
Abstract
Nonsense-mediated messenger RNA (mRNA) decay (NMD) is a quality control mechanism that degrades irregular or faulty mRNAs. NMD mainly degrades mRNAs, which contain a premature termination codon (PTC) and therefore encode a truncated protein. Furthermore, NMD alters the expression of different types of cellular mRNAs, the so-called endogenous NMD substrates. In this review, we focus on the impact of NMD on cellular and molecular physiology. We specify key classes of NMD substrates and provide a detailed overview of the physiological function of gene regulation by NMD. We also describe different mechanisms of NMD substrate degradation and how the regulation of the NMD machinery affects cellular physiology. Finally, we outline the physiological functions of central NMD factors.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The accurate organization and regulation of gene expression is crucial for living cells in order to maintain cellular homeostasis but also to respond to stress, differentiation, or development. NMD is a translation-dependent quality control mechanism that detects erroneous transcripts and thereby ensures the accuracy of gene expression. NMD is best known for degrading messenger RNAs (mRNAs) with a truncated open reading frame (ORF) that harbor a PTC. Nonsense mutations account for approximately 20 % of disease-associated single-base pair substitutions, and most of them are expected to elicit NMD [72]. Thus, NMD is an important modulator of the clinical manifestation of various genetic diseases. On one hand, NMD maintains cellular homeostasis by repressing the production of C-terminally truncated proteins with a potential dominant negative function. On the other hand, truncated proteins might still have residual function, and further reduction of protein levels by NMD can result in an aggravation of the disease phenotype. Hence, it will be important to understand the mechanism underlying NMD and its regulation in order to develop individual treatments for human genetic diseases, especially single gene disorders.
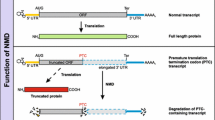
In mammalian cells, termination codons are recognized as premature if they are located more than 50–55 nucleotides (nt) upstream of an exon-exon junction [74]. Exon-exon junctions (i.e., sites of intron removal) are marked by the exon-junction complex (EJC), a multiprotein complex deposited on mRNAs during splicing [50, 51, 101]. When a translating ribosome stalls at a PTC, eukaryotic release factors (eRFs) 1 and 3 bind to the ribosome and eRF3 also interacts with upstream frameshift 1 (UPF1), an ATP-dependent RNA helicase and a central regulator of NMD. A downstream EJC serves as a platform for binding of UPF3B and UPF2, two additional conserved NMD factors. While UPF3B directly binds to the EJC via a short C-terminal motif, UPF2 links UPF3B and UPF1 [19, 25, 42]. The assembly of this complex also recruits the kinase SMG1, which interacts with UPF1 and UPF2 and phosphorylates C-terminal serine and threonine residues of UPF1 [24, 41, 44, 102]. Phosphorylated and nonphosphorylated UPF1 recruits the NMD factor SMG6, which mediates endonucleolytic cleavage of the mRNA substrate in close proximity to the PTC [15, 29, 40, 91]. Alternatively, phosphorylated UPF1 can also bind the SMG5/SMG7 heterodimer, which activates the deadenylation of the target transcript [56, 79] (Fig. 1). Nonetheless, NMD not only destabilizes aberrant transcripts emerging as a result of nonsense mutations or RNA processing errors but also regulates the expression levels of many mRNAs occurring under normal conditions (so-called endogenous or cellular targets). Among other approaches, transcriptome-wide studies revealed that NMD regulates transcripts with upstream open reading frames (uORFs) and transcripts with long 3′ UTRs or with introns downstream of the canonical termination codon [15, 60, 67, 73, 91]. It is currently believed that NMD affects the levels of 3–10 % of all cellular transcripts in addition to its quality control function [67, 110, 116, 126, 130]. NMD activity varies across different cell types and tissues, resulting from differential regulation of individual NMD factors [18, 22]. This suggests that NMD is a highly complex regulated mechanism, involved in a broad spectrum of physiological processes.

Model of PTC-containing transcript degradation by NMD. a Translation of mRNAs by ribosomes removes EJCs from exon-exon junctions (marked in orange) within the ORF. b Stalling of a ribosome at a PTC recruits UPF1 to the ribosome. The downstream EJC serves as a binding platform for UPF3B and UPF2. UPF2 binds to UPF1 as well as the protein kinase SMG1. SMG1 phosphorylates UPF1 at C-terminal serine and threonine residues. c The SMG5/7 heterodimer and the endonuclease SMG6 are recruited to phosphorylated UPF1. SMG6 cleaves the mRNA in close proximity to the PTC, whereas SMG5/SMG7 induces deadenylation of the target transcript (color figure online)
In this review, we describe different classes of endogenous NMD substrates, the mechanisms of their regulation and the consequences for cellular physiology. We outline how NMD activity is regulated by endogenous and exogenous modulators. Finally, we summarize the physiological functions of central NMD factors.
Endogenous targets of nonsense-mediated mRNA decay and their regulation
As described above, NMD is known to regulate the expression levels of many endogenous mRNAs and thereby controls different cellular processes. One of the most intensively studied cellular functions of NMD is alternative splicing-coupled nonsense-mediated decay (AS-NMD). Alternative splicing (AS) occurs in nearly 95 % of mammalian genes [81] and allows for the production of functionally different protein isoforms from an individual gene, thereby increasing the coding capacity of the genome [46]. One third of AS events produces transcripts with NMD activating features [52]. AS can stimulate NMD by including PTC-containing or frameshifting exons or skipping open reading frame-maintaining exons. Intron retention and alternative utilization of the 5′ and 3′ splice sites may also give rise to PTC-containing transcripts.
Regulation and autoregulation of splicing factor expression by alternative splicing-coupled nonsense-mediated decay
SR proteins are abundant splicing regulatory proteins, and their expression is highly regulated [35, 96, 132]. Initially, it has been shown that the inhibition of NMD alters the levels of two alternatively spliced SR proteins in Caenorhabditis elegans [71]. In human cells, all 11 SR genes are regulated by alternative splicing and can produce PTC-containing mRNAs, which are degraded by NMD [52, 77]. This process, also known as regulated unproductive splicing and translation (RUST), allows for regulating the expression of a gene by excluding a fraction of its pre-mRNA from protein production. Splicing factors were shown to utilize RUST to control the expression of their own mRNAs in an autoregulatory manner to maintain constant protein levels. Changing the intron/exon composition of their own pre-mRNA can produce transcripts susceptible to NMD [16, 48, 77, 90]. In general, SR proteins are considered splicing activators that promote exon inclusion through recognition of exonic splicing enhancers (ESEs). In contrast, heterogeneous ribonucleoproteins (hnRNPs) act as splicing repressors [84, 99], which antagonistically regulate splicing through exonic or intronic splicing silencers (ESSs or ISSs) [13, 63]. Expression of splicing repressors (i.e., hnRNP proteins) can also be regulated by RUST. The inclusion of a coding exon results in a functional protein, when the levels of splicing repressors are low. Increased levels of splicing repressors induce exon skipping, which causes translational frameshifting and triggers NMD. Thus, splicing repressor proteins do not only regulate alternative splicing but also balance their own expression in an autoregulatory manner [77] (Fig. 2).

NMD is involved in autoregulation of splicing factor abundance. Splicing repression (exon skipping) of the hnRNP pre-mRNA results in transcripts with frameshifts, which are degraded by NMD (left panel). Splicing activation (exon inclusion) leads to a fully functional mRNA, which is translated to an hnRNP protein. High levels of hnRNP protein autoregulate their abundance by splicing repression. Inclusion of a PTC-containing exon (“poison cassette exon”) during SR protein pre-mRNA splicing (right panel) initiates NMD of the mature transcript, whereas splicing repression (exon skipping) allows the production of functional SR proteins. High abundance of SR proteins promotes the inclusion of poison cassette exons in their own mRNA, thereby regulating SR protein levels
RUST of some SR genes involves an alternative exon with an early in-frame stop codon (“poison cassette exon”). Inclusion of a poison cassette exon leads to transcript degradation by NMD (Fig. 2). SRSF2 (SC35) is a well-described example for splicing factor autoregulation. Overexpression of SRSF2 induces an intron excision as well as an exon inclusion event in its own 3′ UTR, producing a transcript, in which the canonical termination codon is interpreted as PTC [107]. Thereby, SRSF2 regulates its own expression levels by changing the splicing pattern of the SRSF2 pre-mRNA. Unproductive splicing of some SR genes is not only autoregulated by itself, but can also be cross-regulated by other SR or non-SR proteins. SRSF3 (SRp20) regulates its own abundance via unproductive splicing, but additionally induces unproductive splicing of SRSF5 and other splicing factor genes [6]. Likewise, splicing of the SRSF1 (SF2/ASF) mRNA via the RNA-binding protein Sam68 leads to AS-NMD in trans [114]. The SRSF1 mRNA contains an intron in its 3′ UTR, which is usually retained. Intron splicing results in a transcript, in which the canonical termination codon is redefined into a PTC, leading to its degradation via NMD [48]. Splicing of SRSF1 is regulated by Sam68, a RNA binding protein of the signal transduction associated activator of RNA (STAR) family [114]. SRSF1 is an important regulator of the epithelial-to-mesenchymal transition (EMT) [114], a crucial event during embryonic development, wound healing, and epithelial tumor progression [85, 112]. During EMT, Sam68 modulates splicing of the SRSF1 by promoting retention of the 3′ UTR intron which stabilizes the SRSF1 transcript [114]. SRSF1 promotes production of a constitutively active splice variant of the Ron proto-oncogene [34]. Ron encodes the tyrosine kinase receptor for the macrophage stimulating protein, which is involved in the regulation of cell scattering and motility and was shown to trigger EMT [34, 113].
Regulation of other mRNAs by alternative splicing-coupled nonsense-mediated decay
As described above, NMD coupled to AS, can alter the expression of many mRNAs encoding for splicing factors. AS-NMD also targets PTC-containing isoforms of many other transcripts. The expression levels of the encoded protein are determined by the upregulation or downregulation of the isoform(s), which are NMD substrates. Some AS-NMD targeted isoforms are expressed at elevated levels due to the specific inhibition of NMD. By these mechanisms, AS-NMD regulates a variety of physiological processes in the cell, including neuronal or tissue development or mechanisms implicated in oncogenesis [26, 39, 62, 68, 114, 127].
The polypyrimidine-tract-binding proteins 1 and 2 (PTBP1/2) are two RNA-binding proteins of the hnRNP group of proteins with known functions as splicing regulators [45]. Recently, the ratio of PTBP1 and 2 has been shown to be important during neuronal differentiation [16, 62] and to involve AS-NMD. PTBP1 (also known as hnRNP I) upregulation leads to the alternative splicing of its own pre-mRNA and results in skipping of exon 11. The alternatively spliced transcript isoform contains a frameshift, which creates a PTC in exon 12 of the mature transcript, eventually leading to its degradation via NMD [127]. In neuronal progenitor cells, PTBP1 regulates PTBP2 (nPTB), a nervous system-enriched homolog of PTBP1, and induces the skipping of the conserved cassette exon 10 [16, 86]. The resulting PTBP2 isoform is an NMD target. During neuronal differentiation increased expression levels of microRNA 124 (miRNA 124) silence PTBP1, allowing nPTB mRNA and protein to accumulate [62] (Fig. 3a).

Alternative splicing coupled to NMD acting during neuronal differentiation. a Regulation of PTBP1/2 during neuronal differentiation. The polypyrimidine-tract-binding proteins 1 and 2 (PTBP1/2) are two RNA-binding proteins of the hnRNP group of proteins required for neuronal differentiation. In neuronal progenitor cells, low levels of miRNA 124 (miR-124), which lowers the expression of the PTBP1 mRNA, allow the translation of PTBP1 protein (left panel). PTBP1 regulates splicing of its neuronal expressed paralog PTBP2. PTBP1-induced skipping of exon 11 in the PTBP2 pre-mRNA, results in a NMD-sensitive PTBP2 transcript. While neuronal differentiation proceeds (right panel), elevated miR-124 levels silence PTPB1. Functional PTBP2 mRNA and protein accumulate during neuronal differentiation. b ROBO3.2 regulation during neuronal midline crossing of commissural neurons. The roundabout homologue 3 (ROBO3) gene locus encodes for two isoforms (ROBO3.1 and ROBO3.2). Pre-midline crossing (left panel) retention of intron 26 (i26) results in the generation of the PTC-harboring ROBO3.2 splice variant which is susceptible to NMD. No ROBO3.2 protein is produced, but the transcript is stable in this phase, due to translational repression of the ROBO3.2 mRNA. During neuronal midline crossing (middle panel) ROBO3.2 is released from translational repression and translation of ROBO3.2 transcripts starts. Post-midline crossing (right panel), ROBO3.2 transcripts are degraded by NMD leading to lower levels of ROBO3.2 protein
It was shown that the PTB paralog ROD1, a hematopoietic stem cell marker, is also regulated by PTBP1 and 2, which promote inclusion of exon 2 of the ROD1 mRNA [105]. This leads to the formation of an mRNA with a short uORF. Notably, the alternatively spliced RNA is not NMD-sensitive, although it contains a PTC. It has been suggested that the NMD resistance is due to translational re-initiation [105]. However, it remains to be determined if the alternative splicing of ROD1 during hematopoiesis is related to the regulation of PTBP1/2 during neuronal differentiation.
Recently, it was shown that PTBP1 also influences splicing of the mRNA encoding HPS1 [36], a protein involved in the biogenesis of lysosome-related organelles. Mutations in the HPS1 gene are found in patients with Hermansky-Pudlak syndrome, who suffer from prolonged bleeding, lysosomal storage defects, and reduced pigmentation [33, 95, 119]. When PTBP1 was downregulated, an alternative downstream 5′ splice site within exon 18 of HPS1 was preferentially used, producing a transcript susceptible to NMD. In contrast, splicing at the upstream 5′ splice site was preferred in the presence of PTBP1 and results in a fully functional HPS1 protein. Hence, it has been suggested that the correlation of HPS1 and PTBP1 expression across mammalian tissues ensures the proper processing of the HPS1 mRNA and normal expression of the HPS1 protein [36].
Another example of a gene that is regulated by AS-NMD is cysteine rich 61 (CYR61) [39]. The expression of CYR61 is induced in hypoxic cells and the CYR61 protein acts as a proangiogenic factor [68]. Retention of intron 3 of the CYR61 mRNA leads to the production of a NMD-sensitive transcript. CYR61 is considered to be a tumor-promoting factor, and this AS-NMD process was shown to be altered in breast cancer cells, resulting in a transcript that lacks intron 3, but encodes a functional, active protein isoform. In several breast cancer cell lines, hypoxic conditions induced an upregulated expression of the intron 3-lacking CYR61 mRNA. This suggests that hypoxia-mediated changes in alternative splicing patterns might act as a regulatory mechanism for CYR61 expression and its tumor-promoting potential [39].
Roundabout homologue 3 (ROBO3), a receptor for the slit family of guidance cues, regulates midline commissural axon guidance during embryonic development [57, 78, 93]. Guidance of the neurons is orchestrated via two isoforms of ROBO3. ROBO3.2 contains a retained intron, proximal to the pre-mRNA 3′ end, which is able to activate NMD [23]. ROBO3.1 is drastically downregulated before axonal midline crossing, whereas ROBO3.2 is expressed but nor translated during this process, protecting it from NMD [26]. During midline crossing, the translational repression is abrogated and ROBO3.2 mRNA is degraded via NMD, assuring only low ROBO3.2 protein levels in this phase. Controlling ROBO3.2 abundance is crucial during axon guidance and silencing of UPF2 was shown to cause perturbations in commissural neuron migration [26] (Fig. 3b).
Furthermore, Weischenfeldt and colleagues identified several different aberrant splicing events in cells impaired in NMD [122], of which we would like to present two genes, which are striking examples of physiologically important constitutive NMD substrates. Both genes constantly produce isoforms that are NMD substrates and their splicing is not specifically regulated. Hence, these mRNAs do not formally belong to the group of AS-NMD substrates. Acetyl-CoA acetyltransferase 2 (Acat2) is a protein involved in the esterification of cholesterol [21]. Some Acat2 transcript isoforms are known NMD targets, as they can acquire a PTC through exon inclusion. Since NMD silencing induced an upregulation of Acat2 in liver and bone marrow macrophages (BMMs) [122], NMD might exert a regulatory function during cholesterol ester synthesis. The authors also identified transcript isoforms of the natural killer cell triggering receptor (Nktr), in which PTCs are introduced through exon inclusion events. Nktr is required for natural killer (NK) cell effector function as a part of the putative NK target recognition complex. Aberrant isoforms of Nktr are expressed in other cell types than NK cells [100]. Nktr is upregulated upon Upf2 depletion in BMMs and liver, suggesting aberrant Nktr transcripts to be degraded via NMD [122]. These findings imply an important role of NMD in controlling the cell-type-specific expression of Nktr.
Nonsense-mediated decay regulates mRNAs during endoplasmic reticulum stress and the integrated stress response
It has been shown that the inhibition of NMD leads to an upregulation of mRNAs involved in cellular stress responses [31, 67]. Several stress-related transcripts are degraded by NMD under normal (i.e., unstressed) conditions and are stabilized in response to stress-induced NMD inactivation. Similar observations have been made under different conditions, for example during hypoxia, amino acid deprivation, or the activation of the unfolded protein response (UPR) [31, 43, 80, 118]. Sequence features, such as long 3′ UTRs or uORFs, which are known to activate NMD, are found in many of these stress-related mRNAs [43] (Fig. 4).

Function of NMD in the integrated stress response. a NMD regulates transcripts during the unfolded protein response (UPR). Under normal conditions (left panel) transcripts encoding the ER transmembrane sensor inositol requiring enzyme 1 α (IRE1α) and other UPR-related proteins are degraded via NMD, which results in low levels of UPR factors. During ER stress (right panel) global translation and NMD are suppressed, while stress specific transcripts are still translated, which allows the accumulation of UPR factors. b Amino acid starvation, hypoxia, ER stress, oxidative stress and dsRNA recognition trigger the integrated stress response. Different branches of the integrated stress response converge and lead to phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α). Phosphorylation of eIF2α results in the global inhibition of translation initiation, which suppresses NMD. However, stress-specific translation of the central stress-related activating transcription factor 4 (ATF4) is initiated. Under normal (unstressed) conditions translation initiation occurs at one of the five uORFs within ATF4, leading to NMD of the ATF4 mRNA. During the integrated stress response, translation initiation shifts to the main ORF of ATF4, leading to the generation of a functional ATF4 protein. ATF4 then triggers the transcription of stress-related genes. Stress-induced eIF2α phosphorylation also promotes autophagy. Autophagy clears the cell from misfolded and aggregated proteins which accumulate as a consequence of cellular stress signaling. At the same time, autophagy-induced protein degradation provides amino acids to counter amino acid deprivation
The majority of transmembrane proteins are translocated to the lumen of the endoplasmic reticulum (ER) where they fold and mature. When unfolded or misfolded proteins accumulate at the ER, the UPR is activated [117]. It was recently reported that the depletion of UPF3B sensitizes mammalian cells to ER stress [43]. Notably, NMD regulates several mRNAs encoding components of the UPR, for example the protein kinase RNA (PKR)-like ER kinase (PERK), the activating transcription factors (ATFs) 3, 4, and 6 or the ER transmembrane sensor inositol-requiring enzyme 1α (IRE1α) [43]. The IRE1α mRNA contains a long 3′ UTR that is responsive to NMD, leading to an increased expression of IRE1α when NMD is inhibited (Fig. 4a). The overexpression of IRE1α and the depletion of UPF3B show a similar ER-stress-sensitized phenotype. Hence, NMD appears to regulate the UPR via IRE1α [43].
Interestingly, ATF4, a master transcriptional regulator of the UPR, was reported to be upregulated during ER stress signaling. The ATF4 mRNA contains a uORF in its 5′ UTR, which is thought to mediate its regulation via NMD [43, 80]. During stress conditions, translation of ATF4 occurs preferentially via the protein-coding ORF (i.e., skipping the uORF), leading to transcript stabilization. In contrast, translation initiation at the uORF is favored during normal (unstressed) conditions [31, 115, 118]. Furthermore, the UPR and other stress signaling pathways were shown to globally reduce translation, allowing the stabilization of stress-related transcripts, which are usually degraded via NMD [31, 118, 124] (Fig. 4b). The mechanism by which NMD is regulated in response to cellular stress will be described in detail below.
Initial evidence that NMD can be inhibited as a consequence of cellular stress was provided by expression profiling in mammalian cells depleted of UPF1 [67]. Several transcripts involved in amino acid metabolism were identified as NMD targets, and amino acid deprivation itself had an inhibitory effect on NMD. Under conditions of NMD inhibition, elevated transcript levels for the activating transcription factors ATF4 and ATF3 were observed [67, 82]. Both ATF3 and ATF4 are implicated in several cellular stress responses such as amino acid starvation and ER stress signaling [31, 38, 82]. Later, it was shown that NMD inhibition and ATF4 upregulation are common responses to cellular stresses such as hypoxia, oxidative stress, amino acid deprivation, or the detection of double-stranded RNA as an indicator for pathogen infections (together also termed as the integrative stress response) [31, 67, 118, 124]. The ATF4 mRNA contains three uORFs in its 5′ UTR, which are essential for its responsiveness to cellular stress [37, 115]. These uORFs are sufficient to render ATF4 an NMD target [31, 106]. High ATF4 levels, associated to ER stress signaling and the integrated stress response, negatively regulate cell proliferation and survival and are therefore sinister to unstressed cells. This explains why the ATF4 mRNA undergoes rapid degradation via NMD under normal conditions (Fig. 4b).
Recently, Karam and colleagues described NMD as a fine-tuning mechanism for the UPR [43], which mutually regulate each other. On the one hand, UPR components are targeted by the NMD pathway, thereby preventing excessive UPR activation in response to innocuous ER stress. On the other hand, the UPR suppresses NMD to become efficiently activated in the case of bona fide ER stress. Notably, this is not implemented by a downregulation of NMD factors during ER stress, which would be rate-limiting for NMD [43]. The mechanism integrates signals from the different branches of the integrated stress response that lead via specific kinases (e.g., PERK, GCN2, or HRI) to the phosphorylation of the alpha subunit of eukaryotic initiation factor 2 (eIF2α) [104, 123] (Fig. 4b). Phosphorylation of eIF2α has two major effects during stress signaling: (1) inhibition of translation [28] and (2) the paradoxical induction of ATF4 translation [5]. ATF4 in turn activates the transcription of genes involved in cellular stress responses [5, 14, 38]. It has also been shown that eIF2α phosphorylation inhibits NMD in the context of cellular stress and that NMD itself can target components of the integrated stress response [31, 67, 118]. Thereby, NMD inactivation indirectly regulates transcripts through ATF4 stabilization. For example NMD inhibition upregulates mRNA and protein levels of the cysteine/glutamate exchanger SLC7A11, suggesting that cysteine transport and intracellular cysteine levels are regulated by stress-induced NMD inhibition [124].
The eIF2α phosphorylation-dependent inhibition of NMD is not only a central step in ER stress signaling and the integrated stress response but also plays an important role for the induction of autophagy during amino acid starvation [89, 109, 124]. In fact, autophagy is promoted by the NMD-inhibiting branches of the integrated stress response (e.g., hypoxia or ER stress). Autophagy not only serves as a source for amino acids during amino acid deprivation but also clears the cell of misfolded, mutated, or aggregated proteins that result from cellular stress events but also from the NMD inhibition itself (Fig. 4b). Hypoxia, metabolite deprivation, ER stress, and other stress conditions that promote eIF2α phosphorylation and elevated ATF4 levels are common within the tumor microenvironment [124, 129] and contribute to tumor survival and growth. Cells deficient in ATF4 or unable to phosphorylate eIF2α do not form tumors in vivo [12, 129]. This underlines the importance of understanding the mechanisms during stress signaling for possible therapeutic benefits. In this context, Wang et al. showed that elevated expression levels of the c-myc oncogene induce eIF2α phosphorylation via the activation of the stress-associated PERK kinase, resulting in NMD inhibition and upregulation of NMD target transcripts [118].
The exact mechanisms by which NMD suppression via eIF2α phosphorylation is achieved is still not fully understood. As eIF2α phosphorylation inhibits translation [28] and NMD is a strictly translation-dependent process [20], one could conclude that NMD suppression is accomplished through translational shutoff. However, the observation that eIF2a phosphorylation blocks translation only partially and that translation of NMD targets is not completely repressed under stress conditions [31, 118], suggests that NMD inhibition in the context of stress-induced eIF2α phosphorylation occurs independently of translational suppression.
Physiological and pathophysiological regulation of NMD activity
In this second part, we explain how the activity of NMD is regulated by endogenous (e.g., miRNA) and exogenous (e.g., virus infection) modulators. We also describe important physiological functions of central NMD factors.
A microRNA/NMD regulatory circuit during neuronal development
Recently, Bruno and colleagues described miRNA 128 (miR-128), a nervous system-enriched miRNA, as a regulator of NMD during neuronal differentiation [18]. The authors showed that miR-128 targets the 3′ UTR of the central NMD factor UPF1 and the EJC core component MLN51 (also known as Barentsz or CASC3). The direct downregulation of these two NMD factors by miR-128 represses NMD activity in human and mouse cells. Notably, miR-128 is drastically upregulated during brain development and neuronal maturation [9, 92, 103] (Fig. 5). This suggests an important role of miRNA/NMD during the regulation of developmental processes. In this context, miRNAs use their potential to downregulate expression of NMD factors to indirectly stabilize mRNAs that are crucial for neuronal development and maturation.

A microRNA/NMD circuit regulates neuronal development. miR-128 is a neuronal microRNA that can repress translation of the NMD factor UPF1 and the EJC component MLN51 (Barentz). In neuronal progenitor cells (left panel), low miR-128 abundance allows the translation of UPF1 and MLN51 mRNAs at normal levels, which are compatible with the activation of NMD. NMD targets involved in neuronal differentiation are degraded in neuronal progenitors, which promotes an undifferentiated cell state. During neuronal differentiation (right panel) miR-128 expression is upregulated and UPF1 and MLN51 are translationally repressed. NMD targets important for neuronal differentiation are stabilized and neurogenesis is induced
The physiological relevance of the interplay between the NMD machinery and miRNAs during cell differentiation and development was further elucidated by Lou and colleagues [58]. The authors showed that NMD promotes an undifferentiated cell state, because UPF1 is downregulated during murine brain development and the maturation of human neural progenitor cells. In line with this observation, UPF1 depletion led to the upregulation of a series of known neuronal differentiation factors. Rescue of UPF1 expression levels inhibited miR-128 induced differentiation in P19 cells. This suggests that miR-128, at least in part, acts through UPF1 suppression during neural development. The idea of a regulatory feedback loop coupling UPF1 with miR-128 expression was further supported by the observation that UPF1 depletion induced miR-128 upregulation. In addition, Lou and colleagues identified miR-128, miR-9, and miR-124 to target the 3′ UTR of UPF3B. These miRNAs were also found to be upregulated in a UPF1-depleted background [58].
NMD in antiviral immunity
In general, genomes of single stranded RNA viruses are relatively small in size and produce transcripts with all characteristics of mature cellular mRNAs and thus can be subjected to NMD. Exception to this rule are the members of the (+) strand RNA virus group (group IV, Baltimore classification). Often lacking common features of mRNAs, like poly(A) tails or 5′-cap structures, the genomes of these viruses can directly be used for protein production. However, in many cases, after a pilot round of translation, subgenomic mRNAs with short 5′- and long 3′ UTRs are produced, which are known to be targets of NMD [3, 54]. Thus, NMD plays an important role in antiviral immunity and viruses have developed means to escape, counteract, and even utilize the cellular RNA degradation apparatus in order to alleviate viral gene expression and to establish a successful host infection [1, 76, 88].
It was recently shown that NMD protects against positive-stranded RNA virus infections in human and plant cells [10, 30]. In plants, a genetic screen uncovered Upf1 as a restriction factor for viral genomic RNA (gRNA) replication of Potato virus X (PVX) and Turnip crinkle virus (TCV) [30]. This suggests that NMD acts as a defense mechanism upstream of RNA interference (RNAi), which detects the double stranded replication intermediates of viruses and is therefore the major pathway in plants counteracting viral infections. Overexpression of a dominant-negative version of upf1 enhanced PVX infection in Arabidopsis thaliana and Nicotiana clevelandii [30]. In mammalian cells, a siRNA screen revealed that the depletion of UPF1 leads to enhanced replication of Sindbis virus (SINV) and Semliki Forest virus (SFV) as well as the stabilization of SFV gRNA. Furthermore, silencing of the NMD factors SMG5 or SMG7 also increased SFV infection rates [10]. Shortening the long 3′ UTRs in the SFV gRNA did not prevent its decay via NMD. Hence, the mechanisms by which viral RNAs are recognized and degraded by NMD remains to be determined. Taken together, NMD, possibly in addition to RNAi, acts early during positive-stranded RNA virus infection, while the viral gRNA is still accessible and actively translated.
Only the genome of positive-stranded RNA viruses can be targeted by NMD, because their gRNA is directly translated into viral proteins. Nevertheless, retroviruses, DNA and negative-stranded RNA viruses produce mRNAs that can be recognized by the NMD machinery, too. Thus, these viruses have developed strategies to avoid transcript degradation. In the Rous sarcoma virus (RSV), a simple avian virus, a full-length unspliced RNA serves as the genomic template for the production of three mRNAs via alternative splicing. Although the unspliced RNA contains several NMD-inducing features such as uORFs and a long 3′ UTR, it is very stable in host cells [11]. Deletion experiments revealed a cis-acting RNA element within RSV, referred to as RNA stability element (RSE), which protects the full-length transcript from NMD (Fig. 6a). The deletion of the RSE can be rescued by the depletion of UPF1 or by the overexpression of a dominant-negative version of UPF1 [120, 125]. Conversely, it was reported that UPF1, independent of its role in NMD, had positive effects on HIV-1 RNA translatability and that UPF1 overexpression upregulated HIV-1 RNA expression and protein synthesis, possibly via protecting the viral intron-containing RNAs from degradation by the exosome [4].

Viral strategies to inhibit NMD. a The RSE inhibits NMD factor-ribosome association. A cis-acting RNA element, the RNA stability element (RSE), protects the genomic RNA (gRNA) of positive-stranded RNA viruses from degradation by NMD. It is believed that the RSE interferes with NMD factor-ribosome association by either directly binding to NMD factors (e.g. UPF1) or by interacting with ribosome-bound eukaryotic release factor 3 (eRF3). b Tax inhibits translational repression and UPF1 recycling. During NMD, UPF1 interacts with INT6 (also known as EIF3E) a subunit of the eukaryotic translation initiation factor (eIF3) in order to repress further translation of the target transcript. The viral encoded protein Tax interferes with NMD via two mechanisms. On one hand it binds to INT6, which disrupts the eIF3-UPF1 interaction and thereby inhibits translational repression. On the other hand Tax impairs the dephosphorylation of UPF1 by the phosphatase PP2A, a crucial step for UPF1 recycling. c Sequestering PYM1 interferes with EJC recycling. Partner of Y14 and MAGOH (PYM1) facilitates the disassembly of RNA-bound EJCs. When a translating ribosome encounters an EJC, ribosome-associated PYM1 binds to the EJC components Y14 and MAGOH (left panel). EJCs, located at regions in the mRNA that are not translated by the ribosome (e.g., the 3′ UTR), are disassembled by free PYM1 molecules. Disassembled EJCs are translocated to the nucleus for recycling. The hepatitis C virus (HCV) core protein interferes with EJC disassembly and recycling by binding to PYM1 (right panel). EJCs that are encountered by translating ribosomes might still be removed from transcripts, whereas EJCs that exclusively depend on disassembly by PYM1 (e.g., in the 3′ UTR) are retained on the mRNA
The human T-cell leukemia virus type I (HTLV-1) is a delta RNA retrovirus that can cause adult T-cell leukemia. Its genome contains more than ten ORFs, which utilize different mechanisms to ensure proper and coordinated gene expression, including programmed ribosomal frameshifts and alternative splicing. Two independent studies identified virus encoded proteins, Tax and Rex, to interfere with the host NMD machinery to prevent degradation of viral mRNAs and the unspliced full-length genomic RNA [69, 75]. Rex is a high-affinity RNA binding protein that interacts with viral genomic RNA and facilitates its nuclear export [108, 131]. Although it was shown that Rex globally inhibits host cell NMD, the mechanisms underlying the NMD inhibition by Rex are still unclear [27, 75]. The viral Tax protein was found to inhibit NMD via interaction with INT6 [27]. INT6 is also known as EIF3E, a subunit of the eukaryotic translation initiation factor eIF3, which is involved in the NMD-mediated degradation of cellular mRNAs [70]. Tax interacts with both INT6 and UPF1 and inhibits the interaction between these two NMD factors. It also alters the morphology of processing bodies (P bodies, loci where RNAs are accumulating for degradation [97]) and thereby stabilizes viral and also cellular transcripts, which are usually subjected to NMD [69] (Fig. 6b).
A recent study has shed light on how hepatitis C virus (HCV) can interfere with the NMD machinery to escape viral transcript decay [87]. In HCV-infected cells, the viral core protein interacts with the cellular exon junction complex-associated factor PYM homolog 1 (PYM1). This prevents PYM from binding to its interaction partners Y14 and MAGOH, two core components of the EJC (Fig. 6c). EJCs are central factors in NMD and PYM1 helps to disassemble EJCs from cytoplasmic RNAs in order to facilitate the recycling of ECJ components to the nucleus [32]. The knockdown of PYM1 leads to a decreased infection with HCV, indicating that PYM1 plays an important role in the viral life cycle. In contrast, the knockdown of other NMD factors had no effect on viral infection. Interestingly, PYM1 also interacts with capsid proteins of two other members of the Flaviviridae family, dengue, and West Nile virus, suggesting a conserved role of PYM1 within this family. Furthermore, the HCV envelop protein E1 was identified to interact with additional factors of the NMD pathway, including UPF1, UPF3B, Y14, MAGOH, and the transient EJC components ACIN1 and SAP18, but the consequences of these interactions still need to be investigated [87].
In summary, viruses have developed different strategies to inhibit or utilize the NMD machinery to create ideal conditions for the successful infection of and replication within host cells.
Physiological functions of NMD factors
Until today the functions of four mammalian NMD factors (UPF1, UPF2, SMG1, and SMG6) have been investigated using knockout mice [53, 64, 65, 121]. The ablation of any of these factors had dramatic consequences and none of the knockouts was compatible with normal embryonic development. For example, mouse embryos lacking the central NMD factor Upf1 are only viable during the preimplantation period, but not after uterine implantation [65]. Although Upf1-deficient blastocysts were successfully isolated from heterozygous matings, they could only be maintained in culture medium for a few days. After 5 days in culture, UPF1−/− blastocysts showed a strong induction of apoptosis, which eventually led to the regression of the inner cell mass and resulted in only few remaining cells [65]. These observations strongly suggested that UPF1 and NMD are essential for mammalian cellular viability.
The central role of NMD factors for normal embryonic development was further supported by the phenotypes of the Upf2 [121], Smg1 [64], and Smg6 knockout mice [53]. While Smg6−/− embryos do not proceed the blastocyst stage (similar to Upf1−/− mice) [53], Upf2−/− embryos die in utero around embryonic days 3.5–7.5 (E3.5–E7.5) [121]. In contrast, Smg1-deficient mice display a slightly milder phenotype and die by E8.5 with marked developmental defects. NMD-specific changes of the transcriptome were observed in Smg6-, Upf2-, and Smg1-deficient cells with many known and potential NMD targets being upregulated [53, 64, 121].
To what extent are the effects of the ablations of NMD factors attributable to the inhibition of the NMD process? Additional functions beyond NMD have been reported for Upf1, Smg6, and Smg1. For example, Smg6 is involved in telomere maintenance and Upf1 and Smg1 have functions within genotoxic stress and DNA replication [8, 17]. However, we favor the notion that the observed dramatic developmental defects reflect the inhibition of NMD, since Upf1, Smg6, and Smg1 have non-overlapping functions besides NMD.
Taken together, observations from different mouse models indicate that the loss of NMD causes a strong differentiation defect, leading to abnormal embryonic development and impaired cellular viability.
Pathophysiological consequences of NMD factor mutations in humans
Although animal models for NMD factor knockouts existed for many years, only recently a number of human disorders have been found to be caused by mutations in genes encoding NMD factors in humans.
Analysis of pancreatic adenosquamous carcinoma (ASC) tumors has revealed that these tumors frequently harbor somatic point mutations in the UPF1 gene [55]. The UPF1 mutations were specific to the ASC tumors and were not detected in normal pancreatic tissues [55]. Two regions of the UPF1 gene were mostly affected by the point mutations, which were found both, in exons and introns. This suggests that many of the mutations may trigger alternative splicing of the UPF1 pre-mRNA and may lead to the expression of truncated versions of the UPF1 protein. Although none of the mutations were found to completely abrogate normal splicing of the UPF1 pre-mRNA, only little UPF1 expression was detected in ASC tumors [55]. Consequently, the levels of two NMD substrates (ATF3 and MAP3K14) were strongly upregulated in ASC tumors [55]. Although the molecular effects of UPF1 mutations during tumorigenesis remain elusive, it is very likely that they inhibit NMD in the affected tumors.
All eukaryotic genomes contain one copy of the genes encoding UPF1 and UPF2 [7, 66, 83]. While most eukaryotes express one UPF3 protein, mammals express two homologous genes, UPF3A and UPF3B [59, 94]. Although both UPF3 proteins share approximately 60 % sequence identity, UPF3B activates NMD more efficiently and binds with higher affinity to UPF2 than UPF3A [22, 47]. The UPF3A protein requires the interaction with UPF2 for stabilization and is therefore degraded in cells, in which UPF3B is also expressed [22]. Several independent mutations in the UPF3B gene (truncation and point mutations) were identified in different families, in which males were affected by mild to severe X-linked mental retardation [2, 49, 61, 111, 128]. An upregulation of UPF3A protein levels was observed in response to UPF3B mutations. It has been suggested that the degree of UPF3A upregulation correlates with the degree of mental retardation in different patients and may explain the broad range of clinical symptoms associated with UPF3B mutations. The notion that UPF3A represents a UPF3B homolog with only weak NMD activity has been recently challenged. Loss-of-function studies rather implied that UPF3A acts as an NMD inhibitor [98]. The presence of UPF3A stabilizes many NMD substrates and represses NMD activity by preventing the interaction of UPF2 with the EJC. Consequently, in mice lacking UPF3A NMD is hyperactive, leading to defects in embryogenesis and early embryonic death [98]. This suggests that the activity of NMD has to be tightly controlled and not only a lack, but also an excess of NMD can disturb essential processes in mammalian cell.
Summary and outlook
In conclusion, the literature contains many examples of physiologically important transcripts that are either directly or indirectly regulated by NMD. Hence, NMD does not only serve as a cellular quality control mechanism but also plays an important physiological role. Particularly AS-NMD emerges as a principle that determines the expression levels of a large number of mammalian genes, thereby regulating many cellular functions. NMD also represents an important player in cellular stress responses and uses a translational switch to coordinate different stress pathways.
The many different physiological targets of mammalian NMD may explain the severe effects observed in knockout animals. Indeed, it is difficult to imagine that embryos expressing multiple aberrant protein isoforms would show a normal development. However, future studies will be required to dissect the different physiological branches and functions of the NMD machinery in animal models. The recent advances in high-throughput sequencing and genome manipulation will accelerate the progress in this direction and will provide insights into the complex regulation of development, physiology, and disease by NMD.
References
Abernathy E, Glaunsinger B (2015) Emerging roles for RNA degradation in viral replication and antiviral defense. Virology 479–480:600–608. doi:10.1016/j.virol.2015.02.007
Addington AM, Gauthier J, Piton A, Hamdan FF, Raymond A, Gogtay N, Miller R, Tossell J, Bakalar J, Inoff-Germain G, Gochman P, Long R, Rapoport JL, Rouleau GA (2011) A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry 16:238–239. doi:10.1038/mp.2010.59
Ahlquist P (2006) Parallels among positive-strand RNA viruses, reverse-transcribing viruses and double-stranded RNA viruses. Nat Rev Microbiol 4:371–382. doi:10.1038/nrmicro1389
Ajamian L, Abrahamyan L, Milev M, Ivanov PV, Kulozik AE, Gehring NH, Mouland AJ (2008) Unexpected roles for UPF1 in HIV-1 RNA metabolism and translation. RNA 14:914–927. doi:10.1261/rna.829208
Ameri K, Lewis CE, Raida M, Sowter H, Hai T, Harris AL (2004) Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood 103:1876–1882. doi:10.1182/blood-2003-06-1859
Anko ML, Neugebauer KM (2012) RNA-protein interactions in vivo: global gets specific. Trends Biochem Sci 37:255–262. doi:10.1016/j.tibs.2012.02.005
Applequist SE, Selg M, Raman C, Jack HM (1997) Cloning and characterization of HUPF1, a human homolog of the Saccharomyces cerevisiae nonsense mRNA-reducing UPF1 protein. Nucleic Acids Res 25:814–821
Azzalin CM, Lingner J (2006) The human RNA surveillance factor UPF1 is required for S phase progression and genome stability. Curr Biol 16:433–439. doi:10.1016/j.cub.2006.01.018
Bak M, Silahtaroglu A, Moller M, Christensen M, Rath MF, Skryabin B, Tommerup N, Kauppinen S (2008) MicroRNA expression in the adult mouse central nervous system. RNA 14:432–444. doi:10.1261/rna.783108
Balistreri G, Horvath P, Schweingruber C, Zund D, McInerney G, Merits A, Muhlemann O, Azzalin C, Helenius A (2014) The host nonsense-mediated mRNA decay pathway restricts Mammalian RNA virus replication. Cell Host Microbe 16:403–411. doi:10.1016/j.chom.2014.08.007
Barker GF, Beemon K (1994) Rous sarcoma virus RNA stability requires an open reading frame in the gag gene and sequences downstream of the gag-pol junction. Mol Cell Biol 14:1986–1996
Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C (2005) ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. EMBO J 24:3470–3481. doi:10.1038/sj.emboj.7600777
Black DL (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72:291–336. doi:10.1146/annurev.biochem.72.121801.161720
Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG, Bell JC (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24:7469–7482. doi:10.1128/MCB.24.17.7469-7482.2004
Boehm V, Haberman N, Ottens F, Ule J, Gehring NH (2014) 3′ UTR length and messenger ribonucleoprotein composition determine endocleavage efficiencies at termination codons. Cell Rep 9:555–568. doi:10.1016/j.celrep.2014.09.012
Boutz PL, Stoilov P, Li Q, Lin CH, Chawla G, Ostrow K, Shiue L, Ares M Jr, Black DL (2007) A post-transcriptional regulatory switch in polypyrimidine tract-binding proteins reprograms alternative splicing in developing neurons. Genes Dev 21:1636–1652. doi:10.1101/gad.1558107
Brumbaugh KM, Otterness DM, Geisen C, Oliveira V, Brognard J, Li X, Lejeune F, Tibbetts RS, Maquat LE, Abraham RT (2004) The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol Cell 14:585–598. doi:10.1016/j.molcel.2004.05.005
Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, Song HW, Corbett MA, Gifford WD, Gecz J, Pfaff SL, Wilkinson MF (2011) Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell 42:500–510. doi:10.1016/j.molcel.2011.04.018
Buchwald G, Ebert J, Basquin C, Sauliere J, Jayachandran U, Bono F, Le Hir H, Conti E (2010) Insights into the recruitment of the NMD machinery from the crystal structure of a core EJC-UPF3b complex. Proc Natl Acad Sci U S A 107:10050–10055. doi:10.1073/pnas.1000993107
Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF (1995) A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem 270:28995–29003
Cases S, Novak S, Zheng YW, Myers HM, Lear SR, Sande E, Welch CB, Lusis AJ, Spencer TA, Krause BR, Erickson SK, Farese RV Jr (1998) ACAT-2, a second mammalian acyl-CoA:cholesterol acyltransferase. Its cloning, expression, and characterization. J Biol Chem 273:26755–26764
Chan WK, Bhalla AD, Le Hir H, Nguyen LS, Huang L, Gecz J, Wilkinson MF (2009) A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat Struct Mol Biol 16:747–753. doi:10.1038/nsmb.1612
Chen Z, Gore BB, Long H, Ma L, Tessier-Lavigne M (2008) Alternative splicing of the Robo3 axon guidance receptor governs the midline switch from attraction to repulsion. Neuron 58:325–332. doi:10.1016/j.neuron.2008.02.016
Clerici M, Deniaud A, Boehm V, Gehring NH, Schaffitzel C, Cusack S (2014) Structural and functional analysis of the three MIF4G domains of nonsense-mediated decay factor UPF2. Nucleic Acids Res 42:2673–2686. doi:10.1093/nar/gkt1197
Clerici M, Mourao A, Gutsche I, Gehring NH, Hentze MW, Kulozik A, Kadlec J, Sattler M, Cusack S (2009) Unusual bipartite mode of interaction between the nonsense-mediated decay factors, UPF1 and UPF2. EMBO J 28:2293–2306. doi:10.1038/emboj.2009.175
Colak D, Ji SJ, Porse BT, Jaffrey SR (2013) Regulation of axon guidance by compartmentalized nonsense-mediated mRNA decay. Cell 153:1252–1265. doi:10.1016/j.cell.2013.04.056
Desbois C, Rousset R, Bantignies F, Jalinot P (1996) Exclusion of Int-6 from PML nuclear bodies by binding to the HTLV-I Tax oncoprotein. Science 273:951–953
Donnelly N, Gorman AM, Gupta S, Samali A (2013) The eIF2alpha kinases: their structures and functions. Cell Mol Life Sci 70:3493–3511. doi:10.1007/s00018-012-1252-6
Eberle AB, Lykke-Andersen S, Muhlemann O, Jensen TH (2009) SMG6 promotes endonucleolytic cleavage of nonsense mRNA in human cells. Nat Struct Mol Biol 16:49–55. doi:10.1038/nsmb.1530
Garcia D, Garcia S, Voinnet O (2014) Nonsense-mediated decay serves as a general viral restriction mechanism in plants. Cell Host Microbe 16:391–402. doi:10.1016/j.chom.2014.08.001
Gardner LB (2008) Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol 28:3729–3741. doi:10.1128/MCB.02284-07
Gehring NH, Lamprinaki S, Kulozik AE, Hentze MW (2009) Disassembly of exon junction complexes by PYM. Cell 137:536–548. doi:10.1016/j.cell.2009.02.042
Gerondopoulos A, Langemeyer L, Liang JR, Linford A, Barr FA (2012) BLOC-3 mutated in Hermansky-Pudlak syndrome is a Rab32/38 guanine nucleotide exchange factor. Curr Biol 22:2135–2139. doi:10.1016/j.cub.2012.09.020
Ghigna C, Giordano S, Shen H, Benvenuto F, Castiglioni F, Comoglio PM, Green MR, Riva S, Biamonti G (2005) Cell motility is controlled by SF2/ASF through alternative splicing of the Ron protooncogene. Mol Cell 20:881–890. doi:10.1016/j.molcel.2005.10.026
Graveley BR (2000) Sorting out the complexity of SR protein functions. RNA 6:1197–1211
Hamid FM, Makeyev EV (2014) Regulation of mRNA abundance by polypyrimidine tract-binding protein-controlled alternate 5′ splice site choice. PLoS Genet 10, e1004771. doi:10.1371/journal.pgen.1004771
Harding CL, Lloyd DR, McFarlane CM, Al-Rubeai M (2000) Using the Microcyte flow cytometer to monitor cell number, viability, and apoptosis in mammalian cell culture. Biotechnol Prog 16:800–802. doi:10.1021/bp0000813
Harding HP, Zhang Y, Zeng H, Novoa I, Lu PD, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl DF, Bell JC, Hettmann T, Leiden JM, Ron D (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11:619–633
Hirschfeld M, zur Hausen A, Bettendorf H, Jager M, Stickeler E (2009) Alternative splicing of Cyr61 is regulated by hypoxia and significantly changed in breast cancer. Cancer Res 69:2082–2090. doi:10.1158/0008-5472.CAN-08-1997
Huntzinger E, Kashima I, Fauser M, Sauliere J, Izaurralde E (2008) SMG6 is the catalytic endonuclease that cleaves mRNAs containing nonsense codons in metazoan. RNA 14:2609–2617. doi:10.1261/rna.1386208
Ivanov PV, Gehring NH, Kunz JB, Hentze MW, Kulozik AE (2008) Interactions between UPF1, eRFs, PABP and the exon junction complex suggest an integrated model for mammalian NMD pathways. EMBO J 27:736–747. doi:10.1038/emboj.2008.17
Kadlec J, Izaurralde E, Cusack S (2004) The structural basis for the interaction between nonsense-mediated mRNA decay factors UPF2 and UPF3. Nat Struct Mol Biol 11:330–337. doi:10.1038/nsmb741
Karam R, Lou CH, Kroeger H, Huang L, Lin JH, Wilkinson MF (2015) The unfolded protein response is shaped by the NMD pathway. EMBO Rep 16:599–609. doi:10.15252/embr.201439696
Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S (2006) Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 20:355–367. doi:10.1101/gad.1389006
Keppetipola N, Sharma S, Li Q, Black DL (2012) Neuronal regulation of pre-mRNA splicing by polypyrimidine tract binding proteins, PTBP1 and PTBP2. Crit Rev Biochem Mol Biol 47:360–378. doi:10.3109/10409238.2012.691456
Keren H, Lev-Maor G, Ast G (2010) Alternative splicing and evolution: diversification, exon definition and function. Nat Rev Genet 11:345–355. doi:10.1038/nrg2776
Kunz JB, Neu-Yilik G, Hentze MW, Kulozik AE, Gehring NH (2006) Functions of hUpf3a and hUpf3b in nonsense-mediated mRNA decay and translation. RNA 12:1015–1022. doi:10.1261/rna.12506
Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE (2007) Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature 446:926–929. doi:10.1038/nature05676
Laumonnier F, Shoubridge C, Antar C, Nguyen LS, Van Esch H, Kleefstra T, Briault S, Fryns JP, Hamel B, Chelly J, Ropers HH, Ronce N, Blesson S, Moraine C, Gecz J, Raynaud M (2010) Mutations of the UPF3B gene, which encodes a protein widely expressed in neurons, are associated with nonspecific mental retardation with or without autism. Mol Psychiatry 15:767–776. doi:10.1038/mp.2009.14
Le Hir H, Gatfield D, Izaurralde E, Moore MJ (2001) The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. EMBO J 20:4987–4997. doi:10.1093/emboj/20.17.4987
Le Hir H, Izaurralde E, Maquat LE, Moore MJ (2000) The spliceosome deposits multiple proteins 20–24 nucleotides upstream of mRNA exon-exon junctions. EMBO J 19:6860–6869. doi:10.1093/emboj/19.24.6860
Lewis BP, Green RE, Brenner SE (2003) Evidence for the widespread coupling of alternative splicing and nonsense-mediated mRNA decay in humans. Proc Natl Acad Sci U S A 100:189–192. doi:10.1073/pnas.0136770100
Li T, Shi Y, Wang P, Guachalla LM, Sun B, Joerss T, Chen YS, Groth M, Krueger A, Platzer M, Yang YG, Rudolph KL, Wang ZQ (2015) Smg6/Est1 licenses embryonic stem cell differentiation via nonsense-mediated mRNA decay. EMBO J. doi:10.15252/embj.201489947
Li Z, Nagy PD (2011) Diverse roles of host RNA binding proteins in RNA virus replication. RNA Biol 8:305–315
Liu C, Karam R, Zhou Y, Su F, Ji Y, Li G, Xu G, Lu L, Wang C, Song M, Zhu J, Wang Y, Zhao Y, Foo WC, Zuo M, Valasek MA, Javle M, Wilkinson MF, Lu Y (2014) The UPF1 RNA surveillance gene is commonly mutated in pancreatic adenosquamous carcinoma. Nat Med 20:596–598. doi:10.1038/nm.3548
Loh B, Jonas S, Izaurralde E (2013) The SMG5-SMG7 heterodimer directly recruits the CCR4-NOT deadenylase complex to mRNAs containing nonsense codons via interaction with POP2. Genes Dev 27:2125–2138. doi:10.1101/gad.226951.113
Long H, Sabatier C, Ma L, Plump A, Yuan W, Ornitz DM, Tamada A, Murakami F, Goodman CS, Tessier-Lavigne M (2004) Conserved roles for Slit and Robo proteins in midline commissural axon guidance. Neuron 42:213–223
Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, Wilkinson MF (2014) Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep 6:748–764. doi:10.1016/j.celrep.2014.01.028
Lykke-Andersen J, Shu MD, Steitz JA (2000) Human Upf proteins target an mRNA for nonsense-mediated decay when bound downstream of a termination codon. Cell 103:1121–1131
Lykke-Andersen S, Chen Y, Ardal BR, Lilje B, Waage J, Sandelin A, Jensen TH (2014) Human nonsense-mediated RNA decay initiates widely by endonucleolysis and targets snoRNA host genes. Genes Dev 28:2498–2517. doi:10.1101/gad.246538.114
Lynch SA, Nguyen LS, Ng LY, Waldron M, McDonald D, Gecz J (2012) Broadening the phenotype associated with mutations in UPF3B: two further cases with renal dysplasia and variable developmental delay. Eur J Med Genet 55:476–479. doi:10.1016/j.ejmg.2012.03.010
Makeyev EV, Zhang J, Carrasco MA, Maniatis T (2007) The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol Cell 27:435–448. doi:10.1016/j.molcel.2007.07.015
Matlin AJ, Clark F, Smith CW (2005) Understanding alternative splicing: towards a cellular code. Nat Rev Mol Cell Biol 6:386–398. doi:10.1038/nrm1645
McIlwain DR, Pan Q, Reilly PT, Elia AJ, McCracken S, Wakeham AC, Itie-Youten A, Blencowe BJ, Mak TW (2010) Smg1 is required for embryogenesis and regulates diverse genes via alternative splicing coupled to nonsense-mediated mRNA decay. Proc Natl Acad Sci U S A 107:12186–12191. doi:10.1073/pnas.1007336107
Medghalchi SM, Frischmeyer PA, Mendell JT, Kelly AG, Lawler AM, Dietz HC (2001) Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viability. Hum Mol Genet 10:99–105
Mendell JT, Medghalchi SM, Lake RG, Noensie EN, Dietz HC (2000) Novel Upf2p orthologues suggest a functional link between translation initiation and nonsense surveillance complexes. Mol Cell Biol 20:8944–8957
Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC (2004) Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet 36:1073–1078. doi:10.1038/ng1429
Menendez JA, Mehmi I, Griggs DW, Lupu R (2003) The angiogenic factor CYR61 in breast cancer: molecular pathology and therapeutic perspectives. Endocr Relat Cancer 10:141–152
Mocquet V, Neusiedler J, Rende F, Cluet D, Robin JP, Terme JM, Duc Dodon M, Wittmann J, Morris C, Le Hir H, Ciminale V, Jalinot P (2012) The human T-lymphotropic virus type 1 tax protein inhibits nonsense-mediated mRNA decay by interacting with INT6/EIF3E and UPF1. J Virol 86:7530–7543. doi:10.1128/JVI.07021-11
Morris-Desbois C, Bochard V, Reynaud C, Jalinot P (1999) Interaction between the Ret finger protein and the Int-6 gene product and co-localisation into nuclear bodies. J Cell Sci 112(Pt 19):3331–3342
Morrison M, Harris KS, Roth MB (1997) smg mutants affect the expression of alternatively spliced SR protein mRNAs in Caenorhabditis elegans. Proc Natl Acad Sci U S A 94:9782–9785
Mort M, Ivanov D, Cooper DN, Chuzhanova NA (2008) A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat 29:1037–1047. doi:10.1002/humu.20763
Muhlrad D, Parker R (1999) Aberrant mRNAs with extended 3′ UTRs are substrates for rapid degradation by mRNA surveillance. RNA 5:1299–1307
Nagy E, Maquat LE (1998) A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci 23:198–199
Nakano K, Ando T, Yamagishi M, Yokoyama K, Ishida T, Ohsugi T, Tanaka Y, Brighty DW, Watanabe T (2013) Viral interference with host mRNA surveillance, the nonsense-mediated mRNA decay (NMD) pathway, through a new function of HTLV-1 Rex: implications for retroviral replication. Microbes Infect 15:491–505. doi:10.1016/j.micinf.2013.03.006
Narayanan K, Makino S (2013) Interplay between viruses and host mRNA degradation. Biochim Biophys Acta 1829:732–741. doi:10.1016/j.bbagrm.2012.12.003
Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O’Brien G, Shiue L, Clark TA, Blume JE, Ares M Jr (2007) Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev 21:708–718. doi:10.1101/gad.1525507
O’Donnell M, Chance RK, Bashaw GJ (2009) Axon growth and guidance: receptor regulation and signal transduction. Annu Rev Neurosci 32:383–412. doi:10.1146/annurev.neuro.051508.135614
Okada-Katsuhata Y, Yamashita A, Kutsuzawa K, Izumi N, Hirahara F, Ohno S (2012) N- and C-terminal Upf1 phosphorylations create binding platforms for SMG-6 and SMG-5:SMG-7 during NMD. Nucleic Acids Res 40:1251–1266. doi:10.1093/nar/gkr791
Oren YS, McClure ML, Rowe SM, Sorscher EJ, Bester AC, Manor M, Kerem E, Rivlin J, Zahdeh F, Mann M, Geiger T, Kerem B (2014) The unfolded protein response affects readthrough of premature termination codons. EMBO Mol Med 6:685–701. doi:10.1002/emmm.201303347
Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40:1413–1415. doi:10.1038/ng.259
Pan Y, Chen H, Siu F, Kilberg MS (2003) Amino acid deprivation and endoplasmic reticulum stress induce expression of multiple activating transcription factor-3 mRNA species that, when overexpressed in HepG2 cells, modulate transcription by the human asparagine synthetase promoter. J Biol Chem 278:38402–38412. doi:10.1074/jbc.M304574200
Perlick HA, Medghalchi SM, Spencer FA, Kendzior RJ Jr, Dietz HC (1996) Mammalian orthologues of a yeast regulator of nonsense transcript stability. Proc Natl Acad Sci U S A 93:10928–10932
Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G (1988) Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2:215–227
Polyak K, Weinberg RA (2009) Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer 9:265–273. doi:10.1038/nrc2620
Rahman L, Bliskovski V, Kaye FJ, Zajac-Kaye M (2004) Evolutionary conservation of a 2-kb intronic sequence flanking a tissue-specific alternative exon in the PTBP2 gene. Genomics 83:76–84
Ramage HR, Kumar GR, Verschueren E, Johnson JR, Von Dollen J, Johnson T, Newton B, Shah P, Horner J, Krogan NJ, Ott M (2015) A combined proteomics/genomics approach links hepatitis C virus infection with nonsense-mediated mRNA decay. Mol Cell 57:329–340. doi:10.1016/j.molcel.2014.12.028
Rigby RE, Rehwinkel J (2015) RNA degradation in antiviral immunity and autoimmunity. Trends Immunol 36:179–188. doi:10.1016/j.it.2015.02.001
Rouschop KM, van den Beucken T, Dubois L, Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W, Voncken JW, Lambin P, van der Kogel AJ, Koritzinsky M, Wouters BG (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest 120:127–141. doi:10.1172/JCI40027
Saltzman AL, Kim YK, Pan Q, Fagnani MM, Maquat LE, Blencowe BJ (2008) Regulation of multiple core spliceosomal proteins by alternative splicing-coupled nonsense-mediated mRNA decay. Mol Cell Biol 28:4320–4330. doi:10.1128/MCB.00361-08
Schmidt SA, Foley PL, Jeong DH, Rymarquis LA, Doyle F, Tenenbaum SA, Belasco JG, Green PJ (2015) Identification of SMG6 cleavage sites and a preferred RNA cleavage motif by global analysis of endogenous NMD targets in human cells. Nucleic Acids Res 43:309–323. doi:10.1093/nar/gku1258
Sempere LF, Freemantle S, Pitha-Rowe I, Moss E, Dmitrovsky E, Ambros V (2004) Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol 5:R13. doi:10.1186/gb-2004-5-3-r13
Serafini T, Kennedy TE, Galko MJ, Mirzayan C, Jessell TM, Tessier-Lavigne M (1994) The netrins define a family of axon outgrowth-promoting proteins homologous to C. elegans UNC-6. Cell 78:409–424
Serin G, Gersappe A, Black JD, Aronoff R, Maquat LE (2001) Identification and characterization of human orthologues to Saccharomyces cerevisiae Upf2 protein and Upf3 protein (Caenorhabditis elegans SMG-4). Mol Cell Biol 21:209–223. doi:10.1128/MCB.21.1.209-223.2001
Seward SL Jr, Gahl WA (2013) Hermansky-Pudlak syndrome: health care throughout life. Pediatrics 132:153–160. doi:10.1542/peds.2012-4003
Shepard PJ, Hertel KJ (2009) The SR protein family. Genome Biol 10:242. doi:10.1186/gb-2009-10-10-242
Sheth U, Parker R (2006) Targeting of aberrant mRNAs to cytoplasmic processing bodies. Cell 125:1095–1109. doi:10.1016/j.cell.2006.04.037
Shum EY, Jones SH, Shao A, Dumdie J, Krause MD, Chan WK, Lou CH, Espinoza JL, Song HW, Phan MH, Ramaiah M, Huang L, McCarrey JR, Peterson KJ, De Rooij DG, Cook-Andersen H, Wilkinson MF (2016) The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell. doi:10.1016/j.cell.2016.02.046
Silvain J, Collet JP, Nagaswami C, Beygui F, Edmondson KE, Bellemain-Appaix A, Cayla G, Pena A, Brugier D, Barthelemy O, Montalescot G, Weisel JW (2011) Composition of coronary thrombus in acute myocardial infarction. J Am Coll Cardiol 57:1359–1367. doi:10.1016/j.jacc.2010.09.077
Simons-Evelyn M, Young HA, Anderson SK (1997) Characterization of the mouse Nktr gene and promoter. Genomics 40:94–100. doi:10.1006/geno.1996.4562
Singh G, Kucukural A, Cenik C, Leszyk JD, Shaffer SA, Weng Z, Moore MJ (2012) The cellular EJC interactome reveals higher-order mRNP structure and an EJC-SR protein nexus. Cell 151:750–764. doi:10.1016/j.cell.2012.10.007
Singh G, Rebbapragada I, Lykke-Andersen J (2008) A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol 6, e111. doi:10.1371/journal.pbio.0060111
Smirnova L, Grafe A, Seiler A, Schumacher S, Nitsch R, Wulczyn FG (2005) Regulation of miRNA expression during neural cell specification. Eur J Neurosci 21:1469–1477. doi:10.1111/j.1460-9568.2005.03978.x
Sonenberg N, Hinnebusch AG (2009) Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745. doi:10.1016/j.cell.2009.01.042
Spellman R, Llorian M, Smith CW (2007) Crossregulation and functional redundancy between the splicing regulator PTB and its paralogs nPTB and ROD1. Mol Cell 27:420–434. doi:10.1016/j.molcel.2007.06.016
Stockklausner C, Breit S, Neu-Yilik G, Echner N, Hentze MW, Kulozik AE, Gehring NH (2006) The uORF-containing thrombopoietin mRNA escapes nonsense-mediated decay (NMD). Nucleic Acids Res 34:2355–2363. doi:10.1093/nar/gkl277
Sureau A, Gattoni R, Dooghe Y, Stevenin J, Soret J (2001) SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J 20:1785–1796. doi:10.1093/emboj/20.7.1785
Susova O, Gurtsevich VE (2003) The role of region pX in the life cycle of HTLV-I and in carcinogenesis. Mol Biol 37:392–403
Talloczy Z, Jiang W, Virgin HW, Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B (2002) Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 99:190–195. doi:10.1073/pnas.012485299
Tani H, Imamachi N, Salam KA, Mizutani R, Ijiri K, Irie T, Yada T, Suzuki Y, Akimitsu N (2012) Identification of hundreds of novel UPF1 target transcripts by direct determination of whole transcriptome stability. RNA Biol 9:1370–1379. doi:10.4161/rna.22360
Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, O’Meara S, Tofts C, Barthorpe S, Buck G, Cole J, Halliday K, Hills K, Jones D, Mironenko T, Perry J, Varian J, West S, Widaa S, Teague J, Dicks E, Butler A, Menzies A, Richardson D, Jenkinson A, Shepherd R, Raine K, Moon J, Luo Y, Parnau J, Bhat SS, Gardner A, Corbett M, Brooks D, Thomas P, Parkinson-Lawrence E, Porteous ME, Warner JP, Sanderson T, Pearson P, Simensen RJ, Skinner C, Hoganson G, Superneau D, Wooster R, Bobrow M, Turner G, Stevenson RE, Schwartz CE, Futreal PA, Srivastava AK, Stratton MR, Gecz J (2007) Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet 39:1127–1133. doi:10.1038/ng2100
Thiery JP, Sleeman JP (2006) Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol 7:131–142. doi:10.1038/nrm1835
Trusolino L, Comoglio PM (2002) Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat Rev Cancer 2:289–300. doi:10.1038/nrc779
Valacca C, Bonomi S, Buratti E, Pedrotti S, Baralle FE, Sette C, Ghigna C, Biamonti G (2010) Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. J Cell Biol 191:87–99. doi:10.1083/jcb.201001073
Vattem KM, Wek RC (2004) Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A 101:11269–11274. doi:10.1073/pnas.0400541101
Viegas MH, Gehring NH, Breit S, Hentze MW, Kulozik AE (2007) The abundance of RNPS1, a protein component of the exon junction complex, can determine the variability in efficiency of the Nonsense Mediated Decay pathway. Nucleic Acids Res 35:4542–4551. doi:10.1093/nar/gkm461
Walter P, Ron D (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334:1081–1086. doi:10.1126/science.1209038
Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB (2011) Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol 31:3670–3680. doi:10.1128/MCB.05704-11
Wei ML (2006) Hermansky-Pudlak syndrome: a disease of protein trafficking and organelle function. Pigment Cell Res 19:19–42. doi:10.1111/j.1600-0749.2005.00289.x
Weil JE, Beemon KL (2006) A 3′ UTR sequence stabilizes termination codons in the unspliced RNA of Rous sarcoma virus. RNA 12:102–110. doi:10.1261/rna.2129806
Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT (2008) NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev 22:1381–1396. doi:10.1101/gad.468808
Weischenfeldt J, Waage J, Tian G, Zhao J, Damgaard I, Jakobsen JS, Kristiansen K, Krogh A, Wang J, Porse BT (2012) Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol 13:R35. doi:10.1186/gb-2012-13-5-r35
Wek RC, Cavener DR (2007) Translational control and the unfolded protein response. Antioxid Redox Signal 9:2357–2371. doi:10.1089/ars.2007.1764
Wengrod J, Martin L, Wang D, Frischmeyer-Guerrerio P, Dietz HC, Gardner LB (2013) Inhibition of nonsense-mediated RNA decay activates autophagy. Mol Cell Biol 33:2128–2135. doi:10.1128/MCB.00174-13
Withers JB, Beemon KL (2010) Structural features in the Rous sarcoma virus RNA stability element are necessary for sensing the correct termination codon. Retrovirology 7:65. doi:10.1186/1742-4690-7-65
Wittmann J, Hol EM, Jack HM (2006) hUPF2 silencing identifies physiologic substrates of mammalian nonsense-mediated mRNA decay. Mol Cell Biol 26:1272–1287. doi:10.1128/MCB.26.4.1272-1287.2006
Wollerton MC, Gooding C, Wagner EJ, Garcia-Blanco MA, Smith CW (2004) Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol Cell 13:91–100
Xu X, Zhang L, Tong P, Xun G, Su W, Xiong Z, Zhu T, Zheng Y, Luo S, Pan Y, Xia K, Hu Z (2013) Exome sequencing identifies UPF3B as the causative gene for a Chinese non-syndrome mental retardation pedigree. Clin Genet 83:560–564. doi:10.1111/cge.12014
Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C (2010) The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29:2082–2096. doi:10.1038/emboj.2010.81
Yepiskoposyan H, Aeschimann F, Nilsson D, Okoniewski M, Muhlemann O (2011) Autoregulation of the nonsense-mediated mRNA decay pathway in human cells. RNA 17:2108–2118. doi:10.1261/rna.030247.111
Younis I, Green PL (2005) The human T-cell leukemia virus Rex protein. Front Biosci 10:431–445
Zahler AM, Lane WS, Stolk JA, Roth MB (1992) SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev 6:837–847
Acknowledgments
This research was funded by a grant from the Deutsche Forschungsgemeinschaft (GE2014/4-1) to N.H.G.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Ottens, F., Gehring, N.H. Physiological and pathophysiological role of nonsense-mediated mRNA decay. Pflugers Arch - Eur J Physiol 468, 1013–1028 (2016). https://doi.org/10.1007/s00424-016-1826-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00424-016-1826-5