
Abstract
In this paper, we have performed a systematic investigation on structural, magnetic, electronic, mechanical, and thermodynamic properties of BaPuO3 perovskite within density functional theory (DFT) using generalized gradient approximations (GGA), onsite coulomb repulsion (GGA + U), and modified Becke-Johnson (mBJ). The calculated structural parameters were found in good agreement with the experimental results. A large value of magnetic moment equal to integer value of 4 μB was obtained for the compound. The spin-polarized electronic band structure and density of states present 100% of spin polarization at Fermi level, resulting in half-metallic nature for the compound with spin-up states as metallic and spin- down states as a semiconducting. The elastic and mechanical properties have also been predicted. Moreover, we have calculated thermodynamic properties like Debye temperature (𝜃D), specific heat (CV), entropy (S), etc. using quasi-harmonic Debye model.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Recently, perovskites have been investigated with great attention both theoretically as well as experimentally in physics, chemistry, and material science because of their applications in science and technology. The perovskites especially oxides based with general formula ABO3 have attracted the researchers all over the globe as a result of their remarkable properties like multi-ferriocity, superconductivity, thermoelectricity [1], solid-oxide fuel cells [2], half-metallicity, ferromagnetism [3], etc. Investigation of new materials and their properties are key requirements, and among these, electronic, magnetic, mechanical, and thermodynamic properties are of prime interests in material science as to check the behavior of the materials for possible applications. Some of the perovskite materials with B site held by actinide-based plutonium element, MPuO3 (M = Ba and Sr), are mostly obtained from the transmutation of uranium in bulk scale from nuclear reactors. Extensive theoretical investigations on binary or ternary compounds [4,5,6,7], where f-electrons play a crucial role in investigating many physical properties have motivated us to explore these plutonium-based perovskites. There are a number of actinide-based perovskites [2, 3, 8,9,10] which have been investigated, where d-electrons and f-electrons, together with lattice made these perovskites remarkable. On the other side of BaPuO3 perovskite with barium held at A sites is fission products and constituents of many actinide ABO3 (A = Ba and Sr and B = U, Pu, Mo, and Am) perovskites called the grey phase [11].
BaPuO3 is found to be potential candidates for understanding fuel behavior by determining experimental thermodynamic properties [12, 13]. Vaporization and standard molar enthalpies of formation of BaPuO3 have been investigated by high-temperature mass spectrometry in the temperature range of 1400–1900 K [13], where BaPuO3 has been prepared by mixing PuO2 and BaCO3 [14]. Experimental determination of elastic modulus, Debye temperature by longitudinal and sound velocities, and thermal conductivity by measuring density and heat capacity by laser flash method for BaPuO3 has also been reported [15]. The structural properties of BaPuO3 have been experimentally reported in cubic structure with space group Pm-3m (221) [15]. The Wyckoff positions for Ba are 1a (0, 0, 0) O at 3c (0.5, 0.5, 0), (0.5, 0, 0.5), and (0, 0.5, 0.5) positions and Pu at 1b (0.5, 0.5, 0.5) of the cubic unit cell. As far as theoretical investigations on Perovskite family, particularly actinide based are concerned, density functional theory (DFT) has emerged as a powerful tool. DFT successfully explained structural, electronic, and thermal properties of some BaMO3 (M = Pr, Th, U) [8], BaAmO3 [16] SrUO3 [3], and SrAmO3 [10].
In the present work, the structural, electronic, magnetic, elastic, and thermodynamic properties of BaPuO3 have been reported within density functional theory. The electronic and magnetic properties have been obtained within generalized gradient approximation (GGA) [17], (GGA + U) [18], where U is Hubbard term, and also by modified Becke-Johnson (mBJ) [19]. The U term has been incorporated so as to treat the Pu-f electrons properly.
2 Computational Details
The study has been accomplished within the most accurate density functional as implemented in WIEN2K code [20, 21]. The calculations have been performed by structural optimization of the experimental lattice constant followed by electronic, magnetic, and elastic study using full-potential linearized augmented plane wave (FP-LAPW) method [22], which is completely grounded on DFT [20, 21]. The structural optimization was performed within generalized gradient approximation (GGA) scheme of Perdew, Burke, and Ernzerhof (PBE) [17]. The GGA-computed lattice constant has been used to check the magnetic and electronic nature of the compound. Further, to check the effect on our results, on-site coloumb repulsion (GGA + U) [18] and mBJ [19] were also used for electronic and magnetic studies. In order to treat the f-electrons of Pu, GGA + U calculation where used for magnetic and electronic study; for GGA + U, the density matrix was supposed to be slanting and U and J were considered the same for all interactions. There are different ways to incorporate the U term [23, 24]; here, we have made use of self-interaction correction method (SIC) [25] as implemented in WIEN2K. The value of Ueff is set to 1.8 eV and J was set to 0 so as to adjust the Pu-5f in density of states. The muffin tin radii for Ba and Pu in the calculations were selected as 2.40 and 2.30 a.u. while 1.40 a.u. was used for O atoms. The energy convergence function used RMTKmax = 7, where RMT is the smallest atomic radius in the unit cell and Kmax refers to the size of the largest k vector in the plane wave expansion. In the fullpotential scheme the unit cell of the crystal is partitioned into two different regions: (1) atomic spheres and (2) interstitial region. Within the atomic sphere the wave function is extended in atomic-like functions (radial part times spherical harmonics) while in the interstitial region it is extended in a plane wave basis. The energy separation between core and valance states was set to − 6.0 Ry. Inside the sphere the angular momentum Lmax = 10, while the charge density is Fourier expanded up to Gmax = 12 (a.u.)− 1. The self-consistent calculations converge when the difference of the two consecutive iterations is less than 10− 4 Ry. A dense mesh of 1500 K points is used, and the tetrahedral method [26] has been used for the Brillouin zone integration. For elastic constant calculation, the method developed by Charpin [27] as implemented in WIEN2K was used. In this method, the 21 elastic constants are reduced to three independent elastic constants C11, C12, and C44 by applying isotropic, rhombohedra, and tetragonal strains so that there is shape change only.
For the pressure and temperature dependence of thermodynamic parameters, quasi-harmonic Debye model [28,29,30] has been applied. The BirchMurnaghan’s [31] equation generated from GGA optimizations has been used as input to the quasi-harmonic Debye model.
3 Results and Discussion
3.1 Structural, Magnetic, and Electronic Properties
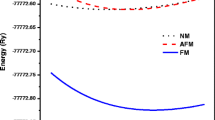
The total energy of BaPuO3 perovskite has been calculated in the framework of density functional theory within FP-LAPW method as implemented in the Wien2k code. BaPuO3 crystallizes in B2 phase space group 221(Pm-3m) with experimental lattice parameters [14] of 4.385 Å. The optimization has been done using GGA scheme and optimized data is tabulated in Table 1. The magnetic stability of the compound is checked by calculating total energy as a function of unit cell volume in nonmagnetic (NM) and ferromagnetic (FM) states as depicted in Fig. 1 It is worth to mention from the E–V curves that the FM state is the most stable state because of its least groundstate energy. The calculated total energies are fitted to BirchMurnaghan’s equation of states [31] to determine the groundstate properties like lattice parameter, bulk modulus and pressure derivatives which are listed in Table 1 together with the available experimental [14] and theoretical results [32,33,34].

Variation of total energy as a function of volume in ferro and nonmagnetic states for BaPuO3
As discussed above the compound has a ferromagnetic ground state. The optimized lattice parameter as obtained from GGA was used to check the magnetic moments of the compound. Further we have made use of the Hubbard term U incorporated with GGA and also mBJ to check the magnetic moments of the compound. The total magnetic moment which arises as the summation of the partial moments of the individual atoms and the interstitial sites is found to be 4 μB in GGA, GGA + U, and mBJ presented in Table 2. The incorporation of the U term and mBJ potentials results in the change of partial moments of Pu atoms; nevertheless, the total magnetic moment is found to be the same in all the potentials.
Band structure and the density of states (DOS) often provide the acceptable description about the electronic potential of a material. Figure 2a–c shows a spin-polarized band structure along high symmetric directions of the Brillouin zone within GGA, GGA + U, and mBJ, respectively. The Fermi level was set to 0 eV, separating the conduction and valance band. The band structures within all the three approximations show a metallic nature in the spin-up states, while a semiconducting one in the spin-down states. For the spin-up state, the Pu-f states lie on the Fermi level, making the compound metallic while as for spin-down states, these f-states of Pu are pulled deep inside the conduction band thereby generating a gap between the valance band maxima (VBM) and conduction band minima (CBM). The whole picture can be understood by taking into consideration Fig. 2a–c; the band structure plots present a metallic nature in the spin-up states while for the spin-down states, the band gap is observed and the gap varies as one changes the correlation potentials. The VBM and CBM are located at two different symmetric points Γ and R; hence, the compound will have the behavior of indirect band gap semiconductors for the spin-down states. The VBM lies at − 2.00, − 1.5, and − 1.4 eV, respectively, in GGA, GGA + U, and mBJ at Γ point, while the CBM lies on the symmetry point R at 1.4, 2.1, and 2.4 eV. The value of band gap is observed as GGA < GGA + U< mBJ. The numerical values of band gap obtained are 3.4, 3.6, and 3.8 eV, respectively, in GGA, GGA + U, and mBJ. Thus, band structure results present 100% of spin polarization at Fermi level with spin-up states as conducting and spin-down states as semiconducting one and hence the overall band structure as half-metallic.

Band structure along the high symmetry directions for spin-up and spin-down states for BaPuO3 within a GGA, b GGA + U, and c mBJ
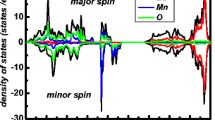
To interpret the origin of bands, the total density of states (TDOS) have been calculated within GGA, GGA + U, and mBJ. In Fig. 3, we have plotted the combined TDOS for BaPuO3 within GGA, GGA + U, and mBJ. As discussed above, the Fermi level remains occupied in the spin-up states, while for the spin-down states, the Fermi level falls in a gap, resulting into a half-metallic nature for the compound. In order to know the elemental or partial contribution of atoms to the TDOS, partial density of states (PDOS) has been evaluated by considering the spin-up and spin-down states within GGA + U as depicted in Fig. 4. From Fig. 4, it is clear that in the case of spin up, Pu-‘f’ states are present at the Fermi level making the compound metallic, while in the case of spin down, the Pu-‘f’ states are pulled inside the conduction band and hence generating a band gap.

Combined total DOS for spin-up and spin-down states for BaPuO3 within GGA, GGA + U, and mBJ

Partial density of states in up and down spins for BaPuO3 within GGA + U
Hence, the band structure and density of states (both total and partial) all present the similar results for the compound, with metallic in spin up (majority spin states) and semiconducting in spin down (minority spin states), and from these plots, we further conclude that the Pu-f states are responsible for the 100% spin polarization in this compound.
3.2 Elastic and Thermodynamic Properties
The estimation of elastic coefficients has remained a vital tool in defining the mechanical stability, nature of bounding forces, ductility, and also various thermodynamic quantities like Debye temperature, specific heat, melting temperature, etc. for a material. The value of the elastic constants Cij (C11, C12, and C44) was estimated from the strain as a function of volume [35, 36] and tabulated in Table 3. In the present study, the elastic constant were calculated by the Charpin method [27] as implemented in the Wien2k code.
For crystals having cubic symmetry, the stability criteria are given as [37]:
From the values of elastic constants (C11, C12, and C44), the mechanical properties like Young’s modulus (E), which deals with the stiffness of the material, Poisson’s ratio (ν) that helps to know the nature of the bonding forces, Bulk modulus (B) which provides the indication about the stiffness of the material, and Shear modulus (G) that helps in knowing the plastic twist of the materials have been calculated using Voigt-Reuss-Hill approximation [38, 39] and presented in Table 3.
where B is the Bulk modulus and GV and GR are Voigt’s and Reuss’s Shear moduli, respectively.
The anisotropy factor A describes the characteristic properties such as electronic and optical properties in different directions of its structure. According to such constraint, a material is isotropic if the value of A factor is less than or equal to unity or otherwise anisotropic. The value of anisotropy is calculated by [40];
The calculated values of A presented in Table 3 is less than unity and thus display anisotropic nature.
Poisson’s ratio (ν) as discussed above, describes the nature of bonding forces between the atoms, whether it is covalent, ionic, or metallic. The range of Poisson’s ratio is 0.25–0.50 [41, 42] for the materials displaying central forces or otherwise directional. The value varies from material to material. For covalent materials, it is 0.1, for ionic materials, it is 0.25, and for metallic materials, it is 0.33. In the present study, our calculated value of Poisson’s ratio is 0.30 for BaPuO3, which are close to 0.33, therefore, suggesting a higher metallic behavior. The B/G ratio provides the evidence on ductility and brittleness of a material. According to Pugh [43], a solid behaves as brittle if the B/G ratio < 1.75 and ductile if B/G ratio > 1.75. The calculated value of B/G ratio is higher than the limit value, thus will show ductile nature. Further the Cauchy pressure (C12–C44) offers the criteria for the ductility and brittleness of a material. Its positive value characterizes a material as ductile and negative value as brittle. Cauchy pressure was found to be positive and hence confirm the ductile nature as presented by B/G ratio
Debye temperature, 𝜃D is one of the key thermodynamic factors, helps to figure out the specific performance of the solids like behavior of heat capacity and thermal expansion and also provides the knowledge about the features of a material on the application of temperature. One of the classic methods to figure out Debye temperature is projected from the average sound velocity, vm by the following equation [44]
where h is the Plank’s constant, KB is the Boltzmann’s constant Va is the average atomic volume, and vm average wave velocity, given by the following relation:
where vl and vt are are longitudinal and transverse elastic wave velocities and determined by the following relations [44]:
The predicted values of Debye temperature for BaPuO3 from the elastic data is 339.74 K. From the information of elastic constants another essential thermodynamic factor namely, melting temperature has also been calculated using (9) [45];
The calculated value of elastic constants and mechanical and thermal properties including Debye temperature are grouped in Table 3.
3.3 Thermodynamic Properties
As discussed in Section 2, the thermodynamic investigation has been carried out within the quasi-harmonic Debye approximation [28,29,30]. Thermodynamic properties help to evaluate some important features of a material like its behavior under temperature. We have calculated thermodynamic properties like specific heat, thermal expansion, entropy, and Debye temperature in the temperature range of 0 to 700 K and pressure range of 0 to 40 GPa. The pressure and temperature dependence of volume for BaPuO3 is illustrated in Fig. 5. From Fig. 5, the volume is found to increases with increasing temperature, while a decrease in volume is observed as the pressure is increased. Such changes in volume with temperature and pressure are almost a common trend in solids. No anomaly is found in the V–T curve during calculations. We have computed specific heat at constant volume (CV) as depicted in Fig. 6. One can see from this figure that CV increases rapidly under the lower temperature range of 0 to 300 K, but above 300 K, a sluggish increase in CV can be observed, which further becomes constant at temperature of 700 K and reaches to Dulong-Petit limit [46]. The calculated value of CV for BaPuO3 at 300 K was found to be 106 J mol− 1 K. The term “entropy (S)” provides an insight into thermal energy of the system. Figure 7 shows the variation of entropy with pressure and temperature. As one can notice, it increases with increasing temperature because as temperature increases, the thermal vibrations increase, on the other hand, a clear decrease in S can be seen as pressure is decreased, with increasing pressure the thermal vibrations tend to seize. The value of entropy for BaPuO3 was calculated to be 102 J mol− 1 K under ambient conditions.

Unit cell volume as a function of temperature and pressure for BaPuO3

Variation of specific heat at constant volume as a function of temperature and pressure for BaPuO3

Entropy as a function of temperature and pressure for BaPuO3
Thermal expansion α, which results due to motion of the atoms or molecules, describes the tendency of expansion of a material on the application of heat. Figure 8 shows the variation of thermal expansion with temperature and pressure for BaPuO3. It is clear from the figure that α increases rapidly up to 300 K and then a tedious increase can be noticed for a particular pressure value. Alternatively, it decreases with increasing pressure at a particular temperature value. The value of α under ambient conditions is calculated to be 1.50 × 10− 5 K− 1.

Variation of thermal expansion co-efficient as a function of temperature and pressure for BaPuO3
Finally, the dependence of Debye temperature on pressure and temperature is depicted in Fig. 9. As discussed in Section 3.1, Debye temperature is the key thermodynamic parameter that describes the specific performance of solids under the application of pressure and temperature. Figure 9 clearly shows that 𝜃D decreases with increasing temperature; nevertheless, an increase in the value of 𝜃D is seen with increasing pressure. The similar variation in Debye temperature under temperature and pressure is found in SrUO3, SrAmO3, and BaAmO3 elsewhere [3, 10, 16]. By considering the above properties of the materials, we can say that our paper may open a scope for future scientific and technological applications for BaPuO3.

Variation of Debye temperature as a function of temperature and pressure for BaPuO3
4 Conclusion
For first time, the electronic, magnetic, mechanical, and thermodynamic properties of BaPuO3 have been carried out within density functional theory. The compound shows 100% spin polarization for electronic studies and hence show half-metallic nature, with spin-up conducting and spin-down semiconducting. The total magnetic moment was found to be 4 μB with a major contribution from the actinide Pu-based element. Further, elastic constants (C11, C12, and C44) and mechanical properties like Young’s modulus, Bulk modulus, etc. have also been reported. The thermodynamic study has been carried within a quasi-harmonic Debye model. The calculated values of Debye temperature from elastic modulus and from Debye approximation are almost similar.
References
Henrich, V.: Rep. Prog. Phys. 11, 1481 (1985)
Ali, Z., Ahmad, I., Amin, B., Maqsood, J., Afaq, A., Maqbool, M., Khan, I., Zahid, M.: Opt. Mat. 33, 553 (2011)
Sahli, B., Bouafia, H., Abidri, B., Abdellaoui, A., Hiadsi, S., Akriche, A., Benkhettou, N., Rached, D.: J. Alloys Compd. 635, 163 (2015)
Szytuła, A.: Mater. Sci. Pol. 24, 01 (2006)
Duan, C.-G., Sabirianov, R.F., Mei, W.N., Dowben, P.A., Jaswal, S.S., Tsymbal, E.Y.: J. Phys. Condens. Matter 19, 315220 (2007)
Srivastava, V., Sanyal, S.: J. Alloys Comp. 366, 15 (2004)
Singh, D.B., Srivasrava, V., Rajagopalan, M., Husain, M., Bandyopadhyay, A.K.: Phys. Rev. B 64, 115110 (2001)
Ali, Z., Ahmad, I., Reshak, A.H.: Physica B 410, 217 (2013)
Lebedev, A.I.: J. Alloys Compd. 580, 487 (2013)
Dar, S.A., Srivastava, V., Sakalle, U.K.: J. Supercond. Nov. Magn. 30, 3055 (2017)
Tanaka, K., Sato, I., Hirosawa, T., Kurosaki, K., Muta, H., Yamanaka, S.: J. Nuc. Sci. Techn. 52, 1285 (2015)
Matsui, T.S.: Thermo. Chimica. Acta 253, 155 (1995)
Nakajime, K., Arai, Y., Suzuki, Y., Yamawaki, M.: J. Mass Spectrom. Soc. 47, 46 (1999)
Russell, L.E., Harrison, J.D.L., Brett, N.H.: J. Nucl. Mater. 2, 310 (1960)
Tanaka, K., Sato, I., Hirosawa, T., Kurosaki, K., Muta, H., Yamanaka, S.: J. Nucl. Mater. 414, 316 (2011)
Dar, S.A., Srivastava, V., Sakalle, U.K., Khandy, S.A., Gupta, D.C.: J. Supercond. Nov. Magn. 31, 141 (2018)
Perdew, J.P., Burke, K., Ernzerhof, M.: Phys. Rev Lett. 77, 3865 (1996)
Dar, S.A., Srivastava, V., Sakalle, U.K., Parey, V., Pagare, G.: Mater. Res. Express 4, 106104 (2017)
Tran, F., Blaha, P.: Phys. Rev. Lett. 102, 226401 (2009)
Blaha, P., Schwarz, K., Madsen, G.K.H., Kuasnicke, D., Luitz, J.: Introduction to WIEN2K, an Augmented Plane Wane Plus Local Orbitals Program for Calculating Crystal Properties. Vienna University of Technology, Vienna (2001)
Schwarz, K., Blaha, P., Madsen, G.K.H.: Comp. Phys. Commun. 147, 71 (2002)
Wu, Z., Cohen, R.E.: Phys. Rev. B 73, 235116 (2006)
Petukhov, A.G., Mazin, I.I.: Phys. Rev. B 67, 153106 (2003)
Novak, P., Kunes, J., Chaput, L., Pickett, W.E.: Phys. Status Solidi B 243, 563 (2006)
Aisimov, V.I., Solovye, I.V., Korotin, M.A., Czyzyk, M.T., Sawatzky, G.A.: Phys. Rev. B 48, 16929 (1993)
Monkhorst, H.J., Pack, J.D.: Phys. Rev. B 13, 5188 (1976)
Charpin, T.: A Package for Calculating Elastic Tensors of Cubic Phases Using WIEN: Laboratory of Geometrix F-75252 (Paris, France) (2001)
Blanco, M.A., Pendas, A.M., Francisco, E.J.: J. Mol. Struct. THEOCHEM 268, 245 (1996)
de la Roza, O., Abbasi-Perez, D., Luaea, V.: Comput. Phys. Commun. 182, 2232 (2011)
de la Roza, O., Luaea, V.: Phys. Rev. B 84, 184103 (2011)
Birch, F.: J. Appl. Phys. 9, 279 (1938)
Verma, A.S., Jindal, V.K.: J. Alloys Comp. 485, 514 (2009)
Verma, A.S., Kumar, A.: J. Alloys Comp. 541, 210 (2012)
Jiang, L.Q., Guo, J.K., Liu, H.B., Zhu, M., Zhou, X., Wu, P., Le, C.H.: J. Phys. Chem. Solids 67, 1531 (2006)
Dar, S.A., Srivastava, V., Sakalle, U.K., Khandy, S.A.: J. Supercond. Nov. Magn. (2017) https://doi.org/10.1007/s10948-017-4365-1
Dar, S.A., Srivastava, V., Sakalle, U.K.: J. Electron. Mater. 46, 6870 (2017)
Sinko, G.V., Smirnov, N.: J. Phys. Condens. Matter 14, 6989 (2002)
Hill, R.: Proc: Phy. Soc. Lond. 65, 349 (1952)
Reuss, A., Angew, Z.: Mater. Phys. 9, 49 (1929)
Tvergaard, V., Hirtchinson, J.W.: J. Am. Ceram. Soc. 71, 157 (1988)
Pertifor, D.G.: Mater. Sci. Technol. 8, 345 (1992)
Haines, J., Leger, J.M., Bocquillon, G.: Annu. Rev. Mater. Sci. 31, 1 (2001)
Pugh, S.F.: Philos. Mag. 45, 823 (1954)
Schreiber, E., Anderson, O.L., Soga, N.: Elastic Constants and Measurements. McGraw-Hill, New York (1973)
Bencherif, K., Yakoubi, A., Della, N., Abid, O.M., Khachai, H., Ahmad, R., et al.: J. Elec. Mater. 45, 3479 (2016)
Petit, A.T., Dulong, P.L.: Ann. Chim. Phys. 10, 395 (1819)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Dar, S.A., Srivastava, V., Sakalle, U.K. et al. Insight into Structural, Electronic, Magnetic, Mechanical, and Thermodynamic Properties of Actinide Perovskite BaPuO3. J Supercond Nov Magn 31, 3201–3208 (2018). https://doi.org/10.1007/s10948-018-4574-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10948-018-4574-2