
Abstract
Molecular markers have revolutionized analyses in population genetics, enabling precise estimates of the amount of genetic variability and how it is distributed within and among populations. The high diversity of bamboos, distributed throughout the world and of high economic relevance, has deserved several studies on molecular characterization. This chapter describes how distinct categories of molecular markers, such as isozymes, RAPD, AFLP, microsatellites, and SNP, have enabled the analysis of population genetic processes, assessments of the genetic diversity, and structure of natural populations and selected cultivars of bamboo species. One important application is their power of phylogenetic inference, enabling the distinction of the diverse set of bamboo species. With the genomic technologies, gene families have been characterized, mainly for Phyllostachys edulis, which has its genome sequenced and deposited to databases, enabling the detection of markers related with environmental constraints. As vegetative propagation is a common mechanism in bamboos and their cultivation relies on this strategy, molecular markers have been important for attesting genetic fidelity to their original source of propagules. Altogether, we provide a panorama of several applications of molecular markers to bamboo conservation and breeding.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
2.1 Introduction
Only a few decades back in the 1960s or even in the 1970s and 1980s, taxonomy involved almost exclusively the use of morphological traits to differentiate one species from another. Genetic variation of populations was also accounted for based on morphological variation. It was in the mid-1960s that Lewontin and Hubby revealed the application of protein electrophoresis to quantify genetic variation in populations of fruit flies (Lewontin and Hubby 1966). Soon enough, isozyme electrophoresis spread throughout the world. Numberless population genetic studies were produced, unraveling genetic systems of a diverse set of species, including plants. A revolution to that began with the use of markers directly at the level of DNA, in the 1980s. Restriction fragment length polymorphisms (RFLP) enabled not only population genetic structure but also the development of linkage maps (Tanksley et al. 1989). The development of polymerase chain reaction accelerated the discovery of molecular markers of various types in a few laboratory steps. Random amplified polymorphic DNA (RAPD) (Williams et al. 1990) and amplified fragment length polymorphisms (AFLP) (Vos et al. 1995) filled in countless issues of scientific journals in the 1990s and early 2000s. Simple sequence repeats (SSR) or microsatellites (Litt and Luty 1989) became more popular and are still being largely employed for population genetic studies that require the distinction of heterozygotes, such as in studies of genetic diversity, structure, paternity and system of mating. Currently, single-nucleotide polymorphisms (SNP) prevail, as those are the most abundant markers in organisms, accounting for the variation in a single base.
As members of the Poaceae family, bamboos comprise up to 1662 species and 121 genera described so far (Canavan et al. 2017), with broad distribution throughout the world. Extensive molecular marker-based surveys have been carried out to constantly improve the resolution of phylogenetic trees of such species (Bamboo Phylogeny Group 2012; Wysocki et al. 2015). Besides taxonomy and molecular phylogenetics, molecular markers have also been employed for the study of bamboos for diverse purposes such as genetic diversity and structure of populations, genetic fidelity assessment in clonal populations, candidate gene search for growth, and developmental processes, among others. The emerging field of epigenomics has also been explored in a few studies so far. It is the aim of this chapter to describe the fundamental applications of molecular markers to bamboos, briefly accounting for the main methods that have been employed, either PCR or gel-based, or obtained directly from sequencing. Up-to-date studies on genomics and perspectives into other “omics,” such as epigenomics and phylogenomics, are also discussed.
2.2 Molecular Markers for Comprehensive Population Genetic Studies in Bamboos
The possibility of inferring genetic diversity and structure of populations with molecular markers allowed unprecedented discoveries in natural populations of several plants, including bamboos. Uncovering phylogenetic bases of the high species diversity of bamboos, in general, has been the target of various other studies employing molecular markers. An increasing number of studies, though, have also been devoted to deciphering intrapopulation genetic variation. Molecular surveys can explain the clonal structure of natural populations and the implications of the rare flowering events in various bamboos.
Bamboos are, in general, clonal plant species, possessing both sexual and asexual propagation systems. Asexual reproduction is advantageous as it enables the successful establishment in novel environments. On the other hand, it does not provide new genetic variation. This is achieved by sexual reproduction (Kitamura and Kawahara 2011). It is frequent that bamboos flower only once in their life cycle and after several decades (Kitamura and Kawahara 2009). A study of Isagi et al. (2004) investigated the flowering pattern of culms regenerated from seeds of Phyllostachys pubescens and that have naturally propagated their genotypes through leptomorphic rhizomes. AFLP screening allowed the identification of distinct genets distributed in the population. The genets had distinct flowering times, suggesting a genetic architecture involved in this trait. Further analysis with microsatellite markers identified the clonal structure of a population of Sasa cernua and revealed an overall synchronism of flowering culms of the same clone, but also that not all culms flowered at once. Culms that flowered died, leaving others of the same clone still able to flower in future events. This may enable novel opportunities for cross-pollination (Kitamura and Kawahara 2009). In Sasamorpha borealis, after vegetative propagation by rhizomes, almost all adults produce flowers, set seeds in large amounts, and die (Lee and Chung 1999). However, dying after producing flowers is not a general rule, as revealed for S. pubiculmis. Among distinct genets that were monitored, one had both flowering and nonflowering patches for 4 years. One genotype can maintain their rhizomes and nonflowering patches alive after mass flowering (Miyazaki et al. 2009).
The rare flowering events in bamboos have important implications in outcrossing rates. The synchronism of flowering within clones (Kitamura and Kawahara 2009) limits the possibility for outcrossing between distinct genotypes. Using microsatellite polymorphisms, Kitamura and Kawahara (2011) studied the mating system of the dwarf bamboo S. cernua. The authors found an overall low outcrossing rate (tm = 0.148). The genotype pairs that presented the highest outcrossing rates also had between 2% and 17% of seeds with homozygote genotypes for the markers. Therefore, high inbreeding coefficients should be expected, but the authors observed some decline toward the stage of seedlings, suggesting some selection against inbred progenies. In P. pubescens, Lin et al. (2014) also detected low outcrossing rates (tm = 0.089) and excess of homozygotes (FIS = 0.195) after genotyping seeds with cDNA SSR markers from three locations separated by at least 100 km. This may be due to an auto compatibility system, such as shown in the Brazilian species Merostachys riedeliana (Guilherme and Ressel 2001).
The characteristics of bamboos are compatible with lower levels of genetic variation within populations and higher genetic differentiation among populations, as revealed by population genetic studies in some species (Table 2.1). However, various species still do not have population genetic surveys published, and high genetic variation is not that rare in data already available. It is not our goal to exhaust the literature on all the studies available for such a purpose, but to provide results and prospects of the use of molecular markers in bamboos.
Although with less polymorphism and as indirect products of gene expression, isozyme markers have been used in bamboo germplasm characterization. The advantage of such markers relies on the possibility of discriminating heterozygotes. Four enzyme systems were screened in accessions of hill bamboo, Sinarundinaria anceps. Out of 12 markers, approximately 75% were polymorphic, and similarities among accessions ranged from 54 to 100% (Tiwari et al. 2019). For isozymes, considerably high genetic variation, measured from the observed heterozygosity (Ho = 0.219), was found in populations of S. borealis from Korea. Relatively moderate genetic differentiation, however, was also identified, as measured by the proportion of genetic variation among populations (GST = 0.310) (Lee and Chung 1999). In 17 populations of Pseudosasa japonica, the mean genetic diversity encountered through 28 isozyme loci (Ho = 0.099) was lower than the expectation under Hardy-Weinberg equilibrium (He = 0.167). A high and significant deficit of heterozygotes was then detected (FIS = 0.457), suggesting considerable inbreeding due to vegetative spread. In this species, most of the genetic variation was found within populations (84.8%) (Huh and Huh 2002).
AFLP, ISSR, and SRAP are dominant markers that, in general, are more suitable for genetic structure analysis rather than genetic diversity, as they do not allow the discrimination of heterozygotes. Sixteen cultivars of P. violascens were analyzed with these three types of markers. AFLP allowed the discrimination of 434 markers, while ISSR (209 bands) and SRAP (222) had a lower number. In general, AFLP showed the highest marker efficiency among other methods, although the polymorphism of AFLP (58.3%) was not higher than ISSR (65.1%) and SRAP (68.5%) (Lin et al. 2011). In the dwarf bamboo Bashania fangiana, AFLP markers showed considerable genetic polymorphism, and most of the genetic diversity was found within populations (GST = 0.057). The study indicated that the two populations investigated were multiclonal and diverse (Ma et al. 2013).
ISSR markers were used to analyze the genetic diversity and structure of seven populations of Melocanna baccifera, a non-clump and evergreen arborescent bamboo in India. Moderate values of Nei’s genetic diversity (H = 0.1639) were detected through the analyses, with 88.4% polymorphic bands. From the hierarchical partition of the genetic diversity, most of it was found within populations (GST = 0.1942) (Nilkanta et al. 2017). Similar differentiation was also detected among populations of Dendrocalamus membranaceus from China. Among all the 155 bands, 153 were polymorphic, and the proportion of genetic differentiation among populations was 25.2% (from GST) and 21.1% based on AMOVA (Yang et al. 2012). Conversely, ISSR proved useful in detecting low genetic diversity (H = 0.0418) and high genetic differentiation (GST = 0.8474) in populations of D. giganteus, one of the largest bamboos in the world. A Mantel’s test suggested that geographic and genetic distances were not significantly correlated, implicating in a high genetic differentiation across populations of the species. Differences in flowering times and limited pollen flow could explain the strong differentiation (Tian et al. 2012). Considerable genetic differentiation (FST = 0.38949) was also found among 13 populations of the Ethiopian lowland bamboo, through ISSR. In this case, considerably high genetic diversity was found at the species level (H = 0.2702) (Oumer et al. 2020).
The multiallelic nature and codominance of microsatellites markers, as well as their transferability to other species, make them important markers for more detailed and precise genetic variation and structure analyses (Table 2.1). Abreu et al. (2011) developed 13 novel microsatellite markers for a Brazilian species of bamboo, Aulonemia aristulata, native to the Atlantic rain forest. In a posterior study, those microsatellites were used to analyze the genetic diversity of two populations. The observed heterozygosity was calculated in saplings and seedlings of both populations, being higher in saplings of the populations (Table 2.1). The fixation indexes varied from 0.43 to 0.84, with higher values in the seedling stage. Inbreeding depression could be the main factor explaining a reduction in density of bamboos in the direction of the sapling stage (Abreu et al. 2014).
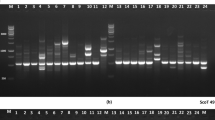
Inbreeding depression is a potential consequence of crossings between individuals identical by descent, as it happens among close relatives or by selfing (Frankham et al. 2010). As discussed before, the clonal structure of bamboos is favored by their limited flowering events, making opportunities from outcrossing scarcely available. Inbreeding depression can be manifested by several characteristics, such as low germination, mortality of seedlings due to factors such as abnormal germination, or lack of the ability to synthesize chlorophyll. Our group collected a source of seeds of Dendrocalamus asper that were obtained after a single flowering event of an individual in an urban area (Data unpublished). Significant mortality and abnormal germination were observed from such seeds. Moreover, several seedlings were not able to grow, as they lacked chlorophyll deposition in their leaves (Fig. 2.1a). Those that possessed partial green areas alternated with white surfaces on their leaves were able to grow normally (Fig. 2.1b). An ISSR profile with a few markers indicated low polymorphism on such materials, such as shown with the universal primer ISSR 827 (Fig. 2.1c).

Plants of Dendrocalamus asper originated from seeds collected from a single plant. (a) Illustration of a few plants after approximately 4 months after germination. The plant circulated in yellow did not survive (white leaves). (b) Plants after 6 months of germination. Plants that have partial ability to accumulate chlorophyll survived. (c) ISSR profile of some of those plants, showing limited polymorphism among them
With microsatellites, genetic diversity and structure of Kuruna debilis, a threatened bamboo species, were determined in populations located in Sri Lanka (Peng et al. 2013). High genetic diversity was found (Ho = 0.708 for all populations), but considerable deficiency of heterozygotes was also computed (FIS = 0.170). The differentiation among populations (FST = 0.113) revealed that most of the diversity was also found within populations (Attigala et al. 2017). P. edulis, the model bamboo which has been sequenced (Peng et al. 2013), was also the object of a microsatellite study in populations of China. Considerable genetic variation, measured from heterozygosity, was measured from 20 novel microsatellite markers developed by Jiang et al. (2013). Nine microsatellites were developed for the Australian endemic species Bambusa arnhemica, revealing an average genetic variation of Ho = 0.360 in four populations. Once again, the deviation from the expected heterozygosity was considerably high (mean of He = 0.690) (Kaneko et al. 2008). In 19 populations of D. hamiltonii from the Himalayas, low genetic variation (Nei’s H = 0.175 at the species level) and moderate genetic differentiation were found (FST = 0.165) (Meena et al. 2019).
From 16 microsatellite markers developed by Dong et al. (2012) for D. sinicus, 8 were screened in another study with natural populations of the species in its distribution range. The data on nuclear microsatellites enabled the discovery of high genetic differentiation among the populations, dividing them into two main subgroups that are consistent with different culm types. With an analysis of chloroplast DNA markers as well, one genetic group encompassed populations with straight culms, while the other genetic cluster grouped the sinuous-culm lineage. Based on both types of molecular markers, the authors concluded that the populations experienced dispersal and long-term isolation, with some dilution due to contemporary gene flow (Yang et al. 2018).
One of the most important food sources to the giant panda, in China, is the bamboo species Fargesia denudata. Therefore, it is straightforward to study the genetic resources available from natural populations of the species. The development of microsatellite markers for F. denudata is an important tool for exploring the genetic diversity of the species. According to a primer note, 14 markers were developed for further population genetic studies (Lv et al. 2016).
Due to the several related species of bamboos, microsatellites can usually be successfully transferred among species for genetic studies. Several of the microsatellites developed for the Brazilian species Aulonemia aristulata were successfully amplified in other species belonging to the genera Bambusa, Dendrocalamus, Gigantochloa, and Guadua (Abreu et al. 2011). Similarly, the microsatellites developed for D. sinicus were transferable to other species within the same genus (Dong et al. 2012). The microsatellites developed for F. denudate were transferable, in different numbers, to F. scabrida, F. rufa, F. ferax, Arundinaria fargesii, and Yushania lineolata (Lv et al. 2016). Bhandawat et al. (2015) dedicated a full study to analyze the crosstransferability between D. latiflorus and other bamboo species.
Another line of studies with molecular markers encompasses comparisons of distinct species and genera of bamboos (Table 2.2). RFLP markers were employed to characterize genetic relationships among several species of the genera Phyllostachys, Bambusa, Arundinaria, Pseudosasa, Sinobambusa, and Sinocalamus (Friar and Kochert 1991). RAPD markers were able to distinguish several bamboo species and genera (Nayak et al. 2003; Ramanayake et al. 2007; Konzen et al. 2017; Annisa et al. 2019). Moreover, RAPD markers were efficiently converted to RFLP markers by enzyme digestion. Those markers were able to distinguish four genera (Bambusa, Dendrocalamus, Guadua, and Phyllostachys) and species (Konzen et al. 2017). ISSR markers were also used in similar approaches (Mukherjee et al. 2010; Amom et al. 2018). Morphological variables and SRAP markers were also combined in a study with several bamboo taxa (Zhu et al. 2014). EST-derived microsatellites have been used in phylogenetic studies of bamboo species (Barkley et al. 2005; Sharma et al. 2009; Cai et al. 2019). EST-based random primers were also useful in the study of Mukherjee et al. (2010). cpDNA markers were the object of distinct studies as well, providing tools for examining genetic differentiation and haplotype diversity within populations (Abreu et al. 2014; Triplett et al. 2010; Yang et al. 2018) and among species (Triplett et al. 2010).
To detect natural hybridization among North American woody bamboos of the genus Arundinaria, AFLP and cpDNA markers were used (Triplett et al. 2010). The distinction of three species (A. gigantea, A. appalachiana, and A. tecta) that were previously recognized based on morphological variation was conducted. Those species were also analyzed for their intraspecies genetic diversity. In general, relatively low levels of genetic diversity were found within each of the species, based on the AFLP data (Triplett et al. 2010). The authors discussed the intergradation of molecular and phenotypic data among the parental species. cpDNA analyses suggested multiple and reciprocal hybridization events (Triplett et al. 2010).
Transposable elements are ubiquitous in eukaryotic genomes, being able to self-replication and insertion to other sites, affecting the stability of genomes. In bamboos, long terminal repeat (LTR) retrotransposons are highly abundant (more than 40% of the genome of P. edulis) (Zhou et al. 2017). Retrotransposon-based markers were used to screen 58 bamboo taxa, including 47 distinct species in the genus Phyllostachys. Those markers behaved as dominant, producing an average of 208.75 bands per primer and average polymorphic information content of 0.327 for the whole set of taxa. After AMOVA, 25% of the genetic variation was detected among the taxa (Li et al. 2020a).
The emerging field of epigenetics deals with heritable variation beyond the level of DNA, such as methylation and histone acetylation and patterns of gene expression. The considerable genomic and transcriptomic data available for bamboo is enabling the discovery of epigenomic variation among taxa. Using methylation-sensitive amplification polymorphism (MSAP), Yuan et al. (2014) encountered distinct patterns of methylation for chronological age of P. edulis. The increase in chronological age was accompanied by an augmented DNA methylation rate. Lu et al. (2012) also detected great differences of DNA methylation rates among developmental stages of P. praecox, based on AFLP using methylation-sensitive enzymes. The rapid increase in sequenced genomes will enable the discovery of methylation patterns across taxa of bamboo, assisting in the clarification of several processes that are accompanied by epigenetic changes.
2.3 SNP Markers and their Application to Conservation and Breeding
Genomic technologies accelerated the discovery of single-nucleotide polymorphisms (SNP), the most abundant type of molecular markers in all organisms. The publication of the genomes of P. edulis (Peng et al. 2013) has opened the field for genomic and transcriptomic studies. Restriction site-associated DNA sequencing (RAD-seq) is a method that enables the detection of thousands of SNP, by a strategy of reduced genomic representation (Baird et al. 2008). SNP markers detected through RAD-seq were used to evaluate phylogenetic relationships in the tribe Arundinarieae. In general, eight lineages were supported by the data, two of them in agreement with previous studies on nuclear markers (Wang et al. 2017a).
After comparing three moso bamboo samples through genome resequencing, 4,700,803 SNP markers were detected (Zhou et al. 2019a). The study also discriminated INDEL markers, accounting for 268,150 distinct positions throughout the genomes. Moreover, copy number variation (CNV = 65,935) and structural variations (SV = 215,297) were also analyzed. The variation detected was present in genes associated with ribosome genesis, caffeine metabolism, nucleotide binding, and ribonucleoside binding, among several other categories, probably involved in adaptation of moso bamboo to their environment (Zhou et al. 2019a).
To date, the number of studies involving SNP markers is yet insipient, but there will be certainly many more publications to come shortly. High-density SNP profiles have provided important information on genetic variation and structure of populations. They also have been largely employed in genome-wide association mapping in plants and other species. Such discoveries enable the design of proper conservation and breeding strategies, as phenotypes can be precisely correlated with SNP and genes that are located throughout genomes. Data on SNP can rapidly be obtained from genotyping by sequencing (GBS) (Elshire et al. 2011), even in species that do not have a reference genome for the alignment of reads (Poland et al. 2012).
2.4 Phylogenomics and its Application to Bamboo Taxonomy
Until a few decades ago, bamboo taxonomy relied exclusively on morphological variation. Moreover, the focus was mainly on vegetative characters. Due to the rare flowering events in bamboos, the morphology-based taxonomy of bamboo has been a challenge (Bhattacharya et al. 2006). The proper distinction between bamboo taxa and comprehension of phylogeographic and phylogenetic relationships are essential to guide conservation and development decisions. The development of molecular marker technologies has provided more consistent phylogenetic trees, improving the taxonomic resolution of bamboos.
Currently, high-throughput sequencing technologies (NGS) allow accessing genome sequences of virtually all species. Through genomic data, researchers can infer species relationships, understand mechanisms of molecular evolution, and control for stochastic events. This intersection between evolution and genomics has been called phylogenomics (Eisen and Fraser 2003; Delsuc et al. 2005; Philippe et al. 2005; Yu et al. 2018). In this topic, we provide a few case studies that used phylogenomics for taxonomical classification and a better understanding of the evolutionary relationships of bamboo species.
The tribe Arundinarieae belongs to the subfamily Bambusoideae (Poaceae), containing more than 1600 species, a highly complicated taxonomy (Bamboo Phylogeny Group 2012; Clark et al. 2015; Canavan et al. 2017). Plastid markers enabled the subdivision of bamboos into 12 major lineages (Zeng et al. 2010; Yang et al. 2013; Zhang et al. 2016). Plastid genome sequencing has also been used to resolve the phylogenetic relationships in Arundinarieae and in obtaining robust relationships among lineages (Ma et al. 2014; Wysocki et al. 2015). Recently, other phylogenomic approaches based on restriction site-associated DNA sequencing (RAD-seq) were used to estimate the phylogenetic relationships among Arundinarieae genera from a nuclear evolutionary trajectory perspective (Wang et al. 2017a).
The phylogenetic relationships of Shibataea, a genus of tribe Arundinarieae and endemic to China, have been reconstructed through a phylogenomic approach based on double digest restriction site-associated DNA sequencing (ddRAD-seq), and whole plastid genomes were generated using genome skimming (Guo et al. 2019). Fargesia, other genus of tribe Arundinarieae, has also been its phylogenetic relationships reconstructed based on complete plastid genome sequences of 26 species from Fargesia (Zhou et al. 2019b).
Phylogenomic approaches based on complete plastid genome sequences have also been used in evolutionary studies of bamboos. Through genome-wide comparative analyses of 76 chloroplast genomes of bamboos, extreme heterogeneity of the evolutionary rate within the bamboos was demonstrated, with the lowest value found in tribe Arundinarieae (Wang et al. 2020). Plastid genome sequences have also been used to estimate phylogenetic relationships among Bambuseae species (do Vieira et al. 2016). Bambuseae is another tribe of subfamily Bambusoideae (Poaceae), containing 66 genera and 812 species (Bamboo Phylogeny Group 2012; Clark et al. 2015).
Whole plastid genomes of Olyreae species were generated using genome-skimming approach and used to strongly support the phylogenetic positions of Froesiochloa and Rehia in the Olyreae (Wang et al. 2018). Olyreae is the smallest tribe of subfamily Bambusoideae (Poaceae), containing 22 genera and 124 species (Bamboo Phylogeny Group 2012; Clark et al. 2015). Therefore, there are few phylogenomic studies involving the Olyreae species.
Finally, all these studies and another show the potential of the phylogenomic approach based on NGS technologies combined with traditional morphological analyses for resolving difficult phylogenetic relationships in the intractable bamboo species, evolutionary, and biogeography studies.
2.5 Protein and Nucleotide Motifs for Evolutionary Studies with Gene Families
So far in this chapter, we have treated molecular markers as tools for comprehending population genetic processes as well as to understand phylogenetic relations among species. By a functional characterization of the allelic variation of genes, sequence motifs affecting phenotyping variation can be identified, those being referred to as functional markers (Andersen and Lübberstedt 2003). Genes that have been functionally characterized may be a reference to annotations of paralogs and orthologs, which enable their categorization in major families. Phylogenetic reconstruction with gene families provides insights into the evolution and adaptation of plants (Neale et al. 2017). Genome-wide searches for gene families use model plants to find homologs in the species of interest, but experiments are required to functionally confirm the inferred genes (Vaattovaara et al. 2019).
Various gene families of transcription factors are implicated in the response of plants to biotic and abiotic stresses. In P. edulis, genome-wide surveys of genes are available for WRKY (responsive to cold and drought) (Li et al. 2017); no apical meristem (NAM); Arabidopsis transcription activation factor (ATAF) and cup-shape cotyledon (CUC), collectively referred to as NAC genes (participate in lignin catabolic process and cellulose biosynthetic process) (Shan et al. 2019), heat shock transcription factors (Hsfs) (participate in shoot and flower development; expression is changed under cold, high temperature, drought and high salinity stresses) (Xie et al. 2019), dehydration responsive element-binding (DREB) (differentially expressed under drought, cold and high salinity) (Wu et al. 2015). Among various other categories of gene families studied in P. edulis are aquaporins (AQP) (Sun et al. 2017), late embryogenesis abundant (LEA) (Huang et al. 2016), amino acid/auxin permease (AAAP) (Liu et al. 2017), plant homeodomain zinc finger (PHD-finger) (Gao et al. 2018), trihelix genes (Gao et al. 2019), SQUAMOSA-promoter binding protein-like (SBP-like) transcription factors (Pan et al. 2017), IQ67-domain (IQD) genes (Wu et al. 2016), and basic leucine zipper domain (bZIP) transcription factors (Pan et al. 2019). In this chapter, we highlight the APETALA2/ethylene-responsive element binding protein (AP2/EREBP) superfamily and aquaporins as examples for the application of protein or nucleotide motifs for evolutionary analyses in bamboos.
The AP2/EREBP superfamily of proteins has a high conserved domain named APETALA2 (AP2), with 55 to 70 amino acids (Jofuku et al. 1994; Okamuro et al. 1997; Konzen et al. 2019). This superfamily is divided into three families that have different numbers of domains and participate in different physiological processes, which are RAV, AP2, and ERF (Sakuma et al. 2002). The RAV family has one AP2 domain and one B3 domain (Kagaya et al. 1999), the AP2 family has two AP2 domains (Okamuro et al. 1997), and the ERF family has only one AP2 domain (Sakuma et al. 2002).
Wu et al. (2016) identified 116 genes from the AP2/EREBP family in P. edulis and divided them into three subfamilies: 28 AP2, 7 RAV, 80 ethylene response factors (ERF), and 1 soloist; the ERF group is divided into two subgroups, ERF and DREB. As an example in this chapter, we selected five amino acid sequences available in the Plant Transcription Factor Database (http://planttfdb.gao-lab.org/) from each group to analyze and identify motifs that can be used as molecular markers. First, we used the Clustal W algorithm for producing an alignment using BioEdit (Hall 1999). Afterward, we constructed a phylogenetic tree by maximum likelihood, using MEGA X (Kumar et al. 2018), and the amino acid substitution model WAG+I + G + F, as determined from Bayesian criterion information, with 500 bootstrap replications. The protein motifs were searched on the MEME-suite website (http://meme-suite.org/). From the phylogeny, two major groups were identified (Fig. 2.2a), one with RAV proteins and the other with AP2, DREB, and ERF, the last two being more similar between them (Data unpublished). Observing the protein motifs, similar motifs are shared within the major groups and according to the subdivision of the superfamily. The protein motifs one and two correspond to the domain AP2, a domain that is present in all sequences with certain differences among the subgroups. The protein motifs 8, 3, and 4 correspond to the domain B3, even though the motif 4 was not identified in the sequence RAV_PH01000581G0530.

Protein and nucleotide motifs as potential phylogenetic markers for the study of gene families in bamboos. (a) Protein motifs searched for some members of the AP2/EREBP superfamily. (b) Nucleotide motifs searched for 12 aquaporin genes These figures were elaborated based on analyses performed by the authors of the chapter, using MEME-suite396 website (http://meme-suite.org/) with sequences available from GenBank/NCBI (https://www.ncbi.nlm.nih.gov/) and the Plant Transcription Factor Database (http://planttfdb.gao-lab.org/)
We also performed a similar analysis of AQP proteins from sequences available in the public database. AQP proteins are channels to water transport along with CO2 and nutrients (Maurel et al. 2015). PeTIP4;1–1, isolated from P. edulis, conferred tolerance to transgenic Arabidopsis thaliana in abiotic stresses caused by drought and high salinity (Sun et al. 2017). PeTIP4;1 and PeTIP4;2 were expressed in the parenchyma and epidermal cells and roots, indicating their role in water absorption and transport (Sun et al. 2018). Sun et al. (2018) classified the aquaporins from P. edulis into four groups: (a) plasma membrane intrinsic proteins 22 (PIP), (b) tonoplast intrinsic proteins 20 (TIP), (c) nodulin 26-like intrinsic proteins 17 (NIP), and (d) small basic intrinsic proteins 4 (SIP); the uncharacterized X intrinsic proteins (XIP) were not found.
We used the 12 sequences from the work of Sun et al. (2018), available from GenBank/NCBI (https://www.ncbi.nlm.nih.gov/) to search for the correspondent nucleotide sequences in A. thaliana with BLASTn. Sequence alignment and phylogenetic reconstruction were conducted similarly to the AP2/EREBP sequences. The maximum likelihood tree, this time, was constructed with the nucleotide substitution model K2 + G, as determined with MEGA X, after 500 bootstrap replications. The tree of aquaporins with the nucleotide sequences from bamboo and A. thaliana was divided into two major groups, the first with SIP and PIP genes and the second with TIP and NIP genes from both species (Fig. 2.2b). Motif 5 was identified in all nucleotide sequences. Motifs 6 and 7 were found only in PIP genes, while motifs 8 and 9 were found in TIP genes, in similar positions. Motifs 10 and 17 were found only in PIP. Motifs 1, 2, and 3 were found in all sequences except SIP. Motif 4 was detected in TIP and PIP genes (Data unpublished).
Therefore, protein or nucleotide motifs can be used as markers to identify and compare gene families, to better understand the evolution and adaptation of plants. With the sequences available for P. edulis, other bamboos may be searched for genes already annotated in this species.
2.6 Functional Gene Markers for Improving Abiotic Stress Tolerance in Bamboos
Drought, salinity, and low and high temperatures are the mains abiotic constraints that negatively affect bamboo growth and development and reduce its industrial productivity (Ramakrishnan et al. 2020). Therefore, understanding the stress responses will increase the ability to improve tolerance in bamboo species. However, the mechanisms of molecular responses in bamboo species under stress conditions are still not completely understood.
The responses to drought, salinity, and low and high temperature stresses are under stronger biochemical and genetic control (Zhu 2016; Haak et al. 2017). As stated before, various gene families have been identified in moso bamboo. In general, no in-depth functional analyses were conducted for those genes. However, there are other gene families in that at least one member was functionally characterized. From these studies, we listed 12 genes that could be used as functional molecular markers in comprehending abiotic stress tolerance (Table 2.3). To select these stress-responsive genes, we considered their expression profile in moso bamboo plants under stress treatments. Moreover, we considered only those genes that were functionally analyzed in at least one model species such as Arabidopsis, rice, and yeast.
Several transcription factors have been identified and associated with drought, salinity, and cold-induced stresses in moso bamboo (Table 2.3). WRKY proteins are important transcription factors involved in different biological processes such as plant growth and abiotic stress responses (Rushton et al. 2010; Chen et al. 2012). A genome-wide survey identified 121 WRKY genes in P. edulis (Li et al. 2017). The authors showed that PheWRKY72–2 is a drought- and cold-inducible gene. Furthermore, the overexpression of PheWRKY72–2 in Arabidopsis enhanced plant tolerance to drought stress by promoting stomatal closure. In another study, PeWRKY83 gene was highly upregulated in moso bamboo seedlings subjected to salinity and drought stress. After ectopic expression in Arabidopsis, it was demonstrated that PeWRKY83 regulates the expression of genes involved in ABA biosynthesis, signaling, and responses to salt and improves salt tolerance (Wu et al. 2017).
The MYB gene family, first described as an oncogene from MYeloBlastosis virus (capital letters highlighting the attributed name of the gene family), also was identified in moso bamboo (Hou et al. 2018). Through in silico and expression analyses, these authors identified a PheMYB4–1 stress-related gene. After ectopic overexpression, PheMYB4–1 promoted improvement in tolerance to cold, drought, and salt stress in Arabidopsis seedlings. Two NAC transcription factors from moso bamboo were identified and functionally characterized. First, the expression levels of PheNAC3 gene increased in leaves of moso bamboo after NaCl treatment. Overexpression in Arabidopsis improved drought and salt tolerance (Xie et al. 2020). Second, drought and salinity also induced the expression of PeSNAC-1 in moso bamboo. Rice plants overexpressing PeSNAC1 gene were more drought- and salt-tolerant (Hou et al. 2020). Moreover, these authors suggested that PesSNAC-1 works as a positive stress regulator in moso bamboo.
The ABA-stress-ripening (ASR) gene family is a small group of chaperone-like proteins and plant-specific transcription factors involved in plant development, senescence, fruit ripening, and abiotic stresses (González and Iusem 2014). After being identified in moso bamboo, ASR genes were found upregulated under drought, NaCl, and ABA. Specifically, the overexpression of PheASR2 in rice improved drought tolerance (Wu et al. 2020b). Stress response-related functions of Teosinte branched1/Cincinnata/proliferating cell factor (TCP) members have been investigated in different plant species including moso bamboo (Liu et al. 2018). A member of this family was characterized in rice and Arabidopsis, and the results showed that PeTCP10 may have positive regulatory functions in drought tolerance (Liu et al. 2020). A drought-induced (Di19) protein, a zinc-finger transcription factor containing two conserved and atypical Cys2/His2 zinc-finger domains, isolated from moso bamboo, has been functionally characterized in Arabidopsis and rice (Wu et al. 2020a). Through transgenic lines, these authors concluded that PheDi19–8 significantly increased drought tolerance. Moreover, complementation analyses showed that PheDi19–8 works as a positive modulator of drought stress tolerance.
Twenty-six DNA binding with one finger (Dof) genes were identified in moso bamboo (Wang et al. 2016). A member of this transcription factor family has been functionally characterized by ectopic expression in Arabidopsis (Liu et al. 2019). These authors showed that the PheDof12–1 gene is highly induced by cold, drought, and salt in moso bamboo and their overexpression promotes early flowering in Arabidopsis. The Arabidopsis lines overexpressing PheDof12–1 gene were not analyzed under those abiotic stresses (Liu et al. 2019).
VQ motif-containing proteins are involved in responses to biotic and abiotic stresses by interaction with WRKY transcription factors (Lai et al. 2011; Hu et al. 2013). VQ genes were identified in moso bamboo genome, and the PheVQ28 gene has been functionally characterized (Wang et al. 2017b; Cheng et al. 2020). PheVQ28 was highly upregulated in moso bamboo seedlings under salt stress, and Arabidopsis plants overexpressing this gene were significantly more tolerant to salt stress (Cheng et al. 2020). Laccases (LAC) are oxidative enzymes involved in flavonoid and lignin biosynthesis (Pourcel et al. 2005; Berthet et al. 2011). Thus, LAC genes are promising for woody, biofuel, and other biotechnological applications. Moreover, LAC genes have also been involved in plant responses to environmental stress (Wang et al. 2015). Laccase gene members have been identified in moso bamboo. PeLAC10 gene was upregulated under ABA and NaCl, and its overexpression in Arabidopsis plants improved drought tolerance (Li et al. 2020b). These results suggest that laccase genes could be potential candidates for the molecular breeding of bamboo to increase the content of lignin and improve their adaptability to drought stresses.
Finally, although all genes list in Table 2.3 requires further functional characterization, they are a potential candidate for the breeding of stress tolerance in bamboo species and other crops through genetic engineering using approaches such as CRISPR (clustered regularly interspaced short palindromic repeats). Using molecular markers for deciphering genetic variation and its functional implications will certainly assist in improving stress tolerance and increase the productivity of bamboos in the climate change scenario that affects the whole planet.
2.7 Molecular Markers for Genetic Fidelity
Molecular markers are also a powerful tool to attest to the genetic fidelity of clonally produced plantlets of bamboos. The use of micropropagation has been a viable alternative for cloning superior genotypes, as well as for the production of seedlings with high quality and free of pathogens. There are strict precautions in micropropagation so that plants can express all their genetic potential, as on the course of in vitro cultivation regenerated plants may not maintain the genetic stability in relation to their mother plant (the donor of propagules) (Kaeppler et al. 2000; Ray and Ali 2017).
Changes in the genotype after micropropagation are referred to as somaclonal variation. Mutations may occur due to the use of growth regulators, the various subcultures of explants, the regeneration by callus induction or protoplasts, and somatic embryogenesis. Therefore, DNA can undergo irreversible and inheritable changes (Ray and Ali 2017). Genotypic changes can occur through chromosomal breaks and rearrangements, changes in the level of ploidy, activation of transposable elements, and DNA methylation (Kaeppler et al. 2000; Krishna et al. 2016). These are examples of genetic and epigenetic changes that may or may not cause phenotypic variations. In fact, the identification of possible phenotypic variation in early stages occurs by observing variations in leaf color or size (albinism, variegation, or dwarfism); however, in most cases, these variations do not become visible requiring analysis by molecular markers to identify somaclonal variants.
The most used markers for the identification of somaclonal variations in bamboo are AFLP, RAPD, ISSR, and SSR (Table 2.4). The principle of the analysis involves the comparison of DNA fingerprint among all propagules with their mother plant. Evidence of somaclonal variation occurs when the fingerprints differ from the original source (Fig. 2.3a). AFLP markers were used to verify the genetic stability of explants from a 2-year subculture of D. hamiltonii in a culture medium containing thidiazuron (TDZ) (Singh et al. 2013a). The combination of the enzymes EcoRI and MseI with 15 selective primers did not reveal any polymorphism between the fragments, which validated the protocol for the species. In another study, nodal segments of Bambusa nutans were used to obtain a somatic embryogenesis protocol in MS culture medium containing 2,4-D (dichlorophenoxyacetic acid) (Mehta et al. 2011). Six AFLP primer combinations produced 407 fragments, 98.8% of which were monomorphic. Therefore, low number of plants showed somaclonal variations. With this high percentage of monomorphism, the technique is efficient for verifying genetic fidelity.

(a) Schematic illustration of the principle of the assessment of genetic fidelity after micropropagation of bamboo. A hypothetical fingerprint shows genetic fidelity vs evidence of somaclonal variation. Although no genetic variation may be present, novel technologies allow the identification of epigenetic mechanisms that might implicate in distinct phenotypic effects. Photo credit: Enéas Ricardo Konzen. (b) Bambusa vulgaris plant originated from micropropagation of mother plant. Photo credit: Siu Mui Tsai
There are also studies that report the success of RAPD markers for bamboo species as well as validating different types of protocols, such as the use of different growth regulators for the in vitro rooting of D. strictus (Goyal et al. 2015), regeneration of axillary buds of B. arundinacea (Kalaiarasi et al. 2014), and validation of commercial materials produced in vitro from D. asper (Singh et al. 2012). For bamboos, ISSR markers have become the most viable alternative for studies of genetic fidelity. The use of ISSR markers to verify the genetic fidelity of B. balcooa, a species of great importance in India, did not show somaclonal variation, even with variations in the concentrations of growth regulators (Rajput et al. 2020). Genetic stability using ISSR markers has also been shown in the in vitro cultivation of G. magna and G. angustifolia (Nogueira et al. 2019).
In D. asper, the use of four different markers, including SSR, did not show polymorphism for the use of nodal segments under different stages of in vitro development, with the use of cytokinin in the multiplication phase, indolebutyric acid, and acetic naphthalene for elongation (Singh et al. 2013b). The authors used 25 SSR primers based on the rice genome, obtaining 164 bands (all monomorphic) observing the absence of somaclonal variation, which shows the efficiency of these markers for studies of genetic fidelity.
The somaclonal variation observed in micropropagated bamboo species may be linked to the use of growth regulators, indirect organogenesis, as well as the number of subcultures. For the identification of variants, ISSR-type markers, which are based on the repetitive portion of eukaryotic DNA, are effective due to their low cost and repeatability. With the evolution of genomic studies in bamboo, other ways of identifying somaclonal variation will allow the selection of true-to-type planting material, such as studies of epigenetic variations, ploidy, and movement of transposable elements. Even if no mutations have occurred at the DNA level, epigenetic changes such as methylation of DNA, acetylation and methylation of histones, and altered gene expression are possible outcomes (Fig. 2.3a), which could implicate in distinct phenotypic effects and performances of the propagules. It is important to ascertain the genetic fidelity to obtain plants with similar performances to their sources (Fig. 2.3b).
2.8 Conclusion and Future Prospects
Molecular markers empowered the distinction of the diversity of bamboo species and enabled comprehensive analyses of their reproduction and population structure. The current genomic technologies will produce numberless novel reports on the genetic diversity and divergence within and among species. Phylogenomics offers novel methods for improving the resolution of phylogenetic trees, reaching a deeper understanding of the evolution of bamboo taxa. Moreover, in the era of systems biology, research is moving forward to integrative approaches for conservation and breeding of such important species. Further comprehension of bamboo DNA variation coupled with the modulation of methylation, acetylation, and gene expression in their environment is necessary to design proper management. From the classical to the ultimate technologies for their obtainment, molecular markers certainly have much more to contribute to future endeavors.
References
Abreu AG, Grombone-Guaratini MT, Monteiro M, Pinheiro JB, Tombolato AFC, Zucchi MI (2011) Development of microsatellite markers for Aulonemia aristulata (Poaceae) and cross-amplification in other bamboo species. Am J Bot 98(4):e90–e92. https://doi.org/10.3732/ajb.1000511
Abreu AG, Grombone-Guaratini MT, Moreira Val T, Zucchi MI (2014) Genetic diversity and age class structure of seedlings and saplings after a mast flowering of bamboo in the Brazilian Atlantic Forest. Int J Plant Sci 175(3):319–327. https://doi.org/10.1086/674448
Amom T, Tikendra L, Rahaman H, Potshangbam A, Nongdam P (2018) Evaluation of genetic relationship between 15 bamboo species of North-East India based on ISSR marker analysis. Mol Biol Res Commun 7(1):7–15. https://doi.org/10.22099/mbrc.2018.28378.1303
Andersen JR, Lübberstedt T (2003) Functional markers in plants. Trends Plant Sci 8(11):554–560. https://doi.org/10.1016/j.tplants.2003.09.010
Annisa HR, Setiawati T, Irawan B, Kusmoro J (2019) Evaluation of RAPD markers for molecular identification of five bamboo genera from Indonesia. Folia For Pol 61(4):255–266. https://doi.org/10.2478/ffp-2019-0025
Attigala L, Gallaher T, Nason J, Clark LG (2017) Genetic diversity and population structure of the threatened temperate woody bamboo Kuruna debilis (Poaceae: Bambusoideae: Arundinarieae) from Sri Lanka based on microsatellite analysis. J Natl Sci Found 45(1):53–65. https://doi.org/10.4038/jnsfsr.v45i1.8038
Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA (2008) Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One 3(10):e3376. https://doi.org/10.1371/journal.pone.0003376
Bamboo Phylogeny Group (2012) An updated tribal and subtribal classification of the bamboos (Poaceae: Bambusoideae). J Am Bamboo Soc 24:1–10
Barkley NA, Newman ML, Wang ML, Hotchkiss MW, Pederson GA (2005) Assessment of the genetic diversity and phylogenetic relationships of a temperate bamboo collection by using transferred EST-SSR markers. Genome 48(4):731–737. https://doi.org/10.1139/g05-022
Berthet S, Demont-Caulet N, Pollet B, Bidzinski P, Cézard L, Le Bris P, Borrega N, Hervé J, Blondet E, Balzergue S, Lapierre C, Jouanin L (2011) Disruption of LACCASE4 and 17 results in tissue-specific alterations to lignification of Arabidopsis thaliana stems. Plant Cell 23(3):1124. https://doi.org/10.1105/tpc.110.082792
Bhandawat A, Sharma V, Sharma H, Sood A, Sharma RK (2015) Development and crosstransferability of functionally relevant microsatellite markers in Dendrocalamus latiflorus and related bamboo species. J Genet 94(S1):48–55. https://doi.org/10.1007/s12041-014-0377-9
Bhattacharya S, Das M, Bar R, Pal A (2006) Morphological and molecular characterization of Bambusa tulda with a note on flowering. Ann Bot 98:529–535. https://doi.org/10.1093/aob/mcl143
Cai K, Zhu L, Zhang K, Li L, Zhao Z, Zeng W, Lin X (2019) Development and characterization of EST-SSR markers from RNA-Seq data in Phyllostachys violascens. Front Plant Sci 10:50. https://doi.org/10.3389/fpls.2019.00050
Canavan S, Richardson DM, Visser V, Roux JJL, Vorontsova MS, Wilson JRU (2017) The global distribution of bamboos: assessing correlates of introduction and invasion. AoB Plants 9:plw078. https://doi.org/10.1093/aobpla/plw078
Chen L, Song Y, Li S, Zhang L, Zou C, Yu D (2012) The role of WRKY transcription factors in plant abiotic stresses. BBA – Gene Regul Mech 819:120–128. https://doi.org/10.1016/j.bbagrm.2011.09.002
Cheng X, Wang Y, Xiong R, Gao Y, Yan H, Xiang Y (2020) A Moso bamboo gene VQ28 confers salt tolerance to transgenic Arabidopsis plants. Planta 251:99. https://doi.org/10.1007/s00425-020-03391-5
Clark LG, Londoño X, Ruiz-Sanchez E (2015) Bamboo taxonomy and habitat BT - bamboo: the plant and its uses. In: Köhl M (ed) Liese W. Springer International Publishing, Cham, pp 1–30
Delsuc F, Brinkmann H, Philippe H (2005) Phylogenomics and the reconstruction of the tree of life. Nat Rev Genet 6:361–375
do Vieira LN, dos Anjos KG, Faoro H, de Fraga HPF, Greco TM, de Pedrosa FO, de Souza EM, Rogalski M, de Souza RF, Guerra MP (2016) Phylogenetic inference and SSR characterization of tropical woody bamboos tribe Bambuseae (Poaceae: Bambusoideae) based on complete plastid genome sequences. Curr Genet 62:443–453. https://doi.org/10.1007/s00294-015-0549-z
Dong Y-R, Zhang Z-R, Yang H-Q (2012) Sixteen novel microsatellite markers developed for Dendrocalamus sinicus (Poaceae), the strongest woody bamboo in the world. Am J Bot 99(9):e347–e349. https://doi.org/10.3732/ajb.1200029
Eisen JA, Fraser CM (2003) Phylogenomics: intersection of evolution and genomics. Science 300(5323):1706–1707. https://doi.org/10.1126/science.1086292
Elshire RJ, Glaubitz JC, Sun Q, Poland JA, Kawamoto K, Buckler ES, Mitchell SE (2011) A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS One 6(5):e19379
Frankham R, Ballou J, Briscoe D (2010) Introduction to conservation genetics, 2nd edn. Cambridge University Press, Cambridge. https://doi.org/10.1017/CBO9780511809002
Friar E, Kochert G (1991) Bamboo germplasm screening with nuclear restriction fragment length polymorphisms. Theor Appl Genet 82(6):697–703. https://doi.org/10.1007/BF00227313
Gao H, Huang R, Liu J, Gao Z, Zhao H, Li X (2019) Genome-wide identification of Trihelix genes in Moso bamboo (Phyllostachys edulis) and their expression in response to abiotic stress. J Plant Growth Regul 38:1127–1140. https://doi.org/10.1007/s00344-019-09918-9
Gao Y, Liu H, Wang Y, Li F, Xiang Y (2018) Genome-wide identification of PHD-finger genes and expression pattern analysis under various treatments in moso bamboo (Phyllostachys edulis). Plant Physiol Biochem 123:378–391. https://doi.org/10.1016/j.plaphy.2017.12.034
González RM, Iusem ND (2014) Twenty years of research on Asr (ABA-stress-ripening) genes and proteins. Planta 239:941–949. https://doi.org/10.1007/s00425-014-2039-9
Gosh S, Waikhom SD, Sen Mandi S, Talukdar NC (2011) Amplified fragment length polymorphism based study of phylogenetic relationship and genetic variability among some edible bamboo species of North-East India. J Plant Mol Biol Biotechnol 2(2):8–15
Goyal AK, Pradhan S, Basistha BC, Sen A (2015) Micropropagation and assessment of genetic fidelity of Dendrocalamus strictus (Roxb.) nees using RAPD and ISSR markers. 3 Biotech 5(4):473–482. https://doi.org/10.1007/s13205-014-0244-7
Guilherme FAG, Ressel K (2001) Biologia floral e sistema de reprodução de Merostachys riedeliana (Poaceae: Bambusoideae). Rev Bras Bot 24(2):205–211. https://doi.org/10.1590/S0100-84042001000200011
Guo C, Guo Z-H, Li D-Z (2019) Phylogenomic analyses reveal intractable evolutionary history of a temperate bamboo genus (Poaceae: Bambusoideae). Plant Divers 41:213–219. https://doi.org/10.1016/j.pld.2019.05.003
Haak DC, Fukao T, Grene R, Hua Z, Ivanov R, Perella G, Li S (2017) Multilevel regulation of abiotic stress responses in plants. Front Plant Sci 8:1564. https://doi.org/10.3389/fpls.2017.01564
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hou D, Cheng Z, Xie L, Li X, Li J, Mu S, Gao J (2018) The R2R3MYB gene family in Phyllostachys edulis: genome-wide analysis and identification of stress or development-related R2R3MYBs. Front Plant Sci 9:738. https://doi.org/10.3389/fpls.2018.00738
Hou D, Zhao Z, Hu Q, Li L, Vasupalli N, Zhuo J, Zeng W, Wu A, Lin X (2020) PeSNAC-1a NAC transcription factor from moso bamboo (Phyllostachys edulis) confers tolerance to salinity and drought stress in transgenic rice. Tree Physiol 40:1792–1806. https://doi.org/10.1093/treephys/tpaa099
Hu Y, Chen L, Wang H, Zhang L, Wang F, Yu D (2013) Arabidopsis transcription factor WRKY8 functions antagonistically with its interacting partner VQ9 to modulate salinity stress tolerance. Plant J 74:730–745. https://doi.org/10.1111/tpj.12159
Huang Z, Zhong X-J, He J, Jin S-H, Guo H-D, Yu X-F, Zhou Y-J, Li X, Ma M-D, Chen Q-B, Long H (2016) Genome-wide identification, characterization, and stress-responsive expression profiling of genes encoding LEA (late embryogenesis abundant) proteins in Moso Bamboo (Phyllostachys edulis). PLoS One 11(11):e0165953. https://doi.org/10.1371/journal.pone.0165953
Huh HW, Huh MK (2002) Genetic diversity and population structure of Pseudosasa japonica (Bambusaceae) in Korea. Bamboo Sci Cult 16:9–17
Isagi Y, Shimada K, Kushima H, Tanaka N, Nagao A, Ishikawa T, OnoDera H, Watanabe S (2004) Clonal structure and flowering traits of a bamboo [Phyllostachys pubescens (Mazel) Ohwi] stand grown from a simultaneous flowering as revealed by AFLP analysis: clonal structure and flowering of bamboo. Mol Ecol 13(7):2017–2021. https://doi.org/10.1111/j.1365-294X.2004.02197.x
Jiang W-X, Zhang W-J, Ding Y-L (2013) Development of polymorphic microsatellite markers for Phyllostachys edulis (Poaceae), an important bamboo species in China. Appl Plant Sci 1(7):1200012. https://doi.org/10.3732/apps.1200012
Jofuku KD, den Boer BGW, Montagu MV, Okamuro JK (1994) Control of Arabidopsis flower and seed development by the homeotic gene APETALA2. Plant Cell 6:1211–1225. https://doi.org/10.1105/tpc.6.9.1211
Kaeppler SM, Kaeppler HF, Rhee Y (2000) Epigenetic aspects of somaclonal variation in plants. In: Plant gene silencing. Springer, Dordrecht, pp 59–68
Kagaya Y, Ohmiya K, Hattori T (1999) RAV1, a novel DNA-binding protein, binds to bipartite recognition sequence through two distinct DNA-binding domains uniquely found in higher plants. Nucleic Acids Res 27(2):470–478. https://doi.org/10.1093/nar/27.2.470
Kalaiarasi K, Sangeetha P, Subramaniam S, Venkatachalam P (2014) Development of an efficient protocol for plant regeneration from nodal explants of recalcitrant bamboo (Bambusa arundinacea Retz. Willd) and assessment of genetic fidelity by DNA markers. Agrofor Syst 88(3):527–537. https://doi.org/10.1007/s10457-014-9716-3
Kaneko S, Franklin DC, Yamasaki N, Isagi Y (2008) Development of microsatellite markers for Bambusa arnhemica (Poaceae: Bambuseae), a bamboo endemic to northern Australia. Conserv Genet 9(5):1311–1313. https://doi.org/10.1007/s10592-007-9467-z
Kitamura K, Kawahara T (2009) Clonal identification by microsatellite loci in sporadic flowering of a dwarf bamboo species, Sasa cernua. J Plant Res 122(3):299–304. https://doi.org/10.1007/s10265-009-0220-1
Kitamura K, Kawahara T (2011) Estimation of outcrossing rates at small-scale flowering sites of the dwarf bamboo species, Sasa cernua. J Plant Res 124(6):683–688. https://doi.org/10.1007/s10265-010-0398-2
Kitamura K, Saitoh T, Matsuo A, Suyama Y (2009) Development of microsatellite markers for the dwarf bamboo species Sasa cernua and Sasa kurilensis (Poaceae) in northern Japan. Mol Ecol Resour 9(6):1470–1472. https://doi.org/10.1111/j.1755-0998.2009.02675.x
Konzen E, Peron R, Ito M, Brondani G, Tsai S (2017) Molecular identification of bamboo genera and species based on RAPD-RFLP markers. Silva Fenn 51:4. https://doi.org/10.14214/sf.1691
Konzen ER, Recchia GH, Cassieri F, Caldas DGG, Berny Mier y Teran JC, Gepts P, Tsai SM (2019) DREB genes from common bean (Phaseolus vulgaris L.) show broad to specific abiotic stress responses and distinct levels of nucleotide diversity. Int J Genomics 2019:1–28. doi:https://doi.org/10.1155/2019/9520642
Krishna H, Alizadeh M, Singh D, Singh U, Chauhan N, Eftekhari M, Sadh RK (2016) Somaclonal variations and their applications in horticultural crops improvement. 3 Biotech 6(1):54. https://doi.org/10.1007/s13205-016-0389-7
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549. https://doi.org/10.1093/molbev/msy096
Lai Z, Li Y, Wang F, Cheng Y, Fan B, Yu J-Q, Chen Z (2011) Arabidopsis sigma factor binding proteins are activators of the WRKY33 transcription factor in plant defense. Plant Cell 23:3824–3841. https://doi.org/10.1105/tpc.111.090571
Lee NW, Chung MG (1999) High levels of genetic variation in Korean populations of Sasamorpha borealis (Poaceae). Bot Bull Acad Sin 40
Lewontin RC, Hubby JL (1966) A molecular approach to the study of genic heterozygosity in natural populations. II. Amount of variation and degree of heterozygosity in natural populations of Drosophila pseudoobscura. Genetics 54(2):595
Li L, Mu S, Cheng Z, Cheng Y, Zang Y, Miao Y, Hou C, Li X, Gao J (2017) Characterization and expression analysis of the WRKY gene family in moso bamboo. Sci Rep 7:6675. https://doi.org/10.1038/s41598-017-06701-2
Li S, Ramakrishnan M, Vinod KK, Kalendar R, Yrjälä K, Zhou M (2020a) Development and deployment of high-throughput retrotransposon-based markers reveal genetic diversity and population structure of Asian bamboo. Forests 11(1):31. https://doi.org/10.3390/f11010031
Li L, Yang K, Wang S, Lou Y, Zhu C, Gao Z (2020b) Genome-wide analysis of laccase genes in moso bamboo highlights PeLAC10 involved in lignin biosynthesis and in response to abiotic stresses. Plant Cell Rep 39:751–763. https://doi.org/10.1007/s00299-020-02528-w
Lin X, Lou Y, Zhang Y, Yuan X, He J, Fang W (2011) Identification of genetic diversity among cultivars of Phyllostachys violascens using ISSR, SRAP and AFLP markers. Bot Rev 77(3):223–232. https://doi.org/10.1007/s12229-011-9078-8
Lin Y, Lu J-J, Wu M-D, Zhou M-B, Fang W, Ide Y, Tang D-Q (2014) Identification, cross-taxon transferability and application of full-length cDNA SSR markers in Phyllostachys pubescens. Springerplus 3(1):486. https://doi.org/10.1186/2193-1801-3-486
Litt M, Luty JA (1989) A hypervariable microsatellite revealed by in vitro amplification of a dinucleotide repeat within the cardiac muscle actin gene. Am J Hum Genet 44(3):397–401
Liu H, Gao Y, Wu M, Shi Y, Wang H, Wu L, Xiang Y (2020) TCP10, a TCP transcription factor in moso bamboo (Phyllostachys edulis), confers drought tolerance to transgenic plants. Environ Exp Bot 172:104002. https://doi.org/10.1016/j.envexpbot.2020.104002
Liu H, Wu M, Zhu D, Pan F, Wang Y, Wang Y, Xiang Y (2017) Genome-wide analysis of the AAAP gene family in moso bamboo (Phyllostachys edulis). BMC Plant Biol 17:29. https://doi.org/10.1186/s12870-017-0980-z
Liu H-L, Wu M, Li F, Gao Y-M, Chen F, Xiang Y (2018) TCP transcription factors in Moso bamboo (Phyllostachys edulis): genome-wide identification and expression analysis. Front Plant Sci 9:1263. https://doi.org/10.3389/fpls.2018.01263
Liu J, Cheng Z, Xie L, Li X, Gao J (2019) Multifaceted role of PheDof12-1 in the regulation of flowering time and abiotic stress responses in moso bamboo (Phyllostachys edulis). Int J Mol Sci 20(2):424. https://doi.org/10.3390/ijms20020424
Loh J, Kiew R, Gan LH, Gan Y-Y (2000) A study of genetic variation and relationships within the bamboo subtribe Bambusinae using amplified fragment length polymorphism. Ann Bot 85(5):607–612. https://doi.org/10.1006/anbo.2000.1109
Lu Y, Wang D, Li H, Jia Q, Wu Z, Lu W (2012) A comparison on DNA methylation levels in bamboo at five developmental stages. J For Res 23(1):157–159. https://doi.org/10.1007/s11676-012-0247-3
Lv Y, Yu T, Lu S, Tian C, Li J, Du FK (2016) Development of microsatellite markers for Fargesia denudata (Poaceae), the staple-food bamboo of the giant panda. Appl Plant Sci 4(6):1600005. https://doi.org/10.3732/apps.1600005
Ma P-F, Zhang Y-X, Zeng C-X, Guo Z-H, Li D-Z (2014) Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst Biol 63:933–950. https://doi.org/10.1093/sysbio/syu054
Ma Q, Song H, Zhou S, Yang W, Li D, Chen J (2013) Genetic structure in dwarf bamboo (Bashania fangiana) clonal populations with different genet ages. PLoS One 8(11):e78784. https://doi.org/10.1371/journal.pone.0078784
Maurel C, Boursiac Y, Luu D-T, Santoni V, Shahzad Z, Verdoucq L (2015) Aquaporins in Plants. Physiol Rev 95:38
Meena RK, Bhandhari MS, Barhwal S, Ginwal HS (2019) Genetic diversity and structure of Dendrocalamus hamiltonii natural metapopulation: a commercially important bamboo species of Northeast Himalayas. 3 Biotech 9(2):60. https://doi.org/10.1007/s13205-019-1591-1
Mehta R, Sharma V, Sood A, Sharma M, Sharma RK (2011) Induction of somatic embryogenesis and analysis of genetic fidelity of in vitro-derived plantlets of Bambusa nutans Wall., using AFLP markers. Eur J For Res 130(5):729–736. https://doi.org/10.1007/s10342-010-0462-4
Miyazaki Y, Ohnishi N, Takafumi H, Hiura T (2009) Genets of dwarf bamboo do not die after one flowering event: evidence from genetic structure and flowering pattern. J Plant Res 122(5):523–528. https://doi.org/10.1007/s10265-009-0241-9
Mukherjee AK, Ratha S, Dhar S, Debata AK, Acharya PK, Mandal S, Panda PC, Mahapatra AK (2010) Genetic relationships among 22 taxa of bamboo revealed by ISSR and EST-based random primers. Biochem Genet 48(11–12):1015–1025. https://doi.org/10.1007/s10528-010-9390-8
Nayak S, Rout GR, Das P (2003) Evaluation of the genetic variability in bamboo using RAPD markers. Plant Soil Environ 49(1):24–28. https://doi.org/10.17221/4085-PSE
Neale DB, Martínez-García PJ, De La Torre AR, Montanari S, Wei X-X (2017) Novel insights into tree biology and genome evolution as revealed through genomics. Annu Rev Plant Biol 68(1):457–483. https://doi.org/10.1146/annurev-arplant-042916-041049
Nilkanta H, Amom T, Tikendra L, Rahaman H, Nongdam P (2017) ISSR marker based population genetic study of Melocanna baccifera (Roxb.) Kurz: a commercially important bamboo of Manipur, North-East India. Scientifica 2017:1–9. https://doi.org/10.1155/2017/3757238
Nogueira JS, Gomes HT, Scherwinski-Pereira JE (2019) Micropropagation, plantlets production estimation and ISSR marker-based genetic fidelity analysis of Guadua magna and G. angustifolia. Pesqui Agropecu Trop 49:e53743. https://doi.org/10.1590/1983-40632019v4953743
Okamuro JK, Caster B, Villarroel R, Van Montagu M, Jofuku KD (1997) The AP2 domain of APETALA2 defines a large new family of DNA binding proteins in Arabidopsis. Proc Natl Acad Sci U S A 94(13):7076–7081. https://doi.org/10.1073/pnas.94.13.7076
Oumer OA, Dagne K, Feyissa T, Tesfaye K, Durai J, Hyder MZ (2020) Genetic diversity, population structure, and gene flow analysis of lowland bamboo [Oxytenanthera abyssinica (A. Rich.) Munro] in Ethiopia. Ecol Evol 2020:1–20. https://doi.org/10.1002/ece3.6762
Pan F, Wang Y, Liu H, Wu M, Chu W, Chen D, Xiang Y (2017) Genome-wide identification and expression analysis of SBP-like transcription factor genes in Moso bamboo (Phyllostachys edulis). BMC Genomics 18:486. https://doi.org/10.1186/s12864-017-3882-4
Pan F, Wu M, Hu W, Liu R, Yan H, Xiang Y (2019) Genome-wide identification and expression analyses of the bZIP transcription factor genes in moso bamboo (Phyllostachys edulis). Int J Mol Sci:20. https://doi.org/10.3390/ijms20092203
Peng Z, Lu Y, Li L, Zhao Q, Feng Q, Gao Z et al (2013) The draft genome of the fast-growing non-timber forest species moso bamboo (Phyllostachys heterocycla). Nat Genet 45(4):456–461
Philippe H, Delsuc F, Brinkmann H, Lartillot N (2005) Phylogenomics. Annu Rev Ecol Evol Syst 36:541–562
Poland JA, Brown PJ, Sorrells ME, Jannink JL (2012) Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One 7(2):e32253
Pourcel L, Routaboul J-M, Kerhoas L, Caboche M, Lepiniec L, Debeaujon I (2005) TRANSPARENT TESTA10 encodes a laccase-like enzyme involved in oxidative polymerization of flavonoids in Arabidopsis seed coat. Plant Cell 17:2966–2980. https://doi.org/10.1105/tpc.105.035154
Rajput BS, Jani M, Ramesh K, Manokari M, Jogam P, Allini VR, Kher MM, Shekhawat MS (2020) Large-scale clonal propagation of Bambusa balcooa Roxb.: An industrially important bamboo species. Ind Crop Prod 157:112905. https://doi.org/10.1016/j.indcrop.2020.112905
Ramakrishnan M, Yrjälä K, Vinod KK, Sharma A, Cho J, Satheesh V, Zhou M (2020) Genetics and genomics of moso bamboo (Phyllostachys edulis): current status, future challenges, and biotechnological opportunities toward a sustainable bamboo industry. Food Energy Secur 2020(00):e229. https://doi.org/10.1002/fes3.229
Ramanayake SMSD, Meemaduma VN, Weerawardene TE (2007) Genetic diversity and relationships between nine species of bamboo in Sri Lanka, using random amplified polymorphic DNA. Plant Syst Evol 269:55–61. https://doi.org/10.1007/s00606-007-0587-1
Ray SS, Ali MN (2017) Factors affecting macropropagation of bamboo with special reference to culm cuttings: a review update. N Z J For Sci 47(1):17. https://doi.org/10.1186/s40490-017-0097-z
Rushton PJ, Somssich IE, Ringler P, Shen QJ (2010) WRKY transcription factors. Trends Plant Sci 15:247–258. https://doi.org/10.1016/j.tplants.2010.02.006
Sakuma Y, Liu Q, Dubouzet JG, Abe H, Shinozaki K, Yamaguchi-Shinozaki K (2002) DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBs, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem Biophys Res Commun 290(3):998–1009. https://doi.org/10.1006/bbrc.2001.6299
Shan, Yang, Xu, Zhu, Gao (2019) Genome-wide investigation of the NAC gene family and its potential association with the secondary cell wall in moso bamboo. Biomol Ther 9(10):609. https://doi.org/10.3390/biom9100609
Sharma V, Bhardwaj P, Kumar R, Sharma RK, Sood A, Ahuja PS (2009) Identification and cross-species amplification of EST derived SSR markers in different bamboo species. Conserv Genet 10(3):721–724. https://doi.org/10.1007/s10592-008-9630-1
Singh SR, Dalal S, Singh R, Dhawan AK, Kalia RK (2012) Micropropagation of Dendrocalamus asper {Schult. & Schult. F.} Backer ex k. Heyne: an exotic edible bamboo. J Plant Biochem Biotechnol 21(2):220–228
Singh SR, Dalal S, Singh R, Dhawan AK, Kalia RK (2013a) Ascertaining clonal fidelity of micropropagated plants of Dendrocalamus hamiltonii Nees et Arn. ex Munro using molecular markers. In Vitro Cell Dev Biol Plant 49(5):572–583. https://doi.org/10.1007/s11627-013-9520-1
Singh SR, Dalal S, Singh R, Dhawan AK, Kalia RK (2013b) Evaluation of genetic fidelity of in vitro raised plants of Dendrocalamus asper (Schult. & Schult. F.) Backer ex K. Heyne using DNA-based markers. Acta Physiol Plant 35(2):419–430. https://doi.org/10.1007/s11738-012-1084-x
Sun H, Li L, Lou Y (2017) The bamboo aquaporin gene PeTIP4;1–1 confers drought and salinity tolerance in transgenic Arabidopsis. Plant Cell Rep 36:597–609. https://doi.org/10.1007/s00299-017-2106-3
Sun H, Wang S, Lou Y, Zhu C, Zhao H, Li Y, Li X, Gao Z (2018) Whole-genome and expression analyses of bamboo aquaporin genes reveal their functions involved in maintaining diurnal water balance in bamboo shoots. Cell 7:195. https://doi.org/10.3390/cells7110195
Tanksley SD, Young ND, Paterson AH, Bonierbale MW (1989) RFLP mapping in plant breeding: new tools for an old science. Nat Biotechnol 7(3):257–264. https://doi.org/10.1038/nbt0389-257
Tian B, Yang H-Q, Wong K-M, Liu A-Z, Ruan Z-Y (2012) ISSR analysis shows low genetic diversity versus high genetic differentiation for giant bamboo, Dendrocalamus giganteus (Poaceae: Bambusoideae), in China populations. Genet Resour Crop Evol 59(5):901–908. https://doi.org/10.1007/s10722-011-9732-3
Tiwari C, Bakshi M, Gupta D, Razjput B (2019) Genetic diversity assessment of Sinarundinaria anceps (Mitf.) Chao and Renvoize accessions through isozyme markers. J Pharmacogn Phytochem 2019:515–519
Triplett JK, Oltrogge KA, Clark LG (2010) Phylogenetic relationships and natural hybridization among the North American woody bamboos (Poaceae: Bambusoideae: Arundinaria). Am J Bot 97(3):471–492
Vaattovaara A, Leppälä J, Salojärvi J, Wrzaczek M (2019) High-throughput sequencing data and the impact of plant gene annotation quality. J Exp Bot 70(4):1069–1076. https://doi.org/10.1093/jxb/ery434
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Friters A, Pot J, Paleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23(21):4407–4414. https://doi.org/10.1093/nar/23.21.4407
Wang J, Feng J, Jia W, Chang S, Li S, Li Y (2015) Lignin engineering through laccase modification: a promising field for energy plant improvement. Biotechnol Biofuels 8:145. https://doi.org/10.1186/s13068-015-0331-y
Wang T, Yue J-J, Wang X-J, Xu L, Li L-B, Gu X-P (2016) Genome-wide identification and characterization of the Dof gene family in moso bamboo (Phyllostachys heterocycla var. pubescens). Genes Genom 38:733–745. https://doi.org/10.1007/s13258-016-0418-2
Wang W, Chen S, Guo W, Li ZX (2020) Tropical plants evolve faster than their temperate relatives: a case from the bamboos (Poaceae: Bambusoideae) based on chloroplast genome data. Biotechnol Biotechnol Equip 34:482–493. https://doi.org/10.1080/13102818.2020.1773312
Wang W, Chen S, Zhang X (2018) Whole-genome comparison reveals divergent IR borders and mutation hotspots in chloroplast genomes of herbaceous bamboos (Bambusoideae: Olyreae). Molecules 23(7):1537. https://doi.org/10.3390/molecules23071537
Wang X, Ye X, Zhao L, Li D, Guo Z, Zhuang H (2017a) Genome-wide RAD sequencing data provide unprecedented resolution of the phylogeny of temperate bamboos (Poaceae: Bambusoideae). Sci Rep 7(1):11546. https://doi.org/10.1038/s41598-017-11367-x
Wang Y, Liu H, Zhu D, Gao Y, Yan H, Xiang Y (2017b) Genome-wide analysis of VQ motif-containing proteins in Moso bamboo (Phyllostachys edulis). Planta 246:165–181. https://doi.org/10.1007/s00425-017-2693-9
Williams JGK, Kubelik AR, Livak KJ, Rafalski JA, Tingey SV (1990) DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res 18(22):6531–6535. https://doi.org/10.1093/nar/18.22.6531
Wu H, Lv H, Li L, Liu J, Mu S, Li X, Gao J (2015) Genome-wide analysis of the AP2/ERF transcription factors family and the expression patterns of DREB genes in Moso Bamboo (Phyllostachys edulis). PLoS One 10(5):e0126657. https://doi.org/10.1371/journal.pone.0126657
Wu M, Li Y, Chen D, Liu H, Zhu D, Xiang Y (2016) Genome-wide identification and expression analysis of the IQD gene family in moso bamboo (Phyllostachys edulis). Sci Rep 6:24520. https://doi.org/10.1038/srep24520
Wu M, Liu H, Gao Y, Shi Y, Pan F, Xiang Y (2020a) The moso bamboo drought-induced 19 protein PheDi19-8 functions oppositely to its interacting partner, PheCDPK22, to modulate drought stress tolerance. Plant Sci 299:110605. https://doi.org/10.1016/j.plantsci.2020.110605
Wu M, Liu H, Han G, Cai R, Pan F, Xiang Y (2017) A moso bamboo WRKY gene PeWRKY83 confers salinity tolerance in transgenic Arabidopsis plants. Sci Rep 7(1):11721. https://doi.org/10.1038/s41598-017-10795-z
Wu M, Liu R, Gao Y, Xiong R, Shi Y, Xiang Y (2020b) PheASR2, a novel stress-responsive transcription factor from moso bamboo (Phyllostachys edulis), enhances drought tolerance in transgenic rice via increased sensitivity to abscisic acid. Plant Physiol Biochem 154:184–194. https://doi.org/10.1016/j.plaphy.2020.06.014
Wysocki WP, Clark LG, Attigala L et al (2015) Evolution of the bamboos (Bambusoideae; Poaceae): a full plastome phylogenomic analysis. BMC Evol Biol 15:50. https://doi.org/10.1186/s12862-015-0321-5
Xie L, Cai M, Li X, Zheng H, Xie Y, Cheng Z, Bai Y, Li J, Mu S, Gao J (2020) Overexpression of PheNAC3 from moso bamboo promotes leaf senescence and enhances abiotic stress tolerance in Arabidopsis. PeerJ 8:e8716. https://doi.org/10.7717/peerj.8716
Xie L, Li X, Hou D, Cheng Z, Liu J, Li J, Mu S, Gao J (2019) Genome-wide analysis and expression profiling of the heat shock factor gene family in Phyllostachys edulis during development and in response to abiotic stresses. Forests 10(2):100. https://doi.org/10.3390/f10020100
Yang H-M, Zhang Y-X, Yang J-B, Li D-Z (2013) The monophyly of Chimonocalamus and conflicting gene trees in Arundinarieae (Poaceae: Bambusoideae) inferred from four plastid and two nuclear markers. Mol Phylogenet Evol 68:340–356. https://doi.org/10.1016/j.ympev.2013.04.002
Yang H-Q, An M-Y, Gu Z-J, Tian B (2012) Genetic diversity and differentiation of Dendrocalamus membranaceus (Poaceae: Bambusoideae), a declining bamboo species in Yunnan, China, as based on inter-simple sequence repeat (ISSR) analysis. Int J Mol Sci 13(4):4446–4457. https://doi.org/10.3390/ijms13044446
Yang JB, Dong YR, Wong KM, Gu ZJ, Yang HQ, Li DZ (2018) Genetic structure and differentiation in Dendrocalamus sinicus (Poaceae: Bambusoideae) populations provide insight into evolutionary history and speciation of woody bamboos. Sci Rep 8(1):16933. https://doi.org/10.1038/s41598-018-35269-8
Yu X, Yang D, Guo C, Gao L (2018) Plant phylogenomics based on genome-partitioning strategies: Progress and prospects. Plant Divers 40:158–164. https://doi.org/10.1016/j.pld.2018.06.005
Yuan J-L, Sun H-M, Guo G-P, Yue J-J, Gu X-P (2014) Correlation between DNA methylation and chronological age of Moso bamboo (Phyllostachys heterocycla var. pubescens). Bot Stud 55(1):4. https://doi.org/10.1186/1999-3110-55-4
Zeng C-X, Zhang Y-X, Triplett JK, Yang J-B, Li D-Z (2010) Large multi-locus plastid phylogeny of the tribe Arundinarieae (Poaceae: Bambusoideae) reveals ten major lineages and low rate of molecular divergence. Mol Phylogenet Evol 56:821–839. https://doi.org/10.1016/j.ympev.2010.03.041
Zhang X-Z, Zeng C-X, Ma P-F, Haevermans T, Zhang Y-X, Zhang L-N, Guo Z-H, Li D-Z (2016) Multi-locus plastid phylogenetic biogeography supports the Asian hypothesis of the temperate woody bamboos (Poaceae: Bambusoideae). Mol Phylogenet Evol 96:118–129. https://doi.org/10.1016/j.ympev.2015.11.025
Zhou M, Hu B, Zhu Y (2017) Genome-wide characterization and evolution analysis of long terminal repeat retroelements in moso bamboo (Phyllostachys edulis). Tree Genet Genomes 13(2):43. https://doi.org/10.1007/s11295-017-1114-3
Zhou M, Wu J, Ramakrishnan M, Meng X, Vinod KK (2019a) Prospects for the study of genetic variation among Moso bamboo wild-type and variants through genome resequencing. Trees (Berl West) 33(2):371–381. https://doi.org/10.1007/s00468-018-1783-z
Zhou Y, Zhang Y-Q, Xing X-C, Zhang J-Q, Ren Y (2019b) Straight from the Plastome: molecular phylogeny and morphological evolution of Fargesia (Bambusoideae: Poaceae). Front Plant Sci 10:981. https://doi.org/10.3389/fpls.2019.00981
Zhu J-K (2016) Abiotic stress signaling and responses in plants. Cell 167:313–324. https://doi.org/10.1016/j.cell.2016.08.029
Zhu S, Liu T, Tang Q, Fu L, Tang S (2014) Evaluation of bamboo genetic diversity using morphological and SRAP analyses. Russ J Genet 50(3):267–273. https://doi.org/10.1134/S1022795414030132
Acknowledgments
We thank the National Council for Scientific and Technological Development (CNPq), Coordination for the Improvement of Higher Education Personnel (CAPES), Minas Gerais Research Funding Foundation (FAPEMIG), and São Paulo Research Foundation (FAPESP) for the financial support and scholarship.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Author Contributions
ERK conceived the general idea of the chapter. ERK, DC, and SMT described the application of molecular markers to bamboo genotyping and population genetic studies. LCP described and analyzed sequence data as potential phylogenetic markers. WFC contributed with the topics of phylogenomics and functional markers. DMSC, SBF, GEB, and DC described the application of molecular markers in studies of genetic fidelity. All authors read and approved the final version of the manuscript.
Author information
Authors and Affiliations
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2021 The Author(s), under exclusive license to Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Konzen, E.R. et al. (2021). Molecular Markers in Bamboos: Understanding Reproductive Biology, Genetic Structure, Interspecies Diversity, and Clonal Fidelity for Conservation and Breeding. In: Ahmad, Z., Ding, Y., Shahzad, A. (eds) Biotechnological Advances in Bamboo. Springer, Singapore. https://doi.org/10.1007/978-981-16-1310-4_2
Download citation
DOI: https://doi.org/10.1007/978-981-16-1310-4_2
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-16-1309-8
Online ISBN: 978-981-16-1310-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)