
Abstract
Epigenetics refers to reversible and hereditary changes in gene expression without alterations in DNA sequences, such as DNA methylation, histone modification and chromatin remodelling. It was first proposed by Waddington in the book Introduction to Modern Genetics in 1939. Autophagy includes at least four processes: autophagy induction, autophagosome formation, autophagosome fusion with lysosomes and lysosomal degradation of cytoplasmic components. The whole process is complex and dynamic, and involves at least 30 autophagy-related proteins. This degradative machinery is regulated by multiple signal molecules. Autophagy was once considered to be a cytoplasmic event; however, in recent years, emerging evidence suggests that nuclear components (transcription factors, histone modification, microRNAs, etc.) also play an important role in autophagy regulation (Baek and Kim 2017). Among them, epigenetic regulation of autophagy has gained much attention. The epigenetic machinery can not only modify autophagy-related genes but also affect some signal molecule genes that regulate autophagy, thus impacting their transcription and subsequent autophagy. This chapter focuses on the role and recent progress in autophagy regulation by DNA methylation and histone modifications. The role of non-coding RNAs such as microRNA in autophagy regulation will be covered in other chapters.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
1 Autophagy Regulation by DNA Methylation
1.1 Introduction to DNA Methylation
DNA methylation refers to the methylation of the fifth carbon atom of cytosine into 5-methylcytosine (5-methylcytosine, 5-mC), which is catalysed by DNA methyltransferase (DNMT) using S-adenosylmethionine as a methyl donor. This modification mainly occurs in the CpG dinucleotide sequence. In most regions of the genome, the appearance frequency of CpG sequences is low. However, in some specific regions, such as the gene promoter, CpG dinucleotides are arranged in high frequency series and called CpG islands. DNA methylation usually results in gene silencing because 5-mC blocks DNA binding to transcription factor complexes. Conversely, DNA demethylation often activates gene transcription. DNA methylation is the earliest identified and most characterized machinery of epigenetic modifications. Notably, the methylated CpG island can be recognized by methyl-CpG-binding protein 2 (MeCP2), which further recruits histone deacetylase (HDAC) and histone methyltransferase (HMT) and other histone-modifying enzymes to alter chromatin structure and subsequently modulate gene transcription. Therefore, it is noteworthy that DNA methylation often interacts with histone modification and thus synergistically regulates gene transcription.
1.2 DNA Methylation of Autophagy-Related Genes
To date, many autophagy-related genes have been found to be methylated and silenced, thus inhibiting the process of autophagy and autophagic flow in some pathological situations. Here are some examples.
1.2.1 ULK Kinase/ATG1
In yeast, autophagy-related protein (Atg) 1, along with Atg13 and Atg17, comprises the ULK1 kinase complex, which plays a critical role in autophagy initiation. The yeast Atg1 has two homologous proteins in mammals, ULK1 and ULK2, collectively known as ULK kinase. Currently, most studies focus on the regulation of ATG1/ULK1 by mTOR and AMPK signalling pathways. In a nutrient-rich environment, activated mTOR phosphorylates and inhibits ULK1 activity. During nutrient deprivation, mTOR activity is inhibited, and thus, its inhibition of ULK1 is released, which activates ULK1 kinase and initiates autophagy activation. Energy depletion, such as glucose deprivation, induces AMPK activation and phosphorylates the ULK1 Ser317 and Ser777 sites, thereby inducing autophagy. Mounting evidence suggests that in addition to modulation by mTOR and AMPK, DNA methylation also exerts an effect on ULK transcription and thus autophagy induction. It was reported that ULK1 and ULK2 expressions were significantly decreased in gliomas and that the promoter region of ULK2 was hypermethylated, whereas the ULK1 promoter was not affected. ULK2 activation inhibited astrocyte transformation and glioma growth by enhancing autophagy activity. This implies that the alteration of ULK2 DNA methylation influences the autophagy activity of glioma cells and thus participates in the formation of glioma.
1.2.2 Beclin1/ATG6
Beclin1 (yeast Atg6 homologous protein) is a multifunctional protein in autophagy. It not only forms a complex by binding to Atg14L and Barkor and regulates autophagy initiation but also interacts with other proteins, such as Vps34 and Bcl-2, to regulate autophagosome maturation. Li et al. found that the Beclin1 mRNA level was decreased in 14 out of 20 breast cancer patients, and its protein expression was also lowered in 13 out of 20 patients. Downregulation of Beclin1 transcription was attributed not only to the decrease in gene copy number but also to the abnormal hypermethylation of the CpG enrichment region in the promoter and intron 2 of the Beclin1 gene. When breast cancer cells were treated with methyltransferase inhibitors, Beclin1 expression was upregulated, accompanied by increased autophagic activity and inhibition of tumour growth, suggesting that DNA methylation of Beclin1 inhibited its transcription and autophagy.
1.2.3 LC3/Atg8
LC3 is a homologue of the yeast autophagy-related gene Atg8. There are many subtypes of LC3 in mammalian species, namely, LC3A, LC3B and LC3C. The amino acid sequences of LC3A and LC3B are highly consistent. Both of these proteins participate in autophagosome formation and are commonly used markers of autophagy. During autophagy, LC3-I covalently binds with phosphatidylethanolamine to form LC3-II under the action of ATG7 and the ATG12–ATG5–ATG16L complex. LC3-II binds to the membrane of autophagosomes. Therefore, the LC3-II/I ratio is a biomarker of autophagosome formation in experimental studies. Previous studies reported that LC3A was silenced due to DNA hypermethylation in a variety of cancer cells, and the DNMT inhibitors could increase LC3A transcription and inhibit the growth of cancer cells. This suggests that hypermethylation of LC3A may inhibit autophagy and promote tumourigenesis. Another study also reported that the DNA methylation inhibitor deoxyazacytidine increased LC3-II levels and autophagy activity in human chronic myeloid leukaemia K-562 and MEG-01 cell lines. All the data indicate that DNA methylation is involved in the regulation of autophagy.
1.2.4 LAMP2
Recent evidence suggests that methylation modification of the lysosomal-associated membrane protein 2 (LAMP2) gene causes autophagy deficiency, which may be involved in the pathogenesis of Danon disease, an X-linked fatal cardiomyopathy. By generating patient-specific induced pluripotent stem cells (iPSCs) and differentiating them into cardiomyocytes (iPSCs-CMs), the researchers reproduced the histological features and autophagy failure of Danon disease. Administration of DNMT inhibitors reactivated the silent LAMP2 alleles in these iPSCs and alleviated the autophagic failure, suggesting that silencing of the LAMP2 gene caused by DNA methylation was responsible for the autophagic failure and was related to the development of Danon disease (Ng et al. 2016).
1.3 DNA Methylation of Autophagy Regulatory Molecules
Apart from the autophagy-related genes mentioned above, DNA methylation can also modify the genes that encode autophagy regulatory signal molecules. Abnormal methylation in some tumour-related genes has been reported to be involved in autophagy regulation.
1.3.1 Nitro Domain-Containing Protein 1 (NOR1)
Nitro domain-containing protein 1 (NOR1) is a putative tumour suppressor gene. Li et al. found that the NOR1 promoter was hypermethylated, which was associated with the downregulation of NOR1 expression in nasopharyngeal carcinoma cells compared with that in normal tissues. DNMT inhibitors reversed the downregulation of NOR1 expression and clone formation of nasopharyngeal carcinoma cells. Meanwhile, the levels of LC3-II/LC3-I and autophagy activity as well as cell survival were suppressed. These results suggest that hypermethylation of the NOR1 promoter enhances the autophagy and survival of nasopharyngeal carcinoma cells. However, the exact molecular mechanism by which NOR1 regulates autophagy has yet to be elucidated.
1.3.2 Death-Associated Protein Kinase (DAPK)
DAPK is a newly identified tumour suppressor gene that is extensively involved in cell proliferation, apoptosis, autophagy and other pathological processes. Hypermethylation of CpG islands in the DAPK promoter, resulting in the silencing of this gene, has been reported in a variety of cancer cells. It correlates with the formation and metastasis of tumours. DAPK interacts with several autophagy-regulated proteins, such as mTOR, Beclin1 and PI3K, thus playing a positive or negative role in autophagy regulation in different conditions. For instance, it was found that following arsenic treatment, the CpG islands in the DAPK promoter were hypermethylated and the transcription of DAPK was suppressed, while the number of autophagosomes and Beclin1 expression were increased in SV-HUC-1 cells. These changes were reversed by the DNMT inhibitor Aza (Chai et al. 2007). These findings suggest that DNA hypermethylation of the DAPK gene is involved in autophagy regulation. However, a recent study found no significant correlation between the transcription of the DAPK gene and its DNA methylation status by comparing the association among the degree of DAPK promoter methylation and its mRNA and protein expression in tumour and adjacent non-tumour tissues from 15 breast cancer patients and matched controls (Streckmann et al. 2018). The difference may be caused by the small sample size of this study. Meanwhile, these results also indicate that the relevance of DNA methylation of the DAPK gene in tumourigenesis has yet to be investigated further.
1.3.3 SOX1
SOX1 belongs to the superfamily of SRY (sex-determining region Y)-box containing transcription factors. It can inhibit Wnt/β-catenin signalling by directly binding to β-catenin, leading to its degradation and function loss. Several studies reported that the SOX1 promoter was hypermethylated in hepatocellular, nasopharyngeal, oesophageal and non-small cell lung cancer (NSCLC) cells, associated with lower SOX1 expression in these tumour cells. Moreover, long-term exposure to cisplatin can also induce methylation of the SOX1 promoter in ovarian cancer cells and participate in cisplatin resistance. A further study demonstrated that the promoter of SOX1 was abnormally hypermethylated, associated with the suppression of SOX1 transcription in NSCLC cells, especially in cisplatin-resistant NSCLC cells. Moreover, silencing SOX1 promoted cisplatin-induced autophagy in NSCLC cells, indicating that DNA hypermethylation of the SOX1 gene participated in autophagy regulation in NSCLC cells.
1.4 Autophagy Regulation by DNA Demethylation
DNA methylation is a dynamic process, and methylated DNA can also be demethylated. It is believed that there are two means of DNA demethylation: active and passive demethylation. Active DNA demethylation is mainly mediated by enzymes that convert 5-mC to unmethylated cytosine. Passive DNA demethylation occurs along with DNA replication, during which the methylated DNA is eliminated due to the semi-reserved mode of DNA replication. As such, methylated CpG is “diluted” with the progress of DNA replication, resulting in passive DNA demethylation. Although active DNA demethylation widely exists in many cells, its molecular mechanism remains controversial. Ten–eleven translocation (TET) proteins, including TET1, TET2 and TET3, are α-ketoglutarate- and Fe2+-dependent dioxygenases that catalyse the hydroxylation of 5mC to 5-hmC. The identification of the TET family reveals a new pathway involved in DNA demethylation. It has emerged as one of the hotspots in epigenetic research in the past decade. A recent study found that TET1 knockout downregulated autophagy in glioma U251 cells, while its overexpression upregulated autophagy, indicating that TET1 plays a role in cancer development by regulating autophagy activity (Fu et al. 2017). However, whether the exact mechanism of autophagy regulation by TET1 is related to demethylation of autophagy-related genes remains to be further studied. Another study reported that TET2 was downregulated during the pathogenesis of atherosclerosis in an ApoE knockout mouse model. The downregulation of TET2 promotes methylation of the Beclin1 promoter, leading to autophagy flux impairment in endothelial cells (Peng et al. 2016). Therefore, these data strongly suggest that DNA demethylation affects autophagy by regulating the transcription of autophagy-related genes. However, research on the relationship between DNA demethylation and autophagy has only begun. At present, little is known about this topic, which deserves further study.
1.5 Conclusion
In summary, the study on autophagy regulation by DNA methylation is still in its infancy. To date, only a few autophagy-related genes and signalling molecules have been reported to undergo DNA methylation and thus affect autophagy activity. Most of the current information regarding autophagy regulation by DNA methylation comes from cancer-related studies. The impact of DNA methylation on autophagy may be tissue- or cell-specific. Whether alterations in DNA methylation contribute to other autophagy-related diseases, such as infection and neurodegeneration, remains to be explored further. In addition, current knowledge on DNA methylation and autophagy comes from the methylation alteration analysis of some genes during DNMT inhibition by pharmacological tools. However, these DNMT inhibitors have non-specific effects. Thus, this still needs to be confirmed with other technical or experimental evidence. The identification of the DNA demethylase TET family offers a new avenue for exploring the correlation between DNA methylation and autophagy in the future.
2 Autophagy Regulation by Histone Modifications
2.1 Introduction to Histone Modifications
Histones are the chief protein components of chromatin, acting as spools around which DNA winds. The structural unit of the histone-DNA complex is called the nucleosome. Five major subtypes of histones exist: H1, H2A, H2B, H3 and H4. Histones H2A, H2B, H3 and H4 are known as the core histones, while H1 is known as the linker between the two nucleosomes. The nucleosome core is formed by two H2A–H2B dimers and an H3–H4 tetramer. The H3 and H4 histones have long tails at the N terminus, where lysine and arginine residues are enriched and often protrude from the nucleosome. The tails can undergo post-translational modifications, including methylation, acetylation, phosphorylation, ubiquitination, SUMOylation and ADP-ribosylation, which often results in changes in chromatin structure and leads to activating or silencing of gene transcription. Post-translational modification of histones provides a readable marker for the binding of other proteins to DNA, which produces synergistic or antagonistic effects to regulate gene transcription and expression. This modification plays a similar role to the DNA codon, so it is also called the “histone code”. Notably, different histone modifications often affect one another. Histone acetylation often interacts with other epigenetic modifications and finely regulates gene transcription. Compelling evidence suggests that histone modifications not only affect autophagy induction but also play a role in the maintenance of long-term autophagy flux during prolonged exposure to autophagic stimuli, which is closely related to the pathogenesis of tumours and neurodegenerative diseases. Histone modification is a research hotspot of epigenetic regulation of autophagy and appears to be the fastest-growing branch of autophagy research in recent years (Fullgrabe et al. 2014a; Shin et al. 2016b).
2.2 Autophagy Regulation by Histone Acetylation
Histone acetylation is a histone modification that mainly occurs at the N-terminal conserved lysine residues of H3 and H4. It is coordinately regulated by histone acetyltransferase (HAT) and histone deacetylase (HDAC). As its name implies, HATs transfer the acetyl groups from acetyl coenzyme A to specific lysine residues at the N-terminal of the histone. They are also known as lysine acetyltransferases (KATs), which mainly include four groups: GCN5-related acetyltransferases (GNAT), MYST-related acetyltransferases, p300/CBP acetylases (KAT3B/KA), and T3A and Rtt109 (KAT11). As a superfamily, 18 subtypes of HDACs have been identified thus far. They can be divided into four categories: type I includes HDAC1–3 and HDAC8, type II includes HDAC4–7 and HDAC9–10, type III includes SIRT1–7 and type IV includes HDAC11. Among them, types I, II and IV are zinc-dependent, while type III catalyses deacetylation with NAD+ as a cofactor. The distribution of HDACs is subcellular specific and may change under different conditions. For example, SIRT1 is mainly expressed in the nucleus under normal conditions, but it can translocate into the cytosol by a variety of stimuli. In the cytosol, SIRT1 catalyses the deacetylation of several autophagy-related proteins and their regulators, such as ATG5, ATG7, LC3, FOXO and E2F1. Interestingly, SIRT1 deacetylates non-histone proteins in the cytoplasm and often activates autophagy, whereas it induces histone deacetylation that inhibits autophagy. In addition to SIRT1, other histone deacetylases, such as HDAC6, also affect the acetylation of non-histones and thus regulate autophagy. Deacetylation of non-histones is one of the hotspots in the field of autophagy-related research.
Relative to non-histone modification, histone acetylation has not received much attention until recently. In 2009, Madeo et al. first proposed the concept that histone modifications regulate autophagy. In ageing yeasts, they found that spermidine-induced autophagy relied on HAT inhibition, which caused global hypoacetylation of histone H3 and inhibited the transcription of many genes. Interestingly, the transcription of some autophagy-related genes (ATG) remained unaffected, and autophagy was still activated during this process. They believe that selective transcriptional activation of ATG is critical for yeast to save “resources” under starvation. In fact, some earlier studies by other groups already suggested that histone acetylation could affect autophagy. In 2004, Shao et al. found that the HDAC inhibitors butyrate and SAHA induced autophagic death in several human cancer cell lines. Notably, HDAC inhibitors also deacetylate non-histone proteins in the cytoplasm. Moreover, autophagy was commonly considered to be a cytoplasmic event, and nuclear events were not recognized as a regulator of this process. Thus, researchers focused on the acetylation of non-histone proteins in the cytoplasm when interpreting the results at that time. In recent years, compelling studies have demonstrated an important role of histone acetylation in sustained autophagy in response to long-term nutritional deprivation or stress (Fullgrabe et al. 2014b). The autophagy-related acetylations of histone 4 at position 16 lysine (H4K16ac) and histone 3 at position 56 lysine (H3K56ac) are the most studied.
2.2.1 H4K16ac
Unlike most histone modifications, the acetylation of H4K16 not only affects the nucleosome level but also has an impact on the chromatin structure; thus, it plays a central role in chromatin remodelling and gene transcription. In humans, hMOF/KAT8 and SIRT1 act as a pair of molecular switches to coordinate the acetylation of H4K16, thereby regulating autophagic activity in cells. Specifically, hMOF catalyses the acetylation modification of H4K16 and upregulates the expression of autophagy-related genes, whereas SIRT1 activation produces antagonistic effects, deacetylating H4K16 and inhibiting basal autophagy under normal conditions. Autophagy activation induced by several stimuli in different cell lines is related to a global reduction of H4K16 acetylation. hMOF/KAT8 is an autophagy substrate. Autophagy-induced degradation of hMOF and the consequent decrease in H4K16 acetylation inhibits the transcription of autophagy-related genes and the persistence of autophagy, which forms a negative feedback preventing autophagic death. It was observed that the autophagy-inducer rapamycin significantly increased cell death in normal somatic cells, HeLa cells and the U1810 cell line, and this was attenuated by blockade of H4K16 deacetylation using a SIRT1-specific inhibitor or hMOF overexpression (Fullgrabe et al. 2013). This suggests that the degree of H4K16 acetylation not only regulates autophagy but also plays a role in cell death or survival decisions. Notably, SIRT1-induced non-histone deacetylation often results in autophagy activation. Conversely, histone deacetylation by nuclear SIRT1 leads to autophagy inhibition. In addition, histone acetylation is often associated with other histone modifications and participates in autophagy regulation. For instance, H4K16ac often cooperates with H3K9me2 to inhibit autophagic flow. In addition, H4K16ac is tightly connected to H4K20me3. During autophagy, the decrease in H4K16ac and the increase in H4K20me3 often collectively inhibit gene expression. Thus, these results suggest that different histone modifications often interact with each other to fine-tune the transcription of autophagy-related genes and autophagy flux. In other words, the alteration of autophagy activity may be the consequence of a variety of histone modification changes. Therefore, when studying the impact of histone modification on autophagy, researchers cannot neglect the potential effect of other histone modifications on autophagy when focusing on one specific modification.
2.2.2 H3K56ac
H3K56 is located in the entry and exit of the nucleosome. Acetylation of H3K56 interrupts the interaction between the histone and DNA and inhibits transcription. Researchers observed an inhibition by rapamycin on H3K56 acetylation in yeast and revealed for the first time a positive regulation of the TOR signalling pathway on H3K56 acetylation. The acetylation of human H3K56 is regulated by EP300/KAT3B/P300 and KAT2A/GCN5. It has been reported that EP300 knockout activates autophagy, while EP300 overexpression inhibits starvation-induced autophagy. However, it remains to be determined whether EP300 affects autophagy merely via the regulation of H3K56 acetylation because EP300 is also known to regulate the acetylation of multiple autophagy-related proteins, including ATG5, ATG7, MAP1LC3 and ATG12. Moreover, the deacetylase of H3K56 is still controversial. Deacetylases, including HDAC1, HDAC2, SIRT1, SIRT2 and SIRT3, have been reported to catalyse the deacetylation of H3K56. It is noteworthy that although these deacetylases regulate autophagy, the role of non-histone deacetylation cannot be ruled out.
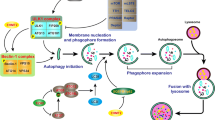
It is noteworthy that intracellular HAT and HDAC cooperatively maintain a dynamic balance between histone and non-histone acetylation and deacetylation (Fig. 11.1). The dysregulation of HAT and/or HDAC functions caused by some pathological factors results in changes in the degree of protein acetylation and upregulation or downregulation of some autophagy-related genes or their regulatory molecules and thus leads to the development of various diseases such as tumours and neurodegenerative disorders. Mounting evidence suggests that HDAC inhibitors promote autophagy by increasing the acetylation of non-histone proteins in the cytoplasm (described in other chapters). Therefore, HDAC inhibitors have become a new direction of drug research and development for these diseases. Currently, researchers have demonstrated that changes in histone acetylation are often correlated with alterations in non-histone acetylation in the cytoplasm. However, several studies show an opposite effect exerted by histone and non-histone acetylation on autophagy activity. Hence, it is highly suggested to use other techniques, such as nucleus/cytoplasm separation, to clarify the net contribution of histone or non-histone acetylation changes in autophagy in addition to their application with pharmacological tools.

Autophagy regulation by histone acetyltransferase (HAT) and histone deacetylase (HDAC)-mediated histone and non-histone acetylation. HATs and HDACs co-regulate and maintain the dynamic balance between histone and non-histone acetylation and deacetylation. This plays a critical role in the different processes of autophagy by promoting or suppressing the expression of autophagy-related genes, their regulatory molecules or affecting the protein–protein interaction. Specifically, the deacetylation of non-histone proteins in the cytoplasm often activates autophagy, while deacetylation of histones in the nucleus inhibits autophagy. Thus, the impact of histone and non-histone acetylation on autophagy activity is inconsistent
2.3 Autophagy Regulation by Histone Methylation
In addition to acetylation, histone methylation is also involved in autophagy regulation. Histone methylation is catalysed by histone methyltransferase (HMT) and usually occurs on lysine (K) and arginine (R) residues. Among them, K4, K9, K27, K36, K79, R2, R17 and R26 of histone H3, and R3 and K20 of H4 can be methylated. Lysine can be monomethylated, dimethylated and trimethylated, while arginine can only be monomethylated and dimethylated. This greatly increases the complexity of histone methylations. Moreover, similar to that of histone acetylation, the process of histone methylation is reversible, and the demethylation of lysine and arginine residues is mediated by specific histone demethylases. Histone arginine demethylases mainly include peptide arginine deimise 4 (PAD4) and JmjC domain-containing protein 6 (JMJD6). Histone lysine demethylases include LSD1, JHDM1, JHDM2 and JMJD2.
The effect of histone lysine methylation on gene transcription depends not only on the modified position and methylation degree but also on the gene region where methylation occurs. For example, methylation of heterochromatin H3K9 often leads to gene silencing, while H3K9 methylation in gene coding regions activates gene transcription. H3K4 methylation is often concentrated in the promoter, serving as a marker of gene transcription activation. Methylation of H3K27 is related to gene transcription inhibition. Histone demethylation has complex effects on gene transcription. For example, the regulation of gene expression by lysine demethylase LSD1 depends on its specific substrate. In summary, histone methylation and demethylation are dynamic and reversible complex processes catalysed by specific enzymes that participate in biological processes, such as gene transcription and autophagy (Fig. 11.2). Here, we list a brief review of histone methylation modifications involved in autophagy.

Histone modifications involved in autophagy regulation. Covalent modification of histone is catalysed or removed by specific enzymes. Accordingly, the enzymes that promote and inhibit histone modification can be called writers and erasers, respectively. Histone modifications involved in autophagy regulation mainly occur on H3 and H4. In contrast to acetylation, histone methylation occurs not only on lysine (K) but also on arginine (R) residues. Ac, acetylation; Me, methylation
2.3.1 H3K4me3
Dimethylation and trimethylation of histone H3 at lysine 4 (H3K4me2/me3) are involved in transcription activation, and the highest H3K4me3 level occurs near the transcription start point of highly expressed genes. H3K4 is methylated by SET1 and the mixed lineage leukaemia family of HMTs (MLL1) and demethylated by KDM1A/LSD1 and KDM5/JARID1. H4K16ac and H3K4me3 often reside in the same nucleosome units in human cells, and this is consistent with the interaction between their corresponding catalytic enzymes KAT8 and KMT2A/MLL1. Similar to the change of H4K16ac, autophagy often results in a decrease in H3K4me3 and leads to global transcription inhibition in multiple cell lines from yeast to human. This may be a conservative mechanism of energy savings in response to persistent starvation. WNT/β-catenin signal activation was reported to inhibit SQSTM1/p62 transcription and autophagy activity, which is related to the increase in H3K4me3 (Petherick et al. 2013). During autophagy, WNT dissociates from the promoter region of SQSTM1 and is degraded, which in turn results in a decrease in H3K4me3. Consequently, this weakens the inhibition by WNT on SQSTM1 transcription and thus increases SQSTM1 transcription and autophagy activity. This process was verified in mixed lineage leukaemia and other cancer cells (Sierra et al. 2006; Wend et al. 2013).
2.3.2 H3K9me3/H3K9me2
H3K9 can not only be mono-, di- or even tri-methylated by histone methylases such as SUV39H1 and EHMT2/G9a but can also be acetylated under the action of histone acetylases such as KAT2A and KAT2B/PCAF. Interestingly, the deacetylation of H3K9 is required for the increase in H3K9 dimethylation. H3K9 methylation is usually involved in gene silencing. It was reported that EHMT/G9a binds to the promoter of several autophagy-related genes, such as LC3B and WIPI1, resulting in H3K9 methylation and silencing of these genes in normal conditions (de Narvajas et al. 2013). During nutrient deprivation-induced autophagy, EHMT2 is released from the promoters of these autophagy-related genes, resulting in subsequent demethylation and acetylation modifications of H3K9 and thus promoting the transcription and expression of autophagy-related proteins. EHMT2 inhibitors were found to increase BNIP3 and LC3 levels. When autophagy is induced by rapamycin along with a persistent inhibition of EHMT2 by specific inhibitors, autophagy appears to be overstimulated, resulting in autophagic death in cells. Therefore, methylation of H3K9 not only regulates autophagy but also has a neglectable impact on cell survival. Notably, several members of the histone lysine demethylase family can catalyse the demethylation of H3K9. For example, KDM2B can demethylate H3K9 and induce autophagy.
2.3.3 H4K20me3
Methylation modification of H4K20 is involved in gene silencing, and H4K20me3 is often distributed in some constitutively expressed heterochromatin regions. H4K20 methylation can be catalysed by several enzymes, including SETD8 and SUV420. Specifically, SETD8 catalyses the monomethylation of H4K20 (H4K20me1), which can be further methylated to H4K20me2 and H4K20me3 by SUV420. PHF8 is identified as an H4K20 demethylase. Serum deprivation-induced autophagy increased the level of H4K20me3, which was accompanied by the alteration of H4K16 acetylation. Studies have shown that the deacetylation of H4K16ac and the increase in H4K20me3 antagonistically regulated 20–30% of human genome expression. The mechanism is related to RNA polymerase II pause (Kapoor-Vazirani et al. 2011). Therefore, the deacetylation of H4K16ac and the methylation of H4K20 during autophagy may work together to fine-tune the pause of RNA polymerase II and repress the transcription of some autophagy-unrelated genes.
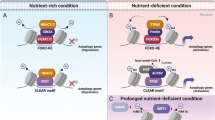
2.3.4 H3R17me2
H3R17 (H3R17me2) dimethylation was reported to enhance the transcription of several autophagy-related genes and transcription factor EB (TFEB), a key transcription factor in autophagy. H3R17me2 is mainly mediated by arginine methyltransferase 1 (CARM1). Under nutrient-rich conditions, the stability of CARM1 is regulated by the E3 ubiquitin ligase SKP2 in the nucleus, not in the cytoplasm. Nutritional starvation and glucose deprivation result in AMP-activated protein kinase (AMPK)-dependent phosphorylation of FOXO3a in the nucleus, which in turn transcriptionally suppresses SKP2. This inhibition led to an increase in CARM1 protein, followed by an increase in histone H3R17me2. Genome-wide analysis showed that this process played a role in transcriptional activation of autophagy-related and lysosome genes (Shin et al. 2016a). This study revealed a new mechanism for the AMPK–SKP2–CARM1 signalling axis in regulating histone modification and autophagy during prolonged nutrition deprivation.
2.3.5 Other Histone Methylations
In addition to the methylation modification of the sites mentioned above, EZH2-mediated H3K27me3 also affects autophagy activity. The expression of EZH2 was increased in human rectal colon cancer tissues. EZH2 knockout or its inhibitor reduced H3K27me3 and activated autophagy, which may be of relevance for the treatment of rectal colon cancer (Wei et al. 2015). However, a recent study found that EZH2-mediated H3K27me3 and DNMT1-induced DNA methylation synergistically inhibited the expression of cystic fibrosis transmembrane conductance regulator (CFTR), which caused autophagy activation and aggravated homocysteine-induced liver injury in mice (Yang et al. 2018). Therefore, EZH2-induced H3K27me3 may have specific effects on autophagy under different pathological conditions. This also confirms the complexity and diversity of histone methylation during autophagy, which should not be ignored in the research.
2.4 Autophagy Regulation by Other Histone Post-translational Modifications
Over the past decade, a large number of studies have revealed interactions between histone methylation and acetylation and autophagy. However, the roles and mechanisms of other covalent modifications of histones, such as phosphorylation, SUMOylation, ubiquitination and ADP ribosylation, in autophagy are still in infancy. A recent study reported a role for H2BK120 monoubiquitination (H2Bub1) in autophagy, which identified a novel epigenetic mechanism. H2Bub1 is critical for maintaining lower autophagy activity under basal conditions. However, when the cells are exposed to nutrient deprivation or starvation, the expression of deubiquitinase USP44 is upregulated, resulting in a decrease in H2Bub1, which may lead to global changes in the expression of genes, especially those autophagy-related genes. These alterations in gene expression eventually contribute to autophagy activation after starvation. It was also observed that this process is accompanied by changes in hMOF expression and the H4K16ac degree (Chen et al. 2017). Notably, other studies show that histone methylation, such as H3K4 and H3K79 methylation, can also be affected by H2Bub1. Therefore, different histone modifications are tightly connected, and the exact role of this interaction in autophagy and autophagy-related diseases needs to be further explored.
3 Conclusion and Prospective
In conclusion, the current knowledge regarding the role of DNA methylation and histone post-translational modifications in autophagy has only revealed the tip of the iceberg of autophagy regulation by the epigenetic machinery. In addition to DNA methylation and histone post-translational modification, great progress has been made in autophagy regulation by non-coding RNA in recent years. This topic is elaborated in other chapters. There are interactions among various epigenetic regulations. Specifically, DNA methylation affects histone acetylation, and different histone modifications, including methylation, acetylation, and even ubiquitination, interact with each other. All of these factors determine the complexity of epigenetic regulation on autophagy. Special attention should be paid to the changes in non-histone acetylation in the cytoplasm, which is an important factor for autophagy regulation. Therefore, when studying the impact of compounds targeting acetylation-modifying enzymes on autophagy, we should comprehensively analyse the changes in both histone and non-histone acetylation with multiple approaches. Finally, epigenetic modification appears to be a dynamic and reversible event in the nucleus, and DNA methylation could have an effect on histone acetylation and other modifications, and vice versa. In addition, the HDACs show potential effects on non-histones in the cytoplasm. Thus, all of these factors contribute to the complexity of epigenetic regulation of autophagy. Therefore, it will be of great significance to strengthen the study on autophagy regulation by epigenetic modifications, which can help to further understand the molecular mechanism of autophagy regulation and its role in the pathophysiological processes of tumours, neurodegenerative diseases and other disorders.
Abbreviations
- CBP:
-
CREB binding protein
- DAPK:
-
Death-associated protein kinase
- DNMT:
-
DNA methyltransferase
- HAT:
-
Histone acetyltransferase
- HDAC:
-
Histone deacetylase
- HMT:
-
Histone methyltransferase
- JMJD6:
-
JmjC domain-containing protein 6
- KAT:
-
Lysine acetyltransferase
- KDM:
-
Histone or lysine demethylase
- LAMP2:
-
Lysosomal-associated membrane protein 2
- 5-mC:
-
5-methylcytosine
- MeCP2:
-
Methyl-CpG-binding protein 2
- NOR1:
-
Nitro domain containing protein 1
- NSCLC:
-
Non-small cell lung cancer
- PAD4:
-
Peptide arginine deiminase 4
References
Baek SH, Kim KI (2017) Epigenetic control of autophagy: nuclear events gain more attention. Mol Cell 65:781–785
Chai CY, Huang YC, Hung WC, Kang WY, Chen WT (2007) Arsenic salts induced autophagic cell death and hypermethylation of DAPK promoter in SV-40 immortalized human uroepithelial cells. Toxicol Lett 173:48–56
Chen S, Jing Y, Kang X, Yang L, Wang DL, Zhang W, Zhang L, Chen P, Chang JF, Yang XM, Sun FL (2017) Histone H2B monoubiquitination is a critical epigenetic switch for the regulation of autophagy. Nucleic Acids Res 45:1144–1158
de Narvajas AAM, Gomez TS, Zhang JS, Mann AO, Taoda Y, Gorman JA, Kim DH (2013) Epigenetic regulation of autophagy by the methyltransferase G9a. Mol Cell Biol 33(20):3983–3993
Fu R, Ding Y, Luo J, Yu L, Li CL, Li DS, Guo SW (2017) TET1 exerts its tumour suppressor function by regulating autophagy in glioma cells. Biosci Rep 37
Fullgrabe J, Lynch-Day MA, Heldring N, Li W, Struijk RB, Ma Q, Hermanson O, Rosenfeld MG, Klionsky DJ, Joseph B (2013) The histone H4 lysine 16 acetyltransferase hMOF regulates the outcome of autophagy. Nature 500:468–471
Fullgrabe J, Heldring N, Hermanson O, Joseph B (2014a) Cracking the survival code: autophagy-related histone modifications. Autophagy 10:556–561
Fullgrabe J, Klionsky DJ, Joseph B (2014b) The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol 15:65–74
Kapoor-Vazirani P, Kagey JD, Vertino PM (2011) SUV420H2-mediated H4K20 trimethylation enforces RNA polymerase II promoter-proximal pausing by blocking hMOF-dependent H4K16 acetylation. Mol Cell Biol 31:1594–1609
Ng KM, Mok PY, Butler AW, Ho JC, Choi SW, Lee YK, Lai WH, Au KW, Lau YM, Wong LY, Esteban MA, Siu CW, Sham PC, Colman A, Tse HF (2016) Amelioration of X-linked related autophagy failure in Danon disease with DNA methylation inhibitor. Circulation 134:1373–1389
Peng J, Yang Q, Li AF, Li RQ, Wang Z, Liu LS, Ren Z, Zheng XL, Tang XQ, Li GH, Tang ZH, Jiang ZS, Wei DH (2016) Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE-/- mice. Oncotarget 7:76423–76436
Petherick KJ, Williams AC, Lane JD, Ordonez-Moran P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik K, Paraskeva C, Greenhough A (2013) Autolysosomal beta-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J 32:1903–1916
Shin HJ, Kim H, Oh S, Lee JG, Kee M, Ko HJ, Kweon MN, Won KJ, Baek SH (2016a) AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 534:553–557
Shin HR, Kim H, Kim KI, Baek SH (2016b) Epigenetic and transcriptional regulation of autophagy. Autophagy 12:2248–2249
Sierra J, Yoshida T, Joazeiro CA, Jones KA (2006) The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev 20:586–600
Streckmann F, Balke M, Lehmann HC, Rustler V, Koliamitra C, Elter T, Hallek M, Leitzmann M, Steinmetz T, Heinen P, Baumann FT, Bloch W (2018) The preventive effect of sensorimotor- and vibration exercises on the onset of Oxaliplatin- or vinca-alkaloid induced peripheral neuropathies—STOP. BMC Cancer 18:62
Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, Hattori N, Wang D, Du Y, Song B, Cao LL, Shen C, Wang L, Yang Y, Xie D, Wang F, Ushijima T, Zhao Y, Zhu WG (2015) Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy 11:2309–2322
Wend P, Fang L, Zhu Q, Schipper JH, Loddenkemper C, Kosel F, Brinkmann V, Eckert K, Hindersin S, Holland JD, Lehr S, Kahn M, Ziebold U, Birchmeier W (2013) Wnt/beta-catenin signalling induces MLL to create epigenetic changes in salivary gland tumours. EMBO J 32:1977–1989
Yang A, Jiao Y, Yang S, Deng M, Yang X, Mao C, Sun Y, Ding N, Li N, Zhang M, Jin S, Zhang H, Jiang Y (2018) Homocysteine activates autophagy by inhibition of CFTR expression via interaction between DNA methylation and H3K27me3 in mouse liver. Cell Death Dis 9:169
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Science Press and Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Hu, LF. (2019). Epigenetic Regulation of Autophagy. In: Qin, ZH. (eds) Autophagy: Biology and Diseases. Advances in Experimental Medicine and Biology, vol 1206. Springer, Singapore. https://doi.org/10.1007/978-981-15-0602-4_11
Download citation
DOI: https://doi.org/10.1007/978-981-15-0602-4_11
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-0601-7
Online ISBN: 978-981-15-0602-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)