
Abstract
The aim of the present study was to evaluate the therapeutic effect of the novel neuroprotective multitarget brain permeable monoamine oxidase inhibitor/iron chelating-radical scavenging drug, VAR10303 (VAR), co-administered with high-calorie/energy-supplemented diet (ced) in SOD1G93A transgenic amyotrophic lateral sclerosis (ALS) mice. Administration of VAR-ced was initiated after the appearance of disease symptoms (at day 88), as this regimen is comparable with the earliest time at which drug therapy could start in ALS patients. Using this rescue protocol, we demonstrated in the current study that VAR-ced treatment provided several beneficial effects in SOD1G93A mice, including improvement in motor performance, elevation of survival time, and attenuation of iron accumulation and motoneuron loss in the spinal cord. Moreover, VAR-ced treatment attenuated neuromuscular junction denervation and exerted a significant preservation of myofibril regular morphology, associated with a reduction in the expression levels of genes related to denervation and atrophy in the gastrocnemius (GNS) muscle in SOD1G93A mice. These effects were accompanied by upregulation of mitochondrial DNA and elevated activities of complexes I and II in the GNS muscle. We have also demonstrated that VAR-ced treatment upregulated the mitochondrial biogenesis master regulator, peroxisome proliferator-activated receptor-γ co-activator 1α (PGC-1α) and increased PGC-1α-targeted metabolic genes and proteins, such as, PPARγ, UCP1/3, NRF1/2, Tfam, and ERRα in GNS muscle. These results provide evidence of therapeutic potential of VAR-ced in SOD1G93A mice with underlying molecular mechanisms, further supporting the importance role of multitarget iron chelators in ALS treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dysregulation of iron homeostasis and the associated oxidative stress are essential pathogenic factors in neurodegenerative diseases, including amyotrophic lateral sclerosis (ALS) (Oshiro et al. 2011; Santillo et al. 2009). Iron has been shown to be accumulated in spinal cord and cerebrospinal fluid in ALS patients (Kasarskis et al. 1995; Kokic et al. 2005), as well as in spinal cord and skeletal muscles of ALS models in rodents (Halon et al. 2014; Jeong et al. 2009; Kupershmidt et al. 2009; Wang et al. 2011; Winkler et al. 2014). Previous studies have shown a deposition of hemoglobin-derived iron within the central nervous system (CNS) and correlation of the pathological deterioration in ALS patients with elevated serum ferritin and microglial iron accumulation in the motor cortex (Ikeda et al. 2012; Kwan et al. 2012; Winkler et al. 2013). Additionally, analyses of postmortem human and cerebrospinal fluid (CSF) in ALS patients have indicated that impairment of blood brain barrier (BBB) and blood spinal cord barrier (BSCB), resulting in accumulation of different plasma proteins, erythrocytes, erythrocyte-derived hemoglobin, iron-containing hemosiderin, and various neurotoxic blood components in the spinal cord and motor cortex, may contribute to early motoneuron degeneration (Zlokovic 2011; Miyazaki et al. 2011; Winkler et al. 2013; Zhao et al. 2015). In support, previous studies demonstrated that treatment with blood coagulation factor, activated protein C (APC) exerted beneficial effects in motoneurons during early and late disease stages in SOD1G93A mice, presumably via repair or preservation of BSCB integrity (Winkler et al. 2014; Zhong et al. 2009).
In addition, it has been reported that intervention in iron accumulation with the use of iron chelators significantly delayed the disease onset and increased life span in transgenic mice expressing ALS-linked SOD1 mutations (Jeong et al. 2009; Winkler et al. 2014). Previously, we have shown that the multifunctional iron chelating/monoamine oxidase (MAO) inhibitory drug, M30 exerted neuroprotective effects in NSC-34 motoneuron cells and SOD1G93A mice (Kupershmidt et al. 2009; Wang et al. 2011). In addition, our recent study demonstrated that the combined administration of M30 with high caloric energy supplemented diet (ced) produced additive effects on motor performance and increased survival of SOD1G93A mice (Golko-Perez et al. 2016). Preliminary observations revealed that M30-ced combined treatment increased messenger RNA (mRNA) expression levels in several mitochondrion-related genes in the gastrocnemius (GNS) muscle in SOD1G93A mice, including peroxisome proliferator-activated receptor-γ (PPARγ), PPARγ co-activator 1 alpha (PGC-1α), and uncoupling protein (UCP) 1 (Golko-Perez et al. 2016).
Another multifunctional member of the novel series of multitarget, non-toxic, and brain permeable drugs, designed and synthesized by our research group, is the iron chelating/MAO inhibitory compound, VAR10303 (VAR). VAR was recently shown to afford iron chelation, iron-induced lipid peroxidation inhibitory potency, and brain-selective MAO-A and -B inhibitory effects (Bar-Am et al. 2015). In addition, VAR was found to exert neuroprotective and neurorescue activities in rat 6-hydroxydomaine (6-OHDA) and mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) Parkinson’s disease models and beneficial effects on age-related alterations in rats (Bar-Am et al. 2015).
In the present study, we applied the multifunctional compound, VAR in combination with ced (VAR-ced) in symptomatic ALS SOD1G93A mice to assess the possible neurorescue effect of the drug. We have determined motor function, motoneuron degeneration and survival, and analyzed some potential protective mechanisms of VAR-ced treatment, in the GNS muscle, including PGC-1α, PGC-1α-related genes, mitochondrial DNA, and complexes I and II activities.
Methods
Materials
The multifunctional iron chelator, VAR10303 [5-(N-methyl-N propargylaminomethyl)-8 hydroxyquinoline] was synthesized and kindly provided by Varinel Inc. (Philadelphia, PA USA). High-fat diet chow was purchased from Research Diets, Inc. Materials for histological analyses: tissue-freezing medium (Triangle Biomedical Sciences, Durham, NC, USA); anti-choline acetyltransferase (ChAT) polyclonal antibody (Chemicon, EMD Millipore, USA); goat anti rabbit IgG antibody (Chemicon, EMD Millipore, USA); Alexa-594-α-bungarotoxin (Invitrogen, USA); anti-neurofilament (SMI-31) (Covance Inc., USA); anti-synaptophysin (Chemicon, EMD Millipore, USA). The following materials were used for real-time RT-PCR: PurfectPure RNA Tissue Kit (5′PRIME Inc., USA); PrimeScript™ RT reagent kit and SYBR Premix Ex Taq (TaKaRa Bio Inc. Korea); primers for gene analyses was purchased from QIAGEN, USA. The following antibodies were used for Western blotting analyses: mouse monoclonal anti-low (L)-ferritin (Santa Cruz Biotechnology, USA); rabbit anti PGC-1α (Epitomics Inc., Burlingame, CA, USA); rabbit monoclonal anti-myocyte enhancer factor 2C (MEF2C), mouse monoclonal anti-glucose transporter type 4 (GluT4), and mouse monoclonal anti-NADH dehydrogenase 1 alpha subcomplex subunit 9 (NDUFA9) (Cell Signaling Technology Inc. Beverly, MA, USA); mouse monoclonal anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Merck Millipore, Germany). C2C12 muscle skeletal myoblasts cells were obtained from American Type Culture Collection (ATCC, Manassas, VA). Mitotracker green FM (Invitrogen, USA); lipophilic cationic probe 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide (JC-1) kit (Immunochemistry Technologies, MN, USA). Other Chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Animal Treatment
All procedures were carried out in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals, and were approved by the Animal Ethics Committee of the Technion, Haifa, Israel. SOD1G93A transgenic familial ALS female mice (high copy number; B6SJL Tg SOD1G93A Gur/J) (Gurney 1997) and littermate (WT; non-Tg) mice were obtained from The Jackson Laboratories (Bar Harbor, ME, USA). The estimation of transgene copy numbers was performed by determining the difference in threshold cycle (∆CT) between the transgene (human SOD1) and a reference gene (mouse interleukin (IL)-2), as previously described (Kupershmidt et al. 2009). Mice were housed at an ambient temperature of 22 °C with a 12-h light/dark cycle and humidity-controlled environment. Food and water were available ad libitum.
SOD1G93A mice (22 mice/group) were administered by oral gavage with VAR (0.5 and 2.5 mg/kg, two times a week) + ced (the diet consisted of regular chow supplemented with 21 % (w/w) fat and 0.15 % (w/w) cholesterol and 1.25 % creatine given in sterile drinking water and replaced by a freshly prepared solution every 2 days). Control mice were given vehicle (no drug, no diet supplementation) and only sterile drinking water by oral gavage. The range of VAR doses used in the experiments were based on our previous studies with VAR (Bar-Am et al. 2015). At 88 days of age, when mice started to lose weight and motor impairments were observed by using various behavioral tests (rotarod performance, screen grasping, tail suspension, balance beam, and gait footprint), compared to WT (non-Tg) mice, VAR-ced, or vehicle were administered to SOD1G93A. At 120 days of age, mice were sacrificed by decapitation, and GNS muscle and spinal cord tissues were dissected, frozen immediately in liquid nitrogen and stored at −80 °C for further biochemical or immunohistochemistry analyses. The time of death was considered when mice could not roll over within 30 s of being pushed on the side (no other cause of death was seen) (Ludolph et al. 2007).
Animal Behavioral Analyses
Behavioral experiments were conducted during the light cycle by a researcher blinded to the group type. Mice were primarily trained in five independent motor behavioral tests, including rotarod performance, screen grasping, tail suspension, balance beam, and gait footprint experiments. After the establishment of stable baselines, animals were assessed at weekly intervals, starting at the week of treatment.
Rotarod Performance Test
Training sessions were conducted to acclimate the mice to the rotarod apparatus (Columbus Instruments, Columbus, OH, USA). Fore- and hindlimb motor coordination and balance were assessed by measuring the length of time at which the mice remain on the rotating rod (12 rpm), as described previously (Azzouz et al. 2000). Three trials were given to each animal, and the longest retention time (maximum of 300 s) was used as the measure of competence at this task. Evaluation scores were as follows: grade 0, >300 s; grade 1, 180–300 s; grade 2, 60–180 s; grade 3, <60; and grade 4, falling off the rod before rotation.
Screen Grasping Assay
This test served as an indicator of general muscle strength (Combs and D’Alecy 1987). The animal was placed on a horizontally positioned screen with grids. The screen was then rotated to the vertical position. Deficit scores were as follows: grade 0, grasping the screen with forepaws for 5 s; grade 1, holding the screen with alternative paws without falling for 5 s; grade 2, holding the screen for less than 5 s; and grade 3, falling off instantaneously.
Tail Suspension Test
The tail suspension test in mice was performed according to the method described previously (Cryan et al. 2005). The mouse was suspended by its tail, and extension of hind limbs was observed. Deficit scores were as follows: grade 0, normal; grade 1, partial hind limb extension of two legs; grade 2, no hind limb extension of one leg; and grade 3, no hind limb extension.
Balance Beam Experiment
Motor coordination and balance of mice were assessed by measuring the ability of the mice to traverse a graded series of narrow beams to reach an enclosed safety platform (Filali et al. 2011; Garbuzova-Davis et al. 2001). The beams consisted of long and fixed stainless steel bar (45-cm long and 0.9 cm in diameter) covered with rough sticker for a comfortable grip, 50 cm above the bench surface. Each mouse was given three trials, and the maximum durations (up to 30 s) at which mice fall off from the bar were scored. Deficit scores were as follows: grade 0, remains on beam 30 s; grade 1, remains on beam 30 s without using four paws; grade 2, remains on beam 11–20 s; grade 3, remains on beam <10 s; and grade 4, unable to maintain grip or balance.
Gait Footprint Test
Gait abnormalities were assessed by analyzing the footprint pattern of mice (Filali et al. 2011). To obtain footprints, the hind- and forefeet of the mice were coated with purple and green non-toxic paints, respectively. The animals were then allowed to walk along a 50-cm-long, 10-cm-wide runway (with 10-cm-high walls). All mice had three training runs and were then given one run per week. The footprint patterns were analyzed for three step parameters (cm): (1) stride length; (2) hind-base width, and (3) front-base width. These values were determined by measuring the perpendicular distance of a given step to a line connecting its opposite preceding and proceeding steps. The mean value of each set of three values was used in subsequent analysis.
Lumbar Spinal Cord Histological Analyses
Spinal cord tissues were post-fixed in 4 % paraformaldehyde in 0.1 M phosphate buffered saline for 24 h in 4 °C. Next, the lumbar spinal cords (L2–L6) were removed and cryoprotected in sucrose gradient (15–30 %) in 0.1 M sodium phosphate buffer (pH 7.4), following embedded in a tissue-freezing medium and sectioned (10-μm thickness) on a cryostat for further immunohistochemical analyses.
Motoneuron Examination
Every fifth section of a serial lumber cross-sections was stained with ChAT polyclonal antibody using immunohistochemical detection with HRP-DAB, according to the manufactory procedure. Briefly, sections were incubated with second goat anti rabbit IgG antibody for 2 h, then in 0.05 % DAB for 10 min and finally with 0.01 % H2O2 for another 5 min, to stop the reaction. Slides were dehydrated in alcohol gradient and coverslipped in a mounted DPX type. The number of surviving motoneurons within the ventral horn of the lumbar spinal cord was assessed by counting the number of ChAT-stained motoneuron, determined by cell body size, using an inverted-phase contrast microscope (Olympus America Inc., Melville, NY) with a digital camera (15–20 sections of ~200 motoneurons). Only large, polygonal neurons with a distinguishable nucleus and nucleolus and clearly identifiable were included in the counts. This method avoided the possibility of counting the same motoneuron in consecutive sections. At least four mice were analyzed from each experimental group. Analysis was performed using the “Fiji” software (ImageJ, NIH, USA).
Perl’s DAB Staining
Iron staining was performed on lumbar serial sections using a Perl’s DAB method, as previously described (Kupershmidt et al. 2012). Iron labeling was observed using an inverted phase contrast microscope (Olympus America Inc., Melville, NY) with a digital camera. The iron burden analysis was performed using the “Image Pro Plus” software (Media Cybernetics, Silver Spring, USA).
GNS Muscle Histological Analysis
Neuromuscular Junction Morphology
Morphological analysis of neuromuscular junction (NMJ) was performed in the GNS muscle, using previously described procedure (Ionescu et al. 2016). GNS muscle was stained with Alexa-594- α-bungarotoxin (red) to visualize post-synaptic acetylcholine receptors (AChR), and double labeled with anti-neurofilament (SMI-31) together with anti-synaptophysin (green) for distal axon and pre-synaptic nerve terminal staining. Immunofluorescence was observed by Zeiss LSM 510 Meta confocal microscopy (Carl Zeiss, Germany) using a 25×/40× objectives. Acquisition and analysis was performed using the “Zeiss Axiovision 4.8” software (Carl Zeiss, Germany). For quantitated analysis, 100 NMJs of three different populations (fully and partial innervated and denervated NMJs) were examined per muscle. NMJs that exhibited an overlap of red and green were considered innervated, while those that exhibited only α-bungarotoxin expression were considered denervated. All NMJs in every 30-μm section were analyzed. The percentage of fully and partial innervated NMJs was determined in each treatment group using previously established counting criteria (Gifondorwa et al. 2007).
Muscle Morphology Determination
GNS muscle was embedded in a tissue-freezing medium and serial cryostat sections were cut at 10 μm and stained by hematoxylin and eosin (H&E). Every fifth slide of 15 sections was examined, by using an inverted phase contrast microscope (Olympus America Inc., Melville, USA) with a digital camera. The number of myofibers in each GNS muscle was assessed by counting the normal myofibers morphology; plump and polygonal or triangular shape with peripherally nuclei. Analysis was performed by using the “Fiji” software (ImageJ, NIH, USA).
Quantitative Real-Time Reverse Transcriptase-PCR
Isolation of total RNA was performed using PurfectPure RNA Tissue Kit, as recommended by the manufacturer. After extraction of total RNA from tissues, the RNA concentrations were determined by NanoDrop spectrophotometer (Thermo Fisher Scientific Inc., Waltham, USA). Reverse transcription (RT) was performed using PrimeScript™ RT reagent kit. The resulting complementary DNA (cDNA) was amplified with specific primers of genes examined in the present study, by using 7500 real-time PCR system (Applied Biosystems, USA). SYBR Premix Ex Taq (Tli RNase H Plus) was performed according the manufacturer’s protocol. Real-time PCR was performed with specific primers for the genes in search and two reference genes: γ-tubulin and GAPDH.
Western Immunoblotting Analysis
For Western blot analyses, the GNS muscle tissues were homogenized in Tris-sucrose buffer pH = 7.4. C2C12 cells were washed with ice-cold PBS and lysed in whole buffer, containing protease inhibitor, as described previously (Kupershmidt et al. 2009). Detection was completed by using Western blotting ECL reagent system (Amersham Pharmacia, UK).
Mitochondrial Respiratory Chain Enzymatic Activities
Mitochondrial respiratory chain enzymatic activity of complex І (NADH ubiquinone oxidoreductase) and complex ІІ (succinate dehydrogenase), from frozen GNS muscle tissues, was based on measuring the ubiquinone/decylubiquinone (DUB) reduction by the electron acceptor after oxidation of NADH/succinate by complexes І and ІІ at 340 mm and 600 nm, respectively, as previously described (Spinazzi et al. 2012). The specific activity of complexes І and ІІ was calculated by subtracting the total activity using specific inhibitors, rotenone (10 μM) and malonate (10 mM), respectively, and expressed as nanomole per minute per milligram.
Cell Culture and MTT Assay
C2C12 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing high glucose (4.5 mg/ml) supplemented with 10 % fetal bovine serum, penicillin/streptomycin (100 U/ml), and incubated at 37 °C in a water-saturated atmosphere of 95 % ambient air and 5 % CO2. For experiments with H2O2, cells were incubated in the cultured medium for 24 h before drug treatment. VAR was added to the culture medium 20 min prior-, or 30 and 60 min post-treatment with H2O2 (1.5 mM) and further incubated for 3 h. Vehicle-treated cultures were used as controls. The cell viability of C2C12 myoblasts was determined by colorimetric MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide) assay, based on conversion of MTT to blue formazan crystals by viable cells, as described previously (Kupershmidt et al. 2009). Cells viability was measured by the absorbance at 570/650 nm.
Measurement of Reactive Oxygen Species Generation
Reactive oxygen species (ROS) generation was measured with the non-fluorescent probe DCF-DA, which passively diffuses into cells and was deacetylated by esterases to form non-fluorescent probe non-fluorescent 2′,7′-dichlorfluorescein (DCFH). In the presence of ROS, DCFH reacts with ROS to form the fluorescent product 2′,7′-dichlorofluorescein (DCF), which was trapped inside the cells. C2C12 cells were pre-incubated with DCF-DA (10 μM) for 30 min, then treated with VAR for 20 min before H2O2 addition in the dark. The fluorescence intensity was measured after 3 h using a MicroPlate reader infinite M200 PRO Tecan (AG, Switzerland) at 485/530 nm.
Mitochondrial Mass Staining and Mitochondrial Membrane Potential (ΔΨM) Determination
The cells were incubated under normal culture conditions for 30 min and then visualized by fluorescence microscopy (Olympus, Tokyo, Japan) for active mitochondrial mass by using Mitotracker green FM, according to manufacturer’s instructions. Mitochondrial membrane potential (ΔΨM) was detected with fluorescent probe lipophilic cationic probe JC-1 kit according to manufacturer’s instructions. For quantitative fluorescence measurements, cells were scanned with a microplate fluorimeter (Tecan, Hombrechtikon, Switzerland) at 485/535 nm excitation and 550/600 nm emission, to measure green and red JC-1 fluorescence, respectively.
Statistical Analysis
Kaplan-Meier survival analysis was used for survival and onset comparisons, using log rank test to compare curves. A two-way repeated measures ANOVA was used to determine significant differences in motor dysfunction and body weight. A one-way ANOVA was used to determine significant differences among means in mRNA expression and mean of survival. When significance occurred (P ≤ 0.05), a Tukey-Kramer post hoc test was used to determine significance. All statistical analyses were completed by using GraphPad Prism program (La Jolla, CA, USA).
Results
VAR-ced Treatment Prolonged Life Span and Improved Motor Behavior of SOD1G93A Mice
The Kaplan-Meyer curve of survival demonstrates that the mean of survival of SOD1G93A mice increased from 128 ± 1.8 days in vehicle-treated to 140 ± 1.8 and 138.5 ± 1.97 days in VAR-ced-treated, 0.5 and 2.5 mg/kg, respectively (Fig. 1).

Effect of VAR-ced treatment on survival of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 and 2.5 mg/kg) two times a week; starting at the 88th day of age and continuing until death. a Kaplan-Meier curve shows the cumulative probability of overall survival (p = 0.0167; log rank Mantel-Cox test) against the respective age of drug-treated SOD1G93A mice. b Histogram represents the mean survival (days) of SOD1G93A mice. Results are expressed as means ± SEM (n = 18–22/experimental group); *p < 0.05 vs. vehicle-treated SOD1G93A mice
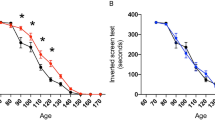
The effect of VAR-ced on overall deficit scores of motor dysfunction was assessed by independent behavioral tests: rotarod performance, screen grasping, tail suspension, and balance beam experiments, performed routinely (1/week), as described in “Methods.” Figure 2a shows that in SOD1G93A mice, the curve of motor dysfunction (scores) was shifted to the right following VAR-ced (0.5 mg/kg) treatment, compared to vehicle-treated animals. Additionally, gait abnormalities were assessed by analyzing the footprint pattern of mice. Indeed, stride length distance is most commonly a gait footprint parameter in mouse models of ALS (Filali et al. 2011). As shown in Fig. 2b, c, a significant increase in stride length distance (cm) was seen at 100–128 days of age in VAR-ced- vs. vehicle-treated SOD1G93A mice.

Effect of VAR-ced treatment on motor deficits in SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. a Total motor dysfunction was determined from four independent tests, including: rotarod performance, screen grasping (grip), tail suspension behavior, and balance beam, as described in “Methods.” Total scores of #14 represent complete loss of motor function. b Gait abnormalities were assessed by analyzing the footprint pattern of mice. Stride length was measured as the average distance (cm) of forward movement between each stride. c A representative walking footprint patterns at day 120. Results are expressed as means ± SEM (n = 18–22/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle-treated SOD1G93A mice
VAR-ced Attenuated Motoneuron Cell Death, Iron Accumulation, and NMJ Denervation in SOD1G93A Mice
Vehicle- and VAR-ced-treated SOD1G93A and WT (non-Tg) mice were evaluated histologically for motoneuron cell number in the lumbar spinal cord. Immunohistochemistry with ChAT antibody was used to visualize motoneuron cell bodies in the ventral horn of the lumbar spinal cord (Fig. 3a). As summarized in Fig. 3b, vehicle-treated SOD1G93A mice had fewer motoneuron cell number (~20 %) in the lumbar spinal cord. In contrast, there was a significant attenuation of motoneuron cell loss in VAR-ced-treated SOD1G93A mice, relative to vehicle-treated SOD1G93A mice (Fig. 3b). VAR-ced treatment also lowered the elevated iron levels in the spinal cord in SOD1G93A mice (Fig. 3c). In addition, L-ferritin expression levels were significantly increased in the GNS muscle of SOD1G93A mice, compared to WT (non-Tg) mice (0.08 ± 0.02 and 3.12 ± 0.50, respectively; p < 0.05), while VAR-ced treatment significantly reduced L-ferritin expression levels (1.38 ± 0.190; p < 0.05).

Effect of VAR-ced treatment on motoneuron cell death and iron accumulation in the lumbar spinal cord of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. The lumbar (L1–L5) spinal cord tissues were collected at symptomatic age of 120 days. a Motoneurons in the ventral horn area (circle) labeling in serial cross-sections, stained with ChAT antibody (arrows). b Motoneuron cell number per horn. c Quantification of iron staining intensities was performed using optical density analysis. Results represent the mean area ± SEM; (n = 4/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle- treated SOD1G93A mice.
Next, we have examined NMJs in the GNS muscle with staining with α-bungarotoxin to visualize post-synaptic AChR, and with anti-neurofilament (SMI-31) and anti-synaptophysin antibodies to identify distal axon and pre-synaptic nerve terminal staining. As illustrated in Fig. 4, NMJs were classified as fully innervated, partially innervated, or denervated based on the extant of overlap of these markers. In WT (non-Tg) mice, motor endplate innervation was ~100 %, while in vehicle-treated SOD1G93A mice, endplate innervation was only ~8 %; in contrast, in VAR-ced-treated SOD1G93A mice, NMJ innervation was significantly increased to ~25 % (Fig. 4b).

Effect of VAR-ced treatment on NMJ innervation in the GNS muscle of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. The GNS muscle tissues were collected at symptomatic age of 120 days. a Fully innervated, partially innervated, or denervated NMJs. The GNS muscle was stained with anti-α-bungarotoxin (red) to stain post-synaptic AChR clusters; axons were identified with neurofilament (SMI-31) and synaptophysin antibodies (green). Results represent percent of total number of NMJ and expressed as means ± SEM; (n = 4/experimental group); *p < 0.05 vs. vehicle-treated SOD1G93A mice. b Representative two-dimensional (2D) images depict fully innervated, partially innervated and denervated NMJs.
Effect of VAR-ced on Myofibril Cell Morphology mRNA Expression of Markers of Denervation and Atrophy in GNS Muscle of SOD1G93A Mice
At 120 days of age, vehicle-treated SOD1G93A mice displayed atrophy of the GNS muscle, as revealed by H&E staining and reduced number of myofibrils with normal morphology of polygonal and triangular shapes, compared to WT (non-Tg) mice (Fig. 5a). In contrast, VAR-ced-treated SOD1G93A mice displayed a significant preservation of myofibril regular morphology (Fig. 5a). In accordance, mRNA expression of markers of muscle denervation: AChRα and AChRγ [two subunits of the nicotinic acetylcholine receptor and muscle-specific tyrosine kinase (MuSK) (Palamiuc et al. 2015)], as well as mRNA expression of muscle atrophy markers: muscle specific ring finger protein 1 (MuRF1) and atrogin-1 (Atg-1) (Chen et al. 2010) were significantly higher in vehicle-treated SOD1G93A mice, compared to WT (non-Tg) mice (Fig. 5b, c). VAR-ced treatment significantly reduced mRNA expression levels of these markers of denervation and atrophy in the GNS muscle in SOD1G93A mice, compared to vehicle- treated SOD1G93A mice (Fig. 5b, c).

Effect of VAR-ced treatment on myofibril cell morphology and mRNA expression levels of denervation and atrophy markers in the GNS muscle of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. The GNS muscle tissues were collected at symptomatic age of 120 days. a The GNS muscle was stained with H&E, for myofibril morphology. Polygonal and triangular shapes with peripherally nuclei (filled arrow) were indicated as normal morphology and round/ shrunk with centrally located nuclei (empty arrow) as irregular morphology. Histogram represents myofibrils with peripherally nucleus (folds). b mRNA expression of markers of denervation, AChR subunits α and γ and MuSK. c mRNA expression of muscle atrophy markers, MuRF1 and Atg-1. Results are expressed as means ± SEM; (n = 4/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle-treated SOD1G93A mice
Effect of VAR-ced Treatment on mtDNA and Mitochondrial Respiratory Chain Complexes I and II Enzymatic Activity in GNS Muscle of SOD1G93A Mice
Given the importance of mtDNA and mitochondrial respiratory chain complex in muscle atrophy (Ripolone et al. 2015), we have measured the levels of mtDNA expression (expressed as the ratio between the mitochondrial encoded gene, cytochrome c oxidase subunit 1 (Cox1), and the nuclear-encoded gene, cyclophilin A (CypA)) and the enzymatic activity of complexes I and II in the GNS muscle of VAR-ced- and vehicle-treated SOD1G93A mice and WT mice). VAR-ced treatment significantly prevented the decrease in mtDNA mRNA expression levels (Fig. 6a) and in complexes I and II enzymatic activity (Fig. 6b) in the GNS muscle of SOD1G93A mice, compared to vehicle-treated SOD1G93A mice.

Effect of VAR-ced treatment on mtDNA and enzymatic activity of complexes I and II in the GNS muscle of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. The GNS muscle tissues were collected at the symptomatic age of 120 days. a mtDNA levels were expressed as the ratio between the mitochondrial encoded gene, Cox1, and nuclear-encoded gene, CypA. mRNA levels were assessed by real-time RT-PCR and normalized to γ-Tubulin and GAPDH. The values represent relative expression vs. WT (non-Tg) controls and results are means ± SEM; (n = 4/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle- treated SOD1G93A mice. b The enzymatic activity was measured by spectrophotometer at 340 and 600 nm for NADH (complex I) and succinate dehydrogenase (complex II), respectively. Results represent the activity (nmol/min/mg) and are expressed as means ± SEM; (n = 4/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle- treated SOD1G93A mice
Effect of VAR-ced on pCREB, PGC-1α, and PGC-1α-Target Genes and Proteins in GNS Muscle of SOD1G93A Mice
We have further analyzed the effect of VAR-ced treatment on levels of the master regulator of mitochondrial biogenesis and energy production PGC-1α (Lin et al. 2002; Wu et al. 1999) in the GNS muscle of SOD1G93A mice. As seen in Fig .7a, PGC-1α mRNA and protein levels were decreased in vehicle-treated SOD1G93A mice, compared to WT (non-Tg) mice, whereas they were significantly increased in VAR-ced- vs. vehicle-treated SOD1G93A mice. This increase in PGC-1α levels was associated with an upregulation of the signaling regulator of PGC-1α, p-cAMP response element-binding protein (CREB) (ser133) (Fig. 7b), and PGC-1α-co-activated gene PPARγ (Fig. 7c).

Effect of VAR-ced treatment on expression levels of PGC-1α and PPARγ and pCREB (ser133) in the GNS muscle of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. The GNS muscle tissues were collected at symptomatic age of 120 days. a mRNA expression and protein levels of PGC-1α; mRNA products were assessed by real-time RT-PCR and normalized to γ-Tubulin and GAPDH; Western blotting analysis was normalized to GAPDH. b Levels of pCREB (ser133) were evaluated by Western blotting analysis; the graph represents densitometry quantification of the lanes, normalized to total CREB and GAPDH. c mRNA expression levels of PPARγ; mRNA products were assessed by real-time RT-PCR and normalized to γ-Tubulin and GAPDH. The values represent relative levels vs. respective controls. Results are expressed as means ± SEM; (n = 4/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle- treated SOD1G93A mice.
In addition, treatment with VAR-ced increased mRNA expression levels of several regulated genes of PGC-1α pathway, including uncoupling protein 3 (UCP3), nuclear respiratory factor 1 (NRF1), NRF2, nuclear-encoded mitochondrial transcription factor A (Tfam), estrogen-related receptor α (ERRα), and vascular endothelial growth factor (VEGF) in the GNS muscle of SOD1G93A mice (Fig. 8a). As shown in Fig. 8b, mRNA and protein levels of the insulin-sensitive glucose transporter 4, Glut4 were reduced in the GNS muscle of vehicle-treated SOD1G93A mice vs. WT (non-Tg). However, VAR-ced administration to SOD1G93A mice significantly upregulated mRNA and protein levels of Glut4 in the GNS muscle, in comparison to vehicle-treated SOD1G93A mice (Fig. 8b). Additionally, VAR-ced increased protein levels of the myocytes-specific enhancer factor MEF2C in the GNS muscles, compared with vehicle-treated SOD1G93A mice (Fig. 8c).

Effect of VAR-ced treatment on PGC-1α-target genes and -proteins in the GNS muscle of SOD1G93A mice. SOD1G93A mice were treated by oral gavage with vehicle or VAR-ced (0.5 mg/kg), as described in Fig. 1. The GNS muscle tissues were collected at symptomatic age of 120 days. a mRNA expression levels of UCP3, NRF1, NRF2, Tfam, ERRα, and VEGF; mRNA products were assessed by real-time RT-PCR and normalized to γ-Tubulin and GAPDH. b mRNA expression and protein levels of GluT4; the amount of mRNA products were assessed by real-time RT-PCR and normalized to γ-Tubulin and GAPDH; Western blotting analysis was normalized to GAPDH. c Western blotting analysis of MEF2C. The graph represents densitometry quantification of the lanes, normalized to GAPDH and the values represent relative levels vs. WT. Results are expressed as means ± SEM; (n = 4/experimental group); #p < 0.05 vs. WT; *p < 0.05 vs. vehicle- treated SOD1G93A mice
The Cytoprotective Effects of VAR Against Oxidative Stress in C2C12 Myoblasts
Functional deficits of the mitochondrial function can cause a major intracellular generation of ROS, such as superoxide and H2O2 (Kiaei et al. 2005). Thus, we have further examined the potential cytoprotective/antioxidant effects of VAR in C2C12 myoblasts, using H2O2 as an oxidative stress inducer. As shown in Fig. 9a, H2O2 (1.5 mM) markedly reduced viability of C2C12 myoblasts, while VAR (25 μM) significantly increased cell survival at pre- (Fig. 9a) and post- (Fig. 9b) H2O2 cytotoxicity treatment, as assessed by MTT test. No effect was seen when the drug was given alone (without H2O2; data not shown). In addition, we determined the intracellular levels of ROS, using the DCF-DA assay. Figure 9c demonstrates that VAR dose-dependently suppressed intracellular ROS generation in H2O2-treated C2C12 myoblasts, indicating a putative antioxidant property of the drug.

Protective/rescue effects of VAR on cell viability and intracellular accumulation of ROS, following H2O2-induced cytotoxicity in C2C12 myoblasts. C2C12 myoblast cells were treated without or with various concentrations of VAR: a 20 min prior- or b 30 and 60 min post-treatment with H2O2 (1.5 mM) and further incubated for 3 h. Cell viability was evaluated by MTT assay; c ROS levels were measured by HPF fluorescence intensity. Data are means ± SEM values and expressed as percentage of control, untreated cells. n = 3; #p < 0.05 vs. untreated cells; *p < 0.05 vs. H2O2 alone
The mitochondrial-specific fluorescent dye, MitoTracker Green was used to assess the effect of VAR on the mitochondrial mass in C2C12 myoblasts following exposure to H2O2. Figure 10a, b show reduced mitochondrial mass in C2C12 myoblasts treated with H2O2 (1.5 mM), whereas VAR significantly and dose-dependently (3.5–25 μM) prevented the loss of mitochondrial abundance.

Protective/rescue effects of VAR on H2O2-induced loss of mitochondrial mass and mitochondrial membrane potential (∆Ψm) in C2C12 myoblasts. C2C12 myoblast cells were treated without or with VAR, 20 min prior H2O2 (1.5 mM) and further incubated for 3 h. a C2C12 myoblasts were stained with MitoTracker Green FM for mitochondrial mass analysis and JC-1 for ∆Ψm analysis, using fluorescence microscope; b Total active mitochondrial mass was detected by MitoTracker Green FM. C2C12 myoblasts were treated without or with VAR, c 20 min prior- and d 30 and 60 min post-treatment with H2O2 (1.5 mM) and further incubated for 3 h. ΔΨm was expressed as the ratio of J-aggregate and JC-1 monomer (red/green) fluorescence intensities. Mitochondrial depolarization (i.e., loss of ΔΨm) manifests itself by a decrease in the red/green fluorescence ratio. Values are means ± SEM of values and expressed as folds of control, untreated cells. n = 3; #p < 0.05 vs. untreated cells; *p < 0.05 vs. H2O2 alone
We have further examined whether Δψm was involved in the protective effect of VAR on H2O2-induced cell death in C2C12 myoblasts, by using the molecular probe JC-1. In living cells, JC-1 accumulates as aggregates in normal hyperpolarized mitochondria, resulting in red fluorescence; while in apoptotic cells, the mitochondrial potential collapses and JC-1 exists in a monomeric form and stains the cytosol in green. Treatment with H2O2 induced a green diffused fluorescence pattern, corresponding to monomers appearing in the cytoplasm, indicating dissipation of the Δψm, while pre- (Fig. 10c) and post- (Fig. 10d) treatment with VAR significantly and dose-dependently showed an increase in red fluorescence, indicating inhibition of mitochondrial transmembrane potential loss in H2O2-induced cells.
Discussion and Conclusions
The concept of complex etiology in neurodegenerative disorders led our group to develop a series of multifunctional, brain permeable, non-toxic compounds with iron chelating and anti-apoptotic properties (Bar-Am et al. 2015; Zheng et al. 2005a, b). Among these novel multitarget drugs, the compound VAR, which amalgamates the propargyl moiety of rasagiline, with the backbone of the iron chelator VK28, was shown to afford iron chelation, iron-induced lipid peroxidation inhibitory potency, and brain MAO-A and -B inhibitory effects (Bar-Am et al. 2015). In this study, we have evaluated the potential therapeutic effects of VAR, combined with ced, in the SOD1G93A transgenic mouse model of ALS. This combination protocol of VAR-ced was based on our recent data, demonstrating additive protective effects of low doses of the multifunctional iron chelator, M30 with ced in SOD1G93A mice (Golko-Perez et al. 2016). At 126 days of age, the overall motor dysfunction scores of the M30-ced group were significantly lower (p < 0.05), compared to the individual treatments, ced, low dose of M30 and vehicle-treated SOD1G93A mice. In addition, the differences in life extension between M30-ced and each of the individual treatments, ced, and low dose of M30 were significantly different (p < 0.05), indicating a superiority of the combination protocol upon the individual treatments (Golko-Perez et al. 2016).
In the current study, the treatment of VAR-ced in SOD1G93A mice was initiated after the appearance of disease symptoms (at day 88), as this regimen is comparable with the earliest time at which drug therapy could start in ALS patients. Using this rescue protocol, here, we demonstrated that VAR-ced provided several beneficial effects in SOD1G93A mice: (1) the treatment increased survival, (2) attenuated spinal cord motoneuron loss, and (3) improved motor performance. We also demonstrated that VAR-ced treatment prevented the upregulation in iron levels in the spinal cord of SOD1G93A mice, indicating that the drug may attenuate the progression of ALS by reducing excessive iron and its redox activity. In addition, VAR-ced treatment reduced expression levels of H-ferritin and L-ferritin in the GNS muscle of SOD1G93A mice. These results are in accordance with previously described iron dysregulation in ALS patients, ALS mouse model, and in culture neurons overexpressing SOD1G93A (Ignjatovic et al. 2012; Imon et al. 1995; Ince et al. 1994; Jeong et al. 2009; Kasarskis et al. 1995; Oba et al. 1993; Winkler et al. 2014). Indeed, it was shown that iron chelators have a potential in delaying disease onset and increasing survival in SOD1G93A mice, with decreasing oxidative stress and glial activation in the spinal cord (Jeong et al. 2009; Winkler et al. 2014; Kupershmidt et al. 2009). In addition, it was shown that iron chelators significantly prevented the decrease in SOD activity in SOD1G93A mice, following 120 days of treatment (Wang et al. 2011).
Although motoneuron loss is the most prominent characteristics of ALS pathogenesis, it has been indicated that the skeletal muscle is a primary target of SOD1G93A mice-induced toxicity (Dobrowolny et al. 2008). It has been proposed that defects in skeletal muscle, leading to muscle cell dysfunction, may contribute to the motoneuron pathology (Capitanio et al. 2012). Recently, Capinio et al. (2012) have characterized protein alteration in hind- and forelimb muscles and identified muscular signatures (e.g., muscle albumin, complexes I and II, PI3 kinase, PGC-1α. rho-associated protein kinase 1 (ROCK1)) of ALS diagnosis and progression in the SOD1G93A mice. Thus, in the current study, we have further evaluated the effects of VAR-ced treatment on the GNS muscle in SOD1G93A mice. The results showed that VAR-ced affected various parameters in the GNS muscle in SOD1G93A mice, including attenuation of NMJ denervation, preservation of myofiber regular morphology, and reduction of mRNA expression levels of markers of muscle denervation and atrophy. Consistent with the involvement of mitochondrial abnormalities in muscles in ALS (Al-Sarraj et al. 2014; Luo et al. 2013; Nefussy and Drory 2010), we also found that VAR-ced treatment prevented the decrease in mRNA expression levels of mtDNA and complexes I and II enzymatic activity in the GNS muscle of SOD1G93A mice.
Based on the analysis of the effect of VAR-ced on mitochondrial dysfunction in skeletal muscles that may precede motoneuron death in SOD1G93A mice, we have further investigated the protective effects of the drug against oxidative stress in muscle cell line. We have used the mouse myoblastic cell line C2C12, which provides a well-established in vitro model for a morpho-functional features of muscle cells (Burattini et al. 2004), as well as common model of oxidative stress-induced muscle cell death to investigate therapeutic implications for ALS (Schoneich et al. 2014; Pansarasa et al. 2014). Our experiments in mouse C2C12 myoblasts demonstrated that VAR markedly and dose-dependently inhibited H2O2-induced cytotoxicity, associated with decreased cellular ROS formation. In addition, VAR treatment attenuated H2O2-induced mitochondrial membrane potential loss, further suggesting that the drug may produce its protective effects via the preservation of mitochondrial function. Indeed, previous studies with M30 and HLA20 demonstrated that both multifunctional iron chelators attenuated H2O2-induced mitochondrial membrane potential loss, decreased the release of cytochrome c into the cytoplasm, and inhibited activation of caspase-3 in pancreatic beta-cells, indicating that these iron cheating drugs may produce cytoprotective effects via the preservation of mitochondrial function (Mechlovich et al. 2010).
In this respect, a potential therapeutic target is the transcriptional co-activator, PGC-1α, which has been identified as a master regulator of mitochondrial biogenesis and also has been shown to regulate proteins involved in angiogenesis and the antioxidant defense in skeletal muscles (Dillon et al. 2012; Kang and Li Ji 2012; Villena 2015). PGC-1α exerts these effects through direct interaction with and co-activation of PPARγ, ERRs, and NRF1 and NRF2 among other transcriptional factors (Finck and Kelly 2006). Studies in various ALS mouse models demonstrated that PGC-1α and PPARγ agonists protected motoneurons and NMJs, altered disease progression, and extended life span in ALS mice (Kiaei et al. 2005; Shibata et al. 2008; Liang et al. 2011). Additionally, assessment of double-transgenic overexpression SOD1G93A and PGC-1α revealed protective effects on motoneuron loss (Liang et al. 2011).
Here, we observed a significant increase in PGC-1α mRNA and protein and phosphorylated levels of CREB at serine 133 in the GNS muscle of VAR-ced-treated SOD1G93A mice, compared with vehicle-treated SOD1G93A mice. This is in accordance with previous findings demonstrating that involvement of pCREB (ser133) in the regulation and activation of PGC-1α (Handschin et al. 2003). In addition, consistent with the activation of PGC-1α pathway, our study demonstrated that VAR-ced could increase mRNA expression levels of several downstream-related factors, involved in regulation of mitochondrial function/biogenesis (e.g., NRF1, NRF2, Tfam); mitochondrial bioenergetic response and angiogenesis (e.g., ERRα, VEGF); oxidation and antioxidants detoxification (e.g., PPARγ, UCP3) and glucose consumption (e.g., MEF2C, GLUT4) in the GNS muscle in SOD1G93A mice.
In summary, we have demonstrated that VAR-ced treatment exerted potent rescue effects on motor/neurological dysfunction and life span in SOD1G93A ALS mice. We have also showed a possible molecular mechanism of VAR-ced treatment in the GNS muscle, including beneficial effects on mitochondrial biogenesis, which may be associated with the observed protective effects on motoneuron degeneration, muscle atrophy, and NMJ destabilization in SOD1G93A mice. These results provide evidence of therapeutic potential of VAR-ced in SOD1G93A mice with underlying molecular mechanisms, further supporting the importance role of multitarget iron chelators in ALS treatment.
References
Al-Sarraj S, King A, Cleveland M, Pradat PF, Corse A, Rothstein JD, Leigh PN, Abila B, Bates S, Wurthner J, Meininger V (2014) Mitochondrial abnormalities and low grade inflammation are present in the skeletal muscle of a minority of patients with amyotrophic lateral sclerosis; an observational myopathology study. Acta Neuropathol Commun 2:165–174. doi:10.1186/s40478-014-0165-z s40478-014-0165-z
Azzouz M, Hottinger A, Paterna JC, Zurn AD, Aebischer P, Bueler H (2000) Increased motoneuron survival and improved neuromuscular function in transgenic ALS mice after intraspinal injection of an adeno-associated virus encoding Bcl-2. Hum Mol Genet 9:803–811
Bar-Am O, Amit T, Kupershmidt L, Aluf Y, Mechlovich D, Kabha H, Danovitch L, Zurawski VR, Youdim MB, Weinreb O (2015) Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol Aging 36:1529–1542. doi:10.1016/j.neurobiolaging.2014.10.026
Burattini S, Ferri P, Battistelli M, Curci R, Luchetti F, Falcieri E (2004) C2C12 murine myoblasts as a model of skeletal muscle development: morpho-functional characterization. Eur J Histochem 48:223–233
Capitanio D, Vasso M, Ratti A, Grignaschi G, Volta M, Moriggi M, Daleno C, Bendotti C, Silani V, Gelfi C (2012) Molecular signatures of amyotrophic lateral sclerosis disease progression in hind and forelimb muscles of an SOD1(G93A) mouse model. Antioxid Redox Signal 17:1333–1350. doi:10.1089/ars.2012.4524
Chen F, Sugiura Y, Myers KG, Liu Y, Lin W (2010) Ubiquitin carboxyl-terminal hydrolase L1 is required for maintaining the structure and function of the neuromuscular junction. Proc Natl Acad Sci U S A 107:1636–1641. doi:10.1073/pnas.0911516107
Combs DJ, D’Alecy LG (1987) Motor performance in rats exposed to severe forebrain ischemia: effect of fasting and 1,3-butanediol. Stroke 18:503–511
Cryan JF, Mombereau C, Vassout A (2005) The tail suspension test as a model for assessing antidepressant activity: review of pharmacological and genetic studies in mice. Neurosci Biobehav Rev 29:571–625
Dillon LM, Rebelo AP, Moraes CT (2012) The role of PGC-1 coactivators in aging skeletal muscle and heart. IUBMB Life 64:231–241. doi:10.1002/iub.608
Dobrowolny G, Aucello M, Molinaro M, Musaro A (2008) Local expression of mIgf-1 modulates ubiquitin, caspase and CDK5 expression in skeletal muscle of an ALS mouse model. Neurol Res 30:131–136. doi:10.1179/174313208X281235
Filali M, Lalonde R, Rivest S (2011) Sensorimotor and cognitive functions in a SOD1(G37R) transgenic mouse model of amyotrophic lateral sclerosis. Behav Brain Res 225:215–221. doi:10.1016/j.bbr.2011.07.034
Finck BN, Kelly DP (2006) PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest 116:615–622. doi:10.1172/JCI27794
Garbuzova-Davis S, Willing AE, Milliken M, Saporta S, Sowerby B, Cahill DW, Sanberg PR (2001) Intraspinal implantation of hNT neurons into SOD1 mice with apparent motor deficit. Amyotroph Lateral Scler Other Motor Neuron Disord 2:175–180. doi:10.1080/14660820152882179
Gifondorwa DJ1, Robinson MB, Hayes CD, Taylor AR, Prevette DM, Oppenheim RW, Caress J, Milligan CE (2007) Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. J Neurosci 27:13173–13180. doi:10.1523/JNEUROSCI.4057-07
Golko-Perez S, Mandel S, Amit T, Kupershmidt L, Youdim MB, Weinreb O (2016) Additive neuroprotective effects of the multifunctional iron chelator M30 with enriched diet in a mouse model of amyotrophic lateral sclerosis. Neurotox Res 29:208–217. doi:10.1007/s12640-015-9574-4
Gurney ME (1997) The use of transgenic mouse models of amyotrophic lateral sclerosis in preclinical drug studies. J Neurol Sci 152(Suppl 1):S67–S73
Halon M, Kaczor JJ, Ziolkowski W, Flis DJ, Borkowska A, Popowska U, Nyka W, Wozniak M, Antosiewicz J (2014) Changes in skeletal muscle iron metabolism outpace amyotrophic lateral sclerosis onset in transgenic rats bearing the G93A hmSOD1 gene mutation. Free Radic Res 48:1363–1370. doi:10.3109/10715762.2014.955484
Handschin C, Rhee J, Lin J, Tarr PT, Spiegelman BM (2003) An autoregulatory loop controls peroxisome proliferator-activated receptor gamma coactivator 1alpha expression in muscle. Proc Natl Acad Sci U S A 100:7111–7116. doi:10.1073/pnas.1232352100
Ignjatovic A, Stevic Z, Lavrnic D, Nikolic-Kokic A, Blagojevic D, Spasic M, Spasojevic I (2012) Inappropriately chelated iron in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Amyotroph Lateral Scler 13:357–362
Ikeda K, Hirayama T, Takazawa T, Kawabe K, Iwasaki Y (2012) Relationships between disease progression and serum levels of lipid, urate, creatinine and ferritin in Japanese patients with amyotrophic lateral sclerosis: a cross-sectional study. Intern Med 51:1501–1508
Imon Y, Yamaguchi S, Yamamura Y, Tsuji S, Kajima T, Ito K, Nakamura S (1995) Low intensity areas observed on T2-weighted magnetic resonance imaging of the cerebral cortex in various neurological diseases. J Neurol Sci 134(Suppl):27–32
Ince PG, Shaw PJ, Candy JM, Mantle D, Tandon L, Ehmann WD, Markesbery WR (1994) Iron, selenium and glutathione peroxidase activity are elevated in sporadic motor neuron disease. Neurosci Lett 182:87–90
Ionescu A, Zahavi EE, Gradus T, Ben-Yaakov K, Perlson E (2016) Compartmental microfluidic system for studying muscle-neuron communication and neuromuscular junction maintenance. Eur J Cell Biol 95:69–88. doi:10.1016/j.ejcb.2015.11.004
Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S (2009) Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis J Neurosci 29:610-619
Kang C, Li Ji L (2012) Role of PGC-1alpha signaling in skeletal muscle health and disease. Ann N Y Acad Sci 1271:110–117. doi:10.1111/j.1749-6632.2012.06738.x
Kasarskis EJ, Tandon L, Lovell MA, Ehmann WD (1995) Aluminum, calcium, and iron in the spinal cord of patients with sporadic amyotrophic lateral sclerosis using laser microprobe mass spectroscopy: a preliminary study. J Neurol Sci 130:203–208
Kiaei M, Kipiani K, Chen J, Calingasan NY, Beal MF (2005) Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp Neurol 191:331–336. doi:10.1016/j.expneurol.2004.10.007
Kokić AN1, Stević Z, Stojanović S, Blagojević DP, Jones DR, Pavlović S, Niketić V, Apostolski S, Spasić MB (2005) Biotransformation of nitric oxide in the cerebrospinal fluid of amyotrophic lateral sclerosis patients. Redox Rep 10:265–270. doi:10.1179/135100005X70242
Kupershmidt L, Amit T, Bar-Am O, Youdim MB, Weinreb O (2012) Neuroprotection by the multitarget iron chelator M30 on age-related alterations in mice. Mech Ageing Dev 133:267–274
Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB (2009) Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 23:3766–3779
Kwan JY, Jeong SY, Van Gelderen P, Deng HX, Quezado MM, Danielian LE, Butman JA, Chen L, Bayat E, Russell J, Siddique T, Duyn JH, Rouault TA, Floeter MK (2012) Iron accumulation in deep cortical layers accounts for MRI signal abnormalities in ALS: correlating 7 Tesla MRI and pathology. PLoS One 7:e35241
Liang H, Ward WF, Jang YC, Bhattacharya A, Bokov AF, Li Y, Jernigan A, Richardson A, Van Remmen H (2011) PGC-1alpha protects neurons and alters disease progression in an amyotrophic lateral sclerosis mouse model. Muscle Nerve 44:947–956. doi:10.1002/mus.22217
Lin J et al (2002) Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature 418:797–801. doi:10.1038/nature00904
Ludolph AC, Bendotti C, Blaugrund E, Hengerer B, Loffler JP, Martin J, Meininger V, Meyer T, Moussaoui S, Robberecht W, Scott S, Silani V, Van Den Berg LH (2007) Guidelines for the preclinical in vivo evaluation of pharmacological active drugs for ALS/MND: report on the 142nd ENMC international workshop. Amyotroph Lateral Scler 8:217–223. doi:10.1080/17482960701292837
Luo G, Yi J, Ma C, Xiao Y, Yi F, Yu T, Zhou J (2013) Defective mitochondrial dynamics is an early event in skeletal muscle of an amyotrophic lateral sclerosis mouse model. PLoS One 8:e82112. doi:10.1371/journal.pone.0082112
Mechlovich D, Amit T, Mandel SA, Bar-Am O, Bloch K, Vardi P, Youdim MB (2010) The novel multifunctional, iron-chelating drugs M30 and HLA20 protect pancreatic beta-cell lines from oxidative stress damage. J Pharmacol Exp Ther 333:874–882
Miyazaki K et al (2011) Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J Neurosci Res 89:718–728. doi:10.1002/jnr.22594
Nefussy B, Drory VE (2010) Moving toward a predictive and personalized clinical approach in amyotrophic lateral sclerosis: novel developments and future directions in diagnosis, genetics, pathogenesis and therapies. EPMA J 1:329–341. doi:10.1007/s13167-010-0027-0
Oba H et al (1993) Amyotrophic lateral sclerosis: T2 shortening in motor cortex at MR imaging. Radiology 189:843–846
Oshiro S, Morioka MS, Kikuchi M (2011) Dysregulation of iron metabolism in Alzheimer’s disease, Parkinson’s disease, and amyotrophic lateral sclerosis. Adv Pharmacol Sci 2011:378278–378286. doi:10.1155/2011/378278
Palamiuc L, Schlagowski A, Ngo ST, Vernay A, Dirrig-Grosch S, Henriques A, Boutillier AL, Zoll J, Echaniz-Laguna A, Loeffler JP, Rene F (2015) A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol Med 7:526–546. doi:10.15252/emmm.2014.04433
Pansarasa O, Rossi D, Berardinelli A, Cereda C (2014) Amyotrophic lateral sclerosis and skeletal muscle: an updates Mol Neurobiol 49:984-990 doi:10.1007/s12035-013-8578-4
Ripolone M, Ronchi D, Violano R, Vallejo D, Fagiolari G, Barca E, Lucchini V, Colombo I, Villa L, Berardinelli A, Balottin U, Morandi L, Mora M, Bordoni A, Fortunato F, Corti S, Parisi D, Toscano A, Sciacco M, DiMauro S, Comi GP, Moggio M (2015) Impaired muscle mitochondrial biogenesis and myogenesis in spinal muscular atrophy. JAMA Neurol 72:666–675. doi:10.1001/jamaneurol.2015.0178
Santillo AF, Skoglund L, Lindau M, Eeg-Olofsson KE, Tovi M, Engler H, Brundin RM, Ingvast S, Lannfelt L, Glaser A, Kilander L (2009) Frontotemporal dementia-amyotrophic lateral sclerosis complex is simulated by neurodegeneration with brain iron accumulation. Alzheimer Dis Assoc Disord 23:298–300
Schoneich C, Dremina E, Galeva N, Sharov V (2014) Apoptosis in differentiating C2C12 muscle cells selectively targets Bcl-2-deficient myotubes. Apoptosis 19:42–57. doi:10.1007/s10495-013-0922-7
Shibata N, Kawaguchi-Niida M, Yamamoto T, Toi S, Hirano A, Kobayashi M (2008) Effects of the PPARgamma activator pioglitazone on p38 MAP kinase and IkappaBalpha in the spinal cord of a transgenic mouse model of amyotrophic lateral sclerosis. Neuropathology 28:387–398. doi:10.1111/j.1440-1789.2008.00890.x
Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C (2012) Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat Protoc 7:1235–1246. doi:10.1038/nprot.2012.058 nprot
Villena JA (2015) New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J 282:647–672. doi:10.1111/febs.13175
Wang Q, Zhang X, Chen S, Zhang X, Zhang S, Youdium M, Le W (2011) Prevention of motor neuron degeneration by novel iron chelators in SOD1G93A transgenic mice of amyotrophic lateral sclerosis. Neurodegener Dis 8:310–321
Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV (2013) Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol 125:111–120. doi:10.1007/s00401-012-1039-8
Winkler EA, Sengillo JD, Sagare AP, Zhao Z, Ma Q, Zuniga E, Wang Y, Zhong Z, Sullivan JS, Griffin JH, Cleveland DW, Zlokovic BV (2014) Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc Natl Acad Sci U S A 111:E1035–E1042. doi:10.1073/pnas.1401595111
Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM (1999) Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator. PGC-1 Cell 98:115–124. doi:10.1016/S0092-8674(00)80611-X
Zhao Z, Nelson AR, Betsholtz C, Zlokovic BV (2015) Establishment and dysfunction of the blood-brain barrier cell 163:1064-1078 doi:10.1016/j.cell.2015.10.067
Zheng H, Youdim MB, Weiner LM, Fridkin M (2005a) Novel potential neuroprotective agents with both iron chelating and amino acid-based derivatives targeting central nervous system neurons. Biochem Pharmacol 70:1642–1652
Zheng H, Youdim MB, Weiner LM, Fridkin M (2005b) Synthesis and evaluation of peptidic metal chelators for neuroprotection in neurodegenerative diseases. J Pept Res 66:190–203
Zhong Z et al (2009) Activated protein C therapy slows ALS-like disease in mice by transcriptionally inhibiting SOD1 in motor neurons and microglia cells. J Clin Invest 119:3437–3449. doi:10.1172/JCI3847638476
Zlokovic BV (2011) Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 12:723–738. doi:10.1038/nrn3114
Acknowledgments
The authors are grateful to Prize 4 Life, Inc. (Berkeley, CA) and Rappaport Family Research, Technion Israel Institute of Technology for their support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
MBH Youdim is the scientific founder of Abital Pharma Pipelines and commercial interest in VAR10303 drug.
Rights and permissions
About this article

Cite this article
Golko-Perez, S., Amit, T., Bar-Am, O. et al. A Novel Iron Chelator-Radical Scavenger Ameliorates Motor Dysfunction and Improves Life Span and Mitochondrial Biogenesis in SOD1G93A ALS Mice. Neurotox Res 31, 230–244 (2017). https://doi.org/10.1007/s12640-016-9677-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-016-9677-6