
Abstract
Genome-wide association studies (GWAS) have facilitated a substantial and rapid rise in the number of confirmed genetic susceptibility variants for type 2 diabetes (T2D) and glycemic traits. Approximately 90 variants for conferring susceptibility to T2D and 80 variants for glycemic traits have been identified until the end of 2016. This success has led to widespread hope that the findings will translate into improved clinical care for the increasing numbers of patients with diabetes. Potential areas or clinical translation include risk prediction and subsequent disease prevention, pharmacogenomics, and the development of novel therapeutics. In contrast, worldwide efforts to identify susceptibility loci to diabetic nephropathy have not been successful so far, and most of heritability for diabetic nephropathy remains to be elucidated. Uncovering the missing heritability is essential to the progress of T2D genetic studies and to the translation of genetic information into clinical practice.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
4.1 GWAS for Type 2 Diabetes
More than 400 million people are affected by diabetes mellitus worldwide, and the number of patents is estimated to rise to more than 600 million by 2040 [1]. Increasing prevalence of diabetes is a serious concern in many countries. Of the total global diabetes rate, 90% are living with type 2 diabetes (T2D), which is characterized by insulin resistance in peripheral tissues and impairments of insulin secretion from pancreatic β-cells. Although the current rise in T2D prevalence is explained mainly by changes in life-style, complex genetic determinants are widely considered to contribute to an inherent susceptibility to this disease [2,3,4,5]. A sibling relative risk of T2D was reported to be approximately 2 [4], and its heritability has been estimated at 30–70% [5]. Like other common diseases, the pathogenesis of T2D is considered polygenic, and the effects of individual genetic factors are modest by themselves [6]. Development of high-throughput genotyping technologies and statistical and computational software has allowed remarkable progress over the past decades in the research fields for genome-wide search to discover novel genetic loci for T2D susceptibility [6]. In 2007, five GWAS for T2D performed by European and American Groups identified robust susceptibility loci to European T2D, and until 2016, more than 90 T2D susceptibility loci have been identified through GWAS in different ethnic groups. The empirical threshold for statistical significance used here is p < 5 × 10−8 unless a different study-wise threshold has been applied and noted. It is also important to remember that loci are labeled by the gene(s) nearest to or functionally plausible for the association signal and that they do not necessarily explain the true functional gene responsible for the signal.
4.1.1 Genetics of T2D: Before the GWAS Era
Prior to the GWAS era, the importance of genetic factors in the etiology of T2D had been well established through family and twin studies [2,3,4,5], and the linkage analysis and candidate-gene association studies were applied as the primary approaches to identify susceptibility loci for diseases or phenotypic traits [7, 8]. Reynisdottir et al. identified segments in chromosomes 5 and 10 with suggestive linkage to T2D [8], and showed that the chromosome 10 region harbored the TCF7L2 [9]. Subsequently, the association of TCF7L2 with T2D was replicated not only in populations of European descent but also in other ethnic groups [10,11,12,13,14,15,16], including the Japanese [17, 18]. Candidate-gene association studies showed that PPARG [19] and KCNJ11 [20] were susceptibility genes for T2D. Both genes encode targets of anti-diabetes medications (thiazolidinediones and sulfonylureas, respectively) and harbor missense variants associated with T2D: P12A in PPARG and E23K in KCNJ11 [19, 20]. The successful identification of these genes encouraged the genetic study of T2D; however, these classical approaches were not recognized as suitable to identify variants that have a smaller effect on disease susceptibility. Therefore, the discovery of novel T2D susceptible loci had been challenging, and a more powerful strategy was needed to overcome this difficulty.
4.1.2 The Initial Phase of GWAS Era of T2D Genetics (2007–2008)
In 2007, GWAS for T2D was conducted in a French population composed of 661 cases and 614 controls, covering 392,935 SNP (single nucleotide polymorphism) loci. This study identified novel association signals at SLC30A8, HHEX, LOC387761, and EXT2 and validated the association at TCF7L2 previously identified through linkage analysis [21]. Shortly after the French GWAS, the Icelandic study group confirmed the association of SLC30A8, HHEX, and the newly identified CDKAL1 with T2D [22]. At the same time, three collaborating groups, the Wellcome Trust Case Control Consortium/United Kingdom Type 2 Diabetes Genetics consortium (WTCCC/UKT2D), the Finland-United States Investigation of NIDDM (FUSION), and the Diabetes Genetics Initiative (DGI), reported the consistent associations of SCL30A8, HHEX, CDKAL1, IGF2BP2, and CDKN2A/B with T2D in European populations [23,24,25]. These novel loci and two previously known variants (PPARG P12A and KCNJ11 E23K) were confirmed by multiple replication studies composed of European and non-European populations with the exception of LOC387761 and EXT2. Thus, the first round of European GWAS confirmed eight T2D susceptibility loci across multiple ethnic groups: TCF7L2, SLC30A8, HHEX, CDKAL1, IGF2BP2, CDKN2A/B, PPARG, and KCNJ11 [21,22,23,24,25]. In addition to these eight loci, the WTCCC/UKT2D study identified a strong association between FTO variants and T2D, although the effect of FTO variants on conferring susceptibility to T2D was mostly mediated through increase in body weight [26].
After the first round of European GWAS, an effort was made to increase sample size so that common variants with smaller effect sizes would be detectable. WTCCC/UKT2D, FUSION, and DGI combined their data to form the Diabetes Genetics Replication and Meta-analysis (DIAGRAM) consortium. Six additional novel loci, JAZF1, CDC123/CAMK1D, TSPAN/LGR5, THADA, ADAMTS9, and NOTCH2, were identified in a genome-wide scan comprising a substantial sample size (4549 cases and 5579 controls) and more than 2.2 million SNPs (either directory genotyped or imputed), followed by replication testing [27].
4.1.3 GWAS in Groups of East Asian Descent (2008–2011)
Over the past decades, many Asian countries have experienced a dramatic increase in the prevalence of T2D. Cumulative evidence suggests that Asians may be more susceptible than populations of European ancestry to insulin resistance and diabetes, which was thought to be due to differences in interethnic genetic inheritance [28]. Many of the association of the T2D loci identified by European GWAS, especially the first round of GWAS, have been confirmed in Japanese populations [6, 29, 30]. However, there are significant interethnic differences in the risk allele frequency or in effect sizes at several loci, which may affect the power to detect associations in these populations. For example, risk allele frequencies of TCF7L2 variants showing the strongest effect on T2D in European populations are very few in the Japanese (~5%) compared to populations of European descent (~40%) [17, 18]. Consequently, the association of TCF7L2 variants and T2D appears statistically less significant in the Japanese [17, 18]. In addition, the effects of some loci identified through European T2D GWAS were not consistent in Japanese populations [6, 29, 30]. Therefore, it is necessary to identify ethnic group-specific T2D susceptibility loci, those have not been captured by the European studies, to explain T2D heritability in populations of Asian descent.
In 2008, two independent Japanese GWAS, conducted by Millennium genome project (MHLW) and BioBankJapan (BBJ), simultaneously identified the KCNQ1 locus as a strong T2D susceptibility locus in the Japanese [31, 32]; this was the first established T2D susceptibility locus through non-European GWAS. Subsequent replication studies performed in different ethnic groups revealed that single nucleotide variants located at intron 15 of KCNQ1 had the strongest effects on conferring susceptibility to T2D in several East Asian populations [33,34,35,36]. The association of the KCNQ1 locus with T2D was replicated in European populations, but the minor allele frequencies in Europeans were considerably lower than those in East Asian populations (~7% in Europeans versus ~40% in East Asians). Thus, in contrast to TCF7L2, the attributable fraction of KCNQ1 on T2D susceptibility was relatively small in European populations. Since the KCNQ1 locus was not captured in the European studies, this finding emphasizes the importance of examining susceptibility loci in different ethnic groups. Although the two Japanese GWAS successfully identified the KCNQ1 locus, these studies had limited sample sizes at the initial stage of the genome-wide scan: MHLW, 187 T2D cases vs. 752 controls [32]; BBJ, 194 T2D cases vs. 1558 controls [31].
A Japanese GWAS of a larger sample size (4470 T2D vs. 3071 controls) discovered additional two T2D susceptibility loci, UBE2E2 and C2CD4A-C2CD4B in 2010 [37]. Associations between these loci and T2D were confirmed in East Asian replication study [37] and large-scale European GWAS afterward [38], suggesting GWAS for T2D using non-European as well as European populations is useful to facilitate identification of both ethnicity-specific and common-susceptibility loci among different ethnic groups.
An effort was made to increase sample size in East Asian population as well as in European combined their data to form Asian Genetic Epidemiology Network (AGEN) consortium [39]. Eight additional novel loci, GLIS3, PEPD, FITM-R3HDML-HNF4A, KCNK16, MAEA, GCC1-PAX4, PSMD6, and ZFAND3, were identified in a genome-wide scan comprising a substantial sample size (6952 cases and 11,865 controls) followed by replication testing (Stage 2 in silico replication analysis 5843 cases and 4574 controls de novo replication analysis 12,284 cases and 13,172 controls) [39].
4.1.4 GWAS with Imputation and Large-Scale Meta-Analyses (2012–)
In order to identify common variants of weaker effects, efforts have been made to increase sample size by combining association data from multiple cohorts by meta-analyses. DIAGRAM consortium has constantly developed the scale of collaboration, incremental meta-analyses (DIAGRAM+ and DIAGRAM v3) [38, 40] adding GWA data from further studies from European descent to DIAGRAM v1 data (DIAGRAM+; total of 8130 cases and 38,097 controls [40], DIAGRAM v3; total of 12,171 cases and 56,862 controls [38]) together with extensive replication have identified additional loci (12 and 8 loci, respectively).
In the meantime, four additional loci (ANK1, MIR129-LEP, GPSM1, and SLC16A11-SLC16A13) have been identified by Japanese GWAS, with increment of the sample size [41] and number of variants examined by the imputation of genotypes [29, 41]. The latest Japanese GWAS meta-analysis has identified seven additional T2D susceptibility loci (CCDC85A, FAM60A, DMRTA1, ASB3, ATP8B2, MIR4686, and INAFM2) in a genome-wide scan comprising the largest sample size in the East Asian population (15,463 cases and 26,183 controls) followed by replication testing (7936 cases and 5539 controls) [30].
Thus, larger GWAS meta-analyses combined multiple cohorts with homogeneous populations have continued to expand the number of T2D loci. In 2014, motivated by a consistency of common variant associations observed across different populations [42, 43], a trans-ethnic GWAS meta-analysis of more than 110,000 individuals, which combined GWAS data in multiple ethnic groups including European, East Asian, South Asian, and Mexican/Mexican American, has been performed [44]. Seven additional new loci for T2D susceptibility were successfully identified by combining GWAS from multiple ancestry groups, which highlighted the benefits of trans-ethnic GWAS [44].
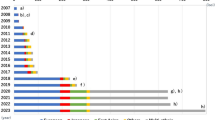
Established susceptibility loci for T2D identified by 2016 are shown in Fig. 4.1.

Established T2D susceptible loci. The x-axis shows the year of publication. Background color indicates ethnic composition of the samples in the discovery GWAS: European (blue), Japanese (red), Chinese (purple), African American (gray), East Asian (orange), South Asian (green), trans-ethnic (yellow), and Inuit (pink)
4.1.5 What Have T2D GWAS Brought About So Far?
4.1.5.1 Identified Loci for T2D Linked More Frequently to β-Cell Function than to Insulin Sensitivity
The etiology of T2D is a combination of β-cell dysfunction and insulin resistance, which is promoted by either genetic or environmental factors (e.g., obesity, Westernized diet, and lifestyle). Interestingly, majority of the known T2D susceptible variants appear to influence insulin secretion rather than insulin resistance. For example, large meta-analysis from DIAGRAM+ demonstrated that of 31 confirmed T2D susceptibility loci, 10 (MTNR1B, SLC30A8, THADA, TCF7L2, KCNQ1, CAMK1D, CDKAL1, IGF2BP2, HNF1B, and CENTD2) were nominally associated with reduced homeostasis model assessment of β-cell function (HOMA-β) which estimates steady-state β-cell function and only 3 (PPARG, FTO, and KLF14) were associated with HOMA of insulin resistance (IR)(HOMA-IR), an indicator for insulin resistance [40]. Consistent result was observed in larger study afterward [45]. Moreover, the loci identified in the early phase of Japanese GWAS, namely, KCNQ1, UBE2E2, C2CD4A-C2CD4B, and ANK1 were shown to be associated with decreased β-cell function in nondiabetic control groups [29, 32, 34, 37]. Prior to the accumulation of GWAS data, a genetic predisposition to insulin resistance had been considered to play dominant roles in development of T2D, especially in populations of European origin [40]. The results obtained from GWAS, however, emphasize the crucial role of the pancreatic β-cell in the onset of T2D, and a genetic predisposition to reduced β-cell function may contribute more to the susceptibility to T2D.
4.1.5.2 Missing Heritability
GWAS have successfully identified novel T2D susceptibility loci that had not been captured by classical approaches. However, it has been estimated that only ~10% of the known T2D heritability could be explained by those T2D susceptibility loci [38, 46]. Because polygenic analyses in the European ancestry GWAS have suggested many more common variant loci not yet reaching genome-wide significance could contribute to the heritability of T2D susceptibility [38, 46], residual genetic variance explained by a long tail of common variant signals of lesser effect could be captured in larger-scale analyses of various individual ethnic populations or trans-ethnic meta-analysis. The rationale of GWAS is based on the “common disease-common variant” hypothesis, and studies have focused on finding common variants associated with the disease; therefore, susceptibility variants having a minor allele frequency (MAF) of less than 1% are frequently missed, with limited exceptions [47, 48]. It has been a matter of considerable debate whether low-frequency risk variants, which could be evaluated by next generation sequencing and may have relatively large effects, could explain the missing heritability. To test this hypothesis, the GoT2D and T2D-GENES consortia performed whole-genome sequencing (WGS, n = 2657) and whole-exome sequencing (WES, n = 12,940) with 26.7 million variants, including 4.16 million low frequency (0.5 <MAF <5%) or 6.26 million rare (MAF <0.5%) variants. The results indicated variants associated with T2D after sequencing were overwhelmingly common (MAF >5%); therefore they concluded that this sequencing analysis did not support the idea that lower-frequency variants have a major role in predisposition to T2D [49], although sample sizes for initial WGS/WES were considered too small for rare variants analyses.
4.1.6 Translation of T2D Genetics into Clinical Practice
4.1.6.1 The Possibility of Disease Prediction and Prevention
One of the most anticipated clinical uses of genetic information is to predict an individual’s risk of developing T2D. Indeed, genetic investigations suggested lifestyle intervention arm of the Diabetes Prevention Program (DPP) attenuated genetic risk defined by carrying TCF7L2 risk allele [10] or GRS constructed with 34 confirmed loci attenuated risk of developing diabetes [50]; these are good examples of the clinical usefulness of genetic testing to allow detection of high-risk individuals with whom physicians should aggressively intervene. Since the discovery of multiple T2D risk genetic variants, genetic risk score (GRS) calculated based on the number of risk alleles in subjects who developed disease has become a common approach to indicate individual’s genetic risk. Our study group examined the utility of GRS based on 49 established T2D loci (GRS-49) in the Japanese (Fig. 4.2) [51]. GRS-49 was significantly associated with T2D risk in a Japanese population, and those with a GRS ≥60 (5.7% of the population examined) were 9.81 times as likely to have type 2 diabetes compared with those with a GRS <46 (4.2% of the population examined) (Fig. 4.2b) [51]. The result suggested even though the impact of each T2D susceptibility locus was very small, accumulation of genetic information was useful to detect a high-risk group for the disease in a population. However, the area under the receiver operating characteristic (ROC) curves for GRS was 0.624, and the effect of adding GRS into clinical factor (age, sex, and BMI) was as small as 0.03 even though the incremental effect was statistically significant (Fig. 4.2c) [51]. The performance of genetic prediction models using GRS has been evaluated in over 30 studies including Asian and European with case-control study sets or prospective cohorts [52]. The results were consistent among these studies including ours [51]: AUCs of genetic information alone for T2D were 0.579–0.641 and incremental predictive performance of T2D using established marker is statistically significant but limited [52]. Insufficient information is available to construct a genetic risk score for T2D because of so-called missing heritability, and it is far from translating into clinical practice at present. Identification of causal variants, epigenetic modifications, gene-gene interactions, and gene-environment interactions as well as uncovering residual T2D susceptible genetic variants may improve the clinical utility of genetic information for T2D prediction [52].

Evaluation of a genetic risk score (GRS) constructed by summing up the number of risk alleles for GWAS-derived 49 single nucleotide variants (SNVs) in 4399 Japanese participants [51]. (a) Distribution of the number of risk alleles in patients with T2D (black bars, n = 2613) and controls (white bars, n = 1786). (b) Odds rates (ORs) for individual groups with different number of T2D risk alleles relative to the reference group having 37–45.5 risk alleles. The vertical bars represent 95% confidence intervals. (c) Receiver Operating Characteristic plot for model 1 containing GRS (black line, area under the curve (AUC) = 0.624); model 2 containing sex, age, and body mass index (BMI) (broken line, AUC = 0.743); and model 3 containing GRS, age, sex, and BMI (gray line; AUC = 0.773)
4.1.6.2 The Possibility of Identifying Novel Biological Mechanisms and Therapeutic Targets
Because GWAS is a biology-agnostic method to detect genetic variations that predispose to a disease, the results may contribute to identify novel biological mechanisms, which may lead to discover novel therapeutic targets for T2D. However, uncovering underlying molecular mechanisms by which the loci contribute to susceptibility to type 2 diabetes has been behind, compared with GWAS discovery. A major obstacle is that the causal variants and molecular mechanisms for diabetes risk are unknown in the most of the identified T2D susceptibility loci. Furthermore, most genetic risk variants are found in the intronic or noncoding regions of genes and are more likely to affect regulation of transcription rather than gene function. Thus, it has been challenging to elicit novel biological insight, which may uncover the disease pathogenesis and guide drug discovery from GWAS derived genetic information.
To identify biological candidate for causal genes at established T2D risk loci systematically, our study group utilized an in silico pipeline, originally developed by Okada et al. [53], using various publicly available bioinformatics methods based on (i) functional annotation, (ii) cis-acting expression quantitative trait loci, (iii) pathway analyses, (iv) genetic overlap with monogenic diabetes, (v) knockout mouse phenotypes, and (vi) PubMed text mining [30]. Seven genes (PPARG, KCNJ11, ABCC8, GCK, KIF11, GSK3B, and JUN) were identified as potential drug targets for T2D treatment by integrating disease-associated variants with diverse genomic and biological datasets and subsequent drug target search (Fig. 4.3) [30]. Of these, PPARG, KCNJ11, and ABCC8 have been well known as targets for the already approved T2D treatment options, and a GCK activator is currently undergoing clinical trials for the T2D treatment. Thus, this in silico pipeline was capable to detect drug target of established T2D treatment suggesting the capability for developing novel T2D treatment. Inhibitors for KIF11, GSK3B, and JUN were under clinical trial for the treatments of cancers (KIF11, GSK3B) or rheumatoid arthritis (JUN); these compounds could also be potential treatments for T2D [30]. Thus, systematic approaches for integrating the findings of genetic, biological, and pharmacological studies could be a useful strategy for developing new T2D treatments.

Discovery of potential drug targets for the treatment of T2D [30]. (a) Strategy for drug targets search based on the genetic information derived from GWAS. Biological T2D risk genes were selected from among the T2D potential risk genes located in any of the established T2D risk loci, using a scoring system by summing up the number of prioritization criteria satisfied. We selected novel T2D therapeutic targets from the overlapping genes between the drug target genes and the biological T2D risk genes or genes those products are in direct PPI with the biological T2D risk gene products. (b) Representative connections between T2D risk SNVs (blue), T2D biological genes (green), drug target genes (purple), and targeted drugs. ∗ Approved compounds for T2D treatment ∗∗Compounds for T2D treatment under clinical trial ∗∗∗ Compounds under clinical trial for treatment against diseases other than T2D
4.2 GWAS of Metabolic Traits
The etiology of T2D is characterized by reduced insulin secretion due to impaired beta-cell dysfunction and the presence of insulin resistance. The heritability of insulin secretion, peripheral insulin action, and nonoxidative glucose metabolism has been investigated in young and old Danish twins and was estimated that 75–84%, 53–55%, and 48–50% were attributed to genetic factor, respectively, showing that there is a strong genetic component in the etiology of these traits [54]. As a result, we would expect to find genetic loci associated with these traits through non-hypothesis-driven GWAS, and see new loci, which we would not have known to be implemented in these traits. As GWAS for type 2 diabetes have been successful in identifying many susceptibility loci (please see the section described above), so has been the case for insulin secretion and action. There are many kinds of metabolic traits such as lipid, adiponectin, and leptin levels that play an important role in the metabolism of type 2 diabetes. Here we will focus on genetic loci reported for fasting glycemic traits, including fasting glucose and insulin, proinsulin, and hemoglobin A1c (HbA1c).
4.2.1 GWAS of Common Variants for Glycemic Traits
4.2.1.1 European Studies
Before the advent of the GWAS era, a few loci were demonstrated to be influencing fasting glucose level in healthy individuals. Using candidate gene approach, association studies identified variants in three genes, GCK, G6PC2, and GCKR [55,56,57,58], unequivocally implemented in fasting glucose level. The first GWAS to report genetic loci for diabetes-related quantitative traits was conducted on HbA1c level. Pare et al. evaluated 337,343 SNPs in 14,618 nondiabetic women of Caucasian ancestry in the Women’s Genome Health Study [59]. In addition to confirming the HbA1c association at GCK and G6PC2, they identified a novel locus at HK1. Another locus, SLC30A8, which was known for its association with T2D reached border-line genome-wide significance (p = 9.8 × 10−8).
The Meta-Analyses of Glucose and Insulin traits Consortium (MAGIC) investigators undertook a series of GWAS on fasting glycemic traits in nondiabetic individuals and succeeded in identifying several genetic loci (Fig. 4.4). By 2011, their efforts led to the discovery of 16 loci for fasting glucose level (known G6PC2, MTNR1B, GCK, GCKR, SLC30A8, TCF7L2; recently reported DGKB-TMEM195; novel ADCY5, MADD, ADRA2A, CRY2, FADS1, GLIS3, SLC2A2, PROX1, and C2CD4B), 2 for fasting insulin level/HOAM-IR (known GCKR and novel IGF1), 5 for postoral glucose tolerance test (OGTT) (GIPR, GCKR, ADCY5, TCF7L2, C2CD4A/B), 10 for proinsulin level (MADD, SLC30A8, TCF7L2, C2CD4A/B, PCSK1, ARAP1, LARP6, SGSM2; body mass index (BMI) adjusted locus SNX7, women-specific locus DDX31), and 10 for HbA1c (known HK1, MTNR1B, GCK, G6PC2; novel SPTA1, FNK3, HFE, TMPRSS6, ANK1, APT11A/TUBGCP3) [60,61,62,63]. This brought the number of loci associated with one or more glycemic traits to 31. These studies highlighted several important biological pathways involved in glucose and insulin metabolism, such as signal transduction, cell proliferation, development, glucose-sense, and circadian regulation. It also demonstrated that on one hand, studying genetics of glycemic trait can help identify T2D risk loci but, on the other hand, that not all loci associated with glycemic traits in healthy population (with glucose level in the “physiological” range) affect the risk of T2D (with glucose level in the “pathological” range), showing that there are un-overlapping mechanisms between fasting glucose elevation and development of T2D.

Schematic view of the >80 established loci for fasting glycemic traits, including fasting glucose, insulin, proinsulin, and HbA1c
MAGIC investigators extended their effort by increasing the sample size for discovery GWAS from 46,186 to 133,010 nondiabetic participants and incorporating Illumina CardioMetabochip, a custom iSELECT array of ~200 k SNPs that covers putative association signals for a wide range of cardiometabolic traits and fine-maps established loci [64]. This approach identified 41 novel loci associated with glycemic traits, raising the number of loci associated with fasting glucose level to 36, fasting insulin to 19 and 2 h postprandial glucose (2hGlu) to 9 (Fig. 4.4). The large increase in the number of insulin-associated loci (from 2 to 19) was partly owing to the incorporation of analyses with and without adjustment for BMI [64]. The authors speculated that because BMI explained more of the variance in fasting insulin level than in fasting glucose (R2 32.6% vs. 8.6%), BMI adjustment provided more opportunity to detect true genetic associations for fasting insulin level by removing the variance in insulin level influenced by BMI. These loci affecting fasting insulin concentration showed association with lipid levels and fat distribution, suggesting impact on insulin resistance. Of the total 53 glycemic loci, 33 were also associated with increased risk of T2D (q < 0.05). Although the overlapping loci between glycemic traits and T2D were increased, the overlap was incomplete and many glycemic loci had no discernible effect on T2D (Fig. 4.5) [64].

Per-allele β coefficients for glucose and insulin concentrations versus ORs for T2D (reproduced from Scott et al. [64]). (a) Fasting glucose concentration versus type 2 diabetes (T2D). (b) Fasting insulin (FI) concentration versus T2D. (c) Fasting insulin concentration adjusted for body mass index (BMI) versus T2D. (d) 2-hour glucose versus T2D. Each locus is color-coded according to the strength of association with T2D as indicated in (a)
From a similar point of view with the BMI adjusted analysis undertaken by MAGIC investigators, Manning et al. implemented a joint meta-analysis approach to test associations with fasting insulin and glucose concentration accounting for variant by BMI interaction on a genome-wide scale [65]. Six previously unknown loci associated with fasting insulin at genome-wide significance were identified (COBLL1-GRB14, IRS1, PPP1R3B, PDGFC, LYPLAL1, and UHRF1BP1).
To characterize the known 37 T2D loci and examine the relationship with indices of proinsulin processing, insulin secretion, and insulin sensitivity, MAGIC investigators combined data on both basal and dynamic measures to perform cluster analysis [45]. This analysis highlighted clusters characterized by (i) primary effects on insulin sensitivity (PPARG, KLF14, IRS1, GCKR), (ii) reduced insulin secretion and fasting hyperglycemia (MTNR1B, GCK), (iii) defects in insulin processing (ARAP1), (iv) influence on insulin processing and secretion without a detectable change in fasting glucose level (TCF7L2, SLC30A8, HHEX/IDE, CDKAL1, CDKN2A/B), and (v) unclassified (20 loci).
4.2.1.2 Studies Conducted in Non-European Population
GWAS on glycemic traits in non-European population was conducted around the world. In 2011, a large-scale GWAS meta-analysis on metabolic traits was conducted in East Asian population, identifying one novel locus for fasting glucose at SIX2-SIX3 [66]. GWAS in African Americans identified novel loci for insulin and insulin resistance assessed by Homeostasis Model Assessment for Insulin Resistance (HOMA-IR) at SC4NOL and TCERG1L [67]. More recent GWAS in East Asians detected several novel loci for glycemic traits: C12orf51, PDK1-RAPGEF4, KANK1, IRS1 for fasting glucose; MYL2, C12orf51, OAS1 for 1-2hGlu; TMEM79, HBS1L/MYB, MYO98, CYBA for HbA1c [68,69,70]. Among these novel loci, IRS1 and C12orf51 were associated with T2D [38, 71]. GWAS in an isolated Inuit population in Greenland has been successful in identifying a common variant in TBC1D4 associated with higher 2hGlu, 2 h-insulin, 2 h-C-peptide, and reduced insulin sensitivity index [72]. This variant was common (minor allele frequency (MAF) 17%) in Greenlandic population, but almost absent in any other population. Homozygous carriers of this TBC1D4 variant had unprecedentedly high risk of T2D (OR = 10.3).
4.2.1.3 Exome-Wide Association Analyses for Glycemic Traits
GWAS for fasting glucose and fasting insulin have identified several common variant loci associated with the traits. However, lead SNPs at GWAS loci have relatively modest effect and explain only a small portion of the variance (4.8% and 1.2%, respectively) [73]. The Illumina HumanExome Beadchip array, a custom array, was designed to facilitate large-scale genotyping of ~250 k mostly rare (MAF <0.5%) and low-frequency (MAF 0.5–5%) protein altering variants selected from sequenced exomes and genomes of ~12,000 individuals. Analyses using this Exomechip have enabled not only to identify novel loci for glycemic traits, but also to clarify the effector transcripts through which the association signals are exerting their effect.
The first report of exome-wide analysis revealed novel loci for low-frequency variants associated with proinsulin level. Low-frequency missense variants in KANK1 (Arg667His, MAF 2.9%) and TBC1D30 (Arg279Cys, MAF 2.0%) were associated with proinsulin level. Missense variants in PAM (Asp563Gly, MAF 5.3%) and the neighboring PPIP5K2 (Ser1228Gly, MAF 5.3%) were associated with insulinogenic index [74]. These two missense variants are significantly associated with T2D and are indistinguishable [75, 76]. Exome array analysis identified two low-frequency missense variants in known GWAS signal for fasting proinsulin concentration, which were independent of the known GWAS SNPs. One was Arg766X (MAF 3.7%) in MADD and the other was Val996Ile (MAF 1.4%) in SGSM2, demonstrating that these two genes were the likely effector transcripts at these loci [74]. Nominal p < 4.46 × 10−8 was used as statistical significance in this analysis, correcting for the number of tests (number of phenotypes multiplied by number of variants tested) conducted [74].
MADD locus was initially identified through GWAS for proinsulin and resides in a region of long-range linkage disequilibrium (LD) that extends >1 Mb in Europeans. Cornes et al. performed targeting deep sequencing of this 11p11.2 locus, encompassing MADD, ACP2, NR1H3, MYBPC3, and SPI1, and conducted association analysis for fasting glucose and insulin concentration using gene-based test (sequence kernel association test (SKAT)) [77]. SKAT is a useful approach to aggregate low-frequency exonic variants to test against phenotype of interest. Gene-based test at 11p11.2 locus demonstrated that 53 rare variants at NRH13 was jointly associated with fasting insulin, suggesting the existence of >2 independent signals at this locus.
Two other exome-array based analyses for fasting glucose and insulin concentration were reported at the same time. Both studies identified a low-frequency missense variant Ala316Thr (MAF 1.5%) at a novel locus GLP1R associated with fasting glucose [73, 78]. The glucose-raising allele of Ala316Thr was associated with lower early insulin secretion, higher 2hGlu concentration and risk of T2D [73]. Multiple low-frequency missense variants at G6PC2/ABCB11, a locus known for its strong association with fasting glucose, were reported in both studies. His 177Tyr, Tyr207Ser, Val219Leu (MAF 0.3%, 0.6%, 45.3%, respectively) in G6PC2 had influence on fasting glucose independently of each other as well as of the known noncoding GWAS common signal [73]. In vitro experiments showed that these missense variants were responsible for the loss of G6PC2 function through proteosomal degradation, and leads to a reduction in fasting glucose level in human [73]. Gene-based SKAT test demonstrated significant association between G6PC2 and fasting glucose level [78]. The two studies used study-wide significance based on the number of variants, genes, and tests performed. For example, one of the studies used p < 3 × 10−7 as significance threshold for single variant analysis and p < 1.6 × 10−6 for gene-based analysis.
Custom Exomechip array contains a certain proportion of noncoding common variants, including known GWAS lead SNPs, in order to facilitate conditional analyses to test evidence for multiple distinct signals at a locus. As a consequence, Exomechip analysis has led to the discovery of several novel loci for glycemic traits with common variants. Exomechip analysis identified additional loci at GPSM1 and HNF1A for Insulinogenic index [74], ABO for insulin action (disposition index) and fasting glucose [73, 74], and URB2 for fasting insulin level [73].
Currently, we are in an exciting time for the discovery of many genetic loci associated with T2D-related quantitative phenotypes. We have summarized >80 loci that have influence on fasting glycemic traits, including fasting glucose, insulin, proinsulin, and HbA1c level (Fig. 4.4). Concurrent approaches using GWAS and Exome array-based analyses have compensatory features to detect these loci. GWAS is widely performed and enables to combine a large number of samples in the meta-analysis. To date, GWAS meta-analysis for fasting glycemic traits are reported on data imputed up to the HapMap reference panel, but ongoing effort to use the latest reference panel for imputation provided by the 1000 Genomes Project will give a better coverage across the low-frequency allele spectrum and is expected to yield many more novel loci for fasting glycemic traits. For exome array-based approach, though it may have limited ability to investigate very rare variants compared to exome sequencing, it is still a cost-effective way and can be more easily performed. Importantly, we have seen proof of principle that exome array genotyping is a powerful way to detect low-frequency variant associations and to enable fine-mapping of the association loci to identify functional variants and effector transcripts through which the association is mediated. The use of these two wheels of analyses is expected to help deciphering the complex picture of the genetics of fasting glycemic traits and its relation with T2D.
4.3 GWAS for Diabetic Nephropathy or Diabetic Kidney Diseases
Diabetic nephropathy is a leading cause of end-stage renal disease (ESRD) in Western countries and Japan. The rising incidence of diabetic nephropathy, especially among patients with type 2 diabetes, is a serious worldwide concern in terms of both poor prognosis and medical costs. Up to now, strict glycemic and/or blood pressure control, protein restriction, or combination of these treatment have been shown to be effective for the prevention of the progression of diabetic nephropathy as well as for reducing cardiovascular events in patients with diabetic nephropathy [79,80,81,82]. Furthermore, remission and/or regression of microalbuminuria have also been reported [83,84,85], and thus the prognosis of subjects with diabetic nephropathy has been significantly improved during the last decade. However, still considerable numbers of subjects were suffered with diabetic nephropathy.
The pathogenesis of diabetic nephropathy appears to be multifactorial, and several environmental and/or genetic factors might be responsible for the development and progression of the disease [86], but precise mechanisms have not been elucidated yet. It has been reported that the cumulative incidence of diabetic retinopathy increased linearly according to the duration of diabetes, whereas the occurrence of nephropathy was almost none after 20–25 years of diabetes duration, and only modest number of individuals with diabetes (~30%) developed diabetic nephropathy [87]. Familial clustering of diabetic nephropathy was also reported both in type 1 [88] and type 2 diabetes [89], From these cumulative evidences, it is suggested that genetic susceptibility plays an important role in the pathogenesis of diabetic nephropathy. Worldwide efforts have been conducted to identify genes conferring susceptibility to diabetic nephropathy, but the efforts by classical approaches, i.e., candidate gene approaches or linkage analyses, have not been successful so far.
GWAS for diabetic nephropathy or diabetic kidney diseases have been performed in European, African American, and Japanese populations. However, the results were not consistent each other, and only a few loci satisfied genome-wide significant level.
4.3.1 GWAS for Diabetic Nephropathy (Diabetic Kidney Disease) in Populations of European Descent
In patients with type 1 diabetes, GWAS for diabetic nephropathy was first conducted by Genetics of Kidneys in Diabetes (GoKinD) study group using 820 cases (284 with proteinuria and 536 with end-stage renal disease) and 885 controls for ~360,000 SNPs, followed by a validation analysis using 1304 participants of the Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Interventions and Complications (EDIC) study, a long-term, prospective investigation of the development of diabetes- associated complications [90]. Four SNP loci were reported to show suggestive associations through the GWAS, rs10868025 near FRMD3 (9q21.32), rs39059 within CHN2 (7p14.3), rs451041 within CARS (11p15.4), and rs1411766/rs1742858 near MYO16/IRS2 (13q33.3). Among the four loci, association of two loci, FRMD3 and CARS, were validated in the DCCT/EDIC study, although the association did not attain genome-wide significant level. The association of the four loci were further evaluated in 66 extended families of European ancestry, the Joslin Study of Genetics of Nephropathy in Type 2 Diabetes Family Collection, the results indicated that FRMD3 locus showed evidence of association with diabetic nephropathy (advanced diabetic nephropathy or advanced diabetic nephropathy plus high microalbuminuria) or with albuminuria (log transformed albumin to creatinine ratio) [91]. The association of FRMD3 locus with diabetic end-stage renal disease was observed in African American patients with type 2 diabetes lacking two MYH9 E1 risk haplotypes, which was well-known strong risk for nondiabetic kidney diseases in African Americans [92]. In Japanese patients with type 2 diabetes, rs1411766 at ch. 13q33.3 was associated with diabetic nephropathy, and the association attained a genome-wide significant level after integration of two data, Japanese type 2 diabetes and Caucasian type 1 diabetes, by a meta-analysis [93].
In 2012, a meta-analysis of diabetic nephropathy for patients with type 1 diabetes in populations of European origin was performed by the Genetics of Nephropathy-an International Effort (GENIE) consortium [94]. The analysis using advanced diabetic nephropathy (4409 overt proteinuria or end-stage renal disease) and 6691 controls identified that rs7588550 within ERBB4 showed suggestive evidence of associated with diabetic nephropathy. In a subsequent sub-analysis for end-stage renal disease, 1786 cases and 8718 controls including patients with overt proteinuria, two loci, rs7583877 in the AFF3 (2q11.2) and rs12437854 in RGMA/MCTP2 locus (15q26.1), were associated with ESRD with a genome-wide significant level. However, these associations were not validated in independent case-control studies [95]. Genotype imputation using directly genotyped data and linkage disequilibrium data in 1000 genomes database for patients with type 1 diabetes was performed in the Finnish Diabetic Nephropathy (FinnDiane) study. The analysis for 11,133,962 tested SNPs and subsequent first and second stage analyses, comprising of 2142 cases and 2494 controls, identified rs1326934 within the SORBS1 as top signal for susceptibility to diabetic nephropathy, but the association did not reach a genome-wide significant level [96]. Sex stratified GWAS for diabetic nephropathy in European patients with type 1 diabetes identified rs4972593 on chromosome 2q31.1 as susceptibility to ESRD only in women, but not in men, and the results were replicated in independent replication studies [97].
In a GWAS meta-analysis for quantitative traits analysis regarding kidney functions in 54,450 individuals, variants within CUBN showed genome-wide significant association with urinary albumin-to-creatinine ratio (UACR), and it was also shown that an effect size on logarithmic UACR values was fourfold larger among 5825 individuals with diabetes (0.19 log[mg/g], p = 2.0 × 10−5) compared with 46,061 individuals without diabetes (0.045 log[mg/g], p = 8.7 × 10−6; p = 8.2 × 10−4 for difference) [98]. In this analysis, rs649529 at RAB38/CTSC locus on chromosome 11q14 and rs13427836 in HS6ST1 on chromosome 2q21 were associated with UACR only in patients with diabetes.
4.3.2 GWAS for Diabetic Nephropathy in the Japanese Population
In order to identify genes conferring susceptibility to diabetic nephropathy, we have performed a GWAS for diabetic nephropathy using 188 Japanese patients with type 2 diabetes [99, 100]. We commenced an association study using SNPs from a Japanese SNP database [101, 102] established prior to the HapMap database. We screened approximately 100,000 gene-based SNP loci, and the genotype and allele frequencies of 94 nephropathy cases, defined as patients with overt proteinuria or ESRD were compared with those of 94 controls defined as patients with normoalbuminuria and diabetic retinopathy. Approximately 80,000 SNP loci were successfully genotyped, and 1615 SNP loci with p < 0.01 were selected, and forwarded to the validation study. These 1615 SNP loci were analyzed further in a greater number of subjects to clarify their statistical significance. As a result, several SNP loci, including the SLC12A3 locus [103], ELMO1 locus [104], and NCALD locus [105] were found to be associated with diabetic nephropathy.
4.3.2.1 Solute Carrier Family 12, Member 3 (SLC12A3)
The SLC12A3, at chromosome 16q13, encodes a thiazide-sensitive Na + -Cl- cotransporter that mediates reabsorption of Na+ and Cl− at the renal distal convoluted tubule; this molecule is the target of thiazide diuretics. Mutations in SLC12A3 are responsible for Gitelman syndrome [106], which is inherited as an autosomal recessive trait characterized by hypokalemia, metabolic alkalosis, hypomagnesemia, hypocalciuria, and volume depletion. A coding SNP in exon 23 of the SLC12A3 (rs11643718, +78 G to A: Arg913Gln) was shown to be associated with diabetic nephropathy (p = 0.00002, odds ratio 2.53 [95% CI 1.64–3.90]). The results implicated that substitution of Arg913 to Gln in the SLC12A3 might reduce the risk to develop diabetic nephropathy. The association of rs11643718 with diabetic nephropathy was replicated in independent case-control studies, including Japanese [107], South Asian [108], and Malaysian subjects [109] with type 2 diabetes. Rs11643718 was associated with end-stage renal disease in Korean patients with type 2 diabetes, but direction of effect was opposite to that in the original report [110]. Rs11643718 did not show significant effect in Caucasian patients with type 2 diabetes (Table 4.1) [111].
4.3.2.2 Engulfment and Cell Motility 1 (ELMO1)
We identified that the ELMO1 was a likely candidate for conferring susceptibility to diabetic nephropathy (rs741301, intron 18 + 9170, GG vs. GA+AA, χ2 = 19.9, p = 0.000008, odds ratio: 2.67, 95%CI 1.71–4.16) [104]. The association of ELMO1 locus with diabetic nephropathy was observed also in African American patients with type 2 diabetes [112], Caucasian patients with type 1 diabetes [113], South Indian patients with type 2 diabetes [108], Chinese patients with type 2 diabetes [114], and American Indian patients with type 2 diabetes [115], although associated SNPs or direction of the effects varied among the individual studies. The ELMO1 gene, on chromosome 7p14, is a known mammalian homologue of the C. elegans gene, ced-12, which is required for engulfment of dying cells and for cell migration [116]. ELMO1 has also been reported to cooperate with CrkII and Dock180, which are homologues of C. elegans ced-2, ced-5, respectively, to promote phagocytosis and cell shape changes [116, 117]. However, until then no evidence had been reported to suggest a role for this gene in the pathogenesis of diabetic nephropathy. By in situ hybridization using the kidney of normal and diabetic mice, we found that ELMO1 expression was weakly detectable mainly in tubular and glomerular epithelial cells in normal mouse kidney, and was clearly elevated in the kidney of diabetic mice. Subsequent in vitro analysis revealed that ELMO1 expression was elevated in cells cultured under high glucose conditions (25 mM) compared to cells cultured under normal glucose conditions (5.5 mM). Furthermore, we identified that the expression of extracellular matrix protein genes, such as Type 1 collagen and fibronectin, were increased in cells that over-expressing ELMO1, whereas the expression of MMPs (matrix metalloproteinase) was decreased [104, 118]. Therefore, it is suggested that persistent excess of ELMO1 in subjects with disease susceptibility allele leads to the overaccumulation of extracellular matrix proteins and to the development and progression of diabetic glomerulosclerosis. It has been also reported that excess of Elmo1 accelerated the progression of renal injury in mouse model of diabetes, whereas Elmo1 depletion protected the renal injury in these mice [119]. In contrast, experiments using zebrafish suggested that elmo1 had a protective role in the progression of renal injury under diabetic conditions [120].
The association of NCALD locus with diabetic nephropathy was not replicated in an independent population, and the association of above mentioned loci identified in Japanese GWAS for diabetic nephropathy did not attain a genome-wide significant level.
4.3.2.3 Acetyl-Coenzyme a Carboxylase Beta Gene (ACACB)
We extended the previous GWAS for diabetic nephropathy to the SNPs with p values between 0.01 and 0.05 and provide evidence that a SNP, rs2268388, within the acetyl-coenzyme A carboxylase beta gene (ACACB; MIM: 601557) contributes to an increased prevalence of proteinuria in patients with type 2 diabetes across different ethnic populations [121].
The frequency of the T allele of rs2268388 was consistently higher among patients with type 2 diabetes with proteinuria (combined meta-analysis gave a p value of 5.35 × 10−8 in the Japanese, 2.3 × 10−9 for all populations). The association of rs2268388 was replicated in patients with type 2 diabetes in different ethnic groups, including Han Chinese [122] and Indians [123].
Expression of ACACB was observed in adipose tissue, heart, and skeletal muscle, and, to a lesser extent, in the kidney. The results of in situ hybridization with normal mouse kidney revealed that Acacb was localized to glomerular epithelial cells and tubular epithelial cells. We also observed the expression of ACACB in cultured human renal proximal tubular epithelial cells (hRPTECs). In cultured hRPTECs, a 29-bp DNA fragments containing the SNP region had significant enhancer activity, and fragments corresponding to the disease susceptibility allele had stronger enhancer activity than those for the major allele [121].
The quantitative real-time PCR (polymerase chain reaction) using glomeruli isolated from these mice revealed that the expression of Acacb was increased in the glomeruli of diabetic db/db mice compared to those of control mice [124]. Furthermore, overexpression of ACACB in hRPTECs resulted in remarkable increase of the expressions of genes encoding pro-inflammatory cytokines, including IL-6, CXCL1, CXCL2, CXCL5, and CXCL6.
Combining these results with the finding in the genetic study, it is suggested that ACACB contributes to conferring susceptibility to diabetic nephropathy at least in part, via the effects of the pro-inflammatory cytokines, and the ACACB-IL-6 or ACACB-CXCLs systems may be considered as new pathways for the development and progression of diabetic nephropathy.
4.3.3 GWAS for Diabetic Nephropathy in Other Ethnic Groups
An African American GWAS for diabetic nephropathy evaluated 965 ESRD patients with type 2 diabetes and control individuals without type 2 diabetes or kidney disease for 832,357 SNP loci, and in addition to MYH9-APOL1 locus, which is already known susceptibility to nondiabetic kidney diseases, several loci, RPS12, LIMK2, SFI1, were associated with ESRD in patients with type 2 diabetes, although any association did not attain a genome-wide significant level [125].
Results of multiethnic GWAS meta-analysis, including African American, American Indian, European, and Mexican, identified significant association of rs955333 at 6q25.2 with diabetic nephropathy [126].
Susceptibility loci for diabetic nephropathy or diabetic kidney disease with genome-wide significant association are listed in Table 4.2.
4.4 Future Perspective
After the human genome (sequencing) project was completed [127, 128], a large body of information on the human genome has been accumulated [129]. Simultaneously, high-throughput genotyping technologies as well as statistical methods and/or tools for handling innumerable datasets have been developed. Then, genome-wide association studies for investigating genes associated with disease susceptibility across the entire human genome have been facilitated, and more than 2000 loci susceptible to various diseases or traits have been discovered [130].
Although this is excellent progress, it has also been recognized that the information obtained from GWAS is still insufficient for clinical application. The focus of ongoing research efforts includes detailed functional characterization of the identified T2D susceptibility variants and the search for missing heritability.
Certain modifications of the GWAS study design will be necessary to uncover the missing heritability. Much larger intra- or trans-ethnic sample sizes will be required to increase the power to detect true signals, which may be conducted in meta-analyses. Examining populations of non-European descent is likely to identify additional T2D loci, and this should be performed more vigorously. Association analyses of low frequency variants for T2D are an additional option. Additionally, it has been shown that the study using small and historically isolated populations may have advantages to identify novel susceptibility to the disease [72]. In this report, GWAS for glycemic traits using a relatively small number of Greenlandic inuits (~2500) identified the nonsense variants in the TBC1D4, which had a striking effect on susceptibility to T2D (OR = ~10). Since similar success was reported to identify novel missense variants within CREBRF associated with obesity in the Samoan population [131], unique variants with a large effect size are conserved in genetically homogeneous populations, and GWAS in these populations, even if its sample size is not so large, are useful to identify novel susceptibility to T2D.
Characterizing disease biology is another relevant goal of genetic studies for T2D, which has been behind compared with GWAS discovery. Recent biological and clinical studies have suggested possible means to increase the translational use of genetic findings through convergence on common resources and workflows, regarding comprehensive gene expression data, epigenomics, PPI networks, and information of cellular and animal models [30, 53, 132]. In order to exploit these trends to advance biological understanding of T2D, it is urgent needs of establishment and effective utilization of publicly available databases including genetic data with large-scale sample size with rich phenotype information, epigenomic and transcriptomic data for diverse tissue types, and comprehensive biological data resource from cellular and animal models.
References
IDF diabetes atlas ver 7. (2015) http://www.diabetesatlas.org/
Poulsen P, Kyvik KO, Vaag A, Beck-Nielsen H (1999) Heritability of type II (non-insulin-dependent) diabetes mellitus and abnormal glucose tolerance – a population-based twin study. Diabetologia 42:139–145
Groop L, Forsblom C, Lehtovirta M, Tuomi T, Karanko S, Nissén M, Ehrnström BO, Forsén B, Isomaa B, Snickars B, Taskinen MR (1996) Metabolic consequences of a family history of NIDDM (the Botnia study): evidence for sex-specific parental effects. Diabetes 45:1585–1593
Hemminki K, Li X, Sundquist K, Sundquist J (2010) Familial risks for type 2 diabetes in Sweden. Diabetes Care 33:293–297
Almgren P, Lehtovirta M, Isomaa B, Sarelin L, Taskinen MR, Lyssenko V, Tuomi T, Groop L, Botnia Study Group (2011) Heritability and familiality of type 2 diabetes and related quantitative traits in the Botnia study. Diabetologia 54:2811–2819
Imamura M, Maeda S (2011) Genetics of type 2 diabetes: the GWAS era and future perspectives. Endocr J 58:723–739
Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Horikawa Y, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26:163–175
Reynisdottir I, Thorleifsson G, Benediktsson R, Sigurdsson G, Emilsson V, Einarsdottir AS, Hjorleifsdottir EE, Orlygsdottir GT, Bjornsdottir GT, Saemundsdottir J, Halldorsson S, Hrafnkelsdottir S, Sigurjonsdottir SB, Steinsdottir S, Martin M, Kochan JP, Rhees BK, Grant SF, Frigge ML, Kong A, Gudnason V, Stefansson K, Gulcher JR (2003) Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am J Hum Genet 73:323–325
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38:320–323
Florez JC, Jablonski KA, Bayley N, Pollin TI, de Bakker PI, Shuldiner AR, Knowler WC, Nathan DM, Altshuler D, Diabetes Prevention Program Research Group (2006) TCF7L2 polymorphisms and progression to diabetes in the diabetes prevention program. N Engl J Med 355:241–250
Groves CJ, Zeggini E, Minton J, Frayling TM, Weedon MN, Rayner NW, Hitman GA, Walker M, Wiltshire S, Hattersley AT, McCarthy MI (2006) Association analysis of 6,736 U.K. subjects provides replication and confirms TCF7L2 as a type 2 diabetes susceptibility gene with a substantial effect on individual risk. Diabetes 55:2640–2644
Zhang C, Qi L, Hunter DJ, Meigs JB, Manson JE, van Dam RM, Hu FB (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene and the risk of type 2 diabetes in large cohorts of U.S. women and men. Diabetes 55:2645–2648
Scott LJ, Bonnycastle LL, Willer CJ, Sprau AG, Jackson AU, Narisu N, Duren WL, Chines PS, Stringham HM, Erdos MR, Valle TT, Tuomilehto J, Bergman RN, Mohlke KL, Collins FS, Boehnke M (2006) Association of transcription factor 7-like 2 (TCF7L2) variants with type 2 diabetes in a Finnish sample. Diabetes 55:2649–2653
Damcott CM, Pollin TI, Reinhart LJ, Ott SH, Shen H, Silver KD, Mitchell BD, Shuldiner AR (2006) Polymorphisms in the transcription factor 7-like 2 (TCF7L2) gene are associated with type 2 diabetes in the Amish: replication and evidence for a role in both insulin secretion and insulin resistance. Diabetes 55:2654–2659
Saxena R, Gianniny L, Burtt NP, Lyssenko V, Giuducci C, Sjögren M, Florez JC, Almgren P, Isomaa B, Orho-Melander M, Lindblad U, Daly MJ, Tuomi T, Hirschhorn JN, Ardlie KG, Groop LC, Altshuler D (2006) Common single nucleotide polymorphisms in TCF7L2 are reproducibly associated with type 2 diabetes and reduce the insulin response to glucose in nondiabetic individuals. Diabetes 55:2890–2895
Cauchi S, Meyre D, Dina C, Choquet H, Samson C, Gallina S, Balkau B, Charpentier G, Pattou F, Stetsyuk V, Scharfmann R, Staels B, Frühbeck G, Froguel P (2006) Transcription factor TCF7L2 genetic study in the French population: expression in human beta-cells and adipose tissue and strong association with type 2 diabetes. Diabetes 55:2903–2908
Hayashi T, Iwamoto Y, Kaku K, Hirose H, Maeda S (2007) Replication study for the association of TCF7L2 with susceptibility to type 2 diabetes in a Japanese population. Diabetologia 50:980–984
Horikoshi M, Hara K, Ito C, Nagai R, Froguel P, Kadowaki T (2007) A genetic variation of the transcription factor 7-like 2 gene is associated with risk of type 2 diabetes in the Japanese population. Diabetologia 50:747–751
Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26:76–80
Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM (2003) Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52:568–572
Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445:881–885
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch-Johnsen K, Jorgensen T, van Vliet-Ostaptchouk JV, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Rotimi C, Gurney M, Chan JC, Pedersen O, Sigurdsson G, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39:770–775
Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Boström K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Råstam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjögren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316:1331–1336
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, Wellcome Trust Case Control Consortium (WTCCC), McCarthy MI, Hattersley AT (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316:1336–1341
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2007) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316:1341–1345
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316:889–894
Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Boström KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jørgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjögren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Wellcome Trust Case Control Consortium, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D (2008) Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40:638–645
Chan JC, Malik V, Jia W, Kadowaki T, Yajnik CS, Yoon KH, Hu FB (2009) Diabetes in Asia: epidemiology, risk factors, and pathophysiology. JAMA 301:2129–2140
Imamura M, Maeda S, Yamauchi T, Hara K, Yasuda K, Morizono T, Takahashi A, Horikoshi M, Nakamura M, Fujita H, Tsunoda T, Kubo M, Watada H, Maegawa H, Okada-Iwabu M, Iwabu M, Shojima N, Ohshige T, Omori S, Iwata M, Hirose H, Kaku K, Ito C, Tanaka Y, Tobe K, Kashiwagi A, Kawamori R, Kasuga M, Kamatani N, Diabetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium, Nakamura Y, Kadowaki T (2012) A single-nucleotide polymorphism in ANK1 is associated with susceptibility to type 2 diabetes in Japanese populations. Hum Mol Genet 21:3042–3049
Imamura M, Takahashi A, Yamauchi T, Hara K, Yasuda K, Grarup N, Zhao W, Wang X, Huerta-Chagoya A, Hu C, Moon S, Long J, Kwak SH, Rasheed A, Saxena R, Ma RC, Okada Y, Iwata M, Hosoe J, Shojima N, Iwasaki M, Fujita H, Suzuki K, Danesh J, Jørgensen T, Jørgensen ME, Witte DR, Brandslund I, Christensen C, Hansen T, Mercader JM, Flannick J, Moreno-Macías H, Burtt NP, Zhang R, Kim YJ, Zheng W, Singh JR, Tam CH, Hirose H, Maegawa H, Ito C, Kaku K, Watada H, Tanaka Y, Tobe K, Kawamori R, Kubo M, Cho YS, Chan JC, Sanghera D, Frossard P, Park KS, Shu XO, Kim BJ, Florez JC, Tusié-Luna T, Jia W, Tai ES, Pedersen O, Saleheen D, Maeda S, Kadowaki T (2016) Genome-wide association studies in the Japanese population identify seven novel loci for type 2 diabetes. Nat Commun 7:10531
Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jørgensen T, Sandbaek A, Lauritzen T, Hansen T, Nurbaya S, Tsunoda T, Kubo M, Babazono T, Hirose H, Hayashi M, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Tai ES, Pedersen O, Kamatani N, Kadowaki T, Kikkawa R, Nakamura Y, Maeda S (2008) SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40:1098–1102
Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, Yamagata K, Hinokio Y, Wang HY, Tanahashi T, Nakamura N, Oka Y, Iwasaki N, Iwamoto Y, Yamada Y, Seino Y, Maegawa H, Kashiwagi A, Takeda J, Maeda E, Shin HD, Cho YM, Park KS, Lee HK, Ng MC, Ma RC, So WY, Chan JC, Lyssenko V, Tuomi T, Nilsson P, Groop L, Kamatani N, Sekine A, Nakamura Y, Yamamoto K, Yoshida T, Tokunaga K, Itakura M, Makino H, Nanjo K, Kadowaki T, Kasuga M (2008) Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat Genet 40:1092–1097
Lee YH, Kang ES, Kim SH, Han SJ, Kim CH, Kim HJ, Ahn CW, Cha BS, Nam M, Nam CM, Lee HC (2008) Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J Hum Genet 53:991–998
Tan JT, Nurbaya S, Gardner D, Ye S, Tai ES, Ng DP (2009) Genetic variation in KCNQ1 associates with fasting glucose and beta-cell function: a study of 3,734 subjects comprising three ethnicities living in Singapore. Diabetes 58:1445–1449
Hu C, Wang C, Zhang R, Ma X, Wang J, Lu J, Qin W, Bao Y, Xiang K, Jia W (2009) Variations in KCNQ1 are associated with type 2 diabetes and beta cell function in a Chinese population. Diabetologia 52:1322–1325
Liu Y, Zhou DZ, Zhang D, Chen Z, Zhao T, Zhang Z, Ning M, Hu X, Yang YF, Zhang ZF, Yu L, He L, Xu H (2009) Variants in KCNQ1 are associated with susceptibility to type 2 diabetes in the population of mainland China. Diabetologia 52:1315–1321
Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, Horikoshi M, Nakamura M, Fujita H, Grarup N, Cauchi S, Ng DP, Ma RC, Tsunoda T, Kubo M, Watada H, Maegawa H, Okada-Iwabu M, Iwabu M, Shojima N, Shin HD, Andersen G, Witte DR, Jørgensen T, Lauritzen T, Sandbæk A, Hansen T, Ohshige T, Omori S, Saito I, Kaku K, Hirose H, So WY, Beury D, Chan JC, Park KS, Tai ES, Ito C, Tanaka Y, Kashiwagi A, Kawamori R, Kasuga M, Froguel P, Pedersen O, Kamatani N, Nakamura Y, Kadowaki T (2010) A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet 42:864–868
Morris AP, Voight BF, Teslovich TM, Ferreira T, Segrè AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Müller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stančáková A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutškov K, Langford C, Leander K, Lindholm E, Lobbens S, Männistö S, Mirza G, Mühleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurðsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvänen AC, Eriksson JG, Peltonen L, Nöthen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L, Wellcome Trust Case Control Consortium, Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC) Investigators, Genetic Investigation of ANthropometric Traits (GIANT) Consortium, Asian Genetic Epidemiology Network–Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njølstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyövälti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jöckel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI, DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44:981–990
Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, Chang YC, Kwak SH, Ma RC, Yamamoto K, Adair LS, Aung T, Cai Q, Chang LC, Chen YT, Gao Y, Hu FB, Kim HL, Kim S, Kim YJ, Lee JJ, Lee NR, Li Y, Liu JJ, Lu W, Nakamura J, Nakashima E, Ng DP, Tay WT, Tsai FJ, Wong TY, Yokota M, Zheng W, Zhang R, Wang C, So WY, Ohnaka K, Ikegami H, Hara K, Cho YM, Cho NH, Chang TJ, Bao Y, Hedman ÅK, Morris AP, McCarthy MI, DIAGRAM Consortium, MuTHER Consortium, Takayanagi R, Park KS, Jia W, Chuang LM, Chan JC, Maeda S, Kadowaki T, Lee JY, Wu JY, Teo YY, Tai ES, Shu XO, Mohlke KL, Kato N, Han BG, Seielstad M (2011) Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 44:67–72
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM, Dupuis J, Qi L, Segrè AV, van Hoek M, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Bengtsson Boström K, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Couper DJ, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jørgensen T, Kao WH, Klopp N, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Perry JR, Petersen AK, Platou C, Proença C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparsø T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van Herpt T, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Mohlke KL, Morris AD, Palmer CN, Pramstaller PP, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Wareham NJ, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly MJ, Hattersley AT, Hu FB, Meigs JB, Pankow JS, Pedersen O, Wichmann HE, Barroso I, Florez JC, Frayling TM, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI, MAGIC investigators, GIANT Consortium (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42:579–589
Hara K, Fujita H, Johnson TA, Yamauchi T, Yasuda K, Horikoshi M, Peng C, Hu C, Ma RC, Imamura M, Iwata M, Tsunoda T, Morizono T, Shojima N, So WY, Leung TF, Kwan P, Zhang R, Wang J, Yu W, Maegawa H, Hirose H, DIAGRAM consortium, Kaku K, Ito C, Watada H, Tanaka Y, Tobe K, Kashiwagi A, Kawamori R, Jia W, Chan JC, Teo YY, Shyong TE, Kamatani N, Kubo M, Maeda S, Kadowaki T (2014) Genome-wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet 23:239–246
Waters KM, Stram DO, Hassanein MT, Le Marchand L, Wilkens LR, Maskarinec G, Monroe KR, Kolonel LN, Altshuler D, Henderson BE, Haiman CA (2010) Consistent association of type 2 diabetes risk variants found in europeans in diverse racial and ethnic groups. PLoS Genet 6:e1001078
Saxena R, Elbers CC, Guo Y, Peter I, Gaunt TR, Mega JL, Lanktree MB, Tare A, Castillo BA, Li YR, Johnson T, Bruinenberg M, Gilbert-Diamond D, Rajagopalan R, Voight BF, Balasubramanyam A, Barnard J, Bauer F, Baumert J, Bhangale T, Böhm BO, Braund PS, Burton PR, Chandrupatla HR, Clarke R, Cooper-DeHoff RM, Crook ED, Davey-Smith G, Day IN, de Boer A, de Groot MC, Drenos F, Ferguson J, Fox CS, Furlong CE, Gibson Q, Gieger C, Gilhuijs-Pederson LA, Glessner JT, Goel A, Gong Y, Grant SF, Grobbee DE, Hastie C, Humphries SE, Kim CE, Kivimaki M, Kleber M, Meisinger C, Kumari M, Langaee TY, Lawlor DA, Li M, Lobmeyer MT, Maitland-van der Zee AH, Meijs MF, Molony CM, Morrow DA, Murugesan G, Musani SK, Nelson CP, Newhouse SJ, O’Connell JR, Padmanabhan S, Palmen J, Patel SR, Pepine CJ, Pettinger M, Price TS, Rafelt S, Ranchalis J, Rasheed A, Rosenthal E, Ruczinski I, Shah S, Shen H, Silbernagel G, Smith EN, Spijkerman AW, Stanton A, Steffes MW, Thorand B, Trip M, van der Harst P, van der A DL, van Iperen EP, van Setten J, van Vliet-Ostaptchouk JV, Verweij N, Wolffenbuttel BH, Young T, Zafarmand MH, Zmuda JM, Look AHEAD Research Group, DIAGRAM consortium, Boehnke M, Altshuler D, McCarthy M, Kao WH, Pankow JS, Cappola TP, Sever P, Poulter N, Caulfield M, Dominiczak A, Shields DC, Bhatt DL, Zhang L, Curtis SP, Danesh J, Casas JP, van der Schouw YT, Onland-Moret NC, Doevendans PA, Dorn GW 2nd, Farrall M, GA FG, Hamsten A, Hegele R, Hingorani AD, Hofker MH, Huggins GS, Illig T, Jarvik GP, Johnson JA, Klungel OH, Knowler WC, Koenig W, März W, Meigs JB, Melander O, Munroe PB, Mitchell BD, Bielinski SJ, Rader DJ, Reilly MP, Rich SS, Rotter JI, Saleheen D, Samani NJ, Schadt EE, Shuldiner AR, Silverstein R, Kottke-Marchant K, Talmud PJ, Watkins H, Asselbergs FW, de Bakker PI, McCaffery J, Wijmenga C, Sabatine MS, Wilson JG, Reiner A, Bowden DW, Hakonarson H, Siscovick DS, Keating BJ (2012) Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet 90:410–425
DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples (T2D-GENES) Consortium, Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MC, Prokopenko I, Saleheen D, Wang X, Zeggini E, Abecasis GR, Adair LS, Almgren P, Atalay M, Aung T, Baldassarre D, Balkau B, Bao Y, Barnett AH, Barroso I, Basit A, Been LF, Beilby J, Bell GI, Benediktsson R, Bergman RN, Boehm BO, Boerwinkle E, Bonnycastle LL, Burtt N, Cai Q, Campbell H, Carey J, Cauchi S, Caulfield M, Chan JC, Chang LC, Chang TJ, Chang YC, Charpentier G, Chen CH, Chen H, Chen YT, Chia KS, Chidambaram M, Chines PS, Cho NH, Cho YM, Chuang LM, Collins FS, Cornelis MC, Couper DJ, Crenshaw AT, van Dam RM, Danesh J, Das D, de Faire U, Dedoussis G, Deloukas P, Dimas AS, Dina C, Doney AS, Donnelly PJ, Dorkhan M, van Duijn C, Dupuis J, Edkins S, Elliott P, Emilsson V, Erbel R, Eriksson JG, Escobedo J, Esko T, Eury E, Florez JC, Fontanillas P, Forouhi NG, Forsen T, Fox C, Fraser RM, Frayling TM, Froguel P, Frossard P, Gao Y, Gertow K, Gieger C, Gigante B, Grallert H, Grant GB, Grrop LC, Groves CJ, Grundberg E, Guiducci C, Hamsten A, Han BG, Hara K, Hassanali N, Hattersley AT, Hayward C, Hedman AK, Herder C, Hofman A, Holmen OL, Hovingh K, Hreidarsson AB, Hu C, Hu FB, Hui J, Humphries SE, Hunt SE, Hunter DJ, Hveem K, Hydrie ZI, Ikegami H, Illig T, Ingelsson E, Islam M, Isomaa B, Jackson AU, Jafar T, James A, Jia W, Jöckel KH, Jonsson A, Jowett JB, Kadowaki T, Kang HM, Kanoni S, Kao WH, Kathiresan S, Kato N, Katulanda P, Keinanen-Kiukaanniemi KM, Kelly AM, Khan H, Khaw KT, Khor CC, Kim HL, Kim S, Kim YJ, Kinnunen L, Klopp N, Kong A, Korpi-Hyövälti E, Kowlessur S, Kraft P, Kravic J, Kristensen MM, Krithika S, Kumar A, Kumate J, Kuusisto J, Kwak SH, Laakso M, Lagou V, Lakka TA, Langenberg C, Langford C, Lawrence R, Leander K, Lee JM, Lee NR, Li M, Li X, Li Y, Liang J, Liju S, Lim WY, Lind L, Lindgren CM, Lindholm E, Liu CT, Liu JJ, Lobbens S, Long J, Loos RJ, Lu W, Luan J, Lyssenko V, Ma RC, Maeda S, Mägi R, Männisto S, Matthews DR, Meigs JB, Melander O, Metspalu A, Meyer J, Mirza G, Mihailov E, Moebus S, Mohan V, Mohlke KL, Morris AD, Mühleisen TW, Müller-Nurasyid M, Musk B, Nakamura J, Nakashima E, Navarro P, Ng PK, Nica AC, Nilsson PM, Njølstad I, Nöthen MM, Ohnaka K, Ong TH, Owen KR, Palmer CN, Pankow JS, Park KS, Parkin M, Pechlivanis S, Pedersen NL, Peltonen L, Perry JR, Peters A, Pinidiyapathirage JM, Platou CG, Potter S, Price JF, Qi L, Radha V, Rallidis L, Rasheed A, Rathman W, Rauramaa R, Raychaudhuri S, Rayner NW, Rees SD, Rehnberg E, Ripatti S, Robertson N, Roden M, Rossin EJ, Rudan I, Rybin D, Saaristo TE, Salomaa V, Saltevo J, Samuel M, Sanghera DK, Saramies J, Scott J, Scott LJ, Scott RA, Segrè AV, Sehmi J, Sennblad B, Shah N, Shah S, Shera AS, Shu XO, Shuldiner AR, Sigurđsson G, Sijbrands E, Silveira A, Sim X, Sivapalaratnam S, Small KS, So WY, Stančáková A, Stefansson K, Steinbach G, Steinthorsdottir V, Stirrups K, Strawbridge RJ, Stringham HM, Sun Q, Suo C, Syvänen AC, Takayanagi R, Takeuchi F, Tay WT, Teslovich TM, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tikkanen E, Trakalo J, Tremoli E, Trip MD, Tsai FJ, Tuomi T, Tuomilehto J, Uitterlinden AG, Valladares-Salgado A, Vedantam S, Veglia F, Voight BF, Wang C, Wareham NJ, Wennauer R, Wickremasinghe AR, Wilsgaard T, Wilson JF, Wiltshire S, Winckler W, Wong TY, Wood AR, Wu JY, Wu Y, Yamamoto K, Yamauchi T, Yang M, Yengo L, Yokota M, Young R, Zabaneh D, Zhang F, Zhang R, Zheng W, Zimmet PZ, Altshuler D, Bowden DW, Cho YS, Cox NJ, Cruz M, Hanis CL, Kooner J, Lee JY, Seielstad M, Teo YY, Boehnke M, Parra EJ, Chambers JC, Tai ES, McCarthy MI, Morris AP (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 46:234–244
Dimas AS, Lagou V, Barker A, Knowles JW, Mägi R, Hivert MF, Benazzo A, Rybin D, Jackson AU, Stringham HM, Song C, Fischer-Rosinsky A, Boesgaard TW, Grarup N, Abbasi FA, Assimes TL, Hao K, Yang X, Lecoeur C, Barroso I, Bonnycastle LL, Böttcher Y, Bumpstead S, Chines PS, Erdos MR, Graessler J, Kovacs P, Morken MA, Narisu N, Payne F, Stancakova A, Swift AJ, Tönjes A, Bornstein SR, Cauchi S, Froguel P, Meyre D, Schwarz PE, Häring HU, Smith U, Boehnke M, Bergman RN, Collins FS, Mohlke KL, Tuomilehto J, Quertemous T, Lind L, Hansen T, Pedersen O, Walker M, Pfeiffer AF, Spranger J, Stumvoll M, Meigs JB, Wareham NJ, Kuusisto J, Laakso M, Langenberg C, Dupuis J, Watanabe RM, Florez JC, Ingelsson E, McCarthy MI, Prokopenko I, MAGIC Investigators (2014) Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 63:2158–2171
Stahl EA, Wegmann D, Trynka G, Gutierrez-Achury J, Do R, Voight BF, Kraft P, Chen R, Kallberg HJ, Kurreeman FA, Diabetes Genetics Replication and Meta-analysis Consortium, Myocardial Infarction Genetics Consortium, Kathiresan S, Wijmenga C, Gregersen PK, Alfredsson L, Siminovitch KA, Worthington J, de Bakker PI, Raychaudhuri S, Plenge RM (2012) Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet 44:483–489
Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, Helgadottir HT, Johannsdottir H, Magnusson OT, Gudjonsson SA, Justesen JM, Harder MN, Jørgensen ME, Christensen C, Brandslund I, Sandbæk A, Lauritzen T, Vestergaard H, Linneberg A, Jørgensen T, Hansen T, Daneshpour MS, Fallah MS, Hreidarsson AB, Sigurdsson G, Azizi F, Benediktsson R, Masson G, Helgason A, Kong A, Gudbjartsson DF, Pedersen O, Thorsteinsdottir U, Stefansson K (2014) Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet 46:294–298
Huyghe JR, Jackson AU, Fogarty MP, Buchkovich ML, Stančáková A, Stringham HM, Sim X, Yang L, Fuchsberger C, Cederberg H, Chines PS, Teslovich TM, Romm JM, Ling H, McMullen I, Ingersoll R, Pugh EW, Doheny KF, Neale BM, Daly MJ, Kuusisto J, Scott LJ, Kang HM, Collins FS, Abecasis GR, Watanabe RM, Boehnke M, Laakso M, Mohlke KL (2013) Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet 45:197–201
Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, Ma C, Fontanillas P, Moutsianas L, McCarthy DJ, Rivas MA, Perry JR, Sim X, Blackwell TW, Robertson NR, Rayner NW, Cingolani P, Locke AE, Fernandez Tajes J, Highland HM, Dupuis J, Chines PS, Lindgren CM, Hartl C, Jackson AU, Chen H, Huyghe JR, van de Bunt M, Pearson RD, Kumar A, Müller-Nurasyid M, Grarup N, Stringham HM, Gamazon ER, Lee J, Chen Y, Scott RA, Below JE, Chen P, Huang J, Go MJ, Stitzel ML, Pasko D, Parker SC, Varga TV, Green T, Beer NL, Day-Williams AG, Ferreira T, Fingerlin T, Horikoshi M, Hu C, Huh I, Ikram MK, Kim BJ, Kim Y, Kim YJ, Kwon MS, Lee J, Lee S, Lin KH, Maxwell TJ, Nagai Y, Wang X, Welch RP, Yoon J, Zhang W, Barzilai N, Voight BF, Han BG, Jenkinson CP, Kuulasmaa T, Kuusisto J, Manning A, Ng MC, Palmer ND, Balkau B, Stancáková A, Abboud HE, Boeing H, Giedraitis V, Prabhakaran D, Gottesman O, Scott J, Carey J, Kwan P, Grant G, Smith JD, Neale BM, Purcell S, Butterworth AS, Howson JM, Lee HM, Lu Y, Kwak SH, Zhao W, Danesh J, Lam VK, Park KS, Saleheen D, So WY, Tam CH, Afzal U, Aguilar D, Arya R, Aung T, Chan E, Navarro C, Cheng CY, Palli D, Correa A, Curran JE, Rybin D, Farook VS, Fowler SP, Freedman BI, Griswold M, Hale DE, Hicks PJ, Khor CC, Kumar S, Lehne B, Thuillier D, Lim WY, Liu J, van der Schouw YT, Loh M, Musani SK, Puppala S, Scott WR, Yengo L, Tan ST, Taylor HA Jr, Thameem F, Wilson G Sr, Wong TY, Njølstad PR, Levy JC, Mangino M, Bonnycastle LL, Schwarzmayr T, Fadista J, Surdulescu GL, Herder C, Groves CJ, Wieland T, Bork-Jensen J, Brandslund I, Christensen C, Koistinen HA, Doney AS, Kinnunen L, Esko T, Farmer AJ, Hakaste L, Hodgkiss D, Kravic J, Lyssenko V, Hollensted M, Jørgensen ME, Jørgensen T, Ladenvall C, Justesen JM, Käräjämäki A, Kriebel J, Rathmann W, Lannfelt L, Lauritzen T, Narisu N, Linneberg A, Melander O, Milani L, Neville M, Orho-Melander M, Qi L, Qi Q, Roden M, Rolandsson O, Swift A, Rosengren AH, Stirrups K, Wood AR, Mihailov E, Blancher C, Carneiro MO, Maguire J, Poplin R, Shakir K, Fennell T, DePristo M, Hrabé de Angelis M, Deloukas P, Gjesing AP, Jun G, Nilsson P, Murphy J, Onofrio R, Thorand B, Hansen T, Meisinger C, Hu FB, Isomaa B, Karpe F, Liang L, Peters A, Huth C, O'Rahilly SP, Palmer CN, Pedersen O, Rauramaa R, Tuomilehto J, Salomaa V, Watanabe RM, Syvänen AC, Bergman RN, Bharadwaj D, Bottinger EP, Cho YS, Chandak GR, Chan JC, Chia KS, Daly MJ, Ebrahim SB, Langenberg C, Elliott P, Jablonski KA, Lehman DM, Jia W, Ma RC, Pollin TI, Sandhu M, Tandon N, Froguel P, Barroso I, Teo YY, Zeggini E, Loos RJ, Small KS, Ried JS, DeFronzo RA, Grallert H, Glaser B, Metspalu A, Wareham NJ, Walker M, Banks E, Gieger C, Ingelsson E, Im HK, Illig T, Franks PW, Buck G, Trakalo J, Buck D, Prokopenko I, Mägi R, Lind L, Farjoun Y, Owen KR, Gloyn AL, Strauch K, Tuomi T, Kooner JS, Lee JY, Park T, Donnelly P, Morris AD, Hattersley AT, Bowden DW, Collins FS, Atzmon G, Chambers JC, Spector TD, Laakso M, Strom TM, Bell GI, Blangero J, Duggirala R, Tai ES, McVean G, Hanis CL, Wilson JG, Seielstad M, Frayling TM, Meigs JB, Cox NJ, Sladek R, Lander ES, Gabriel S, Burtt NP, Mohlke KL, Meitinger T, Groop L, Abecasis G, Florez JC, Scott LJ, Morris AP, Kang HM, Boehnke M, Altshuler D, McCarthy MI (2016) The genetic architecture of type 2 diabetes. Nature 536(7614):41–47
Hivert MF, Jablonski KA, Perreault L, Saxena R, McAteer JB, Franks PW, Hamman RF, Kahn SE, Haffner S, DIAGRAM Consortium, Meigs JB, Altshuler D, Knowler WC, Florez JC, Diabetes Prevention Program Research Group (2011) Updated genetic score based on 34 confirmed type 2 diabetes Loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes 60:1340–1348
Imamura M, Shigemizu D, Tsunoda T, Iwata M, Maegawa H, Watada H, Hirose H, Tanaka Y, Tobe K, Kaku K, Kashiwagi A, Kawamori R, Maeda S (2013) Assessing the clinical utility of a genetic risk score constructed using 49 susceptibility alleles for type 2 diabetes in a Japanese population. J Clin Endocrinol Metab 98:E1667–E1673
Wang X, Strizich G, Hu Y, Wang T, Kaplan RC, Qi Q (2016) Genetic markers of type 2 diabetes: Progress in genome-wide association studies and clinical application for risk prediction. J Diabetes 8:24–35
Okada Y, Wu D, Trynka G, Raj T, Terao C, Ikari K, Kochi Y, Ohmura K, Suzuki A, Yoshida S, Graham RR, Manoharan A, Ortmann W, Bhangale T, Denny JC, Carroll RJ, Eyler AE, Greenberg JD, Kremer JM, Pappas DA, Jiang L, Yin J, Ye L, Su DF, Yang J, Xie G, Keystone E, Westra HJ, Esko T, Metspalu A, Zhou X, Gupta N, Mirel D, Stahl EA, Diogo D, Cui J, Liao K, Guo MH, Myouzen K, Kawaguchi T, Coenen MJ, van Riel PL, van de Laar MA, Guchelaar HJ, Huizinga TW, Dieudé P, Mariette X, Bridges SL Jr, Zhernakova A, Toes RE, Tak PP, Miceli-Richard C, Bang SY, Lee HS, Martin J, Gonzalez-Gay MA, Rodriguez-Rodriguez L, Rantapää-Dahlqvist S, Arlestig L, Choi HK, Kamatani Y, Galan P, Lathrop M, RACI consortium, GARNET consortium, Eyre S, Bowes J, Barton A, de Vries N, Moreland LW, Criswell LA, Karlson EW, Taniguchi A, Yamada R, Kubo M, Liu JS, Bae SC, Worthington J, Padyukov L, Klareskog L, Gregersen PK, Raychaudhuri S, Stranger BE, De Jager PL, Franke L, Visscher PM, Brown MA, Yamanaka H, Mimori T, Takahashi A, Xu H, Behrens TW, Siminovitch KA, Momohara S, Matsuda F, Yamamoto K, Plenge RM (2014) Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506:376–381
Poulsen P, Levin K, Petersen I, Christensen K, Beck-Nielsen H, Vaag A (2005) Heritability of insulin secretion, peripheral and hepatic insulin action, and intracellular glucose partitioning in young and old Danish twins. Diabetes 54:275–283
Weedon MN, Clark VJ, Qian Y, Ben-Shlomo Y, Timpson N et al (2006) A common haplotype of the glucokinase gene alters fasting glucose and birth weight: association in six studies and population-genetics analyses. Am J Hum Genet 79:991–1001
Bouatia-Naji N, Rocheleau G, Van Lommel L, Lemaire K, Schuit F et al (2008) A polymorphism within the G6PC2 gene is associated with fasting plasma glucose levels. Science 320:1085–1088
Sparso T, Andersen G, Nielsen T, Burgdorf KS, Gjesing AP et al (2008) The GCKR rs780094 polymorphism is associated with elevated fasting serum triacylglycerol, reduced fasting and OGTT-related insulinaemia, and reduced risk of type 2 diabetes. Diabetologia 51:70–75
Orho-Melander M, Melander O, Guiducci C, Perez-Martinez P, Corella D et al (2008) A common missense variant in the glucokinase regulatory protein gene (GCKR) is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes 57:3112–3121
Pare G, Chasman DI, Parker AN, Nathan DM, Miletich JP, Zee RY, Ridker PM (2008) Novel association of HK1 with glycated hemoglobin in a non-diabetic population: a genome-wide evaluation of 14, 618 participants in the Women’s Genome Health Study. PLoS Genet 4:e1000312
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N et al (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42:105–116
Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N et al (2009) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41:77–81
Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS et al (2010) Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 42:142–148
Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E et al (2011) Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 60:2624–2634
Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME et al (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 44:991–1005
Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N et al (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44:659–669
Kim YJ, Go MJ, Hu C, Hong CB, Kim et al (2011) Largescale genome-wide association studies in East Asians identify new genetic loci influencing metabolic traits. Nat. Genet 43:990–995
Chen G, Bentley A, Adeyemo A, Shriner D, Zhou J et al (2012) Genome-wide association study identifies novel loci association with fasting insulin and insulin resistance in African Americans. Hum Mol Genet 21:4530–4536
Go MJ, Hwang JY, Kim YJ, Hee Oh J, Kim YJ et al (2013) New susceptibility loci in MYL2, C12orf51 and OAS1 associated with 1-h plasma glucose as predisposing risk factors for type 2 diabetes in the Korean population. J Hum Genet 58:362–365
Hwang JY, Sim X, Wu Y, Liang J, Tabara Y et al (2015) Genome-wide association meta-analysis identifies novel variants associated with fasting plasma glucose in East Asians. Diabetes 64:291–298
Chen P, Takeuchi F, Lee JY, Li H, Wu JY et al (2014) Multiple nonglycemic genomic loci are newly associated with blood level of glycated hemoglobin in East Asians. Diabetes 63:2551–2562
Go MJ, Hwang JY, Park TJ, Kim YJ, Oh JH et al (2014) Genome-wide association study identifies two novel Loci with sex-specific effects for type 2 diabetes mellitus and glycemic traits in a korean population. Diabetes Metab J 38:375–387
Moltke I, Grarup N, Jørgensen ME, Bjerregaard P, Treebak JT, Fumagalli M, Korneliussen TS, Andersen MA, Nielsen TS, Krarup NT, Gjesing AP, Zierath JR, Linneberg A, Wu X, Sun G, Jin X, Al-Aama J, Wang J, Borch-Johnsen K, Pedersen O, Nielsen R, Albrechtsen A, Hansen T (2014) A common Greenlandic TBC1D4 variant confers muscle insulin resistance and type 2 diabetes. Nature 512:190–193
Mahajan A, Sim X, Ng HJ, Manning A, Rivas MA et al (2015) Identification and functional characterization of G6PC2 coding variants influencing glycemic traits define an effector transcript at the G6PC2-ABCB11 locus. PLoS Genet 11:e1004876
Huyghe JR, Jackson AU, Fogarty MP, Buchkovich ML, Stancakova A et al (2013) Exome array analysis identifies new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet 45:197–201
Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N et al (2014) Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet 46:294–298
Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V et al (2016) The genetic architecture of type 2 diabetes. Nature 536:41–47
Cornes BK, Brody JA, Nikpoor N, Morrison AC, Dang HC et al (2014) Association of levels of fasting glucose and insulin with rare variants at the chromosome 11p11.2-MADD locus: Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium Targeted Sequencing Study. Circ Cardiovasc Genet 7:374–382
Wessel J, Chu AY, Willems SM, Wang S, Yaghootkar H et al (2015) Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat Commun 6:5897
The Diabetes Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
UK Prospective Diabetes Study (UKPDS) Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Ohkubo Y, Kishikawa H, Araki E, Miyata T, Isami S, Motoyoshi S, Kojima Y, Furuyoshi N, Shichiri M (1995) Intensive insulin therapy prevents the progression of diabetic microvascular complications in Japanese patients with non-insulin-dependent diabetes mellitus: a randomized prospective 6-year study. Diabetes Res Clin Pract 28:103–117
Gaede P, Vedel P, Larsen N, Jensen GV, Parving HH, Pedersen O (2003) Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Engl J Med 348:383–393
Perkins BA, Ficociello LH, Silva KH, Finkelstein DM, Warram JH, Krolewski AS (2003) Regression of microalbuminuria in type 1 diabetes. N Engl J Med 348:2285–2293
Gaede P, Tarnow L, Vedel P, Parving HH, Pedersen O (2004) Remission to normoalbuminuria during multifactorial treatment preserves kidney function in patients with type 2 diabetes and microalbuminuria. Nephrol Dial Transplant 19:2784–2788
Araki S, Haneda M, Sugimoto T, Isono M, Isshiki K, Kashiwagi A, Koya D (2005) Factors associated with frequent remission of microalbuminuria in patients with type 2 diabetes. Diabetes 54:2983–2987
Brownlee M (2005) The pathobiology of diabetic complications: a unifying mechanism. Diabetes 54:1615–1625
Krolewski AS, Warram JH, Rand LI, Kahn CR (1987) Epidemiologic approach to the etiology of type 1 diabetes mellitus and its complications. N Engl J Med 317:1390–1398
Quinn M, Angelico MC, Warram JH, Krolewski AS (1996) Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia 39:940–945
Fava S, Azzopardi J, Hattersley AT, Watkins PJ (2000) Increased prevalence of proteinuria in diabetic sibs of proteinuric type 2 diabetic subjects. Am J Kidney Dis 35:708–712
Pezzolesi MG, Poznik GD, Mychaleckyj JC, Paterson AD, Barati MT, Klein JB, Ng DP, Placha G, Canani LH, Bochenski J, Waggott D, Merchant ML, Krolewski B, Mirea L, Wanic K, Katavetin P, Kure M, Wolkow P, Dunn JS, Smiles A, Walker WH, Boright AP, Bull SB, DCCT/EDIC Research Group, Doria A, Rogus JJ, Rich SS, Warram JH, Krolewski AS (2009) Genome-wide association scan for diabetic nephropathy susceptibility genes in type 1 diabetes. Diabetes 58:1403–1410
Pezzolesi MG, Jeong J, Smiles AM, Skupien J, Mychaleckyj JC, Rich SS, Warram JH, Krolewski AS (2013) Family-based association analysis confirms the role of the chromosome 9q21.32 locus in the susceptibility of diabetic nephropathy. PLoS One 8(3):e60301
Freedman BI, Langefeld CD, Lu L, Divers J, Comeau ME, Kopp JB, Winkler CA, Nelson GW, Johnson RC, Palmer ND, Hicks PJ, Bostrom MA, Cooke JN, McDonough CW, Bowden DW (2011) Differential effects of MYH9 and APOL1 risk variants on FRMD3 association with diabetic ESRD in African Americans. PLoS Genet 7:e1002150
Maeda S, Araki S, Babazono T, Toyoda M, Umezono T, Kawai K, Imanishi M, Uzu T, Watada H, Suzuki D, Kashiwagi A, Iwamoto Y, Kaku K, Kawamori R, Nakamura Y (2010) Replication study for the association between four Loci identified by a genome-wide association study on European American subjects with type 1 diabetes and susceptibility to diabetic nephropathy in Japanese subjects with type 2 diabetes. Diabetes 59:2075–2079
Sandholm N, Salem RM, McKnight AJ, Brennan EP, Forsblom C, Isakova T, McKay GJ, Williams WW, Sadlier DM, Mäkinen VP, Swan EJ, Palmer C, Boright AP, Ahlqvist E, Deshmukh HA, Keller BJ, Huang H, Ahola AJ, Fagerholm E, Gordin D, Harjutsalo V, He B, Heikkilä O, Hietala K, Kytö J, Lahermo P, Lehto M, Lithovius R, Osterholm AM, Parkkonen M, Pitkäniemi J, Rosengård-Bärlund M, Saraheimo M, Sarti C, Söderlund J, Soro-Paavonen A, Syreeni A, Thorn LM, Tikkanen H, Tolonen N, Tryggvason K, Tuomilehto J, Wadén J, Gill GV, Prior S, Guiducci C, Mirel DB, Taylor A, Hosseini SM, DCCT/EDIC Research Group, Parving HH, Rossing P, Tarnow L, Ladenvall C, Alhenc-Gelas F, Lefebvre P, Rigalleau V, Roussel R, Tregouet DA, Maestroni A, Maestroni S, Falhammar H, Gu T, Möllsten A, Cimponeriu D, Ioana M, Mota M, Mota E, Serafinceanu C, Stavarachi M, Hanson RL, Nelson RG, Kretzler M, Colhoun HM, Panduru NM, Gu HF, Brismar K, Zerbini G, Hadjadj S, Marre M, Groop L, Lajer M, Bull SB, Waggott D, Paterson AD, Savage DA, Bain SC, Martin F, Hirschhorn JN, Godson C, Florez JC, Groop PH, Maxwell AP (2012) New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet 8:e1002921
Maeda S, Imamura M, Kurashige M, Araki S, Suzuki D, Babazono T, Uzu T, Umezono T, Toyoda M, Kawai K, Imanishi M, Hanaoka K, Maegawa H, Uchigata Y, Hosoya T (2013) Replication study for the association of 3 SNP loci identified in a genome-wide association study for diabetic nephropathy in European type 1 diabetes with diabetic nephropathy in Japanese patients with type 2 diabetes. Clin Exp Nephrol 17:866–871
Germain M, Pezzolesi MG, Sandholm N, McKnight AJ, Susztak K, Lajer M, Forsblom C, Marre M, Parving HH, Rossing P, Toppila I, Skupien J, Roussel R, Ko YA, Ledo N, Folkersen L, Civelek M, Maxwell AP, Tregouet DA, Groop PH, Tarnow L, Hadjadj S (2015) SORBS1 gene, a new candidate for diabetic nephropathy: results from a multi-stage genome-wide association study in patients with type 1 diabetes. Diabetologia 58:543–548
Sandholm N, McKnight AJ, Salem RM, Brennan EP, Forsblom C, Harjutsalo V, Mäkinen VP, McKay GJ, Sadlier DM, Williams WW, Martin F, Panduru NM, Tarnow L, Tuomilehto J, Tryggvason K, Zerbini G, Comeau ME, Langefeld CD, FIND Consortium, Godson C, Hirschhorn JN, Maxwell AP, Florez JC, Groop PH, FinnDiane Study Group and the GENIE Consortium (2013) Chromosome 2q31.1 associates with ESRD in women with type 1 diabetes. J Am Soc Nephrol 24:1537–1543
Teumer A, Tin A, Sorice R, Gorski M, Yeo NC, Chu AY, Li M, Li Y, Mijatovic V, Ko YA, Taliun D, Luciani A, Chen MH, Yang Q, Foster MC, Olden M, Hiraki LT, Tayo BO, Fuchsberger C, Dieffenbach AK, Shuldiner AR, Smith AV, Zappa AM, Lupo A, Kollerits B, Ponte B, Stengel B, Krämer BK, Paulweber B, Mitchell BD, Hayward C, Helmer C, Meisinger C, Gieger C, Shaffer CM, Müller C, Langenberg C, Ackermann D, Siscovick D, DCCT/EDIC, Boerwinkle E, Kronenberg F, Ehret GB, Homuth G, Waeber G, Navis G, Gambaro G, Malerba G, Eiriksdottir G, Li G, Wichmann HE, Grallert H, Wallaschofski H, Völzke H, Brenner H, Kramer H, Mateo Leach I, Rudan I, Hillege HL, Beckmann JS, Lambert JC, Luan J, Zhao JH, Chalmers J, Coresh J, Denny JC, Butterbach K, Launer LJ, Ferrucci L, Kedenko L, Haun M, Metzger M, Woodward M, Hoffman MJ, Nauck M, Waldenberger M, Pruijm M, Bochud M, Rheinberger M, Verweij N, Wareham NJ, Endlich N, Soranzo N, Polasek O, van der Harst P, Pramstaller PP, Vollenweider P, Wild PS, Gansevoort RT, Rettig R, Biffar R, Carroll RJ, Katz R, Loos RJ, Hwang SJ, Coassin S, Bergmann S, Rosas SE, Stracke S, Harris TB, Corre T, Zeller T, Illig T, Aspelund T, Tanaka T, Lendeckel U, Völker U, Gudnason V, Chouraki V, Koenig W, Kutalik Z, O’Connell JR, Parsa A, Heid IM, Paterson AD, de Boer IH, Devuyst O, Lazar J, Endlich K, Susztak K, Tremblay J, Hamet P, Jacob HJ, Böger CA, Fox CS, Pattaro C, Köttgen A (2016) Genome-wide association studies identify genetic loci associated with albuminuria in diabetes. Diabetes 65:803–817
Maeda S (2004) Genome-wide search for susceptibility gene to diabetic nephropathy by gene-based SNP. Diabetes Res Clin Pract 66S:S45–S47
Maeda S, Osawa N, Hayashi T, Tsukada S, Kobayashi M, Kikkawa R (2007) Genetic variations associated with diabetic nephropathy and type 2 diabetes in a Japanese population. Kidney Int 72:S43–S48
Haga H, Yamada R, Ohnishi Y, Nakamura Y, Tanaka T (2002) Gene-based SNP discovery as part of the Japanese millennium genome project: identification of 190,562 genetic variation in the human genome. J Hum Genet 47:605–610
Hirakawa M, Tanaka T, Hashimoto Y, Kuroda M, Takagi T, Nakamura Y (2002) JSNP a database of common gene variations in the Japanese population. Nucleic Acids Res 30:158–162
Tanaka N, Babazono T, Saito S, Sekine A, Tsunoda T, Haneda M, Tanaka Y, Fujioka T, Kaku K, Kawamori R, Kikkawa R, Iwamoto Y, Nakamura Y, Maeda S (2003) Association of solute carrier family 12 (sodium/chloride) member 3 with diabetic nephropathy, identified by genome-wide analyses of single nucleotide polymorphisms. Diabetes 52:2848–2853
Shimazaki A, Kawamura Y, Kanazawa A, Sekine A, Saito S, Tsunoda T, Koya D, Babazono T, Tanaka Y, Matsuda M, Kawai K, Iiizumi T, Imanishi M, Shinosaki T, Yanagimoto T, Ikeda M, Omachi S, Kashiwagi A, Kaku K, Iwamoto Y, Kawamori R, Kikkawa R, Nakajima M, Nakamura Y, Maeda S (2005) Genetic variations in the gene encoding ELMO1 are associated with susceptibility to diabetic nephropathy. Diabetes 54:1171–1178
Kamiyama M, Kobayashi M, Araki S, Iida A, Tsunoda T, Kawai K, Imanishi M, Nomura M, Babazono T, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Ng DP, Hansen T, Gaede P, Pedersen O, Nakamura Y, Maeda S (2007) Polymorphisms in the 3′ UTR in the neurocalcin delta gene affect mRNA stability, and confer susceptibility to diabetic nephropathy. Hum Genet 122(3–4):397–407
Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP (1996) Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12:24–30
Nishiyama K, Tanaka Y, Nakajima K, Mokubo A, Atsumi Y, Matsuoka K, Watada H, Hirose T, Nomiyama T, Maeda S, Kawamori R (2005) Polymorphism of the solute carrier family 12 (sodium/chloride transporters) member 3, SLC12A3, gene at exon 23 (+78G/A: Arg913Gln) is associated with elevation of urinary albumin excretion in Japanese patients with type 2 diabetes: a 10-year longitudinal study. Diabetologia 48:1335–1338
Bodhini D, Chidambaram M, Liju S, Revathi B, Laasya D, Sathish N, Kanthimathi S, Ghosh S, Anjana RM, Mohan V, Radha V (2016) Association of rs11643718 SLC12A3 and rs741301 ELMO1 variants with diabetic nephropathy in South Indian population. Ann Hum Genet 80:336–341
Abu Seman N, He B, Ojala JR, Wan Mohamud WN, Östenson CG, Brismar K, Gu HF (2014) Genetic and biological effects of sodium-chloride cotransporter (SLC12A3) in diabetic nephropathy. Am J Nephrol 40:408–416
Kim JH, Shin HD, Park BL, Moon MK, Cho YM, Hwang YH, Oh KW, Kim SY, Lee HK, Ahn C, Park KS (2006) SLC12A3 (solute carrier family 12 member [sodium/chloride] 3) polymorphisms are associated with end-stage renal disease in diabetic nephropathy. Diabetes 55:843–848
Ng DP, Nurbaya S, Choo S, Koh D, Chia KS, Krolewski AS (2008) Genetic variation at the SLC12A3 locus is unlikely to explain risk for advanced diabetic nephropathy in Caucasians with type 2 diabetes. Nephrol Dial Transplant 23:2260–2264
Leak TS, Perlegas PS, Smith SG, Keene KL, Hicks PJ, Langefeld CD, Mychaleckyj JC, Rich SS, Kirk JK, Freedman BI, Bowden DW, Sale MM (2009) Variants in intron 13 of the ELMO1 gene are associated with diabetic nephropathy in African Americans. Ann Hum Genet 73:152–159
Pezzolesi MG, Katavetin P, Kure M, Poznik GD, Skupien J, Mychaleckyj JC, Rich SS, Warram JH, Krolewski AS (2009) Confirmation of genetic associations at ELMO1 in the GoKinD collection supports its role as a susceptibility gene in diabetic nephropathy. Diabetes 58:2698–2702
Wu HY, Wang Y, Chen M, Zhang X, Wang D, Pan Y, Li L, Liu D, Dai XM (2013) Association of ELMO1 gene polymorphisms with diabetic nephropathy in Chinese population. J Endocrinol Investig 36:298–302
Hanson RL, Millis MP, Young NJ, Kobes S, Nelson RG, Knowler WC, DiStefano JK (2010) ELMO1 variants and susceptibility to diabetic nephropathy in American Indians. Mol Genet Metab 101:383–390
Gumienny TL, Brugnera E, Tosello-Trampont AC, Kinchen JM, Haney LB, Nishiwaki K, Walk SF, Nemergut ME, Macara IG, Francis R, Schedl T, Qin Y, Van Aelst L, Hengartner MO, Ravichandran KS (2001) CED-12/ELMO, a novel member of the CrkII/Dock180/Rac pathway, is required for phagocytosis and cell migration. Cell 107:27–41
Brugnera E, Haney L, Grimsley C, Lu M, Walk SF, Tosello-Trampont AC, Macara IG, Madhani H, Fink GR, Ravichandran KS (2002) Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol 4:574–582
Shimazaki A, Tanaka Y, Shinosaki T, Ikeda M, Watada H, Hirose T, Kawamori R, Maeda S (2006) ELMO1 increases expression of extracellular matrix proteins and inhibits cell adhesion to ECMs. Kidney Int 70:1769–1776
Hathaway CK, Chang AS, Grant R, Kim HS, Madden VJ, Bagnell CR Jr, Jennette JC, Smithies O, Kakoki M (2016) High Elmo1 expression aggravates and low Elmo1 expression prevents diabetic nephropathy. Proc Natl Acad Sci U S A 113:2218–2222
Sharma KR, Heckler K, Stoll SJ, Hillebrands JL, Kynast K, Herpel E, Porubsky S, Elger M, Hadaschik B, Bieback K, Hammes HP, Nawroth PP, Kroll J (2016) ELMO1 protects renal structure and ultrafiltration in kidney development and under diabetic conditions. Sci Rep 6:37172
Maeda S, Kobayashi MA, Araki S, Babazono T, Freedman BI, Bostrom MA, Cooke JN, Toyoda M, Umezono T, Tarnow L, Hansen T, Gaede P, Jorsal A, Ng DP, Ikeda M, Yanagimoto T, Tsunoda T, Unoki H, Kawai K, Imanishi M, Suzuki D, Shin HD, Park KS, Kashiwagi A, Iwamoto Y, Kaku K, Kawamori R, Parving HH, Bowden DW, Pedersen O, Nakamura Y (2010) A single nucleotide polymorphism within the acetyl-coenzyme A carboxylase beta gene is associated with proteinuria in patients with type 2 diabetes. PLoS Genet 6:e1000842
Tang SC, Leung VT, Chan LY, Wong SS, Chu DW, Leung JC, Ho YW, Lai KN, Ma L, Elbein SC, Bowden DW, Hicks PJ, Comeau ME, Langefeld CD, Freedman BI (2010) The acetyl-coenzyme A carboxylase beta (ACACB) gene is associated with nephropathy in Chinese patients with type 2 diabetes. Nephrol Dial Transplant 25:3931–3934
Shah VN, Cheema BS, Sharma R, Khullar M, Kohli HS, Ahluwalia TS, Mohan V, Bhansali A (2013) ACACβ gene (rs2268388) and AGTR1 gene (rs5186) polymorphism and the risk of nephropathy in Asian Indian patients with type 2 diabetes. Mol Cell Biochem 372:191–198
Kobayashi MA, Watada H, Kawamori R, Maeda S (2010) Overexpression of acetyl-coenzyme A carboxylase beta increases proinflammatory cytokines in cultured human renal proximal tubular epithelial cells. Clin Exp Nephrol 14:315–324
McDonough CW, Palmer ND, Hicks PJ, Roh BH, An SS, Cooke JN, Hester JM, Wing MR, Bostrom MA, Rudock ME, Lewis JP, Talbert ME, Blevins RA, Lu L, Ng MC, Sale MM, Divers J, Langefeld CD, Freedman BI, Bowden DW (2011) A genome-wide association study for diabetic nephropathy genes in African Americans. Kidney Int 79:563–572
Iyengar SK, Sedor JR, Freedman BI, Kao WH, Kretzler M, Keller BJ, Abboud HE, Adler SG, Best LG, Bowden DW, Burlock A, Chen YD, Cole SA, Comeau ME, Curtis JM, Divers J, Drechsler C, Duggirala R, Elston RC, Guo X, Huang H, Hoffmann MM, Howard BV, Ipp E, Kimmel PL, Klag MJ, Knowler WC, Kohn OF, Leak TS, Leehey DJ, Li M, Malhotra A, März W, Nair V, Nelson RG, Nicholas SB, O'Brien SJ, Pahl MV, Parekh RS, Pezzolesi MG, Rasooly RS, Rotimi CN, Rotter JI, Schelling JR, Seldin MF, Shah VO, Smiles AM, Smith MW, Taylor KD, Thameem F, Thornley-Brown DP, Truitt BJ, Wanner C, Weil EJ, Winkler CA, Zager PG, Igo RP Jr, Hanson RL, Langefeld CD, Family Investigation of Nephropathy and Diabetes (FIND) (2015) Genome-wide association and trans-ethnic meta-analysis for advanced diabetic kidney disease: Family Investigation of Nephropathy and Diabetes (FIND). PLoS Genet 11:e1005352
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, International Human Genome Sequencing Consortium (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X (2001) The sequence of the human genome. Science 291:1304–1351
The 1000 Genomes Project Consortium (2015) A global reference for human genetic variation. Nature 526:68–74
EMBI-EBI GWAS catalog, http://www.ebi.ac.uk/gwas/
Minster RL, Hawley NL, Su CT, Sun G, Kershaw EE, Cheng H, Buhule OD, Lin J, Reupena MS, Viali S, Tuitele J, Naseri T, Urban Z, Deka R, Weeks DE, McGarvey ST (2016) A thrifty variant in CREBRF strongly influences body mass index in Samoans. Nat Genet 48:1049–1054
Flannick J, Florez JC (2016) Type 2 diabetes: genetic data sharing to advance complex disease research. Nat Rev Genet 17:535–549
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Imamura, M., Horikoshi, M., Maeda, S. (2019). Genome-Wide Association Study for Type 2 Diabetes. In: Tsunoda, T., Tanaka, T., Nakamura, Y. (eds) Genome-Wide Association Studies. Springer, Singapore. https://doi.org/10.1007/978-981-13-8177-5_4
Download citation
DOI: https://doi.org/10.1007/978-981-13-8177-5_4
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-8176-8
Online ISBN: 978-981-13-8177-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)