRNAs are known to be regulated by NMD even though they lack an EJC downstream of the stop codon [5]. Furthermore, it has been shown that triosephosphate isomerase (TPI) reporter constructs containing different 3′ UTRs (e.g. SMG5 3′ UTR, UPF3b 3′ UTR, and heterologous GFP coding sequence as a 3′ UTR) are able to elicit NMD [55]. Interestingly, the NMD machinery does not distinguish between the 3′ UTRs of natural NMD targets (SMG5 and UPF3b) but also degrades a TPI reporter with the heterologous GFP coding sequence as a 3′ UTR [55]. This suggests that any long 3′ UTR might be sufficient for NMD activation. Conversely, a different analysis of multiple long 3′ UTR containing reporter constructs revealed that some long 3′ UTRs are targeted by NMD, whereas others are not [56]. This was evident by decreased reporter levels being stabilized by a UPF1 (a central NMD factor, explained in detail below) knockdown [56]. These results imply that the mere length of a 3′ UTR is not enough to determine the fate of a transcript. Instead, they suggest the need for a cis factor binding in close proximity to the termination codon, possibly mediated by the AU-content of the 3′ UTR [56]. An example of a long 3′ UTR containing mRNA is depicted in Fig. 3d (top).
A different approach on long 3′ UTR mediated NMD holds UPF1 occupancy on a transcript responsible as the culprit for NMD activation. It has recently been shown that UPF1 is populating the 3′ UTR of mRNAs in human cells [57–59]. It has been proposed that UPF1 being displaced from mRNA by a translating ribosome is able to increasingly bind to the 3′ UTR where no ribosomal destabilization can occur [58, 59]. This occupancy might differ from transcript to transcript, explaining why some long 3′ UTR containing mRNAs are more susceptible to NMD than others.
The faux 3′ UTR model
Different models of NMD activation have been proposed based on the available data. An early model suggested for NMD in yeast is called the “faux 3′ UTR model”. This model advocates that NMD is activated by an aberrant translation termination event. It has been shown in yeast that a PTC lengthens the distance between a terminating ribosome and the yeast poly(A)-binding protein (Pab1), leading to inefficient translation termination [60]. Furthermore, tethering Pab1 downstream of a PTC suppresses NMD, possibly by restoring normal translation termination [60]. Taking into account that yeast lack EJCs, these data suggest that NMD substrates are defined by 3′ UTR length [61]. This indicates that the distance between a termination codon and Pab1 determines whether NMD in yeast is activated or not. This model was proposed prior to findings linking proper and aberrant translation termination in human cells to NMD activation or suppression.
NMD is linked to active translation
First results indicated that cap-binding protein 80 (CBP80)-bound mRNA was targeted by NMD, suggesting that NMD occurs during the pioneer round of translation [62]. However, it was later reported that NMD is able to occur on eIF4E-bound mRNA, suggesting that NMD is not limited to the first round of translation [63, 64].
It has been well established that NMD requires active translation. Secondary structures in the 5′ UTR inhibiting cytoplasmic translation have been linked to NMD inhibition [36, 65]. Additionally, protein synthesis inhibitors cycloheximide, anisomycin, emetine, pactamycin, and puromycin were able to suppress NMD of T-cell receptor mRNA harboring a PTC [66]. Furthermore, Polio virus infection, which is known to shut down protein translation by inactivating eIF4GI, eIF4GII, and PABP, increased an out-of-frame TCR-β mRNA in HeLa cells indicating NMD suppression [66–68]. Additionally, in-frame start (Met) codons downstream of a PTC in the TPI mRNA and the β-globin mRNA serve as sites of translation re-initiation and have also been shown to suppress mRNA degradation via NMD [69, 70]. This suggests that additional aspects besides active translation and factors recruited to and interacting with a stalling ribosome might be responsible for NMD activation.
Aberrant translation termination
An aberrant translation termination event starts out similar to a normal translation termination, namely by a premature translation termination codon (PTC) entering the A-site of a translating ribosome. However, the subsequent termination cascade has to be different from normal termination, resulting in NMD initiation. Indicative of a substantial difference between normal and premature translation termination are the ribosome profiles at both a normal termination codon (NTC) and a PTC. Toeprinting assays in yeast cell extracts fail to show any signals for ribosomes at NTCs, unless eRF1 was defective [60]. In contrast, ribosomes stalling at PTCs show typical toeprinting signals for a ribosome with an occupied A-site independent of eRF1 inactivation [60]. Human β-globin mRNA with a PTC at position 39 (β-globin NS39) is a known NMD target, which has been identified in patients suffering from beta thalassemia [36, 71, 72]. Toeprinting assays of β-globin NS39 show a signal indicating a stalled ribosome at the PTC, whereas β-globin wild-type mRNA does not [73]. These results are indicative of the aberrant nature of translation termination of PTC-containing mRNA and provide evidence for a mechanistic difference between normal and premature translation termination.
The exact nature of how prematurely terminated translation leads to the degradation of an mRNA is still not entirely clear. Several different sets of data pointing to different models exist. One such model is based on the competition between UPF1 and PABPC1 for eRF3 binding. As described above, PABPC1 is known to interact with eRF3a, stimulating peptide release and ribosome recycling. UPF1 has been shown to interact with eRF3 as well; however, the interaction between PABPC1 and eRF3 seems favored over the UPF1–eRF3 interaction [5, 74]. The fact that both PABPC1 and UPF1 can bind eRF3 but PABPC1 binding to eRF3 is preferred leads to the idea that PABPC1 might be able to antagonize the interaction between eRF3 and UPF1. Indeed, it has been shown that increasing amounts of PABPC1 can prevent the interaction between UPF1 and eRF3 in vitro [5]. This finding suggests that when a translating ribosome stalls at a PTC the interaction between PABPC1 and eRF3 is outcompeted by UPF1, possibly due to the large physical distance between PABPC1 and the termination complex. This model is further supported by the fact that a MS2-tagged PABPC1 tethered downstream but in close proximity to a PTC of a β-globin NS39 reporter mRNA prevented NMD [3, 5]. Tethering PABPC1 close to the PTC shortens the distance between PABPC1 and eRF3, which suggests that PABPC1 efficiently outcompetes UPF1 for eRF3 binding. Supporting evidence for this competition model has been shown by detailing two distinct roles for PABPC1 and UPF1 in efficient or aberrant translation termination, respectively. A readthrough assay with a reporter containing the ORF of Renilla luciferase upstream of the ORF of firefly luciferase, with a termination codon in between, showed decreased readthrough upon UPF1 knockdown for all three termination codons [3]. This suggests that UPF1 is able to decrease the efficiency of translation termination.
Another possible mechanism, besides the competition between PABPC1 and UPF1, for NMD suppression involves both ribosome recycling as well as translation initiation. It is known that NMD can target mRNAs with a long 3′ UTR and tethering MS2-tagged PABPC1 at the beginning of a long 3′ UTR can suppress NMD [2, 5, 75, 76]. Additionally, it has been shown that MS2-eIF4G tethered downstream of a termination codon and upstream of a long 3′ UTR or downstream of a PTC is able to suppress NMD in a similar manner as PABPC1 [2, 4]. Point mutations of PABPC1 that abolish the interaction between PABPC1 and eIF4G lose their ability to suppress NMD [2]. In contrast, PABPC1 lacking the ability to interact with eRF3 can still suppress NMD when tethered downstream of a termination codon and upstream of a long 3′ UTR as well as downstream of a PTC [2, 4]. The fact that PABPC1 does not require the interaction with eRF3 to suppress NMD contradicts the findings that PABPC1 and UPF1 compete for eRF3 binding during translation termination at a PTC and, thereby, either suppress or activate NMD, respectively. Additionally, these findings also implicate eIF4G and, therefore, the eIF4F complex in NMD inhibition. This suggests that the interaction cascade involving eRF3, PABPC1, and eIF4G is required for NMD suppression. These interactions promote a normal translation termination event, followed by efficient ribosome release and recycling, essentially antagonizing NMD [1, 2].
Further support for a model that links ribosome recycling and translation initiation to NMD suppression was given by the fact that the eukaryotic initiation complex 3 (eIF3) was shown to be involved in NMD suppression [4]. Although tethering subunits of eIF3 to an NMD-susceptible reporter did not antagonize NMD, knockdown of eIF3 subunits eIF3f and eIF3 h reduced the reporter mRNA levels of an NMD reporter stabilized by tethered eIF4G [4]. Overall, this leads to the paradoxical situation that efficient translation re-initiation after ribosome recycling suppresses NMD, while at the same time efficient translation of a substrate mRNA is required to activate NMD.
Additional evidence implicates the proximity of PABPC1 to a termination codon in NMD suppression. A reporter carrying several PTCs shows gradually less efficient NMD for 5′ as well as 3′ PTCs [76]. The more 5′ PTCs resistant to NMD implicate the closed loop structure of a transcript in translation termination. Since 5′ PTCs are not efficiently degraded it can be suggested, that PABPC1 is in close proximity to these PTCs by looping of the mRNA and, thereby, signaling a normal translation termination event. A similar experiment uses a reporter construct that harbors two complementary sequences to fold the poly(A) tail back into close proximity of a PTC. This foldback brings PABPC1 in close proximity of the PTC and is able to antagonize NMD even without the depletion of UPF1 [76].
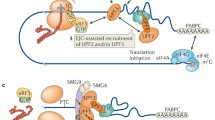
Figure 2 shows a current model of NMD of a PTC-carrying transcript. Still, many links between ribosome recycling, translation initiation, and NMD are still missing and need to be unveiled in future research.

Model for degradation of PTC carrying transcripts. Step 1 Translation is initiated at the AUG start codon. The ribosome starts translation in the 5′ to 3′ direction. Step 2 When the translating ribosome stalls at a PTC, eRF1 and eRF3 interact with and bind to the ribosome. The physical distance between poly(A)-bound PABPC1 and the stalled ribosome is too large for efficient interaction and subsequent translation termination. Step 3 Since the interaction between PABPC1 and eRF3 is inefficient in this scenario, UPF1 is able to interact with eRF3 instead. Furthermore, the EJC serves as a binding platform for UPF3b and UPF2. Additionally, SMG1 is recruited to UPF1. SMG1 not only interacts with UPF1 and UPF2 but also phosphorylates UPF1. Step 4 Phosphorylated UPF1 recruits the SMG5/7 heterodimer as well as the endonuclease SMG6. The stalling ribosome has been removed from the transcript by this point. SMG6 cleaves the transcript in close proximity of the PTC, whereas SMG5/7 recruit the catalytic subunit of the CCR4-NOT deadenylase complex POP2. Step 5 The NMD factors are removed from the cleaved transcript and decapping and deadenylation commences. As a last step, the exonuclease XRN1 is recruited and degrades the transcript in 5′ to 3′ direction, and the exosome supposedly mediates 3′ to 5′ degradation
Various cellular transcripts are NMD substrates
Selenoproteins are a class of polypeptides containing the trace element Selenium (Se) and are characterized by the incorporation of the unusual amino acid selenocysteine (Sec) [77]. Selenoprotein-encoding transcripts are believed to be degraded via NMD since the codon UGA, more prominently known for being a stop codon, also codes for Sec when it is accompanied by specific sequence elements (SECIS) [78]. It has been shown that the stability of selenoprotein mRNAs is dependent on Se concentrations [79, 80]. Interestingly, about half of all selenoprotein-encoding mRNAs are regulated by NMD, whereas the other half is not [81]. This occurrence is believed to be related to the location of the termination codon in the mRNA of selenoproteins. Sec codons in the last exon are NMD resistant, while Sec codons in any other exon are subjected to NMD [81].
As described previously, many NMD targets have an EJC downstream of a termination codon. This also fits for many transcripts carrying upstream open reading frames (uORFs), which are usually followed by a long main ORF with several splice sites. Microarray analysis in HeLa cells showed that several uORF transcripts are upregulated after UPF1 knockdown [82]. This indicates that transcripts with a uORF can be endogenous targets for the NMD pathway. It is of interest to note that not all uORF containing transcripts are degraded via NMD. Thrombopoietin (TPO) is such an example. TPO contains seven uORFs and has been shown to be unaffected by knockdown of UPF1 [83]. This leads to the current understanding that many uORF transcripts are in fact targeted by NMD, but a uORF can by no means be generally considered a NMD target.
Alternative splicing is responsible for modulation of gene expression by generating different mRNA transcript isoforms [84]. It has been shown that alternatively spliced pre-mRNAs can be targets of NMD, since many of the transcript variants will carry a PTC [85]. An interesting example is the splicing factor SRSF2 (SC35). SRSF2 is known to regulate its own mRNA expression by altering its splicing pattern [86].
Another subset of NMD targets are so-called long non-coding RNAs (lncRNAs). A study employed growth arrest factor 5 (GAS5) as an exemplary lncRNA to examine if lncRNAs are indeed degraded via NMD [87]. The study showed that GAS5 transcript levels were increased upon UPF1 depletion, which indicates that NMD is responsible for the degradation of GAS5 mRNA [87]. It is surprising that lncRNAs are NMD targets since NMD is known to require actively translated mRNA. However, a recent study showed that many lncRNAs are indeed protein coding and therefore actively translated [88]. Figure 3 depicts a graphical overview of possible NMD targets and Table 1 lists examples for each class of NMD substrate.

Examples of NMD substrates. a Alternative splicing of a pre-mRNA can lead to PTC formation by, for example, exon-skipping. The PTC-containing transcript isoform is degraded in an EJC-dependent manner, whereas regularly spliced transcripts without a PTC will not be targeted by NMD. b An error during gene expression can lead to a nonsense mutation in the ORF of a transcript. If the PTC that arises due to this nonsense mutation is upstream of the last exon–exon junction, an EJC will be present downstream of the PTC and the transcript will be degraded via NMD. c Some long ncRNAs are known to harbor snoRNAs within their introns. The snoRNAs are released when the introns are spliced. snoRNA host genes do not encode a protein, but instead have only a short ORF with a PTC, which will lead to activation of NMD followed by subsequent degradation of the transcript. d Top Many transcripts with long 3′ UTRs are known targets of NMD. Middle Some mRNAs can host one or multiple uORFs, which will lead to EJC-mediated NMD, since the EJCs will not be displaced by a translating ribosome. Bottom Selenoprotein-encoding transcripts are a class of NMD targets that incorporate the unusual amino acid selenocysteine (Sec) at UGA codons. With low concentrations of Sec present, the UGA codon is decoded as a classical stop codon and, depending on its position, is treated as a PTC
NMD factors and their role in substrate recognition and degradation
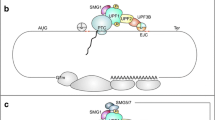
The number of identified proteins involved in NMD, especially in metazoan cells, has tremendously increased over the past two decades. The first NMD factors were described more than 30 years ago in S. cerevisiae. At that time, genetic screening was done in a yeast strain expressing a HIS4 transcript with a +1 frameshift, which leads to premature translation termination and decreased stability of the mRNA. The screen aimed to identify mutations, which suppress this frameshift and result in increased mRNA stability [89]. Several upf (up–frameshift) mutants were characterized later and the mutated genes were termed UPF1, UPF2 and UPF3 [90–92]. They represent the central set of NMD factors. The UPF proteins have been found in all late-branching eukaryotes, ranging from yeast over nematodes and fruit flies to mammals and plants, whereas the set is incomplete in certain protists [93–96]. Additional factors in the NMD pathway of higher metazoans were found in nonsense suppression screens in C. elegans and were termed smg1-7 (suppressor with morphogenetic effect on genitalia). Mutation of these genes did not only abolish NMD, but also caused phenotypical abnormalities in the male bursa and hermaphrodite vulva [97–99]. Smg2-4 represent homologs of the conserved UPF1-3; therefore, the extended NMD core machinery comprises UPF1-3, SMG1, and SMG5-7 [100–106]. An overview of the central characteristics of these core NMD factors can be found in Table 2. Figure 4 depicts the molecular architecture of the core NMD factors and summarizes their functions and interaction sites, both of which are discussed in detail below. A number of additional NMD factors have been identified in different organisms, including SMG8, SMG9, PNRC2, DHX34, NBAS, RUVBL1, RUVBL2, MOV10, GNL2 and SEC13 for the human system [107–113].

Overview of the core NMD factors UPF1, UPF2, UPF3b/a, the kinase SMG1, and the decay inducing factors SMG5, SMG6, and SMG7. The domain architecture for all factors, as well as the NMD-specific functions of important regions are depicted here (for details, see text). The most studied isoforms of NMD factors were chosen for representation and match, except for SMG1, those indicated in Table 2 (see footnote b for SMG1 discrepancy). UPF1 phosphorylation sites (SQ and TQ motifs) verified by various experimental approaches (ultradeep HeLa cell phosphoproteome [222], in vitro phophorylation assay with UPF1 peptides [106] or full length UPF1 [154] ) are indicated. The phosphosites connected to specific recruiting functions are highlighted specifically
UPF1 is the central nexus of the NMD machinery
UPF1 is considered the central NMD factor, since it acts as an interaction hub for other NMD factors, is involved in all decisive stages of the NMD process, and is essential for NMD in all investigated organisms [91, 114–117]. The central part of UPF1 is highly conserved between species with more than 40 % sequence identity between the yeast and human homolog. It consists of two functional domains: an N-terminal zinc knuckle cystidine-histidine-rich CH domain followed by a central helicase domain [94]. UPF1 is classified based on several functional features. The helicase domain of UPF1 is formed by two RecA-like domains belonging to the superfamily 1Bα (SF1Bα), which uses ATP hydrolysis to unwind double stranded nucleic acids in 5′-3′ direction [118–121]. The helicase domain also mediates the direct binding of UPF1 to the RNA and, according to the current model, RNA-bound UPF1 is required for NMD factor assembly [118, 122]. Whereas the presence of ATP, or nucleotide analogs that mimic certain transition states in the hydrolysis reaction, reduce the RNA binding affinity of the helicase domain in vitro, these nucleotide-induced conformational changes still allow concomitant ATP and RNA binding [119, 122–124]. Initially it was proposed that UPF1 is recruited to the NMD target by the terminating ribosome via the interaction with the release factors eRF1 and eRF3. This implies that UPF1 recruitment on NMD targets is directly linked to translation termination and hence occurs in the proximity of the termination codon [74]. However, individual nucleotide resolution UV cross-linking and immunoprecipitation (iCLIP) experiments showed that UPF1 directly binds mostly spliced transcripts, regardless of whether they are NMD targets or not [59]. Thus, UPF1 abundance on an mRNA does not correlate directly with its NMD susceptibility. As mentioned above, UPF1 preferentially occupies the 3′ UTR region of mRNAs due to displacement from the 5′ UTR and coding region by scanning and translating ribosomes, respectively [57–59]. The overall importance of a functional UPF1 helicase domain is represented by the fact that the ATPase activity and direct RNA binding ability are both required for NMD [124–126]. Once associated with the mRNA, the helicase domain of UPF1 is believed to utilize the hydrolysis of ATP to remodel the downstream mRNP, which is essential to facilitate recycling of NMD factors [119, 121]. This would allow efficient progression of exonucleolytic degradation of the mRNA once initial decay steps have taken place [127]. Alternatively, it was proposed that UPF1 is able to translocate on, or thread in, the mRNA to bridge the physical distance to downstream mRNP components, such as the EJC [128]. This early “licensing” step would explain why a distance-independent activation of NMD is observed, no matter how far away the EJC is located from the upstream terminating ribosome [55]. However, it was shown that both the CH domain as well as a C-terminal region of UPF1 (SQ region) can regulate the helicase activity, which ensures that UPF1 clamps to the RNA and does not translocate in the earlier stages of NMD [123, 129]. More specifically, the CH domain directly interacts with the RecA2 domain, which results in conformational changes that promote more extensive RNA binding and thereby repress the helicase activity [123]. In order to initiate the unwinding activity of UPF1, the CH domain has to be removed from the helicase core, which is achieved by the interaction with the C-terminal UPF1-binding domain (U1BD) of UPF2 [122, 123]. This means in turn that if UPF1 needs to use its helicase activity in the early steps of NMD, for example for finding a downstream EJC, UPF2 already needs to be bound to UPF1 or the CH domain has to be pulled away by a as of yet unknown mechanism or factor.
Multiple functions define the role of UPF2
The domain architecture of UPF2 consists of three tandem MIF4G domains (middle portion of eIF4G), followed by the U1BD [130–132]. Proteins containing MIF4G domains are commonly involved in general mRNA metabolism, such as the components of the nuclear or cytoplasmic cap-binding complex (CBC) CBP80 and eIF4G, or the spliceosomal protein CWC22 [130, 133–136]. Consistent with the role of MIF4G domains to provide the surface for critical interactions, the MIF4G-3 domain of UPF2 is required for the interaction with UPF3 [137, 138]. This interaction establishes a linear cascade ranging from UPF1 over UPF2 to UPF3 [122]. Whereas the exact function of the first two MIF4G domains is not known, a structural role was proposed in which these domains are required to form a ring-like scaffolding structure required for NMD factor assembly [131, 139]. Moreover, conserved residues on the surface of the N-terminal helices of MIF4G-1 of the S. cerevisiae Upf2 were shown to be essential for NMD [140]. Although potential interaction partners were identified, the function of these interactions in the molecular pathway of NMD remains unclear [140]. However, this finding strengthens the possibility that the MIF4G-1 and -2 domains of UPF2 play a role that goes beyond providing the correct structural architecture for NMD.
UPF3 bridges decay inducing elements and NMD factors
Higher eukaryotes contain two UPF3 paralogs with high sequence similarity, UPF3a and UPF3b, the latter being expressed from the X chromosome in mammals [103, 138]. In contrast, only one UPF3 protein exists in yeast and other invertebrates. UPF3b was found to be the dominant NMD factor of both paralogs. However, a cross-regulatory circuit was described, in which the stability of UPF3a is regulated as a consequence of the competition of both UPF3 proteins for binding to UPF2 [53, 141, 142]. Besides providing robustness to NMD by functional redundancy, the advantage of having two UPF3 paralogs becomes evident when the expression of one paralog is cell-specifically down regulated. Meiotic male germ cells are one such example, since they inactivate the X chromosome transcriptionally and, therefore, shut down UPF3b expression [141, 143]. UPF3b is a nucleocytoplasmic shuttling protein and contains a conserved N-terminal RRM [103, 138]. The N-terminal RRM domain is the binding site for the MIF4G-3 domain of UPF2, but does not mediate RNA binding as the name suggests [137]. A short linear motif termed EJC-binding motif (EBM) at the C-terminus of UPF3b is responsible for the interaction with a composite binding site of the EJC formed by the core components eIF4A3, MAGOH and Y14 [53, 122, 144, 145]. The exact molecular function of UPF3 in NMD remains elusive. The proposed role of UPF3b in mammals was the bridging of the EJC and UPF1-UPF2; however, this does not explain the function of UPF3 in EJC-independent NMD [122, 139, 146]. This is especially interesting in organisms that do not employ EJC-enhanced NMD as the standard pathway, but still rely on UPF3 for NMD. Examples are yeast, flies, and worms, which either contain a very small number of spliced transcripts, lack EJC proteins and the EBM in the C-terminus of UPF3, or do not require EJC core components for NMD [53, 94, 111, 147–149].
Regulation and mechanism of UPF1 phosphorylation
It was first observed in C. elegans that the phosphorylation status of the phosphoprotein UPF1/SMG2 is regulated by other core NMD factors. Of these, SMG1, UPF2/SMG3 and UPF3/SMG4 are required for phosphorylation, whereas SMG5, SMG6, and SMG7 are involved in the dephosphorylation of UPF1/SMG2, respectively [105]. SMG1 is a member of the phosphatidylinositol (PI) 3-kinase-related kinase (PIKK) family and was confirmed to be the kinase responsible for UPF1 phosphorylation [102, 105, 106, 150]. The 410 kDa human SMG1 has quite a complex domain structure with N-terminal helical HEAT repeats, followed by the FRAP, ATM, and TRRAP (FAT) domain, a FKBP12-rapamycin-binding (FRB) domain, the catalytic PIKK domain, and the very C-terminal FATC domain [151]. Cryo-EM structures of SMG1 showed a thinner arm formed by the HEAT repeat region and a globular head consisting of the remaining C-terminal domains [151, 152]. SMG8 and SMG9, additional NMD factors and components of the SMG1 complex (SMG1C), interact with the HEAT repeat region, and regulate the kinase activity of SMG1 by inducing conformational changes [113, 152, 153]. More specifically, SMG9 is required for SMG8 interaction with SMG1, which leads to a motion of the SMG1 arm and a concomitant conformational change of the head region. This conformational change has been shown to repress kinase activity [113, 152]. Although it was previously observed that the C-terminal domain of SMG1 can interact with UPF1 and UPF2, recent structural and biochemical studies refined this observation. It has been shown that UPF1 interacts with the PIKK domain of SMG1 via its helicase domain, whereas UPF2 binds the FRB domain via its MIF4G-3 domain [131, 151]. The concurrent interaction of SMG1 with a UPF2-UPF3b dimer is possible, pointing to different interaction sites within the MIF4G-3 domain of UPF2 for both interaction partners [131]. UPF2 binding to the FRB domain of SMG1 is believed to modulate and positively stimulate the kinase activity [3, 74]. Although UPF2 is also phosphorylated preferentially at S1046 by SMG1 in vitro, the functional relevance of this phosphorylation site is unclear as it is not essential for NMD [131].
Phosphorylated UPF1 recruits decay inducing factors
SQ and TQ motifs in the extended and unstructured N- and C-terminus of UPF1 are the preferred motifs for phosphorylation by SMG1 [105, 106, 154]. Even though phosphorylation was also reported for yeast Upf1, the mechanism and the responsible kinase are different. Yeast Upf1 lacks most of the clustered SQ and TQ motifs in the C-terminus and no ortholog of SMG1 has been found [155, 156]. The phosphorylation sites in mammalian UPF1 act as recruitment platforms for the remaining core NMD factors, SMG5, SMG6, and SMG7. The three proteins share one common domain feature, a 14-3-3-like domain which folds similar to 14-3-3 proteins and is able to interact with phosphorylated peptides [157]. SMG5 and SMG7 interact with their N-terminal 14-3-3-like domains in a perpendicular back-to-back orientation in order to form a heterodimer. This heterodimer exhibits an uncommon arrangement compared to the normal head-to-head interaction found in most 14-3-3 dimers [158–160]. The 14-3-3-like domain of SMG7 is mostly responsible for the phosphorylation-dependent interaction between phosphorylated amino acids (e.g. S1096) in the C-terminus of UPF1 and the heterodimer SMG5-SMG7 [154, 157, 159, 161]. The 14-3-3-like domain of SMG5, which by itself is not able to interact with UPF1, is postulated to provide additional binding strength and specificity [159, 161].
Initiation of exonucleolytic degradation
Early work showed that tethering of full length SMG7 or the C-terminal proline-rich (PC) region to a reporter mRNA induces mRNA degradation in a position-independent and XRN1-/DCP2-dependent manner [162]. Recently, the direct interaction of the PC region of SMG7 with POP2, the catalytic subunit of the CCR4-NOT deadenylase complex has been shown [163]. Therefore, SMG7 recruitment to phospho-UPF1 induces deadenylation followed by DCP2-mediated decapping and XRN1-catalyzed degradation of the mRNA in the 5′-3′ direction [163]. Early reports showed that UPF1 can associate with decapping proteins like DCP2, the catalytic subunit of the decapping complex. However, it was unclear if this interaction is direct or mediated by another factor [164–167]. Recent studies have identified the proline-rich nuclear receptor coregulatory protein 2 (PNRC2) as an additional NMD factor. PNRC2 interacts with UPF1 and the decapping complex component DCP1, thereby providing a link for deadenylation-independent decapping during NMD [168, 169]. Additionally, PNRC2 was reported to form a functional complex with SMG5, which is devoid of SMG6 or SMG7 and initiates NMD in a UPF1-dependent manner when tethered to a reporter mRNA [170]. Recent data, however, contradicts these observations, as no interaction between PNRC2 and SMG5 could be detected and SMG5-mediated degradation was reported to be strictly SMG7-dependent. Therefore, the existence and contribution of a PNRC2-SMG5 complex to NMD remains unclear [159].
Dephosphorylation of UPF1 is initiated by decay factors
NMD is impaired under conditions where UPF1 accumulates in the hyper- or hypo-phosphorylated form, suggesting that a cycle of UPF1 phosphorylation and dephosphorylation is essential for NMD activity [104–106, 150, 161]. Protein phosphatase 2A (PP2A) was identified as the specific phosphatase essential for the dephosphorylation of UPF1. PP2A associates with the SMG5-SMG7 heterodimer via a direct interaction with SMG5 [104, 171]. SMG5 contains a C-terminal PilT N-terminus (PIN) domain, which is potentially involved in the interaction with PP2A. Deletion of the very C-terminal amino acids or the replacement of a conserved aspartate at position 860 in this domain increased phosphorylation of UPF1 [104]. PIN domains are commonly found in proteins exhibiting endonuclease activity. However, the catalytic triad normally consisting of three aspartate residues is mutated in the SMG5 PIN domain and no endocleavage activity was observed in vivo or in vitro [172–174]. Interestingly, D860 is the one remaining aspartate residue in the active site of SMG5, which was implicated in the regulation of UPF1 phosphorylation status [104]. It is worth mentioning that SMG6 associates with the PP2A complex as well, suggesting that, in line with initial observations in C. elegans, all three SMG5-7 proteins mediate UPF1 dephosphorylation by recruiting phosphatases [175].
Endonucleolytic cleavage is executed by SMG6
Studies to elucidate the degradation pathway of PTC-containing mRNA in D. melanogaster S2 cells showed that the knockdown of exonucleolytic machineries catalyzing deadenylation (CCR4, CAF1, PAN2, PAN3), decapping (DCP1, DCP2, LSM1), 3′-5′ (CSL4, RRP4, RRP6 and SKI2), and 5′-3′ degradation (XRN1, RAT1) could not stabilize reporter mRNA levels [176]. However, evidence for PTC-dependent endonucleolytic cleavage was found due to the accumulation of 3′ and 5′ fragments upon depletion of the major 5′-3′ exonuclease XRN1 and components of the 3′-5′ degrading exosome complex, respectively [176]. In metazoans, SMG6 was identified as the endonuclease responsible for cleavage of the NMD targets in the vicinity of the PTC [176–178]. SMG6 contains a C-terminal PIN domain similar to SMG5. In contrast to SMG5, however, all catalytically important residues are present in the active site and the SMG6 PIN domain exhibits endonucleolytic activity in vitro [174]. Mutations of any of the catalytic aspartate residues, which are required to coordinate divalent metal ions for the nucleophilic attack of H2O on the phosphodiester bond of the RNA, renders the protein inactive and abolishes endonucleolytic degradation of NMD targets [55, 145, 174, 177–179]. Like SMG5 and SMG7, SMG6 contains a 14-3-3-like domain, which is located centrally in the protein [157]. Interestingly, the SMG6 14-3-3-like domain was found to be monomeric as it neither forms homodimers nor heterodimers with either SMG5 or SMG7 14-3-3-like domains [154]. This domain was also suggested to bind phosphorylated UPF1 and biochemical analysis showed that mutation of the residues in the phosphopeptide binding pocket abolished the interaction with UPF1 [161]. Similarly, alanine exchange of T28 in the N-terminus of UPF1 greatly reduced the interaction with SMG6, suggesting that the 14-3-3-like domain of SMG6 interacts with the phosphorylated N-terminus of UPF1 [161]. The phospho-dependent interaction of phosphorylated UPF1 with SMG5-SMG7 was confirmed by recently reported in vitro experiments [154]. However, no interaction of the isolated 14-3-3-like domain of SMG6 with hyperphosphorylated UPF1 was observed [154]. This is in line with recent data showing that phosphorylated UPF1 preferentially occupies the 3′ UTR of NMD targets in a complex with SMG5 and SMG7, but not SMG6 [180]. However, the unstructured region preceding the 14-3-3-like domain of SMG6 was observed to bind UPF1 in a phospho-independent manner in vitro [154]. This observation was supported by functional studies of SMG6 tethering and UPF1 complementation assays performed in another recent publication [179]. Additionally, two EBMs were characterized in the very N-terminus of SMG6, which, similarly to UPF3b, mediate the interaction with the EJC [145]. Despite initial observation that these motifs are crucial for NMD, recent data suggests that they are dispensable for endocleavage [55, 145]. Taken together, the exact mechanisms by which SMG6 is recruited to a target mRNA remain elusive, although at least three potentially redundant or inter-dependent mechanisms exist: (1) recruitment to the EJC via the EBMs, (2) interaction with various regions of UPF1 in a phospho-independent manner, (3) phospho-dependent binding of the 14-3-3-like domain to the N-terminus of UPF1.
Interplay between exo- and endonucleolytic decay during NMD
Recent high-throughput sequencing experiments showed that SMG6-mediated endocleavage is the preferred NMD degradation pathway compared to SMG7-mediated degradation [181, 182]. However, other studies showed that the knockdown of either SMG6 or SMG7 alone is not sufficient to achieve NMD inhibition. Consequently, both proteins need to be depleted, thereby shutting down both degradation pathways, to substantially increase reporter mRNA level [159]. It is still unknown, whether both pathways (initiated by SMG6 or SMG5/7) operate independently, or are somehow connected and regulate each other. As the factors required for the initiation of degradation share the majority of their binding sites (N- and C-terminus of UPF1), it is conceivable that there is a cross-talk between SMG5-7, PNRC2 and/or DCP1/2. Interestingly, in vitro binding studies showed that due to the phosphorylation-independent interaction of SMG6 with UPF1, both SMG6 and SMG5/7 can in principle be accommodated simultaneously on phosphorylated UPF1 [154].
Physiological functions of NMD factors
In lower eukaryotes, such as S. cerevisiae or C. elegans, an NMD factor deficiency has only very mild effects at the organismal level. In contrast, several mammalian NMD factors are essential for normal embryonic development and knockout mice display striking phenotypes [183]. For example, murine Upf1 is essential for embryonic development. Mouse embryos lacking Upf1 are only viable in the pre-implantation period and die due to massive apoptosis soon after uterine implantation [115]. Furthermore, Upf1-deficient blastocysts could be maintained in cell culture for 5 days, but ultimately regressed showing massive apoptosis. Likewise, homozygous targeting of Upf1 in ES cell lines was unsuccessful and Upf1 null cells were never observed [115]. The importance of NMD for normal embryonic development is underscored by data demonstrating that mice deficient of Upf2 die in utero around E3.5-E7.5 [184], whereas Smg1-deficient mice die before embryonic day 12.5 [185]. The use of conditional Upf2 alleles enabled testing of the function of Upf2 in adult animals [184, 186]. Induced loss of Upf2 is highly detrimental to a steady state adult liver, but only a relatively minor phenotype is observed in Upf2 null fetal livers. However, Upf2 null fetal livers do not undergo terminal differentiation, which is incompatible with postnatal life [186]. Furthermore, ablation of Upf2 in the hematopoietic system results in a complete extinction of hematopoietic stem cells and subsequent death of the affected organisms [184]. It was suggested that NMD plays a particularly important role in proliferating cells, because differentiated cells were only mildly affected by the Upf2 knockout [184].
Very recently, knockout mice lacking Smg6 have been reported to show embryonic lethality at the blastocyst stage [187]. However, a floxed Smg6 allele could be used to delete the Smg6 locus in cultured embryonic stem cells (ESC) and to establish Smg6∆/∆ ESCs, which were morphologically indistinguishable from control ESCs and proliferated normally. While Smg6 appears to be dispensable for normal viability and self‐renewal of ESCs, its deletion blocks ESC differentiation in vitro and in vivo in a c-Myc-dependent manner. Although it has been previously suggested that the knockdown of NMD factors compromises the survivability of mouse ESCs, the NMD factors Smg1, Smg5, Upf1, and Upf2 could be knocked down successfully using shRNA vectors [187]. However, these ESCs showed a differentiation defect similar to that observed for Smg6, indicating that NMD plays a general role during ESC differentiation.
UPF1, SMG1, and SMG6 have reported functions beyond NMD and it is difficult to disentangle to what extent the dramatic effects of the genetic knockout can be attributed to the inhibition of NMD [188, 189]. However, non-overlapping moonlighting functions of these factors argue that the majority of the observed phenotypes indeed reflect their role in NMD itself. Furthermore, it was shown that SMG1- and UPF2-deficient cells displayed NMD-specific changes in their transcriptome and many potential NMD targets appear to be upregulated [184, 185]. In summary, according to the prevailing view in the field, the regulation of gene expression by NMD has an important role during normal embryonic development and cellular viability.
Human disorders associated with NMD factor mutations
Upf3b
Mutations in the UPF3B gene were described in males with mild to severe X-linked mental retardation [190]. Until now, ten families carrying UPF3B mutations have been analyzed and seven truncation mutations and three missense mutations in the UPF3B gene have been identified [190–194]. A broad range of clinical symptoms, including autism, schizophrenia, and facial dysmorphism have been observed in patients with UPF3B mutations. The different degree of mental retardation and of the other phenotypes of patients with UPF3B mutations depend on the amount of UPF3a, which is upregulated in response to UPF3b deficiency [190]. However, the clinical manifestations of UPF3B mutations demonstrate that the degree of UPF3a up regulation is insufficient to completely compensate for the lack of UPF3b. Hence, UPF3a only partially rescues NMD in the absence of functional UPF3b and is a potential modifier of the clinical phenotype of UPF3B patients [195, 196].
Upf2
The idea that misregulation of NMD predisposes for neuro-developmental disorders is supported by an association with heterozygous deletions of a genomic region that include UPF2 [197]. In addition, a de novo missense mutation in UPF2 has been identified in a patient with schizophrenia [198].
Upf1
Recently, somatic mutations in the UPF1 gene have been described in pancreatic adenosquamous carcinoma (ASC) tumors [199]. All mutations were somatic in origin and not detected in normal pancreatic tissues from the same patients. ASC-specific point mutations clustered in two regions of the UPF1 gene and many seem to trigger alternative splicing of the UPF1 pre- mRNA, leading to the expression of truncated UPF1 proteins. UPF1 mutations appear to represent a signature of many pancreatic ASC tumors, since no UPF1 mutations were found in non-ASC pancreatic tumors and lung squamous cell carcinomas. Furthermore, no mutations in other NMD genes (UPF2, UPF3A and UPF3B) were detected in ASC tumors. Although the molecular effects of UPF1 mutations remain to be elucidated, it is very likely that they alter the efficiency of NMD. This is supported by the observation that a PTC-containing splice variant of p53, representing an endogenous NMD substrate, was only detectably expressed in tumor tissue.
Y14
TAR syndrome (thrombocytopenia with absent radius) is a rare genetic disorder with 55 cases being described until today [200]. Patients with TAR syndrome have low numbers of megakaryocytes, leading to a dramatically reduced platelet count (hypomegakaryocytic thrombocytopenia). In all cases the radius bone in the forearm is absent, but the skeletal abnormalities show a high degree of variation, from absence of radii to virtual absence of upper limbs. The lower limbs, the gastrointestinal as well as the cardiovascular systems may also show defects [201]. In most cases, TAR syndrome manifests in patients having one allele with a reduced expression of the RBM8A gene (encoding the Y14 component of the EJC) in combination with a heterozygous small deletion on chromosome 1q21.1 containing the RBM8A gene [200]. The reduced expression of the RBM8A gene is caused by low-frequency SNPs either in its 5′ UTR, or in the first intron. Two patients with TAR syndrome did not have the chromosome 1q21.1 deletion. In these cases the 5′UTR SNP was found in combination with a 4-bp frameshift insertion at the start of the fourth exon of the RBM8A gene and a nonsense mutation in the last exon of RBM8A. Hence, the compound inheritance of a null allele (deletion, frameshift-, or nonsense mutation) and low-frequency noncoding SNPs in RBM8A is the genetic cause of TAR syndrome [200]. Why certain tissues require higher levels of Y14 and are therefore specifically affected by its reduced expression is currently unclear. While these patients likely have reduced amounts of EJCs due to the insufficiency of the Y14 protein, the effects on NMD will require further analysis.
eIF4A3
In 1992 Richieri-Costa and Pereira described a new autosomal-recessive syndrome characterized by mandibular median cleft associated with other craniofacial anomalies and severe limb defects [202]. This syndrome, now referred to as Richieri-Costa-Pereira Syndrome (RCPS), almost exclusively affects families from Brazil. Different non-coding expansions in the 5′UTR of the mRNA encoding the EJC core component eIF4A3 have been found in patients [203]. The expansions are located in a region with several repeat motifs. Homozygosity as well as compound heterozygosity of different repeat expansions and a missense mutation have been described. Notably, the presence of the expansions does not seem to influence processing of the pre-mRNA, but rather reduces the abundance of the eIF4A3 transcript by 30–40 % in patient cells compared to control cells [203]. A similar reduction of the expression of eIF4A3 by injecting specific morpholinos in zebrafish embryos led to developmental defects in several craniofacial cartilage and bone structures [203]. Nonetheless, it is currently unclear how the partial loss-of-function of eIF4A3 leads to the pleiotropic phenotype of RCPS.
Effect of NMD on human disease
Hereditary disorder
Many inherited disorders and several cancers have been suggested to be caused by nonsense or frameshift mutations [204]. More than 2400 genetic disorders have at least one causative nonsense allele [205]. A recent meta-analysis showed that nonsense mutations account for approximately 11 % of all described gene lesions causing human inherited disease. Furthermore, they represent approximately 20 % of disease-associated single-basepair substitutions [206]. For many diseases caused by nonsense mutations, NMD acts as a modifier of the clinical phenotype and eliminates mutated transcripts, which encode C-terminally truncated proteins [207]. Such truncated proteins may have dominant-negative effects and, therefore, may be deleterious to the cell. This observation has been made for β-thalassemia, which is caused in many cases by nonsense mutations in the first or second exon of the β-globin gene. Due to the activity of NMD only very low levels of mutant β-globin mRNA are present in red blood cells of heterozygous individuals, which are clinically usually asymptomatic. This can be explained by sufficient amounts of β-globin that are produced from the normal allele. In contrast, nonsense mutations in the last exon of β-globin escape NMD and high levels of mutant mRNA are translated into truncated β globin. The proteolytic system of the red blood cells fails to degrade these truncated β chains, causing a clinical phenotype in the heterozygote called thalassemia intermedia [208]. A similar effect has been documented in many other diseases [207, 209].
In recent years it has become clear that the protective function of NMD is only one side of the coin. Indeed, NMD can worsen the clinical manifestation of genetic disorders, in which the reduced expression of partially functional proteins leads to haploinsufficiency. For example, nonsense mutations in the dystrophin gene, which activate NMD and preclude the synthesis of truncated dystrophin protein, cause a severe form of Duchenne muscular dystrophy (DMD). In contrast, some mutations in the dystrophin gene escape NMD and produce partially functional C-terminally truncated dystrophin protein. These mutations are usually associated with a clinically less severe disease, termed Becker muscular dystrophy [210].
Treatment of genetic disorders by targeting NMD
A large number of patients are affected by nonsense mutations, but only a limited amount of therapeutic treatments are available. In many cases, disease-causing nonsense mutations exert two effects, namely accelerated mRNA degradation due to NMD and translation of a truncated ORF. Hence, potential treatment strategies would require translational read-through by nonsense suppression, inhibition of NMD, or both. Since it may not be necessary to restore normal gene expression levels in order to eliminate the disease [211], translational read-through at stop codons without inhibiting NMD is currently assumed to be the favorable approach. Notably, a drug that is able to induce translational read-though at a given nonsense mutation may potentially be used for the treatment of other nonsense mutations and could, therefore, be used to treat many different diseases caused by nonsense mutations [205].
Currently only a few approaches have been specifically tested to treat diseases caused by nonsense mutations. Originally, aminoglycosides were used due to their ability to cause read-through of translation termination codons by recognition of a near-cognate tRNA and misincorporation of an amino acid [212]. After initial tests of aminoglycosides in cystic fibrosis (CF) cell culture models [213], clinical trials with gentamycin were carried out in patients suffering from CF or DMD. These trials were in principle successful and confirmed that the administration of aminoglycosides in vivo is capable of restoring protein function. However, the high doses of gentamycin required for a prolonged effect may have adverse effects, and therefore limits the application as treatment for patients [214].
An alternative for aminoglycosides is the drug Ataluren, a small-molecule compound formerly known as PTC124 [205]. Ataluren has been reported to selectively promote the read-through of premature termination codons, while not affecting normal stop codons [215]. However, the molecular mechanism of the compound has recently been challenged [216, 217] and its future prospects will largely depend on its efficacy in currently ongoing clinical trials.
Concluding remarks
NMD plays a pivotal role during mammalian gene expression. It controls not only the fidelity of mRNA expression, but also regulates the expression of many genes at a post-transcriptional level. NMD employs a sophisticated machinery of conserved factors, which act at different steps (nuclear and cytoplasmic) of gene expression. Factors involved in NMD are essential for embryonic development in mammals and regulate the symptoms of inherited and acquired genetic diseases. However, a large portion of the NMD pathway and its specific activation remains elusive and needs to be addressed in future studies.
References
Brogna S, Wen J (2009) Nonsense-mediated mRNA decay (NMD) mechanisms. Nat Struct Mol Biol 16:107–113. doi:10.1038/nsmb.1550
Fatscher T, Boehm V, Weiche B, Gehring NH (2014) The interaction of cytoplasmic poly(A)-binding protein with eukaryotic initiation factor 4G suppresses nonsense-mediated mRNA decay. RNA 20:1579–1592. doi:10.1261/rna.044933.114
Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE (2008) Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J 27:736–747. doi:10.1038/emboj.2008.17
Joncourt R, Eberle AB, Rufener SC, Muhlemann O (2014) Eukaryotic initiation factor 4G suppresses nonsense-mediated mRNA decay by two genetically separable mechanisms. PLoS One 9:e104391. doi:10.1371/journal.pone.0104391
Singh G, Rebbapragada I, Lykke-Andersen J (2008) A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol 6:e111. doi:10.1371/journal.pbio.0060111
Janzen DM, Geballe AP (2004) The effect of eukaryotic release factor depletion on translation termination in human cell lines. Nucleic Acids Res 32:4491–4502. doi:10.1093/nar/gkh791
Chauvin C, Salhi S, Le Goff C, Viranaicken W, Diop D, Jean-Jean O (2005) Involvement of human release factors eRF3a and eRF3b in translation termination and regulation of the termination complex formation. Mol Cell Biol 25:5801–5811. doi:10.1128/MCB.25.14.5801-5811.2005
Hoshino S, Imai M, Mizutani M, Kikuchi Y, Hanaoka F, Ui M, Katada T (1998) Molecular cloning of a novel member of the eukaryotic polypeptide chain-releasing factors (eRF) - Its identification as eRF3 interacting with eRF1. J Biol Chem 273:22254–22259. doi:10.1074/jbc.273.35.22254
Hoshino S, Miyazawa H, Enomoto T, Hanaoka F, Kikuchi Y, Kikuchi A, Ui M (1989) A human homolog of the yeast Gst1-gene codes for a Gtp-binding protein and is expressed in a proliferation-dependent manner in mammalian-cells. EMBO J 8:3807–3814
Chavatte L, Frolova L, Kisselev L, Favre A (2001) The polypeptide chain release factor eRF1 specifically contacts the s(4)UGA stop codon located in the A site of eukaryotic ribosomes. Eur J Biochem 268:2896–2904
Frolova L, Le Goff X, Rasmussen HH, Cheperegin S, Drugeon G, Kress M, Arman I, Haenni AL, Celis JE, Philippe M et al (1994) A highly conserved eukaryotic protein family possessing properties of polypeptide chain release factor. Nature 372:701–703. doi:10.1038/372701a0
Frolova LY, Tsivkovskii RY, Sivolobova GF, Oparina NY, Serpinsky OI, Blinov VM, Tatkov SI, Kisselev LL (1999) Mutations in the highly conserved GGQ motif of class 1 polypeptide release factors abolish ability of human eRF1 to trigger peptidyl-tRNA hydrolysis. RNA 5:1014–1020. doi:10.1017/s135583829999043x
Song H, Mugnier P, Das AK, Webb HM, Evans DR, Tuite MF, Hemmings BA, Barford D (2000) The crystal structure of human eukaryotic release factor eRF1—mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell 100:311–321
Zhouravleva G, Frolova L, Le Goff X, Le Guellec R, Inge-Vechtomov S, Kisselev L, Philippe M (1995) Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. EMBO J 14:4065–4072
Salas-Marco J, Bedwell DM (2004) GTP hydrolysis by eRF3 facilitates stop codon decoding during eukaryotic translation termination. Mol Cell Biol 24:7769–7778. doi:10.1128/MCB.24.17.7769-7778.2004
Frolova L, LeGoff X, Zhouravleva G, Davydova E, Philippe M, Kisselev L (1996) Eukaryotic polypeptide chain release factor eRF3 is an eRF1- and ribosome-dependent guanosine triphosphatase. RNA 2:334–341
Alkalaeva EZ, Pisarev AV, Frolova LY, Kisselev LL, Pestova TV (2006) In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell 125:1125–1136. doi:10.1016/j.cell.2006.04.035
Pisarev AV, Skabkin MA, Pisareva VP, Skabkina OV, Rakotondrafara AM, Hentze MW, Hellen CU, Pestova TV (2010) The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell 37:196–210. doi:10.1016/j.molcel.2009.12.034
Barthelme D, Dinkelaker S, Albers SV, Londei P, Ermler U, Tampe R (2011) Ribosome recycling depends on a mechanistic link between the FeS cluster domain and a conformational switch of the twin-ATPase ABCE1. Proc Natl Acad Sci USA 108:3228–3233. doi:10.1073/pnas.1015953108
Becker T, Franckenberg S, Wickles S, Shoemaker CJ, Anger AM, Armache JP, Sieber H, Ungewickell C, Berninghausen O, Daberkow I, Karcher A, Thomm M, Hopfner KP, Green R, Beckmann R (2012) Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature 482:501–506. doi:10.1038/nature10829
Adam SA, Nakagawa T, Swanson MS, Woodruff TK, Dreyfuss G (1986) mRNA polyadenylate-binding protein: gene isolation and sequencing and identification of a ribonucleoprotein consensus sequence. Mol Cell Biol 6:2932–2943
Sachs AB, Bond MW, Kornberg RD (1986) A single gene from yeast for both nuclear and cytoplasmic polyadenylate-binding proteins—domain-structure and expression. Cell 45:827–835. doi:10.1016/0092-8674(86)90557-X
Deo RC, Bonanno JB, Sonenberg N, Burley SK (1999) Recognition of polyadenylate RNA by the poly(A)-binding protein. Cell 98:835–845
Cosson B, Berkova N, Couturier A, Chabelskaya S, Philippe M, Zhouravleva G (2002) Poly(A)-binding protein and eRF3 are associated in vivo in human and Xenopus cells. Biol Cell 94:205–216. doi:10.1016/S0248-4900(02)01194-2
Kozlov G, Trempe JF, Khaleghpour K, Kahvejian A, Ekiel I, Gehring K (2001) Structure and function of the C-terminal PABC domain of human poly(A)-binding protein. Proc Natl Acad Sci USA 98:4409–4413. doi:10.1073/pnas.071024998
Si Hoshino, Imai M, Kobayashi T, Uchida N, Katada T (1999) The eukaryotic polypeptide chain releasing factor (eRF3/GSPT) carrying the translation termination signal to the 3′-Poly(A) tail of mRNA. Direct association of eRF3/GSPT with polyadenylate-binding protein. J Biol Chem 274:16677–16680. doi:10.1074/jbc.274.24.16677
Uchida N, Hoshino S, Imataka H, Sonenberg N, Katada T (2002) A novel role of the mammalian GSPT/eRF3 associating with poly(A)-binding protein in Cap/Poly(A)-dependent translation. J Biol Chem 277:50286–50292. doi:10.1074/jbc.M203029200
Kozlov G, Gehring K (2010) Molecular basis of eRF3 recognition by the MLLE domain of poly(A)-binding protein. PLoS One 5:e10169. doi:10.1371/journal.pone.0010169
Tarun SZ Jr, Sachs AB (1996) Association of the yeast poly(A) tail binding protein with translation initiation factor eIF-4G. EMBO J 15:7168–7177
Safaee N, Kozlov G, Noronha AM, Xie J, Wilds CJ, Gehring K (2012) Interdomain allostery promotes assembly of the poly(A) mRNA complex with PABP and eIF4G. Mol Cell 48:375–386. doi:10.1016/j.molcel.2012.09.001
Marcotrigiano J, Gingras AC, Sonenberg N, Burley SK (1997) Cocrystal structure of the messenger RNA 5′ cap-binding protein (eIF4E) bound to 7-methyl-GDP. Cell 89:951–961
Amrani N, Ghosh S, Mangus DA, Jacobson A (2008) Translation factors promote the formation of two states of the closed-loop mRNP. Nature 453:1276–1280. doi:10.1038/nature06974
Kahvejian A, Svitkin YV, Sukarieh R, M’Boutchou MN, Sonenberg N (2005) Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev 19:104–113. doi:10.1101/gad.1262905
Wells SE, Hillner PE, Vale RD, Sachs AB (1998) Circularization of mRNA by eukaryotic translation initiation factors. Mol Cell 2:135–140
Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23:198–199. doi:10.1016/S0968-0004(98)01208-0
Thermann R, Neu-Yilik G, Deters A, Frede U, Wehr K, Hagemeier C, Hentze MW, Kulozik AE (1998) Binary specification of nonsense codons by splicing and cytoplasmic translation. EMBO J 17:3484–3494. doi:10.1093/emboj/17.12.3484
Zhang J, Sun X, Qian Y, LaDuca JP (1998) At least one intron is required for the nonsense-mediated decay of triosephosphate isomerase mRNA: a possible link between nuclear splicing and cytoplasmic translation. Mol Cell Biol 18:5272–5283
Zhang J, Sun XL, Qian YM, Maquat LE (1998) Intron function in the nonsense-mediated decay of beta-globin mRNA: indications that pre-mRNA splicing in the nucleus can influence mRNA translation in the cytoplasm. RNA 4:801–815. doi:10.1017/S1355838298971849
Wang J, Gudikote JP, Olivas OR, Wilkinson MF (2002) Boundary-independent polar nonsense-mediated decay. EMBO Rep 3:274–279. doi:10.1093/embo-reports/kvf036
Brocke KS, Neu-Yilik G, Gehring NH, Hentze MW, Kulozik AE (2002) The human intronless melanocortin 4-receptor gene is NMD insensitive. Hum Mol Genet 11:331–335. doi:10.1093/hmg/11.3.331
Maquat LE, Li XJ (2001) Mammalian heat shock p70 and histone H4 transcripts, which derive from naturally intronless genes, are immune to nonsense-mediated decay. RNA 7:445–456. doi:10.1017/S1355838201002229
Neu-Yilik G, Gehring NH, Thermann R, Frede U, Hentze MW, Kulozik AE (2001) Splicing and 3′ end formation in the definition of nonsense-mediated decay-competent human beta-globin mRNPs. EMBO J 20:532–540. doi:10.1093/emboj/20.3.532
Dostie J, Dreyfuss G (2002) Translation is required to remove Y14 from mRNAs in the cytoplasm. Curr Biol 12:1060–1067
Lejeune F, Ishigaki Y, Li X, Maquat LE (2002) The exon junction complex is detected on CBP80-bound but not eIF4E-bound mRNA in mammalian cells: dynamics of mRNP remodeling. EMBO J 21:3536–3545. doi:10.1093/emboj/cdf345
Le Hir H, Izaurralde E, Maquat LE, Moore MJ (2000) The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon–exon junctions. EMBO J 19:6860–6869. doi:10.1093/emboj/19.24.6860
Steckelberg A-LL, Boehm V, Gromadzka AM, Gehring NH (2012) CWC22 connects pre-mRNA splicing and exon junction complex assembly. Cell Rep 2:454–461. doi:10.1016/j.celrep.2012.08.017
Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW (2009) Disassembly of exon junction complexes by PYM. Cell 137:536–548. doi:10.1016/j.cell.2009.02.042
Ballut L, Marchadier B, Baguet A, Tomasetto C, Seraphin B, Le Hir H (2005) The exon junction core complex is locked onto RNA by inhibition of eIF4AIII ATPase activity. Nat Struct Mol Biol 12:861–869. doi:10.1038/nsmb990
Bono F, Ebert J, Lorentzen E, Conti E (2006) The crystal structure of the exon junction complex reveals how it maintains a stable grip on mRNA. Cell 126:713–725. doi:10.1016/j.cell.2006.08.006
Andersen CB, Ballut L, Johansen JS, Chamieh H, Nielsen KH, Oliveira CL, Pedersen JS, Seraphin B, Le Hir H, Andersen GR (2006) Structure of the exon junction core complex with a trapped DEAD-box ATPase bound to RNA. Science 313:1968–1972. doi:10.1126/science.1131981
Kim VN, Kataoka N, Dreyfuss G (2001) Role of the nonsense-mediated decay factor hUpf3 in the splicing-dependent exon–exon junction complex. Science 293:1832–1836. doi:10.1126/science.1062829
Le Hir H, Gatfield D, Izaurralde E, Moore MJ (2001) The exon–exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J 20:4987–4997. doi:10.1093/emboj/20.17.4987
Gehring NH, Neu-Yilik G, Schell T, Hentze MW, Kulozik AE (2003) Y14 and hUpf3b form an NMD-activating complex. Mol Cell 11:939–949. doi:10.1016/S1097-2765(03)00142-4
Buhler M, Steiner S, Mohn F, Paillusson A, Muhlemann O (2006) EJC-independent degradation of nonsense immunoglobulin-mu mRNA depends on 3′ UTR length. Nat Struct Mol Biol 13:462–464. doi:10.1038/nsmb1081
Boehm V, Haberman N, Ottens F, Ule J, Gehring NH (2014) 3′ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep 9:555–568. doi:10.1016/j.celrep.2014.09.012
Toma KG, Rebbapragada I, Durand S, Lykke-Andersen J (2015) Identification of elements in human long 3′ UTRs that inhibit nonsense-mediated decay. RNA 21:887–897. doi:10.1261/rna.048637.114
Hurt JA, Robertson AD, Burge CB (2013) Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res 23:1636–1650. doi:10.1101/gr.157354.113
Kurosaki T, Maquat LE (2013) Rules that govern UPF1 binding to mRNA 3′ UTRs. Proc Natl Acad Sci USA 110:3357–3362. doi:10.1073/pnas.1219908110
Zund D, Gruber AR, Zavolan M, Muhlemann O (2013) Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol 20:936–943. doi:10.1038/nsmb.2635
Amrani N, Ganesan R, Kervestin S, Mangus DA, Ghosh S, Jacobson A (2004) A faux 3′-UTR promotes aberrant termination and triggers nonsense-mediated mRNA decay. Nature 432:112–118. doi:10.1038/nature03060
Muhlrad D, Parker R (1999) Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5:1299–1307
Ishigaki Y, Li X, Serin G, Maquat LE (2001) Evidence for a pioneer round of mRNA translation: mRNAs subject to nonsense-mediated decay in mammalian cells are bound by CBP80 and CBP20. Cell 106:607–617
Durand S, Lykke-Andersen J (2013) Nonsense-mediated mRNA decay occurs during eIF4F-dependent translation in human cells. Nat Struct Mol Biol 20:702–709. doi:10.1038/nsmb.2575
Rufener SC, Muhlemann O (2013) eIF4E-bound mRNPs are substrates for nonsense-mediated mRNA decay in mammalian cells. Nat Struct Mol Biol 20:710–717. doi:10.1038/nsmb.2576
Belgrader P, Cheng J, Maquat LE (1993) Evidence to implicate translation by ribosomes in the mechanism by which nonsense codons reduce the nuclear level of human triosephosphate isomerase mRNA. Proc Natl Acad Sci USA 90:482–486
Carter MS, Doskow J, Morris P, Li SL, Nhim RP, Sandstedt S, Wilkinson MF (1995) A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in-vivo is reversed by protein-synthesis inhibitors in-vitro. J Biol Chem 270:28995–29003
Gradi A, Svitkin YV, Imataka H, Sonenberg N (1998) Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc Natl Acad Sci USA 95:11089–11094
Kuyumcu-Martinez NM, Joachims M, Lloyd RE (2002) Efficient cleavage of ribosome-associated poly(A)-binding protein by enterovirus 3C protease. J Virol 76:2062–2074
Zhang J, Maquat LE (1997) Evidence that translation reinitiation abrogates nonsense-mediated mRNA decay in mammalian cells. EMBO J 16:826–833. doi:10.1093/emboj/16.4.826
Neu-Yilik G, Amthor B, Gehring NH, Bahri S, Paidassi H, Hentze MW, Kulozik AE (2011) Mechanism of escape from nonsense-mediated mRNA decay of human beta-globin transcripts with nonsense mutations in the first exon. RNA 17:843–854. doi:10.1261/rna.2401811
Baserga SJ, Benz EJ Jr (1988) Nonsense mutations in the human beta-globin gene affect mRNA metabolism. Proc Natl Acad Sci USA 85:2056–2060
Pergolizzi R, Spritz RA, Spence S, Goossens M, Kan YW, Bank A (1981) Two cloned beta thalassemia genes are associated with amber mutations at codon 39. Nucleic Acids Res 9:7065–7072
Peixeiro I, Inacio A, Barbosa C, Silva AL, Liebhaber SA, Romao L (2012) Interaction of PABPC1 with the translation initiation complex is critical to the NMD resistance of AUG-proximal nonsense mutations. Nucleic Acids Res 40:1160–1173. doi:10.1093/nar/gkr820
Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S (2006) Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20:355–367. doi:10.1101/gad.1389006
Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Muhlemann O (2011) Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 17:2108–2118. doi:10.1261/rna.030247.111
Eberle AB, Stalder L, Mathys H, Orozco RZ, Muhlemann O (2008) Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol 6:e92. doi:10.1371/journal.pbio.0060092
Lee BJ, Worland PJ, Davis JN, Stadtman TC, Hatfield DL (1989) Identification of a selenocysteyl-tRNA(Ser) in mammalian cells that recognizes the nonsense codon, UGA. J Biol Chem 264:9724–9727
Berry MJ, Banu L, Harney JW, Larsen PR (1993) Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J 12:3315–3322
Bermano G, Nicol F, Dyer JA, Sunde RA, Beckett GJ, Arthur JR, Hesketh JE (1995) Tissue-specific regulation of selenoenzyme gene expression during selenium deficiency in rats. Biochem J 311(Pt 2):425–430
Hadley KB, Sunde RA (2001) Selenium regulation of thioredoxin reductase activity and mRNA levels in rat liver. J Nutr Biochem 12:693–702
Seyedali A, Berry MJ (2014) Nonsense-mediated decay factors are involved in the regulation of selenoprotein mRNA levels during selenium deficiency. RNA 20:1248–1256. doi:10.1261/rna.043463.113
Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36:1073–1078. doi:10.1038/ng1429
Stockklausner C, Breit S, Neu-Yilik G, Echner N, Hentze MW, Kulozik AE, Gehring NH (2006) The uORF-containing thrombopoietin mRNA escapes nonsense-mediated decay (NMD). Nucleic Acids Res 34:2355–2363. doi:10.1093/nar/gkl277
Brett D, Hanke J, Lehmann G, Haase S, Delbruck S, Krueger S, Reich J, Bork P (2000) EST comparison indicates 38% of human mRNAs contain possible alternative splice forms. FEBS Lett 474:83–86
Lewis BP, Green RE, Brenner SE (2003) Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci USA 100:189–192. doi:10.1073/pnas.0136770100
Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J (2001) SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20:1785–1796. doi:10.1093/emboj/20.7.1785
Tani H, Torimura M, Akimitsu N (2013) The RNA degradation pathway regulates the function of GAS5 a non-coding RNA in mammalian cells. PLoS One 8:e55684. doi:10.1371/journal.pone.0055684
Chew GL, Pauli A, Rinn JL, Regev A, Schier AF, Valen E (2013) Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development 140:2828–2834. doi:10.1242/dev.098343
Culbertson MR, Underbrink KM, Fink GR (1980) Frameshift suppression Saccharomyces cerevisiae. II. Genetic properties of group II suppressors. Genetics 95:833–853
Cui Y, Hagan KW, Zhang S, Peltz SW (1995) Identification and characterization of genes that are required for the accelerated degradation of mRNAs containing a premature translational termination codon. Genes Dev 9:423–436
Leeds P, Peltz SW, Jacobson A, Culbertson MR (1991) The product of the yeast UPF1 gene is required for rapid turnover of mRNAs containing a premature translational termination codon. Genes Dev 5:2303–2314
Leeds P, Wood JM, Lee BS, Culbertson MR (1992) Gene products that promote mRNA turnover in Saccharomyces cerevisiae. Mol Cell Biol 12:2165–2177
Behm-Ansmant I, Kashima I, Rehwinkel J, Sauliere J, Wittkopp N, Izaurralde E (2007) mRNA quality control: an ancient machinery recognizes and degrades mRNAs with nonsense codons. FEBS Lett 581:2845–2853. doi:10.1016/j.febslet.2007.05.027
Culbertson MR, Leeds PF (2003) Looking at mRNA decay pathways through the window of molecular evolution. Curr Opin Genet Dev 13:207–214
Chen YH, Su LH, Sun CH (2008) Incomplete nonsense-mediated mRNA decay in Giardia lamblia. Int J Parasitol 38:1305–1317. doi:10.1016/j.ijpara.2008.02.006
Kadlec J, Guilligay D, Ravelli RB, Cusack S (2006) Crystal structure of the UPF2-interacting domain of nonsense-mediated mRNA decay factor UPF1. RNA 12:1817–1824. doi:10.1261/rna.177606
Cali BM, Kuchma SL, Latham J, Anderson P (1999) smg-7 is required for mRNA surveillance in Caenorhabditis elegans. Genetics 151:605–616
Hodgkin J, Papp A, Pulak R, Ambros V, Anderson P (1989) A new kind of informational suppression in the nematode Caenorhabditis elegans. Genetics 123:301–313
Pulak R, Anderson P (1993) mRNA surveillance by the Caenorhabditis elegans smg genes. Genes Dev 7:1885–1897
Applequist SE, Selg M, Raman C, Jack HM (1997) Cloning and characterization of HUPF1, a human homolog of the Saccharomyces cerevisiae nonsense mRNA-reducing UPF1 protein. Nucleic Acids Res 25:814–821
Aronoff R, Baran R, Hodgkin J (2001) Molecular identification of smg-4, required for mRNA surveillance in C. elegans. Gene 268:153–164
Denning G, Jamieson L, Maquat LE, Thompson EA, Fields AP (2001) Cloning of a novel phosphatidylinositol kinase-related kinase: characterization of the human SMG-1 RNA surveillance protein. J Biol Chem 276:22709–22714. doi:10.1074/jbc.C100144200
Lykke-Andersen J, Shu MD, Steitz JA (2000) Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121–1131
Ohnishi T, Yamashita A, Kashima I, Schell T, Anders KR, Grimson A, Hachiya T, Hentze MW, Anderson P, Ohno S (2003) Phosphorylation of hUPF1 induces formation of mRNA surveillance complexes containing hSMG-5 and hSMG-7. Mol Cell 12:1187–1200
Page MF, Carr B, Anders KR, Grimson A, Anderson P (1999) SMG-2 is a phosphorylated protein required for mRNA surveillance in Caenorhabditis elegans and related to Upf1p of yeast. Mol Cell Biol 19:5943–5951
Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S (2001) Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev 15:2215–2228. doi:10.1101/gad.913001
Casadio A, Longman D, Hug N, Delavaine L, Vallejos Baier R, Alonso CR, Caceres JF (2015) Identification and characterization of novel factors that act in the nonsense-mediated mRNA decay pathway in nematodes, flies and mammals. EMBO Rep 16:71–78. doi:10.15252/embr.201439183
Hug N, Caceres JF (2014) The RNA helicase DHX34 activates NMD by promoting a transition from the surveillance to the decay-inducing complex. Cell Rep 8:1845–1856. doi:10.1016/j.celrep.2014.08.020
Izumi N, Yamashita A, Ohno S (2012) Integrated regulation of PIKK-mediated stress responses by AAA+ proteins RUVBL1 and RUVBL2. Nucleus 3:29–43. doi:10.4161/nucl.18926
Longman D, Hug N, Keith M, Anastasaki C, Patton EE, Grimes G, Caceres JF (2013) DHX34 and NBAS form part of an autoregulatory NMD circuit that regulates endogenous RNA targets in human cells, zebrafish and Caenorhabditis elegans. Nucleic Acids Res 41:8319–8331. doi:10.1093/nar/gkt585
Longman D, Plasterk RH, Johnstone IL, Caceres JF (2007) Mechanistic insights and identification of two novel factors in the C. elegans NMD pathway. Genes Dev 21:1075–1085. doi:10.1101/gad.417707
Gregersen LH, Schueler M, Munschauer M, Mastrobuoni G, Chen W, Kempa S, Dieterich C, Landthaler M (2014) MOV10 Is a 5′ to 3′ RNA helicase contributing to UPF1 mRNA target degradation by translocation along 3′ UTRs. Mol Cell 54:573–585. doi:10.1016/j.molcel.2014.03.017
Yamashita A, Izumi N, Kashima I, Ohnishi T, Saari B, Katsuhata Y, Muramatsu R, Morita T, Iwamatsu A, Hachiya T, Kurata R, Hirano H, Anderson P, Ohno S (2009) SMG-8 and SMG-9, two novel subunits of the SMG-1 complex, regulate remodeling of the mRNA surveillance complex during nonsense-mediated mRNA decay. Genes Dev 23:1091–1105. doi:10.1101/gad.1767209
Riehs-Kearnan N, Gloggnitzer J, Dekrout B, Jonak C, Riha K (2012) Aberrant growth and lethality of Arabidopsis deficient in nonsense-mediated RNA decay factors is caused by autoimmune-like response. Nucleic Acids Res 40:5615–5624. doi:10.1093/nar/gks195
Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC (2001) Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet 10:99–105
Wittkopp N, Huntzinger E, Weiler C, Sauliere J, Schmidt S, Sonawane M, Izaurralde E (2009) Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survival. Mol Cell Biol 29:3517–3528. doi:10.1128/MCB.00177-09
Avery P, Vicente-Crespo M, Francis D, Nashchekina O, Alonso CR, Palacios IM (2011) Drosophila Upf1 and Upf2 loss of function inhibits cell growth and causes animal death in a Upf3-independent manner. RNA 17:624–638. doi:10.1261/rna.2404211
Bhattacharya A, Czaplinski K, Trifillis P, He F, Jacobson A, Peltz SW (2000) Characterization of the biochemical properties of the human Upf1 gene product that is involved in nonsense-mediated mRNA decay. RNA 6:1226–1235
Cheng Z, Muhlrad D, Lim MK, Parker R, Song H (2007) Structural and functional insights into the human Upf1 helicase core. EMBO J 26:253–264. doi:10.1038/sj.emboj.7601464
Fairman-Williams ME, Guenther UP, Jankowsky E (2010) SF1 and SF2 helicases: family matters. Curr Opin Struct Biol 20:313–324. doi:10.1016/j.sbi.2010.03.011
Singleton MR, Dillingham MS, Wigley DB (2007) Structure and mechanism of helicases and nucleic acid translocases. Annu Rev Biochem 76:23–50. doi:10.1146/annurev.biochem.76.052305.115300
Chamieh H, Ballut L, Bonneau F, Le Hir H (2008) NMD factors UPF2 and UPF3 bridge UPF1 to the exon junction complex and stimulate its RNA helicase activity. Nat Struct Mol Biol 15:85–93. doi:10.1038/nsmb1330
Chakrabarti S, Jayachandran U, Bonneau F, Fiorini F, Basquin C, Domcke S, Le Hir H, Conti E (2011) Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol Cell 41:693–703. doi:10.1016/j.molcel.2011.02.010
Weng Y, Czaplinski K, Peltz SW (1996) Genetic and biochemical characterization of mutations in the ATPase and helicase regions of the Upf1 protein. Mol Cell Biol 16:5477–5490
Mendell JT, ap Rhys CM, Dietz HC (2002) Separable roles for rent1/hUpf1 in altered splicing and decay of nonsense transcripts. Science 298:419–422. doi:10.1126/science.1074428
Weng Y, Czaplinski K, Peltz SW (1996) Identification and characterization of mutations in the UPF1 gene that affect nonsense suppression and the formation of the Upf protein complex but not mRNA turnover. Mol Cell Biol 16:5491–5506
Franks TM, Singh G, Lykke-Andersen J (2010) Upf1 ATPase-dependent mRNP disassembly is required for completion of nonsense- mediated mRNA decay. Cell 143:938–950. doi:10.1016/j.cell.2010.11.043
Shigeoka T, Kato S, Kawaichi M, Ishida Y (2012) Evidence that the Upf1-related molecular motor scans the 3′-UTR to ensure mRNA integrity. Nucleic Acids Res 40:6887–6897. doi:10.1093/nar/gks344
Fiorini F, Boudvillain M, Le Hir H (2013) Tight intramolecular regulation of the human Upf1 helicase by its N- and C-terminal domains. Nucleic Acids Res 41:2404–2415. doi:10.1093/nar/gks1320
Ponting CP (2000) Novel eIF4G domain homologues linking mRNA translation with nonsense-mediated mRNA decay. Trends Biochem Sci 25:423–426
Clerici M, Deniaud A, Boehm V, Gehring NH, Schaffitzel C, Cusack S (2014) Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res 42:2673–2686. doi:10.1093/nar/gkt1197
Aravind L, Koonin EV (2000) Eukaryote-specific domains in translation initiation factors: implications for translation regulation and evolution of the translation system. Genome Res 10:1172–1184
Alexandrov A, Colognori D, Shu MD, Steitz JA (2012) Human spliceosomal protein CWC22 plays a role in coupling splicing to exon junction complex deposition and nonsense-mediated decay. Proc Natl Acad Sci USA 109:21313–21318. doi:10.1073/pnas.1219725110
Barbosa I, Haque N, Fiorini F, Barrandon C, Tomasetto C, Blanchette M, Le Hir H (2012) Human CWC22 escorts the helicase eIF4AIII to spliceosomes and promotes exon junction complex assembly. Nat Struct Mol Biol 19:983–990. doi:10.1038/nsmb.2380
Buchwald G, Schussler S, Basquin C, Le Hir H, Conti E (2013) Crystal structure of the human eIF4AIII-CWC22 complex shows how a DEAD-box protein is inhibited by a MIF4G domain. Proc Natl Acad Sci USA 110:E4611–E4618. doi:10.1073/pnas.1314684110
Steckelberg AL, Boehm V, Gromadzka AM, Gehring NH (2012) CWC22 connects pre-mRNA splicing and exon junction complex assembly. Cell Rep 2:454–461. doi:10.1016/j.celrep.2012.08.017
Kadlec J, Izaurralde E, Cusack S (2004) The structural basis for the interaction between nonsense-mediated mRNA decay factors UPF2 and UPF3. Nat Struct Mol Biol 11:330–337. doi:10.1038/nsmb741
Serin G, Gersappe A, Black JD, Aronoff R, Maquat LE (2001) Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein (Caenorhabditis elegans SMG-4). Mol Cell Biol 21:209–223. doi:10.1128/MCB.21.1.209-223.2001
Melero R, Buchwald G, Castano R, Raabe M, Gil D, Lazaro M, Urlaub H, Conti E, Llorca O (2012) The cryo-EM structure of the UPF-EJC complex shows UPF1 poised toward the RNA 3′ end. Nat Struct Mol Biol 19(498–505):S491–S492. doi:10.1038/nsmb.2287
Fourati Z, Roy B, Millan C, Coureux PD, Kervestin S, van Tilbeurgh H, He F, Uson I, Jacobson A, Graille M (2014) A highly conserved region essential for NMD in the Upf2 N-terminal domain. J Mol Biol 426:3689–3702. doi:10.1016/j.jmb.2014.09.015
Chan WK, Bhalla AD, Le Hir H, Nguyen LS, Huang L, Gecz J, Wilkinson MF (2009) A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat Struct Mol Biol 16:747–753. doi:10.1038/nsmb.1612
Kunz JB, Neu-Yilik G, Hentze MW, Kulozik AE, Gehring NH (2006) Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA 12:1015–1022. doi:10.1261/rna.12506
Turner JM (2007) Meiotic sex chromosome inactivation. Development 134:1823–1831. doi:10.1242/dev.000018
Buchwald G, Ebert J, Basquin C, Sauliere J, Jayachandran U, Bono F, Le Hir H, Conti E (2010) Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc Natl Acad Sci USA 107:10050–10055. doi:10.1073/pnas.1000993107
Kashima I, Jonas S, Jayachandran U, Buchwald G, Conti E, Lupas AN, Izaurralde E (2010) SMG6 interacts with the exon junction complex via two conserved EJC-binding motifs (EBMs) required for nonsense-mediated mRNA decay. Genes Dev 24:2440–2450. doi:10.1101/gad.604610
Metze S, Herzog VA, Ruepp MD, Muhlemann O (2013) Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA 19:1432–1448. doi:10.1261/rna.038893.113
Gatfield D, Unterholzner L, Ciccarelli FD, Bork P, Izaurralde E (2003) Nonsense-mediated mRNA decay in Drosophila: at the intersection of the yeast and mammalian pathways. EMBO J 22:3960–3970. doi:10.1093/emboj/cdg371
Spingola M, Grate L, Haussler D, Ares M Jr (1999) Genome-wide bioinformatic and molecular analysis of introns in Saccharomyces cerevisiae. RNA 5:221–234
Wen J, Brogna S (2010) Splicing-dependent NMD does not require the EJC in Schizosaccharomyces pombe. EMBO J 29:1537–1551. doi:10.1038/emboj.2010.48
Grimson A, O’Connor S, Newman CL, Anderson P (2004) SMG-1 is a phosphatidylinositol kinase-related protein kinase required for nonsense-mediated mRNA decay in Caenorhabditis elegans. Mol Cell Biol 24:7483–7490. doi:10.1128/MCB.24.17.7483-7490.2004
Melero R, Uchiyama A, Castano R, Kataoka N, Kurosawa H, Ohno S, Yamashita A, Llorca O (2014) Structures of SMG1-UPFs complexes: SMG1 contributes to regulate UPF2-dependent activation of UPF1 in NMD. Structure 22:1105–1119. doi:10.1016/j.str.2014.05.015
Arias-Palomo E, Yamashita A, Fernandez IS, Nunez-Ramirez R, Bamba Y, Izumi N, Ohno S, Llorca O (2011) The nonsense-mediated mRNA decay SMG-1 kinase is regulated by large-scale conformational changes controlled by SMG-8. Genes Dev 25:153–164. doi:10.1101/gad.606911
Fernandez IS, Yamashita A, Arias-Palomo E, Bamba Y, Bartolome RA, Canales MA, Teixido J, Ohno S, Llorca O (2011) Characterization of SMG-9, an essential component of the nonsense-mediated mRNA decay SMG1C complex. Nucleic Acids Res 39:347–358. doi:10.1093/nar/gkq749
Chakrabarti S, Bonneau F, Schussler S, Eppinger E, Conti E (2014) Phospho-dependent and phospho-independent interactions of the helicase UPF1 with the NMD factors SMG5-SMG7 and SMG6. Nucleic Acids Res 42:9447–9460. doi:10.1093/nar/gku578
Lasalde C, Rivera AV, Leon AJ, Gonzalez-Feliciano JA, Estrella LA, Rodriguez-Cruz EN, Correa ME, Cajigas IJ, Bracho DP, Vega IE, Wilkinson MF, Gonzalez CI (2014) Identification and functional analysis of novel phosphorylation sites in the RNA surveillance protein Upf1. Nucleic Acids Res 42:1916–1929. doi:10.1093/nar/gkt1049
Wang W, Cajigas IJ, Peltz SW, Wilkinson MF, Gonzalez CI (2006) Role for Upf2p phosphorylation in Saccharomyces cerevisiae nonsense-mediated mRNA decay. Mol Cell Biol 26:3390–3400. doi:10.1128/MCB.26.9.3390-3400.2006
Fukuhara N, Ebert J, Unterholzner L, Lindner D, Izaurralde E, Conti E (2005) SMG7 is a 14-3-3-like adaptor in the nonsense-mediated mRNA decay pathway. Mol Cell 17:537–547. doi:10.1016/j.molcel.2005.01.010
Gardino AK, Smerdon SJ, Yaffe MB (2006) Structural determinants of 14-3-3 binding specificities and regulation of subcellular localization of 14-3-3-ligand complexes: a comparison of the X-ray crystal structures of all human 14-3-3 isoforms. Semin Cancer Biol 16:173–182. doi:10.1016/j.semcancer.2006.03.007
Jonas S, Weichenrieder O, Izaurralde E (2013) An unusual arrangement of two 14-3-3-like domains in the SMG5-SMG7 heterodimer is required for efficient nonsense-mediated mRNA decay. Genes Dev 27:211–225. doi:10.1101/gad.206672.112
Obsil T, Obsilova V (2011) Structural basis of 14-3-3 protein functions. Semin Cell Dev Biol 22:663–672. doi:10.1016/j.semcdb.2011.09.001
Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S (2012) N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res 40:1251–1266. doi:10.1093/nar/gkr791
Unterholzner L, Izaurralde E (2004) SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol Cell 16:587–596. doi:10.1016/j.molcel.2004.10.013
Loh B, Jonas S, Izaurralde E (2013) The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev 27:2125–2138. doi:10.1101/gad.226951.113
He F, Jacobson A (1995) Identification of a novel component of the nonsense-mediated mRNA decay pathway by use of an interacting protein screen. Genes Dev 9:437–454
Lykke-Andersen J (2002) Identification of a human decapping complex associated with hUpf proteins in nonsense-mediated decay. Mol Cell Biol 22:8114–8121
He F, Jacobson A (2001) Upf1p, Nmd2p, and Upf3p regulate the decapping and exonucleolytic degradation of both nonsense-containing mRNAs and wild-type mRNAs. Mol Cell Biol 21:1515–1530. doi:10.1128/MCB.21.5.1515-1530.2001
Lejeune F, Li X, Maquat LE (2003) Nonsense-mediated mRNA decay in mammalian cells involves decapping, deadenylating, and exonucleolytic activities. Mol Cell 12:675–687
Cho H, Kim KM, Kim YK (2009) Human proline-rich nuclear receptor coregulatory protein 2 mediates an interaction between mRNA surveillance machinery and decapping complex. Mol Cell 33:75–86. doi:10.1016/j.molcel.2008.11.022
Lai T, Cho H, Liu Z, Bowler MW, Piao S, Parker R, Kim YK, Song H (2012) Structural basis of the PNRC2-mediated link between mrna surveillance and decapping. Structure 20:2025–2037. doi:10.1016/j.str.2012.09.009
Cho H, Han S, Choe J, Park SG, Choi SS, Kim YK (2013) SMG5-PNRC2 is functionally dominant compared with SMG5-SMG7 in mammalian nonsense-mediated mRNA decay. Nucleic Acids Res 41:1319–1328. doi:10.1093/nar/gks1222
Anders KR, Grimson A, Anderson P (2003) SMG-5, required for C.elegans nonsense-mediated mRNA decay, associates with SMG-2 and protein phosphatase 2A. EMBO J 22:641–650. doi:10.1093/emboj/cdg056
Clissold PM, Ponting CP (2000) PIN domains in nonsense-mediated mRNA decay and RNAi. Curr Biol 10:R888–R890
Schoenberg DR (2011) Mechanisms of endonuclease-mediated mRNA decay. Wiley Interdiscip Rev RNA 2:582–600. doi:10.1002/wrna.78
Glavan F, Behm-Ansmant I, Izaurralde E, Conti E (2006) Structures of the PIN domains of SMG6 and SMG5 reveal a nuclease within the mRNA surveillance complex. EMBO J 25:5117–5125. doi:10.1038/sj.emboj.7601377
Chiu SY, Serin G, Ohara O, Maquat LE (2003) Characterization of human Smg5/7a: a protein with similarities to Caenorhabditis elegans SMG5 and SMG7 that functions in the dephosphorylation of Upf1. RNA 9:77–87
Gatfield D, Izaurralde E (2004) Nonsense-mediated messenger RNA decay is initiated by endonucleolytic cleavage in Drosophila. Nature 429:575–578. doi:10.1038/nature02559
Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E (2008) SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14:2609–2617. doi:10.1261/rna.1386208
Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH (2009) SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16:49–55. doi:10.1038/nsmb.1530
Nicholson P, Josi C, Kurosawa H, Yamashita A, Muhlemann O (2014) A novel phosphorylation-independent interaction between SMG6 and UPF1 is essential for human NMD. Nucleic Acids Res 42:9217–9235. doi:10.1093/nar/gku645
Kurosaki T, Li W, Hoque M, Popp MW, Ermolenko DN, Tian B, Maquat LE (2014) A post-translational regulatory switch on UPF1 controls targeted mRNA degradation. Genes Dev 28:1900–1916. doi:10.1101/gad.245506.114
Lykke-Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH (2014) Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28:2498–2517. doi:10.1101/gad.246538.114
Schmidt SA, Foley PL, Jeong DH, Rymarquis LA, Doyle F, Tenenbaum SA, Belasco JG, Green PJ (2014) Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res. doi:10.1093/nar/gku1258
Hwang J, Maquat LE (2011) Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr Opin Genet Dev 21:422–430. doi:10.1016/j.gde.2011.03.008
Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT (2008) NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22:1381–1396. doi:10.1101/gad.468808
McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW (2010) Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci USA 107:12186–12191. doi:10.1073/pnas.1007336107
Thoren LA, Norgaard GA, Weischenfeldt J, Waage J, Jakobsen JS, Damgaard I, Bergstrom FC, Blom AM, Borup R, Bisgaard HC, Porse BT (2010) UPF2 is a critical regulator of liver development, function and regeneration. PLoS One 5:e11650. doi:10.1371/journal.pone.0011650
Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, Chen YS, Groth M, Krueger A, Platzer M, Yang YG, Rudolph KL, Wang ZQ (2015) Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. doi:10.15252/embj.201489947
Isken O, Maquat LE (2008) The multiple lives of NMD factors: balancing roles in gene and genome regulation. Nat Rev Genet 9:699–712. doi:10.1038/nrg2402
Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Muhlemann O (2010) Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci 67:677–700. doi:10.1007/s00018-009-0177-1
Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, O’Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Hills K, Jones D, Mironenko T, Perry J, Varian J, West S, Widaa S, Teague J, Dicks E, Butler A, Menzies A, Richardson D, Jenkinson A, Shepherd R, Raine K, Moon J, Luo Y, Parnau J, Bhat SS, Gardner A, Corbett M, Brooks D, Thomas P, Parkinson-Lawrence E, Porteous ME, Warner JP, Sanderson T, Pearson P, Simensen RJ, Skinner C, Hoganson G, Superneau D, Wooster R, Bobrow M, Turner G, Stevenson RE, Schwartz CE, Futreal PA, Srivastava AK, Stratton MR, Gecz J (2007) Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet 39:1127–1133. doi:10.1038/ng2100
Laumonnier F, Shoubridge C, Antar C, Nguyen LS, Van Esch H, Kleefstra T, Briault S, Fryns JP, Hamel B, Chelly J, Ropers HH, Ronce N, Blesson S, Moraine C, Gecz J, Raynaud M (2010) Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol Psychiatry 15:767–776. doi:10.1038/mp.2009.14
Addington AM, Gauthier J, Piton A, Hamdan FF, Raymond A, Gogtay N, Miller R, Tossell J, Bakalar J, Inoff-Germain G, Gochman P, Long R, Rapoport JL, Rouleau GA (2011) A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry 16:238–239. doi:10.1038/mp.2010.59
Lynch SA, Nguyen LS, Ng LY, Waldron M, McDonald D, Gecz J (2012) Broadening the phenotype associated with mutations in UPF3B: two further cases with renal dysplasia and variable developmental delay. Eur J Med Genet 55:476–479. doi:10.1016/j.ejmg.2012.03.010
Xu X, Zhang L, Tong P, Xun G, Su W, Xiong Z, Zhu T, Zheng Y, Luo S, Pan Y, Xia K, Hu Z (2013) Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin Genet 83:560–564. doi:10.1111/cge.12014
Nguyen LS, Jolly L, Shoubridge C, Chan WK, Huang L, Laumonnier F, Raynaud M, Hackett A, Field M, Rodriguez J, Srivastava AK, Lee Y, Long R, Addington AM, Rapoport JL, Suren S, Hahn CN, Gamble J, Wilkinson MF, Corbett MA, Gecz J (2012) Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol Psychiatry 17:1103–1115. doi:10.1038/mp.2011.163
Chan WK, Huang L, Gudikote JP, Chang YF, Imam JS, MacLean JA 2nd, Wilkinson MF (2007) An alternative branch of the nonsense-mediated decay pathway. EMBO J 26:1820–1830. doi:10.1038/sj.emboj.7601628
Nguyen LS, Kim HG, Rosenfeld JA, Shen Y, Gusella JF, Lacassie Y, Layman LC, Shaffer LG, Gecz J (2013) Contribution of copy number variants involving nonsense-mediated mRNA decay pathway genes to neuro-developmental disorders. Hum Mol Genet 22:1816–1825. doi:10.1093/hmg/ddt035
Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, Rippey C, Shahin H, Consortium on the Genetics of S, Group PS, Nimgaonkar VL, Go RC, Savage RM, Swerdlow NR, Gur RE, Braff DL, King MC, McClellan JM (2013) Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell 154:518–529. doi:10.1016/j.cell.2013.06.049
Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, Zhu J, Wang Y, Zhao Y, Foo WC, Zuo M, Valasek MA, Javle M, Wilkinson MF, Lu Y (2014) The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 20:596–598. doi:10.1038/nm.3548
Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, Jolley JD, Cvejic A, Kostadima M, Bertone P, Breuning MH, Debili N, Deloukas P, Favier R, Fiedler J, Hobbs CM, Huang N, Hurles ME, Kiddle G, Krapels I, Nurden P, Ruivenkamp CA, Sambrook JG, Smith K, Stemple DL, Strauss G, Thys C, van Geet C, Newbury-Ecob R, Ouwehand WH, Ghevaert C (2012) Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet 44(435–439):S431–S432. doi:10.1038/ng.1083
Greenhalgh KL, Howell RT, Bottani A, Ancliff PJ, Brunner HG, Verschuuren-Bemelmans CC, Vernon E, Brown KW, Newbury-Ecob RA (2002) Thrombocytopenia-absent radius syndrome: a clinical genetic study. J Med Genet 39:876–881
Richieri-Costa A, Pereira SC (1992) Short stature, Robin sequence, cleft mandible, pre/postaxial hand anomalies, and clubfoot: a new autosomal recessive syndrome. Am J Med Genet 42:681–687. doi:10.1002/ajmg.1320420511
Favaro FP, Alvizi L, Zechi-Ceide RM, Bertola D, Felix TM, de Souza J, Raskin S, Twigg SR, Weiner AM, Armas P, Margarit E, Calcaterra NB, Andersen GR, McGowan SJ, Wilkie AO, Richieri-Costa A, de Almeida ML, Passos-Bueno MR (2014) A noncoding expansion in EIF4A3 causes Richieri-Costa-Pereira syndrome, a craniofacial disorder associated with limb defects. Am J Hum Genet 94:120–128. doi:10.1016/j.ajhg.2013.11.020
Culbertson MR (1999) RNA surveillance. Unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet 15:74–80
Peltz SW, Morsy M, Welch EM, Jacobson A (2013) Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med 64:407–425. doi:10.1146/annurev-med-120611-144851
Mort M, Ivanov D, Cooper DN, Chuzhanova NA (2008) A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat 29:1037–1047. doi:10.1002/humu.20763
Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE (2010) NMD: RNA biology meets human genetic medicine. Biochem J 430:365–377. doi:10.1042/BJ20100699
Hall GW, Thein S (1994) Nonsense codon mutations in the terminal exon of the beta-globin gene are not associated with a reduction in beta-mRNA accumulation: a mechanism for the phenotype of dominant beta-thalassemia. Blood 83:2031–2037
Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE (2004) Nonsense-mediated decay approaches the clinic. Nat Genet 36:801–808. doi:10.1038/ng1403
Kerr TP, Sewry CA, Robb SA, Roberts RG (2001) Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay? Hum Genet 109:402–407. doi:10.1007/s004390100598
Kerem E (2004) Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pulm Med 10:547–552
Hermann T (2007) Aminoglycoside antibiotics: old drugs and new therapeutic approaches. Cell Mol Life Sci 64:1841–1852. doi:10.1007/s00018-007-7034-x
Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ (1997) Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med 3:1280–1284
Kovesi TA, Swartz R, MacDonald N (1998) Transient renal failure due to simultaneous ibuprofen and aminoglycoside therapy in children with cystic fibrosis. N Engl J Med 338:65–66. doi:10.1056/NEJM199801013380115
Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, Paushkin S, Patel M, Trotta CR, Hwang S, Wilde RG, Karp G, Takasugi J, Chen G, Jones S, Ren H, Moon YC, Corson D, Turpoff AA, Campbell JA, Conn MM, Khan A, Almstead NG, Hedrick J, Mollin A, Risher N, Weetall M, Yeh S, Branstrom AA, Colacino JM, Babiak J, Ju WD, Hirawat S, Northcutt VJ, Miller LL, Spatrick P, He F, Kawana M, Feng H, Jacobson A, Peltz SW, Sweeney HL (2007) PTC124 targets genetic disorders caused by nonsense mutations. Nature 447:87–91. doi:10.1038/nature05756
Auld DS, Thorne N, Maguire WF, Inglese J (2009) Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc Natl Acad Sci USA 106:3585–3590. doi:10.1073/pnas.0813345106
McElroy SP, Nomura T, Torrie LS, Warbrick E, Gartner U, Wood G, McLean WH (2013) A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol 11:e1001593. doi:10.1371/journal.pbio.1001593
Chang JC, Kan YW (1979) Beta 0 thalassemia, a nonsense mutation in man. Proc Natl Acad Sci USA 76:2886–2889
Moriarty PM, Reddy CC, Maquat LE (1998) Selenium deficiency reduces the abundance of mRNA for Se-dependent glutathione peroxidase 1 by a UGA-dependent mechanism likely to be nonsense codon-mediated decay of cytoplasmic mRNA. Mol Cell Biol 18:2932–2939
Schmidt SA, Foley PL, Jeong DH, Rymarquis LA, Doyle F, Tenenbaum SA, Belasco JG, Green PJ (2015) Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res 43:309–323. doi:10.1093/nar/gku1258
Jones RB, Wang F, Luo Y, Yu C, Jin C, Suzuki T, Kan M, McKeehan WL (2001) The nonsense-mediated decay pathway and mutually exclusive expression of alternatively spliced FGFR2IIIb and -IIIc mRNAs. J Biol Chem 276:4158–4167. doi:10.1074/jbc.M006151200
Sharma K, D’Souza RC, Tyanova S, Schaab C, Wisniewski JR, Cox J, Mann M (2014) Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep 8:1583–1594. doi:10.1016/j.celrep.2014.07.036
Acknowledgments
This research was funded by grants from the Fritz Thyssen Stiftung and the Deutsche Forschungsgemeinschaft (SFB635, project B06) to N.H.G.
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Fatscher and V. Boehm contributed equally to this work.
Rights and permissions
About this article

Cite this article
Fatscher, T., Boehm, V. & Gehring, N.H. Mechanism, factors, and physiological role of nonsense-mediated mRNA decay. Cell. Mol. Life Sci. 72, 4523–4544 (2015). https://doi.org/10.1007/s00018-015-2017-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-2017-9