
Abstract
The current paper is about the properties of barium chalcogenides compounds, using the first-principle total energy calculations within linear combination of atomic orbital method. The work is basically built up using density functional theory with the coordination of CRYSTAL coding. Becke and PBE scheme was used as the exchange-correlation potential for constructing Kohn–Sham Hamiltonian. Several structural and electronic properties, such as lattice constant (a), bulk modulus (B0) and bandgap (Eg), have been studied. All the results obtained were found to be in good agreement with the earlier works.
Access provided by Autonomous University of Puebla. Download conference paper PDF
Similar content being viewed by others

Keywords
1 Introduction
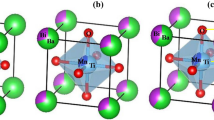
A compound semiconductor is made up of two different materials from two dissimilar groups of the periodic table. The BaX compounds belong to II-VI group. Barium chalcogenides are inorganic crystalline solid and are colourless. The present bandgap for BaS is 3.88 eV, for BaSeis 3.58 eV, and for BaTe is 3.08 eV. At ideal temperature and pressure, these compounds exhibit closed-shell ionic structural system. Also, they have shown their stability in rock salt (B1) structure.
In the past, as there was only the high production of silicon as a semiconductor material, (Earlier, silicon was largely produced as a semiconductor material while) compound semiconductors were not commercialized in a wider way. However, in recent years, semiconductor prices are reduced. Presently semiconductor compounds are commercialized in a wide manner.
Also, those compounds are having great importance through their fundamental properties, and overcome the gap between silicon and recent days’ semiconductor compounds.
As barium chalcogenides compounds like BaS, BaSe, BaTe and their various alloys, are having a short wavelength, those are used optoelectronic applications. Now it is important to understand their structural and electronic properties for their usage in all types of electronic displays.
The article is organized as various sections: In Sect. 2, literature have been explored. In Sect. 3, computational methods and in Further in segment 4, results and discussions for structural properties are presented. Finally, the conclusion has been presented in Sect. 5.
2 Literature Review
The electronic and structural properties of barium chalcogenides compounds have been calculated by Bouhemadou et al. [1]. In this work, they have two approximations i.e. Local Density Approximation (LDA) and Generalised Gradient approximation (GGA). The calculated bulk modulus and lattice constant at a stable state agree with the previous computational and experimental data. They concluded that their calculated band gaps are in the best approximation with other computational values but slightly differ with experimental ones. The electronic properties of barium chalcogenides were systematically observed by Lin et al. [2] using the correlation with density functional theory. In this paper, Cambridge serial total energy package (CASTEP) has been used for stimulation. The properties of an electronic system of the semiconductor compounds having oxygen atoms within it always differ from the compounds having no oxygen atoms within it. Their results predict that energy bandgap can be adjustable by emerging the oxygen atom into the lattice system. Tuncel et al. [3] used DFT as inbuilt in SIESTA code to determine the elastic, structural, lattice dynamical and thermodynamic properties of barium chalcogenides in rock salt and CsCl structure. In this work, the bulk modulus, the pressure derivative of bulk moduli, lattice constants, transition pressures, elastic constants, etc. have been determined and the outcoming values are, usually, closely in tune with existing experimental values and some computational data. The elastic as well as structural properties of barium chalcogenides under a high-pressure condition have been calculated by Arya et al. [4] using interionic potential of two-body problem approaches with a modified charge of ion. In B1 structural phase to B2 structural phase for the barium chalcogenides compounds, the equation of states has been predicted and observed that at a particular pressure phase transition occurs. They concluded that the determined values of transitional pressure from one phase to another phase, the percentage of volume collapsed, cohesive energies and lattice constant of the BaX compounds are in excellent agreement. It has also been shown that the cohesive energy decreases as X changes from S to Te. It suggests that BaTe is more compressible than BaS. Bhardwaj et al. [5] have calculated structural phase transition under high pressure for the BaO, BaSe, and BaTe compounds with a help of ionic interaction potential approach of a three-body system, which is a MTBIP approach and also it has been revised by integrating the covalency effects. They have assumed a zero-point energy effect. This zero-point energy can be defined by the ground state energy in which the compounds can make some band. Using some parameters from the standard model, they have obtained that the values for the phase transition pressures obtained from the TBIP approach show an excellent contract with experimental values. Also, with the variation of cation–anion radii ratio, the pressure for the phase transition from NaCl to CsCl structure varies inversely. Drablia et al. [6] have used FP-LAPW method. In this work, they have used both LDA and GGA. They have determined the various properties of barium chalcogenides compounds and in chalcogenides compound series they have used O, S, Se, Te and Po elements in the stable phase. The electronic band structure calculations predict that except BaO compound among the above compounds all are having the bandgap of indirect type, whereas the bandgap of BaO is direct. The dielectric function is having an imaginary part. That part has shown various certain interband transitions. The reflectivity has been also calculated. And the obtained values are in good contract with other previously done computational data. First-principles calculations have been used by Haj Hassan and Akbarzadeh [7] to determine the bond structures and properties related to elastic constants of the BaX compounds. This study has been done for those compounds at stable phase as well as at the phase where high pressure applied Full potential-linearized augmented plane wave (FP-LAPW) method has been performed, which is a density functional study with WEIN2K code. They have used LDA first with the combination of GGA and then again without the combination of GGA for total energy optimization with exchange-correlation functions.
BaX compounds have shown a transition from B1 phase to B2 phase under some standard condition. For investigating the properties they have utilized LDA in and without a combination of GGA. At certain different values of unit cell’s volumes, the total energy calculations have been done at the stable phase and the results were fitted with the Brich–Murnaghan’s equation of state. For the BaX compounds, the optical and electronic properties have been calculated with the DFT by Pourghazi and Dadsetani [8]. They have studied mainly NaCl (B1) crystal structure. FP-LAPW method has been used to obtain the electronic band structures of the barium chalcogenides compounds. For constructing the Hamiltonian equation, the WIEN2K code has been used. GGA scheme is used for the exchange-correlation potential treatment. The spin–orbit coupling was taking place to calculate the optical and electronic properties and then the same process had been followed without the spin–orbit coupling also to find the same properties. Also, some important parameter like dielectric function, the reflectivity, the optical absorption coefficient, and the energy loss function has been calculated. Their calculations revealed that the inclusion of the spin–orbit coupling decreases the energy band gap by about 0.27 eV, 0.24 eV and 0.16 eV for BaTe, BaSe and BaS, respectively. Benamrani et al. [9] have performed energy optimization with first-principle study within the pseudo-potential plane wave method as inbuilt in ABINIT to investigate the properties of BaX in B1 as well as B2 structures. The vibrational, elastic and dynamical properties have been determined within the LDA. By changing the volume of unit cells the dependence of the pressure with the structural, vibrational and lattice dynamical parameters have been calculated simultaneously. It was concluded in this work that the vibrational and elastic properties and also the lattice dynamics are determined with the help of DFT approach and the results for elastic constant values are in an excellent agreement with another observed data using LDA-FP-LAPW method.
3 Computational Method
Linear combination of atomic orbital, a first-principle approach has been done to determine the structural and electronic properties of barium chalcogenides compounds in this paper. For the calculations, the Kohn–Sham Hamiltonian is solved in the DFT as present in the CRYSTAL06 code. The Kohn–Sham Hamiltonian built taking into an account the Becke exchange program and PBE correlation scheme. To calculate the structural properties of barium chalcogenides the energy optimization was done by minimizing the total energy with respect to the unit cell volume for each crystal structure. The calculated results are verified with the existing results. The self-consistent calculations are used taking into account 84 k points of the Brillion zone irreducible with sufficient tolerance level. The self-consistency has been achieved by 55% mixing within nine cycles.
3.1 Result and Discussion
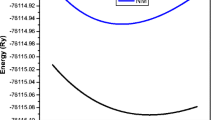
In the structural properties lattice constant and bulk modulus of BaX compounds in B1 structure as well as in B2 structure have been computed by the total energy optimization with the variation of volume of primitive cell of the crystal by LCAO method. Figures 1, 2, 3, 4, 5 and 6 represent the energy versus volume curve for the XH. In these figures, the dots represent the calculated energies through DFT and the curves represent the fitted energies to the Brich–Murnaghan equation of state, which is represent mathematically by

Energy versus volume plot (B1)

Energy versus volume plot for BaS (B2)

Energy versus volume plot for BaSe (B1)

Energy versus volume plot for BaSe (B2)

Energy versus volume plot for BaTe (B1)

Energy versus volume plot for BaTe (B2)
Here, E(V) is energy corresponds to a particular volume, Eo is equilibrium energy, Vo is the corresponding volume and Bo is bulk modulus and \(B_{\text{o}}^{{\prime }}\) is the pressure derivative of bulk modulus.
In Table 1 structural parameters are shown.
The electronic band structures of barium chalcogenides compounds have been examined in the stable state of lattice constant and are shown in Figs. 7, 8 and 9. In the Brillouin zone, W(0.5, 0.25, 0.75), L(0.5, 0.5, 0.5), ┌(0.0, 0.0, 0.0), X(0.5, 0.0, 0,0), W(0.5, 0.25, 0.75) and K(0.375, 0.375, 0.75) are the various symmetry points considered.

Plot for energy band gap of BaS

Plot for energy band gap of BaSe

Plot for energy band gap of BaTe
It has been observed from the figures that BaS, BaSe and BaTe have indirect (┌-X) bandgap.
These values are consistent with the previous calculations. The calculated results of band gaps for barium chalcogenides are given in Table 2.
4 Conclusion
In summary, LCAO method has been used to perform first-principle calculations to investigate the structural and electronic properties of the BaX compounds. It has been observed that according to total calculated energy the B1 phase for barium chalcogenides is more stable as compare to B2 phase. The calculated energy band gap for BaX are 3.72, 3.52 and 3.02 eV. The calculated structural and electronic properties for barium chalcogenides are in excellent contract with previous studies.
References
Bouhemadu, A., Khenata, R., Zegrar, F., Sahnoun, M., Baltache, H., Reshak, A.H.: Ab initio study of structural, electronic, elastic and high pressure properties of barium chalcogenides. Comput. Mater. Sci. 38, 263–270 (2006)
Lin, G.Q., Gong, Hao, Ping, Wu: Electronic properties of barium chalcogenides from first-principles calculations: tailoring wide-band-gap II-VI semiconductor. Phys. Rev. B 71, 085203 (2005)
Tuncel, E., Colakoglu, K., Deligoz, E., Ciftci, Y.O.: A first-principles study on the structural, elastic, vibrational, and thermo dynamical properties of BaX (X = S, Se, and Te). J. Phys. Chem. Solids 70, 371–378 (2009)
Arya, Balwant S., Aynyas, Mahendra, Sanyal, Sankar P.: High pressure study of structural and elastic properties of barium chalcogenides. Indian J. Pure Appl. Phys. 46, 722–726 (2008)
Bhardwaj, P., Singh, S., Gaur, N.K.: Structural and elastic properties of barium chalcogenides (BaX, X = O, Se, Te) under high pressure. Cent. Eur. J. Phys. 6(2), 223–229 (2008)
Drablia, S., Meradji, H., Ghemid, S., Boukhris, N., Bouhafsand, B., Nouet, G.: Electronic and optical properties BaO, BaS, BaSe, BaTe and BaPo compounds under hydrostatic pressure. Mod. Phys. Lett. B 23, 065–3079 (2009)
El Haj Hassan, F., Akbarzadeh, H.: First-principles elastic and bonding properties of barium chalcogenides. Comput. Mater. Sci. 38, 362–368 (2006)
Pourghazi, A., Dadsetani, M.: Electronic and optical properties of BaTe, BaSe and BaS from first principles. Phys. B 370, 35–45 (2005)
Benamrani, A., Kassali, K., Bouamama, Kh: Pseudopotential study of barium chalcogenides under hydrostatic pressure. High Press. Res. 30, 207–218 (2010)
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Singapore Pte Ltd.
About this paper

Cite this paper
Das Majumdar, A., Munjal, N., Kamboj, U., Dogra, K. (2020). Investigation of Structural and Electronic Properties of BaX (X = S, Se, and Te): A DFT Study. In: Prakash, C., Singh, S., Krolczyk, G., Pabla, B. (eds) Advances in Materials Science and Engineering. Lecture Notes in Mechanical Engineering. Springer, Singapore. https://doi.org/10.1007/978-981-15-4059-2_23
Download citation
DOI: https://doi.org/10.1007/978-981-15-4059-2_23
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-15-4058-5
Online ISBN: 978-981-15-4059-2
eBook Packages: EngineeringEngineering (R0)