
Abstract
Genome-wide association (GWA) studies represent the single most effective technique for identifying genetic risk loci causing complex diseases. Since the publication of the first GWA studies for type 2 diabetes (T2D) in 2007, nearly 90 statistically robust risk loci have been identified. The T2D risk loci identified by GWA studies contained several genes that are targets of current diabetic therapies; however, the majority of genes in these loci had not previously been implicated in the pathophysiology of T2D. Mechanistic insights about the physiological role of T2D loci in the disease predisposition have been gained from investigation of their contribution into glycemic trait variability in nondiabetic individuals. Current efforts to identify the causative genetic mutations in these loci and the molecular mechanisms through which they exert their effects have the potential to make far-reaching contributions to our understanding of molecular basis of T2D and the development of novel strategies for patient care.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Glycemic Traits
- Nondiabetic Individuals
- Meta-Analyses Of Glucose And Insulin-related Traits Consortium (MAGIC)
- Transcription Factor 7-like 2 (TCF7L2)
- Glucokinase Regulator (GCKR)
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
1 Introduction
Type 2 diabetes (T2D) is a common, chronic disorder whose prevalence is increasing rapidly across the globe. Like other complex diseases, T2D represents a challenge for genetic studies aiming to uncover the underlying pathophysiological mechanisms. It is predicted that T2D will affect 592 million individuals by 2035 (Federation 2013) in developed and low- and middle-income countries. While the recent increase in T2D prevalence has been attributed to a sedentary “westernized” lifestyle and changes in diet, a significant proportion of heritable factors also contribute to individual susceptibility (Hu 2011).
The strong family clustering and heritability of T2D and related glycemic traits have motivated a large number of studies to identify genetic factors that cause this disease (Permutt et al. 2005; Stumvoll et al. 2005); despite much effort, by late 2006 only three genetic loci had been reproducibly shown to increase T2D risk [reviewed in Majithia and Florez (2009), McCarthy (2008), Stolerman and Florez (2009)]. The earliest attempts to discover T2D-associated genes used either position- or function-based strategies. In a position-based search, genes are identified within families by studying the co-inheritance of the disease with a set of polymorphic markers whose genomic positions are known. Such “linkage studies” usually identify a genomic region (~10 Mbp) that confers genetic risk; the disease-causative mutations are identified by sequencing transcribed and functional elements of all genes in the target region. In function-based approaches, risk association is tested for common genetic variants in candidate genes involved in T2D pathophysiology. In these studies, variants identified in a small number of patients and control subjects are genotyped in larger case-control samples. Both these approaches are characterized by a number of limitations. Linkage analysis is underpowered to detect low penetrance variants, expected to contribute to T2D susceptibility, given its high population prevalence. Candidate gene studies usually were conducted in samples of insufficient size and their findings had low reproducibility as well as difficulty to select good biological candidates.
Positional strategies have identified putative T2D loci in several large chromosome regions (McCarthy 2003) and in a number of specific genes (Horikawa et al. 2000; Meyre et al. 2005; Silander et al. 2004; Hara et al. 2002); however, none of these associations have been convincingly replicated. The candidate-gene approach generated a large number of positive reports, two of which have been confirmed in independent studies (Table 2.2) (Gloyn and McCarthy 2001). The Pro12Ala variant in the peroxisome proliferator-activated receptor gamma (PPARG) gene (Deeb et al. 1998; Altshuler et al. 2000; Lohmueller et al. 2003) and the Glu23Lys variant in the potassium inwardly rectifying channel, subfamily J, member 11 (KCNJ11) gene were shown to contribute to T2D risk in multiple studies (Gloyn et al. 2003; Laukkanen et al. 2004). Each of these two common variants contributes only modestly (increasing T2D risk by 15–20 % for each susceptibility allele) to the risk of developing common form of diabetes, while rare variants in both these genes cause monogenic diseases such as familial partial lipodystrophy and neonatal diabetes. Interestingly, these variants occur within pharmacological targets for the thiazolidinedione (PPARG) and sulfonylurea compounds (KCNJ11) used to treat T2D.
2 Common Variants: The First Steps Toward Large-Scale Association Mapping
Given the inefficient progress of early T2D gene discovery, the application of genome-wide association (GWA) studies to identify risk loci for T2D and glycemic traits represented a major advance in complex trait genetics. GWA studies are observational epidemiological studies in which genetic risk exposure is measured using hundreds of thousands of genotyping assays. The critical difference between GWAs and other observational epidemiological studies lies in the large number of genetic tests performed to assess exposure in each individual patient. On the one hand, the success of GWA studies relies on the development of technologies capable of screening a large number of polymorphisms (predominantly represented by single-nucleotide polymorphisms, SNPs), as the prior probability that any individual polymorphism will be associated with disease is small. On the other hand, the polymorphisms studied using commercial microarray platforms are not genetically independent and display complex linkage structures that may extend over tens or hundreds of thousands of base pairs. As a result, the success of a GWA study relies on achieving an adequate marker density to model local linkage structures across the genome.
The potential benefits of using GWA studies to discover complex disease risk loci were first demonstrated in a seminal paper by Risch and Merikangas that showed that the analysis of one million variants in the sample of unrelated individuals had greater statistical power than a linkage analysis with a few hundred markers (Risch and Merikangas 1996). In this context, Reich and Lander suggested a theoretical population-genetics model for a relatively simple distribution of susceptibility variants at a disease locus and rephrased the common disease common variant hypothesis (CDCV) to propose that high-frequency variants with low penetrance at disease loci contribute to the largest proportion of disease risk in a population (Reich and Lander 2001). Their theoretical demonstration of the CDCV hypothesis did not provide any expectation about the number of disease loci or their effect sizes in establishing complex disease risk.
The majority of GWA studies performed to identify T2D risk loci have used a case-control study design (Table 2.1), with retrospective longitudinal studies being primarily reserved for validation of previously identified loci. Alternate study designs to detect T2D risk associations are far less common and have included populations with early-onset diabetes (taken as a proxy for more severe illness), longitudinal studies in at-risk populations, and studies in isolated populations. Affected individuals in the genetic discovery cohorts are typically selected carefully using diagnostic criteria established by the American Diabetes Association or World Health Organization that are based solely on blood glucose levels. In contrast, selection of control subjects has been more problematic, with most discovery cohorts including patients based on a single normal blood glucose measurement and absent medical history of glucose intolerance. Many discovery cohorts have excluded patients with monogenic diabetes based on a suggestive family history or specific genetic tests and patients with autoimmune diabetes based on specific serum markers. In many studies, the case and control samples differ significantly in age, in order to avoid selecting individuals who will develop diabetes later in life as control subjects. Comparable environmental exposures are used as basis for selection of both, T2D cases and controls; and in addition to matching affected and control individuals for general ethnic background, ethnic outliers are excluded from analyses. Diabetic individuals usually are of comparable body mass index (BMI) in respect to controls in large genetic studies.
The first successful “hypothesis-free” demonstration of T2D association came from the discovery of an intronic SNP in the transcription factor 7-like-2 (TCF7L2) gene, which confers the largest effect on T2D risk reported to date among common variants (Fig. 2.1; Tables 2.1 and 2.2) (Weedon 2007; Grant et al. 2006). These association studies were motivated by the group’s earlier demonstration of microsatellite associations in a linkage region on chromosome 10 (Reynisdottir et al. 2003) rather than by functional criteria. In fact, TCF7L2 encodes a transcription factor within the Wnt signaling pathway whose involvement in T2D pathogenesis remained elusive for many years following the initial genetic studies. Despite this, detailed physiological studies have now demonstrated the importance of the TCF7L2 locus in β-cell function and insulin secretion in human cohorts (da Silva Xavier et al. 2009, 2012; Dupuis et al. 2010; Dimas et al. 2014) and as a critical regulator of β-cell mass and function (Takamoto et al. 2014) and hepatic carbohydrate metabolism (Boj et al. 2012) in mouse models (see Chap. 15 for details).

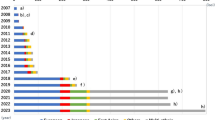
Effect sizes of established T2D-susceptibility loci. Established T2D-susceptibility variants have only modest individual effects. The x-axis gives the per-allele odds ratio estimated for European-descent samples or for the ethnic group of discovery, if association was reported after mid-2012 (Table 2.1) for each locus listed on the y-axis. Loci are sorted by descending order of per-allele effect size within each year. Colors highlight the discovery study approach: red, candidate gene; yellow, large-scale association; blue, genome-wide association; dark blue, genome-wide association meta-analysis; sky blue, genome-wide meta-analysis with Metabochip follow-up; green, genome-wide meta-analysis of glycemic traits; pink, genome-wide sex-differentiated meta-analysis with larger effects in women; brown, genome-wide sex-differentiated meta-analysis with larger effects in men; hacky, genome-wide meta-analysis in lean/obese; gray, whole-exome sequencing. For loci with sex differentiation, the effect size for the sex with larger effect is presented. X-axis lists loci names, labeled by the gene names within region. Y-axis shows odds ratio for T2D observed at a given locus. Loci are split by the year of discovery and are ordered from top to bottom by the decreasing OR on T2D risk within each year. Shadow is used for loci from studies with discovery including non-European individuals
3 Loci Established Through T2D GWA Studies
The capacity to undertake efficient, large-scale association analyses using hypothesis-free approach through genome-wide studies opened a new wave of discoveries in T2D genetics. Four GWA studies published in 2007 (Diabetes Genetics Initiative of Broad Institute of H et al. 2007; Scott et al. 2007b; Sladek et al. 2007; Zeggini et al. 2007) (Table 2.1) confirmed the strongest association at TCF7L2, two previously established signals at PPARG and KCNJ11 and identified six novel loci, at HHEX/IDE, CDKAL1, IGF2BP2, CDKN2A/2B, SLC30A8, and FTO (Frayling et al. 2007; Freathy et al. 2008; Fall et al. 2013). Although it is conventionally used to name the loci by the most credible regional candidate (e.g., SLC30A8) rather than the tag SNP showing the strongest association (e.g., rs13266634), these assignments are used as a matter of convenience and do not imply that a mechanistic link has been proven. The association signals found in GWA studies require further investigation through extensive fine mapping and functional characterization to establish causal variants and determine their impact on T2D pathogenesis at a molecular level (Prokopenko et al. 2008).
The first round of published T2D GWA studies has provided both the identification of novel associated loci and the landscape of T2D susceptibility across the whole genome, the latter providing a point of reference for the previous equivocal findings accumulated through candidate-gene and linkage studies (Parikh and Groop 2004). Thus, controversial evidence for variants in Calpain-10 (CAPN10) and the insulin (INS) gene in T2D has not been confirmed by the GWA approach. Similarly, while the 1q chromosome region (30-Mb region near to centromere) contained a number of highly plausible candidates supported by genome-wide linkage analyses in multiple ethnicities, none have been confirmed through association studies and large-scale GWA meta-analyses to date (Prokopenko et al. 2009b; Morris et al. 2012; Replication and Meta-analysis 2014).
Simultaneously with the advent of GWA studies, large-scale replication efforts confirmed two loci highlighted by the candidate-pathway pre-GWA studies. Variants within Wolfram syndrome 1 (WFS1) gene and common variants in hepatocyte nuclear factor 1-b (HNF1B, also known as TCF2) were confirmed as associated with T2D (Franks et al. 2008; Sandhu et al. 2007; Winckler et al. 2007). These variants, along with KCNJ11 and PPARG, provide interesting examples of the convergence between Mendelian and polygenic causes of diabetes, as coding variants in these genes had previously been isolated in families with autosomal dominant inheritance of diabetes (Maturity Onset Diabetes of the Young, MODY) and as part of the multisystem Wolfram syndrome (McCarthy and Hattersley 2008).
As anticipated, GWA studies, by testing hundreds of thousands of genetic variants in parallel, have identified loci with modest effects (Manolio et al. 2009). To contend with the stringent significance thresholds that account for the number of independent tests performed across the genome, identification of additional T2D susceptibility loci required larger population samples, which was achieved by combining existing GWA studies in meta-analyses. The Diabetes Genetics Replication And Meta-analysis (DIAGRAM, http://www.diagram-consortium.org/) consortium carried out the first meta-analysis for T2D (Zeggini et al. 2008) of three GWA studies of European-descent individuals, including ~4500 cases and 5500 controls. Differences in the genotyping platforms used for individual GWA studies were overcome by imputation using a common variant set based on haplotype structure of densely characterized reference samples in HapMap (Consortium IH 2005) and extended the analysis to ~2.2 million SNPs across the genome (Scott et al. 2007b; Zeggini et al. 2007; Diabetes Genetics Initiative of Broad Institute of H et al. 2007). Following a 2-stage replication with genotyping of selected SNPs in ~75,500 individuals, the DIAGRAM study identified six novel loci (Tables 2.1 and 2.2), including only one reasonable biological candidate gene (NOTCH2, Notch homologue 2, Drosophila), which is involved in pancreatic development.
The DIAGRAM consortium published two further meta-analyses, each based on increasingly larger case-control samples from European populations. The first combined discovery data from 21 GWA studies in up to 8130 individuals with T2D and 38,987 controls all imputed to a HapMap 2 reference panel, followed by large-scale replication in 34,412 cases and 59,925 controls where 13 (11 novel) out of 23 autosomal signals were confirmed (Tables 2.1 and 2.2) (Voight et al. 2010). This meta-analysis was the first to examine T2D associations on chromosome X (taking X-inactivation into account) and identified an association at DUSP9 with a large effect on T2D risk (OR = 1.27, Table 2.2; Fig. 2.1) (Voight et al. 2010). The second meta-analysis, in addition to dramatically increasing the sample size (34,840 cases and 114,981 controls), implemented a novel cost-effective strategy for large-scale replication based on the CardioMetabochip (Metabochip), an Illumina iSelect genotyping array. Metabochip, which was designed through collaboration between six GWA consortia studying metabolic and atherosclerotic/cardiovascular diseases and traits (Voight et al. 2012), permitted follow-up of ~66,000 putative signals for cardiometabolic phenotypes (~5000 of which were selected for T2D) (Morris et al. 2012). The Metabochip array also contained approximately 120,000 SNP probes to fine map 257 established loci in an attempt to identify causal T2D susceptibility variants. The DIAGRAM meta-analysis with Metabochip follow-up established T2D associations at 10 loci (Tables 2.1 and 2.2), including two at CCND2 and GIPR with larger effects on T2D risk in males and females, respectively (Morris et al. 2012). Among previously established T2D loci, sex differentiation in effect size has been shown for KCNQ1, DGKB, and BCL11A (larger effects in males) and GRB14 (larger effects in females).
A separate DIAGRAM GWA meta-analysis of the effects of obesity on T2D risk, performed in Europeans through GWA meta-analysis of lean (BMI < 25 kg/m2) and obese (BMI ≥ 30 kg/m2) T2D diabetics with ~54,000 controls, identified associations with lean diabetic participants at LAMA1 and with obese subjects at HMG20A (Perry et al. 2012). A GWA meta-analysis in >8000 T2D cases and >10,870 controls in Europeans with large replication, including several additional datasets with de novo genotyping and the DIAGRAM discovery meta-analysis data in silico, reported association at RBMS1 (Tables 2.1 and 2.2) (Qi et al. 2010).
In parallel to studies in European populations, T2D GWA studies in Asian ethnic groups (representing Japanese, Chinese, Punjabi Sikhs, Indians, South Asian, and East Asian subjects) have established T2D associations at 27 loci (Table 2.1). These studies have generally followed a design based on a GWA study with large-scale replication in an individual ethnic group, frequently undertaken in multistage fashion. In addition, several groups have combined efforts to complete a recent East Asian GWA meta-analysis in up to 6952 T2D cases and 11,865 controls (with imputation based on the East Asian HapMap 2 reference panel) and identified eight novel loci, including GLIS3, PEPD, FITM2-R3HDML-HNF4A, KCNK16, MAEA, GCC1-PAX4, PSMD6, and ZFAND3 (Cho et al. 2012). A second meta-analysis of Chinese samples (with imputation based on the 1000 Genomes Project JPT (Japanese in Tokyo) and CHB (Han Chinese in Beijing) reference panels) has described T2D association with a common variant in the PAX4 gene , which is expressed in early pancreatic endocrine cells. The association, which was confirmed in a multiethnic analysis including European and five East Asian populations (Ma et al. 2013), adds another example of common variant associations with T2D at a MODY locus as heterozygous mutations in PAX4 have been identified as a cause of MODY9 (omim.org/entry/606391). Therefore, while rare coding mutations severely impair islet function and cause rare monogenic forms of diabetes, common variants can act through the same genes, but with smaller effects, to increase an individual’s risk of developing a more common form of diabetes.
A small number of GWA studies have been reported for other ethnic groups. Studies in Mexican individuals reported associations at several established loci (Parra et al. 2011) and a novel association at SLC16A11/SLC16A13 where the haplotype carriers had amino acid substitutions in SLC16A11 (Consortium et al. 2014b). The locus is thought to affect triacylglycerol metabolism and shows stronger association in leaner and younger people. While common in Native Americans and Asians, risk variants at this locus are rare in European and African individuals and have introgressed into modern humans through admixture with Neanderthals. A second study in American Pima Indians confirmed associations for a set of previously established loci while reaching study-wise significance (P-value = 6.6 × 10−8) at the DNER gene (Hanson et al. 2014). Finally, an African American GWA study has provided evidence for association at RND3/RBM43 (Palmer et al. 2012).
Methodological development in to combine data from multiple ancestry groups by accounting for heterogeneous allelic effects (Morris 2011) has enabled performing meta-analysis across different ethnicities. For example, combining European, East Asian, South Asian, and Mexican and Mexican-American GWA meta-analyses in up to 26,488 T2D cases and 83,964 controls has identified seven novel T2D susceptibility loci TMEM154, SSR1-RREB1, FAF1, POU5F1-TCF19, LPP, ARL15, and MPHOSPH9 (Replication et al. 2014). Importantly, the study demonstrated an overwhelming concordance of allelic effects across ethnicities, even at loci with only weak evidence of association, supporting the hypothesis that T2D risk variants predate migration of humans out of Africa and arguing against the “synthetic association ” hypothesis, which predicts that associations at common variants are driven by unobserved lower frequency causal alleles with large effects (Dickson et al. 2010).
It has long been suggested that the high prevalence of metabolic disorders related to impaired glucose homeostasis may be a result of selective evolutionary advantage of T2D and obesity-risk variants during periods of scarce food resources, which resulted in an increase in their frequency at the population level (thrifty gene hypothesis) (Neel 1962, 1999). Given that food intake is known to act as a trigger for insulin release, it has also been hypothesized that a positive selection may have operated in particular on those loci associated with T2D through an influence on β-cell function (Ayub et al. 2014). Some evidence of directional population differentiation and nominal positive selection at individual T2D risk loci, including TCF7L2, THADA, and NOTCH2, has been reported (Chen et al. 2012; Corona et al. 2013; Klimentidis et al. 2011). The collective analysis of all T2D-associated variants along with stratified by their impact on β-cell function or insulin resistance has to date found no support for global or differential positive selection at T2D loci, thus offering little support for the thrifty gene hypothesis (Ayub et al. 2014; Southam et al. 2009).
4 Common Variants with Modest Effect Sizes
Most GWA study designs are based on common variant genotyping arrays, which have determined the allele spectrum of the resulting T2D-associated variants (Table 2.2). The 88 known T2D risk loci (Table 2.2) show only modest effects (OR = 1.1–1.2), with TCF7L2 being the only locus showing larger effects in European populations (OR ~1.40, Fig. 2.1) (Morris et al. 2012). While this has led to an intense search for additional rare and common variants (particularly for causal variants which are expected to have larger effects), the early search for rare coding variants has had limited success (Table 2.2) (Steinthorsdottir et al. 2014; Albrechtsen et al. 2013). Additionally, studies in non-Europeans have recently provided support for a number of novel T2D susceptibility loci that show low allele frequencies in European populations (Unoki et al. 2008; Hanson et al. 2014; Consortium et al. 2014b). While this provides a challenge to validating these loci in European populations, the high concordance of the direction of effects across ethnicities for T2D risk variants (Replication et al. 2014) suggests that additional common T2D risk variants with consistent and modest effects across ethnic groups remain to be described. Their identification will require larger sample sizes and combined efforts of many studies and research centers (Morris et al. 2012).
The discriminatory capacity of genetic variants for T2D risk prediction and patient stratification has been assessed in longitudinal studies by examining whether inclusion of genetic risk scores (GRS) in predictive models increases the area under the receiver-operating-characteristic curve compared to predictive models including only clinical parameters. Early studies suggested that inclusion of GRS provided little improvement in T2D risk prediction compared to clinical risk factors and family history alone (Lyssenko et al. 2008; Meigs et al. 2008; Balkau et al. 2008; Talmud et al. 2010; de Miguel-Yanes et al. 2011). More recent studies, incorporating increasing numbers of T2D risk variants into the GRS, have also had mixed results (Hivert et al. 2011; Muhlenbruch et al. 2013; Vaxillaire et al. 2014). For example, while a recent study incorporating 43 T2D associated variants showed little improvement in T2D prediction, inclusion of the GRS in predictive models improved the receiver-operating-characteristic curve for subgroups of subjects at increased risk of T2D, including obese subjects, older participants, and those with a family history of diabetes (Muhlenbruch et al. 2013). Similarly, Hivert et al. have shown that a GRS with 34 variants was significantly associated with increased risk of progression to T2D in high-risk individuals, as well as a reduced effect of lifestyle interventions on genetic risk (Hivert et al. 2011).
A recent study comparing the discriminative capacity of GRSs including 65 T2D-associated loci and 36 FG-associated loci FG showed modest but significant improvement in T2D reclassification rates in models including a GRS incorporating T2D risk loci and modestly improved reclassification rates of incident and non-incident T2D and impaired fasting glucose (IFG) using the GRS incorporating both T2D risk and FG loci, suggesting that inclusion of risk loci associated with glycemic traits may be beneficial for intermediate phenotypes such as IFG (Vaxillaire et al. 2014). Further studies using GRS based on new loci and causative variants will help to improve insight into the longitudinal impact of genetic variants associated with glycemic traits on T2D risk of and disease trajectories.
5 Understanding Relationship with Other Phenotypes
Two critical processes leading to T2D development are β-cell dysfunction and insulin resistance in peripheral tissues including fat, muscle, liver, and elsewhere (Prokopenko et al. 2008). Beginning long before the clinical diagnosis of T2D, these processes are hallmarks of prediabetes; following which, progressive deterioration of β-cell function reaches a point when they are no longer able to meet the increased insulin demands from peripheral tissues, leading to the development of diabetes. In parallel to T2D GWA meta-analyses, a number of large-scale association studies have been successful in identifying genetic loci that influence quantitative glycemic traits, including fasting and postprandial glucose and serum insulin levels. These studies take advantage of the increased power that can be obtained when similarly sized cohorts studied for continuous traits compared to dichotomous outcomes; their success relies on the hypothesis that genes influencing blood levels in normal subjects will also increase diabetes risk. Significantly, while the genetic risk loci identified for T2D overlap to some degree with quantitative trait loci for blood glucose and insulin, several genes have shown association only with glycemic traits or only with increased T2D risk (Fig. 2.1). While it’s possible that this discordance may reflect the statistical power of the studies completed to date, the milder phenotypes observed in patients with glucokinase mutations compared to patients with other forms of MODY (McDonald and Ellard 2013) suggest that it is important to distinguish two overlapping but distinct groups of GWAs loci that are associated with altered glucose homeostasis on the one hand and the progressive of metabolic decompensation that leads to T2D on the other.
The only association with FG established before the GWA study era was at the glucokinase (GCK) locus (Weedon et al. 2005, 2006), a gene in which rare mutations cause MODY2 (Froguel et al. 1992). GCK catalyzes the first step in glycolysis and is one of the principal regulators of FG concentration and of β-cell secretory activity. An indicative association at the glucokinase regulator (GCKR) locus (rs780094) with FG, as well as an association at the same variant with serum triglyceride levels, was described by the DGI T2D GWA study, which however was not powered enough to detect an effect on T2D (Diabetes Genetics Initiative of Broad Institute of H et al. 2007). The product of GCKR regulates GCK activity and is a highly plausible candidate involved in T2D pathogenesis (see Chap. 16). The GCK and GCKR loci have since been associated with FG/HOMA-B (homeostasis model assessment of β-cell function) and FG/FI/HOMA-IR (homeostasis model assessment of insulin resistance), respectively, and with T2D (Dupuis et al. 2010; Manning et al. 2012). These findings prompted further interest in well-powered GWA studies for glycemic traits to detect reliable genetic associations which may be relevant to T2D pathogenesis (see Chap. 3).
In 2009, the collaborative Meta-Analyses of Glucose and Insulin-related traits Consortium (MAGIC, http://www.magicinvestigators.org/) was established to consolidate the efforts of many groups working on glycemic trait genetics, in order to understand the variation of these traits within the physiological range and investigate their impact on T2D risk and other cardiometabolic traits (Prokopenko et al. 2009a). The first effort of MAGIC confirmed the association at GCK and G6PC2 loci and identified a novel signal at the melatonin receptor 1B (MTNR1B) locus for higher FG and lower insulin secretion. The inverse correlation between the levels of the neurohormone melatonin, secreted by the pineal gland, and insulin has long been known. However, few studies had investigated the relationship between melatonin signaling in pancreatic islets and metabolic disease (Peschke et al. 2007), prior to publication of large-scale association studies (Prokopenko et al. 2009a; Bouatia-Naji et al. 2009; Chambers et al. 2009; Go et al. 2013; Lyssenko et al. 2009). Association with T2D at MTNR1B locus was subsequently confirmed at genome-wide significance (Prokopenko et al. 2009a; Dupuis et al. 2010; Voight et al. 2010; Lyssenko et al. 2009).
To extend the first MAGIC study, a new, larger, whole GWA meta-analysis (21 studies, up to 46,186 nondiabetic individuals) was performed (Dupuis et al. 2010). It increased the number of glycemic trait loci to 16 and reported novel effects on T2D from a large-scale analysis at five of the FG/FI-associated loci (ADCY5, GCK, GCKR, DGKB, PROX1), thus highlighting that the overlap between the genetic variation influencing glucose homeostasis and risk of T2D is only partial (Fig. 2.1). Four of these loci contributed to impaired β-cell function as measured by HOMA-B and one (GCKR) was associated with insulin resistance.
Three MAGIC GWA meta-analyses for additional glycemic traits provided further insights into pathophysiology of T2D. A study focusing on 2-hour postprandial glucose (2hGlu) levels (15,234 nondiabetic individuals in discovery and up to 30,620 in replication) identified five associated loci (GIPR, VPS13C, ADCY5, GCKR, TCF7L2), including the novel locus GIPR (rs10423928) containing the gene encoding the GIP receptor for the insulin-response stimulating hormone GIP (glucose-dependent insulinotropic polypeptide) in pancreatic islet β-cells (Saxena et al. 2010) and in linkage disequilibrium (LD, HapMap CEU r 2 = 0.78) with BMI-associated rs2287019 (Speliotes et al. 2010). Genome-wide meta-analysis of HbA1c, including study in 46,368 in nondiabetic individuals by the MAGIC investigators, identified 10 genetic loci, of which MTNR1B and GCK also increase T2D risk, suggesting that their effect on hyperglycemia (as measured by FG) extends an effect on average glycemia over a 2- to 3-month period (as detected through HbA1c) and is related to T2D pathogenesis, while ANK1 maps close to T2D risk variant (Soranzo et al. 2010). Variants at VPS13C/C2CD4A/B and GIPR were subsequently associated with T2D, the latter showing larger effects in women, but both were in weak LD with glycemic trait variants (Yamauchi et al. 2010; Morris et al. 2012). Similarly, the ANK1 HbA1c locus variant rs4737009 identified by Soranzo et al. is not in LD with the T2D risk variant (rs516946, HapMap CEU r 2 < 0.01) (Morris et al. 2012). The genetic architecture at these three loci is complex and requires further investigation to dissect the relationships between genetic effects on the associated glycemic phenotypes.
Large-scale studies of glycemic traits using Metabochip have discovered additional common variant loci with small effects on FG/FI/2hGlu loci trait variability and further increased the overlap with T2D risk loci. In this study, 39 FG-raising alleles were related to increased T2D risk, although only 20 (>60 %) of them showed at least nominal significance (P < 0.05) for T2D. Similarly, 13 of the 19 FI loci were nominally associated with T2D and all but TCF7L2. Similarly, 13 of the 19 FI loci were nominal association with T2D and all, but TCF7L2, FI/insulin resistance-increasing alleles were associated with higher T2D risk and showed an impaired lipid profile (Fig. 2.2) (Scott et al. 2012).

Effects of established T2D loci: (a) on cardiometabolic phenotypes and (b) on glycemic traits. (a) Among a total 88 T2D loci, only 49 do not overlap with other cardiometabolic traits. The independent loci were defined by physical distance more than 500 kb from each other and by CEU LD r 2 > 0.01. (b) Among a total 88 T2D loci, only 27 overlap with fasting glucose or fasting insulin levels. The independent loci were defined by physical distance more than 500 kb from each other and by CEU LD r 2 > 0.01
FG-associated loci from GWAS studies have also helped define the relationship between T2D and abnormal insulin processing and secretion in β-cells. Among other glycemic trait analyses by the MAGIC, nine genome-wide significant loci were described for corrected insulin response (CIR), seven of which were previously associated with both T2D and other glycemic traits (MTNR1B, GCK, HHEX/IDE, CDKAL1, CDKN2A/2B, ANK1, C2CD4A/B) (Prokopenko et al. 2014). Two other loci included G6PC2 associated with glycemic trait variability in nondiabetic individuals and the novel GRB10 association, which showed potential tissue-specific methylation and parental imprinting that might mask its association with T2D). Meta-analysis of GWA studies by MAGIC for fasting proinsulin levels adjusted for FI identified eight loci, of which four demonstrated that both proinsulin-raising (for TCF7L2, SLC30A8, and VPS13C/C2CD4A/B) and proinsulin-lowering alleles (for ARAP1) influenced T2D risk through a decrease in insulin secretion caused by distal or proximal impairment of proinsulin conversion, respectively (Strawbridge et al. 2011). Similarly, Dimas and colleagues described associations at the HHEX/IDE and MTNR1B loci with defects in early insulin secretion through reduced insulinogenic index for the T2D risk allele and showed that the T2D risk allele at ARAP1 was related to defects in the first steps of insulin production, through association with 32,33 split proinsulin (Dimas et al. 2014).
These large-scale discovery efforts in nondiabetic individuals have provided genetic markers that may provide mechanistic insights into the pathogenesis of T2D and possibly to classify disease mechanisms that are active in individual patients. For example, physiological characterization of the effects of glycemic and T2D loci on quantitative glycemic traits has revealed a clear separation of hyperglycemic loci (MTNR1B and GCK) which are associated with reduced basal and stimulated β-cell secretion and consequent fasting hyperglycemia without large effects on T2D risk from β-cell loci that show an effect on insulin processing and secretion that only modestly change FG but exert much stronger effects on T2D risk (TCF7L2, SLC30A8, HHEX/IDE, CDKAL1, CDKN2A/2B, THADA, DGKB, PROX1, ADCY5) (Dimas et al. 2014).
Loci with effects on insulin sensitivity represent a much smaller proportion of T2D variants. Physiological characterization of T2D loci grouped variants with primary effects on insulin sensitivity in basal and stimulated state (IRS1, GCKR, PPARG, KLF14); in addition, weak effects on insulin sensitivity have also been suggested for HMGA2 (Voight et al. 2010). Insulin sensitivity indices showed consistently decreased effects for T2D risk alleles only for loci with known effects on insulin resistance at basal measures (HOMA-IR) (Dimas et al. 2014). In many cases, these loci may exert widespread biochemical changes affecting cardiometabolic risk (Fig. 2.2): some FI-associated loci can alter BMI and body fat distribution, while most loci associated with higher insulin levels are also associated with lower HDL cholesterol and higher triglyceride levels (Manning et al. 2012). For example, variants within the fat mass and obesity-associated (FTO) gene and at melanocortin-4 receptor (MC4R) exert their T2D effect through a primary impact on BMI (Frayling et al. 2007; Loos et al. 2008; Morris et al. 2012). In contrast, effects of IRS1 and PPARG on insulin resistance and T2D are independent from obesity (Scott et al. 2012; Kilpelainen et al. 2011; Rung et al. 2009). For a number of loci, the association with lipids and T2D (HNF4A, CILP2, KLF14, HNF1A/TCF1, MC4R) and additionally with FI (GRB14, GCKR, FTO, PEPD, ANKRD55, IRS1, ARL15) has been reported independently for each phenotype, underlying the close relationship between increased lipids/adiposity and increased insulin (Fig. 2.2) (Scott et al. 2012). This picture is consistent with the first stages of diabetes, where high adiposity in peripheral tissues causes insulin resistance, which is complemented by an increase in β-cell insulin production.
Several T2D loci appear to have an effect on complex diseases whose pathogenesis is not commonly associated with changes in metabolic fitness : pleiotropy could be a probable mechanism for these effects, since the correlation between the associated disease outcomes is low for them to be considered as comorbidities. Thus, variants at ~20 T2D loci, including CDKN2A/2B, JAZF1, HNF1B, THADA, CCND2, ZMIZ1, and IGF2, have a role in cancer susceptibility (Gudmundsson et al. 2007; Thomas et al. 2008; Finkel et al. 2007). Interestingly, T2D risk alleles at THADA, TSPAN8, and HNF1B are protective against prostate cancer, an inverse relationship that supports epidemiological observations. The genetic links between diabetes and cancer point to a set of shared biological pathways, including opposing roles in regulation of cell cycle and common signaling pathways.
6 What Is Next in T2D GWA Studies?
Despite the success of GWA studies in identification of common variant associations, the largest heritable component of T2D susceptibility remains unexplained. Rapid development and reduced costs of exome sequencing approaches has opened wide opportunities in both sequencing of large numbers of individuals and generation of large reference panels for imputation of rare variants from resequencing [e.g., those from the 1000 Genomes Project (Genomes Project et al. 2010)]. Population-based studies have also benefited from sequencing through implementation of population-specific next-generation sequencing-based reference panels, including deCODE Icelandic and Genome of the Netherlands (GoNL) reference panels (Boomsma et al. 2014; Steinthorsdottir et al. 2014). To date, these sequencing studies have not succeeded in identifying a large number of novel risk loci. For example, whole-exome sequencing at 8× depth in a Danish sample of 1000 T2D cases and 1000 controls hasn’t produced evidence of association with T2D at rare exomic variants, but has confirmed associations with T2D at common variants in COBLL1 and MACF1 (Tables 2.1 and 2.2) (Albrechtsen et al. 2013). While the sample size used in the study was small and the variant calling accuracy was not optimal for detecting small indels or changes in copy number, the results are consistent with previous regional resequencing studies which suggest that most causative variants linked to the GWA risk loci will not alter protein coding sequences. A recent whole-exome sequencing study in 3756 Latinos with an average depth 67.17× has identified a rare missense variant in HNF1A (c.1522G>A [p.E508K], odds ratio [OR] = 5.48) (Consortium et al. 2014a). As a result, there is considerable interest in pursuing whole-genome and whole-exome sequencing studies, particularly in cohorts that have sufficient statistical power to detect epistatic interactions that may confer additional T2D risk. Several international T2D collaborations have recently focused their efforts on large-scale sequencing projects, including the GoT2D (genomics of T2D) consortium that has undertook whole-genome (low-pass 4–6×) and deep whole-exome sequencing for ~2800 T2D case and control individuals from Northern Europe and the T2D Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples (T2D-GENES ) consortium that has undertook trans-ethnic deep whole-exome sequencing in ~10,000 individuals distributed equally between five ethnic groups (McCarthy 2010).
GWA studies have provided an excellent springboard for large-scale T2D studies through international collaborative efforts focused on Europeans and being widely extended to other ethnic groups. Improved sequencing technologies and variant calling algorithms will extend the variant set to other types of genetic variability, including copy number variation, which may have significant impact on the dissection of T2D susceptibility. These collaborations will enable well-powered fine-mapping studies and identification and functional characterization of disease-causing variants. Overall, identifying causative genetic variants and discovering the molecular mechanisms linking them to the development of prediabetic changes will be essential in understanding the pathophysiology of T2D. This in turn may lead to rational drug development and suggest therapies that can be applied appropriately and early to those most at risk of developing T2D (Tuomilehto and Lindstrom 2003). This outcome is potentially feasible as genes that have already been associated with diabetes have also acted as targets for its treatment : while this is best demonstrated by the use of sulfonylureas to treat neonatal diabetes associated with inactivating mutations of the Sur1 protein (Gloyn et al. 2004), the same family of drugs have also provided a mainstay for treating adults with polygenic T2D for many years.
References
Albrechtsen A, Grarup N, Li Y, Sparso T, Tian G, Cao H, Jiang T, Kim SY, Korneliussen T, Li Q, Nie C, Wu R, Skotte L, Morris AP, Ladenvall C, Cauchi S, Stancakova A, Andersen G, Astrup A, Banasik K, Bennett AJ, Bolund L, Charpentier G, Chen Y, Dekker JM, Doney AS, Dorkhan M, Forsen T, Frayling TM, Groves CJ, Gui Y, Hallmans G, Hattersley AT, He K, Hitman GA, Holmkvist J, Huang S, Jiang H, Jin X, Justesen JM, Kristiansen K, Kuusisto J, Lajer M, Lantieri O, Li W, Liang H, Liao Q, Liu X, Ma T, Ma X, Manijak MP, Marre M, Mokrosinski J, Morris AD, Mu B, Nielsen AA, Nijpels G, Nilsson P, Palmer CN, Rayner NW, Renstrom F, Ribel-Madsen R, Robertson N, Rolandsson O, Rossing P, Schwartz TW, Group DESIRS, Slagboom PE, Sterner M, Consortium D, Tang M, Tarnow L, Tuomi T, Van’t Riet E, van Leeuwen N, Varga TV, Vestmar MA, Walker M, Wang B, Wang Y, Wu H, Xi F, Yengo L, Yu C, Zhang X, Zhang J, Zhang Q, Zhang W, Zheng H, Zhou Y, Altshuler D, Thart LM, Franks PW, Balkau B, Froguel P, McCarthy MI, Laakso M, Groop L, Christensen C, Brandslund I, Lauritzen T, Witte DR, Linneberg A, Jorgensen T, Hansen T, Wang J, Nielsen R, Pedersen O (2013) Exome sequencing-driven discovery of coding polymorphisms associated with common metabolic phenotypes. Diabetologia 56(2):298–310. doi:10.1007/s00125-012-2756-1
Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, Nemesh J, Lane CR, Schaffner SF, Bolk S, Brewer C, Tuomi T, Gaudet D, Hudson TJ, Daly M, Groop L, Lander ES (2000) The common PPARgamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nat Genet 26(1):76–80. doi:10.1038/79216
Ayub Q, Moutsianas L, Chen Y, Panoutsopoulou K, Colonna V, Pagani L, Prokopenko I, Ritchie GR, Tyler-Smith C, McCarthy MI, Zeggini E, Xue Y (2014) Revisiting the thrifty gene hypothesis via 65 loci associated with susceptibility to type 2 diabetes. Am J Hum Genet 94(2):176–185. doi:10.1016/j.ajhg.2013.12.010
Balkau B, Lange C, Fezeu L, Tichet J, de Lauzon-Guillain B, Czernichow S, Fumeron F, Froguel P, Vaxillaire M, Cauchi S, Ducimetiere P, Eschwege E (2008) Predicting diabetes: clinical, biological, and genetic approaches: data from the Epidemiological Study on the Insulin Resistance Syndrome (DESIR). Diabet Care 31(10):2056–2061. doi:10.2337/dc08-0368
Boj SF, van Es JH, Huch M, Li VS, Jose A, Hatzis P, Mokry M, Haegebarth A, van den Born M, Chambon P, Voshol P, Dor Y, Cuppen E, Fillat C, Clevers H (2012) Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 151(7):1595–1607. doi:10.1016/j.cell.2012.10.053
Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, Ye K, Guryev V, Vermaat M, van Dijk F, Francioli LC, Hottenga JJ, Laros JF, Li Q, Li Y, Cao H, Chen R, Du Y, Li N, Cao S, van Setten J, Menelaou A, Pulit SL, Hehir-Kwa JY, Beekman M, Elbers CC, Byelas H, de Craen AJ, Deelen P, Dijkstra M, den Dunnen JT, de Knijff P, Houwing-Duistermaat J, Koval V, Estrada K, Hofman A, Kanterakis A, Enckevort D, Mai H, Kattenberg M, van Leeuwen EM, Neerincx PB, Oostra B, Rivadeneira F, Suchiman EH, Uitterlinden AG, Willemsen G, Wolffenbuttel BH, Wang J, de Bakker PI, van Ommen GJ, van Duijn CM (2014) The genome of the Netherlands: design, and project goals. Eur J Hum Genet 22(2):221–227. doi:10.1038/ejhg.2013.118
Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, Sparso T, Holmkvist J, Marchand M, Delplanque J, Lobbens S, Rocheleau G, Durand E, De Graeve F, Chevre JC, Borch-Johnsen K, Hartikainen AL, Ruokonen A, Tichet J, Marre M, Weill J, Heude B, Tauber M, Lemaire K, Schuit F, Elliott P, Jorgensen T, Charpentier G, Hadjadj S, Cauchi S, Vaxillaire M, Sladek R, Visvikis-Siest S, Balkau B, Levy-Marchal C, Pattou F, Meyre D, Blakemore AI, Jarvelin MR, Walley AJ, Hansen T, Dina C, Pedersen O, Froguel P (2009) A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet 41(1):89–94. doi:10.1038/ng.277
Chambers JC, Zhang W, Zabaneh D, Sehmi J, Jain P, McCarthy MI, Froguel P, Ruokonen A, Balding D, Jarvelin MR, Scott J, Elliott P, Kooner JS (2009) Common genetic variation near melatonin receptor MTNR1B contributes to raised plasma glucose and increased risk of type 2 diabetes among Indian Asians and European Caucasians. Diabetes 58(11):2703–2708. doi:10.2337/db08-1805
Chen R, Corona E, Sikora M, Dudley JT, Morgan AA, Moreno-Estrada A, Nilsen GB, Ruau D, Lincoln SE, Bustamante CD, Butte AJ (2012) Type 2 diabetes risk alleles demonstrate extreme directional differentiation among human populations, compared to other diseases. PLoS Genet 8(4):e1002621. doi:10.1371/journal.pgen.1002621
Cho YS, Chen CH, Hu C, Long J, Ong RT, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, Chang YC, Kwak SH, Ma RC, Yamamoto K, Adair LS, Aung T, Cai Q, Chang LC, Chen YT, Gao Y, Hu FB, Kim HL, Kim S, Kim YJ, Lee JJ, Lee NR, Li Y, Liu JJ, Lu W, Nakamura J, Nakashima E, Ng DP, Tay WT, Tsai FJ, Wong TY, Yokota M, Zheng W, Zhang R, Wang C, So WY, Ohnaka K, Ikegami H, Hara K, Cho YM, Cho NH, Chang TJ, Bao Y, Hedman AK, Morris AP, McCarthy MI, Consortium D, Mu TC, Takayanagi R, Park KS, Jia W, Chuang LM, Chan JC, Maeda S, Kadowaki T, Lee JY, Wu JY, Teo YY, Tai ES, Shu XO, Mohlke KL, Kato N, Han BG, Seielstad M (2012) Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 44(1):67–72. doi:10.1038/ng.1019
Consortium IH (2005) A haplotype map of the human genome. Nature 437(7063):1299–1320. doi:10.1038/nature04226
Consortium STD, Estrada K, Aukrust I, Bjorkhaug L, Burtt NP, Mercader JM, Garcia-Ortiz H, Huerta-Chagoya A, Moreno-Macias H, Walford G, Flannick J, Williams AL, Gomez-Vazquez MJ, Fernandez-Lopez JC, Martinez-Hernandez A, Centeno-Cruz F, Mendoza-Caamal E, Revilla-Monsalve C, Islas-Andrade S, Cordova EJ, Soberon X, Gonzalez-Villalpando ME, Henderson E, Wilkens LR, Le Marchand L, Arellano-Campos O, Ordonez-Sanchez ML, Rodriguez-Torres M, Rodriguez-Guillen R, Riba L, Najmi LA, Jacobs SB, Fennell T, Gabriel S, Fontanillas P, Hanis CL, Lehman DM, Jenkinson CP, Abboud HE, Bell GI, Cortes ML, Boehnke M, Gonzalez-Villalpando C, Orozco L, Haiman CA, Tusie-Luna T, Aguilar-Salinas CA, Altshuler D, Njolstad PR, Florez JC, MacArthur DG (2014a) Association of a low-frequency variant in HNF1A with type 2 diabetes in a Latino population. JAMA 311(22):2305–2314. doi:10.1001/jama.2014.6511
Consortium STD, Williams AL, Jacobs SB, Moreno-Macias H, Huerta-Chagoya A, Churchhouse C, Marquez-Luna C, Garcia-Ortiz H, Gomez-Vazquez MJ, Burtt NP, Aguilar-Salinas CA, Gonzalez-Villalpando C, Florez JC, Orozco L, Haiman CA, Tusie-Luna T, Altshuler D (2014b) Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature 506(7486):97–101. doi:10.1038/nature12828
Corona E, Chen R, Sikora M, Morgan AA, Patel CJ, Ramesh A, Bustamante CD, Butte AJ (2013) Analysis of the genetic basis of disease in the context of worldwide human relationships and migration. PLoS Genet 9(5):e1003447. doi:10.1371/journal.pgen.1003447
da Silva XG, Loder MK, McDonald A, Tarasov AI, Carzaniga R, Kronenberger K, Barg S, Rutter GA (2009) TCF7L2 regulates late events in insulin secretion from pancreatic islet beta-cells. Diabetes 58(4):894–905. doi:10.2337/db08-1187
da Silva XG, Mondragon A, Sun G, Chen L, McGinty JA, French PM, Rutter GA (2012) Abnormal glucose tolerance and insulin secretion in pancreas-specific Tcf7l2-null mice. Diabetologia 55(10):2667–2676. doi:10.1007/s00125-012-2600-7
de Miguel-Yanes JM, Shrader P, Pencina MJ, Fox CS, Manning AK, Grant RW, Dupuis J, Florez JC, D’Agostino RB Sr, Cupples LA, Meigs JB, Investigators M, Investigators D (2011) Genetic risk reclassification for type 2 diabetes by age below or above 50 years using 40 type 2 diabetes risk single nucleotide polymorphisms. Diabet Care 34(1):121–125. doi:10.2337/dc10-1265
Deeb SS, Fajas L, Nemoto M, Pihlajamaki J, Mykkanen L, Kuusisto J, Laakso M, Fujimoto W, Auwerx J (1998) A Pro12Ala substitution in PPARgamma2 associated with decreased receptor activity, lower body mass index and improved insulin sensitivity. Nat Genet 20(3):284–287. doi:10.1038/3099
Diabetes Genetics Initiative of Broad Institute of H, Mit LU, Novartis Institutes of BioMedical R, Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, Hughes TE, Groop L, Altshuler D, Almgren P, Florez JC, Meyer J, Ardlie K, Bengtsson Bostrom K, Isomaa B, Lettre G, Lindblad U, Lyon HN, Melander O, Newton-Cheh C, Nilsson P, Orho-Melander M, Rastam L, Speliotes EK, Taskinen MR, Tuomi T, Guiducci C, Berglund A, Carlson J, Gianniny L, Hackett R, Hall L, Holmkvist J, Laurila E, Sjogren M, Sterner M, Surti A, Svensson M, Svensson M, Tewhey R, Blumenstiel B, Parkin M, Defelice M, Barry R, Brodeur W, Camarata J, Chia N, Fava M, Gibbons J, Handsaker B, Healy C, Nguyen K, Gates C, Sougnez C, Gage D, Nizzari M, Gabriel SB, Chirn GW, Ma Q, Parikh H, Richardson D, Ricke D, Purcell S (2007) Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science 316(5829):1331–1336. doi:10.1126/science.1142358
DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium, Asian Genetic Epidemiology Network Type 2 Diabetes (AGEN-T2D) Consortium, South Asian Type 2 Diabetes (SAT2D) Consortium, Mexican American Type 2 Diabetes (MAT2D) Consortium, Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in multi-Ethnic Samples (T2D-GENES) Consortium, Mahajan A et al (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 46(3):234–244
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB (2010) Rare variants create synthetic genome-wide associations. PLoS Biol 8(1):e1000294. doi:10.1371/journal.pbio.1000294
Dimas AS, Lagou V, Barker A, Knowles JW, Magi R, Hivert MF, Benazzo A, Rybin D, Jackson AU, Stringham HM, Song C, Fischer-Rosinsky A, Boesgaard TW, Grarup N, Abbasi FA, Assimes TL, Hao K, Yang X, Lecoeur C, Barroso I, Bonnycastle LL, Bottcher Y, Bumpstead S, Chines PS, Erdos MR, Graessler J, Kovacs P, Morken MA, Narisu N, Payne F, Stancakova A, Swift AJ, Tonjes A, Bornstein SR, Cauchi S, Froguel P, Meyre D, Schwarz PE, Haring HU, Smith U, Boehnke M, Bergman RN, Collins FS, Mohlke KL, Tuomilehto J, Quertemous T, Lind L, Hansen T, Pedersen O, Walker M, Pfeiffer AF, Spranger J, Stumvoll M, Meigs JB, Wareham NJ, Kuusisto J, Laakso M, Langenberg C, Dupuis J, Watanabe RM, Florez JC, Ingelsson E, McCarthy MI, Prokopenko I, Investigators M (2014) Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes 63(6):2158–2171. doi:10.2337/db13-0949
Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU, Wheeler E, Glazer NL, Bouatia-Naji N, Gloyn AL, Lindgren CM, Magi R, Morris AP, Randall J, Johnson T, Elliott P, Rybin D, Thorleifsson G, Steinthorsdottir V, Henneman P, Grallert H, Dehghan A, Hottenga JJ, Franklin CS, Navarro P, Song K, Goel A, Perry JR, Egan JM, Lajunen T, Grarup N, Sparso T, Doney A, Voight BF, Stringham HM, Li M, Kanoni S, Shrader P, Cavalcanti-Proenca C, Kumari M, Qi L, Timpson NJ, Gieger C, Zabena C, Rocheleau G, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Payne F, Roccasecca RM, Pattou F, Sethupathy P, Ardlie K, Ariyurek Y, Balkau B, Barter P, Beilby JP, Ben-Shlomo Y, Benediktsson R, Bennett AJ, Bergmann S, Bochud M, Boerwinkle E, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, Brunner E, Bumpstead SJ, Charpentier G, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Cornelis M, Crawford G, Crisponi L, Day IN, de Geus EJ, Delplanque J, Dina C, Erdos MR, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Fox CS, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Groves CJ, Grundy S, Gwilliam R, Gyllensten U, Hadjadj S, Hallmans G, Hammond N, Han X, Hartikainen AL, Hassanali N, Hayward C, Heath SC, Hercberg S, Herder C, Hicks AA, Hillman DR, Hingorani AD, Hofman A, Hui J, Hung J, Isomaa B, Johnson PR, Jorgensen T, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Lyssenko V, Mahley R, Mangino M, Manning AK, Martinez-Larrad MT, McAteer JB, McCulloch LJ, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Morken MA, Mukherjee S, Naitza S, Narisu N, Neville MJ, Oostra BA, Orru M, Pakyz R, Palmer CN, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rathmann W, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Roden M, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Scott LJ, Seedorf U, Sharp SJ, Shields B, Sigurethsson G, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvanen AC, Tanaka T, Thorand B, Tichet J, Tonjes A, Tuomi T, Uitterlinden AG, van Dijk KW, van Hoek M, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Walters GB, Ward KL, Watkins H, Weedon MN, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zeggini E, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Consortium D, Consortium G, Global BC, Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Hattersley AT, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Rios M, Morris AD, Lind L, Palmer LJ, Hu FB, Franks PW, Ebrahim S, Marmot M, Kao WH, Pankow JS, Sampson MJ, Kuusisto J, Laakso M, Hansen T, Pedersen O, Pramstaller PP, Wichmann HE, Illig T, Rudan I, Wright AF, Stumvoll M, Campbell H, Wilson JF, Anders Hamsten on behalf of Procardis C, Investigators M, Bergman RN, Buchanan TA, Collins FS, Mohlke KL, Tuomilehto J, Valle TT, Altshuler D, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Frayling TM, Ferrucci L, Kong A, Thorsteinsdottir U, Stefansson K, van Duijn CM, Aulchenko YS, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Abecasis GR, Wareham NJ, Sladek R, Froguel P, Watanabe RM, Meigs JB, Groop L, Boehnke M, McCarthy MI, Florez JC, Barroso I (2010) New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet 42(2):105–116. doi:10.1038/ng.520
Fall T, Hagg S, Magi R, Ploner A, Fischer K, Horikoshi M, Sarin AP, Thorleifsson G, Ladenvall C, Kals M, Kuningas M, Draisma HH, Ried JS, van Zuydam NR, Huikari V, Mangino M, Sonestedt E, Benyamin B, Nelson CP, Rivera NV, Kristiansson K, Shen HY, Havulinna AS, Dehghan A, Donnelly LA, Kaakinen M, Nuotio ML, Robertson N, de Bruijn RF, Ikram MA, Amin N, Balmforth AJ, Braund PS, Doney AS, Doring A, Elliott P, Esko T, Franco OH, Gretarsdottir S, Hartikainen AL, Heikkila K, Herzig KH, Holm H, Hottenga JJ, Hypponen E, Illig T, Isaacs A, Isomaa B, Karssen LC, Kettunen J, Koenig W, Kuulasmaa K, Laatikainen T, Laitinen J, Lindgren C, Lyssenko V, Laara E, Rayner NW, Mannisto S, Pouta A, Rathmann W, Rivadeneira F, Ruokonen A, Savolainen MJ, Sijbrands EJ, Small KS, Smit JH, Steinthorsdottir V, Syvanen AC, Taanila A, Tobin MD, Uitterlinden AG, Willems SM, Willemsen G, Witteman J, Perola M, Evans A, Ferrieres J, Virtamo J, Kee F, Tregouet DA, Arveiler D, Amouyel P, Ferrario MM, Brambilla P, Hall AS, Heath AC, Madden PA, Martin NG, Montgomery GW, Whitfield JB, Jula A, Knekt P, Oostra B, van Duijn CM, Penninx BW, Davey Smith G, Kaprio J, Samani NJ, Gieger C, Peters A, Wichmann HE, Boomsma DI, de Geus EJ, Tuomi T, Power C, Hammond CJ, Spector TD, Lind L, Orho-Melander M, Palmer CN, Morris AD, Groop L, Jarvelin MR, Salomaa V, Vartiainen E, Hofman A, Ripatti S, Metspalu A, Thorsteinsdottir U, Stefansson K, Pedersen NL, McCarthy MI, Ingelsson E, Prokopenko I, European Network for G, Genomic Epidemiology C (2013) The role of adiposity in cardiometabolic traits: a Mendelian randomization analysis. PLoS Med 10(6):e1001474. doi:10.1371/journal.pmed.1001474
Federation ID (2013) IDF diabetes atlas, 6th edn. International Diabetes Federation, Brussels
Finkel T, Serrano M, Blasco MA (2007) The common biology of cancer and ageing. Nature 448(7155):767–774. doi:10.1038/nature05985
Franks PW, Rolandsson O, Debenham SL, Fawcett KA, Payne F, Dina C, Froguel P, Mohlke KL, Willer C, Olsson T, Wareham NJ, Hallmans G, Barroso I, Sandhu MS (2008) Replication of the association between variants in WFS1 and risk of type 2 diabetes in European populations. Diabetologia 51(3):458–463. doi:10.1007/s00125-007-0887-6
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, Shields B, Harries LW, Barrett JC, Ellard S, Groves CJ, Knight B, Patch AM, Ness AR, Ebrahim S, Lawlor DA, Ring SM, Ben-Shlomo Y, Jarvelin MR, Sovio U, Bennett AJ, Melzer D, Ferrucci L, Loos RJ, Barroso I, Wareham NJ, Karpe F, Owen KR, Cardon LR, Walker M, Hitman GA, Palmer CN, Doney AS, Morris AD, Smith GD, Hattersley AT, McCarthy MI (2007) A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 316(5826):889–894. doi:10.1126/science.1141634, 1141634 [pii]
Freathy RM, Timpson NJ, Lawlor DA, Pouta A, Ben-Shlomo Y, Ruokonen A, Ebrahim S, Shields B, Zeggini E, Weedon MN, Lindgren CM, Lango H, Melzer D, Ferrucci L, Paolisso G, Neville MJ, Karpe F, Palmer CN, Morris AD, Elliott P, Jarvelin MR, Smith GD, McCarthy MI, Hattersley AT, Frayling TM (2008) Common variation in the FTO gene alters diabetes-related metabolic traits to the extent expected given its effect on BMI. Diabetes 57(5):1419–1426. doi:10.2337/db07-1466, db07-1466 [pii]
Froguel P, Vaxillaire M, Sun F, Velho G, Zouali H, Butel MO, Lesage S, Vionnet N, Clement K, Fougerousse F et al (1992) Close linkage of glucokinase locus on chromosome 7p to early-onset non-insulin-dependent diabetes mellitus. Nature 356(6365):162–164. doi:10.1038/356162a0
Genomes Project C, Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA (2010) A map of human genome variation from population-scale sequencing. Nature 467(7319):1061–1073. doi:10.1038/nature09534
Gloyn AL, McCarthy MI (2001) The genetics of type 2 diabetes. Best Pract Res Clin Endocrinol Metab 15(3):293–308. doi:10.1053/beem.2001.0147
Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, McCarthy MI, Hattersley AT, Frayling TM (2003) Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 52(2):568–572
Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, Edghill EL, Frayling TM, Temple IK, Mackay D, Shield JP, Sumnik Z, van Rhijn A, Wales JK, Clark P, Gorman S, Aisenberg J, Ellard S, Njolstad PR, Ashcroft FM, Hattersley AT (2004) Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. N Engl J Med 350(18):1838–1849. doi:10.1056/NEJMoa032922
Go MJ, Hwang JY, Kim YJ, Hee Oh J, Kim YJ, Heon Kwak S, Soo Park K, Lee J, Kim BJ, Han BG, Cho MC, Cho YS, Lee JY (2013) New susceptibility loci in MYL2, C12orf51 and OAS1 associated with 1-h plasma glucose as predisposing risk factors for type 2 diabetes in the Korean population. J Hum Genet 58(6):362–365. doi:10.1038/jhg.2013.14
Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, Helgason A, Stefansson H, Emilsson V, Helgadottir A, Styrkarsdottir U, Magnusson KP, Walters GB, Palsdottir E, Jonsdottir T, Gudmundsdottir T, Gylfason A, Saemundsdottir J, Wilensky RL, Reilly MP, Rader DJ, Bagger Y, Christiansen C, Gudnason V, Sigurdsson G, Thorsteinsdottir U, Gulcher JR, Kong A, Stefansson K (2006) Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet 38(3):320–323. doi:10.1038/ng1732, ng1732 [pii]
Gudmundsson J, Sulem P, Steinthorsdottir V, Bergthorsson JT, Thorleifsson G, Manolescu A, Rafnar T, Gudbjartsson D, Agnarsson BA, Baker A, Sigurdsson A, Benediktsdottir KR, Jakobsdottir M, Blondal T, Stacey SN, Helgason A, Gunnarsdottir S, Olafsdottir A, Kristinsson KT, Birgisdottir B, Ghosh S, Thorlacius S, Magnusdottir D, Stefansdottir G, Kristjansson K, Bagger Y, Wilensky RL, Reilly MP, Morris AD, Kimber CH, Adeyemo A, Chen Y, Zhou J, So WY, Tong PC, Ng MC, Hansen T, Andersen G, Borch-Johnsen K, Jorgensen T, Tres A, Fuertes F, Ruiz-Echarri M, Asin L, Saez B, van Boven E, Klaver S, Swinkels DW, Aben KK, Graif T, Cashy J, Suarez BK, van Vierssen TO, Frigge ML, Ober C, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Palmer CN, Rotimi C, Chan JC, Pedersen O, Sigurdsson G, Benediktsson R, Jonsson E, Einarsson GV, Mayordomo JI, Catalona WJ, Kiemeney LA, Barkardottir RB, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) Two variants on chromosome 17 confer prostate cancer risk, and the one in TCF2 protects against type 2 diabetes. Nat Genet 39(8):977–983. doi:10.1038/ng2062
Hanson RL, Muller YL, Kobes S, Guo T, Bian L, Ossowski V, Wiedrich K, Sutherland J, Wiedrich C, Mahkee D, Huang K, Abdussamad M, Traurig M, Weil EJ, Nelson RG, Bennett PH, Knowler WC, Bogardus C, Baier LJ (2014) A genome-wide association study in American Indians implicates DNER as a susceptibility locus for type 2 diabetes. Diabetes 63(1):369–376. doi:10.2337/db13-0416
Hara K, Boutin P, Mori Y, Tobe K, Dina C, Yasuda K, Yamauchi T, Otabe S, Okada T, Eto K, Kadowaki H, Hagura R, Akanuma Y, Yazaki Y, Nagai R, Taniyama M, Matsubara K, Yoda M, Nakano Y, Tomita M, Kimura S, Ito C, Froguel P, Kadowaki T (2002) Genetic variation in the gene encoding adiponectin is associated with an increased risk of type 2 diabetes in the Japanese population. Diabetes 51(2):536–540
Hara K, Fujita H, Johnson TA, Yamauchi T, Yasuda K, Horikoshi M, Peng C, Hu C, Ma RC, Imamura M, Iwata M, Tsunoda T, Morizono T, Shojima N, So WY, Leung TF, Kwan P, Zhang R, Wang J, Yu W, Maegawa H, Hirose H, Consortium D, Kaku K, Ito C, Watada H, Tanaka Y, Tobe K, Kashiwagi A, Kawamori R, Jia W, Chan JC, Teo YY, Shyong TE, Kamatani N, Kubo M, Maeda S, Kadowaki T (2014) Genome-wide association study identifies three novel loci for type 2 diabetes. Hum Mol Genet 23(1):239–246. doi:10.1093/hmg/ddt399
Hivert MF, Jablonski KA, Perreault L, Saxena R, McAteer JB, Franks PW, Hamman RF, Kahn SE, Haffner S, Consortium D, Meigs JB, Altshuler D, Knowler WC, Florez JC, Diabetes Prevention Program Research G (2011) Updated genetic score based on 34 confirmed type 2 diabetes loci is associated with diabetes incidence and regression to normoglycemia in the diabetes prevention program. Diabetes 60(4):1340–1348. doi:10.2337/db10-1119
Horikawa Y, Oda N, Cox NJ, Li X, Orho-Melander M, Hara M, Hinokio Y, Lindner TH, Mashima H, Schwarz PE, del Bosque-Plata L, Horikawa Y, Oda Y, Yoshiuchi I, Colilla S, Polonsky KS, Wei S, Concannon P, Iwasaki N, Schulze J, Baier LJ, Bogardus C, Groop L, Boerwinkle E, Hanis CL, Bell GI (2000) Genetic variation in the gene encoding calpain-10 is associated with type 2 diabetes mellitus. Nat Genet 26(2):163–175. doi:10.1038/79876
Hu FB (2011) Globalization of diabetes: the role of diet, lifestyle, and genes. Diabet Care 34(6):1249–1257. doi:10.2337/dc11-0442
Kilpelainen TO, Zillikens MC, Stancakova A, Finucane FM, Ried JS, Langenberg C, Zhang W, Beckmann JS, Luan J, Vandenput L, Styrkarsdottir U, Zhou Y, Smith AV, Zhao JH, Amin N, Vedantam S, Shin SY, Haritunians T, Fu M, Feitosa MF, Kumari M, Halldorsson BV, Tikkanen E, Mangino M, Hayward C, Song C, Arnold AM, Aulchenko YS, Oostra BA, Campbell H, Cupples LA, Davis KE, Doring A, Eiriksdottir G, Estrada K, Fernandez-Real JM, Garcia M, Gieger C, Glazer NL, Guiducci C, Hofman A, Humphries SE, Isomaa B, Jacobs LC, Jula A, Karasik D, Karlsson MK, Khaw KT, Kim LJ, Kivimaki M, Klopp N, Kuhnel B, Kuusisto J, Liu Y, Ljunggren O, Lorentzon M, Luben RN, McKnight B, Mellstrom D, Mitchell BD, Mooser V, Moreno JM, Mannisto S, O’Connell JR, Pascoe L, Peltonen L, Peral B, Perola M, Psaty BM, Salomaa V, Savage DB, Semple RK, Skaric-Juric T, Sigurdsson G, Song KS, Spector TD, Syvanen AC, Talmud PJ, Thorleifsson G, Thorsteinsdottir U, Uitterlinden AG, van Duijn CM, Vidal-Puig A, Wild SH, Wright AF, Clegg DJ, Schadt E, Wilson JF, Rudan I, Ripatti S, Borecki IB, Shuldiner AR, Ingelsson E, Jansson JO, Kaplan RC, Gudnason V, Harris TB, Groop L, Kiel DP, Rivadeneira F, Walker M, Barroso I, Vollenweider P, Waeber G, Chambers JC, Kooner JS, Soranzo N, Hirschhorn JN, Stefansson K, Wichmann HE, Ohlsson C, O’Rahilly S, Wareham NJ, Speliotes EK, Fox CS, Laakso M, Loos RJ (2011) Genetic variation near IRS1 associates with reduced adiposity and an impaired metabolic profile. Nat Genet 43(8):753–760. doi:10.1038/ng.866
Klimentidis YC, Abrams M, Wang J, Fernandez JR, Allison DB (2011) Natural selection at genomic regions associated with obesity and type-2 diabetes: East Asians and sub-Saharan Africans exhibit high levels of differentiation at type-2 diabetes regions. Hum Genet 129(4):407–418. doi:10.1007/s00439-010-0935-z
Kooner JS, Saleheen D, Sim X, Sehmi J, Zhang W, Frossard P, Been LF, Chia KS, Dimas AS, Hassanali N, Jafar T, Jowett JB, Li X, Radha V, Rees SD, Takeuchi F, Young R, Aung T, Basit A, Chidambaram M, Das D, Grundberg E, Hedman AK, Hydrie ZI, Islam M, Khor CC, Kowlessur S, Kristensen MM, Liju S, Lim WY, Matthews DR, Liu J, Morris AP, Nica AC, Pinidiyapathirage JM, Prokopenko I, Rasheed A, Samuel M, Shah N, Shera AS, Small KS, Suo C, Wickremasinghe AR, Wong TY, Yang M, Zhang F, Diagram MT, Abecasis GR, Barnett AH, Caulfield M, Deloukas P, Frayling TM, Froguel P, Kato N, Katulanda P, Kelly MA, Liang J, Mohan V, Sanghera DK, Scott J, Seielstad M, Zimmet PZ, Elliott P, Teo YY, McCarthy MI, Danesh J, Tai ES, Chambers JC (2011) Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat Genet 43(10):984–989. doi:10.1038/ng.921
Laukkanen O, Pihlajamaki J, Lindstrom J, Eriksson J, Valle TT, Hamalainen H, Ilanne-Parikka P, Keinanen-Kiukaanniemi S, Tuomilehto J, Uusitupa M, Laakso M, Finnish Diabetes Prevention Study G (2004) Polymorphisms of the SUR1 (ABCC8) and Kir6.2 (KCNJ11) genes predict the conversion from impaired glucose tolerance to type 2 diabetes. The Finnish Diabetes Prevention Study. J Clin Endocrinol Metab 89(12):6286–6290. doi:10.1210/jc.2004-1204
Li H, Gan W, Lu L, Dong X, Han X, Hu C, Yang Z, Sun L, Bao W, Li P, He M, Sun L, Wang Y, Zhu J, Ning Q, Tang Y, Zhang R, Wen J, Wang D, Zhu X, Guo K, Zuo X, Guo X, Yang H, Zhou X, Consortium D, Consortium A-TD, Zhang X, Qi L, Loos RJ, Hu FB, Wu T, Liu Y, Liu L, Yang Z, Hu R, Jia W, Ji L, Li Y, Lin X (2013) A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes 62(1):291–298. doi:10.2337/db12-0454
Lohmueller KE, Pearce CL, Pike M, Lander ES, Hirschhorn JN (2003) Meta-analysis of genetic association studies supports a contribution of common variants to susceptibility to common disease. Nat Genet 33(2):177–182. doi:10.1038/ng1071
Loos RJ, Lindgren CM, Li S, Wheeler E, Zhao JH, Prokopenko I, Inouye M, Freathy RM, Attwood AP, Beckmann JS, Berndt SI, Jacobs KB, Chanock SJ, Hayes RB, Bergmann S, Bennett AJ, Bingham SA, Bochud M, Brown M, Cauchi S, Connell JM, Cooper C, Smith GD, Day I, Dina C, De S, Dermitzakis ET, Doney AS, Elliott KS, Elliott P, Evans DM, Sadaf Farooqi I, Froguel P, Ghori J, Groves CJ, Gwilliam R, Hadley D, Hall AS, Hattersley AT, Hebebrand J, Heid IM, Lamina C, Gieger C, Illig T, Meitinger T, Wichmann HE, Herrera B, Hinney A, Hunt SE, Jarvelin MR, Johnson T, Jolley JD, Karpe F, Keniry A, Khaw KT, Luben RN, Mangino M, Marchini J, McArdle WL, McGinnis R, Meyre D, Munroe PB, Morris AD, Ness AR, Neville MJ, Nica AC, Ong KK, O’Rahilly S, Owen KR, Palmer CN, Papadakis K, Potter S, Pouta A, Qi L, Randall JC, Rayner NW, Ring SM, Sandhu MS, Scherag A, Sims MA, Song K, Soranzo N, Speliotes EK, Syddall HE, Teichmann SA, Timpson NJ, Tobias JH, Uda M, Vogel CI, Wallace C, Waterworth DM, Weedon MN, Willer CJ, Wraight YX, Zeggini E, Hirschhorn JN, Strachan DP, Ouwehand WH, Caulfield MJ, Samani NJ, Frayling TM, Vollenweider P, Waeber G, Mooser V, Deloukas P, McCarthy MI, Wareham NJ, Barroso I, Kraft P, Hankinson SE, Hunter DJ, Hu FB, Lyon HN, Voight BF, Ridderstrale M, Groop L, Scheet P, Sanna S, Abecasis GR, Albai G, Nagaraja R, Schlessinger D, Jackson AU, Tuomilehto J, Collins FS, Boehnke M, Mohlke KL (2008) Common variants near MC4R are associated with fat mass, weight and risk of obesity. Nat Genet 40(6):768–775. doi:10.1038/ng.140
Lyssenko V, Jonsson A, Almgren P, Pulizzi N, Isomaa B, Tuomi T, Berglund G, Altshuler D, Nilsson P, Groop L (2008) Clinical risk factors, DNA variants, and the development of type 2 diabetes. N Engl J Med 359(21):2220–2232. doi:10.1056/NEJMoa0801869
Lyssenko V, Nagorny CL, Erdos MR, Wierup N, Jonsson A, Spegel P, Bugliani M, Saxena R, Fex M, Pulizzi N, Isomaa B, Tuomi T, Nilsson P, Kuusisto J, Tuomilehto J, Boehnke M, Altshuler D, Sundler F, Eriksson JG, Jackson AU, Laakso M, Marchetti P, Watanabe RM, Mulder H, Groop L (2009) Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet 41(1):82–88. doi:10.1038/ng.288
Ma RC, Hu C, Tam CH, Zhang R, Kwan P, Leung TF, Thomas GN, Go MJ, Hara K, Sim X, Ho JS, Wang C, Li H, Lu L, Wang Y, Li JW, Wang Y, Lam VK, Wang J, Yu W, Kim YJ, Ng DP, Fujita H, Panoutsopoulou K, Day-Williams AG, Lee HM, Ng AC, Fang YJ, Kong AP, Jiang F, Ma X, Hou X, Tang S, Lu J, Yamauchi T, Tsui SK, Woo J, Leung PC, Zhang X, Tang NL, Sy HY, Liu J, Wong TY, Lee JY, Maeda S, Xu G, Cherny SS, Chan TF, Ng MC, Xiang K, Morris AP, Consortium D, Keildson S, Mu TC, Hu R, Ji L, Lin X, Cho YS, Kadowaki T, Tai ES, Zeggini E, McCarthy MI, Hon KL, Baum L, Tomlinson B, So WY, Bao Y, Chan JC, Jia W (2013) Genome-wide association study in a Chinese population identifies a susceptibility locus for type 2 diabetes at 7q32 near PAX4. Diabetologia 56(6):1291–1305. doi:10.1007/s00125-013-2874-4
Majithia AR, Florez JC (2009) Clinical translation of genetic predictors for type 2 diabetes. Curr Opin Endocrinol Diabetes Obes 16(2):100–106
Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H, Rybin D, Liu CT, Bielak LF, Prokopenko I, Amin N, Barnes D, Cadby G, Hottenga JJ, Ingelsson E, Jackson AU, Johnson T, Kanoni S, Ladenvall C, Lagou V, Lahti J, Lecoeur C, Liu Y, Martinez-Larrad MT, Montasser ME, Navarro P, Perry JR, Rasmussen-Torvik LJ, Salo P, Sattar N, Shungin D, Strawbridge RJ, Tanaka T, van Duijn CM, An P, de Andrade M, Andrews JS, Aspelund T, Atalay M, Aulchenko Y, Balkau B, Bandinelli S, Beckmann JS, Beilby JP, Bellis C, Bergman RN, Blangero J, Boban M, Boehnke M, Boerwinkle E, Bonnycastle LL, Boomsma DI, Borecki IB, Bottcher Y, Bouchard C, Brunner E, Budimir D, Campbell H, Carlson O, Chines PS, Clarke R, Collins FS, Corbaton-Anchuelo A, Couper D, de Faire U, Dedoussis GV, Deloukas P, Dimitriou M, Egan JM, Eiriksdottir G, Erdos MR, Eriksson JG, Eury E, Ferrucci L, Ford I, Forouhi NG, Fox CS, Franzosi MG, Franks PW, Frayling TM, Froguel P, Galan P, de Geus E, Gigante B, Glazer NL, Goel A, Groop L, Gudnason V, Hallmans G, Hamsten A, Hansson O, Harris TB, Hayward C, Heath S, Hercberg S, Hicks AA, Hingorani A, Hofman A, Hui J, Hung J, Jarvelin MR, Jhun MA, Johnson PC, Jukema JW, Jula A, Kao WH, Kaprio J, Kardia SL, Keinanen-Kiukaanniemi S, Kivimaki M, Kolcic I, Kovacs P, Kumari M, Kuusisto J, Kyvik KO, Laakso M, Lakka T, Lannfelt L, Lathrop GM, Launer LJ, Leander K, Li G, Lind L, Lindstrom J, Lobbens S, Loos RJ, Luan J, Lyssenko V, Magi R, Magnusson PK, Marmot M, Meneton P, Mohlke KL, Mooser V, Morken MA, Miljkovic I, Narisu N, O’Connell J, Ong KK, Oostra BA, Palmer LJ, Palotie A, Pankow JS, Peden JF, Pedersen NL, Pehlic M, Peltonen L, Penninx B, Pericic M, Perola M, Perusse L, Peyser PA, Polasek O, Pramstaller PP, Province MA, Raikkonen K, Rauramaa R, Rehnberg E, Rice K, Rotter JI, Rudan I, Ruokonen A, Saaristo T, Sabater-Lleal M, Salomaa V, Savage DB, Saxena R, Schwarz P, Seedorf U, Sennblad B, Serrano-Rios M, Shuldiner AR, Sijbrands EJ, Siscovick DS, Smit JH, Small KS, Smith NL, Smith AV, Stancakova A, Stirrups K, Stumvoll M, Sun YV, Swift AJ, Tonjes A, Tuomilehto J, Trompet S, Uitterlinden AG, Uusitupa M, Vikstrom M, Vitart V, Vohl MC, Voight BF, Vollenweider P, Waeber G, Waterworth DM, Watkins H, Wheeler E, Widen E, Wild SH, Willems SM, Willemsen G, Wilson JF, Witteman JC, Wright AF, Yaghootkar H, Zelenika D, Zemunik T, Zgaga L, Replication DIG, Meta-analysis C, Multiple Tissue Human Expression Resource C, Wareham NJ, McCarthy MI, Barroso I, Watanabe RM, Florez JC, Dupuis J, Meigs JB, Langenberg C (2012) A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet 44(6):659–669. doi:10.1038/ng.2274
Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TF, McCarroll SA, Visscher PM (2009) Finding the missing heritability of complex diseases. Nature 461(7265):747–753. doi:10.1038/nature08494, nature08494 [pii]
McCarthy MI (2003) Growing evidence for diabetes susceptibility genes from genome scan data. Curr Diab Rep 3(2):159–167
McCarthy MI (2008) Casting a wider net for diabetes susceptibility genes. Nat Genet 40(9):1039–1040. doi:10.1038/ng0908-1039
McCarthy MI (2010) Genomics, type 2 diabetes, and obesity. N Engl J Med 363(24):2339–2350. doi:10.1056/NEJMra0906948
McCarthy MI, Hattersley AT (2008) Learning from molecular genetics: novel insights arising from the definition of genes for monogenic and type 2 diabetes. Diabetes 57(11):2889–2898. doi:10.2337/db08-0343
McDonald TJ, Ellard S (2013) Maturity onset diabetes of the young: identification and diagnosis. Ann Clin Biochem 50(Pt 5):403–415. doi:10.1177/0004563213483458
Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, Manning AK, Florez JC, Wilson PW, D’Agostino RB Sr, Cupples LA (2008) Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med 359(21):2208–2219. doi:10.1056/NEJMoa0804742
Meyre D, Bouatia-Naji N, Tounian A, Samson C, Lecoeur C, Vatin V, Ghoussaini M, Wachter C, Hercberg S, Charpentier G, Patsch W, Pattou F, Charles MA, Tounian P, Clement K, Jouret B, Weill J, Maddux BA, Goldfine ID, Walley A, Boutin P, Dina C, Froguel P (2005) Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet 37(8):863–867. doi:10.1038/ng1604
Morris AP (2011) Transethnic meta-analysis of genomewide association studies. Genet Epidemiol 35(8):809–822. doi:10.1002/gepi.20630
Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V, Strawbridge RJ, Khan H, Grallert H, Mahajan A, Prokopenko I, Kang HM, Dina C, Esko T, Fraser RM, Kanoni S, Kumar A, Lagou V, Langenberg C, Luan J, Lindgren CM, Muller-Nurasyid M, Pechlivanis S, Rayner NW, Scott LJ, Wiltshire S, Yengo L, Kinnunen L, Rossin EJ, Raychaudhuri S, Johnson AD, Dimas AS, Loos RJ, Vedantam S, Chen H, Florez JC, Fox C, Liu CT, Rybin D, Couper DJ, Kao WH, Li M, Cornelis MC, Kraft P, Sun Q, van Dam RM, Stringham HM, Chines PS, Fischer K, Fontanillas P, Holmen OL, Hunt SE, Jackson AU, Kong A, Lawrence R, Meyer J, Perry JR, Platou CG, Potter S, Rehnberg E, Robertson N, Sivapalaratnam S, Stancakova A, Stirrups K, Thorleifsson G, Tikkanen E, Wood AR, Almgren P, Atalay M, Benediktsson R, Bonnycastle LL, Burtt N, Carey J, Charpentier G, Crenshaw AT, Doney AS, Dorkhan M, Edkins S, Emilsson V, Eury E, Forsen T, Gertow K, Gigante B, Grant GB, Groves CJ, Guiducci C, Herder C, Hreidarsson AB, Hui J, James A, Jonsson A, Rathmann W, Klopp N, Kravic J, Krjutskov K, Langford C, Leander K, Lindholm E, Lobbens S, Mannisto S, Mirza G, Muhleisen TW, Musk B, Parkin M, Rallidis L, Saramies J, Sennblad B, Shah S, Sigurethsson G, Silveira A, Steinbach G, Thorand B, Trakalo J, Veglia F, Wennauer R, Winckler W, Zabaneh D, Campbell H, van Duijn C, Uitterlinden AG, Hofman A, Sijbrands E, Abecasis GR, Owen KR, Zeggini E, Trip MD, Forouhi NG, Syvanen AC, Eriksson JG, Peltonen L, Nothen MM, Balkau B, Palmer CN, Lyssenko V, Tuomi T, Isomaa B, Hunter DJ, Qi L, Wellcome Trust Case Control C, Meta-Analyses of G, Insulin-Related Traits Consortium I, Genetic Investigation of ATC, Asian Genetic Epidemiology Network-Type 2 Diabetes C, South Asian Type 2 Diabetes C, Shuldiner AR, Roden M, Barroso I, Wilsgaard T, Beilby J, Hovingh K, Price JF, Wilson JF, Rauramaa R, Lakka TA, Lind L, Dedoussis G, Njolstad I, Pedersen NL, Khaw KT, Wareham NJ, Keinanen-Kiukaanniemi SM, Saaristo TE, Korpi-Hyovalti E, Saltevo J, Laakso M, Kuusisto J, Metspalu A, Collins FS, Mohlke KL, Bergman RN, Tuomilehto J, Boehm BO, Gieger C, Hveem K, Cauchi S, Froguel P, Baldassarre D, Tremoli E, Humphries SE, Saleheen D, Danesh J, Ingelsson E, Ripatti S, Salomaa V, Erbel R, Jockel KH, Moebus S, Peters A, Illig T, de Faire U, Hamsten A, Morris AD, Donnelly PJ, Frayling TM, Hattersley AT, Boerwinkle E, Melander O, Kathiresan S, Nilsson PM, Deloukas P, Thorsteinsdottir U, Groop LC, Stefansson K, Hu F, Pankow JS, Dupuis J, Meigs JB, Altshuler D, Boehnke M, McCarthy MI, Replication DIG, Meta-analysis C (2012) Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet 44(9):981–990. doi:10.1038/ng.2383
Muhlenbruch K, Jeppesen C, Joost HG, Boeing H, Schulze MB (2013) The value of genetic information for diabetes risk prediction - differences according to sex, age, family history and obesity. PloS One 8(5):e64307. doi:10.1371/journal.pone.0064307
Neel JV (1962) Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14:353–362
Neel JV (1999) The “thrifty genotype” in 1998. Nutr Rev 57(5 Pt 2):S2–S9
Palmer ND, McDonough CW, Hicks PJ, Roh BH, Wing MR, An SS, Hester JM, Cooke JN, Bostrom MA, Rudock ME, Talbert ME, Lewis JP, Consortium D, Investigators M, Ferrara A, Lu L, Ziegler JT, Sale MM, Divers J, Shriner D, Adeyemo A, Rotimi CN, Ng MC, Langefeld CD, Freedman BI, Bowden DW, Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM, Dupuis J, Qi L, Segre AV, van Hoek M, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Bostrom KB, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Couper DJ, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jorgensen T, Kao WH, Klopp N, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Perry JR, Petersen AK, Platou C, Proenca C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparso T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van Herpt T, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Mohlke KL, Morris AD, Palmer CN, Pramstaller PP, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Wareham NJ, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly MJ, Hattersley AT, Hu FB, Meigs JB, Pankow JS, Pedersen O, Wichmann HE, Barroso I, Florez JC, Frayling TM, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI, Soranzo N, Wheeler E, Glazer NL, Bouatia-Naji N, Magi R, Randall J, Johnson T, Elliott P, Rybin D, Henneman P, Dehghan A, Hottenga JJ, Song K, Goel A, Egan JM, Lajunen T, Doney A, Kanoni S, Cavalcanti-Proenca C, Kumari M, Timpson NJ, Zabena C, Ingelsson E, An P, O’Connell J, Luan J, Elliott A, McCarroll SA, Roccasecca RM, Pattou F, Sethupathy P, Ariyurek Y, Barter P, Beilby JP, Ben-Shlomo Y, Bergmann S, Bochud M, Bonnefond A, Borch-Johnsen K, Bottcher Y, Brunner E, Bumpstead SJ, Chen YD, Chines P, Clarke R, Coin LJ, Cooper MN, Crisponi L, Day IN, de Geus EJ, Delplanque J, Fedson AC, Fischer-Rosinsky A, Forouhi NG, Frants R, Franzosi MG, Galan P, Goodarzi MO, Graessler J, Grundy S, Gwilliam R, Hallmans G, Hammond N, Han X, Hartikainen AL, Hayward C, Heath SC, Hercberg S, Hicks AA, Hillman DR, Hingorani AD, Hui J, Hung J, Jula A, Kaakinen M, Kaprio J, Kesaniemi YA, Kivimaki M, Knight B, Koskinen S, Kovacs P, Kyvik KO, Lathrop GM, Lawlor DA, Le Bacquer O, Lecoeur C, Li Y, Mahley R, Mangino M, Manning AK, Martinez-Larrad MT, McAteer JB, McPherson R, Meisinger C, Melzer D, Meyre D, Mitchell BD, Mukherjee S, Naitza S, Neville MJ, Oostra BA, Orru M, Pakyz R, Paolisso G, Pattaro C, Pearson D, Peden JF, Pedersen NL, Perola M, Pfeiffer AF, Pichler I, Polasek O, Posthuma D, Potter SC, Pouta A, Province MA, Psaty BM, Rayner NW, Rice K, Ripatti S, Rivadeneira F, Rolandsson O, Sandbaek A, Sandhu M, Sanna S, Sayer AA, Scheet P, Seedorf U, Sharp SJ, Shields B, Sijbrands EJ, Silveira A, Simpson L, Singleton A, Smith NL, Sovio U, Swift A, Syddall H, Syvanen AC, Tanaka T, Tonjes A, Uitterlinden AG, van Dijk KW, Varma D, Visvikis-Siest S, Vitart V, Vogelzangs N, Waeber G, Wagner PJ, Walley A, Ward KL, Watkins H, Wild SH, Willemsen G, Witteman JC, Yarnell JW, Zelenika D, Zethelius B, Zhai G, Zhao JH, Zillikens MC, Borecki IB, Loos RJ, Meneton P, Magnusson PK, Nathan DM, Williams GH, Silander K, Salomaa V, Smith GD, Bornstein SR, Schwarz P, Spranger J, Karpe F, Shuldiner AR, Cooper C, Dedoussis GV, Serrano-Rios M, Lind L, Palmer LJ, Franks PW, Ebrahim S, Marmot M, Kao WH, Pramstaller PP, Wright AF, Stumvoll M, Hamsten A, Buchanan TA, Valle TT, Rotter JI, Siscovick DS, Penninx BW, Boomsma DI, Deloukas P, Spector TD, Ferrucci L, Cao A, Scuteri A, Schlessinger D, Uda M, Ruokonen A, Jarvelin MR, Waterworth DM, Vollenweider P, Peltonen L, Mooser V, Sladek R (2012) A genome-wide association search for type 2 diabetes genes in African Americans. PloS One 7(1):e29202. doi:10.1371/journal.pone.0029202
Parikh H, Groop L (2004) Candidate genes for type 2 diabetes. Rev Endocr Metab Disord 5(2):151–176. doi:10.1023/B:REMD.0000021437.46773.26
Parra EJ, Below JE, Krithika S, Valladares A, Barta JL, Cox NJ, Hanis CL, Wacher N, Garcia-Mena J, Hu P, Shriver MD, Diabetes Genetics R, Meta-analysis C, Kumate J, McKeigue PM, Escobedo J, Cruz M (2011) Genome-wide association study of type 2 diabetes in a sample from Mexico City and a meta-analysis of a Mexican-American sample from Starr County, Texas. Diabetologia 54(8):2038–2046. doi:10.1007/s00125-011-2172-y
Permutt MA, Wasson J, Cox N (2005) Genetic epidemiology of diabetes. J Clin Invest 115(6):1431–1439. doi:10.1172/JCI24758
Perry JR, Voight BF, Yengo L, Amin N, Dupuis J, Ganser M, Grallert H, Navarro P, Li M, Qi L, Steinthorsdottir V, Scott RA, Almgren P, Arking DE, Aulchenko Y, Balkau B, Benediktsson R, Bergman RN, Boerwinkle E, Bonnycastle L, Burtt NP, Campbell H, Charpentier G, Collins FS, Gieger C, Green T, Hadjadj S, Hattersley AT, Herder C, Hofman A, Johnson AD, Kottgen A, Kraft P, Labrune Y, Langenberg C, Manning AK, Mohlke KL, Morris AP, Oostra B, Pankow J, Petersen AK, Pramstaller PP, Prokopenko I, Rathmann W, Rayner W, Roden M, Rudan I, Rybin D, Scott LJ, Sigurdsson G, Sladek R, Thorleifsson G, Thorsteinsdottir U, Tuomilehto J, Uitterlinden AG, Vivequin S, Weedon MN, Wright AF, Magic CD, Consortium G, Hu FB, Illig T, Kao L, Meigs JB, Wilson JF, Stefansson K, van Duijn C, Altschuler D, Morris AD, Boehnke M, McCarthy MI, Froguel P, Palmer CN, Wareham NJ, Groop L, Frayling TM, Cauchi S (2012) Stratifying type 2 diabetes cases by BMI identifies genetic risk variants in LAMA1 and enrichment for risk variants in lean compared to obese cases. PLoS Genet 8(5):e1002741. doi:10.1371/journal.pgen.1002741
Peschke E, Stumpf I, Bazwinsky I, Litvak L, Dralle H, Muhlbauer E (2007) Melatonin and type 2 diabetes - a possible link? J Pineal Res 42(4):350–358. doi:10.1111/j.1600-079X.2007.00426.x
Prokopenko I, McCarthy MI, Lindgren CM (2008) Type 2 diabetes: new genes, new understanding. Trends Genet 24(12):613–621. doi:10.1016/j.tig.2008.09.004
Prokopenko I, Langenberg C, Florez JC, Saxena R, Soranzo N, Thorleifsson G, Loos RJ, Manning AK, Jackson AU, Aulchenko Y, Potter SC, Erdos MR, Sanna S, Hottenga JJ, Wheeler E, Kaakinen M, Lyssenko V, Chen WM, Ahmadi K, Beckmann JS, Bergman RN, Bochud M, Bonnycastle LL, Buchanan TA, Cao A, Cervino A, Coin L, Collins FS, Crisponi L, de Geus EJ, Dehghan A, Deloukas P, Doney AS, Elliott P, Freimer N, Gateva V, Herder C, Hofman A, Hughes TE, Hunt S, Illig T, Inouye M, Isomaa B, Johnson T, Kong A, Krestyaninova M, Kuusisto J, Laakso M, Lim N, Lindblad U, Lindgren CM, McCann OT, Mohlke KL, Morris AD, Naitza S, Orru M, Palmer CN, Pouta A, Randall J, Rathmann W, Saramies J, Scheet P, Scott LJ, Scuteri A, Sharp S, Sijbrands E, Smit JH, Song K, Steinthorsdottir V, Stringham HM, Tuomi T, Tuomilehto J, Uitterlinden AG, Voight BF, Waterworth D, Wichmann HE, Willemsen G, Witteman JC, Yuan X, Zhao JH, Zeggini E, Schlessinger D, Sandhu M, Boomsma DI, Uda M, Spector TD, Penninx BW, Altshuler D, Vollenweider P, Jarvelin MR, Lakatta E, Waeber G, Fox CS, Peltonen L, Groop LC, Mooser V, Cupples LA, Thorsteinsdottir U, Boehnke M, Barroso I, Van Duijn C, Dupuis J, Watanabe RM, Stefansson K, McCarthy MI, Wareham NJ, Meigs JB, Abecasis GR (2009a) Variants in MTNR1B influence fasting glucose levels. Nat Genet 41(1):77–81. doi:10.1038/ng.290
Prokopenko I, Zeggini E, Hanson RL, Mitchell BD, Rayner NW, Akan P, Baier L, Das SK, Elliott KS, Fu M, Frayling TM, Groves CJ, Gwilliam R, Scott LJ, Voight BF, Hattersley AT, Hu C, Morris AD, Ng M, Palmer CN, Tello-Ruiz M, Vaxillaire M, Wang CR, Stein L, Chan J, Jia W, Froguel P, Elbein SC, Deloukas P, Bogardus C, Shuldiner AR, McCarthy MI, International Type 2 Diabetes 1q C (2009b) Linkage disequilibrium mapping of the replicated type 2 diabetes linkage signal on chromosome 1q. Diabetes 58(7):1704–1709. doi:10.2337/db09-0081
Prokopenko I, Poon W, Magi R, Prasad BR, Salehi SA, Almgren P, Osmark P, Bouatia-Naji N, Wierup N, Fall T, Stancakova A, Barker A, Lagou V, Osmond C, Xie W, Lahti J, Jackson AU, Cheng YC, Liu J, O’Connell JR, Blomstedt PA, Fadista J, Alkayyali S, Dayeh T, Ahlqvist E, Taneera J, Lecoeur C, Kumar A, Hansson O, Hansson K, Voight BF, Kang HM, Levy-Marchal C, Vatin V, Palotie A, Syvanen AC, Mari A, Weedon MN, Loos RJ, Ong KK, Nilsson P, Isomaa B, Tuomi T, Wareham NJ, Stumvoll M, Widen E, Lakka TA, Langenberg C, Tonjes A, Rauramaa R, Kuusisto J, Frayling TM, Froguel P, Walker M, Eriksson JG, Ling C, Kovacs P, Ingelsson E, McCarthy MI, Shuldiner AR, Silver KD, Laakso M, Groop L, Lyssenko V (2014) A central role for GRB10 in regulation of islet function in man. PLoS Genet 10(4):e1004235. doi:10.1371/journal.pgen.1004235
Qi L, Cornelis MC, Kraft P, Stanya KJ, Linda Kao WH, Pankow JS, Dupuis J, Florez JC, Fox CS, Pare G, Sun Q, Girman CJ, Laurie CC, Mirel DB, Manolio TA, Chasman DI, Boerwinkle E, Ridker PM, Hunter DJ, Meigs JB, Lee CH, Hu FB, van Dam RM, Meta-Analysis of G, Insulin-Related Traits C, Diabetes Genetics R, Meta-Analysis C (2010) Genetic variants at 2q24 are associated with susceptibility to type 2 diabetes. Hum Mol Genet 19(13):2706–2715. doi:10.1093/hmg/ddq156
Reich DE, Lander ES (2001) On the allelic spectrum of human disease. Trends Genetics 17(9):502–510
Replication DIG, Meta-analysis C (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 46(3):234–244. doi:10.1038/ng.2897
Replication DIG, Meta-analysis C, Asian Genetic Epidemiology Network Type 2 Diabetes C, South Asian Type 2 Diabetes C, Mexican American Type 2 Diabetes C, Type 2 Diabetes Genetic Exploration by Next-generation sequencing in multi-Ethnic Samples C, Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, Horikoshi M, Johnson AD, Ng MCY, Prokopenko I, Saleheen D, Wang X, Zeggini E, Abecasis GR, Adair LS, Almgren P, Atalay M, Aung T, Baldassarre D, Balkau B, Bao Y, Barnett AH, Barroso I, Basit A, Been LF, Beilby J, Bell GI, Benediktsson R, Bergman RN, Boehm BO, Boerwinkle E, Bonnycastle LL, Burtt N, Cai Q, Campbell H, Carey J, Cauchi S, Caulfield M, Chan JCN, Chang L-C, Chang T-J, Chang Y-C, Charpentier G, Chen C-H, Chen H, Chen Y-T, Chia K-S, Chidambaram M, Chines PS, Cho NH, Cho YM, Chuang L-M, Collins FS, Cornelis MC, Couper DJ, Crenshaw AT, van Dam RM, Danesh J, Das D, de Faire U, Dedoussis G, Deloukas P, Dimas AS, Dina C, Doney ASF, Donnelly PJ, Dorkhan M, van Duijn C, Dupuis J, Edkins S, Elliott P, Emilsson V, Erbel R, Eriksson JG, Escobedo J, Esko T, Eury E, Florez JC, Fontanillas P, Forouhi NG, Forsen T, Fox C, Fraser RM, Frayling TM, Froguel P, Frossard P, Gao Y, Gertow K, Gieger C, Gigante B, Grallert H, Grant GB, Groop LC, Groves CJ, Grundberg E, Guiducci C, Hamsten A, Han B-G, Hara K, Hassanali N, Hattersley AT, Hayward C, Hedman AK, Herder C, Hofman A, Holmen OL, Hovingh K, Hreidarsson AB, Hu C, Hu FB, Hui J, Humphries SE, Hunt SE, Hunter DJ, Hveem K, Hydrie ZI, Ikegami H, Illig T, Ingelsson E, Islam M, Isomaa B, Jackson AU, Jafar T, James A, Jia W, Jockel K-H, Jonsson A, Jowett JBM, Kadowaki T, Kang HM, Kanoni S, Kao WHL, Kathiresan S, Kato N, Katulanda P, Keinanen-Kiukaanniemi SM, Kelly AM, Khan H, Khaw K-T, Khor C-C, Kim H-L, Kim S, Kim YJ, Kinnunen L, Klopp N, Kong A, Korpi-Hyovalti E, Kowlessur S, Kraft P, Kravic J, Kristensen MM, Krithika S, Kumar A, Kumate J, Kuusisto J, Kwak SH, Laakso M, Lagou V, Lakka TA, Langenberg C, Langford C, Lawrence R, Leander K, Lee J-M, Lee NR, Li M, Li X, Li Y, Liang J, Liju S, Lim W-Y, Lind L, Lindgren CM, Lindholm E, Liu C-T, Liu JJ, Lobbens S, Long J, Loos RJF, Lu W, Luan J, Lyssenko V, Ma RCW, Maeda S, Magi R, Mannisto S, Matthews DR, Meigs JB, Melander O, Metspalu A, Meyer J, Mirza G, Mihailov E, Moebus S, Mohan V, Mohlke KL, Morris AD, Muhleisen TW, Muller-Nurasyid M, Musk B, Nakamura J, Nakashima E, Navarro P, Ng P-K, Nica AC, Nilsson PM, Njolstad I, Nothen MM, Ohnaka K, Ong TH, Owen KR, Palmer CNA, Pankow JS, Park KS, Parkin M, Pechlivanis S, Pedersen NL, Peltonen L, Perry JRB, Peters A, Pinidiyapathirage JM, Platou CGP, Potter S, Price JF, Qi L, Radha V, Rallidis L, Rasheed A, Rathmann W, Rauramaa R, Raychaudhuri S, Rayner NW, Rees SD, Rehnberg E, Ripatti S, Robertson N, Roden M, Rossin EJ, Rudan I, Rybin D, Saaristo TE, Salomaa V, Saltevo J, Samuel M, Sanghera DK, Saramies J, Scott J, Scott LJ, Scott RA, Segre AV, Sehmi J, Sennblad B, Shah N, Shah S, Shera AS, Shu XO, Shuldiner AR, Sigursson G, Sijbrands E, Silveira A, Sim X, Sivapalaratnam S, Small KS, So WY, Stancakova A, Stefansson K, Steinbach G, Steinthorsdottir V, Stirrups K, Strawbridge RJ, Stringham HM, Sun Q, Suo C, Syvanen A-C, Takayanagi R, Takeuchi F, Tay WT, Teslovich TM, Thorand B, Thorleifsson G, Thorsteinsdottir U, Tikkanen E, Trakalo J, Tremoli E, Trip MD, Tsai FJ, Tuomi T, Tuomilehto J, Uitterlinden AG, Valladares-Salgado A, Vedantam S, Veglia F, Voight BF, Wang C, Wareham NJ, Wennauer R, Wickremasinghe AR, Wilsgaard T, Wilson JF, Wiltshire S, Winckler W, Wong TY, Wood AR, Wu J-Y, Wu Y, Yamamoto K, Yamauchi T, Yang M, Yengo L, Yokota M, Young R, Zabaneh D, Zhang F, Zhang R, Zheng W, Zimmet PZ, Altshuler D, Bowden DW, Cho YS, Cox NJ, Cruz M, Hanis CL, Kooner J, Lee J-Y, Seielstad M, Teo YY, Boehnke M, Parra EJ, Chambers JC, Tai ES, McCarthy MI, Morris AP (2014) Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 46(3):234–244. doi:10.1038/ng.2897, http://www.nature.com/ng/journal/v46/n3/abs/ng.2897.html#supplementary-information
Reynisdottir I, Thorleifsson G, Benediktsson R, Sigurdsson G, Emilsson V, Einarsdottir AS, Hjorleifsdottir EE, Orlygsdottir GT, Bjornsdottir GT, Saemundsdottir J, Halldorsson S, Hrafnkelsdottir S, Sigurjonsdottir SB, Steinsdottir S, Martin M, Kochan JP, Rhees BK, Grant SF, Frigge ML, Kong A, Gudnason V, Stefansson K, Gulcher JR (2003) Localization of a susceptibility gene for type 2 diabetes to chromosome 5q34-q35.2. Am J Hum Genet 73(2):323–335. doi:10.1086/377139
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273(5281):1516–1517
Rung J, Cauchi S, Albrechtsen A, Shen L, Rocheleau G, Cavalcanti-Proenca C, Bacot F, Balkau B, Belisle A, Borch-Johnsen K, Charpentier G, Dina C, Durand E, Elliott P, Hadjadj S, Jarvelin MR, Laitinen J, Lauritzen T, Marre M, Mazur A, Meyre D, Montpetit A, Pisinger C, Posner B, Poulsen P, Pouta A, Prentki M, Ribel-Madsen R, Ruokonen A, Sandbaek A, Serre D, Tichet J, Vaxillaire M, Wojtaszewski JF, Vaag A, Hansen T, Polychronakos C, Pedersen O, Froguel P, Sladek R (2009) Genetic variant near IRS1 is associated with type 2 diabetes, insulin resistance and hyperinsulinemia. Nat Genet 41(10):1110–1115. doi:10.1038/ng.443
Sandhu MS, Weedon MN, Fawcett KA, Wasson J, Debenham SL, Daly A, Lango H, Frayling TM, Neumann RJ, Sherva R, Blech I, Pharoah PD, Palmer CN, Kimber C, Tavendale R, Morris AD, McCarthy MI, Walker M, Hitman G, Glaser B, Permutt MA, Hattersley AT, Wareham NJ, Barroso I (2007) Common variants in WFS1 confer risk of type 2 diabetes. Nat Genet 39(8):951–953. doi:10.1038/ng2067, ng2067 [pii]
Saxena R, Hivert MF, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, Lyssenko V, Bouatia-Naji N, Dupuis J, Jackson AU, Kao WH, Li M, Glazer NL, Manning AK, Luan J, Stringham HM, Prokopenko I, Johnson T, Grarup N, Boesgaard TW, Lecoeur C, Shrader P, O’Connell J, Ingelsson E, Couper DJ, Rice K, Song K, Andreasen CH, Dina C, Kottgen A, Le Bacquer O, Pattou F, Taneera J, Steinthorsdottir V, Rybin D, Ardlie K, Sampson M, Qi L, van Hoek M, Weedon MN, Aulchenko YS, Voight BF, Grallert H, Balkau B, Bergman RN, Bielinski SJ, Bonnefond A, Bonnycastle LL, Borch-Johnsen K, Bottcher Y, Brunner E, Buchanan TA, Bumpstead SJ, Cavalcanti-Proenca C, Charpentier G, Chen YD, Chines PS, Collins FS, Cornelis M, JC G, Delplanque J, Doney A, Egan JM, Erdos MR, Firmann M, Forouhi NG, Fox CS, Goodarzi MO, Graessler J, Hingorani A, Isomaa B, Jorgensen T, Kivimaki M, Kovacs P, Krohn K, Kumari M, Lauritzen T, Levy-Marchal C, Mayor V, McAteer JB, Meyre D, Mitchell BD, Mohlke KL, Morken MA, Narisu N, Palmer CN, Pakyz R, Pascoe L, Payne F, Pearson D, Rathmann W, Sandbaek A, Sayer AA, Scott LJ, Sharp SJ, Sijbrands E, Singleton A, Siscovick DS, Smith NL, Sparso T, Swift AJ, Syddall H, Thorleifsson G, Tonjes A, Tuomi T, Tuomilehto J, Valle TT, Waeber G, Walley A, Waterworth DM, Zeggini E, Zhao JH, Consortium G, Investigators M, Illig T, Wichmann HE, Wilson JF, van Duijn C, Hu FB, Morris AD, Frayling TM, Hattersley AT, Thorsteinsdottir U, Stefansson K, Nilsson P, Syvanen AC, Shuldiner AR, Walker M, Bornstein SR, Schwarz P, Williams GH, Nathan DM, Kuusisto J, Laakso M, Cooper C, Marmot M, Ferrucci L, Mooser V, Stumvoll M, Loos RJ, Altshuler D, Psaty BM, Rotter JI, Boerwinkle E, Hansen T, Pedersen O, Florez JC, McCarthy MI, Boehnke M, Barroso I, Sladek R, Froguel P, Meigs JB, Groop L, Wareham NJ, Watanabe RM (2010) Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet 42(2):142–148. doi:10.1038/ng.521
Saxena R, Elbers CC, Guo Y, Peter I, Gaunt TR, Mega JL, Lanktree MB, Tare A, Castillo BA, Li YR, Johnson T, Bruinenberg M, Gilbert-Diamond D, Rajagopalan R, Voight BF, Balasubramanyam A, Barnard J, Bauer F, Baumert J, Bhangale T, Bohm BO, Braund PS, Burton PR, Chandrupatla HR, Clarke R, Cooper-DeHoff RM, Crook ED, Davey-Smith G, Day IN, de Boer A, de Groot MC, Drenos F, Ferguson J, Fox CS, Furlong CE, Gibson Q, Gieger C, Gilhuijs-Pederson LA, Glessner JT, Goel A, Gong Y, Grant SF, Grobbee DE, Hastie C, Humphries SE, Kim CE, Kivimaki M, Kleber M, Meisinger C, Kumari M, Langaee TY, Lawlor DA, Li M, Lobmeyer MT, Maitland-van der Zee AH, Meijs MF, Molony CM, Morrow DA, Murugesan G, Musani SK, Nelson CP, Newhouse SJ, O’Connell JR, Padmanabhan S, Palmen J, Patel SR, Pepine CJ, Pettinger M, Price TS, Rafelt S, Ranchalis J, Rasheed A, Rosenthal E, Ruczinski I, Shah S, Shen H, Silbernagel G, Smith EN, Spijkerman AW, Stanton A, Steffes MW, Thorand B, Trip M, van der Harst P, van Der AD, van Iperen EP, van Setten J, van Vliet-Ostaptchouk JV, Verweij N, Wolffenbuttel BH, Young T, Zafarmand MH, Zmuda JM, Look ARG, Consortium D, Boehnke M, Altshuler D, McCarthy M, Kao WH, Pankow JS, Cappola TP, Sever P, Poulter N, Caulfield M, Dominiczak A, Shields DC, Bhatt DL, Zhang L, Curtis SP, Danesh J, Casas JP, van der Schouw YT, Onland-Moret NC, Doevendans PA, Dorn GW 2nd, Farrall M, FitzGerald GA, Hamsten A, Hegele R, Hingorani AD, Hofker MH, Huggins GS, Illig T, Jarvik GP, Johnson JA, Klungel OH, Knowler WC, Koenig W, Marz W, Meigs JB, Melander O, Munroe PB, Mitchell BD, Bielinski SJ, Rader DJ, Reilly MP, Rich SS, Rotter JI, Saleheen D, Samani NJ, Schadt EE, Shuldiner AR, Silverstein R, Kottke-Marchant K, Talmud PJ, Watkins H, Asselbergs FW, de Bakker PI, McCaffery J, Wijmenga C, Sabatine MS, Wilson JG, Reiner A, Bowden DW, Hakonarson H, Siscovick DS, Keating BJ (2012) Large-scale gene-centric meta-analysis across 39 studies identifies type 2 diabetes loci. Am J Hum Genet 90(3):410–425. doi:10.1016/j.ajhg.2011.12.022
Saxena R, Saleheen D, Been LF, Garavito ML, Braun T, Bjonnes A, Young R, Ho WK, Rasheed A, Frossard P, Sim X, Hassanali N, Radha V, Chidambaram M, Liju S, Rees SD, Ng DP, Wong TY, Yamauchi T, Hara K, Tanaka Y, Hirose H, McCarthy MI, Morris AP, Diagram MT, Agen BA, Barnett AH, Katulanda P, Matthews D, Mohan V, Wander GS, Singh JR, Mehra NK, Ralhan S, Kamboh MI, Mulvihill JJ, Maegawa H, Tobe K, Maeda S, Cho YS, Tai ES, Kelly MA, Chambers JC, Kooner JS, Kadowaki T, Deloukas P, Rader DJ, Danesh J, Sanghera DK (2013) Genome-wide association study identifies a novel locus contributing to type 2 diabetes susceptibility in Sikhs of Punjabi origin from India. Diabetes 62(5):1746–1755. doi:10.2337/db12-1077
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2007a) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316(5829):1341–1345
Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, Prokunina-Olsson L, Ding CJ, Swift AJ, Narisu N, Hu T, Pruim R, Xiao R, Li XY, Conneely KN, Riebow NL, Sprau AG, Tong M, White PP, Hetrick KN, Barnhart MW, Bark CW, Goldstein JL, Watkins L, Xiang F, Saramies J, Buchanan TA, Watanabe RM, Valle TT, Kinnunen L, Abecasis GR, Pugh EW, Doheny KF, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2007b) A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science 316(5829):1341–1345. doi:10.1126/science.1142382
Scott RA, Lagou V, Welch RP, Wheeler E, Montasser ME, Luan J, Magi R, Strawbridge RJ, Rehnberg E, Gustafsson S, Kanoni S, Rasmussen-Torvik LJ, Yengo L, Lecoeur C, Shungin D, Sanna S, Sidore C, Johnson PC, Jukema JW, Johnson T, Mahajan A, Verweij N, Thorleifsson G, Hottenga JJ, Shah S, Smith AV, Sennblad B, Gieger C, Salo P, Perola M, Timpson NJ, Evans DM, Pourcain BS, Wu Y, Andrews JS, Hui J, Bielak LF, Zhao W, Horikoshi M, Navarro P, Isaacs A, O’Connell JR, Stirrups K, Vitart V, Hayward C, Esko T, Mihailov E, Fraser RM, Fall T, Voight BF, Raychaudhuri S, Chen H, Lindgren CM, Morris AP, Rayner NW, Robertson N, Rybin D, Liu CT, Beckmann JS, Willems SM, Chines PS, Jackson AU, Kang HM, Stringham HM, Song K, Tanaka T, Peden JF, Goel A, Hicks AA, An P, Muller-Nurasyid M, Franco-Cereceda A, Folkersen L, Marullo L, Jansen H, Oldehinkel AJ, Bruinenberg M, Pankow JS, North KE, Forouhi NG, Loos RJ, Edkins S, Varga TV, Hallmans G, Oksa H, Antonella M, Nagaraja R, Trompet S, Ford I, Bakker SJ, Kong A, Kumari M, Gigante B, Herder C, Munroe PB, Caulfield M, Antti J, Mangino M, Small K, Miljkovic I, Liu Y, Atalay M, Kiess W, James AL, Rivadeneira F, Uitterlinden AG, Palmer CN, Doney AS, Willemsen G, Smit JH, Campbell S, Polasek O, Bonnycastle LL, Hercberg S, Dimitriou M, Bolton JL, Fowkes GR, Kovacs P, Lindstrom J, Zemunik T, Bandinelli S, Wild SH, Basart HV, Rathmann W, Grallert H, Replication DIG, Meta-analysis C, Maerz W, Kleber ME, Boehm BO, Peters A, Pramstaller PP, Province MA, Borecki IB, Hastie ND, Rudan I, Campbell H, Watkins H, Farrall M, Stumvoll M, Ferrucci L, Waterworth DM, Bergman RN, Collins FS, Tuomilehto J, Watanabe RM, de Geus EJ, Penninx BW, Hofman A, Oostra BA, Psaty BM, Vollenweider P, Wilson JF, Wright AF, Hovingh GK, Metspalu A, Uusitupa M, Magnusson PK, Kyvik KO, Kaprio J, Price JF, Dedoussis GV, Deloukas P, Meneton P, Lind L, Boehnke M, Shuldiner AR, van Duijn CM, Morris AD, Toenjes A, Peyser PA, Beilby JP, Korner A, Kuusisto J, Laakso M, Bornstein SR, Schwarz PE, Lakka TA, Rauramaa R, Adair LS, Smith GD, Spector TD, Illig T, de Faire U, Hamsten A, Gudnason V, Kivimaki M, Hingorani A, Keinanen-Kiukaanniemi SM, Saaristo TE, Boomsma DI, Stefansson K, van der Harst P, Dupuis J, Pedersen NL, Sattar N, Harris TB, Cucca F, Ripatti S, Salomaa V, Mohlke KL, Balkau B, Froguel P, Pouta A, Jarvelin MR, Wareham NJ, Bouatia-Naji N, McCarthy MI, Franks PW, Meigs JB, Teslovich TM, Florez JC, Langenberg C, Ingelsson E, Prokopenko I, Barroso I (2012) Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat Genet 44(9):991–1005. doi:10.1038/ng.2385
Shu XO, Long J, Cai Q, Qi L, Xiang YB, Cho YS, Tai ES, Li X, Lin X, Chow WH, Go MJ, Seielstad M, Bao W, Li H, Cornelis MC, Yu K, Wen W, Shi J, Han BG, Sim XL, Liu L, Qi Q, Kim HL, Ng DP, Lee JY, Kim YJ, Li C, Gao YT, Zheng W, Hu FB (2010) Identification of new genetic risk variants for type 2 diabetes. PLoS Genet 6(9):e1001127. doi:10.1371/journal.pgen.1001127
Silander K, Scott LJ, Valle TT, Mohlke KL, Stringham HM, Wiles KR, Duren WL, Doheny KF, Pugh EW, Chines P, Narisu N, White PP, Fingerlin TE, Jackson AU, Li C, Ghosh S, Magnuson VL, Colby K, Erdos MR, Hill JE, Hollstein P, Humphreys KM, Kasad RA, Lambert J, Lazaridis KN, Lin G, Morales-Mena A, Patzkowski K, Pfahl C, Porter R, Rha D, Segal L, Suh YD, Tovar J, Unni A, Welch C, Douglas JA, Epstein MP, Hauser ER, Hagopian W, Buchanan TA, Watanabe RM, Bergman RN, Tuomilehto J, Collins FS, Boehnke M (2004) A large set of Finnish affected sibling pair families with type 2 diabetes suggests susceptibility loci on chromosomes 6, 11, and 14. Diabetes 53(3):821–829
Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Balkau B, Heude B, Charpentier G, Hudson TJ, Montpetit A, Pshezhetsky AV, Prentki M, Posner BI, Balding DJ, Meyre D, Polychronakos C, Froguel P (2007) A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature 445(7130):881–885. doi:10.1038/nature05616, nature05616 [pii]
Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, Dupuis J, Bouatia-Naji N, Langenberg C, Prokopenko I, Stolerman E, Sandhu MS, Heeney MM, Devaney JM, Reilly MP, Ricketts SL, Stewart AF, Voight BF, Willenborg C, Wright B, Altshuler D, Arking D, Balkau B, Barnes D, Boerwinkle E, Bohm B, Bonnefond A, Bonnycastle LL, Boomsma DI, Bornstein SR, Bottcher Y, Bumpstead S, Burnett-Miller MS, Campbell H, Cao A, Chambers J, Clark R, Collins FS, Coresh J, de Geus EJ, Dei M, Deloukas P, Doring A, Egan JM, Elosua R, Ferrucci L, Forouhi N, Fox CS, Franklin C, Franzosi MG, Gallina S, Goel A, Graessler J, Grallert H, Greinacher A, Hadley D, Hall A, Hamsten A, Hayward C, Heath S, Herder C, Homuth G, Hottenga JJ, Hunter-Merrill R, Illig T, Jackson AU, Jula A, Kleber M, Knouff CW, Kong A, Kooner J, Kottgen A, Kovacs P, Krohn K, Kuhnel B, Kuusisto J, Laakso M, Lathrop M, Lecoeur C, Li M, Li M, Loos RJ, Luan J, Lyssenko V, Magi R, Magnusson PK, Malarstig A, Mangino M, Martinez-Larrad MT, Marz W, McArdle WL, McPherson R, Meisinger C, Meitinger T, Melander O, Mohlke KL, Mooser VE, Morken MA, Narisu N, Nathan DM, Nauck M, O’Donnell C, Oexle K, Olla N, Pankow JS, Payne F, Peden JF, Pedersen NL, Peltonen L, Perola M, Polasek O, Porcu E, Rader DJ, Rathmann W, Ripatti S, Rocheleau G, Roden M, Rudan I, Salomaa V, Saxena R, Schlessinger D, Schunkert H, Schwarz P, Seedorf U, Selvin E, Serrano-Rios M, Shrader P, Silveira A, Siscovick D, Song K, Spector TD, Stefansson K, Steinthorsdottir V, Strachan DP, Strawbridge R, Stumvoll M, Surakka I, Swift AJ, Tanaka T, Teumer A, Thorleifsson G, Thorsteinsdottir U, Tonjes A, Usala G, Vitart V, Volzke H, Wallaschofski H, Waterworth DM, Watkins H, Wichmann HE, Wild SH, Willemsen G, Williams GH, Wilson JF, Winkelmann J, Wright AF, Wtccc, Zabena C, Zhao JH, Epstein SE, Erdmann J, Hakonarson HH, Kathiresan S, Khaw KT, Roberts R, Samani NJ, Fleming MD, Sladek R, Abecasis G, Boehnke M, Froguel P, Groop L, McCarthy MI, Kao WH, Florez JC, Uda M, Wareham NJ, Barroso I, Meigs JB (2010) Common variants at 10 genomic loci influence hemoglobin A(1)(C) levels via glycemic and nonglycemic pathways. Diabetes 59(12):3229–3239. doi:10.2337/db10-0502
Southam L, Soranzo N, Montgomery SB, Frayling TM, McCarthy MI, Barroso I, Zeggini E (2009) Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes- and obesity-susceptibility variants? Diabetologia 52(9):1846–1851. doi:10.1007/s00125-009-1419-3
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segre AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpelainen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, Campbell H, Cappuccio FP, Cavalcanti-Proenca C, Chen YD, Chen CM, Chines PS, Clarke R, Coin L, Connell J, Day IN, den Heijer M, Duan J, Ebrahim S, Elliott P, Elosua R, Eiriksdottir G, Erdos MR, Eriksson JG, Facheris MF, Felix SB, Fischer-Posovszky P, Folsom AR, Friedrich N, Freimer NB, Fu M, Gaget S, Gejman PV, Geus EJ, Gieger C, Gjesing AP, Goel A, Goyette P, Grallert H, Grassler J, Greenawalt DM, Groves CJ, Gudnason V, Guiducci C, Hartikainen AL, Hassanali N, Hall AS, Havulinna AS, Hayward C, Heath AC, Hengstenberg C, Hicks AA, Hinney A, Hofman A, Homuth G, Hui J, Igl W, Iribarren C, Isomaa B, Jacobs KB, Jarick I, Jewell E, John U, Jorgensen T, Jousilahti P, Jula A, Kaakinen M, Kajantie E, Kaplan LM, Kathiresan S, Kettunen J, Kinnunen L, Knowles JW, Kolcic I, Konig IR, Koskinen S, Kovacs P, Kuusisto J, Kraft P, Kvaloy K, Laitinen J, Lantieri O, Lanzani C, Launer LJ, Lecoeur C, Lehtimaki T, Lettre G, Liu J, Lokki ML, Lorentzon M, Luben RN, Ludwig B, Manunta P, Marek D, Marre M, Martin NG, McArdle WL, McCarthy A, McKnight B, Meitinger T, Melander O, Meyre D, Midthjell K, Montgomery GW, Morken MA, Morris AP, Mulic R, Ngwa JS, Nelis M, Neville MJ, Nyholt DR, O’Donnell CJ, O’Rahilly S, Ong KK, Oostra B, Pare G, Parker AN, Perola M, Pichler I, Pietilainen KH, Platou CG, Polasek O, Pouta A, Rafelt S, Raitakari O, Rayner NW, Ridderstrale M, Rief W, Ruokonen A, Robertson NR, Rzehak P, Salomaa V, Sanders AR, Sandhu MS, Sanna S, Saramies J, Savolainen MJ, Scherag S, Schipf S, Schreiber S, Schunkert H, Silander K, Sinisalo J, Siscovick DS, Smit JH, Soranzo N, Sovio U, Stephens J, Surakka I, Swift AJ, Tammesoo ML, Tardif JC, Teder-Laving M, Teslovich TM, Thompson JR, Thomson B, Tonjes A, Tuomi T, van Meurs JB, van Ommen GJ, Vatin V, Viikari J, Visvikis-Siest S, Vitart V, Vogel CI, Voight BF, Waite LL, Wallaschofski H, Walters GB, Widen E, Wiegand S, Wild SH, Willemsen G, Witte DR, Witteman JC, Xu J, Zhang Q, Zgaga L, Ziegler A, Zitting P, Beilby JP, Farooqi IS, Hebebrand J, Huikuri HV, James AL, Kahonen M, Levinson DF, Macciardi F, Nieminen MS, Ohlsson C, Palmer LJ, Ridker PM, Stumvoll M, Beckmann JS, Boeing H, Boerwinkle E, Boomsma DI, Caulfield MJ, Chanock SJ, Collins FS, Cupples LA, Smith GD, Erdmann J, Froguel P, Gronberg H, Gyllensten U, Hall P, Hansen T, Harris TB, Hattersley AT, Hayes RB, Heinrich J, Hu FB, Hveem K, Illig T, Jarvelin MR, Kaprio J, Karpe F, Khaw KT, Kiemeney LA, Krude H, Laakso M, Lawlor DA, Metspalu A, Munroe PB, Ouwehand WH, Pedersen O, Penninx BW, Peters A, Pramstaller PP, Quertermous T, Reinehr T, Rissanen A, Rudan I, Samani NJ, Schwarz PE, Shuldiner AR, Spector TD, Tuomilehto J, Uda M, Uitterlinden A, Valle TT, Wabitsch M, Waeber G, Wareham NJ, Watkins H, Wilson JF, Wright AF, Zillikens MC, Chatterjee N, McCarroll SA, Purcell S, Schadt EE, Visscher PM, Assimes TL, Borecki IB, Deloukas P, Fox CS, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, Mohlke KL, O’Connell JR, Peltonen L, Schlessinger D, Strachan DP, van Duijn CM, Wichmann HE, Frayling TM, Thorsteinsdottir U, Abecasis GR, Barroso I, Boehnke M, Stefansson K, North KE, McCarthy MI, Hirschhorn JN, Ingelsson E, Loos RJ (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42(11):937–948. doi:10.1038/ng.686
Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, Baker A, Snorradottir S, Bjarnason H, Ng MC, Hansen T, Bagger Y, Wilensky RL, Reilly MP, Adeyemo A, Chen Y, Zhou J, Gudnason V, Chen G, Huang H, Lashley K, Doumatey A, So WY, Ma RC, Andersen G, Borch-Johnsen K, Jorgensen T, van Vliet-Ostaptchouk JV, Hofker MH, Wijmenga C, Christiansen C, Rader DJ, Rotimi C, Gurney M, Chan JC, Pedersen O, Sigurdsson G, Gulcher JR, Thorsteinsdottir U, Kong A, Stefansson K (2007) A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat Genet 39(6):770–775
Steinthorsdottir V, Thorleifsson G, Sulem P, Helgason H, Grarup N, Sigurdsson A, Helgadottir HT, Johannsdottir H, Magnusson OT, Gudjonsson SA, Justesen JM, Harder MN, Jorgensen ME, Christensen C, Brandslund I, Sandbaek A, Lauritzen T, Vestergaard H, Linneberg A, Jorgensen T, Hansen T, Daneshpour MS, Fallah MS, Hreidarsson AB, Sigurdsson G, Azizi F, Benediktsson R, Masson G, Helgason A, Kong A, Gudbjartsson DF, Pedersen O, Thorsteinsdottir U, Stefansson K (2014) Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet 46(3):294–298. doi:10.1038/ng.2882
Stolerman ES, Florez JC (2009) Genomics of type 2 diabetes mellitus: implications for the clinician. Nat Rev Endocrinol 5(8):429–436. doi:10.1038/nrendo.2009.129
Strawbridge RJ, Dupuis J, Prokopenko I, Barker A, Ahlqvist E, Rybin D, Petrie JR, Travers ME, Bouatia-Naji N, Dimas AS, Nica A, Wheeler E, Chen H, Voight BF, Taneera J, Kanoni S, Peden JF, Turrini F, Gustafsson S, Zabena C, Almgren P, Barker DJ, Barnes D, Dennison EM, Eriksson JG, Eriksson P, Eury E, Folkersen L, Fox CS, Frayling TM, Goel A, Gu HF, Horikoshi M, Isomaa B, Jackson AU, Jameson KA, Kajantie E, Kerr-Conte J, Kuulasmaa T, Kuusisto J, Loos RJ, Luan J, Makrilakis K, Manning AK, Martinez-Larrad MT, Narisu N, Nastase Mannila M, Ohrvik J, Osmond C, Pascoe L, Payne F, Sayer AA, Sennblad B, Silveira A, Stancakova A, Stirrups K, Swift AJ, Syvanen AC, Tuomi T, Hooft FM, Walker M, Weedon MN, Xie W, Zethelius B, Consortium D, Consortium G, Mu TC, Consortium CA, Consortium CD, Ongen H, Malarstig A, Hopewell JC, Saleheen D, Chambers J, Parish S, Danesh J, Kooner J, Ostenson CG, Lind L, Cooper CC, Serrano-Rios M, Ferrannini E, Forsen TJ, Clarke R, Franzosi MG, Seedorf U, Watkins H, Froguel P, Johnson P, Deloukas P, Collins FS, Laakso M, Dermitzakis ET, Boehnke M, McCarthy MI, Wareham NJ, Groop L, Pattou F, Gloyn AL, Dedoussis GV, Lyssenko V, Meigs JB, Barroso I, Watanabe RM, Ingelsson E, Langenberg C, Hamsten A, Florez JC (2011) Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 60(10):2624–2634. doi:10.2337/db11-0415
Stumvoll M, Goldstein BJ, van Haeften TW (2005) Type 2 diabetes: principles of pathogenesis and therapy. Lancet 365(9467):1333–1346. doi:10.1016/S0140-6736(05)61032-X, S0140-6736(05)61032-X [pii]
Tabassum R, Chauhan G, Dwivedi OP, Mahajan A, Jaiswal A, Kaur I, Bandesh K, Singh T, Mathai BJ, Pandey Y, Chidambaram M, Sharma A, Chavali S, Sengupta S, Ramakrishnan L, Venkatesh P, Aggarwal SK, Ghosh S, Prabhakaran D, Srinath RK, Saxena M, Banerjee M, Mathur S, Bhansali A, Shah VN, Madhu SV, Marwaha RK, Basu A, Scaria V, McCarthy MI, Diagram, Indico, Venkatesan R, Mohan V, Tandon N, Bharadwaj D (2013) Genome-wide association study for type 2 diabetes in Indians identifies a new susceptibility locus at 2q21. Diabetes 62(3):977–986. doi:10.2337/db12-0406
Takamoto I, Kubota N, Nakaya K, Kumagai K, Hashimoto S, Kubota T, Inoue M, Kajiwara E, Katsuyama H, Obata A, Sakurai Y, Iwamoto M, Kitamura T, Ueki K, Kadowaki T (2014) TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia 57(3):542–553. doi:10.1007/s00125-013-3131-6
Talmud PJ, Hingorani AD, Cooper JA, Marmot MG, Brunner EJ, Kumari M, Kivimaki M, Humphries SE (2010) Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. Br Med J 340:b4838. doi:10.1136/bmj.b4838
Thomas G, Jacobs KB, Yeager M, Kraft P, Wacholder S, Orr N, Yu K, Chatterjee N, Welch R, Hutchinson A, Crenshaw A, Cancel-Tassin G, Staats BJ, Wang Z, Gonzalez-Bosquet J, Fang J, Deng X, Berndt SI, Calle EE, Feigelson HS, Thun MJ, Rodriguez C, Albanes D, Virtamo J, Weinstein S, Schumacher FR, Giovannucci E, Willett WC, Cussenot O, Valeri A, Andriole GL, Crawford ED, Tucker M, Gerhard DS, Fraumeni JF Jr, Hoover R, Hayes RB, Hunter DJ, Chanock SJ (2008) Multiple loci identified in a genome-wide association study of prostate cancer. Nat Genet 40(3):310–315. doi:10.1038/ng.91
Tsai FJ, Yang CF, Chen CC, Chuang LM, Lu CH, Chang CT, Wang TY, Chen RH, Shiu CF, Liu YM, Chang CC, Chen P, Chen CH, Fann CS, Chen YT, Wu JY (2010) A genome-wide association study identifies susceptibility variants for type 2 diabetes in Han Chinese. PLoS Genet 6(2):e1000847. doi:10.1371/journal.pgen.1000847
Tuomilehto J, Lindstrom J (2003) The major diabetes prevention trials. Curr Diab Rep 3(2):115–122
Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jorgensen T, Sandbaek A, Lauritzen T, Hansen T, Nurbaya S, Tsunoda T, Kubo M, Babazono T, Hirose H, Hayashi M, Iwamoto Y, Kashiwagi A, Kaku K, Kawamori R, Tai ES, Pedersen O, Kamatani N, Kadowaki T, Kikkawa R, Nakamura Y, Maeda S (2008) SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat Genet 40(9):1098–1102. doi:10.1038/ng.208
Vaxillaire M, Yengo L, Lobbens S, Rocheleau G, Eury E, Lantieri O, Marre M, Balkau B, Bonnefond A, Froguel P (2014) Type 2 diabetes-related genetic risk scores associated with variations in fasting plasma glucose and development of impaired glucose homeostasis in the prospective DESIR study. Diabetologia. doi:10.1007/s00125-014-3277-x
Voight BF, Scott LJ, Steinthorsdottir V, Morris AP, Dina C, Welch RP, Zeggini E, Huth C, Aulchenko YS, Thorleifsson G, McCulloch LJ, Ferreira T, Grallert H, Amin N, Wu G, Willer CJ, Raychaudhuri S, McCarroll SA, Langenberg C, Hofmann OM, Dupuis J, Qi L, Segre AV, van Hoek M, Navarro P, Ardlie K, Balkau B, Benediktsson R, Bennett AJ, Blagieva R, Boerwinkle E, Bonnycastle LL, Bengtsson Bostrom K, Bravenboer B, Bumpstead S, Burtt NP, Charpentier G, Chines PS, Cornelis M, Couper DJ, Crawford G, Doney AS, Elliott KS, Elliott AL, Erdos MR, Fox CS, Franklin CS, Ganser M, Gieger C, Grarup N, Green T, Griffin S, Groves CJ, Guiducci C, Hadjadj S, Hassanali N, Herder C, Isomaa B, Jackson AU, Johnson PR, Jorgensen T, Kao WH, Klopp N, Kong A, Kraft P, Kuusisto J, Lauritzen T, Li M, Lieverse A, Lindgren CM, Lyssenko V, Marre M, Meitinger T, Midthjell K, Morken MA, Narisu N, Nilsson P, Owen KR, Payne F, Perry JR, Petersen AK, Platou C, Proenca C, Prokopenko I, Rathmann W, Rayner NW, Robertson NR, Rocheleau G, Roden M, Sampson MJ, Saxena R, Shields BM, Shrader P, Sigurdsson G, Sparso T, Strassburger K, Stringham HM, Sun Q, Swift AJ, Thorand B, Tichet J, Tuomi T, van Dam RM, van Haeften TW, van Herpt T, van Vliet-Ostaptchouk JV, Walters GB, Weedon MN, Wijmenga C, Witteman J, Bergman RN, Cauchi S, Collins FS, Gloyn AL, Gyllensten U, Hansen T, Hide WA, Hitman GA, Hofman A, Hunter DJ, Hveem K, Laakso M, Mohlke KL, Morris AD, Palmer CN, Pramstaller PP, Rudan I, Sijbrands E, Stein LD, Tuomilehto J, Uitterlinden A, Walker M, Wareham NJ, Watanabe RM, Abecasis GR, Boehm BO, Campbell H, Daly MJ, Hattersley AT, Hu FB, Meigs JB, Pankow JS, Pedersen O, Wichmann HE, Barroso I, Florez JC, Frayling TM, Groop L, Sladek R, Thorsteinsdottir U, Wilson JF, Illig T, Froguel P, van Duijn CM, Stefansson K, Altshuler D, Boehnke M, McCarthy MI, Investigators M, Consortium G (2010) Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet 42(7):579–589. doi:10.1038/ng.609
Voight BF, Kang HM, Ding J, Palmer CD, Sidore C, Chines PS, Burtt NP, Fuchsberger C, Li Y, Erdmann J, Frayling TM, Heid IM, Jackson AU, Johnson T, Kilpelainen TO, Lindgren CM, Morris AP, Prokopenko I, Randall JC, Saxena R, Soranzo N, Speliotes EK, Teslovich TM, Wheeler E, Maguire J, Parkin M, Potter S, Rayner NW, Robertson N, Stirrups K, Winckler W, Sanna S, Mulas A, Nagaraja R, Cucca F, Barroso I, Deloukas P, Loos RJ, Kathiresan S, Munroe PB, Newton-Cheh C, Pfeufer A, Samani NJ, Schunkert H, Hirschhorn JN, Altshuler D, McCarthy MI, Abecasis GR, Boehnke M (2012) The metabochip, a custom genotyping array for genetic studies of metabolic, cardiovascular, and anthropometric traits. PLoS Genet 8(8):e1002793. doi:10.1371/journal.pgen.1002793
Weedon MN (2007) The importance of TCF7L2. Diabet Med 24(10):1062–1066. doi:10.1111/j.1464-5491.2007.02258.x
Weedon MN, Frayling TM, Shields B, Knight B, Turner T, Metcalf BS, Voss L, Wilkin TJ, McCarthy A, Ben-Shlomo Y, Davey Smith G, Ring S, Jones R, Golding J, Byberg L, Mann V, Axelsson T, Syvanen AC, Leon D, Hattersley AT (2005) Genetic regulation of birth weight and fasting glucose by a common polymorphism in the islet cell promoter of the glucokinase gene. Diabetes 54(2):576–581
Weedon MN, Clark VJ, Qian Y, Ben-Shlomo Y, Timpson N, Ebrahim S, Lawlor DA, Pembrey ME, Ring S, Wilkin TJ, Voss LD, Jeffery AN, Metcalf B, Ferrucci L, Corsi AM, Murray A, Melzer D, Knight B, Shields B, Smith GD, Hattersley AT, Di Rienzo A, Frayling TM (2006) A common haplotype of the glucokinase gene alters fasting glucose and birth weight: association in six studies and population-genetics analyses. Am J Hum Genet 79(6):991–1001. doi:10.1086/509517
Winckler W, Weedon MN, Graham RR, McCarroll SA, Purcell S, Almgren P, Tuomi T, Gaudet D, Bostrom KB, Walker M, Hitman G, Hattersley AT, McCarthy MI, Ardlie KG, Hirschhorn JN, Daly MJ, Frayling TM, Groop L, Altshuler D (2007) Evaluation of common variants in the six known maturity-onset diabetes of the young (MODY) genes for association with type 2 diabetes. Diabetes 56(3):685–693. doi:10.2337/db06-0202
Yamauchi T, Hara K, Maeda S, Yasuda K, Takahashi A, Horikoshi M, Nakamura M, Fujita H, Grarup N, Cauchi S, Ng DP, Ma RC, Tsunoda T, Kubo M, Watada H, Maegawa H, Okada-Iwabu M, Iwabu M, Shojima N, Shin HD, Andersen G, Witte DR, Jorgensen T, Lauritzen T, Sandbaek A, Hansen T, Ohshige T, Omori S, Saito I, Kaku K, Hirose H, So WY, Beury D, Chan JC, Park KS, Tai ES, Ito C, Tanaka Y, Kashiwagi A, Kawamori R, Kasuga M, Froguel P, Pedersen O, Kamatani N, Nakamura Y, Kadowaki T (2010) A genome-wide association study in the Japanese population identifies susceptibility loci for type 2 diabetes at UBE2E2 and C2CD4A-C2CD4B. Nat Genet 42(10):864–868. doi:10.1038/ng.660
Zeggini E, Weedon MN, Lindgren CM, Frayling TM, Elliott KS, Lango H, Timpson NJ, Perry JR, Rayner NW, Freathy RM, Barrett JC, Shields B, Morris AP, Ellard S, Groves CJ, Harries LW, Marchini JL, Owen KR, Knight B, Cardon LR, Walker M, Hitman GA, Morris AD, Doney AS, McCarthy MI, Hattersley AT (2007) Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science 316(5829):1336–1341. doi:10.1126/science.1142364, 1142364 [pii]
Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jorgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjogren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Wellcome Trust Case Control C, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D (2008) Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 40(5):638–645. doi:10.1038/ng.120
Acknowledgments
We would like to thank Dr. Letizia Marullo (University of Ferrara, Italy) and Dr. Julia Sarah El-Sayed Moustafa (Imperial College London, UK) for their help in the context of manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Sladek, R., Prokopenko, I. (2016). Genome-Wide Association Studies of Type 2 Diabetes. In: Florez, J. (eds) The Genetics of Type 2 Diabetes and Related Traits. Springer, Cham. https://doi.org/10.1007/978-3-319-01574-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-01574-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-01573-6
Online ISBN: 978-3-319-01574-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)