
Abstract
Currently, we still lack effective measures to modify disease progression in neurodegenerative diseases. Iron-containing proteins play an essential role in many fundamental biological processes in the central nervous system. In addition, iron is a redox-active ion and can induce oxidative stress in the cell. Although the causes and pathology hallmarks of different neurodegenerative diseases vary, iron dyshomeostasis, oxidative stress and mitochondrial injury constitute a common pathway to cell death in several neurodegenerative diseases. MRI is capable of depicting iron content in the brain, and serves as a potential biomarker for early and differential diagnosis, tracking disease progression and evaluating the effectiveness of neuroprotective therapy. Iron chelators have shown their efficacy against neurodegeneration in a series of animal models, and been applied in several clinical trials. In this review, we summarize recent developments on iron dyshomeostasis in Parkinson’s disease, Alzheimer’s disease, Friedreich ataxia, and Huntington’s disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
As the population is aging worldwide, neurodegenerative diseases, especially Parkinson’s disease (PD) and Alzheimer’s disease (AD) are becoming a main challenge to health care professionals (Brookmeyer et al. 2007; Dorsey et al. 2007). Unfortunately, so far we have no cure or effective intervention to slow down the progression of these neurodegenerative diseases. Although PD and AD have different symptoms and pathological changes, several processes are common in their pathogenesis and cell death: iron accumulation, excess oxidative stress, and mitochondrial dysfunction (Ward et al. 2014; Crichton et al. 2011; Parker et al. 1994; Deibel et al. 1996; Connor et al. 1992; Dexter et al. 1989; Riederer et al. 1989; Devi et al. 2008; Jenner et al. 1992). Moreover, iron overload has direct interplays with the key components of pathological hallmarks, α-synuclein in PD, β-amyloid and tau protein in AD (Ostrerova-Golts et al. 2000; Golts et al. 2002; Ortega et al. 2015; Becerril-Ortega et al. 2014; Bodovitz et al. 1995; Everett et al. 2014; Yamamoto et al. 2002). Besides, in several hereditary neurodegenerative diseases such as Huntington’s disease, and Friedreich ataxia, iron dyshomeostasis also plays a critical role, and these diseases share a common core mechanism of neurodegeneration with PD and AD (Babcock et al. 1997; Rotig et al. 1997; Hilditch-Maguire et al. 2000; Bulteau et al. 2004; Bartzokis et al. 2007; Mena et al. 2015). Neuroimaging examination, especially MRI, is a good measure to detect iron accumulation early in the disease process, monitor iron overload along disease progression, and evaluate the effect of treatment (Ward et al. 2014; Apple et al. 2014; He et al. 2015; Wieler et al. 2015; Devos et al. 2014). Iron chelation is demonstrated effective in several animal models of the above diseases, and promising in clinical trials of PD and Friedreich’s ataxia (Pandolfo et al. 2014; Dexter et al. 2011, 2014; Grolez et al. 2015; Devos et al. 2014; Kaur et al. 2003). In this review, we mainly discuss iron dyshomeostasis and its role in the pathogenesis of PD, the application of imaging tools for iron detection in research and clinical practice, and the prospect of iron chelation therapy in PD. Additionally, we will give a brief overview of iron dysregulation in AD, Friedreich ataxia, and Huntington’s disease.
Regulation of cellular iron in the brain
Regulations of cellular iron in different cell types of the brain varied. In microglia, oligodendrocytes, and most neurons, the majority of the iron is stored in ferritin as a non-reactive but bio-available form (Connor et al. 1994; Hansen et al. 1999). However, in dopaminergic neurons of the substantia nigra (SN), neuromelanin serves as the main non-reactive iron storage. In the study by Zecca et al. (2001), iron, ferritin, and neuromelanin in the SN of normal subjects all increase with age. Since the second decade, neuromelanin is the predominant molecule reserving iron, and its proportion in total iron content increases with age. In subjects aged 80–90 years, the concentration of total iron, ferritin and neuromelanin are 109–199, 300–400, and 3500 ng/mg, respectively (Zecca et al. 2001). We need to note that the authors did not discriminate the iron, ferritin and neuromelanin distributions between different cell types, but we can infer the mainstay of neuromelanin in storing iron in dopaminergic neurons because of the significant predominance of neuromelanin in SN. Studies on Macaca arctoides and rat brains reported that astrocytes lack ferritin (Connor et al. 1994; Hansen et al. 1999). However, researches using cultured rat astrocytes showed that stimulations with iron together with TNF-α, iron oxide nanoparticles, or ferric ammonium citrate induced a significant increase of ferritin in astrocytes (Hoepken et al. 2004; Rathore et al. 2012; Geppert et al. 2012). These studies suggest that astrocytes can store iron into ferritin to reduce oxidative stress under the above stimulations. In addition to iron stored in ferritin or neuromelanin as non-reactive forms, a small proportion (less than 5 %) of iron in the cell is in the labile cell pool, where the iron is redox-active, chelatable and exchangeable. Labile cell iron is maintained within a range of 0.5–1.5 µM physiologically (Cabantchik 2014). Iron-sulfur clusters and heme are two crucial iron-containing prosthetic groups, which are essential elements of the mitochondrial electron transport chain (Gille and Reichmann 2011; Zhang et al. 1998; Beinert et al. 1997; Lin et al. 1982).
Normally, iron homeostasis is strictly controlled by a series of regulators. In the plasma, iron is transported by transferrin (Tf). To enter the central nervous system, iron must cross the blood–brain barrier and the blood–cerebrospinal fluid barrier, and there are transferrin receptors (TfR) on the luminal surface of the capillary endothelial cells. Firstly, transferrin with Fe3+ and TfR form a Tf–TfR complex on the luminal surface of the endothelial cells, and then this complex is taken up into the endothelia by endocytosis (Visser et al. 2004). How iron is expelled out of the abluminal surface of the endothelia and into the brain interstitial fluid is still controversial. Some studies showed that iron was segregated from Tf in the endothelia and then released to the brain interstitial fluid (Moos et al. 2006). Afterwards, iron is mainly incorporated with Tf in the interstitial fluid. Neurons can import iron by endocytosis of Tf–TfR complex (Leitner and Connor 2012; Moos et al. 1998), then divalent metal ion transporter 1 (DMT1) helps iron transport from the endosomes to the cell cytoplasm (Moos and Morgan 2004), where some of the iron is sent to ferritin with the help of the chaperone poly-r(C)-binding protein 1 (PCBP) family (Leidgens et al. 2013), some imported into mitochondria for heme and iron-sulfur cluster (ISC) synthesis, and few of the iron stays in the labile iron pool (Lane et al. 2015). How iron is transported into mitochondria is still not fully elucidated, and mitoferrin is a putative mitochondrial iron importer in neuronal cells (Carroll et al. 2011; Lane et al. 2015). Mastroberardino et al. (2009) demonstrated a Tf–TfR2 pathway importing iron into mitochondria in the neurons in SN. As neuromelanin is the main protein storing iron in dopaminergic cells in SN, it is synthesized in the process of dopamine oxidation (Zucca et al. 2015). Excess iron is transported by ferroportin out of the neurons, with the help of ferroxidases such as ceruloplasmin (CP) (De Domenico et al. 2007). The homeostasis of cellular iron is kept by two iron regulatory proteins (IRP1 and IRP2), which regulate the translations of the mRNAs of proteins involved in iron storage, influx, and efflux (Rouault 2006; Klausner et al. 1993).
The iron transport related to the glia is less clear than that in the neurons. Virtually no DMT1 or transferrin receptor can be detected in quiescent astrocytes, microglia, and oligodendrocytes (Moos and Morgan 2004; Pelizzoni et al. 2013; Skjorringe et al. 2015; Moos 1996). Ferroportin was not detected in astrocytes and resting microglia (Moos and Rosengren Nielsen 2006). Microglia iron uptake is performed via phagocytosis of ferritin (Leitner and Connor 2012), and oligodendrocytes also obtain iron from ferritin (Todorich et al. 2008). Astrocytes acquire iron through the resident transient receptor potential (TRP) channels in the quiescent state, and the de novo expressed DMT1 in activated state (Pelizzoni et al. 2013).
Interplay between mitochondrial injury, iron dyshomeostasis, and oxidative stress
Mitochondria provide energy for the cell via oxidative phosphorylation. This makes mitochondria a source of hydrogen peroxide and superoxide, which can react with iron. In addition, heme and ISC are synthesized in mitochondria, so iron is actively transported into and within mitochondria (Lill et al. 2006; Heinemann et al. 2008; Nilsson et al. 2009). Through Fenton and Haber–Weiss reactions, the most reactive oxygen species (ROS) hydroxyl radicals are produced (Wardman and Candeias 1996; Kehrer 2000). Therefore, iron overload can lead to increased oxidative stress, which can induce lipid peroxidation, damage DNA, and oxidize proteins (especially protein carbonylation) (Catala 2009; Stadtman 2006; Keyer and Imlay 1996). In particular, mitochondrial DNA is vulnerable to oxidative damage because of the absence of protection from histones (Shokolenko et al. 2009). Mitochondrial injury leads to reduced synthesis of ISCs and heme, then decreased ISCs causes IRPs activation and further exacerbates iron accumulation and related oxidative stress. Thus mitochondrial injury, iron accumulation, and oxidative stress form a vicious cycle that can lead to cell death (Mena et al. 2015). Moreover, this process discussed above can trigger inflammation response by activating microglia, adding oxidative stress in the vicious circle (Urrutia et al. 2014).
Iron and PD
Iron overload in the substantia nigra in PD
The Lewy body is the pathological hallmark of PD, and SN is an especially vulnerable area. Neurons in the SN progressively decreased in PD, which is responsible for the disabling motor symptoms. Total iron in SN is demonstrated to be increased by multiple post-mortem examinations (such as inductively coupled plasma spectroscopy, atomic absorption spectroscopy), MRI, and transcranial sonography in PD (Dexter et al. 1989; Riederer et al. 1989; Zecca et al. 2005; Michaeli et al. 2007; Rossi et al. 2013; Martin et al. 2008). Furthermore, ferritin and neuromelanin are reported to be decreased in the SN of PD patients (Dexter et al. 1990; Connor et al. 1995; Zecca et al. 2002). Considering the increase of total iron, and the decrease of iron-binding proteins, the labile iron pool of the cells in SN of PD patients is probably enlarged. Riederer et al. reported that Fe(III) was significantly increased in SN in PD, while Fe(II) remained unchanged (Riederer et al. 1989). Iron dyshomeostasis is caused by increased expression of the iron import transporter DMT1, decreased expression of the iron export protein ferroportin and CP activity (Salazar et al. 2008; Song et al. 2010; Ayton et al. 2013). Moreover, IRP is up-regulated in PD, rather than down-regulated to keep iron homeostasis (Wong and Duce 2014; Faucheux et al. 2002; Jiang et al. 2010). The shift of IRP may be partially caused by increased oxidative stress. So far, it is unclear what is the primary drive for the above mechanism of iron excess in SN of patients with PD, but it is suggested that α-synuclein aggregation, oxidative stress, and mitochondrial dysfunction might be involved. In addition, these factors and iron accumulation compose a vicious circle leading to neuroinflammation and neurodegeneration (Mena et al. 2015; Urrutia et al. 2014).
Iron involvement in the pathogenesis of PD
The vicious circle of mitochondrial injury, oxidative stress, iron dyshomeostasis and neuroinflammation has close interactions with several factors in PD. Firstly, dopamine metabolism creates highly reactive species in SN, and co-localization of iron and dopamine in SN raises the risk of oxidative stress (Hare et al. 2014). Secondly, as oxidative stress can induce protein carbonylation, Münch and colleagues suggested that the products of protein carbonylation could induce α-synuclein crosslinking and Lewy body formation (Munch et al. 2000). Thirdly, ferric iron may directly catalyze the formation of α-synuclein oligomers, and α-synuclein overexpression can exacerbate iron accumulation (Ostrerova-Golts et al. 2000; Golts et al. 2002; Ortega et al. 2015). In turn, aggregated α-synuclein can impair mitochondria, enhance oxidative stress and iron dyshomeostasis, thus intimately participate into the positive feedback loop (Davies et al. 2011; Devi et al. 2008; Funke et al. 2013). Moreover, neuromelanin released by dying dopaminergic neurons contains large amounts or iron, which can lead to the activation of adjacent microglia. The activated microglia induces inflammation and aggravates the vicious circle of oxidative stress, mitochondrial injury and cell death. Then more neuromelanin can be released from the demised neurons, and form a positive feedback of neuroinflammation and neurodegeneration (Zucca et al. 2014).
Imaging modalities for brain iron detection in PD
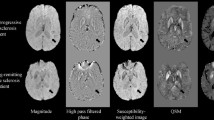
MRI can display the morphological changes of SN and evaluate the iron content in SN. MR imaging of SN is based on its iron components. Generally, SN has a high level of iron, so it appears as low intensity in T2WI, T2*, and susceptibility weighted imaging (SWI) (Lehericy et al. 2014; Jin et al. 2011). The iron level can be quantitatively assessed by R2*, SWI phase values, quantitative susceptibility mapping (QSM) and similar techniques (Rossi et al. 2013; Jin et al. 2011; He et al. 2015). Recently, it was recognized that neuromelanin had a T1 shortening effect, and could be well demonstrated by neuromelanin MR imaging (Lehericy et al. 2014; Blazejewska et al. 2013). Studies using 1.5 Tesla and 3.0 Tesla MRI have shown that the volume of SN is decreased, while iron load of SN is increased in PD. Furthermore, iron elevation of SN is correlated with disease severity and disease duration (He et al. 2015; Rossi et al. 2013). Due to the better spatial resolution and contrast, 7 Tesla MRI can reliably differentiate SN pars reticulata (SNr) and SN pars compacta (SNc), and in particular recognize the nigrosome-1 with high confidence. That is because SNr has abundant iron while SNc is rich in neuromelanin (Lehericy et al. 2014; Kwon et al. 2012; Blazejewska et al. 2013). Research using 7 Tesla MRI suggests that the main abnormalities in SN in PD are: loss of nigrosome-1 hyperintensity, abnormal SN contours, and volume changes (Lehericy et al. 2014; Kwon et al. 2012; Blazejewska et al. 2013). MRI can also be used for early diagnosis (even presymptomatic), differential diagnosis, monitoring disease progression and assessing the effect of iron chelation treatment, as well as exploring the pathophysiology of iron toxicity (Jin et al. 2011; Ward et al. 2014; Pyatigorskaya et al. 2015; Boelmans et al. 2012; Devos et al. 2014).
SN hyperechogenicity in transcranial sonography is detected in approximately 90 % of the patients with PD (Berg et al. 2001; Berg 2011). The source of SN hyperechogenicity may be increased iron content and microglia activation (Berg 2011; Zecca et al. 2005). About 10 % of the healthy people also have SN hyperechogenicity, and longitudinal studies showed that those healthy people with SN hyperechogenicity had a significantly higher risk to develop PD (Becker et al. 1995; Behnke et al. 2007; Berg 2011). On one hand, SN hyperechogenicity can present early in the disease course, even before motor symptoms occur (Haehner et al. 2007; Miyamoto and Miyamoto 2013). On the other hand, it does not change during disease progression, and is poorly correlated with striatal FP-CIT uptake (Li et al. 2015). Therefore, it may be an appropriate tool for early diagnosis. Although in patients with hyposmia or rapid eye movement sleep behavior disorder, the sensitivity and specificity of SN hyperechogenicity for predicting future PD is not satisfactory, combining other biomarkers may improve the ability of future PD prediction (Miyamoto and Miyamoto 2013; Haehner et al. 2007). In addition, transcranial sonography can provide help in differentiating PD from other Parkinsonian disorders (Berg 2011; Tsai et al. 2007).
Iron chelation therapy in PD
Iron chelation therapy in PD is still an expanding field of research. Genetic (overexpression of ferritin) and medical iron chelation treatments showed neuroprotective affects in various PD animal models, including 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA), mouse/rat models of PD (Dexter et al. 2011; Kaur et al. 2003; Shachar et al. 2004; Devos et al. 2014). Devos et al. (2014) had demonstrated that deferiprone reduced iron overload in SN, decreased neuronal labile iron, increased levels of glutathione, diminished oxidation products of lipid and DNA, reduced dopamine depletion and improve motor symptoms in the MPTP mouse model. It showed that deferiprone could stop the vicious cycle of iron accumulation, mitochondrial injury and oxidative stress, then decrease neuronal destruction (Devos et al. 2014). Moreover, the authors showed deferiprone can slow down the motor symptom progression and decrease iron accumulation in SN in PD patients (Devos et al. 2014). Additionally, Grolez et al. (2015) showed CP activity might play a role in the therapeutic mechanism of deferiprone, and PD patients with lower CP responded better to iron chelation therapy. The authors pointed out insights for the prospect of future pharmacological modulation of CP activity in PD (Grolez et al. 2015). In recent years, Youdim and colleagues have developed a series of multifunctional iron chelators, such as M30, HLA20, and VAR. These drugs can both chelate iron and inhibit monoamine oxidase (MAO) activity. In a recent study by Bar-Am et al. (2015), VAR not only chelated iron, and alleviated oxidative stress induced lipid peroxidation, but also inhibited MAO-A and MAO-B, and increased dopamine and 5-Hydroxytryptophan (5-HT) levels. In 6-OHDA and MPTP rat models, VAR could increase dopamine and 5-HT, as well as improve motor function (Bar-Am et al. 2015). Since PD patients often have accompanying depression which is related to 5-HT (Goetz 2010), VAR is a promising drug that can slow disease progression, improve motor symptoms, and alleviate depression in PD. Another novel chelator M30, has the ability of chelating iron, up-regulating hypoxia-inducible factor (HIF)-1α and its downstream proteins such as vascular endothelial growth factor, erythropoietin, enolase-1, inducing the expression of a series of neurotrophic factors like brain-derived neurotrophic factor, glial cell-derived neurotrophic factor and antioxidant enzymes including catalase, superoxide dismutase, and glutathione peroxidase in the brain, and regulating several factors involved in pro-survival signaling pathways such as phosphorylated protein kinase C, ERK1/2, -Akt and GSK-3β, in the CNS. Therefore, M30 has an extensive effect of neuroprotection (Kupershmidt et al. 2011).
Iron and other neurodegenerative diseases
Iron and AD
Excessive iron is deposited in multiple regions of the brain in AD, especially in the hippocampus. Iron also accumulates in senile plaques and neurofibrillary tangles, which are pathologic hallmarks of AD (Connor et al. 1992; Deibel et al. 1996; Good et al. 1992). The changes of iron regulating proteins drive iron overload, iron importer DMT1 is increased, while ferroportin 1 and CP that are responsible for iron exportation are decreased (Zheng et al. 2009; Crespo et al. 2014; Raha et al. 2013; Connor et al. 1993; Guerreiro et al. 2015; Wan et al. 2011). Similar as in other neurodegenerative diseases, iron also induces oxidative stress and mitochondrial dysfunction in AD (Wan et al. 2011; Mena et al. 2015). Moreover, amyloid β (Aβ), its precursor amyloid precursor protein (APP) and hyperphosphorylated tau have close interactions with excess iron in AD (Mena et al. 2015; Crichton et al. 2011; Everett et al. 2014; Yamamoto et al. 2002; Bodovitz et al. 1995). Normally, APP is cleaved by α- and γ-secretase. This process produces neuroprotective extracellular soluble sAβPPsα fragments and avoids Aβ formation. On the contrary, in AD, APP is cleaved by β- and γ-secretase, and this pathway leads to Aβ production and aggregation. Whether APP is firstly cleaved by α- or β- secretase is regulated by iron via furin. Iron overload can decrease furin expression, and as a result promote Aβ accumulation (Crichton et al. 2011; Bodovitz et al. 1995; Silvestri and Camaschella 2008; Silvestri et al. 2008). Besides, APP expression is modulated by IRP. Thus excessive iron can enhance APP production, and this will further increase Aβ formation (Rogers et al. 2002). In turn, Aβ can impair mitochondrial function, reduce ferric iron into a redox-active ferrous state, induce oxidative stress, and then exacerbate iron overload, thus aggravating the common pathway of neurodegenerative diseases (Mena et al. 2015; Everett et al. 2014; Wang et al. 2008; Smith et al. 1998). Iron can also interplay with hyperphosphorylated tau and induce the formation of neurofibrillary tangles (Castellani et al. 2012; Sayre et al. 2000; Yamamoto et al. 2002).
MRI is capable of evaluating iron accumulation in AD, and is promising for assisting diagnosis, as well as monitoring the efficacy of iron chelation and disease progression (Ward et al. 2014; van Rooden et al. 2015). In addition, a recent post-mortem study using 7 Tesla MRI demonstrated activated iron-containing microglia in the hippocampus of patients with AD (Zeineh et al. 2015). The advent of 7 Tesla MRI may provide more information on the role of iron in AD.
In recent years, clinical trials in AD targeting Aβ have failed one after another (Mangialasche et al. 2010; Salloway et al. 2014; Doody et al. 2014). Part of the reasons is that the pathophysiology of AD is complex, and contains a self-propagating vicious circle of iron accumulation, oxidative stress, and mitochondrial injury. Only cutting off the upstream factors such as Aβ cannot stop this vicious circle. On the other hand, iron chelation therapy may provide some hope. Iron chelation treatments using deferoxamine, clioquinol, and PBT2 can improve cognition, reduce Aβ accumulation and tau phosphorylation in animal models (Guo et al. 2013a, b; Grossi et al. 2009; Adlard et al. 2008). A clinical trial by Crapper McLachlan showed desferrioxamine significantly slowed down the decline of daily living skills in AD (Crapper McLachlan et al. 1991). More recently, the multi-target iron chelators M30 and HLA20 improved cognition of sporadic AD rat models. Additionally, chronic M30 therapy completely restored streptozotocin induced tau hyperphosphorylation in the hippocampus of those rats (Salkovic-Petrisic et al. 2015). Incorporating iron chelation in AD treatments is a promising approach in the future.
Iron and Friedreich ataxia
Friedreich ataxia is the most prevalent hereditary ataxia, and most of the patients with Friedreich ataxia are caused by an expanded GAA trinucleotide repeat in Frataxin (FXN) gene. This mutation decreases the level of FXN protein. FXN is an iron chaperone in iron–sulfur cluster and heme synthesis, and plays a critical role in keeping iron homeostasis and reducing oxidative stress (Gille and Reichmann 2011; Bulteau et al. 2004). Therefore, reduced expression of FXN leads to mitochondrial dysfunction, oxidative stress, and mitochondrial iron dyshomeostasis (Gille and Reichmann 2011; Babcock et al. 1997; Rotig et al. 1997). Whether overall iron is increased in the dorsal root ganglia and cerebellum is still controversial, and recent research showed iron was relocated from degenerated neurons to peripheral glial cells (Martelli and Puccio 2014; Koeppen et al. 2009, 2012, 2013). A quite recent phase 2, multicenter clinical trial on the safety and efficacy of iron chelator deferiprone did not find a significant improvement in clinical outcomes. However, subgroup analysis implied that deferiprone might be effective in less severe patients (Pandolfo et al. 2014). The effectiveness of iron chelation therapy in Friedreich ataxia warrant further investigation.
Iron and Huntington’s disease
Huntington’s disease is caused by a CAG trinucleotide repeat expansion in the huntingtin gene. Then the mutant huntingtin protein leads to multiple detrimental outcomes such as mitochondrial dysfunction, oxidative stress, abnormal transcription of multiple genes, calcium dyshomeostasis, activation of proteolytic enzymes, and microglia activation (Muller and Leavitt 2014). As a redox-active metal, iron is closely involved in the mutant huntingtin-induced pathological cascade (Firdaus et al. 2006; Muller and Leavitt 2014). In addition, iron homeostasis is influenced in this process, and iron accumulation in multiple brain regions has been demonstrated by MRI and post-mortem examinations (Bartzokis et al. 1999, 2007). MRI examination incorporating iron-sensitive techniques (such as T2*, SWI, R2*, SWI phase values, and QSM) is promising to assist presymptomatic diagnosis and monitor disease progression (Bartzokis et al. 2007; Apple et al. 2014; Macerollo et al. 2014; Dominguez et al. 2015; Sanchez-Castaneda et al. 2015). So far, there is scarce evidence on iron chelation treatments in Huntington’s disease. Recently, Chen and colleagues demonstrated the neuroprotective efficacy of deferoxamine in a mouse model (Chen et al. 2013). The effectiveness of iron chelation therapy in Huntington’s disease animal models warrants further exploration.
Conclusions
Although the above neurodegenerative diseases have distinct causes and pathology features, iron dyshomeostasis, oxidative stress and mitochondrial dysfunction form a vicious circle and play a crucial role in their pathogenesis. The mechanisms of abnormal iron metabolism in those neurodegenerative diseases are to be further elucidated. MRI is a helpful tool in revealing iron accumulation in the brain, and is increasingly used in early and differential diagnosis, tracking disease progression, and evaluating the efficacy of chelation therapy. Iron chelation is promising and has already exhibited its effectiveness in several clinical studies.
References
Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI (2008) Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59(1):43–55. doi:10.1016/j.neuron.2008.06.018
Apple AC, Possin KL, Satris G, Johnson E, Lupo JM, Jakary A, Wong K, Kelley DA, Kang GA, Sha SJ, Kramer JH, Geschwind MD, Nelson SJ, Hess CP (2014) Quantitative 7T phase imaging in premanifest Huntington disease. AJNR Am J Neuroradiol 35(9):1707–1713. doi:10.3174/ajnr.A3932
Ayton S, Lei P, Duce JA, Wong BX, Sedjahtera A, Adlard PA, Bush AI, Finkelstein DI (2013) Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann Neurol 73(4):554–559. doi:10.1002/ana.23817
Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, Pandolfo M, Kaplan J (1997) Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 276(5319):1709–1712
Bar-Am O, Amit T, Kupershmidt L, Aluf Y, Mechlovich D, Kabha H, Danovitch L, Zurawski VR, Youdim MB, Weinreb O (2015) Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol Aging 36(3):1529–1542. doi:10.1016/j.neurobiolaging.2014.10.026
Bartzokis G, Cummings J, Perlman S, Hance DB, Mintz J (1999) Increased basal ganglia iron levels in Huntington disease. Arch Neurol 56(5):569–574
Bartzokis G, Lu PH, Tishler TA, Fong SM, Oluwadara B, Finn JP, Huang D, Bordelon Y, Mintz J, Perlman S (2007) Myelin breakdown and iron changes in Huntington’s disease: pathogenesis and treatment implications. Neurochem Res 32(10):1655–1664. doi:10.1007/s11064-007-9352-7
Becerril-Ortega J, Bordji K, Freret T, Rush T, Buisson A (2014) Iron overload accelerates neuronal amyloid-beta production and cognitive impairment in transgenic mice model of Alzheimer’s disease. Neurobiol Aging 35(10):2288–2301. doi:10.1016/j.neurobiolaging.2014.04.019
Becker G, Seufert J, Bogdahn U, Reichmann H, Reiners K (1995) Degeneration of substantia nigra in chronic Parkinson’s disease visualized by transcranial color-coded real-time sonography. Neurology 45(1):182–184
Behnke S, Double KL, Duma S, Broe GA, Guenther V, Becker G, Halliday GM (2007) Substantia nigra echomorphology in the healthy very old: correlation with motor slowing. Neuroimage 34(3):1054–1059. doi:10.1016/j.neuroimage.2006.10.010
Beinert H, Holm RH, Munck E (1997) Iron-sulfur clusters: nature’s modular, multipurpose structures. Science 277(5326):653–659
Berg D (2011) Hyperechogenicity of the substantia nigra: pitfalls in assessment and specificity for Parkinson’s disease. J Neural Trans 118(3):453–461. doi:10.1007/s00702-010-0469-5
Berg D, Siefker C, Becker G (2001) Echogenicity of the substantia nigra in Parkinson’s disease and its relation to clinical findings. J Neurol 248(8):684–689
Blazejewska AI, Schwarz ST, Pitiot A, Stephenson MC, Lowe J, Bajaj N, Bowtell RW, Auer DP, Gowland PA (2013) Visualization of nigrosome 1 and its loss in PD: pathoanatomical correlation and in vivo 7 T MRI. Neurology 81(6):534–540. doi:10.1212/WNL.0b013e31829e6fd2
Bodovitz S, Falduto MT, Frail DE, Klein WL (1995) Iron levels modulate alpha-secretase cleavage of amyloid precursor protein. J Neurochem 64(1):307–315
Boelmans K, Holst B, Hackius M, Finsterbusch J, Gerloff C, Fiehler J, Munchau A (2012) Brain iron deposition fingerprints in Parkinson’s disease and progressive supranuclear palsy. Mov Disord 27(3):421–427. doi:10.1002/mds.24926
Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM (2007) Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement 3(3):186–191. doi:10.1016/j.jalz.2007.04.381
Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI (2004) Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 305(5681):242–245. doi:10.1126/science.1098991
Cabantchik ZI (2014) Labile iron in cells and body fluids: physiology, pathology, and pharmacology. Front Pharmacol 5:45. doi:10.3389/fphar.2014.00045
Carroll CB, Zeissler ML, Chadborn N, Gibson K, Williams G, Zajicek JP, Morrison KE, Hanemann CO (2011) Changes in iron-regulatory gene expression occur in human cell culture models of Parkinson’s disease. Neurochem Int 59(1):73–80. doi:10.1016/j.neuint.2011.05.006
Castellani RJ, Moreira PI, Perry G, Zhu X (2012) The role of iron as a mediator of oxidative stress in Alzheimer disease. BioFactors 38(2):133–138. doi:10.1002/biof.1010
Catala A (2009) Lipid peroxidation of membrane phospholipids generates hydroxy-alkenals and oxidized phospholipids active in physiological and/or pathological conditions. Chem Phys Lipids 157(1):1–11. doi:10.1016/j.chemphyslip.2008.09.004
Chen J, Marks E, Lai B, Zhang Z, Duce JA, Lam LQ, Volitakis I, Bush AI, Hersch S, Fox JH (2013) Iron accumulates in Huntington’s disease neurons: protection by deferoxamine. PLoS ONE 8(10):e77023. doi:10.1371/journal.pone.0077023
Connor JR, Menzies SL, St Martin SM, Mufson EJ (1992) A histochemical study of iron, transferrin, and ferritin in Alzheimer’s diseased brains. J Neurosci Res 31(1):75–83. doi:10.1002/jnr.490310111
Connor JR, Tucker P, Johnson M, Snyder B (1993) Ceruloplasmin levels in the human superior temporal gyrus in aging and Alzheimer’s disease. Neurosci Lett 159(1–2):88–90
Connor JR, Boeshore KL, Benkovic SA, Menzies SL (1994) Isoforms of ferritin have a specific cellular distribution in the brain. J Neurosci Res 37(4):461–465. doi:10.1002/jnr.490370405
Connor JR, Snyder BS, Arosio P, Loeffler DA, LeWitt P (1995) A quantitative analysis of isoferritins in select regions of aged, parkinsonian, and Alzheimer’s diseased brains. J Neurochem 65(2):717–724
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337(8753):1304–1308
Crespo AC, Silva B, Marques L, Marcelino E, Maruta C, Costa S, Timoteo A, Vilares A, Couto FS, Faustino P, Correia AP, Verdelho A, Porto G, Guerreiro M, Herrero A, Costa C, de Mendonca A, Costa L, Martins M (2014) Genetic and biochemical markers in patients with Alzheimer’s disease support a concerted systemic iron homeostasis dysregulation. Neurobiol Aging 35(4):777–785. doi:10.1016/j.neurobiolaging.2013.10.078
Crichton RR, Dexter DT, Ward RJ (2011) Brain iron metabolism and its perturbation in neurological diseases. J Neural Transm 118(3):301–314. doi:10.1007/s00702-010-0470-z
Davies P, Moualla D, Brown DR (2011) Alpha-synuclein is a cellular ferrireductase. PLoS ONE 6(1):e15814. doi:10.1371/journal.pone.0015814
De Domenico I, Ward DM, di Patti MC, Jeong SY, David S, Musci G, Kaplan J (2007) Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J 26(12):2823–2831. doi:10.1038/sj.emboj.7601735
Deibel MA, Ehmann WD, Markesbery WR (1996) Copper, iron, and zinc imbalances in severely degenerated brain regions in Alzheimer’s disease: possible relation to oxidative stress. J Neurol Sci 143(1–2):137–142
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK (2008) Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem 283(14):9089–9100. doi:10.1074/jbc.M710012200
Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, Jonneaux A, Ryckewaert G, Garcon G, Rouaix N, Duhamel A, Jissendi P, Dujardin K, Auger F, Ravasi L, Hopes L, Grolez G, Firdaus W, Sablonniere B, Strubi-Vuillaume I, Zahr N, Destee A, Corvol JC, Poltl D, Leist M, Rose C, Defebvre L, Marchetti P, Cabantchik ZI, Bordet R (2014) Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid Redox Signal 21(2):195–210. doi:10.1089/ars.2013.5593
Dexter DT, Wells FR, Lees AJ, Agid F, Agid Y, Jenner P, Marsden CD (1989) Increased nigral iron content and alterations in other metal ions occurring in brain in Parkinson’s disease. J Neurochem 52(6):1830–1836
Dexter DT, Carayon A, Vidailhet M, Ruberg M, Agid F, Agid Y, Lees AJ, Wells FR, Jenner P, Marsden CD (1990) Decreased ferritin levels in brain in Parkinson’s disease. J Neurochem 55(1):16–20
Dexter DT, Statton SA, Whitmore C, Freinbichler W, Weinberger P, Tipton KF, Della Corte L, Ward RJ, Crichton RR (2011) Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson’s disease after peripheral administration. J Neural Trans 118(2):223–231. doi:10.1007/s00702-010-0531-3
Dexter DT, Martin-Bastida A, Kabba C, Piccini P, Sharp D, Ward R, Newbold R (2014) A pilot 6 months efficacy and safety study utilising the iron chelator deferiprone in early stage Parkinson’s disease [abstract]. Mov Disord 29(Suppl 1):633
Dominguez DJ, Ng AC, Poudel G, Stout JC, Churchyard A, Chua P, Egan GF, Georgiou-Karistianis N (2015) Iron accumulation in the basal ganglia in Huntington’s disease: cross-sectional data from the IMAGE-HD study. J Neurol Neurosurg Psychiatry. doi:10.1136/jnnp-2014-310183
Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, Kieburtz K, Raman R, Sun X, Aisen PS, Siemers E, Liu-Seifert H, Mohs R, Alzheimer’s Disease Cooperative Study Steering C, Solanezumab Study G (2014) Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med 370(4):311–321. doi:10.1056/NEJMoa1312889
Dorsey ER, Constantinescu R, Thompson JP, Biglan KM, Holloway RG, Kieburtz K, Marshall FJ, Ravina BM, Schifitto G, Siderowf A, Tanner CM (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68(5):384–386. doi:10.1212/01.wnl.0000247740.47667.03
Everett J, Cespedes E, Shelford LR, Exley C, Collingwood JF, Dobson J, van der Laan G, Jenkins CA, Arenholz E, Telling ND (2014) Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1–42). J R Soc Interface 11(95):20140165. doi:10.1098/rsif.2014.0165
Faucheux BA, Martin ME, Beaumont C, Hunot S, Hauw JJ, Agid Y, Hirsch EC (2002) Lack of up-regulation of ferritin is associated with sustained iron regulatory protein-1 binding activity in the substantia nigra of patients with Parkinson’s disease. J Neurochem 83(2):320–330
Firdaus WJ, Wyttenbach A, Giuliano P, Kretz-Remy C, Currie RW, Arrigo AP (2006) Huntingtin inclusion bodies are iron-dependent centers of oxidative events. FEBS J 273(23):5428–5441. doi:10.1111/j.1742-4658.2006.05537.x
Funke C, Schneider SA, Berg D, Kell DB (2013) Genetics and iron in the systems biology of Parkinson’s disease and some related disorders. Neurochem Int 62(5):637–652. doi:10.1016/j.neuint.2012.11.015
Geppert M, Hohnholt MC, Nurnberger S, Dringen R (2012) Ferritin up-regulation and transient ROS production in cultured brain astrocytes after loading with iron oxide nanoparticles. Acta Biomater 8(10):3832–3839. doi:10.1016/j.actbio.2012.06.029
Gille G, Reichmann H (2011) Iron-dependent functions of mitochondria—relation to neurodegeneration. J Neural Transm 118(3):349–359. doi:10.1007/s00702-010-0503-7
Goetz CG (2010) New developments in depression, anxiety, compulsiveness, and hallucinations in Parkinson’s disease. Mov Disord 25(Suppl 1):S104–S109. doi:10.1002/mds.22636
Golts N, Snyder H, Frasier M, Theisler C, Choi P, Wolozin B (2002) Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J Biol Chem 277(18):16116–16123. doi:10.1074/jbc.M107866200
Good PF, Perl DP, Bierer LM, Schmeidler J (1992) Selective accumulation of aluminum and iron in the neurofibrillary tangles of Alzheimer’s disease: a laser microprobe (LAMMA) study. Ann Neurol 31(3):286–292. doi:10.1002/ana.410310310
Grolez G, Moreau C, Sablonniere B, Garcon G, Devedjian JC, Meguig S, Gele P, Delmaire C, Bordet R, Defebvre L, Cabantchik IZ, Devos D (2015) Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol 15:74. doi:10.1186/s12883-015-0331-3
Grossi C, Francese S, Casini A, Rosi MC, Luccarini I, Fiorentini A, Gabbiani C, Messori L, Moneti G, Casamenti F (2009) Clioquinol decreases amyloid-beta burden and reduces working memory impairment in a transgenic mouse model of Alzheimer’s disease. J Alzheimers Dis 17(2):423–440. doi:10.3233/JAD-2009-1063
Guerreiro C, Silva B, Crespo AC, Marques L, Costa S, Timoteo A, Marcelino E, Maruta C, Vilares A, Matos M, Couto FS, Faustino P, Verdelho A, Guerreiro M, Herrero A, Costa C, de Mendonca A, Martins M, Costa L (2015) Decrease in APP and CP mRNA expression supports impairment of iron export in Alzheimer’s disease patients. Biochim Biophys Acta 1852(10 Pt A):2116–2122. doi:10.1016/j.bbadis.2015.07.017
Guo C, Wang P, Zhong ML, Wang T, Huang XS, Li JY, Wang ZY (2013a) Deferoxamine inhibits iron induced hippocampal tau phosphorylation in the Alzheimer transgenic mouse brain. Neurochem Int 62(2):165–172. doi:10.1016/j.neuint.2012.12.005
Guo C, Wang T, Zheng W, Shan ZY, Teng WP, Wang ZY (2013b) Intranasal deferoxamine reverses iron-induced memory deficits and inhibits amyloidogenic APP processing in a transgenic mouse model of Alzheimer’s disease. Neurobiol Aging 34(2):562–575. doi:10.1016/j.neurobiolaging.2012.05.009
Haehner A, Hummel T, Hummel C, Sommer U, Junghanns S, Reichmann H (2007) Olfactory loss may be a first sign of idiopathic Parkinson’s disease. Mov Disord 22(6):839–842. doi:10.1002/mds.21413
Hansen TM, Nielsen H, Bernth N, Moos T (1999) Expression of ferritin protein and subunit mRNAs in normal and iron deficient rat brain. Brain Res Mol Brain Res 65(2):186–197
Hare DJ, Lei P, Ayton S, Roberts BR, Grimm R, George JL, Bishop DP, Beavis AD, Donovan SJ, McColl G, Volitakis I, Masters CL, Adlard PA, Cherny RA, Bush AI, Finkelstein DI, Doble PA (2014) An iron-dopamine index predicts risk of parkinsonian neurodegeneration in the substantia nigra pars compacta. Chem Sci 5(6):2160–2169. doi:10.1039/c3sc53461h
He N, Ling H, Ding B, Huang J, Zhang Y, Zhang Z, Liu C, Chen K, Yan F (2015) Region-specific disturbed iron distribution in early idiopathic Parkinson’s disease measured by quantitative susceptibility mapping. Hum Brain Mapp. doi:10.1002/hbm.22928
Heinemann IU, Jahn M, Jahn D (2008) The biochemistry of heme biosynthesis. Arch Biochem Biophys 474(2):238–251. doi:10.1016/j.abb.2008.02.015
Hilditch-Maguire P, Trettel F, Passani LA, Auerbach A, Persichetti F, MacDonald ME (2000) Huntingtin: an iron-regulated protein essential for normal nuclear and perinuclear organelles. Hum Mol Genet 9(19):2789–2797
Hoepken HH, Korten T, Robinson SR, Dringen R (2004) Iron accumulation, iron-mediated toxicity and altered levels of ferritin and transferrin receptor in cultured astrocytes during incubation with ferric ammonium citrate. J Neurochem 88(5):1194–1202
Jenner P, Dexter DT, Sian J, Schapira AH, Marsden CD (1992) Oxidative stress as a cause of nigral cell death in Parkinson’s disease and incidental Lewy body disease. The Royal Kings and Queens Parkinson’s Disease Research Group. Ann Neurol 32(Suppl):S82–S87
Jiang H, Song N, Xu H, Zhang S, Wang J, Xie J (2010) Up-regulation of divalent metal transporter 1 in 6-hydroxydopamine intoxication is IRE/IRP dependent. Cell Res 20(3):345–356. doi:10.1038/cr.2010.20
Jin L, Wang J, Zhao L, Jin H, Fei G, Zhang Y, Zeng M, Zhong C (2011) Decreased serum ceruloplasmin levels characteristically aggravate nigral iron deposition in Parkinson’s disease. Brain 134(Pt 1):50–58. doi:10.1093/brain/awq319
Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK (2003) Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson’s disease. Neuron 37(6):899–909
Kehrer JP (2000) The Haber–Weiss reaction and mechanisms of toxicity. Toxicology 149(1):43–50
Keyer K, Imlay JA (1996) Superoxide accelerates DNA damage by elevating free-iron levels. Proc Natl Acad Sci USA 93(24):13635–13640
Klausner RD, Rouault TA, Harford JB (1993) Regulating the fate of mRNA: the control of cellular iron metabolism. Cell 72(1):19–28
Koeppen AH, Morral JA, Davis AN, Qian J, Petrocine SV, Knutson MD, Gibson WM, Cusack MJ, Li D (2009) The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol 118(6):763–776. doi:10.1007/s00401-009-0589-x
Koeppen AH, Ramirez RL, Yu D, Collins SE, Qian J, Parsons PJ, Yang KX, Chen Z, Mazurkiewicz JE, Feustel PJ (2012) Friedreich’s ataxia causes redistribution of iron, copper, and zinc in the dentate nucleus. Cerebellum 11(4):845–860. doi:10.1007/s12311-012-0383-5
Koeppen AH, Kuntzsch EC, Bjork ST, Ramirez RL, Mazurkiewicz JE, Feustel PJ (2013) Friedreich ataxia: metal dysmetabolism in dorsal root ganglia. Acta Neuropathol Commun 1:26. doi:10.1186/2051-5960-1-26
Kupershmidt L, Weinreb O, Amit T, Mandel S, Bar-Am O, Youdim MB (2011) Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 189:345–358. doi:10.1016/j.neuroscience.2011.03.040
Kwon DH, Kim JM, Oh SH, Jeong HJ, Park SY, Oh ES, Chi JG, Kim YB, Jeon BS, Cho ZH (2012) Seven-Tesla magnetic resonance images of the substantia nigra in Parkinson disease. Ann Neurol 71(2):267–277. doi:10.1002/ana.22592
Lane DJ, Merlot AM, Huang ML, Bae DH, Jansson PJ, Sahni S, Kalinowski DS, Richardson DR (2015) Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim Biophys Acta 1853(5):1130–1144. doi:10.1016/j.bbamcr.2015.01.021
Lehericy S, Bardinet E, Poupon C, Vidailhet M, Francois C (2014) 7 Tesla magnetic resonance imaging: a closer look at substantia nigra anatomy in Parkinson’s disease. Mov Disord 29(13):1574–1581. doi:10.1002/mds.26043
Leidgens S, Bullough KZ, Shi H, Li F, Shakoury-Elizeh M, Yabe T, Subramanian P, Hsu E, Natarajan N, Nandal A, Stemmler TL, Philpott CC (2013) Each member of the poly-r(C)-binding protein 1 (PCBP) family exhibits iron chaperone activity toward ferritin. J Biol Chem 288(24):17791–17802. doi:10.1074/jbc.M113.460253
Leitner DF, Connor JR (2012) Functional roles of transferrin in the brain. Biochim Biophys Acta 1820(3):393–402. doi:10.1016/j.bbagen.2011.10.016
Li DH, Zhang LY, Hu YY, Jiang XF, Zhou HY, Yang Q, Kang WY, Liu J, Chen SD (2015) Transcranial sonography of the substantia nigra and its correlation with DAT-SPECT in the diagnosis of Parkinson’s disease. Parkinsonism Relat Disord 21(8):923–928. doi:10.1016/j.parkreldis.2015.05.024
Lill R, Dutkiewicz R, Elsasser HP, Hausmann A, Netz DJ, Pierik AJ, Stehling O, Urzica E, Muhlenhoff U (2006) Mechanisms of iron-sulfur protein maturation in mitochondria, cytosol and nucleus of eukaryotes. Biochim Biophys Acta 1763(7):652–667. doi:10.1016/j.bbamcr.2006.05.011
Lin CI, Gollub EG, Beattie DS (1982) Synthesis of the proteins of complex III of the mitochondrial respiratory chain in heme-deficient cells. Eur J Biochem 128(2–3):309–313
Macerollo A, Perry R, Stamelou M, Batla A, Mazumder AA, Adams ME, Bhatia KP (2014) Susceptibility-weighted imaging changes suggesting brain iron accumulation in Huntington’s disease: an epiphenomenon which causes diagnostic difficulty. Eur J Neurol 21(2):e16–e17. doi:10.1111/ene.12298
Mangialasche F, Solomon A, Winblad B, Mecocci P, Kivipelto M (2010) Alzheimer’s disease: clinical trials and drug development. Lancet Neurol 9(7):702–716. doi:10.1016/S1474-4422(10)70119-8
Martelli A, Puccio H (2014) Dysregulation of cellular iron metabolism in Friedreich ataxia: from primary iron-sulfur cluster deficit to mitochondrial iron accumulation. Front Pharmacol 5:130. doi:10.3389/fphar.2014.00130
Martin WR, Wieler M, Gee M (2008) Midbrain iron content in early Parkinson disease: a potential biomarker of disease status. Neurology 70(16 Pt 2):1411–1417. doi:10.1212/01.wnl.0000286384.31050.b5
Mastroberardino PG, Hoffman EK, Horowitz MP, Betarbet R, Taylor G, Cheng D, Anderson M (2009) A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson's disease. Neurobiol dis 34(3):417–431
Mena NP, Urrutia PJ, Lourido F, Carrasco CM, Nunez MT (2015) Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 21:92–105. doi:10.1016/j.mito.2015.02.001
Michaeli S, Oz G, Sorce DJ, Garwood M, Ugurbil K, Majestic S, Tuite P (2007) Assessment of brain iron and neuronal integrity in patients with Parkinson’s disease using novel MRI contrasts. Mov Disord 22(3):334–340. doi:10.1002/mds.21227
Miyamoto M, Miyamoto T (2013) Neuroimaging of rapid eye movement sleep behavior disorder: transcranial ultrasound, single-photon emission computed tomography, and positron emission tomography scan data. Sleep Med 14(8):739–743. doi:10.1016/j.sleep.2013.03.004
Moos T (1996) Immunohistochemical localization of intraneuronal transferrin receptor immunoreactivity in the adult mouse central nervous system. J Comp Neurol 375(4):675–692. doi:10.1002/(SICI)1096-9861(19961125)375:4<675:AID-CNE8>3.0.CO;2-Z
Moos T, Morgan EH (2004) The significance of the mutated divalent metal transporter (DMT1) on iron transport into the Belgrade rat brain. J Neurochem 88(1):233–245
Moos T, Rosengren Nielsen T (2006) Ferroportin in the postnatal rat brain: implications for axonal transport and neuronal export of iron. Semin Pediatr Neurol 13(3):149–157. doi:10.1016/j.spen.2006.08.003
Moos T, Oates PS, Morgan EH (1998) Expression of the neuronal transferrin receptor is age dependent and susceptible to iron deficiency. J Comp Neurol 398(3):420–430
Moos T, Skjoerringe T, Gosk S, Morgan EH (2006) Brain capillary endothelial cells mediate iron transport into the brain by segregating iron from transferrin without the involvement of divalent metal transporter 1. J Neurochem 98(6):1946–1958. doi:10.1111/j.1471-4159.2006.04023.x
Muller M, Leavitt BR (2014) Iron dysregulation in Huntington’s disease. J Neurochem 130(3):328–350. doi:10.1111/jnc.12739
Munch G, Luth HJ, Wong A, Arendt T, Hirsch E, Ravid R, Riederer P (2000) Crosslinking of alpha-synuclein by advanced glycation endproducts—an early pathophysiological step in Lewy body formation? J Chem Neuroanat 20(3–4):253–257
Nilsson R, Schultz IJ, Pierce EL, Soltis KA, Naranuntarat A, Ward DM, Baughman JM, Paradkar PN, Kingsley PD, Culotta VC, Kaplan J, Palis J, Paw BH, Mootha VK (2009) Discovery of genes essential for heme biosynthesis through large-scale gene expression analysis. Cell Metab 10(2):119–130. doi:10.1016/j.cmet.2009.06.012
Ortega R, Carmona A, Roudeau S, Perrin L, Ducic T, Carboni E, Bohic S, Cloetens P, Lingor P (2015) Alpha-synuclein over-expression induces increased iron accumulation and redistribution in iron-exposed neurons. Mol Neurobiol. doi:10.1007/s12035-015-9146-x
Ostrerova-Golts N, Petrucelli L, Hardy J, Lee JM, Farer M, Wolozin B (2000) The A53T alpha-synuclein mutation increases iron-dependent aggregation and toxicity. J Neurosci 20(16):6048–6054
Pandolfo M, Arpa J, Delatycki MB, Le Quan Sang KH, Mariotti C, Munnich A, Sanz-Gallego I, Tai G, Tarnopolsky MA, Taroni F, Spino M, Tricta F (2014) Deferiprone in Friedreich ataxia: a 6-month randomized controlled trial. Ann Neurol 76(4):509–521. doi:10.1002/ana.24248
Parker WD Jr, Parks J, Filley CM, Kleinschmidt-DeMasters BK (1994) Electron transport chain defects in Alzheimer’s disease brain. Neurology 44(6):1090–1096
Pelizzoni I, Zacchetti D, Campanella A, Grohovaz F, Codazzi F (2013) Iron uptake in quiescent and inflammation-activated astrocytes: a potentially neuroprotective control of iron burden. Biochim Biophys Acta 1832(8):1326–1333. doi:10.1016/j.bbadis.2013.04.007
Pyatigorskaya N, Sharman M, Corvol JC, Valabregue R, Yahia-Cherif L, Poupon F, Cormier-Dequaire F, Siebner H, Klebe S, Vidailhet M, Brice A, Lehericy S (2015) High nigral iron deposition in LRRK2 and Parkin mutation carriers using R2* relaxometry. Mov Disord 30(8):1077–1084. doi:10.1002/mds.26218
Raha AA, Vaishnav RA, Friedland RP, Bomford A, Raha-Chowdhury R (2013) The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun 1:55. doi:10.1186/2051-5960-1-55
Rathore KI, Redensek A, David S (2012) Iron homeostasis in astrocytes and microglia is differentially regulated by TNF-alpha and TGF-beta1. Glia 60(5):738–750. doi:10.1002/glia.22303
Riederer P, Sofic E, Rausch WD, Schmidt B, Reynolds GP, Jellinger K, Youdim MB (1989) Transition metals, ferritin, glutathione, and ascorbic acid in parkinsonian brains. J Neurochem 52(2):515–520
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR (2002) An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem 277(47):45518–45528. doi:10.1074/jbc.M207435200
Rossi M, Ruottinen H, Soimakallio S, Elovaara I, Dastidar P (2013) Clinical MRI for iron detection in Parkinson’s disease. Clin Imaging 37(4):631–636. doi:10.1016/j.clinimag.2013.02.001
Rotig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, Munnich A, Rustin P (1997) Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet 17(2):215–217. doi:10.1038/ng1097-215
Rouault TA (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol 2(8):406–414. doi:10.1038/nchembio807
Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M, Duyckaerts C, Sazdovitch V, Zhao L, Garrick LM, Nunez MT, Garrick MD, Raisman-Vozari R, Hirsch EC (2008) Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson’s disease. Proc Natl Acad Sci USA 105(47):18578–18583. doi:10.1073/pnas.0804373105
Salkovic-Petrisic M, Knezovic A, Osmanovic-Barilar J, Smailovic U, Trkulja V, Riederer P, Amit T, Mandel S, Youdim MB (2015) Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer’s disease. Life Sci. doi:10.1016/j.lfs.2015.06.026
Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Ferris S, Reichert M, Ketter N, Nejadnik B, Guenzler V, Miloslavsky M, Wang D, Lu Y, Lull J, Tudor IC, Liu E, Grundman M, Yuen E, Black R, Brashear HR, Bapineuzumab, Clinical Trial I (2014) Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370(4):322–333. doi:10.1056/NEJMoa1304839
Sanchez-Castaneda C, Squitieri F, Di Paola M, Dayan M, Petrollini M, Sabatini U (2015) The role of iron in gray matter degeneration in Huntington’s disease: a magnetic resonance imaging study. Hum Brain Mapp 36(1):50–66. doi:10.1002/hbm.22612
Sayre LM, Perry G, Harris PL, Liu Y, Schubert KA, Smith MA (2000) In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: a central role for bound transition metals. J Neurochem 74(1):270–279
Shachar DB, Kahana N, Kampel V, Warshawsky A, Youdim MB (2004) Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 46(2):254–263
Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF (2009) Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 37(8):2539–2548. doi:10.1093/nar/gkp100
Silvestri L, Camaschella C (2008) A potential pathogenetic role of iron in Alzheimer’s disease. J Cell Mol Med 12(5A):1548–1550. doi:10.1111/j.1582-4934.2008.00356.x
Silvestri L, Pagani A, Camaschella C (2008) Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood 111(2):924–931. doi:10.1182/blood-2007-07-100677
Skjorringe T, Burkhart A, Johnsen KB, Moos T (2015) Divalent metal transporter 1 (DMT1) in the brain: implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front Mol Neurosci 8:19. doi:10.3389/fnmol.2015.00019
Smith MA, Hirai K, Hsiao K, Pappolla MA, Harris PL, Siedlak SL, Tabaton M, Perry G (1998) Amyloid-beta deposition in Alzheimer transgenic mice is associated with oxidative stress. J Neurochem 70(5):2212–2215
Song N, Wang J, Jiang H, Xie J (2010) Ferroportin 1 but not hephaestin contributes to iron accumulation in a cell model of Parkinson’s disease. Free Radic Biol Med 48(2):332–341. doi:10.1016/j.freeradbiomed.2009.11.004
Stadtman ER (2006) Protein oxidation and aging. Free Radic Res 40(12):1250–1258. doi:10.1080/10715760600918142
Todorich B, Zhang X, Slagle-Webb B, Seaman WE, Connor JR (2008) Tim-2 is the receptor for H-ferritin on oligodendrocytes. J Neurochem 107(6):1495–1505. doi:10.1111/j.1471-4159.2008.05678.x
Tsai CF, Wu RM, Huang YW, Chen LL, Yip PK, Jeng JS (2007) Transcranial color-coded sonography helps differentiation between idiopathic Parkinson’s disease and vascular parkinsonism. J Neurol 254(4):501–507. doi:10.1007/s00415-006-0403-9
Urrutia PJ, Mena NP, Nunez MT (2014) The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front Pharmacol 5:38. doi:10.3389/fphar.2014.00038
van Rooden S, Doan NT, Versluis MJ, Goos JD, Webb AG, Oleksik AM, van der Flier WM, Scheltens P, Barkhof F, Weverling-Rynsburger AW, Blauw GJ, Reiber JH, van Buchem MA, Milles J, van der Grond J (2015) 7T T(2)*-weighted magnetic resonance imaging reveals cortical phase differences between early- and late-onset Alzheimer’s disease. Neurobiol Aging 36(1):20–26. doi:10.1016/j.neurobiolaging.2014.07.006
Visser CC, Voorwinden LH, Crommelin DJ, Danhof M, de Boer AG (2004) Characterization and modulation of the transferrin receptor on brain capillary endothelial cells. Pharm Res 21(5):761–769
Wan L, Nie G, Zhang J, Luo Y, Zhang P, Zhang Z, Zhao B (2011) beta-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic Biol Med 50(1):122–129. doi:10.1016/j.freeradbiomed.2010.10.707
Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, Casadesus G, Zhu X (2008) Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci USA 105(49):19318–19323. doi:10.1073/pnas.0804871105
Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L (2014) The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol 13(10):1045–1060. doi:10.1016/S1474-4422(14)70117-6
Wardman P, Candeias LP (1996) Fenton chemistry: an introduction. Radiat Res 145(5):523–531
Wieler M, Gee M, Martin WR (2015) Longitudinal midbrain changes in early Parkinson’s disease: iron content estimated from R2*/MRI. Parkinsonism Relat Disord 21(3):179–183. doi:10.1016/j.parkreldis.2014.11.017
Wong BX, Duce JA (2014) The iron regulatory capability of the major protein participants in prevalent neurodegenerative disorders. Front Pharmacol 5:81. doi:10.3389/fphar.2014.00081
Yamamoto A, Shin RW, Hasegawa K, Naiki H, Sato H, Yoshimasu F, Kitamoto T (2002) Iron(III) induces aggregation of hyperphosphorylated τ and its reduction to iron(II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J Neurochem 82(5):1137–1147
Zecca L, Gallorini M, Schunemann V, Trautwein AX, Gerlach M, Riederer P, Vezzoni P, Tampellini D (2001) Iron, neuromelanin and ferritin content in the substantia nigra of normal subjects at different ages: consequences for iron storage and neurodegenerative processes. J Neurochem 76(6):1766–1773
Zecca L, Fariello R, Riederer P, Sulzer D, Gatti A, Tampellini D (2002) The absolute concentration of nigral neuromelanin, assayed by a new sensitive method, increases throughout the life and is dramatically decreased in Parkinson’s disease. FEBS Lett 510(3):216–220
Zecca L, Berg D, Arzberger T, Ruprecht P, Rausch WD, Musicco M, Tampellini D, Riederer P, Gerlach M, Becker G (2005) In vivo detection of iron and neuromelanin by transcranial sonography: a new approach for early detection of substantia nigra damage. Mov Disord 20(10):1278–1285. doi:10.1002/mds.20550
Zeineh MM, Chen Y, Kitzler HH, Hammond R, Vogel H, Rutt BK (2015) Activated iron-containing microglia in the human hippocampus identified by magnetic resonance imaging in Alzheimer disease. Neurobiol Aging 36(9):2483–2500. doi:10.1016/j.neurobiolaging.2015.05.022
Zhang Z, Huang L, Shulmeister VM, Chi YI, Kim KK, Hung LW, Crofts AR, Berry EA, Kim SH (1998) Electron transfer by domain movement in cytochrome bc1. Nature 392(6677):677–684. doi:10.1038/33612
Zheng W, Xin N, Chi ZH, Zhao BL, Zhang J, Li JY, Wang ZY (2009) Divalent metal transporter 1 is involved in amyloid precursor protein processing and Abeta generation. FASEB J 23(12):4207–4217. doi:10.1096/fj.09-135749
Zucca FA, Basso E, Cupaioli FA, Ferrari E, Sulzer D, Casella L, Zecca L (2014) Neuromelanin of the human substantia nigra: an update. Neurotox Res 25(1):13–23. doi:10.1007/s12640-013-9435-y
Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L (2015) Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol. doi:10.1016/j.pneurobio.2015.09.012
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Li, K., Reichmann, H. Role of iron in neurodegenerative diseases. J Neural Transm 123, 389–399 (2016). https://doi.org/10.1007/s00702-016-1508-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00702-016-1508-7