
Abstract
This chapter reviews the recent introductory of the micronutrient iron and its association with brain degenerative diseases and oxidative stress. The essential sources, forms, and human requirements of iron were highlighted. The detailed information of iron’s bioactivity, metabolism, absorption, excretion, and regulation were also discussed. In addition, the proposed mechanisms behind the absorption, uptake, metabolism and transportation of iron were delineated. This chapter also described the current available knowledge regarding selective cases of brain degenerative diseases and their interplay with iron-induced oxidative stress.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Iron as an Essential Micronutrient
1.1 Introduction
Iron is an essential micromineral needed by most living organisms, including humans (Bao et al., 2012; Abbaspour et al., 2014; Wang & Babitt, 2019). Despite being found in small quantities in the human body and hence required in amounts less than100 mg/day in the adult diet, iron is a vital key player involved in a wide range of fundamental cellular and organismal metabolic processes that take place continuously in the body (Sizer & Whitney, 2013). These dynamic metabolic processes include oxygen transportation, cellular proliferation, energy production, host defense, neurotransmitter synthesis and function, and other oxidation-reduction reactions (Gupta, 2014; Samaniego-Vaesken et al., 2017; Gao et al., 2019). Therefore, decreased quantities or absence of iron in the body or its intakes from diet/ supplements hampers the proper function(s) of these vital processes resulting in the onset of various health symptoms and diseases. Indeed, iron deficiency and in it severe states; iron deficiency anemia is to date, a worldwide health concern recognized and highlighted by the World Health Organization in both developed and developing nations (WHO, 2004; Abbaspour et al., 2014; Samaniego-Vaesken et al., 2017). Excess iron, on the other hand, is also implicated with the manifestation of many other ailments; resulted from the destructive damage occurring to the body’s lipids, proteins, and DNA upon the elevation in the production of iron-induced free radicals (Kohgo et al., 2008; Wang & Babitt, 2019). Taking the above-mentioned notes which delineate the importance of iron and its precise homeostasis for the wellbeing of the human body; this chapter tackles important issues related to the mineral iron in the human body from iron’s dietary sources, its various forms present in the diet, the daily recommended intakes, to the overall ingestion to excretion pathway(s) of iron, iron function(s) in various parts of the body particularly in the brain, the body mechanisms for iron homeostasis and their crosstalk with oxidative stress, and finally the consequences of impaired iron homeostasis and brain diseases.
1.2 Dietary Sources, Forms and Requirements of Iron
1.2.1 Iron Dietary Sources
Being essential for proper body functioning and, at the same time, the incapability of the body to synthesize it for daily’s requirements, iron must be provided continuously by the diet/ supplements in considerable amounts (Sizer & Whitney, 2013). Meat, poultry, eggs, seafood/ fish, nuts, cereals, and vegetables are considered iron’s richest food sources. The major iron form in foods of animal origin is found centered in the hemoglobin and myoglobin molecules, while the common iron forms present exclusively in plant sources are low molecular weight compounds (e.g., ferric citrate) and high molecular weight compounds (e.g., ferritin). Moreover, iron is found in certain fortified foods and is available as pharmaceutical and dietary supplements. Iron as lactoferrin is found in infant formula, while fortified food and supplements of iron generally contain various forms of iron such as ferrous sulfate, ferric chloride, and ferrous gluconate, among others (Shayeghi et al., 2005; Vandevijvere et al., 2013; Zielińska-Dawidziak, 2015; Skolmowska & Głąbska, 2019).
1.2.2 Iron Forms
Absorbable iron found in the diet can be categorized into two main distinctive forms: heme iron (ferrous protoporphyrin IX) and non-heme iron (organic and inorganic freely metal). These two forms differ in their chemical structures, absorption and uptake processes, bioavailability, and food sources (Abbaspour et al., 2014; Ems & Huecker, 2020).
Heme iron is found as iron (II)-centered porphyrin (ferrous ion, Fe+2) that forms the prosthetic group within many proteins and is mainly derived from the diet upon the breakdown of the two major abundant proteins found only in animal sources: hemoglobin and myoglobin (West & Oates, 2008; De Carli et al., 2018). Due to its unique chemical structure being incorporated and protected by the protein structure carrying it and the presence of specific heme transporters (e.g., heme carrier protein 1, HCP1) that facilitate its movement across the cell membranes and into the bloodstream, heme iron is highly bioavailable and can reach up to 40% being absorbed from the diet during an iron deficiency state (Abbaspour et al., 2014; Shubham et al., 2020). Moreover, dietary components such as phytates, oxalates, and polyphenols, have no effect on the absorption of this form of iron. High pH in the small intestine renders the solubility of certain inorganic iron forms, which in turn reduces their absorption, but has minimal effect on the heme iron uptake and absorption (Pizarro et al., 2003; Wang & Babitt, 2019).
Non-heme iron, on the other hand, is found purely in plant-based, fortified food products, and as a mixer alongside the heme iron in animal products. This form is commonly found as freely iron metal in the oxidized or reduced forms, with the inorganic ferric ion (Fe+3) as the main non-heme iron form present in plants (Abbaspour et al., 2014; Moris et al., 2021). Owing to their chemical structures, the absorption of the non-heme iron, excluding plant ferritin, is usually less uniform, and certain limiting factors (e.g., quantities in the diet, duodenal pH, presence or absence of enhancers /inhibitors factors) reduces their bioavailability, reaching to merely an average of 1–15% being absorbed from the diet (Young et al., 2018; Ems & Huecker, 2020). Nonetheless, non-heme iron quantities in the diet are greater by folds than that of the heme iron, making it the chief contributor for dietary iron (West & Oates, 2008; Samaniego-Vaesken et al., 2017).
1.2.3 Human Requirements of Iron
Human daily requirements for iron differ according to age, gender, health status, food source, and body’s iron status (Samaniego-Vaesken et al., 2017; Ems & Huecker, 2020). Any disruption in the iron’s homeostats in the body can cause either iron deficiency or iron excess, which in turn can cause serious adverse health effects and concerns. Hence, specific requirement values were set for iron alongside other vital nutrients to support the body’s needs to accommodate the various demographic variance among individuals and establish proper iron homeostasis. As a result, the Food and Nutritional Board (FNB) of the National Academies (NASEM), US, developed a term known as “Dietary References Intakes” (DRI), in which it sets reference values for nutrient intakes used in planning and assessing diets of healthy individuals (National Institutes of Health, 2016).
In general, infants in their early stages get their iron requirements fully from their mothers’ milk. However, after 4–6 months of birth, mother’s milk becomes insufficient, and the demand for iron increases drastically. As age increases, the DRI of iron increases reaching approximately 8 mg/day at 13 years old. During the adolescent’s period, iron requirements again increase due to the rapid body growth and puberty spurt, with females requiring a DRI of 15 mg/day with an additional 2.9 mg/day over that value and males requiring an additional 1.1 mg/day with their regular DRI value of 11 mg/day. The significant increment of iron DRI value for adolescent females compared to adolescent males compensates for the iron loss via blood during menstruation. As for the adulthood period, the DRI for adult men and postmenopausal women is set at 8 mg/day, whereas fertile adult women who require more are set with DRI of 18 mg/day. Pregnant women also require higher iron intake for the rapid growth of the placenta, fetus, and maternal red blood cells requiring almost 27 mg/ day of iron intake (Samaniego-Vaesken et al., 2017; National Institutes of Health, 2016).
Gastrointestinal (GI) disorders such as inflammatory bowel disease, celiac disease, Heliobacter infections, and other malabsorption-related diseases reduce iron uptake by the body, causing iron deficiency. Therefore, patients with these conditions require extra iron DRI values than those required by healthy individuals (Usman et al., 2019; Montoro-Huguet et al., 2021).
As discussed earlier, plant-based foods contain only the non-heme form of iron which is generally characterized by low bioavailability due to its chemical structure that is greatly affected by the body’s pH and the presence of other plant components that interfere with its absorption (Samaniego-Vaesken et al., 2017; Young et al., 2018). Thus, logically, vegetarians, especially vegan individuals who rely strictly on plants for their diet, require higher iron DRI requirements than those who consume significant amounts of animal products. Indeed, the iron needs for vegetarians particularly vegan, may reach up to 1.8 times more than those required by non-vegetarians (Pawlak & Bell, 2017; National Institutes of Health, 2016).
It is well-known that regardless of the type of iron (heme or non-heme) and the dietary factors that promote or hamper iron absorption, the determinant factor that regulates the uptake and absorption rate of this essential mineral relies on the concept of the systematic iron need of the body. This means that regardless of the richness of the diet with iron, the absorption rate increases or decreases based on the iron status of the body. A significant increment in the absorption rate of iron is noticed upon the onset of iron deficiency state, whereas reduced absorption rate takes place when iron depots in the body are replenished and filled up (Samaniego-Vaesken et al., 2017). This mechanism, alongside other body pathways, helps maintain the homeostasis of iron inside the body, compensating for any iron losses as well as preventing toxicity and iron overloading in vital body organs (Kohgo et al., 2008; Loncar et al., 2021).
1.3 Iron Bioavailability
Generally, the onset of micronutrient deficiencies is due to the low dietary intake represented by taking diets with low to null micronutrient composition. Logically, increasing their intakes by consuming diets rich in these nutrients or the ingestion of their purified pharmaceutical supplementation should replenish the shortage and subside their deficiency’s symptoms. However, this is not the case in iron deficiency, indicating the presence of other determinantal factors (West & Oates, 2008; Abbaspour et al., 2014). Bioavailability, defined as the amount of ingested nutrient being absorbed and available for body’s utilization, is one undeniable factor influencing the dietary availability of many vital micronutrients including iron. Indeed, dietary enhancer and inhibitory factors profoundly influence the availability of iron for absorption. These diet-related factors exert different action according to the type of iron found in the diet (De Carli et al., 2018; Young et al., 2018; Ems & Huecker, 2020). Due to its chemical structures (soluble Fe+2 and predominantly the insoluble Fe+3), dietary non-heme iron show low bioavailability that is profoundly influenced by the dietary components (promoters or inhibitors), pH, and oxidation activity. Phytates, phenolic compounds, and calcium, among others, are major inhibitors of iron absorption that function as either: (1) chelator agents converting non-heme iron into insoluble complexes, (2) binding agents affecting iron’s uptake and transport, or (3) competitors binding with iron’s absorption sites. The alkaline pH present in the lumen and the oxidation capacity also renders the non-heme iron bioavailability. High pH readily oxidizes the soluble Fe+2 to insoluble Fe+3 increasing by that the percentage of unavailable non-heme iron for absorption. Meanwhile, dietary enhancers such as ascorbic acid and meat proteins promote acidity condition, the ideal environment for non-heme iron solubility and absorption, maximizing by that the bioavailability of non-heme iron. Dietary heme iron, on the other hand, being in a protective chemical structure is unaffected by the above-mentioned factors (both inhibitors or promoter factors), and therefore show high bioavailability rates (Hunt, 2005; West & Oates, 2008; Abbaspour et al., 2014; Dasa & Abera, 2018).
1.4 Iron Metabolism
The human body contains around 3–5 g of iron on average (around 55 mg and 44 mg/kg of body weight for men and women, respectively). Due to its vast utility and necessity and if overload, toxicity in the body, these quantities of iron in the body must be kept within an optimal physiological range (Gkouvatsos et al., 2012; Abbaspour et al., 2014; Ems & Huecker, 2020). The body maintains such balanced iron quantities via strict and tight iron metabolism pathways, which controls the mechanisms of absorption, transport, storage, utilization, excretion, and the systematic regulations of iron in the body (Frazer & Anderson, 2014; Gao et al., 2019; Wang & Babitt, 2019).
1.4.1 Iron Uptake, Absorption, and Transportation
The absorption of iron (heme or non-heme) predominantly occurs in the duodenum and the upper jejunum. For heme iron, Wyllie and Kaufman (1982) and Gräsbeck et al. (1982) described two possible mechanisms for its uptake by the enterocyte: (1) Pinocytosis-like uptake mechanism (Wyllie & Kaufman, 1982) or (2) Binding with a specific heme transporter; the HCP1 (Gräsbeck et al., 1982). Recent evidence has shown that HCP1 is also coupled with another protein carrier know as proton-coupled folate transporter (PCFT) (Qiu et al., 2006; Antileo et al., 2013). Despite being primarily allocated for folate transportation, PCFT/HCP1 also has a notable role in heme iron transportation. Once inside the enterocyte, heme containing proteins are catabolized by both heme oxygenase and biliverdin reductase enzymes, releasing inorganic iron and bilirubin, respectively. The form of released inorganic iron from these heme proteins is the non-heme iron, Fe+2 (Pizarro et al., 2003; Moris et al., 2021).
Non-heme iron, on the contrary, requires more elaborated absorption mechanisms than that of the heme iron (Pizarro et al., 2003; Moris et al., 2021). Typically, non-heme iron absorption occurs mainly in the apical membrane of the enterocyte with the aid of a specific transporter called the divalent-metal transporter-1 (DMT-1). This protein transporter selectively transports the soluble iron Fe+2 rapidly than the insoluble form of iron, Fe+3. Since the predominantly non-heme iron form found in the diet is the Fe+3, and the rapid oxidization of available plant’s Fe+2 to the Fe+3 at the body’s physiological pH, the bioavailability of these non-heme iron forms is significantly influenced by the pH co-existing in the duodenum and the presence of other dietary constituents (Abbaspour et al., 2014; Moris et al., 2021). The pH dilemma is somehow overcomed by the action of the body’s gastric acid, which lowers the pH of the proximal duodenum allowing for a ferric reductase enzyme, duodenal cytochrome B (Dcytb), to reduce Fe+3 to Fe+2, facilitating DMT-1 function to readily uptake and transport the soluble iron Fe+2 into the enterocyte. Hence, impairment in the gastric acid production can substantially reduce the non-heme iron absorption percentage by the body (Kohgo et al., 2008; Frazer & Anderson, 2014). Similar to body’s pH condition, the presence of dietary enhancers/ inhibitors that either augment the solubility or hinder the absorption process profoundly affects the absorption of these non-heme iron forms from the diet (Ems & Huecker, 2020). These dietary enhancers/ inhibitors of iron will be further explained later in the bioavailability section.
An independent mechanism has also been reported by Theil et al. (2012) regarding a second possible absorption pathway for the non-heme, yet high bioavailable iron form; the plant ferritin. The clathrin-dependent endocytic mechanism, which is still under investigation; hypothesizes that intact plant ferritin that escapes the digestive enzymes binds to the surface of the enterocytes, and then is internalized within the enterocytic cells. The fate of ferritin inside the enterocytic cells remains unclear (Kalgaonkar & Lönnerdal, 2009; Theil et al., 2012). Regardless the presence or absence of this unique absorption mechanism, it is crucial to highlight the fact that the bulk of plant ferritins seem to be subjected to degradation upon food preparation/processing and digestion, thereby freeing the iron within it as inorganic non-heme iron, which in turn is absorbed through the normal DMT-1 pathway (Abbaspour et al., 2014; Gao et al., 2019).
The absorption of iron in the duodenum and the upper jejunum is the first and, by most, the key step in iron metabolism and homeostasis. This means, that even with adequate and sufficient intakes of iron via diet or supplements, the efficiency of iron absorption is governed mainly by the status of iron’s stores in the body. An inverse association is found between iron absorption and the body’s iron stores which is directly correlated with the erythropoiesis rate. This explains the wide observed variations in iron absorption among healthy individuals ingesting almost the same quantities of dietary/ supplements of iron (Frazer & Anderson, 2014; Moris et al., 2021). Nevertheless, factors such as food types, food-food interactions, food-drug interactions, the timing of food consumption, and gastrointestinal tract’s pH can also solely or jointly modulate the extraction of the iron from the diet, and therefore should not be neglected (Pizarro et al., 2003; De Carli et al., 2018).
As mentioned above, the fate of iron inside the enterocytes depends on the iron status in the body. When the body cells are saturated with iron, the excess iron released by the enterocytes binds to apoferritin forming ferritin molecules, cellular storage form of iron. At the end of enterocytes’ lifecycle, these cells are shed into the intestinal lumen (exfoliation of intestinal epithelium), and the unused stored iron in ferritin is lost (Abbaspour et al., 2014; Frazer & Anderson, 2014). On the other hand, when there is a demand on iron, free internalized Fe+2 in enterocytes (the converted absorbable form of heme and non-heme iron coming from diet) is transported across the basolateral membrane and into blood circulation via iron efflux protein ferroportin 1 (FPN1). Excreted freely Fe+2 is then re-oxidized into Fe+3 by either a membrane-bound copper dependent ferroxidase hephaestin or circulating caeruloplasmin, before actively binding with the serum glycoprotein transferrin (Tf), which serves as the major delivery vehicle for iron to the body’s utilization/ storage sites (Petrak & Vyoral, 2005; Gao et al., 2019; Thirupathi & Chang, 2019).
1.4.2 Iron Utilization, Circulation, Reutilization, and Storage
The approximately 3–5 g of iron found in the human body are mainly incorporated in the red blood cells’ (RBCs) pigment (2.5 g), the hemoglobin, while around 3–4 mg of body’s iron exist as a circulating pool of Fe- transferrin (Fe-Tf). The remaining portion is stored as ferritin complexes in all cells and major storage organs (Garrick & Garrick, 2009; Thirupathi & Chang, 2019). Iron enters body’s cells by binding with the transferrin receptor 1 (TfR1) found on cells’ surface, undergoes endocytosis releasing the iron as Fe+3 from Tf. Ferrireductase six-transmembrane epithelial antigen of prostate 3 (STEAP3) reduces Fe+3 to Fe+2, which is then easily transported to cell’s cytoplasm via DMT1. Intracellular iron can be either transported to the cell’s mitochondria via mitoferrin 1 and/or 2,5-dihydroxybenzoic acid for vital metabolic utilization. Cells can either use iron for the synthesis of hemoglobin, non-hemoglobin hemoproteins (e.g. myoglobin, cytochromes, catalases), and iron sulfur clusters (Fe-S), store it as cytosolic ferritin, or export iron from cells via FPN1 when exceeding cell’s needs. Minute and tightly regulated amounts of Fe+2 are kept as liable ions in cells, participating in the oxidation-reduction reactions, promoting cell proliferation, and facilitating cell signaling (Abbaspour et al., 2014; Anderson & Frazer, 2017; Galaris et al., 2019; Gao et al., 2019).
Normally and under physiological condition, merely 1 mg of dietary iron is being absorbed by the human intestine daily, which also accounts for the same amount of iron lost from the body on a daily basis. Hence, the body’s iron requirements rely momentously on the recycling process of the intracellular iron, and to a lesser extent on the iron provided by diet/ supplements (Abbaspour et al., 2014; Gao et al., 2019). The bulk of the body’s iron content found in RBCs is reutilized with the help of the reticuloendothelial macrophages. Effete RBCs (normally after 120 days) are scavenged and subjected to phagocytosis by the resident macrophages present in bone marrow, spleen, and liver. Inside the macrophages, senescent RBCs are dismantled, and the resulted heme is further catabolized by the action of heme oxygenase-1 (HO-1) releasing the iron (Fe+2) within. Free Fe+2 goes through the normal route of being circulated via FPN1, re-oxidized to Fe+3 by either hephaestin or circulating caeruloplasmin, loaded to Tf, and finally is reutilized for erythropoiesis and/ or other cellular processes, or stored in many body’s organs, mainly the liver, spleen, and bone marrow (Kohgo et al., 2008; Garrick & Garrick, 2009). The exact molecular mechanisms of iron reutilization in the body to date have many unknowns, but it is crystal clear that these mechanisms are considered the major suppliers of the essential iron for the body.
Excess iron in the body is bonded and stored in two major storage proteins namely: ferritin and hemosiderin (Anderson & Frazer, 2017; Galaris et al., 2019). Ferritin is the major body’s iron-storage protein characterized with a high binding capacity (holding up to approximately 4500 atoms of iron) and an easily and readily releasing ability of iron for all body cells when needed. Under steady conditions, serum ferritin concentrations reflect the total body’s iron stores, commonly used as a laboratory indicator for the body’s iron content. Hemosiderin, on the other hand, is a complex iron storage protein that results from the degradation of iron-overloaded ferritin molecules via lysosomes. This storage form is prominent in macrophages upon the degradation of erythrocytes and in patients with iron-load diseases. Unlike ferritin, hemosiderin is poorly available to supply iron for body’s needs. Despite the differences between two iron storage proteins, ferritin and hemosiderin play an important role in iron homeostasis, for both don’t merely serve as vital iron reservoir needed to sustain body’s various metabolic pathways, but also act as protective mechanisms by which the body sequesters toxic iron ions (Fe+2 and Fe+3) in a nontoxic insoluble forms. Moreover, both proteins reflect the body’s iron concentrations in physiological and pathological conditions (Abbaspour et al., 2014; Gao et al., 2019).
1.4.3 Iron Excretion and Regulation
Minute losses of iron of around 1 mg/day are resulted from the physiological exfoliation of cells from the epithelial surfaces. Substantial losses of iron from the body can be due to hemorrhage, menstruation, or pregnancy. Apart from that, there are no active or regulated iron elimination and excretion mechanisms from the body (Petrak & Vyoral, 2005; Abbaspour et al., 2014).
Maintaining iron homeostasis in the body is vital for both iron deficiency and iron excess are equally detrimental to the body’s cells. Therefore, body’s iron homeostasis is controlled by tight regulatory mechanisms at both cellular and systemic levels (Petrak & Vyoral, 2005; Carocci et al., 2018; Katsarou & Pantopoulos, 2020). At the cellular level, iron homeostasis is governed by iron regulatory proteins 1 and 2 (IRP1 and IRP2) which modulate the expression of proteins involved in iron uptake, traffic, utilization, and storage. IRPs regulate the expressions of iron proteins namely, DMT1, FPN1, TfR1, and ferritin on the mRNAs level via binding to their iron responsive elements (IRE). Combined, IRP-IRE system responds to cellular iron deficiency by increasing iron import via the enhancement of DMT1 mRNA expression, while obstructing the iron storage proteins’ uptake, storage, and export through decreasing the stability of TfR1, ferritin, and FPN1 mRNAs. The opposite occurs when an excess in cellular iron is detected, DMT1 mRNA expression is reduced and TfR1, ferritin, and FPN1 mRNAs expressions are stimulated (Gao et al., 2019; Hermann et al., 2021). At the systemic level, iron homeostasis is maintained by keeping a strict balance between iron intake and iron utilization and egress. As body lacks an active excretion mechanism for iron, this regulation is mainly controlled on the absorption sites. A peptide hormone known as hepcidin plays a central role in the iron homeostasis at the systemic level. Synthesized mainly by the liver, and to a lower level expressed in macrophages, adipocytes, and brain, hepcidin negatively regulates body’s iron, inhibiting both intestinal absorption and reticuloendothelial release of iron (Kohgo et al., 2008; Abbaspour et al., 2014; Frazer & Anderson, 2014; Wang & Babitt, 2019). When iron levels are elevated beyond cellular needs under normal physiological conditions or in pathological cases such as inflammation, hepcidin kicks in and binds to FPN1 found on intestinal duodenum cells and other targeted cells, causing FPN1 internalization and degradation in lysosomes. The loss of FPN1 from targeted cells prevents iron absorption and its entry into the body’s plasma. This in turn lowers Tf saturation of iron and hence lessen cellular uptake of iron. In addition to the degradation action of FPN1, hepcidin blocks iron efflux from this protein, minimizing by that the release of unwanted iron. Conversely, the release of hepcidin from liver is reduced upon physiological condition of normal cellular iron deficiency or under pathological conditions such as the genetic disorder of erythropoietic protoporphyria. The absence of the inhibitory activity of hepcidin maximizes the absorption of dietary iron by the intestinal duodenum cells to replenish the body’s iron content (Galaris et al., 2019; Nemeth & Ganz, 2009). Aside the individual’s internal iron status controlled by tight cellular and systemic mechanisms, it is important to remember that iron’s absorption is also influenced by external factors such as dietary composition, pH, iron form, and oxidation activity that affect the bioavailability of iron at the absorption sites (West & Oates, 2008; Dasa & Abera, 2018; Gao et al., 2019).
1.5 Brain Iron
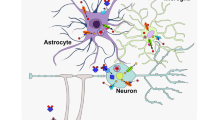
The amount of iron in the brain accounts for merely 2% or less of that of the total body iron content, yet the homeostasis of this minute quantity is vital for the brain’s normal physiological functions (Gupta, 2014; Carocci et al., 2018; Ems & Huecker, 2020). Like its fundamental functions in other body cells, iron acts as a prosthetic group in various key enzymes crucial in oxygen transportation, high mitochondrial respiration, and vital redox signalling in brain cells. In addition, iron present in brain is involved in the neurotransmitters, myelin sheets, and DNA synthesis. Iron, particularly the non-heme iron tyrosine hydroxylase, is also needed for the myelination formation and the synthesis of the dopamine, norepinephrine, and serotonin neurotransmitters (Singh, 2014; Carocci et al., 2018; Ferreira et al., 2019). Disruption of the iron homeostasis (deficiency or excess) in the brain results in severe defects of brain’s motor, cognitive, and behavioral functions. Due to the high vulnerability of brain cells and their life determining roles, brain rests behind a physical barrier known as the blood-brain barrier (BBB) that regulates the exchange of materials with the circulating blood. BBB acts as a selective fence that protects brain from harmful molecules found in the circulating blood and maintains the optimal microenvironment ions content including iron needed for neurological signaling (Benarroch, 2009; Ferreira et al., 2019). Plasma Fe-Tf selectively enters brain cells across BBB’s apical (blood side) membrane that resides between brain vascular endothelial cells (BVECs). There, BVECs uptake iron via two main mechanisms: (1) Tf/ TfR1 pathway and (2) non-transferrin bound iron (NTBI). The BVECs’ Tf/ TfR1 pathway follows the same process used in most body cells, starting with the binding of Fe-Tf with TfR1, Fe-Tf-TfR1complex endocytosis, acidification and release of Fe+3, reduction of Fe+3 to Fe+2, movement of Fe+2 across endosomal membrane, and lastly into various brain cells (Singh, 2014; McCarthy & Kosman, 2015; Duck et al., 2017; Qian & Ke, 2019). Alternatively, the NTBI pathway allows iron uptake directly by brain cells through apical DMT1, H-ferritin (iron storage protein with high capacity), lactoferrin, melanotransferrin (p97), transcytosis of Tf/ TfR1 complexes pathways, and others. The exact mechanisms of these pathways are still poorly understood and heavily investigated to date (Mills et al., 2010; McCarthy & Kosman, 2015). After being released into brain’s interstitial fluid, iron is distributed and absorbed unevenly by various brain cells in different brain regions. Thus, different brain regions/ structures have different iron concentrations, with substantia nigra, dentate gyrus, and red nucleus containing the highest iron levels. Such variance supports the hypothesis of the high susceptibility to oxidative damage and speedy denaturation rates exhibited by certain brain regions/ structures compared to others (McCarthy & Kosman, 2015; Carocci et al., 2018; Yu & Chang, 2019). Neuroglia (non-neurons) and neurons (nerve cells), the two type cells found in brain, possess different iron uptake mechanisms. Each of neuroglia’s three cell types, astrocytes, microglia, and oligodendrocytes, possesses a unique iron uptake mechanism. Astrocytes acquire iron via NTBI different pathways such as DMT1, citrate, and ATP. Microglia, on the other hand, acquire iron via either Tf/ TfR1 pathway or NTBI pathways, based on the presence of certain stimuli such as inflammation. Meanwhile, mature oligodendrocytes which are the brain cells with the highest iron content due to their active metabolic needs for myelination, acquire iron via Tf/ TfR1 pathway and/ or the NTBI pathway; H-ferritin. Classical Tf/ TfR1 pathway or NTBI pathway (DMT1) are the mechanisms by which brain neurons acquire iron (West & Oates, 2008; Antileo et al., 2013; Yu & Chang, 2019). Iron fate, depending on brain-iron status, can be either mobilized for various neuroglia and neurons’ metabolism, stored as cytosolic ferritin and/ or as labile iron pool if found in excess, or when overloaded, it is released by FPN1 and is effluxed to the interstitial fluid, where iron is sent back to the blood circulation via Tf/ TfR1 pathway and/ or the NTBI pathways (Duck et al., 2017; Carocci et al., 2018; Qian & Ke, 2019; Yu & Chang, 2019). Iron homeostasis in the brain is tightly controlled by regulators both at the cellular level via IRP-IRE system and at the systemic level via hepcidin (Abbaspour et al., 2014; Gao et al., 2019; Thirupathi & Chang, 2019).
Observing the significant role(s) of iron for normal brain activities and the tight regulation systems aimed to maintain its homeostasis within brain cells, brain neuropathies are expected upon the diminish or excess of this essential iron in brain cells (Mills et al., 2010; Duck et al., 2017). Iron deficiency at early life stages hampers brain cells division, growth, and development. In addition, the absence of iron causes defects in the functional properties of brain cells. Iron anemia and progressive mental retardation are the early and late manifestations of iron deficiency. Iron overload, on the other hand, has been detected in various neurodegenerative conditions and is considered one major culprit in the onset and progression of these diseases via the initiation of the detrimental oxidative stress (Carocci et al., 2018; Galaris et al., 2019). Moreover, defects of several iron regulatory proteins found in brain were further detected in brain cells of individuals with neurodegenerative disorders. Hence, the basic understanding of the concept of oxidative stress, iron homeostasis and redox signalling, and the crosstalk between iron imbalance and the initiation of oxidative stress are of vital clinical relevance for degenerative diseases affecting the brain (Benarroch, 2009; Liguori et al., 2018; Galaris et al., 2019).
2 Oxidative Stress, Iron Overload, and Brain Degenerative Diseases
2.1 The Concept of Oxidative Stress and Redox Signaling
Oxidative stress, coined by Helmut Sies in 1985, is best defined as the balance disturbance occurring between the formation levels of the so-called reactive species (free radicals) and their cellular clearance levels from cells via defense mechanisms (antioxidants) (Pizzino et al., 2017; Sharma et al., 2018). Reactive species/ free radicals namely; reactive oxygen (ROS) and reactive nitrogen species (RNS); are the normal by-products of various bodily metabolic processes. Mitochondria, being the core of most metabolic pathways (e.g. TCA cycle, B-oxidation, urea cycle, oxidative phosphorylation), continuously generates these reactive species/free radicals as by-products, as well as molecules needed for other various body functions. Differentiation, apoptosis, immunity, protein phosphorylation, activation and expression of transcriptional factors, among others, require certain levels of cellular ROS and RNS for proper functioning (Gutteridge & Halliwell, 2018; Ifeanyi, 2018; Alkadi, 2020). Meanwhile, inflammation, cancer, stress, overload of heavy metals, and neurogenerative disorders are the pathological endogenous sources of these notorious ROS and RNS. The physiological or pathological nature of ROS/RNS; is profoundly governed by the so-called “redox homeostatic balance”. Specific body sensors detect variations in the redox balance, resulting in the initiation of a cascade signalling ending with the activation of antioxidant defensive system. Comprising of enzymatic and non-enzymatic components, this antioxidant system via various mechanisms maintains the levels of ROS/RNS within the normal safe threshold (Collins et al., 2012; Liguori et al., 2018). However, dysfunctional of the defensive antioxidant systems and/ or other bodily metabolic processes that disturb the balance between ROS/RNS and the antioxidant system favouring the former, result in the onset of damaging cascade oxidative events that ends with the oxidation and denaturation of body’s proteins, lipids, and DNA. Exogenous sources of ROS/RNS such as cigarettes, pollutants, certain dietary components, drugs, cooking methods, and radiation can also trigger the overproduction of ROS/RNS in the body, promoting harmful oxidative stress status and its deleterious consequences (Galaris et al., 2019; Ferreira et al., 2019; Mahmoud et al., 2020).
2.2 The Crosstalk Between Iron and Redox Signaling and the Consequences of Iron Homeostasis Impairment
The exact chemical properties that contribute to iron’s essentiality to the human body are also implicated to the onset of various oxidative-induced diseases (Anderson & Frazer, 2017; Pizzino et al., 2017; Thirupathi & Chang, 2019). The iron’s ability to donate electrons is needed for the functioning of various bodily redox cycles. However, once iron levels exceed normal physiological levels, this essential ability of exchanging electrons dramatically shift to the negative activity of ROS/RNS overproduction, initiating the damaging oxidative stress condition (Grubman et al., 2014; Carocci et al., 2018; Gutteridge & Halliwell, 2018). It is important to note that iron overload itself is not always toxic per se. Some clinical cases detected the occurrence of iron overload in body organs, yet these organs did not show any injury (Carocci et al., 2018). Furthermore, the onset of iron-overload diseases such as neurodegenerative diseases do not occur instantly upon iron accumulation but rather require longer periods, indicating the involvement of other possible factors. However, it is important to note that one important factor influencing iron overload toxicity it’s the ability to produce the damaging oxidative condition within cells. Based on this notion, the extent to which iron overload can reach to a toxicity level variers partially or totally with the: (1) biochemical form of cellular iron, (2) localization of the iron deposits, and (3) presence/ absence of cellular defensive mechanisms (Carocci et al., 2018; Mahmoud et al., 2020). Iron in living organisms is mostly found in sequestered forms, incorporated within vital cellular components such as haemoglobin, myoglobin, and ferritin. Being in such forms deter their interaction with cellular reactive species such as superoxide anion (O2− ˙) and hydrogen peroxidase (H2O2) and further reduce iron’s presence for harmful microorganisms (Carocci et al., 2018; Gutteridge & Halliwell, 2018). However, minute quantities of iron can be also found as reduced (Fe+2) and oxidized (Fe+3) liable forms. Being able to interchangeably transform between these two forms and denote electrons to oxygen molecules, Fe+2 and Fe+3 are potential endogenic producers of ROS (directly) and RNS (indirectly), the species involved in numerous oxidative-induced diseases (Chiueh, 2001; Thirupathi & Chang, 2019; Alkadi, 2020). Fe+2 including Fe+2-citrate complexes interact with cellular oxygen, generating a continuous flux of the reactive species O2− ˙, H2O2, and hydroxyl radicals (OH ˙) via Fenton reaction or Haber-Weiss reaction. Moreover, the reaction between Fe+2 and the reactive specie O2− ˙ convert this iron form to a more prooxidative state, Fe+3. Fe+3, in turn, reacts with H2O2 to produce the highly reactive OH ˙ via Fenton reaction. Nitric oxide (NO ˙) radicals (most common cellular RNS) are also produced by the oxidation of specific protein’s amino acid by iron generated ROS. These radicals, in turn reacts with O2− ˙ generating other harmful reactive species (e.g. peroxynitrite anions; ONOO−). Highly reactive peroxyl (ROO ˙) and alkoxyl (RO ˙) radicals are too generated by the interaction of excess iron with oxidized lipid via Fe+2-dependent lipid peroxidation. These lipid’s by-products possess longer half-lives than O2− ˙, H2O2, and OH ˙ radicals, hence, causing chronic toxicity and severe DNA damage (Gutteridge & Halliwell, 2018; Ifeanyi, 2018; Mahmoud et al., 2020). Non-enzymatic oxidation of dopamine facilitated by iron redox-reaction generates the reactive species of semiquinones and H2O2 at the electron transport chain within the mitochondria. This mitochondrial insult further initiates and propagates the iron release as Fe+2 and Fe+3 ions from non-toxic cellular iron forms (e.g. iron-sulphur clusters, myoglobin). Collectively, these iron-induced reactive species attack the vital cellular macromolecules, the lipids, proteins, and DNA. In addition to their denaturation and loss of function, the oxidation of these cellular macromolecules forms another positive loop of generated reactive species that exacerbates the detrimental oxidative stress effect which ultimately ends with cell’s demise (Benarroch, 2009; Mills et al., 2010). The toxicity of iron overload also depends on its subcellular localization (cytosolic, lysosomal, and mitochondrial) within the body cells, including brain cells. The pH co-existing within these organelles and their cellular functions may deter or enhance the prooxidant activity of iron. Most iron transport proteins are designed for the soluble, less prooxidant iron form, Fe+2 which is maintained in that form in acidic environment. While high pH oxidizes Fe+2 to the lesser soluble, more prooxidant iron form, Fe+3. Despite the ability of both iron forms, Fe+2 and Fe+3 in forming harmful ROS/RNS, the different pH found in the lysosomes (acidic pH), cytoplasm (near to neutral pH), and mitochondria (neutral to alkaline pH) may further aggravate or render this prooxidant activity (Kohgo et al., 2008; Ferreira et al., 2019). Lysosome acidity keeps circulated iron as Fe+2 to be sequestered via binding with other cellular components, reducing its ability to participate in harmful oxidation processes. Mitochondria, on the other hand, keeps circulated iron as prooxidant Fe+3 required for respiration and oxidative phosphorylation. Moreover, being the metabolic central of the cell and a site for endogenous generation of ROS, mitochondria with excess iron deposit, may result in the fast and severe adverse iron toxicity compared to the iron accumulation in either lysosome or cytoplasm (Galaris et al., 2019). Hence, the localization of the iron overload determines the toxicity of this metal and should not be overlooked.
Normally, both free prooxidative iron forms, Fe+2 and Fe+3, are sequestered in the body into inert unactive forms, unavailable for redox-inducing activities. In addition to the different body processes designed to maintain Fe+2 and Fe+3 as inert unactive forms (e.g. ferritin), body’s defensive/ antioxidant systems kick in once levels of ROS/ RNS induced by iron overload, are beyond the physiological threshold. Indeed, the presence or absence of a precise antioxidant system(s) determine the level of iron overload toxicity imposed on the cells. The presence of BBB is considered the first brain defensive mechanism against excess levels of iron. BBB acts as a selective barrier that regulates the uptake of circulated iron from plasma to brain cells. On another level, within different brain cells, an integrated antioxidant system made of the major antioxidant enzymes superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (Gpx); low molecular weight reductants ascorbate (vitamin C), α-tocopherol (vitamin E), and glutathione (GSH); and metal chelators ceruloplasmin and metallothionein co-exists (Wilson, 1997; Grubman et al., 2014; Berndt & Lillig, 2017; Golko-Perez et al., 2017; Ifeanyi, 2018). The antioxidant enzyme copper/zinc SOD and magnesium SOD react with O2− ˙ and change it to H2O2, which in turn, is converted by the antioxidant enzymes CAT and Gpx to nontoxic water molecules. Vitamin C and E act as direct radical scavengers, interacting with O2− ˙, H2O2, and other lipid peroxidases, stopping further oxidation of vital cellular components (especially membrane lipids) and thus, terminating the cycle of reactive species generation. Meanwhile, GSH protects brain cells from the oxidative stress via multiple modes. Similar to vitamin C and E, GSH coupled with its generator by-product glutathione disulfide (GSSG) act as a direct reactive species’ scavenger. GSH is also important for iron homeostasis with its role in the biosynthesis of Fe-S clusters (Devos et al., 2014; Berndt & Lillig, 2017; Carocci et al., 2018; Galaris et al., 2019; Alkadi, 2020; Dwivedi et al., 2020). Metal chelators; ceruloplasmin and metallothionein; protect brain cells form oxidative stress by reducing the iron overload. On one hand, ceruloplasmin, a multicopper enzyme with ferroxidase activity, regulates iron egress from brain cells by oxidizing Fe+2 and loading it on Tf (Musci et al., 2014). On the other hand, metallothionein is a cysteine-rich metal chelator that binds with various metals including iron, preventing iron overload and the consequence of iron-induced oxidative status (Ullio et al., 2015). Together, these defensive systems protect brain cells from iron-induced oxidative stress. Nevertheless, the increased permeability of BBB or/and the defect/ alteration of the antioxidant systems in brain cells upon aging process, genetic disorders (e.g. hemochromatosis), and/ or introduction of exogenous iron (e.g. heavy metal pollutants) may lead to imbalance in the iron homeostasis favouring iron accumulation, triggering the onset of the damaging oxidative stress in brain, and occurrence of iron-induce brain neuropathies (Wilson, 1997; Duck et al., 2017).
2.3 Iron Overload, Oxidative Stress, and Brain Degenerative Diseases (Selective Cases)
Pathological conditions describing brain iron overload coupled with the onset of oxidative stress are well-recognized and are designated as iron-induced neurodegenerative diseases. They are characterized by the overproduction of various reactive species, oxidation and dysfunction of vital cellular macromolecules, neurological toxicity, and brain cells’ death (Benarroch, 2009; Mills et al., 2010; Grubman et al., 2014; Pizzino et al., 2017; Carocci et al., 2018). Overproduced ROS/RNS promotes the oxidative stress situation characterized by glutathione consumption and depletion, protein denaturation and aggregation, phospholipids remodelling and destabilization, and DNA alteration and destruction (Carocci et al., 2018; Dwivedi et al., 2020). Classically, iron-overload diseases can be either primary (genetic) or secondary iron-overload diseases. Primary iron-overload diseases are the result of genetic defects in iron transport or regulation mechanisms. Hemochromatosis, a primary (genetic) iron-overload disease, is characterised by reduced to null expression of hepcidin, making the body unable to limit dietary iron intake at the absorption sites in the intestine, hence creating the iron-overload condition. The iron-overload toxicity associated with this genetic disease produces severe clinical manifestations throughout multiple body’s organs including the brain (Carocci et al., 2018; Ferreira et al., 2019). On the contrary, secondary iron-overload diseases are mainly due to long-term blood transfusion treatment used for severe anaemia condition (e.g. thalassemia). Iron overload here occurs from the excess exogenous iron from blood transfusion treatment coupled with the ineffective body usage of iron for the synthesis of RBCs (erythropoiesis). Again, this secondary iron-overload disease promotes the dysfunction and death of multiple body organs’ cells including brain cells (Kohgo et al., 2008; Anderson & Frazer, 2017). In addition, normal ageing process or inflammation may result in the uncontrollable accumulation of iron,- producing age-related or inflammation-related neurodegenerative diseases such as Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Piperno, 1998; Grubman et al., 2014; Thirupathi & Chang, 2019). To date, the exact role of iron-overloading in neurodegenerative diseases as either the primary etiology causing these diseases or as a secondary consequence resulted upon the occurrence of neurodegenerative diseases remains unclear. Nevertheless, it is evident that AD, PD, Amyotrophic lateral sclerosis (ALS), and other destructive neuropathies, all have one prevailing hallmark; the iron-overload and its induced oxidative stress impact (Mills et al., 2010; Grubman et al., 2014). It also important to note that the brain’s unique structure with specific features, makes it more prone to oxidative stress than other organs. Brain is highly vulnerable to oxidative stress, due to being a highly oxygen consumer organ with rich unsaturated fatty acids content, the main component of the neurons’ myelin sheets and the mediator for impulse transmission. Furthermore, the high levels of prooxidants (iron and vitamin C) concomitant with the lower repair capacity of brain cells, cause the damage by the oxidative stress fuelled by iron-overload calamitous and irreversible (Walker et al., 2016; Salim, 2017; Carocci et al., 2018).
AD is the most frequent type of the neurodegenerative disease with an early symptom of lost memory with a decline in cognitive and motor functions, that ends with dementia (Pratico, 2008; Pizzino et al., 2017; Ferreira et al., 2019). Despite the obfuscate etiology causing AD, oxidative stress by high ROS flux in brain cells mediated by iron has been implicated as one major culprit in its pathophysiology. Supporting such notion was the numerous reports of (1) high iron content detected in amyloid β plaques (as iron-amyloid complex), neurofibrillary tangles, and even injured neurons of post-mortem brains from AD’s patients, (2) amplified oxidative stress observed in AD brain cells by iron-amyloid complexes through the production of H2O2 radicals, (3) presence of OH ˙ radicals produced by the help of iron via Fenton reaction that causes the aggregation of amyloid β in AD patients, (4) altered expressions of iron proteins responsible for iron’s storage, transport, and regulation found in AD patients, and finally (5) iron accumulation reduction in various brain regions of AD patients by drugs acting as iron chelator (e.g. deferiprone) alleviating some AD symptoms. These observations indicate the implication of iron overload in the pathogenesis of AD; however, iron-induce oxidation in AD mechanism(s) bears many uncertainties and requires more investigations (Zecca et al., 2004; Jiang et al., 2009; Telling et al., 2017; Carocci et al., 2018).
PD, on the other hand, is the second most frequent type of neurodegenerative diseases after AD, with early symptoms of tremor, loss of posture, bradykinesia, changes in writing and speech skills, with late stages symptoms of insomnia, depression, and progressive cognitive and motor abnormalities. Again, neurological studies have pointed out the undeniable impact of iron overload coupled with oxidative stress in the pathogenesis of PD (Mills et al., 2010; Grubman et al., 2014; Hernandez-Baltazar et al., 2018). Confirming the implication of iron overload in PD was through the following observations: (1) Elevated iron levels were recorded in the examined substantia nigra of PD patients, (2) α-synuclein aggregates (high in iron deposits) were found to alter brain’s mitochondria activity, triggering overproduction of ROS and RNS, and severe depletion of cellular antioxidant reductant GSH, (3) iron auto-oxidation of dopamine and production of lethal ROS were documented in PD patients, (4) accumulation of ferritin in the macrophage microglial cells forming ferritin-reactive microglia was also observed in the degenerative PD brain cells, and lastly (5) administration of pharmacological or genetic iron chelation increased the survival rate of dopaminergic neurons, thus delaying the PD progression (Carocci et al., 2018; Kausar et al., 2018; Varešlija et al., 2020). Jointly, these observations indicate the implication of iron overload in PD. However, the exact mechanism(s) by which iron overload affect the brain and its role in PD still needs further illustration.
ALS, known also as Charcot’s disease, is a fatal sporadic or familial neurodegenerative disease with pronounced deterioration and death of the motor neurons of the spinal cord and brain (Grubman et al., 2014; Zhang et al., 2020). Iron overload has been also implicated with ALS pathogenesis evident by: (1) irregulates of various mitochondrial iron proteins discerned in both in vitro and in vivo ALS studies, (2) prevalence of mutated hemochromatosis gene (act as an intracellular iron sensor when binding to Tf) leading to iron homeostasis disturbance and consequence oxidative damage, and lastly (3) usage of iron chelator prolonged the life span of ALS animal models. Similar to AD and PD, the primary or secondary iron overload mechanisms in ALS is still unclear and warrants additional studies (Grubman et al., 2014; Golko-Perez et al., 2017; Carocci et al., 2018; Gao et al., 2019; Zhang et al., 2020).
Other neurodegenerative diseases such as Friedrich’s ataxia (FRDA), Huntington’s disease (HD), and neurodegeneration with brain iron accumulation (NBIA), demonstrated a strong association between their onsets and/or progression with iron accumulation in various brain regions. Despite the unclarity in which initiates the other, iron accumulation or the destructive oxidation condition detected in these neurodegeneration disorders, one thing is confirmed; the initiation of both iron overload and the oxidative stress are bidirectional and their uncontrolled presences in brain cells have profound degenerative impacts (Grubman et al., 2014; Galaris et al., 2019; Gao et al., 2019).
3 Conclusion
The cruciality of the micronutrient iron to the lives of all living organisms is undeniable and is evident by multitudinous research and scientific investigations. Hence, the need to identify and understand all biochemical aspects related to this mineral deemed logical and necessary. Iron is found in various food items at different concentrations and in various chemical forms, causing diversity in its bioavailability for bodily usage and consequently the fluctuating needs for this mineral. Besides its bioavailability differences, iron’s quantity also differs throughout the human life cycle, imposing distinct iron daily requirements based on age and gender. The delineated overall metabolic pathways of iron starting from ingestion to excretion and its functions in various parts of the body particularly in the brain, further enforces the essentiality of this mineral for maintaining a healthy body. Nevertheless, the tight regulation governing iron’s cellular concentrations indicates the presence of a harmful and destructive side of iron. Indeed, mounted evidence has shown significant correlation between iron overload and various neurodegenerative diseases including brain degenerative diseases. This iron-induced degenerative activity is concomitant with the onset of the destructive oxidative stress status, affecting vital cellular biomolecules including brain’s cells. Together, these notes show that iron behaves as a two-edged sword, where both iron’s deficiency and excess are equally harmful to the body, hence, delicate regulations of iron concentrations in the body are absolutely needed.
References
Abbaspour, N., Hurrell, R., & Kelishadi, R. (2014). Review on iron and its importance for human health. Journal of Research in Medical Sciences, 19(2), 164.
Alkadi, H. (2020). A review on free radicals and antioxidants. Infectious Disorders-Drug Targets (Formerly Current Drug Targets-Infectious Disorders), 20(1), 16–26.
Anderson, G. J., & Frazer, D. M. (2017). Current understanding of iron homeostasis. The American Journal of Clinical Nutrition, 106(suppl_6), 1559S–1566S.
Antileo, E., Garri, C., Tapia, V., Muñoz, J. P., Chiong, M., Nualart, F., et al. (2013). Endocytic pathway of exogenous iron-loaded ferritin in intestinal epithelial (Caco-2) cells. American Journal of Physiology-Gastrointestinal and Liver Physiology, 304(7), G655–G661.
Bao, W., Rong, Y., Rong, S., & Liu, L. (2012). Dietary iron intake, body iron stores, and the risk of type 2 diabetes: A systematic review and meta-analysis. BMC Medicine, 10(1), 1–13.
Benarroch, E. E. (2009). Brain iron homeostasis and neurodegenerative disease. Neurology, 72(16), 1436–1440.
Berndt, C., & Lillig, C. H. (2017). Glutathione, glutaredoxins, and iron. Antioxidants & Redox Signaling, 27(15), 1235–1251.
Carocci, A., Catalano, A., Sinicropi, M. S., & Genchi, G. (2018). Oxidative stress and neurodegeneration: The involvement of iron. Biometals, 31(5), 715–735.
Chiueh, C. C. (2001). Iron overload, oxidative stress, and axonal dystrophy in brain disorders. Pediatric Neurology, 25(2), 138–147.
Collins, Y., Chouchani, E. T., James, A. M., Menger, K. E., Cochemé, H. M., & Murphy, M. P. (2012). Mitochondrial redox signalling at a glance. Journal of Cell Science, 125(4), 801–806.
Dasa, F., & Abera, T. (2018). Factors affecting iron absorption and mitigation mechanisms: A review. International Journal of Agricultural Science and Food Technology, 4(2), 024–030.
De Carli, E., Dias, G. C., Morimoto, J. M., Marchioni, D. M. L., & Colli, C. (2018). Dietary iron bioavailability: Agreement between estimation methods and association with serum ferritin concentrations in women of childbearing age. Nutrients, 10(5), 650.
Devos, D., Moreau, C., Devedjian, J. C., Kluza, J., Petrault, M., Laloux, C., et al. (2014). Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxidants & Redox Signaling, 21(2), 195–210.
Duck, K. A., Simpson, I. A., & Connor, J. R. (2017). Regulatory mechanisms for iron transport across the blood-brain barrier. Biochemical and Biophysical Research Communications, 494(1–2), 70–75.
Dwivedi, D., Megha, K., Mishra, R., & Mandal, P. K. (2020). Glutathione in brain: Overview of its conformations, functions, biochemical characteristics, quantitation and potential therapeutic role in brain disorders. Neurochemical Research, 45(7), 1461–1480.
Ems, T., & Huecker, M. R. (2020). Biochemistry, iron absorption. StatPearls [Internet].
Ferreira, A., Neves, P., & Gozzelino, R. (2019). Multilevel impacts of iron in the brain: The cross talk between neurophysiological mechanisms, cognition, and social behavior. Pharmaceuticals, 12(3), 126.
Frazer, D. M., & Anderson, G. J. (2014). The regulation of iron transport. BioFactors, 40(2), 206–214.
Galaris, D., Barbouti, A., & Pantopoulos, K. (2019). Iron homeostasis and oxidative stress: An intimate relationship. Biochimica et Biophysica Acta (BBA)-Molecular Cell Research, 1866(12), 118535.
Gao, G., Li, J., Zhang, Y., & Chang, Y. Z. (2019). Cellular iron metabolism and regulation. Brain Iron Metabolism and CNS Diseases, 21–32.
Garrick, M. D., & Garrick, L. M. (2009). Cellular iron transport. Biochimica et Biophysica Acta (BBA)-General Subjects, 1790(5), 309–325.
Gkouvatsos, K., Papanikolaou, G., & Pantopoulos, K. (2012). Regulation of iron transport and the role of transferrin. Biochimica et Biophysica Acta (BBA)-General Subjects, 1820(3), 188–202.
Golko-Perez, S., Amit, T., Bar-Am, O., Youdim, M. B., & Weinreb, O. (2017). A novel iron chelator-radical scavenger ameliorates motor dysfunction and improves life span and mitochondrial biogenesis in SOD1 G93A ALS mice. Neurotoxicity Research, 31(2), 230–244.
Gräsbeck, R., Majuri, R., Kouvonen, I., & Tenhunen, R. (1982). Spectral and other studies on the intestinal haem receptor of the pig. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology, 700(2), 137–142.
Grubman, A., White, A. R., & Liddell, J. R. (2014). Mitochondrial metals as a potential therapeutic target in neurodegeneration. British Journal of Pharmacology, 171(8), 2159–2173.
Gupta, C. P. (2014). Role of iron (Fe) in body. IOSR Journal of Applied Chemistry, 7(11), 38–46.
Gutteridge, J. M., & Halliwell, B. (2018). Mini-review: Oxidative stress, redox stress or redox success? Biochemical and Biophysical Research Communications, 502(2), 183–186.
Hermann, E., Oliveira, T., & Montgomery, M. (2021). The contributions of iron regulatory proteins 1 and 2 to ferroptosis activation and ferroptotic cell death. Cancer Research, 81(13_Supplement), 2006-2006.
Hernandez-Baltazar, D., Nadella, R., Rovirosa-Hernandez, M. J., Zavala-Flores, L. M., & Rosas-Jarquin, C. J. (2018). Animal model of Parkinson disease: Neuroinflammation and apoptosis in the 6-hydroxydopamine-induced model. Experimental animal models of human diseases [internet]. Rijeka: InTech, 375–393.
Hunt, J. R. (2005). Dietary and physiological factors that affect the absorption and bioavailability of iron. International Journal for Vitamin and Nutrition Research, 75(6), 375–384.
Ifeanyi, O. E. (2018). A review on free radicals and antioxidants. International Journal of Current Research on Medicinal Science, 4(2), 123–133.
Jiang, D., Li, X., Williams, R., Patel, S., Men, L., Wang, Y., & Zhou, F. (2009). Ternary complexes of iron, amyloid-β, and nitrilotriacetic acid: Binding affinities, redox properties, and relevance to iron-induced oxidative stress in Alzheimer’s disease. Biochemistry, 48(33), 7939–7947.
Kalgaonkar, S., & Lönnerdal, B. (2009). Receptor-mediated uptake of ferritin-bound iron by human intestinal Caco-2 cells. The Journal of Nutritional Biochemistry, 20(4), 304–311.
Katsarou, A., & Pantopoulos, K. (2020). Basics and principles of cellular and systemic iron homeostasis. Molecular Aspects of Medicine, 75, 100866.
Kausar, S., Wang, F., & Cui, H. (2018). The role of mitochondria in reactive oxygen species generation and its implications for neurodegenerative diseases. Cell, 7(12), 274.
Kohgo, Y., Ikuta, K., Ohtake, T., Torimoto, Y., & Kato, J. (2008). Body iron metabolism and pathophysiology of iron overload. International Journal of Hematology, 88(1), 7–15.
Liguori, I., Russo, G., Curcio, F., Bulli, G., Aran, L., Della-Morte, D., et al. (2018). Oxidative stress, aging, and diseases. Clinical Interventions in Aging, 13, 757.
Loncar, G., Obradovic, D., Thiele, H., von Haehling, S., & Lainscak, M. (2021). Iron deficiency in heart failure. ESC heart failure.
Mahmoud, I. F., Kanthimathi, M. S., & Aziz, A. A. (2020). ROS/RNS-mediated apoptosis in HT-29 colorectal cancer cells by methanolic extract of Tamarindus indica seeds. European Journal of Integrative Medicine, 40, 101244.
McCarthy, R. C., & Kosman, D. J. (2015). Mechanisms and regulation of iron trafficking across the capillary endothelial cells of the blood-brain barrier. Frontiers in Molecular Neuroscience, 8, 31.
Mills, E., Dong, X. P., Wang, F., & Xu, H. (2010). Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Medicinal Chemistry, 2(1), 51–64.
Montoro-Huguet, M. A., Belloc, B., & Domínguez-Cajal, M. (2021). Small and large intestine (I): Malabsorption of nutrients. Nutrients, 13(4), 1254.
Moris, W., Verhaegh, P. L., Verbeek, J., Swinkels, D. W., Laarakkers, C. M., Masclee, A. A., … & van Deursen, C. T. B. (2021). Absorption of non-heme iron during gastric acid suppression in patients with hereditary hemochromatosis and healthy controls. American Journal of Physiology-Gastrointestinal and Liver Physiology.
Musci, G., Polticelli, F., & di Patti, M. C. B. (2014). Ceruloplasmin-ferroportin system of iron traffic in vertebrates. World Journal of Biological Chemistry, 5(2), 204.
National Institutes of Health. (2016). Nutrient recommendations: Dietary Reference Intakes (DRIs).
Nemeth, E., & Ganz, T. (2009). The role of hepcidin in iron metabolism. Acta Haematologica, 122(2–3), 78–86.
Pawlak, R., & Bell, K. (2017). Iron status of vegetarian children: A review of literature. Annals of Nutrition and Metabolism, 70(2), 88–99.
Petrak, J., & Vyoral, D. (2005). Hephaestin—A ferroxidase of cellular iron export. The International Journal of Biochemistry & Cell Biology, 37(6), 1173–1178.
Piperno, A. (1998). Classification and diagnosis of iron overload. Haematologica, 83(5), 447–455.
Pizarro, F., Olivares, M., Hertrampf, E., Mazariegos, D. I., & Arredondo, M. (2003). Heme-iron absorption is saturable by heme-iron dose in women. The Journal of Nutrition, 133(7), 2214–2217.
Pizzino, G., Irrera, N., Cucinotta, M., Pallio, G., Mannino, F., Arcoraci, V., et al. (2017). Oxidative stress: Harms and benefits for human health. Oxidative Medicine and Cellular Longevity, 2017.
Pratico, D. (2008). Evidence of oxidative stress in Alzheimer's disease brain and antioxidant therapy: Lights and shadows. Annals of the New York Academy of Sciences, 1147(1), 70–78.
Qian, Z. M., & Ke, Y. (2019). Brain iron transport. Biological Reviews, 94(5), 1672–1684.
Qiu, A., Jansen, M., Sakaris, A., Min, S. H., Chattopadhyay, S., Tsai, E., et al. (2006). Identification of an intestinal folate transporter and the molecular basis for hereditary folate malabsorption. Cell, 127(5), 917–928.
Salim, S. (2017). Oxidative stress and the central nervous system. Journal of Pharmacology and Experimental Therapeutics, 360(1), 201–205.
Samaniego-Vaesken, M., Partearroyo, T., Olza, J., Aranceta-Bartrina, J., Gil, Á., González-Gross, M., et al. (2017). Iron intake and dietary sources in the Spanish population: Findings from the ANIBES study. Nutrients, 9(3), 203.
Sharma, G. N., Gupta, G., & Sharma, P. (2018). A comprehensive review of free radicals, antioxidants, and their relationship with human ailments. Critical Reviews™ in Eukaryotic Gene Expression, 28, 2.
Shayeghi, M., Latunde-Dada, G. O., Oakhill, J. S., Laftah, A. H., Takeuchi, K., Halliday, N., et al. (2005). Identification of an intestinal heme transporter. Cell, 122(5), 789–801.
Shubham, K., Anukiruthika, T., Dutta, S., Kashyap, A. V., Moses, J. A., & Anandharamakrishnan, C. (2020). Iron deficiency anemia: A comprehensive review on iron absorption, bioavailability and emerging food fortification approaches. Trends in Food Science & Technology, 99, 58–75.
Singh, N. (2014). The role of iron in prion disease and other neurodegenerative diseases. PLoS Pathogens, 10(9), e1004335.
Sizer, F., & Whitney, E. (2013). Nutrition: Concepts and controversies. Cengage Learning.
Skolmowska, D., & Głąbska, D. (2019). Analysis of heme and non-heme iron intake and iron dietary sources in adolescent menstruating females in a national polish sample. Nutrients, 11(5), 1049.
Telling, N. D., Everett, J., Collingwood, J. F., Dobson, J., van der Laan, G., Gallagher, J. J., et al. (2017). Iron biochemistry is correlated with amyloid plaque morphology in an established mouse model of Alzheimer's disease. Cell Chemical Biology, 24(10), 1205–1215.
Theil, E. C., Chen, H., Miranda, C., Janser, H., Elsenhans, B., Núñez, M. T., et al. (2012). Absorption of iron from ferritin is independent of heme iron and ferrous salts in women and rat intestinal segments. The Journal of Nutrition, 142(3), 478–483.
Thirupathi, A., & Chang, Y. Z. (2019). Brain iron metabolism and CNS diseases. Brain Iron Metabolism and CNS Diseases, 1–19.
Ullio, C., Brunk, U. T., Urani, C., Melchioretto, P., Bonelli, G., Baccino, F. M., & Autelli, R. (2015). Autophagy of metallothioneins prevents TNF-induced oxidative stress and toxicity in hepatoma cells. Autophagy, 11(12), 2184–2198.
Usman, M., Mehmood, H. O., Iqbal, N., Babar, S., & Ghazanfer, S. (2019). Malabsorption; acase of Microcytic Anemia in Pakistan. Baqai Journal of Health Sciences, 2(2).
Vandevijvere, S., Michels, N., Verstraete, S., Ferrari, M., Leclercq, C., Cuenca-García, M., et al. (2013). Intake and dietary sources of haem and non-haem iron among European adolescents and their association with iron status and different lifestyle and socio-economic factors. European Journal of Clinical Nutrition, 67(7), 765–772.
Varešlija, D., Tipton, K. F., Davey, G. P., & McDonald, A. G. (2020). 6-hydroxydopamine: A far from simple neurotoxin. Journal of Neural Transmission, 127(2), 213–230.
Walker, T., Michaelides, C., Ekonomou, A., Geraki, K., Parkes, H. G., Suessmilch, M., et al. (2016). Dissociation between iron accumulation and ferritin upregulation in the aged substantia nigra: Attenuation by dietary restriction. Aging (Albany NY), 8(10), 2488.
Wang, C. Y., & Babitt, J. L. (2019). Liver iron sensing and body iron homeostasis. Blood, The Journal of the American Society of Hematology, 133(1), 18–29.
West, A. R., & Oates, P. S. (2008). Mechanisms of heme iron absorption: Current questions and controversies. World Journal of Gastroenterology: WJG, 14(26), 4101.
Wilson, J. X. (1997). Antioxidant defense of the brain: A role for astrocytes. Canadian Journal of Physiology and Pharmacology, 75(10–11), 1149–1163.
World Health Organization. (2004). Focusing on anaemia: Towards an integrated approach for effective anaemia control. World Health Organization.
Wyllie, J. C., & Kaufman, N. (1982). An electron microscopic study of heme uptake by rat duodenum. Laboratory Investigation, 47(5), 471–476.
Young, I., Parker, H. M., Rangan, A., Prvan, T., Cook, R. L., Donges, C. E., et al. (2018). Association between haem and non-haem iron intake and serum ferritin in healthy young women. Nutrients, 10(1), 81.
Yu, P., & Chang, Y. Z. (2019). Brain iron metabolism and regulation. Brain Iron Metabolism and CNS Diseases, 33–44.
Zecca, L., Youdim, M. B., Riederer, P., Connor, J. R., & Crichton, R. R. (2004). Iron, brain ageing and neurodegenerative disorders. Nature Reviews Neuroscience, 5(11), 863–873.
Zhang, Q. Q., Jiang, H., Li, C. Y., Liu, Y. L., & Tian, X. Y. (2020). H63D CG genotype of HFE is associated with increased risk of sporadic amyotrophic lateral sclerosis in a single population. Journal of Integrative Neuroscience, 19(3), 495–499.
Zielińska-Dawidziak, M. (2015). Plant ferritin—A source of iron to prevent its deficiency. Nutrients, 7(2), 1184–1201.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Mahmoud, I.F., Alhaj, O.A., Jahrami, H.A. (2023). The Interplay between Iron and Oxidative Stress in Brain Neurodegenerative Diseases. In: Mohamed, W., Brogazzi, N.L., Kostrzewa, R.M. (eds) Brain-Iron Cross Talk. Nutritional Neurosciences. Springer, Singapore. https://doi.org/10.1007/978-981-19-7327-7_2
Download citation
DOI: https://doi.org/10.1007/978-981-19-7327-7_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-19-7326-0
Online ISBN: 978-981-19-7327-7
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)