
Abstract
Hepatocellular adenoma (HCA) is a benign tumor derived from hepatocytes, the incidence of which is 3–4/100,000 in Europe and North America; most of the patients are female with administration of oral contraceptives, and women account for 85% of all the patients [1]. In Western countries, contraceptives have been considered as a key cause for HCA, and studies have shown that HCA will decrease in sizes in patients who have a history of taking some of the oral contraceptives after they stop them [2]. Androgen is another potential cause for HCA [3]. Hereditary metabolic diseases, hepato-glycogenosis [4], maturity-onset diabetes of young type 3 (MODY3) [5], familial adenomatous polyposis (FAP), and hemoglobin deposition are all associated with the onset of HCA. Several researches have proposed abroad that obesity is also a key factor with an impact on the genesis of HCA [6, 7]. In addition, the relationship between alcohol and HCA should also be paid more attention. However, the epidemiological situation of the disease in China is different from that in the Western countries. According to the statistics from the Department of Pathology, East Hepatobiliary Surgery Hospital, Second Military Medical University, among the 189 cases with surgical resection of HCA, middle-aged male patients account for 70% of all the patients, and 50% of the patients are overweight or obese, while female patients rarely have a history of oral contraceptive administration [8], suggesting the difference in the pathogenesis and prevalence populations of HCA between China and Occidental countries which deserves further study.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others



Keywords
These keywords were added by machine and not by the authors. This process is experimental and the keywords may be updated as the learning algorithm improves.
6.1 Hepatocellular Tumors
6.1.1 Hepatocellular Adenoma
6.1.1.1 Pathogenesis and Mechanism
Hepatocellular adenoma (HCA) is a benign tumor derived from hepatocytes, the incidence of which is 3–4/100,000 in Europe and North America; most of the patients are female with administration of oral contraceptives, and women account for 85% of all the patients [1]. In Western countries, contraceptives have been considered as a key cause for HCA, and studies have shown that HCA will decrease in sizes in patients who have a history of taking some of the oral contraceptives after they stop them [2]. Androgen is another potential cause for HCA [3]. Hereditary metabolic diseases, hepato-glycogenosis [4], maturity-onset diabetes of young type 3 (MODY3) [5], familial adenomatous polyposis (FAP), and hemoglobin deposition are all associated with the onset of HCA. Several researches have proposed abroad that obesity is also a key factor with an impact on the genesis of HCA [6, 7]. In addition, the relationship between alcohol and HCA should also be paid more attention. However, the epidemiological situation of the disease in China is different from that in the Western countries. According to the statistics from the Department of Pathology, East Hepatobiliary Surgery Hospital, Second Military Medical University, among the 189 cases with surgical resection of HCA, middle-aged male patients account for 70% of all the patients, and 50% of the patients are overweight or obese, while female patients rarely have a history of oral contraceptive administration [8], suggesting the difference in the pathogenesis and prevalence populations of HCA between China and Occidental countries which deserves further study.
6.1.1.2 Clinical Features
In China, HCA is discovered predominantly in males with a male to female ratio of about 2:1 and an average age of 37.9 (ranged 13–71) years old. Most of the female patients have no history of long-term oral contraceptives. Most HCA patients manifest no clinical symptoms, while a few of them have abdominal pain, abdominal distention, nausea, abdominal mass, abnormal serum liver function tests, etc. with AFP levels within the normal range. Rupture of the tumor can be found in 20–25% of the patients which may lead to shock, and as tumor increases in sizes to be >5 cm, the risk of tumor rupture and hemorrhage increases. It has been reported that 7% of the HCA cases will develop into HCC in the literature [9].
HCA imaging is corresponding to its histological classification, including three types as following:
-
1.
HNF1α-inactivated HCA: Intratumoral hemorrhage or much fat content in HCA exhibits typical hyperintensity on T1-weighted magnetic resonance imaging and iso-, hypo-, or hyperintensity on T2-weighted images [10], but shows enhancement to a certain extent in arterial phase and no enhancement in venous or delayed phase after venous administration of gadolinium chelate. HCA with diffused fatty degeneration is difficult to be differentiated from benign nodular fatty degeneration and HCC containing large amounts of fat based on the imaging results.
-
2.
β-Catenin-activated HCA: The performance of the lesions shows homogeneous or heterogeneous mass with rich blood supply, without any identifiable intratumoral fat. And heterogeneous intensity is shown on MRI T2-weighted images which can be iso-, hypo-, or hyperintensity.
-
3.
Inflammatory HCA: The lesions demonstrate liver masses with rich blood vessels with continuous enhancement in venous and delayed phase, and MRI T2-weighted images show obvious hyperintensity. A few cases (11%) exhibit tiny fat infiltration in the lesion. The sensitivity, specificity, positive and negative predictive values of T2-weighted hyperintensity, and enhanced scan in delayed phase for the diagnosis of HCA are 85.2%, 87.5%, 88.5%, and 84%, respectively.
6.1.1.3 Gross Features
HCA is usually a mass with a clear boundary (Fig. 6.1), and a few HCA cases involve multiple lesion, with the size of the lesion varying from several millimeters to 26 cm in diameter and most lesions encapsulated in a thin or no fibrous envelope, near to which can observe supplying vessels. The section of the mass is often gray-red, soft, and homogeneous in texture (Fig. 6.1a), while cholestasis (Fig. 6.1b), congestion, hemorrhage, necrosis, fibrosis (Fig. 6.1c), etc. can also be found. The boundary between the lesion and the surrounding liver tissue is distinct, while the latter often appears in no pathological changes. Fatty degeneration and glycogen storage are found in a few cases (Fig. 6.1d).

(a) The section of a huge mass is gray-red, soft, and homogeneous in texture. (b) A huge mass with a gray-green section. (c) HCA with hemorrhage. (d) Mutiple HCA of a case of glycogen storage; the section of surrounding liver tissue is yellow
6.1.1.4 Microscopic Features
HCA is a type of benign tumor due to proliferation of hepatocytes, and the tumor cells are similar to normal liver cells without obvious atypia that karyoplasmic ratio is not increased and nuclear division is rarely seen under the microscope. Fatty degeneration and hyaline degeneration can be found in the tumor cells, the cytoplasm of which contains lipofuscin and bile particle. The disorderly trabecular arrangement of the tumor cells is often 1–2 hepatocytes thick, with visible pseudoglandular structure occasionally and interstitial thin-walled veins; orphan arterioles have no concomitant bile ducts. The lesions are often accompanied by bleeding, necrosis, fibrosis, purpura, and other changes. The surrounding liver tissue is without cirrhosis, with fatty degeneration in some cases. Glycogen storage appears in a few cases with a pale hepatocellular cytoplasm, a clear membrane, and a lucent nuclear.
HCA can be divided into four different molecular types according to WHO classification, and each molecular type of HCA has corresponding clinical, imaging, pathological, and moleculobiological characteristics.
-
1.
HNF1α mutation-inactivated HCA (H-HCA). H-HCA accounts for 35–40% of all the HCA cases. HNF1α (hepatocyte nuclear factor 1 α) gene is a tumor suppressor gene located on chromosome 12q24, encoding HNF1 protein, associated with the differentiation of the liver cells. H-HCA mutations include somatic type (90%) and germ line cell type (10%), both resulting in the production of a nonfunctional protein HNF11α which facilitates fatty degeneration and hepatocellular proliferation. Approximately 90% of the patients are young females with a history of oral contraceptives, while germ line cell type H-HCA are primarily seen in patients complicated by MODY-3 or young patients with familial adenomatous polyposis rather than a history of oral contraceptives. From the perspective of the characteristics of morphological pathology, H-HCA generally is a lobulated mass, with single or multiple lesions. Under the microscope, the boundary is clear and the arrangement of the liver plates is regular. The liver cells are enlarged with a small and anachromasis nucleus, obvious fatty degeneration, and no atypia or infiltration of inflammatory cells (Fig. 6.2). No dilation of the hepatic sinusoid. Some cases showed abundant lipofuscin (Fig. 6.3) with normal or fatty degenerated liver cells around the tumor. FABP is a gene positively regulated by HNF1A, encoding liver fatty acid-binding protein (L-FABP) positively expressed in normal liver tissue but markedly decreased in H-HCA. Immunohistochemical staining showed β-catenin stained on the cell membrane, almost complete negative or low expression of L-FABP, and intensive positive expression in the surrounding normal liver cells. L-FABP is a diagnostic marker of H-HCA with both 100% specificity and sensitivity. HCA does not express C-reactive protein (CRP) or serum amyloid A (SAA). GS stained the perivascular region, while focal GS positive expression can be found in the surrounding liver tissue. Focal expression of CD34 is also observed (Fig. 6.4) [11]. The risk of malignant transformation for H-HCA is lower than that of other types of HCA.
Fig. 6.2 
HNF1α mutation-inactivated HCA (H-HCA). The boundary is clear, hepatic cells with obvious fatty degeneration and no atypia, without infiltration of inflammatory cells
Fig. 6.3 
HNF1α mutation-inactivated HCA (H-HCA) abundant lipofuscin in the tumor tissue
Fig. 6.4 
Immunohistochemistry expression of CD34. Focal-patchy-diffused positive, negative in some area



-
2.
β-Catenin mutant-activated HCA (B-HCA). B-HCA often occurs in males, which is associated with glycogen storage disease and male hormone use. This type accounts for 10–15% of all the HCA cases with malignant potential. To date, the Wnt pathway associated with beta-catenin takes part in the genesis of adenomas. No HNF1α mutation has been found in B-HCA, but it can have both mutations of gp130 and GNAS, and about 50% of β-catenin mutant adenomas are inflammatory ones. B-HCA is often solitary nodules, while in cases with glycogen deposition disease, the multiple lesions are observed. Histologically, B-HCA often had no fatty degeneration or infiltration of inflammatory cells (Figs. 6.5 and 6.6), while the liver cells are atypia, enlarged cellular volumes, and irregular nuclei, and the liver plates thicken to form pseudoglandular structure, which sometimes makes it difficult to distinguish it from well-differentiated HCC . Immunohistochemical staining showed high expression of β-catenin and glutamine synthetase (GS) in B-HCA. β-Catenin was nucleus positive and often observed in only a small number of nuclei which needs careful investigation. GS was diffuse and homogeneous positive in the cytoplasm (Fig. 6.7), which is different from the map-like expression pattern of FNH . Though the combination of β-catenin and GS antibodies would increase the specificity for the diagnosis of B-HCA (100%), its sensitivity is relatively poor (75%–85%) [12]. Considering potential malignant transformation of B-HCA, molecular pathological screening of β-catenin mutation is necessary in HCA cases with no overexpression of β-catenin target genes or HNF1α mutation.
Fig. 6.5 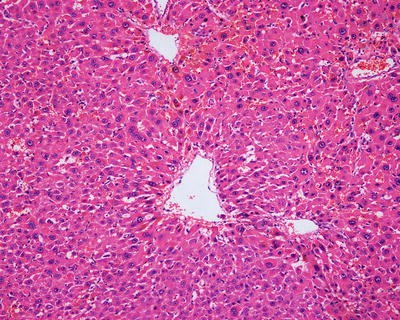
β-Catenin mutant-activated HCA (B-HCA). B-HCA had no fatty degeneration or infiltration of inflammatory cells, while the liver cells are atypia
Fig. 6.6 
β-Catenin mutant-activated HCA (B-HCA). B-HCA had no fatty degeneration or infiltration of inflammatory cells, while the liver cells are atypia
Fig. 6.7 
β-Catenin mutant-activated HCA (B-HCA). GS was diffuse and homogeneous positive in the cytoplasm



-
3.
Inflammatory hepatocellular adenoma (I-HCA). I-HCA is the most common type of HCA, accounting for 40–55% of all HCA cases and also known as vascular expansion type of hepatic adenoma, which is closely related to obesity, alcohol consumption, and liver steatosis, and almost all I-HCA cases are found in young and middle-aged women with a history of oral contraceptives. The tumor is vulnerable to rupture and hemorrhage, generally with no nuclear atypia and low risk of malignant transformation, which can be divided into two subtypes.
-
(1)
Inflammation with β-catenin activation: Sixty percent of the inflammation-type HCA cases contain gp130 (IL-6ST) somatic frameshift mutation, which is responsible for binding to IL-6 to activate the downstream inflammatory signal pathway of STAT3. And when gp130 mutation occurs, it will automatically activate the downstream inflammatory signaling pathways leading to inflammation without the ligand IL-6 [13]. β-Catenin staining is positive or GS staining shows diffusely positive.
-
(2)
Inflammation without β-catenin activation: A small part (10%) of inflammation-type HCA may contain both β-catenin gene and gp130 mutations with mild malignant potential [14]. β-Catenin staining shows positive stained cell membrane and GS stains perivascular region.
-
(1)
Fifty percent of the patients demonstrate elevated serum C-reactive protein and accelerated erythrocyte sedimentation rate, and fever and anemia can be observed in rare cases which can be eliminated after resection of the tumors. In general, I-HCA can be solitary or multiple with clear boundaries to the surrounding tissue. The tumor is soft and its cross section is heterogeneous, with no central scar. Histological characteristics of I-HCA include proliferation of liver cells, significant expansion or purpura of the liver blood sinus, focal or diffuse infiltration of inflammatory cells in the tumor (Figs. 6.8 and 6.9), visible fiber interval, dysplasia of the bile ducts, and solitary arteries (Fig. 6.10). In addition, various degrees of the fatty degeneration can be found in the liver cells. Immunohistochemistry showed overexpression of CRP (Fig. 6.11) or SAA. The proliferated bile ducts can sometimes be distinguished with expression of CK7 (Fig. 6.12) or CK19 immunohistochemical staining.

Inflammatory hepatocellular adenoma (I-HCA) proliferation of liver cells, significant expansion of the liver blood sinus, and focal infiltration of inflammatory cells in the tumor

Inflammatory hepatocellular adenoma (I-HCA). High power of Fig. 6.8

Inflammatory hepatocellular adenoma (I-HCA). Visible fiber interval and dysplasia of the bile ducts in the tumor

Inflammatory hepatocellular adenoma (I-HCA). Immunohistochemistry showed overexpression of CRP

Inflammatory hepatocellular adenoma (I-HCA). CK7 showed the proliferated bile ducts
-
4.
Undifferentiated HCA. This type of HCA accounts for less than 10% of all the cases, without any mutations or histological or immunohistochemical characteristics involved in the previously discussed types, and no explicit predisposing factors or clinical features have been determined currently.
It is worth noting that molecular pathological changes of Chinese HCA patients can be different from that in the Western countries. In the Department of Pathology of East Hepatobiliary Surgery Hospital of Second Military Medical University, we studied three HCA marker genes including HNF1α, β-catenin, and gp130 in 36 cases of HCA [15], and the gene sequencing showed that all HCA contained HNF1α mutation with a significantly higher mutation rate by 35–40% than that reported in Europe. Furthermore, we also found several new hot spots of HNF1 gene mutation which were not included in the European reports. However, no mutations of β-catenin or gp130 genes in the HCA tissues have been found in our experience with negative β-catenin nuclear staining in immunohistochemical investigation. Varying incidences of inflammation type of HCA with β-catenin activation have been reported abroad, ranged 0–17%. In addition, the canceration rate of Chinese HCA cases is reported lower than that in Western countries [17]. Therefore, we should put more efforts into the researches on the tumor molecular pathology of Chinese HCA cases to determine molecular markers for molecular typing.
6.1.1.5 Differential Diagnosis
Differential diagnosis of HCA and focal nodular hyperplasia and fibrolamellar hepatocellular carcinoma is shown in Table 6.1.
6.1.1.6 Treatment and Prognosis
The major complications of HCA are tumor rupture and malignant transformation. The occurrence of rupture and hemorrhage is directly related with tumor size. For adenomas larger than >5 cm in diameter, the risk of rupture and bleeding is higher. The risk of malignant transformation of hepatic adenomas is about 7%, predominantly found in males with β-catenin mutation. So far, no uniform standards for the treatment of hepatic adenoma have been determined. Nauh et al. [19] proposed the principles for the diagnosis and treatment of hepatic adenoma and suggested indications for surgery, including: ➀ adenoma with a maximum diameter > 5 cm, ➁ male, and ➂ female, adenoma with a maximum diameter <5 cm, with manifestation of B-HCA. Indications for follow-up by MRI include: female, MRI shows typical features of H-HCA or I-HCA and adenoma in small volumes. And if MRI shows no typical images of H-HCA or I-HCA, a biopsy should be conducted for screening of β-catenin mutation to evaluate the risk of malignant transformation.
6.1.2 Hepatic Adenomatosis
6.1.2.1 Pathogenesis and Mechanism
Hepatic adenomatosis was first described by Flejou et al. [20] in 1985, and no consistent standard for the tumor number of hepatic adenomatosis has been determined. Some scholars suggested that hepatic adenomatosis can be diagnosed according to the number of more than ten and the involvement of left and ring lobes of the liver without any history of steroid medication or glycogen storage disease [21, 22]. However, we support the diagnostic criteria proposed by Wesley et al. [23] in 2008 including more than four tumor lesions because of the scarcity of more than four tumors in hepatic adenomatosis. Despite the low incidence, there have been 177 reported cases of hepatic adenomatosis [24]. Many studies suggested the major difference of hepatic adenomatosis from hepatocellular adenoma that the former is mainly found in young or middle-aged females with oral contraceptive medication.
6.1.2.2 Clinical Features
The disease is more common in young and middle-aged women with an average age of 32 years old. However, Babaoglu et al. [25] reported one case of a 7-year-old female patient who had about 20 nodules with varying sizes, the maximum one was 7 cm in diameter, which were confirmed as hepatic adenomatosis via liver biopsy. She was the youngest patient that has ever been reported. Most cases exhibit no clinical symptoms, though abdominal pain and fever can be found in cases with intratumoral hemorrhage, and severe pain, shock, and even death can be observed in cases with tumor rupture into the abdominal cavity. Imaging often shows multiple intrahepatic lesions consistent to the nature of hepatic adenoma.
6.1.2.3 Pathological Features
Histopathology of single nodule in hepatic adenomatosis is identical to that of simple hepatocellular adenoma , the relevant description of which has been discussed in the first section of this chapter.
6.1.2.4 Molecular Pathology
Iwen et al. [26] reported one case associated with juvenile patient with diabetes type 3 and hepatic adenomatosis who was found to have Q495X mutation, which was suggested to repress the expression of wild HNF1A gene, resulting in the genesis of the disease. His father had the same mutation but only suffered from impaired glucose tolerance, suggesting the necessity of liver examination for diabetes patients without autoimmune antibodies and oral glucose tolerance test (OGTT) for adenomatosis patients without diabetes. Furthermore, OGTT screening is beneficial in the early diagnosis of diabetes in the patients’ lineal relatives with HNF1A mutation.
6.1.2.5 Differential Diagnosis
Hepatic adenomatosis should be differentiated from multiple hepatic focal nodular hyperplasia , liver cirrhosis, liver metastasis of tumor, liver dysplastic nodules , and liver multiple well-differentiated hepatocellular carcinoma .
6.1.2.6 Treatment and Prognosis
Hepatic adenomatosis belongs to benign tumors with severe potential complications, such as malignant transformation, tumor rupture, and hemorrhage. At present, the treatment of the disease is still in discussion with much controversy. For patients in whom the tumor cannot be completely excised, surgical resection of huge lesion can be conducted to remove part of the lesions, relieve symptoms, and prevent complications, followed by close follow-up subsequently. And in suspected malignant cases or cases with significant symptoms, surgical resection can be adopted. There have been cases in which liver transplantation was adopted, with good therapeutic effects in most of these cases, while reports of deaths due to postsurgical complications or HCC in transplanted liver have also been discovered [27].
6.2 Bile Duct Tumors
Intrahepatic bile duct benign tumors and tumorlike lesions account for 1.9–2.4% of bile duct epithelial tumors. Although benign tumors of bile duct are relatively rare, they are occasionally cancerous and should be differentiated from malignant and metastatic tumors both clinically and pathologically. Therefore, it is necessary to give enough attention to them.
6.2.1 Bile Duct Adenoma
6.2.1.1 Pathogenesis and Mechanism
Intrahepatic bile duct adenoma (BDA ), also known as benign cholangioma, is a type of low-incidence bile duct benign lesions. However, the biological characteristics of BDA are still not clear. Currently, it is believed that BDA is a post-damage repair process, including inflammation due to injury of liver cells and reactive hyperplasia of small bile ducts, and it is not relevant to bile duct hamartoma or hepatic cysts. Another hypothesis of the genesis of BDA involves peribiliary glands and thus can be called peribiliary gland hamartoma, because both of them have the same immunohistochemistry phenotype. However, due to the finding of K-ras mutation in BDA cases, it is also regarded as a true-type tumor.
6.2.1.2 Clinical Features
The average age of the patients was 55 years old (1.5–99 years), with men accounting for 59.39% and without specific symptoms or signs, most of which are found by chance in examinations or abdominal surgeries for other diseases. Enhanced MRI images demonstrate mild arterial enhancement and continuous enhancement in portal venous phase, and the apparent diffusion coefficient shown on diffusion-weighted MRI is more than twice that of the background of liver parenchyma [28]. Eight cases of BDA diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, with an average age of the patients of 56 years old (37–74 years), including six males, and four accidental discoveries of BDA lesions were made in HCC surgeries which received resection during the operations.
6.2.1.3 Gross Features
The tumors are often solitary small nodules, accounting for 82.9%, while about 10% of the cases involve multiple lesions, predominantly located in the subcapsular region or can also be present in the deep liver parenchyma, white or gray, round or oval in shape, hard in texture, with clear boundaries and no true envelopes. The average diameter of the nodules in about 90% cases is 0.6 (0.5–1) cm or occasionally even up to 9.2 cm. The eight cases of BDA diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, were all solitary lesions with an average diameter of 0.98 (0.2–3.8) cm [29] (Figs. 6.13 and 6.14).

Intrahepatic bile duct adenoma . A yellow round lesion with clear boundary and no envelope

Intrahepatic bile duct adenoma . A gray-white round lesion with clear boundary, it is a canceration case
6.2.1.4 Microscopic Features
The lesions of BDA is composed of small bile ducts with similar sizes and narrow lumens, in a curved shape or a solid cords, mostly containing no concentrated bile (Fig. 6.15), showing no cystic cavity or dilation. The epithelial cells of the bile ducts are cuboidal or low columnar with no atypia and rich cytoplasm which is slightly basophilic, and the nuclei are small and round in uniform sizes showing no mitosis, even with visible focal mucinous metaplasia of glandular epithelium and moderate chronic inflammation mainly containing fibrous tissue and lymphocytes rather than ovarian interstitial tissue, and the fibrous interstitial tissue was loose and edematous or with hyaline degeneration. Residual normal bile ducts can be seen around the proliferated bile ductules in the portal area (Fig. 6.16), and no capsules are found surrounding the lesions while the boundary is clear showing no invasion into the surrounding liver tissue varying amounts of fat.

Intrahepatic bile duct adenoma . Dense prolification of small bile ducts, remaining portal area, and focal lymphocyte infiltration (yellow arrow); the boundary is clear

Intrahepatic bile duct adenoma . High power of Fig. 6.16
Albores-Saavedra J et al. [32] and Wu et al. [30] reported four cases of clear cell-type BDA , 0.8–2.8 cm in diameter, with abundant clear cytoplasm in the tumor cells which arranged in shapes of small tubules, fine beam cable, or small nests, and more than 99% of the tumor cell cytoplasm was translucent, hyperchromatic nucleus were round or oval, and the substitution processes of normal bile duct epithelium by hyaline cells can been seen (Fig. 6.17).

Clear cell-type BDA . Substitution of normal bile duct epithelium by hyaline cells (yellow arrow)
Three cases of BDA with acidophilic degeneration in tumor cells have been reported [31, 33, 34], with the tumor diameter of 0.3–1.8 cm. Acidophilic degeneration refers to the generation of abundant fine granular eosinophilic cytoplasm in epithelial cells due to increase in the number of mitochondria under the microscope [33] (Fig. 6.18). The scarce bile duct tumors with acidophilic degeneration are mainly seen in intrahepatic bile duct papillary tumors and intrahepatic biliary cystadenocarcinoma , so whether BDA with acidophilic degeneration is a type of precancerous lesions for the aforementioned bile duct tumors should be discussed further. And comprehensive consideration on the nature of BDA should be made according to the size and cellular and nuclear morphology of the lesions.

BDA , with acidophilic degeneration. A,B: H&E staining showed abundant eosinophilic cytoplasm
Among the eight cases of BDA diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, four cases have concomitant HCC , one case with high-grade intraepithelial neoplasia, and one case canceration with transition between cancerous tissue and BDA (Fig. 6.19).

Canceration with transition between cancerous tissue (right) and BDA (left)
6.2.1.5 Immunohistochemistry
EMA, CEA, CK, and CK19 (Fig. 6.20) staining are positive, while CK20 and hepatic cell marker staining should be negative. BDA also expresses CD10 and 1F6 antigen as peripheral glands do, and 1F6 is not expressed in the normal interlobular bile ducts. CD10 staining exhibits continuous stains on the luminal surface of the apical cells in the clear cell-type BDA and outlines the irregularly shaped lumen (Fig. 6.21).

CK19 immunohistochemical staining in BDA . CK19-positive expression in tumor gland duct (blue arrow), the normal interlobular bile ducts (red arrow)

Clear cell-type BDA . CD10 staining exhibits continuous stains on the luminal surface of the apical cells in the clear cell-type BDA
6.2.1.6 Differential Diagnosis
BDA in liver parenchyma should mainly be differentiated from well-differentiated intrahepatic cholangiocarcinoma and metastatic adenocarcinoma, needing careful identification especially in the frozen biopsy. The author has encountered one case of a 71-year-old male patient receiving a surgical resection of gastric cardia adenocarcinoma, and a 0.2 cm gray-white nodule was found in the left lobe of the liver which was suspected as metastasis and excised during the operation. Later pathological examination confirmed is an intrahepatic BDA . Studies have shown that EZH2 and p16 (INK4a) can be used as markers of immunohistochemistry to distinguish ICC and BDA , because EZH2 is expressed in all ICC but not expressed in BDA , while p16 (INK4a) is expressed in 12% ICC and 81% BDA [36]. CD10 is expressed in benign tumors of bile duct but not expressed in malignant tumors, indicating that CD10 can be adopted as a marker for the identification of the nature of intrahepatic bile duct transparent cell tumors to tell whether a tumor is malignant or benign [37]. In addition, BDA and bile duct hamartoma are both benign intrahepatic bile duct proliferative lesions which are difficult to differentiate [35]. Main points of the differentiation for them are shown in Table 6.2.
6.2.1.7 Treatment and Prognosis
BDA grows slowly and may undergo hyaline degeneration to form scars. Despite the nature of benign lesions, canceration of BDA has already been reported in some cases. To date, BDA is recognized as precancerous lesions which may develop into bile duct carcinoma, so once diagnosed, surgical resection should be considered. For distal bile duct adenoma , endoscopic resection and argon plasma coagulation can be conducted for the treatment. And endoscopic resection in situ is suitable for the treatment of common bile duct adenoma .
6.2.2 Biliary Cystadenoma
6.2.2.1 Pathogenesis and Mechanism
Biliary cystadenoma (BCA) was reported by Hueter in 1887 for the first time, and Keen reported the first BCA resection in 1892. An epidemiological study published in 1993 has shown that 5–10% of the population may have cystic lesions in the liver, and the occurrence rate of bile duct cystic lesions was less than 5% the rate of intrahepatic cysts. And 90% bile duct cystic lesions arise in the intrahepatic while only 10% occur in extrahepatic biliary system.
The etiology of BCA remains unclear; Wheeler and Edmondson speculated that it may be a tumor formed during the development of ectopic bile ducts in the embryo. The patients may have hepatic cysts, polycystic liver abnormalities, or abnormal hepatic bile ducts. Comparison between mucous membrane of the gallbladder in adult BCA and fetus showed that the columnar epithelium in the former was similar to that in the embryonic development both under light microscope and electron microscope, mesenchymal vimentin staining was positive, and the latter only appeared in the fetal gallbladder tissue at 15th week of gestation, suggesting that BCA may derive from the ectopic embryonic gallbladder tissue. Another hypothesis for the genesis involves ectopic ovarian tissue or residual tissue of congenital embryonic foregut. In addition, due to endocrine cells found in about 50% of bile duct cysts, it has been also suggested that BCA may originate from intrahepatic peritubular glands.
6.2.2.2 Clinical Features
More than 85–90% of the patients are middle-aged or elderly women. An American report by Devaney et al. (1984) concerned a group of 52 cases of BCA patients, aged from 2 to 87 years old, and the average age was 45 years old, including 51 cases of mucinous type. Seventy-eight cases of BCA have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military University, with 55 cases of females (70.5%). The average age was 49.3 (26–77) years old, and the majority of the patients had no symptoms or only manifested nonspecific symptoms such as abdominal pain. Some patients came to the hospital because of abdominal tumor rupture and bleeding, secondary infection, obstructive jaundice, etc., and some had ascites as the first symptom due to rupture of BCA. Cases of BCA with pleural effusion have also been reported [48].
More than half of the patients have varying degrees of elevated serum CA19-9 [(838.4 + 2485.7) U/ml, reference value of 0–37 U/ml], mild elevation of serum alkaline phosphatase (ALP), γ-glutamyl transfer peptidase (γ-GT) and CEA, and negative serum AFP detection, all of which can turn normal after complete resection of the tumors. Despite controversy, cystic fluid obtained via puncture for tests of CA19-9 and CEA level were reported to be adopted to differentiate bile duct cystic lesions from cysts and abscess. However, due to the risk of rupture of the lesions leading to ascites or pleural effusion and the major difficulty in differentiation of benign tumor and malignant ones, cyst puncture biopsy should be conducted with much cautiousness.
ERCP cholangiopancreatography often shows filling defects [51]. B-ultrasound examination shows irregular thickening of the cyst wall and several thickened septa inside the cavity, with papillary projections and nodules on the inner wall of the cyst. CT images display several hypodense circular regions inside the cystic mass. And MRI images show typical irregular thick-walled multilocular lesions.
6.2.2.3 Gross Features
BCA occurs mainly in the liver, which can be located in the liver parenchyma, underneath the capsule, or even into the bile duct in the form of polyp, or involves both intra- and extrahepatic bile ducts, characterized by solitary multilocular cystic lesions (Fig. 6.22), and in some cases BCA are unilocular cysts with a smooth inner wall, while cases of multiple cysts are rare. Mucoid BCA contains transparent mucus or has a soapy section, and serous BCA is microcystic similar to cystadenoma of the pancreas. Both types of BCA can occur with intraductal papillary tumors. Among the 78 cases of BCA diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 60 cases are multilocular, with an average diameter of 8.4 cm, and the maximum diameter was 34 cm in one case (Fig. 6.23). Sixty-nine cases concerned the intrahepatic bile duct, five cases extrahepatic bile duct, and four cases hilar bile duct, while one case involved both the left lobe of the liver and extrahepatic bile ducts. In cases with cystic papillary hyperplasia on the inner cystic wall, gray-white granular papilla can be observed, with clear, yellow-brown mucinous, jelly or bloody cystic fluid and complete fibrous capsules.

Biliary cystadenoma . The tumor was huge, cystic, and solid

Biliary cystadenoma . A multilocular cystic lesion
6.2.2.4 Microscopic Features
-
1.
Mucinous BCA. Columnar, cuboidal, or flattened epithelial cells are lined on the basement membrane of the cyst wall, which secretes mucus, with pale eosinophilic cytoplasm, nucleus located in the basal part, and no atypia (Fig. 6.24). Sometimes there will be focal intestinal epithelial metaplasia or absence of the lining epithelium (Fig. 6.25). The tumor cells were arranged into tubules. Bile duct papillary cystadenoma can be diagnosed according to the existence of papillary hyperplasia. Borderline lesions are indicated if dysplasia of epithelial cells is observed, such as increased nuclear volume, increased chromatin, multilayer arrangement, absence of cellular polarity, and mitotic activity. Lesions with highly graded atypical hyperplasia have a tendency of canceration, and junctional zone of normal mucosal epithelium-highly graded atypical lesion-cystadenocarcinoma can be found in canceration cases. Among the 78 cases of BCA diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 30 cases involved low- or high-grade intraepithelial neoplasia, 3 cases involved canceration, and statistical analysis showed patients with portal or extrahepatic bile duct tumors, or male patients had higher proportions of intraepithelial neoplasia or canceration (Table 6.3). It is worth noting that two cases of hilar BCA had canceration. The cystic wall of BCA was composed of dense fibrous tissue with potential secondary changes, such as macrophages containing foam or pigments, hyaline degeneration, hemosiderin deposition, cholesterol fracture, focal calcification, etc. Spindle cells with round nucleus are found between the basement membrane and the lower part of the outer layer of fibrous connective tissue, which can go on differentiation to smooth muscle cells, fibroblasts, or adipose tissue and are termed as ovarian-type stroma. About 85% of mucinous BCA cases can have ovarian-type stroma, but only in females, while BCA cases with absence of ovarian-type stroma often concern male patients and only a few females. And among the 78 cases diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 37 cases of ovarian-type stroma are all women.
Fig. 6.24 
Intrahepatic biliary cystadenoma . Cuboidal epithelial cells are lined on the surface of the cyst wall; the lower layer was the ovarian-type stroma
Fig. 6.25 
Intrahepatic biliary cystadenoma . Intestinal epithelial metaplasia of the lining cells, ovarian-type stroma, and few hemorrhages were seen in the cyst wall
Table 6.3 Relationship of location and gender with intraepithelial neoplasia or carcinogenesis


-
2.
Serous BCA. The cyst wall is lined with a single layer of cuboidal epithelium with bright cytoplasm because of the rich glycogen, and mucous staining shows negative. The lower layer below the basement membrane is connective tissue with hyaline degeneration, and the interstitial tissue is composed of varying amounts of smooth muscle, fat, mucous glands, capillaries, and inflammatory cells rather than spindle cells.
6.2.2.5 Immunohistochemistry
Glandular epithelium was positive for CA19-9, low molecular weight CK, CEA, and EMA, similar to that of the normal bile duct and the pancreatic and ovarian tumors. Approximately 5% of BCA may have neuroendocrine differentiation; immunohistochemistry shows positive for synaptophysin (Syn) and chromogranin A (CgA) staining. Ovarian-type stroma was positive for vimentin, actin, desmin, and SMA, suggesting differentiation of smooth muscle, and expression of ER and PR, which may explain the reason of female predomination in BCA patients. Reports demonstrated that BCA often occurred in patients with hormone therapy, and tumor volume increased during pregnancy [53, 54].
6.2.2.6 Differential Diagnosis
-
1.
Differentiation should be made between BCA and various kinds of cystic lesions in the liver, such as bile duct hamartoma, cystic mesenchymal hamartoma , liver hydatid cysts, post-traumatic cysts, liver abscess, polycystic liver disease, bleeding cysts, embryonal sarcoma, HCC with cystic necrosis, metastatic tumors, biliary cystadenocarcinoma , teratoma , etc., all of which are without overlying epithelial ovarian-type stroma. Serum CA19-9 is synthesized by normal pancreatic and biliary ductal epithelium and thus will increase mildly in BDC which may be mistaken as a malignant tumor.
-
2.
Mucinous papillary BCA should be particularly distinguished from intraductal papillary mucinous tumors. Females account for the majority of all BCA patients, often with multilocular cysts and the mucous confined in the cystic cavity, while IPNB has no obvious gender difference in incidence, and the mucus can be found in all bile ducts. Both tumors are intraductal papillary neoplasms, demonstrating papillary protruding growth of epithelial cells from the cyst wall under the microscope; however, BCA cysts do not have communication with the normal bile duct system, while IPNB may involve multiple bile ducts and exhibit transition of bile duct epithelial cells. Microscopically, ovarian-like stroma can be found in BCA but not in IPNB . Furthermore, both have malignant potential, and IPNB is more prone to recurrence and residue due to crisp nature and easy defluvium of the tumor tissue and larger involvement [52].
-
3.
BCA and biliary cystadenocarcinoma are both rare liver-occupying lesions, but are common bile duct cystic lesions, with similar imaging manifestations and clinical manifestation which are difficult to distinguish. According to the studies by Sang et al. [49]. (2011) and Wang et al. [50], some clinical indicators can be used for the differentiation (Table 6.4).
Table 6.4 Clinical indicators for differentiation of BCA from bile duct cystadenocarcinoma
6.2.2.7 Treatment and Prognosis
BCA grows slowly; however, up to 25% of the BCA may develop into biliary cystadenocarcinoma in a few years as pointed out by some scholars, and 33% of mucinous cystadenocarcinoma tissues contain residual benign cystadenoma tissue or transition of severe atypical epithelial hyperplasia and carcinoma tissue. Therefore, BCA is considered to be a precancerous lesion for biliary cystadenocarcinoma and should receive active surgical resection. In addition, ovarian-like stroma may also develop into cystadenocarcinoma or sarcoma [55,56,57,58,59,60].
The patients with complete resection of BCA have a good prognosis, but partial resection is potentially complicated by recurrent or residual cancer. Patients of about 132 cases reported in English literature during the recent 20 years of follow-up underwent surgical resection, with only 3 cases of recurrence after operation. There are reports on increased risks of recurrence in incomplete resection cases. Sun Zengpeng [61] reported 15 cases of BCA, with no recurrence in 12 cases receiving complete resection during the follow-up period, while patients with partial resection in 3 cases had recurrence 5 months, 12 months, and 18 months after the operations, respectively. Shen Hua [62] reported 11 cases of BCA, with 2 cases of canceration. One patient received puncture and drainage, developed recurrence and metastasis 2 years later, and died 3 months later, while the other patient was without recurrence. Therefore, puncture of liver cystic masses which is unclear in nature is with a considerable risk. Romagnoli et al. [63] reported one case of BCA with injury of intrahepatic bile duct, during the surgery due to the deep location of the tumor, and it was then treated via liver transplantation, obtaining satisfactory curative effect.
6.2.3 Intraductal Papillary Neoplasms of the Bile Ducts
According to the fourth edition of the WHO’s Neoplasms of Digestive System in 2010, intraductal papillary neoplasms of the bile ducts (IPNB ) include papilloma or papillary adenoma and papillomatosis, among which papillary tumors with mucous secretion are known as intraductal papillary mucinous biliary neoplasm (IPMN) , and multiple bile duct papillary tumors are called biliary papillomatosis.
6.2.3.1 Pathogenesis and Mechanism
With the gradually deepening knowledge on IPNB in recent years, more and more reports on the disease have been found, and there are more than 100 cases reported in the literature both abroad and at home since 1958 when the first case was reported by Caroli.
Studies from Taiwan, China, and South Korea showed that cholelithiasis and clonorchiasis are two major risk factors of this disease, which has highly malignant tendency. It takes 6–8 years for bile duct stones to develop into IPNB , while it takes 1–2 years for the development of a carcinoma into invasive cancer, the benign-to-malignant development of which is consistent with the general canceration process of adenoma, similar to the process of colorectal adenoma-adenocarcinoma. In addition, this tumor also involves inflammatory reaction, cell damage and repair, and dysplasia [64, 65]. IPNB often shares gene mutation spectrum with cholangiocarcinoma , including KRAS, TP53, p16, and SMAD4. It has been reported in the literature that 40–80% IPNB contain components of tubular adenocarcinoma, mucinous adenocarcinoma, or invasive carcinoma, indicating a high malignant risk for IPNB canceration. A total of 29 cases of IPNB have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, among which 27 patients are with low- or high-grade intraepithelial neoplasia or carcinogenesis.
The histogenesis of IPNB is still unknown and there are a variety of views. Nakanuma et al. [67] suggested that IPNB probably originated from peribiliary gland cells, while Cardinale et al. considered periductal stem or progenitor cells surrounding the bile duct as the origination, and the genesis includes a sequence of processes including reaction to risk factors such as inflammation, mutations in stem or progenitor cells, and gradual development of dysplasia into invasive cancer.
6.2.3.2 Clinical Features
According to an epidemiological survey in 2012 [66], the male to female ratio of IPNB patients was 1.06:1, with an average age of 60 (ranged 40–77) years old, and the majority of the patients are 50–70 years old. The most common clinical symptoms are recurrent abdominal pain, jaundice, and fever, and acute cholangitis is commonly seen in mucoid cases, while patients with non-mucoid IPNB are often asymptomatic. Some patients are complicated by hepatolithiasis, ulcerative colitis, Caroli disease, choledochal cyst, and polyposis coli. Twenty-nine cases have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, concerning 13 males and 16 females, with the average age of 59.7 (38–79) years old, including five cases with biliary stones and one case of colorectal cancer and liver metastasis. Imaging study demonstrates intraductal masses and dilatation of bile duct, and dilatation of both proximal and distal bile ducts has certain significance in diagnosis of IPNB . Laboratory examination results are often indicative of the general performance of the bile duct obstruction, such as an increase in ALT, AST, total bilirubin, conjugated bilirubin, γ-glutamyl transferase, and alkaline phosphatase. About 74.4% of the patients have elevated total serum bilirubin, 40–80% have elevated serum levels of CA19-9 which is much higher in mucous cases, and about 25% of patients have increased serum CEA.
6.2.3.3 Gross Features
IPNB usually consists of diffuse or multicentric papillary clusters which are ecru in color, connected to the bile duct mucosa via peduncles and mostly found in the left hepatic lobe, involving the whole biliary tree or even the extrahepatic bile ducts, cystic duct, and main pancreatic duct. The tumor, crisp, soft, and easy to fall off, mainly generated from epithelial growth on the surface, protruding into the lumen, and may cause complete obstruction of the bile ducts. The surrounding liver tissue is green in color due to cholestasis or presents biliary cirrhosis. Among the 29 cases of IPNB diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 19 cases involved intrahepatic bile ducts, and 10 cases involved the hilar or extrahepatic bile ducts. The cross section of the tumors exhibited obvious dilatation of the bile ducts filled with gray-white or gray-red floccule, tumor thrombus-like tissue (Fig. 6.26), bile duct polyp, or cauliflower mass in the lumen.

Intraductal papillary neoplasms of the bile ducts. The bile ducts filled with gray-white polyp tissue
6.2.3.4 Microscopic Features
Histologically, IPNB shows multiple papillary-villous hyperplasia of biliary epithelium, which grows in branches from fibrous vascular axis protruding into the lumen and lined with cuboidal or columnar epithelium on the surface. The cells are with pale eosinophilic cytoplasm (Figs. 6.27 and 6.28) and arranged in multilayers with darker staining nuclei during dysplasia (Figs. 6.29 and 6.30). And as the degree of dysplasia increases, the cell layers and cells with mitosis multiply, with a visible transition between the lesion and the epithelium of normal bile duct (Fig. 6.31). The bile ducts dilate significantly, surrounded by normal bile ducts with acute or chronic inflammation of epithelium, which may detach due to ulcer, and duct wall thickens because of fibrous tissue hyperplasia. Most of the lesions protrude into the lumen which may be filled up, and infiltration into basal bile duct wall can be observed when canceration occurs (Fig. 6.32). Mucus secretion is found in one third cases of IPNB (IPMN ). IPNB is prone to be malignant; thus, multiple sections are needed for careful examination. According to different degrees of dysplasia and infiltration, it can be divided into four stages: IPNB with low-grade intraepithelial neoplasia; IPNB with high-grade intraepithelial neoplasia; intraductal carcinoma of bile duct in situ, T1; and infiltrating carcinoma of the bile duct, T2 or higher. The 29 cases of IPNB diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, included 8 cases with low-grade intraepithelial neoplasias, 14 cases with high-grade intraepithelial neoplasia, and 5 cases with canceration.

Intraductal papillary neoplasms of the bile ducts. IPNB shows papillary hyperplasia, which grows in branches from fibrous vascular axis protruding into the lumen

Intraductal papillary neoplasms of the bile ducts

Intraductal papillary neoplasms of the bile ducts with high-grade intraepithelial neoplasia. The tumor cells are arranged in multilayers with darker staining nuclei. High power of Fig. 6.28

Intraductal papillary neoplasms of the bile ducts with high-grade intraepithelial neoplasia. The small bile duct branch cells are arranged in multilayers with darker staining nuclei; the lumen is narrow with necrosis

Intraductal papillary neoplasms of the bile ducts. Transition between the lesion and the epithelium of normal bile duct

Intraductal papillary neoplasms of the bile ducts with canceration. Papillary hyperplasia of biliary epithelium infiltration into basal bile duct wall
According to the tumor tissues and the morphological characteristics, IPNB can be divided into the following four types:
-
1.
Pancreatobiliary type. The tumor is similar to the pancreas duct and ductular epithelium with columnar cells which have basophilic cytoplasm and round nuclei (Fig. 6.33). Immunohistochemistry staining often shows MUC1 positive and MUC2 negative. It is the most common type which tends to undergo malignant transformation to form tubular adenocarcinoma.
Fig. 6.33 
Intraductal papillary neoplasms, pancreatobiliary type. The tumor is similar to the pancreas duct and ductular epithelium with columnar cells

-
2.
Intestinal type. The tumor has similarity to the features of intestinal villous tumors (Fig. 6.34), and immunohistochemistry staining shows that the tumor cells stably expressed MUC2 and MUC5AC, while no expression of MUC1 was detected. It is less likely to undergo malignant transformation which results in colloid (mucinous) invasive carcinoma.
Fig. 6.34 
Intraductal papillary neoplasms, intestinal type. Columnar tumor cells have similarity to the features of intestinal villous tumors

-
3.
Oncocytic type. The tumor cells have rich cytoplasm which is strongly eosinophilic (Fig. 6.35), and immunohistochemistry staining shows stable expression of MUC5AC and focal expression of MUC1 and (or) MUC2 in the cells. It has less tendency of malignant transformation.
Fig. 6.35 
Intraductal papillary neoplasms, oncocytic type. The tumor cells have rich cytoplasm which is strongly eosinophilic

-
4.
Gastric type. The tumor consists of columnar epithelial cells, similar to the gastric pit structure (Fig. 6.36), and immunohistochemistry staining shows MUC5AC expression and no expression of MUC1 or MUC2.
Fig. 6.36 
Intraductal papillary neoplasms, gastric type. The tumor consists of columnar epithelial cells, similar to the gastric pit structure

Papillomatosis was reported by Caroli in 1959 for the first time, and more than 150 cases have been reported till now. The lesions grow in multiple foci, with involvement of the whole intrahepatic biliary tree in severe cases, and both intra- and extrahepatic bile ducts can be involved. Furthermore, lesions in different regions may be at different stages in the developmental course and can be divided into mucus biliary papillomatosis (MBP) and non-mucus biliary papillomatosis (NMBP). Generally, it is believed that BP has a highly malignant tendency and is an important type of precancerous lesions . Its canceration rate is as high as 41–83%, and radical surgical resection or liver transplantation should be preferred for the treatment [68].
6.2.3.5 Immunohistochemistry
Almost all of the IPNB lesions express biomarkers of bile duct epithelium and gastrointestinal epithelium, such as CK7, CK20, and MUC5AC, suggesting the combination of its retaining bile duct epithelial immunophenotypes and obtaining MUC1 immunophenotypes of the gastrointestinal epithelium in the genesis of IPNB . The expression of MUC1 usually indicates the infiltrating carcinoma, while mucinous tumors in IPMN are MUC1 negative and MUC2 positive. Sasaki et al. [69] demonstrated that IPNB overexpressed EZH2 and MUC1, and the low expression of MUC6 may indicate the malignant biological behavior of the tumor. Tubulin β-III (TUBB3) is negative in IPNB but positive in about half the number of extrahepatic bile duct carcinoma. Murakami et al. [70] found P53, CD133, and mucus staining were positive in primary tumors and cell line (KBDC)-11 of IPNB .
6.2.3.6 Differential Diagnosis
IPNB should be distinguished from intrahepatic bile duct stones, bile duct cystadenoma or carcinoma, cholangiocarcinoma , etc.
-
1.
Intrahepatic bile duct stones. The clinical symptoms of intrahepatic bile duct stones are similar to that of IPNB , and the latter can be complicated by bile duct stones, while intrahepatic bile duct stones often present simple dilatation of the bile duct, active proliferation of the peripheral glands, interstitial infiltration of lots of inflammatory cells, no papillary hyperplasia of epithelium, and little mucus in the lumen under the microscope. They are benign lesions, but intraepithelial neoplasia and cancer can also be found occasionally. Therefore, careful sample drawing is needed to prevent missed diagnosis.
-
2.
Biliary cystadenoma . IPNB is without ovarian-like stroma, and its cystic dilatation is communicated with bile ducts.
-
3.
Cholangiocarcinoma . Cholangiocarcinoma exhibits no cholangitis or biliary stones in most cases, and visible mucus is not common in it, while the above three manifestations are common in IPNB . Enhancement of IPNB is noted in outflow phase of enhanced CT, rather than persistent or progressive enhancement as in cholangiocarcinoma .
6.2.3.7 Treatment and Prognosis
IPNB may develop into intrahepatic papillary carcinoma or mucinous cholangiocarcinoa and is a defined type of precancerous lesions . Focal resection is for limited localized lesions in one side of the hepatic lobe. However, lesions that are widely distributed cannot be easily removed, and they are often crisp and easy to fall off, resulting in high relapse rate after surgery and poor prognosis. Papillomatosis can also cause a variety of complications, including intrahepatic duct stones, biliary hemorrhage, common bile duct stones, and gallbladder stones. Other common and fatal complications include bacterial cholangitis, obstructive jaundice, sepsis, hepatic failure, and malignant transformation, which need liver transplantation when necessary.
Generally, the growth of IPNB is mainly limited inside the bile duct, and it has a better prognosis than that of common intrahepatic cholangiocarcinoma . Mucous IPNB accounts for 83% of all the cases, and the average postoperative survival period of non-mucinous IPNB is reported longer than that of mucous IPNB , 52.27 + 6.72 months and 30.84 + 8.36 months, respectively. However, biological behaviors of IPNB lesions differ from each other due to their different stages. Rocha et al. [71] demonstrated that different infiltration depths and different canceration degrees of the tumors are influencing factors for patients’ survival time. Kim et al. [72] suggested that the biological behavior of pancreatobiliary type was different from other types with a higher risk of lymph node metastasis and recurrence.
6.2.4 Biliary Adenofibroma
6.2.4.1 Pathogenesis and Mechanism
Biliary adenofibroma (BAF) is a very rare tumor, first reported by Tsui et al. in 1993 [73], and is considered as a benign tumor with potential of malignant transformation originating from the bile duct. Only six cases of BAF have been reported in the Chinese and foreign literature (Table 6.5), and one case was diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University. Concentrated bile was found in the duct of BAF indicating the connection to the bile duct system, which is a supportive evidence that it derives from hamartomatous bile ducts. Parada et al. [74] detected the chromosome mutation of one case of BAF using cytogenetic methods, finding abnormal chromosome 22, which is common in benign mesenchymal tumors, suggesting that BAF is originated from mesenchymal tissue rather than epithelium.
6.2.4.2 Clinical Features
The average age of the patients is 56.5 (25–79) years old, and the first symptom is often dull pain in the right upper abdomen, with normal laboratory examination and hypervascular masses in CT scanning. The case diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, concerned a 51-year-old patient, who was admitted into the hospital because of upper abdominal pain for 5 months and an occupying lesion in the right lobe of the liver found in CT examination, with normal laboratory examination and no history of hepatitis.
6.2.4.3 Gross Features
The tumor is a spherical mass with an average diameter of 9.9 (3.5–20) cm, and the section shows multiple closely distributed oval thin-walled cavities with a diameter of 1–5 mm accounting for three fourths of the volume of the tumor. The cystic cavity was divided into lobules by slender septa throughout the tumor, and the remaining part of the tumor is composed of relatively dense stroma. The tumor has a clear boundary with no capsule and paler than the surrounding liver tissue in color, while the latter may exhibit atrophy. One case of BAF was diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, the size of which was 3 cm ×2.4 cm × 1.3 cm, the cross section gray and grayish yellow, and hard in texture, with visible fibrous bands, no hemorrhage or necrosis, multiple small cavities in the lesion, fiber bundles stretching out along the duct wall, no complete capsule, and a clear boundary (Fig. 6.37).

Biliary adenofibroma. The cross section gray and grayish yellow, and hard in texture, with visible fibrous bands
6.2.4.4 Microscopic Features
The tumor consists of small vesicles and tubular or cystic cavity-like structures of varying sizes in loose and separated arrangement, embedded in abundant fibrous stroma (Fig. 6.38). The major cells of the tumor are cuboidal and columnar epithelial cells containing abundant cytoplasm with no atypia, and the cytoplasm of some epithelial cells is both eosinophilic and basophilous. The nucleus is small and round or oval, with small and clear nucleolus. The dilated ducts can be cystic with branches or bending, or small papillary protrusions formed on the ductal wall, and a few ducts contain cell debris, bile embolus, or thin eosinophilic fluid. The epithelial cells do not contain mucus, but can go through apocrine glands which changes occasionally on the inner layer of the ductal wall (Fig. 6.39), or are arranged in multilayers with darker nuclear staining and mild to moderate atypical hyperplasia. The interstitial fibrous tissue contains moderate numbers of cells which are myofibroblast-like and spindle shaped, with sparse and red-dye cytoplasm. The local interstitial tissue can exhibit inflammatory reaction mainly composed of lymphocytes, and densely distributed glandules can be found in sparse interstitial regions, similar to the features of BDA . Residual liver cell islands can be observed in the tumor tissue, with no envelope surrounding the tumor, a clear boundary, and no invasion into the surrounding liver tissue.

Biliary adenofibroma. The tumor consists of proliferated small bile duct and abundant fibrous stroma

Biliary adenofibroma. Abundant bile ducts and glands hyperplasia, with abundant collagen fibrous stroma
6.2.4.5 Immunohistochemistry
The epithelial cells are CK5.2, CK7, CK19, EMA, CEA and D10 positive, 1F6, desmin, SMA, mucus staining negative, and interstitial VI and SMA positive. P53 staining positive suggests potential for malignant transformation. Interstitial fibrous tissue staining demonstrates positive VI and SMA and negative NSE and S-100.
6.2.4.6 Differential Diagnosis
-
1.
Bile duct adenoma parts of the lesions may be similar to BDA , but BDA does not contain any bile, and it often presents tubular lumen of bile ducts rather than cystic cavities, which is narrow, closely arranged, and compressed by the matrix. BDA is usually 1–20 mm in diameter, and positive D10 and 1F6 are shown in immunohistochemistry staining positive.
-
2.
Biliary cystadenoma (BCA) is often a huge multilocular cystic mass, and the BAF sac and BCA are similar, but a BCA lesion is generally a solid one, less than 5 mm in diameter. And epithelial mucus staining is positive in BCA but negative in BAF, and the latter does not contain ovarian-like stroma.
-
3.
Well-differentiated cholangiocarcinoma , congenital choledochal cysts, liver benign cystic mesothelioma, and biliary hamartoma should also be taken into consideration during differentiation.
6.2.4.7 Treatment and Prognosis
BAF with positive p53 staining has been reported, presenting tetraploid and S phase block, suggesting that it may belong to the precancerous lesions . And if untreated, the epithelial cells in BAF can go on malignant transformation, especially in regions with multilayers of epithelial cells and darker staining. Therefore, complete surgical resection is the recommended treatment. Lesions with no atypia and negative p53 indicate benign and inert biological behaviors; however, there was one reported case of BAF with epithelial malignant components. Therefore, it is necessary to pay attention to the disease.
6.3 Vascular and Lymphatic Tumors
6.3.1 Cavernous Hemangioma
6.3.1.1 Pathogenesis and Mechanism
Cavernous hemangioma is a benign vascular tumor composed of thin-walled honeycomb vascular cavities and is the most common benign tumor of the liver, with an incidence rate of 1–7%, accounting for about 74% of benign hepatic tumors. The tumor usually occurs in childhood and is often diagnosed in adulthood, regarded as a type of congenital lesions and related to vascular developmental loss of embryonic liver, or an acquired disease induced by steroids, contraceptive, pregnancy, etc. Recent studies have shown that accumulation of mast cells may be associated with its occurrence. From January 1982 to July 2014, 2732 cases of cavernous hemangioma have been diagnosed in the Pathology Department of East Hepatobiliary Surgery Hospital of Second Military Medical University.
6.3.1.2 Clinical Features
Among 172 cases of excised liver hemangioma , diagnosed in the East Hepatobiliary Surgery Hospital of Second Military Medical University from 2004 to 2006, the ratio of male to female ratio was 1:2.4, with an average age of 44.5 years old. Among them, 83 cases had symptoms, including 68 cases of abdominal pain, 16 cases of back and shoulder discomfort, and 4 cases of abdominal distension. The tumors can be palpated in the hypochondrium region in 25 patients, and the size of tumor increased significantly in 84 cases during the course of the disease, with an average increase of 4.4 cm. Most liver hemangiomas did not cause obvious symptoms or signs despite huge volume and were found only in the physical examination. Some of these patients may have symptoms, mainly chronic abdominal dull pain and postprandial fullness, which were occasionally mistaken as hepatic neoplasms. B-ultrasound examination showed small hemangioma with low to medium echo, and CT images showed hypodensity lesions, while MRI T2-weighted images showed high signal density, the so-called bulb sign.
6.3.1.3 Gross Features
Among 172 cases of excised liver hemangioma , diagnosed in the East Hepatobiliary Surgery Hospital of Second Military Medical University from 2004 to 2006, solitary lesions were found in 94 cases [79], two lesions in 40 cases, and three or more lesions in 38 cases. The tumors were 4–32 cm in diameter, averaged at 10.5 cm. A giant hepatic cavernous hemangioma was found and excised by Mengchao Wu and colleagues in one case with a tumor of 63 cm * 48.5 cm * 40 cm in size and 18 kg in weight. The cavernous hemangioma exhibited expansive growth, lobulated surface, and purple-red or dark red in color, soft, with streak-shaped fibrous coating. The cross section showed spongy or cellular lacunae containing blood components, often accompanied by varying sizes of gray-white fibrous sclerosis nodules. A few cases of hemangioma were found with degeneration, thrombosis, or further fibrosis and calcification, with gray fibrosclerosis nodules, and the hemangioma was called sclerosed hemangioma. Liver cirrhosis in the surrounding liver tissue was also found in patients with chronic hepatitis (Figs. 6.40 and 6.41).

Cavernous hemangioma . The tumor is purple-red in color and soft

Cavernous hemangioma . The cross section showed spongy, accompanied by gray-white fibrous sclerosis nodules
6.3.1.4 Microscopic Features
Lesions of cavernous hemangioma are often uniform, composed of varying sizes of communicating vascular cavities, lined with flattened epithelial cells supported by fibrous tissue on the inner wall of the vessels, and filled with blood, and the vascular wall is often thickened to varying degrees due to fibrosis, also known as sclerosed hemangioma (Figs. 6.42, 6.43, and 6.44). The surrounding liver tissue is often distributed with scattered hemangioma foci or highly dilated vascular clusters.

Cavernous hemangioma . The tumor was composed of highly dilated thin-walled vessels, filled with blood

Sclerosed hemangioma

Cavernous hemangioma , sclerosed type. The vascular wall is thickened to varying degrees due to fibrosis
Hepatic hemangioma includes the following rare subtypes. ① Capillary hemangioma, diagnosed in a 58-year-old woman with a surgical resection of a tumor in the left lobe of the liver in our experience, 2.3 cm × 2 cm in size with a dark red section (Fig. 6.45), was clustered with immature capillaries (Fig. 6.46), lined by a monolayer flattened endothelial cells (Fig. 6.47) under the microscope, and CD34 staining showed the immature microvascular lumens (Fig. 6.48). In addition, visible thrombosis and arteries with thickened vascular wall and stenosis can also be seen. ② Infantile hemangioma (IH), also known as infantile hemangioendothelioma, often occurs in 10-day-old to 3-month-old infants after birth, with multiple lesions of infantile skin hemangiomatosis, 0.8–21 cm in diameter, and can be divided into focal (single lesion), multifocal (4–20 lesions), and diffuse type (>20 lesions). The morphological characteristics are similar to those of juvenile capillary hemangioma in the soft tissue. In the early stage of the disease, the neocapillaries may contain no or only a few cavities, and the hypertrophic endothelial cells are arranged in the shape of solid plates, while in maturity stage they are similar to adult capillary hemangioma with intensive expression of glucose transporter (GLUT1) and formation of angiosarcoma in a small number of cases [80], and the solitary lesions can be surgically excised, while multiple lesions can be treated by liver transplantation. ③ Hepatic vascular malformation with capillary proliferation (HVMCP), with lesions of 5–11 cm in diameter, typically exhibits as major infarction and hemorrhage center and strip-shaped arrangement of congested blood vessels with thick walls, lined with flat endothelial cells, surrounded by proliferation of peripheral blood capillaries and interstitial mucinous degeneration. Negative GLUT1 staining is shown in vascular endothelial cells and major blood vessels. The prognosis of the tumor which receives surgical resection is good [81].

Capillary hemangioma . The cross section showed dark red with clear boundary

Capillary hemangioma . The tumor was clustered with capillaries; vascular lumens were narrow

Capillary hemangioma . Immature small vessels lined by endothelial cells showed microvascular lumens

Capillary hemangioma . CD34 staining showed the immature microvascular lumens
6.3.1.5 Immunohistochemistry
CD34 and F-VIII positive.
6.3.1.6 Differential Diagnosis
Pathological diagnosis of the disease is not difficult, but misdiagnosis from vascular lesions, such as angiolymphoma, angiosarcoma , and peliosis hepatis, should be avoided.
6.3.1.7 Treatment and Prognosis
Cavernous hemangioma grows slowly with no malignant tendency. Lesions >5 cm in diameter or manifests by obvious symptoms can be treated with surgical resection or hepatic artery ligation, and conservative treatment or observation is suited for small or asymptomatic lesions, while selective hepatic portal occlusion and anatomic technique are extremely important when treating hemangioma located at complex and difficult sites [82]. Successful liver transplantation was reported in a case of giant hepatic cavernous hemangioma (40 cm×30 cm×30 cm) by Zhongshan Medical University. In recent years, interventional technique and radiofrequency ablation have also been applied to the treatment of this disease which shows good curative effects in surgery. The prognosis is good, with no recurrence, and hepatic cavernous hemangioma associated with medication may disappear spontaneously or stop growing or after drug withdrawal.
6.3.2 Hemangioblastoma
Hemangioblastoma, also called as haemangioblastomas and composed of large thick-walled blood vessels and stromal cells with rich lipid droplets, is a benign vascular tumor originated from mesoderm and commonly found in patients with von Hippel-Lindau disease (VHL). VHL is an autosomal dominant cancer predisposition syndrome, mainly involving multiple organs and systems and a variety of hereditary neoplastic syndromes, and hemangioblastoma of central nervous system is the most common tumor, often found in the cerebellum, cerebrum, spinal cord, brain stem, and retina and rarely seen in the liver. Only three cases of liver hemangioblastoma have been reported in the English literature, all of which were combined with VHL [83,84,85].
Previous reports of VHL involving the liver were often adenoma, cysts, and hemangioma , while liver hemangioblastoma is extremely rare. Ultrasound examination shows a solid hypoechoic mass, and CT shows a hypodense mass, while angiography shows a mass with rich blood vessels. Microscopic observation shows the similarity of the lesion to hemangioblastoma of the central nervous system, consisting of vascular endothelial cells, pericytes, and stromal cells which form atypical capillaries with expansion of vessels and a small amount of red blood cells in the lumen. The stromal cells are rich in adipose tissue, which may form bubbles or bubble-like structures, and increased nuclear chromatin and enlarged nuclei are occasionally found in the stromal cells. When VHL patients are accompanied with liver occupation, hemangioblastoma should be first excluded. Surgical treatment for intracranial hemangioblastomas shows good therapeutic effects, and complete resection is a radical therapy. However, there are also many reported cases concerning hemangioblastoma in the liver or lung after multiple surgeries.
6.3.3 Infantile Hemangioendothelioma
6.3.3.1 Pathogenesis and Mechanism
Infantile hemangioendothelioma (IHE) is the most common benign vascular tumor in the liver; almost all cases concern infants under 1 year old after birth, derived from abnormal development of the central vein and portal vein system. From January 1982 to December 2012, six cases of IHE were diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University, accounting for 0.019% of all liver primary malignant tumors during the same period. IHE is similar to skin capillary hemangioma that if regression occurs after proliferation, maturation, and degradation stages, liver-occupying lesions cannot be formed. This disease can also be a part of Kasabach-Merritt syndrome, characterized by cutaneous and visceral angiomatosis, thrombocytopenia syndrome, and disseminated intravascular coagulation, or accompanied by certain congenital diseases, such as congenital heart disease, 21 trisomy syndrome, deletions in chromosome 6q, and heterotopic left liver in the thoracic cavity. Rapini et al. [86] conducted karyotype analysis in one 3-year-old child with IHE (tumor size, 57 mm × 56 mm × 70 mm), finding interstitial deletion of 13q13.3 to 21.32 on the long arm of chromosome 13, with a deletion range of 27.87 MB containing the RB1 gene, and the patient manifested developmental delay and poor growth.
6.3.3.2 Clinical Features
The patients of IHE include infants within 6 months after birth accounting for 86% and children under the age of 3 years old accounting for 99%. However, there are also reports of adult IHE. Among the six cases of IHE in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University, the male to female ratio was 5:1, and the average age was 38.8 years (5–61). About 30% of the children cases had angioendothelioma and at the same time in the skin, lymph node, spleen, gastrointestinal tract, pleura, prostate, lung, and bone. Riley et al. [87] reported two cases of IHE with biliary atresia, acute liver failure, and some other congenital diseases, such as diaphragmatic hernia, Down syndrome, large artery transposition, and multi-finger deformity. Forty percent of the patients were primarily diagnosed when their mother manifested abdominal masses. Related clinical manifestations include hepatomegaly, nausea, vomiting, gastrointestinal bleeding, jaundice, hemolytic anemia, thrombocytopenia, liver failure, or even death caused by high-output heart failure because of intrahepatic arteriovenous shunt due to tumor compression. Mild to moderate increase of serum AFP levels can be found in the children patients, which is limited for reference in younger children, because its level in normal newborns reaches 2500 ng/ml and decreases to normal level in adults at 6 months after birth. The performance of the lesions on CT shows hypodensity with a clear boundary, and scattered calcification in the center can be found in 50% cases, while obvious calcification demonstrates wide distribution inside the mass-forming granular clusters. And early filling of the lesions in SPECT scans is of diagnostic value.
6.3.3.3 Gross Features
Intrahepatic solitary or multiple lesions are found in liver IHE. In our hospital, all the six cases diagnosed as IHE concerned solitary lesions of 1.2–6.3 cm in diameter. The cross section of the tumors was brownish-red capillary cavities with rich blood contents or yellow white in cases with necrosis. The boundary was not completely clear between the tumor and the surrounding liver tissue, and local infiltration can be observed (Fig. 6.49).

Infantile hemangioendothelioma. The cross section was yellow white with unclear boundary
6.3.3.4 Microscopic Features
IHE can be divided into two pathological types:
Type I accounts for about 80% of the tumors. The surrounding structure was composed of dense proliferation of irregular thin-walled capillary cavities, lined with monolayer of fat or flat endothelial cells (Figs. 6.50 and 6.51), which are consistent in morphology and contain small- or medium-sized nuclei and eosinophilic nucleoli, with little nuclear fission. The cavities contain red blood cells, and fewer tumorous interstitial components can be found with visible residue of hepatocytes, bile duct, extramedullary hematopoiesis regions, and no fibrous envelope. The infiltration border was interspersed in the liver between the plates (Fig. 6.52). Moreover, hemorrhage, necrosis, and calcification can also be found in the tumor. Type I IHE can undergo canceration.

Infantile hemangioendothelioma, type I. Capillary cavities lined with flat endothelial cells

Infantile hemangioendothelioma, type II. Irregular capillary cavities, lined with monolayer of cubic endothelial cells

Infantile hemangioendothelioma, type I. The tumor was composed of dense proliferation of irregular thin-walled capillary cavities, no fibrous envelope and infiltration border
Type II accounts for about 20% of the tumors. And five type I and one type II of IHE were found among the six cases diagnosed in our hospital. The latter mainly demonstrated obvious proliferation of vascular endothelial cells, occurrence of pleomorphic endothelial cells arranged in a single layer of nail dendritic shape or multiple layers, or even protrudes to the cavity in tufted morphology. No lumen or ambiguous tubular structure can be found, and some of them may form papillary structures. The cells are with significant nuclear atypia, hyperchromatic nuclei, and mitosis. The nature of this type IHE is angiosarcoma , with strong infiltrating ability into the surrounding liver tissue.
6.3.3.5 Immunohistochemistry
Vascular endothelial cells were positive in vimentin, CD34 (Fig. 6.53), CD31, UEA-1, and F-VIII staining and negative in CK staining, and SMA staining displays positive smooth muscle cells within the vascular basement membrane.

CD34 staining showed active hyperplasia microvessels and “infiltrated border”
6.3.3.6 Differential Diagnosis
According to the age of the patients and characteristics of tumor growth, correct diagnosis can be made, with cautions of differentiation from angiosarcoma , epithelioid hemangioendothelioma, and malignant hemangiopericytoma (Table 6.6). Other tumors that should be differentiated from are primary or secondary tumors of the liver, such as hepatoblastoma , neuroblastoma with liver metastasis, hepatic mesenchymal hamartoma , cavernous hemangioma of the liver, and teratoma . The final diagnosis depends on the pathological examination.
6.3.3.7 Treatment and Prognosis
Surgical resection is usually preferred. This group of IHE patients with surgical resection had a good prognosis. Sondhi et al. [88] treated one case of huge multiple liver IHE with TACE, finding volume reduction of the primary tumor, after which surgical resection was conducted and the residual tumor was treated via chemotherapy, but this was of poor efficacy and replaced by metronomic therapy of cyclophosphamide and tamoxifen, with complete disappearance of liver lesions.
6.3.4 Lymphangioma and Lymphangiomatosis
6.3.4.1 Pathogenesis and Mechanism
Lymphangioma is a benign tumor consisting of dilated lymphatic vessels containing lymph, once called cavernoma lymphaticum, lymphatic endothelioma, etc. and derived from congenital dysplasia of embryonic lymphatic system, or abnormal formation of lymphatic vessels, resulting in blocked lymphatic flow. Lymphangioma can be involved in the spleen, lung, bone, kidney, gastrointestinal tract, and other parts. Multiple lymphangiomas are known as lymphangiomatosis. Hepatic lymphangioma and lymphangiomatosis are very rare with more than ten cases reported so far, both in the domestic and foreign literature, and seven cases have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University.
6.3.4.2 Clinical Features
The patients include infants and the elderly as reported in the literature, but children and youth are more common, with a male and female ratio of 1:2 [89]. Seven patients of liver lymphangioma were diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, with five males and two females aged 10–65 years old, and the average age was 31.9 years old. The symptoms and signs were associated with the number and location of the involving organs, generally abdominal distention, hepatosplenomegaly, pleural effusion, ascites, and organ dysfunction. Imaging showed a multilocular cystic mass, with clear or pale red liquid content which can be obtained via puncture. MRI examination is more effective, which can demonstrate lymphangioma as irregular lesions with ambiguous boundaries, and part of them is surrounded by vessels which is called vascular package, producing mass-occupying effects. Liquid-liquid plane can be observed in cystic lymphangioma lesions. Mild or no enhancement is shown in enhanced scan which can be differentiated from the images of other hepatic tumors [90].
6.3.4.3 Gross Features
Solitary lymphangioma is large in volume, often > 10 cm in diameter. The lesions in seven cases of liver lymphangioma, diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, were 3.2–24 cm in diameter with an average of 11.8 cm. The tumor section was gray-white (Fig. 6.54), honeycomb, or multilocular cavities with varying sizes, and local solid fibrous scars can be found with transparent slurry or chylous fluid in the lumen. Lymphangiomatosis contains multiple lesions distributed in the whole liver, and the numbers of lesions vary in cases.

Lymphangioma. Cystic and solid tumor, 14 cm in diameter
6.3.4.4 Microscopic Features
The lesions are characterized by numerous lymphatic cavities of varying sizes with cystic dilatation in the liver parenchyma, containing eosinophilic lymphatic fluid (serous fluid and small lymphocytes) (Fig. 6.55), surrounded by small bubbles. Red cells can be found in the cavities when there is bleeding (Fig. 6.56), and the walls of them are lined with a single layer of flat endothelial cells (Fig. 6.57) with consistent nuclei and no atypia. Fibrous hyperplasia can be seen in part of tumor tissue forming a more solid region (Fig. 6.58), and the surrounding patches of fibrous connective tissue with residual hepatocyte isles can be observed (Fig. 6.59).

Lymphangioma. Lymphatic cavities with cystic dilatation are lined with a single layer of flat endothelial cells, containing light dye lymphatic fluid

Lymphangioma. Interstitial hemorrhage, red cells can be found in the cavities

Lymphangioma. Lymphatic cavities lined with a single layer of flat cells

Lymphangioma. Interstitial fibrous hyperplasia with hemorrhage

Lymphangioma. The surrounding patches of residual hepatocytes
6.3.4.5 Immunohistochemistry
Endothelial cells express CD31, CD34, and F-VIII, and lymphatic marker of podoplanin staining is positive.
6.3.4.6 Differential Diagnosis
Note that it should be differentiated from mesenchymal hamartoma of the liver and hepatic hemangioma .
6.3.4.7 Treatment and Prognosis
Lymphangioma is a benign tumor, but it can cause significant damage to the surrounding liver tissue when active proliferation occurs. Ra et al. [91] reported a case of lymphangiomatosis in the liver, which was treated by liver transplantation with recurrence 19 years later. Speculation has been made that it is related to the lymph tube endothelial progenitor cells which were released into the peripheral blood circulation. Thus, it is considered as a low-grade tumor or with metastatic potential. In general, surgical resection is the preferred treatment method, and liver transplantation is another choice. It is worth noting that the use of immunosuppressive agents may result in progression of extrahepatic lymphangiom if lymphangioma involves both the liver and extrahepatic organs [92, 93].
6.4 Muscular, Fibrous, and Adipose Tumors
6.4.1 Leiomyoma
6.4.1.1 Pathogenesis and Mechanism
Leiomyoma is more accepted to originate from visceral or vascular muscular layer, commonly found in the urinary tract and digestive tract. Demel reported the first case of primary liver leiomyoma in 1926, and a total of about 30 cases have been reported in English literature so far. One case has been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University. Possible origination of liver leiomyoma is in the blood vessels or biliary tree of the liver, and primary hepatic leiomyoma can be found in posttransplantation patients using immunosuppressant and patients with AIDS. Researches have also shown that EBV infection might be related to the occurrence of the disease.
6.4.1.2 Clinical Features
Primary hepatic leiomyoma is more commonly seen in adult females, and the male to female ratio is about 1:1.8, with an average age of 41 (1.5–71) years old. No early discomfort is found with no obvious clinical symptoms, and it is often discovered in physical examination. When the tumor grows to a size sufficient to cause oppression or pull on the adjacent organs, symptoms appear such as abdominal fullness, discomfort, or pain. Imaging findings include: ① large tumors with clear boundaries and visible patchy necrosis, ② significant enhancement in arterial phase and more significant enhancement in portal vein phase on CT enhanced scan, ③ multiple abnormal blood vessels which are thickened and circuitous around the tumor, and ④ MR T2WI showed solid and low signals indicating tumors which is the feature for leiomyoma [94].
6.4.1.3 Gross Features
The average diameter of the tumors was 6.8 (2–12) cm, which are solid masses with cystic degeneration in some cases. The section shows gray-white bundles and the capsule is complete.
6.4.1.4 Microscopic Features
The tumor is composed of spindle-shaped smooth muscle cells with pale staining cytoplasm, rod-shaped nucleus, normal nucleocytoplasmic ratio, no nuclear atypia, and no or occasional nuclear division. The tumor tissue was shown in interwoven bundles or plexiform arrangement (Fig. 6.60). Interstitial tissue may contain mucous secretion, forming myxoid leiomyoma , with a complete capsule and no invasion into the surrounding liver tissue.

Leiomyoma . The tumor is composed of spindle-shaped smooth muscle cells and shown in interwoven bundle arrangement
6.4.1.5 Immunohistochemistry
Vimentin, SMA, and desmin expressions are positive, and CD-117, CD34, DOG-1, and other GIST markers are negative, which should also be combined with clinical findings and the detection of EBV infection.
6.4.1.6 Differential Diagnosis
It should be differentiated from gastrointestinal stromal tumors , neurofibroma, leiomyosarcoma, and other hepatic tumors.
6.4.1.7 Treatment and Prognosis
There is no report on the recurrence of leiomyoma after surgical resection.
6.4.2 Solitary Fibrous Tumor
6.4.2.1 Pathogenesis and Mechanism
Solitary fibrous tumor (SFT) is a kind of dendritic cell neoplasm expressing CD34, first reported by Klemperer and Rabin, and is more commonly found in the pleura, also known as localized fibrous mesothelioma previously. The occurrence sites for extrapleural SFT include the upper respiratory tract, nasal cavity, mediastinum, lung, orbit, pelvic peritoneum, mesenterium, perididymis, liver, breast, spinal cord, kidney, and adrenal gland. Primary hepatic SFT is rare, and more than 40 cases have been reported by 2013. The origin of the tumor is inconclusive and is more accepted that it originates from the liver submesothelial tissue. For a long period, whether the lesions are benign or malignant remains controversial. Liu et al. [95] regarded it as a borderline tumor, while Fisher [96] classified it into intermediate fibroblastic and myofibroblastic tumors in Atlas of Soft Tissue Tumor Pathology published in 2013, which is not defined as benign fibrous tumor. And in the 2010 WHO’s Histological Classification Guideline of Digestive System Tumors, it is grouped into benign hepatic tumors derived from mesenchymal tissue [97].
6.4.2.2 Clinical Features
Two cases of hepatic SFT were diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, from January 2005 to June 2014, including a 49-year-old male and 51-year-old female, both of them had no history of hepatitis. Combined with the reports of 61 cases of primary hepatic SFT in the literature, the patients of the disease are often adults aged from 16 to 85 years old with an average of 55.6 years old, and most of them are females with a male to female ratio of 1:1.47. Nonspecific clinical manifestations mainly involving the digestive system and accounting for 77% include epigastric fullness, nausea and vomiting, abdominal pain, diarrhea, back pain, abdominal distention, painless abdominal mass, hepatomegaly, lower limb edema, and weight loss (or increase). No abnormality was found in laboratory examinations, with normal liver function. Mild to moderate elevation of ALP and (or) γ-GT is found in a few cases, and tumor markers including AFP, CA19–9, and CEA are generally normal. Ultrasound examination shows heterogeneous masses, and CT images demonstrate clearly bounded hypodense lesions, with necrosis and calcification in some cases, and cyclic centripetal progressive enhancement is shown in enhanced CT. MRI shows low signals on T1WI and high signals or mixed signals on T2WI, and flaky, arc, or ring enhancement is shown in gadolinium contrast enhancement MRI. PET-CT displays elevated SUV values in the tumor region.
6.4.2.3 Gross Features
The tumors are often solitary, while multiple lesions are rare, and it is located mainly in the left lobe of the liver, or ligamentum teres hepatis as reported, with a mean diameter of 16.6 (0.5–32) cm. In two cases of SFT in the liver, diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, the diameters were 8.7 and 8.1 cm, and the lesions are located in the left and the right lobe, respectively. The tumor body in one case was completely dissociated from the liver parenchyma, with only two thin pedicles connected to the hepatic capsule. The surface of the tumors was smooth, and the tumor capsule is complete, and continuity with liver capsule was observed with good tenacity. The section is gray or grayish yellow, showing interwoven fiber bundles (Fig. 6.61), mucoid degeneration, and varying numbers and sizes of cystic cavities.

Solitary fibrous tumor. The section is gray or grayish yellow, showing interwoven fiber bundles
6.4.2.4 Microscopic Features
Most of the hepatic SFT is benign, composed of spindle fibroblasts with a small amount of eosinophilic cytoplasm, and transparent eosinophilic bodies can be visible in part of the cytoplasm. The nuclei were spindle-like with thin ends, obsolete nucleolus, and no nuclear atypia. The tumor cells were arranged in short seat patterns or disordered distribution in abundant collagen fibrous interstitial tissue. Some tumor cells are found around the expanded vessels forming a hemangiopericytoma-like structure. Cells with nuclear division (< 3/10HPF) and increased nuclear atypia indicate a malignant potential. The WHO classification of soft tissue tumors suggested the characteristics of SFT, including large lesions (≥5 cm or 10 cm), no tumor pedicle, infiltrative borders, abundant cells, moderate or high degree of nuclear atypia, mitotic activity (> 4/10HPF), hemorrhage, and necrosis (Fig. 6.62).

Solitary fibrous tumor. Spindle cells with small rod-shaped and no atypia nuclei and no mitosis
6.4.2.5 Immunohistochemistry
Most of the tumor cells were intensively positive for CD34, Bcl-2 (Figs. 6.63 and 6.64), and vimentin; negative for CK, S-100, HMB45, and EMA and SMA; and partially positive for CD99 and desmin, while malignant tumor cells may lose expression of CD34.

Solitary fibrous tumor. CD34, positive

Solitary fibrous tumor. BCL-2, positive
6.4.2.6 Specific Staining
Masson trichrome staining shows dense collagen bundles, and reticular fiber staining shows reticular fibrous septa surrounding tumor cell bundles.
6.4.2.7 Differential Diagnosis
-
1.
Hepatic leiomyoma expresses SMA and desmin, but no CD34.
-
2.
Malignant mesothelioma contains cells with rich polymorphism, active nuclear division, and CD34 negative.
-
3.
Fibrosarcoma contains cells in woven-like arrangement, with obvious nuclear atypia, active mitosis, and invasive growth.
-
4.
Hepatic inflammatory pseudotumor contains a large number of mixed inflammatory cells and fibrous tissue.
-
5.
Gastrointestinal stromal tumors were positive for CD117 staining.
-
6.
Malignant SFT has been reported in five cases of primary malignant liver SFT with postoperative recurrence and extrahepatic multiple organ metastasis since 2000, seriously worsening the prognosis [98,99,100,101,102].
-
7.
Metastatic SFT in the liver is often derived from extrahepatic organs such as the mesenterium, kidney, and spinal cord.
-
8.
Other tumors which should be differentiated from include hemangioperithelioma, synovial sarcoma, and peripheral nerve sheath tumors.
6.4.2.8 Treatment and Prognosis
The prognosis of benign hepatic SFT treated with resection is good.
6.4.3 Angiomyolipoma
6.4.3.1 Pathogenesis and Mechanism
Angiomyolipoma ( AML ) was first reported by Ishak with a case of hepatic AML in 1976, and Cong Wen Ming and colleagues began to report the constitution of thick-walled blood vessels, smooth muscle, and fat in different proportions in AML in 1992 [103]. Once known as hamartoma, it is now known as a true neoplasm, and it derives from perivascular epithelioid cells (PEC), belonging to the family of perivascular epithelioid cell tumors (PEComas). These cells are usually epithelioid, with transparent or eosinophilic red cytoplasm, PAS staining positive, not resistant to diastase digestion, and small nucleoli are often visible, with differentiation characteristics of both melanoma cells and muscular cells. The etiology of hepatic AML is not clear, and there are reports on AML with tuberous sclerosis complex (TSC), accompanied by hamartoma in the kidney or other organs or skin nodules [104, 105].
6.4.3.2 Clinical Features
A total of 244 cases of surgically excised hepatic AML were diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, from January 2001 to November 2012, with 68 males (27.87%) and 176 females (72.13%), with an average age of 44.3 (23–79) years old, and the male to female ratio was 1:2.6 (Fig. 6.65). The reported male to female ratio of hepatic AML patients was about 1:2–5, and the average age was 41.7 (10–86) years old in the literature, and the age of onset was mainly concentrated in the range of 35–55 years old. Hepatic AML cases are mostly solitary, 60% located in the right lobe, 30% located in the left lobe, 20% in both the left and right leaves, and 8% in the caudate lobe.

Angiomyolipoma . Surgically treated hepatic AML patients were diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital
Most of the patients with hepatic AML are asymptomatic, usually discovered in physical examination. A few patients with larger tumors have upper abdominal discomfort or pain, and other rare manifestations include abdominal mass, hematemesis and melena, fever, fatigue, lack of appetite, and weight loss. If accompanied by renal AML , the patients may manifest corresponding symptoms such as lumbago, and those with nodular sclerosis may have mental retardation and developmental delay [106]. The difference between renal and hepatic AML includes that about half of renal AML are associated with tuberous sclerosis and about 1.15% of the hepatic AML cases are associated with tuberous sclerosis [107]. Wang Jue-ru et al. [108] reviewed the clinical data of 523 cases of hepatic AML and found that most patients had no history of hepatitis suggesting irrelevance with hepatitis, and female patients are without long-term oral contraceptives indicating hepatic AML is not associated with a contraceptive history. No obvious abnormalities are found in biochemical or liver function tests.
Imaging results of AML vary according to different proportions of the three components, blood vessels, vascular smooth muscle, and fat. Ultrasound examination shows high or mixed echoes, and when the mass reflects a strong echo, indicating fat or fatlike component, while a weak echo is reflected, strong patchy or strip-shaped echoes can be detected inside the lesion which is similar to the echo of fat [109, 110]. Signs of blood vessels in fat shown on CT scan are of diagnostic value [111], and enhanced scan shows enhancement in patterns such as “fast in and fast out,” “fast in and slow out,” or delayed enhancement. The enhancement amplitude of the tumor is often weaker than that of the surrounding normal liver parenchyma in portal venous/delay phase, and some large tumors oppressing the surrounding normal liver parenchyma to form a pseudocapsule show delayed enhancement [112, 113]. MRI images of hepatic AML show high or low signals on T1-weighted image and heterogeneous high signals on T2-weighted image. The definition of fat signals on MRI imaging of hepatic AML with much fat is an evidence to differentiate from the majority of liver cancers [114].
6.4.3.3 Gross Features
The tumors often have no capsule, and the diameter is 0.8–36 cm with an average of > 5 cm. The gross appearance of the tumors often varies according to different proportions of the components. Tumors with a majority of muscular cells are often gray-white or gray-brown (Fig. 6.66), accompanied by varying amounts of hemorrhage. Tumors with major content of fat are similar to the appearance of lipoma which is gray in color. And those composed mainly of blood vessels are grayish red or grayish brown on the cross section, soft in texture. The tumors have clear boundaries with the surrounding liver tissue, and the latter often shows no sclerosis [115].

Angiomyolipoma , muscle cell-based type
6.4.3.4 Microscopic Features
Hepatic angiomyolipoma (HAML) contains curved thick-walled blood vessels, muscle cells, and fat cells in different proportions, and much difference is found in different tumors or different regions of the same tumor. According to the varying proportions of the composition, HAML is divided into four types [116–118]:
-
1.
Classical type. Solid patches of myoid cells mixed with slices of fat cells, interspersed with irregular thick-walled blood vessels (Fig. 6.67).
Fig. 6.67 
Angiomyolipoma , classical type
-
2.
Muscle cell type. Mainly composed of muscle cells. And according to various morphological features of muscle cells, this type of HAML can be divided into five subtypes as some scholar suggested: epithelioid cells (Fig. 6.68), intermediate cell type, spindle cell type, monomorphic cell type/eosinophilic cell type, and polymorphic cell type [119] (Fig. 6.69). And epithelioid angiomyolipoma accounts for most of hepatic AML .
Fig. 6.68 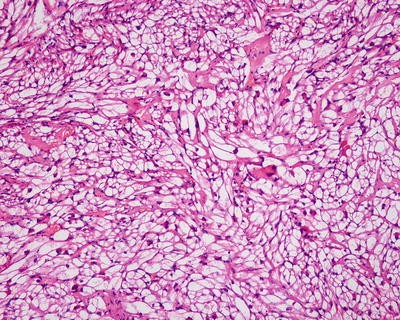
Angiomyolipoma , epithelioid cell type. No vessels and adipocytes
Fig. 6.69 
Angiomyolipoma , polymorphic cell type
-
3.
Fat cell type. The tumor is mainly composed of mature fat cells, which interlace with intermediate muscle cells to form a network (Fig. 6.70).
Fig. 6.70 
Angiomyolipoma , fat cell type
-
4.
Angiomatous type. The vascular components consist of varying amounts of bending thick-walled blood vessels, often without elastic layer, and the muscle cells are locally distributed. Epithelioid cells and spindle myoid cells often surround the blood vessels to form peripheral vascular sheaths, especially evident in the periphery region of the tumor, and many thin-walled veins or blood vessels diffuse in the whole parenchyma, similar to purpura [120].




The tumors often have no capsule with clear boundaries with the surrounding liver tissue, and “invasive boundary” can also be found (Fig. 6.71), which is not a sign of malignant transformation.

Angiomyolipoma “invasive boundary”
6.4.3.5 Immunohistochemistry
Epithelioid cells of hepatic AML have the ability to synthesize melanin, so they express HMB45 (Fig. 6.72) and A103, but did not express CD34, VIII factor, EMA, keratin, myoglobin, etc. [122]. Stronger positive HMB45 and A103 are found in the epithelioid and pleomorphic muscle cell type than those in spindle cell type, which often express VI, SMA (Fig. 6.73), and desmin. Muscle cells in intermediate cell type also express vimentin, SMA, and desmin, the positive degrees of which are between those of epithelioid and spindle cell type. Recent reports suggested that cathepsin K demonstrated a higher positive rate in the perivascular epithelioid cell tumors (PEComas), as well as higher specificity [121].

Angiomyolipoma . HMB45 positive

Angiomyolipoma . SMA positive
6.4.3.6 Differential Diagnosis
When the tumor is mainly composed of fat, especially in cases with visible lipoblasts, it can be easily misdiagnosed as well-differentiated liposarcoma, and positive HMB45 and SMA detected by immunohistochemistry are very helpful in diagnosis. FISH detection of MDM2 gene status is a choice when necessary. In AML with major spindle-shaped cells, which is easy to confuse with tumors derived from smooth muscle or nongastrointestinal ST, the finding of thick-walled blood vessels and fat is a good indication for AML . AML with epithelioid cells arranged in trabecular structure containing eosinophilic or transparent cytoplasm and pleomorphic cells with visible nucleoli can be easily misdiagnosed as hepatocellular carcinoma . And HMB45 and SMA are beneficial during differential diagnosis. It should also be differentiated from malignant melanoma, metastatic carcinoma, and angiosarcoma of renal cell tumor identification.
6.4.3.7 Treatment and Prognosis
The preoperative diagnosis accuracy of hepatic AML is low with a high misdiagnosis rate, so most scholars advocate surgical treatment for the patients as soon as possible. Small tumors (e.g., < 5 cm) can be observed during follow-up; however, large tumors (> 5 cm) or small tumors in rapid growth should be treated with surgical resection as soon as possible. It has been reported in the literature that with the passage of time, the tumor size increases to form a giant hepatic tumor, easily affecting liver function, and is at risk of rupture or canceration [122]. You J et al. [106] report one case of multiple hepatic AML , renal AML associated with tuberous sclerosis treated by superselective arterial embolization with a good curative effect. And foreign reports suggested that hepatic AML contains estrogen and progesterone receptors; thus, tamoxifen can be adopted for the treatment and has obtained certain curative effects [123]. Gennatas [124] reported a case of PEComas treated with the mTOR inhibitor everolimus with good therapeutic effects. All these case reports provide valuable experience for the treatment of hepatic AML .
Hepatic AML has long been considered as a benign tumor, but with the development of researches and the accumulation of cases including reports on malignant hepatic AML , hepatic AML is a potentially malignant tumor [125, 126]. However, so far, it lacks reliable morphological criteria to determine whether it is benign or malignant. Malignant hepatic AML described in the literature are often separate cases. To date, the common features of malignant hepatic AML suggested by Folpe [127] include: tumor diameter > 10 cm, tumor necrosis, cellular atypia, visible epithelioid cells, and increased mitosis (Fig. 6.74). Immunohistochemistry shows elevated Ki-67 proliferation index, positive p53, and negative CD117. However, simple invasive growth, vascular deformity, and visible nucleoli are shared by both benign and malignant AML , which are no hints of biological behaviors [128].

Angiomyolipoma . Mitosis is visible
In summary, hepatic AML is a type of tumor with malignant potential originating from perivascular epithelioid cells, often contributing no clinical manifestations or specific imaging features. The preoperative diagnostic accurate is relatively low; therefore, early surgical treatment should be recommended once the tumor is discovered. During postoperative pathological diagnosis, careful observation and identification of the morphology of muscle cells should be made, as well as the varying kinds of immunophenotype of different muscle cell types. After surgery, long-term follow-up to observe recurrence or metastasis for the patients is recommended.
6.4.4 Lipoma
6.4.4.1 Pathogenesis and Mechanism
Ramchand reported the first case of hepatic lipoma in 1970, and over 20 cases have been reported in the literature up to now. Thirteen cases of surgically excised hepatic lipoma were diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University from 1982 to 2013. Hepatic lipoma is composed of mature adipose cells, suggesting its potential origin of pluripotent stem cells with lipid-storing ability surrounding the hepatic sinusoids. Martin-Benitez et al. [129] suggested the relationship of hepatic lipoma and fatty liver, and insulin resistance may be the possible mechanism of the disease.
6.4.4.2 Clinical Features
The 13 cases of hepatic lipoma excised surgically in our hospital concerned three males and ten females, and the average age was 41.4 (29–58) years old. The patients almost manifested no obvious symptoms, and the lesions were often discovered occasionally during physical examination.
6.4.4.3 Gross Features
The tumor was 6.8 (2–12) cm in diameter, mostly pale yellow or gray-white in a few cases, soft in texture, part of which are encapsulated and lobulated, with a greasy section and a clear boundary (Fig. 6.75).

Lipoma. The tumor was yellow and white, with a greasy section and a clear boundary
6.4.4.4 Microscopic Features
The tumor was composed of mono-differentiation mature fat cells, which are consistent in size, and the karyoplasmic ratio is normal, with no mitotic figures (Fig. 6.76).

Lipoma. The tumor was composed of mono-differentiation mature fat cells
6.4.4.5 Differential Diagnosis
It should sometimes be differentiated from focal hepatic steatosis, well-differentiated liposarcoma, and lipid-rich type of hepatic angiomyolipoma .
6.4.4.6 Treatment and Prognosis
Focal resection is the main treatment for hepatic lipoma. And for small lipomas with definite diagnosis, temporary observation can be adopted, and surgical resection can be applied when the tumors increase significantly. Aggressive surgical resection if the optimal therapeutic scheme for those cases is difficult to identify from malignant tumors, such as liposarcoma.
6.4.5 Myelolipoma
6.4.5.1 Pathogenesis and Mechanism
Myelolipoma is a benign lipoma mixed with extramedullary hematopoietic tissue, often found in the adrenal cortex. The cause is unknown; however, there are several viewpoints concerning the pathogenesis of myelolipoma, including development and differentiation of residual embryonic mesenchymal cells, transposition of adrenal myelolipoma, plantation and metaplasia of bone marrow emboli, etc. In present, gene analysis has been conducted on only one patient, finding translocation of chromosomes (3; 21) (q25; p11), suggesting that the tumor belongs to neoplastic lesions [130]。. Allison et al. [131] suggested the relation between myelolipoma and obesity, gene mutation, and hormone use. Reports on hepatic myelolipoma have been published since the 1970s, and over ten cases both abroad and at home were found in the literature. One case of hepatic myelolipoma was diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, and no association of the hepatic myelolipoma with metabolic and endocrine diseases has been found. However, there are reports of myelolipoma in surgically excised hepatocellular carcinoma .
6.4.5.2 Clinical and Pathological Features
Hepatic myelolipoma is more common in people older than 40 years old in both genders, mostly with no clinical symptoms and found during physical examination, surgery, or autopsy, while some patients manifest recurrent abdominal distension, decreased appetite, hepatomegly, or even rupture and hemorrhage in cases with larger tumors. Individual patients may have a history of up to 10 years’ hepatic masses which gradually increased in size. CT examination demonstrates hypodensity which indicates adipose tissue [132]. The tumor is commonly seen in the right lobe of the liver with a diameter of 1–16 cm, light yellow or brown, often with no capsule, exhibiting gray or grayish red and circular nodules on the section with or without hemorrhage and necrosis. The boundary is clear and the tumor is generally solitary lesions, and multiple hepatic myelolipomas have also been reported [133]. The case diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, concerned a 60-year-old male with the tumor located in the right lobe of the liver, and the diameter was 2.3 cm. Microscopic observation showed that the tumor was mainly composed of mature fat cells and hematopoietic cells which were similar to the bone marrow. The latter included granulocyte, erythroid, and megakaryocytic components at different mature stages; however, they are different from the cells in the bone marrow because of the lack of sinusoid in the bone marrow with no capsule in the surrounding area, and tumor tissue and liver tissue are staggered into each other to form an “infiltration border” (Figs. 6.77 and 6.78). Attention should be paid to the differential diagnosis from angiomyolipoma , lipoma, liposarcoma, etc.

Myelolipoma. Microscopic observation showed that the tumor was mainly composed of mature fat cells and hematopoietic cells

Myelolipoma. With no capsule in the surrounding area, and tumor tissue forming an “infiltration border”
6.4.5.3 Treatment and Prognosis
Myelolipomas are benign tumors, and surgical resection is recommended with good effects, especially when symptoms or complications appear. Generally, no other auxiliary treatments are necessary, and there is no report of malignant transformation of myelolipoma.
6.5 Neurological and Endocrinic Tumors
6.5.1 Neurilemmoma/Schwannoma
6.5.1.1 Pathogenesis and Mechanism
Neurilemmoma is a benign tumor derived from the nerve sheath cells (Schwann cells), also known as schwannoma or perineural fibroblastomas. In 1920, Antoni divided the tumor into cell rich zone (Antoni A type) and loose myxoid zone (Antoni B type). The patients with neurofibromatosis type I account for 20–50%, and neurilemmoma has been suggested to be a subtype of gastrointestinal stromal tumors , but inconsistency exists in immunohistochemical staining results for CD117. Neurilemmoma is mostly found in the head, neck, upper and lower limbs, and other parts of the body surface, and mediastinum is the major location in vivo, while abdominal cavity is less involved. Pereira et al. first reported hepatic neurilemmoma in 1978, and more than 20 cases have been reported in the literature by December 2013 [134, 135]. Hepatic neurilemmoma originates from various sympathetic nerve and parasympathetic nerve branches distributed in the connective tissue of the hepatic lobules or along the portal vein branches.
6.5.1.2 Clinical Features
Hepatic neurilemmoma is more common in elderly female patients, and the male to female ratio is 1: 4.5 with a mean age of 54.6 (35–70) years old. The main clinical manifestations include abdominal pain or back pain, and the course of the disease can be as long as 20 years due to slow growth of the tumor. Increased serum alkaline phosphatase and gamma GT can be detected, while AFP and CEA are normal. And elevation of serum collagenase type IV level was the important marker for peripheral neurilemmoma. B-ultrasound and CT images display hepatic nodular masses, and huge cystic occupying lesions with multiple calcification foci can be easily mistaken as parasitic cysts of the liver. Kim et al. [136] reported a case of hepatic neurilemmoma with positive finding on FDG-PE imaging, which is difficult to distinguish the nature of the tumor.
6.5.1.3 Gross Features
The tumors were more commonly located in the left lobe of the liver, often huge in size with an average diameter of 13 (2.3–24) cm and can be multinodular. The lesions appear in long fusiform, nodular, or lobulated shapes with a complete capsule, and the section is gray-white or pale yellow in color and crisp in nature, and interwoven structure can sometimes be observed (Fig. 6.79). Hemorrhage, necrosis, or cystic change can be found in cases with large lesions.

Neurilemmoma. With a complete capsule and the section is pale yellow, crisp in nature
6.5.1.4 Microscopic Features
Hepatic neurilemmoma owns the mass morphology with neurilemmomas in other parts of the body, mainly consisting of Antoni A zone (bundle or compact type) and Antoni B zone (reticular type) in different proportions.
Antoni A zone contains abundant cells, fusiform or oval in shape with ambiguous cellar boundaries, pale staining cytoplasm, loosely arranged, and finely granular chromatin, and visible slender fiber bundles can sometimes be seen at both ends of the nucleus, with no nuclear atypia, small or unclear nucleolus, and little karyokinesis. Nuclei of the tumor cells are arranged in a spiral or parallel palisade shape, between which is the slender homogeneous nuclear-free zone formed by cytoplasmic processes, and this is characteristic of Verocay bodies (Fig. 6.80).

Neurilemmoma. Antoni A zone: the tumor cells are arranged in a palisade shape, formed “Verocay bodies”
Antoni B zone contains loosely arranged stellate tumor cells, and the cytoplasmic processes are connected to each other to form a network with grids expanded to be a small sac containing transparent matrix. There are three forms of the tumor cells, namely, lymphocyte-like cells, spindle cells, and stellate cells, the former of which are the most common type of cells with naked nucleus similar to lymphocyte. Antoni B zone contains numerous small thin-walled blood vessels, and piles of foam and yellow tumor cells are sometimes observed.
Neurilemmomas can be divided into several subtypes. The tumor with abundant cells and majorly composed by Antoni A zones is called cell-type neurilemmoma. The tumor mainly consisting of Antoni B zones and myxoid stroma is called myxoid neurilemmoma. And if the tumor contains significant melanin and positive HMB45, it is called melanin-type neurilemmoma, and if it is rich in small thin-walled blood vessels, the tumor is called angiomatous neurilemmoma, while for the tumor with cytoplasm containing eosinophilic granules, positive PAS, and anti-diastase digestion, it is called granulosa cell-type neurilemmoma (Fig. 6.81).

Neurilemmoma. Antoni B zone: the tumor cells are loosely arranged and form a network
6.5.1.5 Immunohistochemistry
The tumors are NSE, S-100 (Fig. 6.82), CD57, glial fiber acidic protein (GFAP), and vimentin positive and SMA, desmin, and CK negative, with Ki-67 index < 5%.

Neurilemmoma. S-100 positive
6.5.1.6 Differential Diagnosis
-
1.
Malignant neurilemmoma. Neurilemmoma with abundant cells should be identified from a malignant neurilemmoma. The former has a clear boundary even in the absence of a capsule, with no or mild atypia, and karyokinesis is rarely seen. The latter is often large in volume with an incomplete capsule, abundant cells, and obvious atypia, losing typical structures of Antoni A and B zoned, lack of Verocay bodies, and significant karyokinesis (> 5/10HPF), and tumor cells are distributed surrounding blood vessels forming a peripheral vascular sheath.
-
2.
Neurofibroma and neurofibromatosis. There are no Verocay bodies, nuclei arranged in palisade shape or glass-like thickness of vascular wall, and neurofibromatosis also has corresponding genetic characteristics.
-
3.
Leiomyoma . The tumor cells were arranged in a wave shape, with palisading arranged nuclei in some cases, abundant and eosinophilic cytoplasm, and visible myofilaments, and the nuclei are larger than those of the neurilemmoma cells, with both nuclear blunt ends, and no Antoni A and B zones. VG staining shows yellow green, rather than orange. And immunohistochemistry examination shows negative S-100 and positive actin and SMA.
-
4.
Gastrointestinal stromal tumors . They are S-100 positive, as well as CD117 and CD34 positive, which can be distinguished from neurilemmomas. Moreover, it should be differentiated from fibrosarcoma and leiomyosarcoma.
6.5.1.7 Treatment and Prognosis
Surgical resection is a proper treatment for neurilemmoma with a good prognosis, and occasionally postoperative recurrence is related to incomplete resection of the tumor. And about 2–3% of the neurofibromatosis will evolve into malignant neurilemmomas.
6.5.2 Neurofibroma
6.5.2.1 Pathogenesis and Mechanism
Neurofibroma is a local or solitary benign tumor originating from neurilemma cells, consisting of proliferated Schwann cells and fibroblasts, mainly found in the skin, neck, mediastinum, etc. and related to type I neurofibromatosis (NF1). Hepatic neurofibroma is extremely rare, and only two cases have been reported in the English literature. Five cases have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University.
6.5.2.2 Clinical Features
All the five cases of hepatic neurofibroma, diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University, were female patients (Table 6.7), with an average age of 53.5 (3.5–84) years old. The tumor growth is slow, and the patients have no symptoms or only general gastrointestinal, local compression symptoms or radioactive pain. Elevated levels of serum alkaline phosphatase and γ-GT and normal AFP and CEA can be found. B-ultrasound and CT scan show intrahepatic solid lesions.
6.5.2.3 Gross Features
The tumors were mainly located in the left lobe of the liver, and the average diameter of the tumors was 6.2 (4–13.5) cm. The gross morphology of neurofibromas varies greatly in different cases (Fig. 6.83), with no capsule or incomplete capsules and soft or hard and mildly flexible in texture. The section appears gray or grayish yellow, moist, and semitranslucent with luster and bundle or spiral structures (Fig. 6.84). The tumors are often solid with no obvious hemorrhage, or necrosis, and little cystic degeneration can be found.

Neurofibroma. The section appears grayish yellow, hard in texture

Neurofibroma. The section appears grayish white, hard in texture
6.5.2.4 Microscopic Features
Neurofibromas are composed by a proliferation mixture of all components of peripheral nerve, including neurilemma cells, axons, perineurium cells, and fibroblasts, and neurilemma cells are the major cellular component. The tumors are characterized by nerve fibers scattered within the tumor. And the tumor cells and nuclei were long wave shaped, with both tip ends of the nuclei and rare mitosis (Fig. 6.85). There is no Antoni A or Antoni B zone and the interstitial tissue is rich in collagen fibers with mucoid degeneration. The characteristics of neurofibromas developing into malignant neurilemmoma include increased cell density, polymorphism, nuclear mitosis, and vascular sheath formed by tumor cells.

Neurofibroma. The tumor cells and nuclei were long wave shaped, with both tip ends of the nuclei
6.5.2.5 Immunohistochemistry
S-100 and vimentin staining are positive, while CD34 and CD117 are negative.
6.5.2.6 Differential Diagnosis
The pathological diagnosis of neurofibromatosis is generally of no difficulties, and it should be differentiated from neurilemmoma which contains Antoni A and Antoni B zones.
6.5.2.7 Treatment and Prognosis
Recurrence after surgical resection of the tumor is rare; however, malignancy (malignant change rate of 3–13%) or involvement of other organ has been found; thus, the patients after surgical resection should be closely followed up.
6.5.3 Plexiform Neurofibroma
6.5.3.1 Pathogenesis and Mechanism
Plexiform neurofibroma is characterized by masses consisting of nerve axons, nerve sheath cells, and fibroblasts in collagen or myxoid stroma, which mainly occurred in regions rich in innervation, and hepatic plexiform neurofibroma is rare. Zacharia et al. [137] reported that hepatic plexiform neurofibromas account for about 2.3% of the total number of abdominal and pelvic plexiform neurofibromas. And less than 20 cases have been reported in the literature. Hepatic plexiform neurofibromas are also common in NF1 patients. And plexiform neurofibromatosis is an autosomal dominant genetic disease with several cases of hepatic plexiform neurofibromatosis reported.
6.5.3.2 Clinical Features
Hepatic plexiform neurofibromas are common in adults and rare in children. The patients usually manifest no symptoms or only general gastrointestinal symptoms, local compression symptoms, or radioactive pain, and the severity of these symptoms varies depending on the location and size of the tumor. Some cases involving the hepatic hilus, small intestine, and biliary obstruction have been reported. The liver function in the patients is generally normal. MRI image characteristics are basically identical to the same tumors in other parts, and it is showed that all tumors are distributed along the intrahepatic portal tracts, sometimes involving the pancreas and gall bladder [138, 139]. Hepatic plexiform neurofibromas develop slowly, and stable condition of the tumor has been observed during years of follow-up.
6.5.3.3 Gross Features
Hepatic neoplasms can be huge with extensive involvement of the liver. The tumors are irregularly cylindrical, wormlike cords, spindle shaped, or spherically enlarged, often forming multiple nodules or beads (i.e., “plexiform”). Sometimes a tumor grows into a huge mass. The section of the tumor is often grayish white, soft in texture with elasticity, and a jelly part can be observed in some region of the tumor, while cystic change is rarely seen.
6.5.3.4 Microscopic Features
The lesions are characterized by a mixture of plexiform structure, peripheral proliferated collagen fibers, and stroma with mucoid degeneration, which is formed by all components of peripheral nerves, including axons, neurilemma cells, fibroblasts, and perineurium cells, and neurilemma cells are the main components, which are of spindle shape and loosely arranged, with light-stained cytoplasm, unclear cellular boundaries, long or wave-shaped nuclei with both tip ends, and dark nuclear chromatin. Generally, tumors which are > 5 cm in diameter, with infiltration into the surrounding liver tissue, tumor necrosis, calcification, and disappearance of target signs which is often central low signals in the tumor on MRI T2-weighted images, suggest potential malignancy [140].
6.5.3.5 Immunohistochemistry
S-100 staining shows neurilemma cells, and EMA staining can display the perineurium cells, while NSE and NFP (neurofilament) staining shows the axons.
6.5.3.6 Differential Diagnosis
The pathological diagnosis is not difficult, but we should pay attention to the identification whether it is one of the concomitant lesions of the visceral neurofibromatosis.
6.5.3.7 Treatment and Prognosis
The lifetime risk of malignancy for plexiform neurofibromas to develop into malignant peripheral neurilemmomas (malignant peripheral nerve sheath tumors (MPNSTs)) is 7–13%, and surgical resection is the optimal choice. Robertson et al. reported that treatment with imatinib mesylate facilitates the reduction of the size of plexiform neurofibroma [141].
6.5.4 Neurofibromatosis
6.5.4.1 Pathogenesis and Mechanism
Neurofibromatosis is an autosomal dominant genetic disease with onset of the disease in every generation of the family. According to the different pathogenic genes, it is divided into type I (NF1) and type II (NF2). NF1 was first described by Von Recklinghausen in 1882, characterized by peripheral neurofibroma, also known as Von Recklinghausen disease (VRD). NF2 was characterized by multiple tumors of the central nervous system, such as bilateral acoustic neuroma, spinal cord astrocytomas, meningiomas, retinal hamartoma, and ependymoma. The similarities and differences between NF1 and NF2 are summarized in Table 6.8. In addition, there are NF3–NF7 subtypes. The pathogenesis of NF1 is associated with multiple gene variants including NF1 gene, and 3–18% of the NF1 patients have the risk of other malignant tumors, including plexiform neurofibroma, glioma, pheochromocytoma, malignant schwannoma, carcinoid tumors, rhabdomyosarcoma, osteosarcoma, Wilms’ tumor, and medulloblastoma. More than ten cases of neurofibromatosis with hepatic involvement have been reported so far, among which are cases of NF1 patients with hepatoblastoma [142], hepatic plexiform neurofibromas, liver plexiform neurofibroma and hepatic angiosarcoma [143], malignant hepatic neurilemmoma complicated with hepatic angiosarcoma [144], hepatic neurofibroma [145], as well as NF2 patients with hepatic angiomyolipoma [146]. Furthermore, one case of NF1 in a 44-year-old patient after liver transplantation treated with immunosuppressant has also been reported [147].
6.5.4.2 Clinical Features
The patients with hepatic neurofibromatosis are often young people, including children aged 4–17. The American National Institutes of Health (NIH) proposed the diagnostic standards for NF1 in 1988 which is still in use till now, including:
-
1.
Prepuberty, the number of cafe-au-lait spots ≥ 6 with the maximum diameter ≥ 5 mm; postpuberty, the number of cafe-au-lait spots ≥ 6 with the maximum diameter ≥15 mm
-
2.
The number of neurofibroma ≥2 or one plexiform neurofibroma
-
3.
Visible nevoid lentigo in the armpit or groin area
-
4.
Optic glioma
-
5.
More than two iris pigment hamartoma (Lisch nodules)
-
6.
Defined damage to the bone, such as sphenoid dysplasia or cortical thinning of long bones, with or without pseudoarthrosis
-
7.
The first-degree relatives of NF1
Patients with two or more than two items of the above criteria can be diagnosed as NF1, and for those who do not meet the criteria but are highly suspected of NF1, genetic analysis could be considered.
6.5.4.3 Pathological Diagnosis
According to the corresponding histological features of hepatic tumors, the diagnosis can be made. And genetic testing can also provide an important evidence for the diagnosis. Mutations of NF1 gene located around the center of 17q11.2 are an important cause of NF1; therefore, PCR detection for microsatellite heterozygosity deletion near the related genes is useful, such as heterozygous deletions of microsatellites D22S929 and D22S1169 in NF2 patients, suggesting NF2 gene deletion in the tumor [148].
6.5.4.4 Differential Diagnosis
Patients with NF1 are the most common, and the differential diagnosis of tumors with no genetic factors according to the diagnostic criteria of NF1 should be paid much attention to.
6.5.4.5 Treatment and Prognosis
Surgery is the major treatment. And gene therapy is the most promising and the most important technique in the future. For instance, it has been discussed to introduce the normal NF1 gene into the diseased cells, so that it encodes normal fibrin to treat NF1 [149].
6.5.5 Paraganglioma
6.5.5.1 Pathogenesis and Mechanism
Paraganglioma is a kind of neoplasm, derived from the sympathetic-adrenal neuroendocrine system and originated from the embryonic neural crest cells, usually referring to tumors of sympathetic and parasympathetic ganglion at sites except adrenal glands, and those in adrenal medulla are specifically known as pheochromocytoma. About 30% of patients with pheochromocytoma or paraganglioma have familial predisposition, and the pathogenesis involves mutations of VHL, MEN1, RET, and NF1. Hepatic paraganglioma is rare, and 16 cases have been reported so far [150].
6.5.5.2 Clinical Features
Among the16 cases of hepatic paraganglioma reported in the literature, the male to female ratio is 1:1.3, and the average age of the patients was 43 (ranged 14–71) years old. Hepatic paraganglioma is often nonfunctional due to the metabolism of catecholamine in liver cells; therefore, it generally manifests no clinical symptoms such as increased peripheral blood and urinary catecholamine concentrations, hypertension, heart palpitations, dizziness, and metabolic disorders. CT images display round or oval soft tissue masses in the liver, and tumors are lobulated, heterogeneous in density, clearly bounded, and irregularly hypodense, and marked enhancement in non-cystic parts can be found in cases with cystic change. High positive rate has been reported in radiation scanning with 131I metaiodobenzylguanidine, but cases of false-negative results are found in a few cases.
6.5.5.3 Gross Features
Of the reported 16 cases of hepatic paraganglioma, 14 cases concerned intrahepatic solitary tumors, 2 cases exhibited multiple masses, and most of the lesions were located in the right lobe of the liver. The tumors are often large in volume with an average diameter of 8.2 (1.2–18) cm, round or lobulated in shape, and the sections are grayish white or brown in color, solid in texture, and hard and flexible, and brown-black hemorrhage and cystic cavities can be found inside the tumors. Koh et al. [151] reported one case of huge hepatic paraganglioma, 12 cm × 18 cm × 18 cm in size, which was treated by surgical resection.
6.5.5.4 Microscopic Features
The histological features of hepatic paraganglioma are similar to those of the same kind of tumor in other parts, including large amounts of capillaries which separate the tumor to form nests, acinar, or pseudorosette shapes. The cell nests are wrapped by flat supporting cells and surrounded by rich blood sinus, forming organ-like structures. The main cellular components are chief cells and supporting cells, and the former are polygonal, round, or oval shaped, densely arranged with abundant cytoplasm, rich in eosinophilic and basophilic or amphophilic particles of varying sizes (Figs. 6.86 and 6.87). And eosinophilic hyaline bodies can sometimes be found in the cytoplasm, which are positive for PAS staining after diastase digestion. The nuclei of the tumor cells are round or oval, with obvious nucleolus and intranuclear pseudoinclusion-like structures due to invagination of the cytoplasm. Cells in parts of the tumor are with atypia, abnormal nuclei, and less nuclear division. The supporting cells are spindle and distributed surrounding the tumor cell nests or interspersed between the chief cells. And no invasion of the tumor into the surrounding liver tissue has been found.

Paraganglioma. The tumor arranged in a nest or interlaced shape; the tumor cells are polygonal shaped, with abundant cytoplasm

Paraganglioma// pheochromocytoma?? The tumor arranged in a nest or interlaced shape; the tumor cells are spindle shaped, with abundant vessels
6.5.5.5 Immunohistochemistry
The chief cells of the ganglioma are positive for NSE, CgA (Fig. 6.88), Syn, and GFAP, and the supporting cells are positive for S-100, while both cells are negative for CD10, nestin, Bcl-2, SMA, EMA, etc.

Paraganglioma. CgA positive
6.5.5.6 Differential Diagnosis
1. Metastatic paraganglioma. The diagnosis of primary hepatic pheochromocytoma should first rule out metastasis of adrenal pheochromocytoma, with the bone and liver as the most common sites for metastasis. Three cases of hepatic metastatic paraganglioma have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, which were wrapped by a thick fibrous capsule.
2. Malignant paraganglioma. The identification of benign and malignant nature of a paraganglioma is of certain difficulties, and about 10% of all the cases were malignant. The histological findings may be inconsistent with the biologic behaviors. Of the reported 16 cases both at home and abroad, 2 cases with multiple hepatic masses were diagnosed as malignant lesions. Generally, when the tumor is larger than 6 cm in diameter, breaks through the capsule, and invades the adjacent liver tissue, with vascular infiltration, formation of tumor thrombus, abnormal increase of pathologic mitosis, metastasis, and diagnosis of malignant paraganglioma can be made.
6.5.5.7 Treatment and Prognosis
Surgical resection is the preferred treatment for paraganglioma. Due to the lower incidence, there are fewer reports of the tumor. The biological characteristics of this disease are not clear, and it has been reported that paraganglioma in the pancreas was a tumor with potential function and malignancy; thus, close follow-up should be carried out for a long term after surgery [152]. According to the reports on cases with no recurrence or metastasis during the 3-year follow-up after surgical resection, hepatic ganglioma treated with surgical removal is of good prognosis.
6.5.6 Adrenal Rest Tumor
6.5.6.1 Pathogenesis and Mechanism
Adrenal rest tumor was first reported by Hyams in 1960, and nine cases have been reported so far in the literature. The tumor is composed of adrenocortical cells, and its pathogenesis may involve ectopic location of the primordium or the whole organ of the right adrenal gland inside the hepatic capsule during embryonic development.
6.5.6.2 Clinical Features
Hepatic residual adrenal tumors can be both nonfunctional or functional, with the male to female ratio of 1:1, and the onset age in patients ranges 1–62 years old. Nonfunctional ones are hepatic pseudo-residual adrenal tumors, most without clinical symptoms, while patients with functional residual adrenal tumors manifest endocrine disorders such as Cushing syndrome or virilization or elevated serum levels of steroid hormones in some cases. Angiography shows the tumors in the liver parenchyma which are homogeneous in texture and rich in blood vessels. B-ultrasound examination shows round, clearly bounded, heterogeneous solid masses. And CT images display hypervascular densities of fat or soft tissue masses [153].
6.5.6.3 Gross Features
The tumors are mainly located in the hepatic capsule of the right lobe, which is associated with the blood supply of the tumor by hepatic artery. The tumors are 0.7–2.5 cm in diameter, and the cross sections are gray or yellowish gray, with or without calcification and cystic degeneration, and tumor often oppresses the adjacent liver tissue resulting in the formation of a thin pseudofibrous capsule.
6.5.6.4 Microscopic Features
The tumor exhibits expansive growth resulting in oppression on the adjacent liver tissue and the formation of thin pseudofibrous capsules. The tumor cells, resembling the pale cells in the adrenal cortex, are polygonal containing eosinophilic particles in the cytoplasm, oval nucleus, and no nucleolus and are arranged in wide trabecula or acinar shape, separated by slender fibrovascular septa. Some cells in certain regions are pleomorphic with transparent cytoplasm. Visible adrenal tissue can be sometimes found at the margins of the liver tissues. Tumors with huge sizes and obvious atypia of the tumor cells may be malignant. The tumor cells are positive in Sudan III staining (frozen section).
6.5.6.5 Immunohistochemistry
Steroid hormone contained in the tumor tissue is a key evidence for the diagnosis of the tumor. And the tumor cells are stained positive for adrenal 4-binding protein (Ad4BP) and CgA, but negative for EMA, S-100, CK, and vimentin.
6.5.6.6 Differential Diagnosis
-
1.
Hepatic adenoma. Cholestasis and Mallory bodies can be found, and the tumors often contain small thin-walled veins or arteries with dilated lumen in diffuse distribution. PAS staining is positive in the tumor cells, while adrenal rest tumor shows negative result in PAS staining.
-
2.
Metastatic adrenocortical carcinoma. The key point is to determine completeness and existence of tumor in the right adrenal gland.
-
3.
Metastasis of renal cell carcinoma. Metastatic tumors in the liver of renal cell carcinoma are multiple nodular lesions, with transparent or eosinophilic cytoplasm of the tumor cells, positive staining for glycogen CD10. While adrenal rest tumor is often a solitary nodule and its glycogen staining and expression test of CD10 are both negative.
-
4.
Hepatocellular carcinoma . HCC has a background of hepatitis or cirrhosis, and the cancer cells contain bile in the cytoplasm, with obvious nuclear atypia, and positive pCEA and Hep Par 1 result in immunohistochemistry.
-
5.
Metastatic melanoma. The tumor cells contain melanin granules and are positive for HMB45 staining.
6.5.6.7 Treatment and Prognosis
Recurrence of tumors treated by surgical resection is rarely seen. And effective ketoconazole treatment for hepatic adrenal rest tumor has been reported.
6.5.7 Pancreatic Rest Tumor
Ectopic pancreatic tissue is more common in the stomach (27.5%), duodenum (25.5%), and jejunum (15.9%), while intrahepatic pancreatic rest tumor or heterotopic pancreas is extremely rare, with less than ten reported cases. Early in 1909, Heinrich et al. classified the tumor into three types according to the histological characteristics of ectopic pancreatic tissues. Type I contains acini, ducts, and endocrine islets, similar to normal pancreatic tissue; type II contains a large number of acini and small amount of ducts, with no islets; and type III contains several ducts, a small amount of acini and no islet.
The major clinical and pathological significance of ectopic pancreas lies in two aspects. One is repeated reports of canceration of the heterotopic pancreas, and the other is the differential diagnosis from other tumors. Yan et al. [154] first reported a 45-year-old woman with a 3.0 cm×2.0 cm×2.0 cm lesion near the portal area of the left lobe in the liver, which was surgically excised, and microscopic observation showed acinar cells containing abundant eosinophilic granules, centroacinar cells, and pancreatic ducts in the pancreatic tissue. The adenocarcinoma derived from ectopic pancreatic tissue and grew outward resulting in invasion of nerve tissue, and the pathological diagnosis was moderately differentiated adenocarcinoma originating from hepatic heterotopic pancreas. Guillou et al. [155] proposed three criteria for the diagnosis of adenocarcinoma originating from ectopic pancreatic tissue, including: ① the tumors should be located within or around the ectopic pancreatic tissue; ② there is a transition between the pancreatic tissue and cancer tissue; ③ the nontumor pancreatic tissue should contain acini, epithelium, and ducts.
6.5.8 Gastrinoma
6.5.8.1 Pathogenesis and Mechanism
Gastrinoma is a neuroendocrine tumor of the gastrointestinal tract and the pancreas, derived from G cells distributed in mucous membrane of the gastric antrum, duodenum, and proximal jejunum and characterized by the secretion of gastrin. Around 20–30% of the gastrinomas belong to multiple endocrine neoplasia type 1 (MEN1), and it is an autosomal dominant syndrome involving multiple endocrine organs. Related cancer genes for the pathogenesis of gastrinoma include c-Myc and HER2/neu (ElbB-2), and tumor suppressor genes include MEN1 and P16 (INK4a). Sporadic gastrinomas (75–90%) were often solitary, while MEN1-type gastrinomas (10–25%) are often multiple lesions and can be complicated by lesions in the parathyroid glands, pituitary, islet, and adrenal cortex. Gastrinomas located in “gastrinoma triangle” (below the junction of the cystic duct and common bile duct, above the junction of the descending and cross parts of the duodenum, and out of the junction of the neck and body of the pancreas) are the most common, accounting for up to 80–90%, among which the pancreas (21–65%) and duodenum (6–32%) are more common sites for gastrinomas than other locations, such as lymph nodes, stomach, mesentery, liver, ovary, kidney, and heart. So far, less than 30 cases of primary hepatic gastrinoma have been reported in the literature, and the current suggestion of the potential origin for hepatic gastrinomas concerns the uptake of amine precursor in the biliary tree and APUD cells [157,158,159].
6.5.8.2 Clinical Features
It is reported that hepatic gastrinoma occurs mainly in children and the elderly (28–77 years old), involving more men than women. Most of the patients manifest Zollinger-Ellison syndrome (ZES), including refractory ulcers, watery diarrhea, stomach burning sensation, and esophageal reflux. Fasting serum gastrin levels are more than 1000 pg/ml, and 123I-octreotide somatostatin receptor scintigraphy (SRS) demonstrating high expression of somatostatin in the tumor is of diagnostic value. And gastrin stimulation test is helpful in tumor location. CT images show intrahepatic masses with clear boundaries, a number of septa, and no enhancement in enhanced scan. Ogawa et al. [160] suggested that selective arterial calcium infusion (SACI) in insulin stimulation test helps to exclude gastrinomas in the “gastrinoma triangle.” Ga-68-DOTATOC PET-CT examination is beneficial to exclude gastrinomas out of the “gastrinoma triangle” and metastasis.
6.5.8.3 Gross Features
According to the literature, hepatic gastrinoma is often a small solitary nodule, but multiple lesions can also be found, with the diameter of 1–5 cm and a clear boundary, with or without a capsule, and the tumor section was pink-white.
6.5.8.4 Microscopic Features
Consistent with the histological morphology of gastrinomas in other locations, hepatic gastrinoma is often arranged in the shape of a nest, cord, or acinar structure. The tumor cells are often small or medium in size, round, oval, polygonal, cuboidal, or columnar, consistent in morphology and size, without obvious atypia, and visible mitosis is rare. Ki-67 labeling index is not high, and abundant blood vessels can be found in the interstitial tissue. Whether the tumor is benign or malignant is difficult to determine based on the histological examination, and the basic evidences for a malignant tumor include invasive growth into the surrounding tissue and metastasis into lymph nodes and other organs.
6.5.8.5 Immunohistochemistry
The tumor is positive for gastrin staining, and the tumor cells may also express a variety of hormones, including glucagon, vasoactive intestinal peptide (VIP), somatostatin and its receptors, insulin, CgA, Syn, and NSE.
6.5.8.6 Differential Diagnosis
The liver is the most common metastatic site for gastrinomas, so the first step for differential diagnosis is to exclude hepatic metastasis of gastrinomas from the pancreas and other parts.
6.5.8.7 Treatment and Prognosis
Gastrinomas with malignancy and metastasis account for 60–90% of all the gastrinomas. Based on the reports in the literature, surgery is the preferred treatment method for hepatic gastrinomas. Naoe et al. [156] reported one case of a 77-year-old woman with persistent diarrhea for 8 months, weight loss, and serum gastrin levels >4000 pg/ml, and CT showed two low-attenuation rounded nodules in the right lobe of the liver, 1.6 cm and 1.9 cm in diameter, respectively. The tumors were treated by resection, and immunohistochemistry was positive for gastrin and Ki-67 index >20%. Cranial MRI and thyroid ultrasound examinations suggested no abnormalities. FDG PET showed no extrahepatic lesions, excluding MEN1. And the pathological diagnosis was primary hepatic gastrinoma. Postoperative follow-up of 12 months revealed the disappearance of Zollinger-Ellison syndrome and decrease of serum gastrin levels to normal, indicating that the surgery cured the patient.
6.5.9 Vasoactive Intestinal Polypeptide-Secreting Tumor
Vasoactive intestinal polypeptide-secreting tumor (VIP tumor) is a rare neuroendocrine tumor , which often derives from D1 cells in the pancreatic islets, and more than 60% of the VIP tumors are malignant. VIP tumors outside the pancreas are generally neurogenic. Since the first case of primary hepatic VIP tumor reported by Ayub et al. [161] in 1993, only five cases have been found in the literature. The patients are often middle-aged men, aged 35–41 years old, with elevated serum levels of VIP due to secretion of large amounts of VIP, by D1 cells, resulting in periodic intractable watery diarrhea, hypokalemia caused by massive discharge of potassium via the intestinal tract, and lack of gastric acid due to inhibition by VIP. All of the above manifestations are called Verner-Morrison syndrome.
Primary hepatic VIP tumor is generally located in the right lobe of the liver, large in size with a maximum diameter of 10 cm. Microscopic observation shows well-differentiated tumor in nest or acinar-like structures, similar to the morphology of islet cell tumor. Immunohistochemistry demonstrates positive results for vasoactive intestinal peptide (VIP), CgA, Syn, and NSE and negative for gastrin. VIP tumors are of high malignancy, and more than 70% of the patients are found to have metastasis at the time of diagnosis, so exclusion of metastatic tumor in the liver derived from VIP tumors of pancreas or digestive tract. Dohmen et al. reported one case of a 33-year-old male patient with primary VIP tumor in the pancreatic tail, which was small in volume; however, two large metastatic nodules were found in the liver. It should also be differentiated from primary hepatic neuroendocrine tumors . Surgical resection is the first choice for the treatment and can be combined with radiofrequency and somatostatin therapies. After the tumor is excised, the clinical symptoms will be improved or completely disappear, and serum VIP levels will decrease to normal. Hachicha et al. [162] reported one case of primary hepatic VIP tumor which was treated with surgical resection, and no abnormality was found during the 42-month follow-up.
6.5.10 Somatostatinoma
Somatostatinoma is often found in the pancreas and small intestine, and over 200 cases have been reported globally. Primary hepatic somatostatinoma is rare, with only two cases found in the literature, and its origin is not clear. However, it is found that the cells containing somatostatin exist in the branches of the biliary tree, suggesting these endocrine cells may be the origin for hepatic somatostatinoma. Patients with somatostatinomas in the pancreas or duodenum often manifest “inhibition syndrome,” including diabetes, diarrhea, abdominal pain, and cholelithiasis, due to the release of a large number of somatostatin. And laboratory examination showing elevated levels of serum somatostatin and calcitonin is helpful for the diagnosis [163].
Among the two reports on hepatic somatostatinoma, one case concerned a 56-year-old male patient with NF1 and multiple small lesions in the liver, which were confirmed as hepatic somatostatinomas via liver biopsy [164]. The other one concerned a 48-year-old woman with a huge abdominal mass as the first clinical manifestation, which was 20 cm×20 cm×10 cm in size and surgically excised, but recurrence occurred 5 years later and a hepatic mass of 11 cm×10 cm×10 cm was excised [165]. On gross appearance, the tumor is round with a thin capsule, and its section is grayish yellow, with no bleeding or necrosis, and the boundary is clear. Microscopic observation shows cuboidal tumor cells of medium sizes, with abundant and red-stained cytoplasm containing fine granules, round or oval nuclei, clear nucleolus, with mild atypia and mitosis. Tumor tissues are in trabecular, nest, banded, or glandular arrangement, surrounded by fibrovascular stroma. Most of the tumor cells are positive stained for somatostatin (SS), NSE, Syn, and CgA and negative for VIP, insulin, and gastrin (GAS) in immunohistochemistry.
The liver is the most common organ for metastasis of somatostatinoma, and metastatic hepatic somatostatinoma should be first excluded at the time of diagnosis. It is a high differentiated tumor with slow growth and low degree of malignancy, and the natural course can be several years. However, the discovery of hepatic somatostatinoma is usually complicated with metastasis, and invasion and metastasis into the surrounding liver tissue and blood vessels indicate malignancy. The treatment for extrahepatic somatostatinomas can be divided into two methods. For cases with no liver metastasis, radical resection of the primary lesions and dissection of lymph nodes are recommended. While surgical resection can also be applied in cases with liver metastasis [166], for unresectable lesions, liver transplantation is a proper choice which can improve the postoperative survival rates at 5 years and 10 years. For patients with somatostatinoma and expression of somatostatin receptors, injection of octreotide can help decrease the plasma somatostatin level and further improve symptoms such as diabetes and diarrhea [167].
6.6 Other Benign Tumors in the Liver
6.6.1 Teratoma
6.6.1.1 Pathogenesis and Mechanism
Teratoma is a tumor associated with abnormal embryonic tissue growth and development, originating from pluripotent germ cells, with the potential to differentiate into all three germinal layers to produce a variety of tissues and organs. The predilection sites for teratoma are distributed along the center line of the body, namely, the embryonic notochord line, including the ovary, testis, anterior mediastinum, retroperitoneal region, presacral space, and tail and pineal gland, sequenced by frequency. Teratomas can be divided into benign (mature) and malignant (immature) teratoma according to the degree of differentiation. To date, more than 30 cases of hepatic teratoma have been reported, which may derive from ectopic primordial germ cells, developed from residual primordial germ cells in the liver during the early development of embryos (blastocysts or morula) [168,169,170,171]. Three cases of hepatic teratoma have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University.
6.6.1.2 Clinical Features
Patients are often female infants younger than 1 year old, among whom 25% of them are diagnosed at birth. The patients of the three cases of hepatic teratoma diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, of Second Military Medical University, were a 7-and-a-half-month female infant, a 32-year-old woman, and a 65-year-old man. Hepatic teratoma is often a benign tumor with no specific clinical manifestations, and the main symptoms include right upper abdominal pain, nausea, and vomiting due to tumor compression on the adjacent organs. And some patients go to hospital because of accidental discovery of abdominal mass with normal serum AFP. Imaging shows cystic lesions in the liver, which should be differentiated from liver abscess and liver parasitic cyst. CT shows cystic fat dilution in the liver with teeth and calcification. Cases of simple teratoma or mature teratoma have normal serum AFP and β-HCG levels, which are often significantly increased in cases of malignant teratoma or immature teratoma, and AFP can be used as a biomarker in distinguishing benign and malignant teratomas. The serum level of CA19–9 > 570 U/ml in one case in our hospital was significantly decreased after tumor resection.
6.6.1.3 Gross Features
The three cases of hepatic teratoma diagnosed in our hospital varied in the structure and morphology. These tumors were 13–25 cm in diameter, cystic or cystic-solid, with an uneven surface, soft or hard in texture, and solitary or multiple. Some lesions could grow into a huge mass, even occupying half of the liver. Structures such as cysts, bone, cartilage, hair, and teeth can be observed on the sections (Figs. 6.89 and 6.90), and cystic teratoma contains fetal fatlike or jellylike liquid in the sac.

Teratoma. An uneven surface, soft or hard in texture; hair can be observed on the sections

Teratoma. Cystic-solid tumor, yellow and white
6.6.1.4 Microscopic Features
Teratoma is composed of three layers of mesodermal tissues. Ectodermal tissues include the skin, hair, sebaceous glands, sweat glands, and nerve tissue (Fig. 6.91); the mesoderm contains bone, cartilage, bone marrow, skeletal muscle, lymph tissue, and connective tissue; and endodermal tissues contain the epithelium of the digestive tract (Fig. 6.92), fallopian tube, thyroid tissue, and respiratory epithelium. And extramedullary hematopoiesis in the liver can be found in some cases. Hepatic teratoma can also be complicated by yolk sac tumor and hepatocellular carcinoma .

Teratoma. Mature cartilage (left) and nerve tissue (right)

Teratoma. Secreting epithelium of the digestive tract
6.6.1.5 Differential Diagnosis
-
1.
Malignant teratoma . It is also known as immature teratoma. And when teratomas contain yolk sac carcinoma, embryonal carcinoma, or choriocarcinoma, the level of serum alpha-fetoprotein and chorionic gonadotropin will increase, which can be used as biomarkers for malignant teratoma.
-
2.
Metastatic teratoma . The first step in diagnosis of primary hepatic teratoma is to exclude metastatic teratoma derived from predilection sites such as the ovary and testis.
-
3.
Mesenchymal hamartoma of the liver. It is mainly myxoid matrix containing stellate and spindle primitive mesenchymal cells, thick-walled blood vessels, proliferated bile ducts, and liver cell clusters, the component of which are simple.
6.6.1.6 Treatment and Prognosis
Surgery is the first choice for the treatment of teratoma , and once the diagnosis is made, early surgical resection should be conducted and the prognosis is good. Follow-up and regular detection of AFP and β-HCG are recommended after the surgery.
6.6.2 Mesothelioma
6.6.2.1 Pathogenesis and Mechanism
Miller first reported mesothelioma in 1908, which occurs mainly in locations with mesepithelium covering, such as the pleura, peritoneum, and pericardium. The relation between the occurrence of some pleural and peritoneal mesotheliomas and inhalation of asbestos dusts has been defined. And patients with long-term use of immunosuppressants after organ transplantation have been found to develop peritoneal mesothelioma. Primary hepatic benign mesothelioma is rare, and Rout et al. [172] reported one case of hepatic mesothelioma in 1999, and Flemming et al. [173] reported one case of a 51-year-old woman with a 8 cm×6.5 cm×6 cm cystic mass in the left lobe of the liver, which was pathologically confirmed as a cystic mesothelioma after resection. Wang Jinbo et al. [174] reported one case of a 24-year-old male patient with surgically resected hepatic fibrous mesothelioma located in the right lobe underneath the hepatic capsule, and the tumor was 7 cm×6 cm×6 cm in size, gray or grayish yellow on the section, with swirling texture and greasy feeling. Hepatic mesothelioma may be derived from the mesothelial cells in the inner layer of the Glisson capsule.
6.6.2.2 Clinical Features
The majority of the patients are females aged 22–83 years old, and the major clinical manifestations include discomfort or pain in the liver zone, hypoglycemia-related symptoms, and elevation in serum CA19-9 levels. The X-ray or CT images show homogeneous densities in the liver which can be solitary, multinodular, or lobulated with clear boundaries. And qualitative examination depends on postsurgical pathological investigation.
6.6.2.3 Gross Features
The tumors are round and nodular with clear boundaries and complete capsules, and parts of the lesions are lobulated. The sections are gray or grayish yellow. The tumor is often large in size, even up to 15 cm × 9 cm × 8 cm and weighing up to 3800 g, moderate to hard in texture, and giant tumors may be complicated by mucinous degeneration, cystic degeneration, punctate hemorrhage, and necrosis. Multicystic mesothelioma contains sac cavities or cystic structures.
6.6.2.4 Microscopic Features
Benign mesothelioma can be divided into the following histological types:
-
1.
Fibrous mesothelioma. The tumor tissue is mainly constituted by fibroblasts and epithelial spindle cells, with no nuclear atypia, covered by an intact layer of mesothelial cells, and parenchymal components of the liver can be bundled into the tumor. Immunohistochemistry shows positive CK, EMA, and vimentin and negative CD117 and CD34, while positive actin can be observed in a few tumors. The tumor tends to be recurrent with rare malignant transformation.
-
2.
Epithelial mesothelioma. The tumor is papillary or tubular, lined with a single layer of cuboidal or low columnar cells with consistency in size, no obvious atypia, and rare mitosis. The tumor cells are positive for CK, CK7, CA-125, calretinin, D2–40, thrombomodulin, WT-1, and mesothelin (a kind of anti mesothelial cell antibody). And well-differentiated papillary mesothelioma is a tumor with weak malignancy.
-
3.
Multicystic mesothelioma. It is known as a benign or indolent tumor, consisting of cystic cavities in varying sizes and round or irregular shapes. The cystic walls are lined with a single layer of well-differentiated flat or cuboidal epithelioid cells which protrude into the lumen in hobnail or papillary shapes. Positive CK, EMA and HBME-1, and negative F-VII are shown in immunohistochemistry tests. Recurrence has been observed in some cases with no metastasis, and a second resection can be conducted due to the malignant potential of the tumor.
-
4.
Differentiated papillary mesothelioma. The tumor consists of diffuse papillary hyperplasia which appears as thick branched tubular papilla with fibrous tissue and blood vessel in the center. It is a borderline tumor with a benign course, while recurrence may be found several years after resection in a few cases, and development for malignant mesothelioma and distant metastasis will occur in individual cases.
6.6.2.5 Differential Diagnosis
Benign mesothelioma is rare, and malignant epithelial mesothelioma contains obvious hemorrhage and necrosis foci with abundant tumor cells in diffuse distribution and marked nuclear atypia with mitosis figures > 5/10HPF. It tends to metastasize to lymph nodes, and the tumor cells are negatively stained for desmin and positive in p53 and Ki-67 staining. In addition, it should be differentiated from other cystic tumors in the liver, such as parasitic cysts, biliary cystadenoma , or cystadenocarcinoma .
6.6.2.6 Treatment and Prognosis
Surgical resection is the first choice for the treatment of benign mesothelioma with a good prognosis when complete resection has been demonstrated.
6.6.3 Myxoma
Myxoma is a benign mesenchymal tumor characterized by myxoid matrix which is rich in acid mucopolysaccharides and glycoproteins, generally with no progression. Myxoma is often discovered in the myocardium, bone, skin, and urogenital system, while hepatic myxoma is rare. One case of hepatic myxoma has been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, and the patient was a 19-year-old male who complained of a gradually increased mass in the right upper abdomen for 2 years, and his main symptoms included an upper abdominal mass, abdominal distension, and abdominal pain. He was generally in a good condition, with no history of hepatitis, and his serum AFP detection was negative, and the liver function was normal. A lobulated tumor in the right lobe of the liver was surgically excised, and it was 15 cm×12 cm×6.5 cm in size. Zhang Jianping et al. [175] reported one case of hepatic myxoma which was 19 cm×13 cm in size, and the section was gray and hard with no obvious hemorrhage or necrosis, with an intact capsule and no cirrhosis in the surrounding liver tissue (Fig. 6.93). Under the microscope, tumor tissue contained large amounts of mucus (Fig. 6.94) with varying numbers of fusiform and star-shaped mucous cells. The round or spindle nuclei of the tumor cells contain dense and dark-stained chromatin, no visible nucleolus, with no obvious atypia. The spindle-shaped tumor cells were positive for vimentin and SMA staining, and the abundant myxoid stroma was positive in Alcian blue staining. The adjacent liver tissue was compressed by the expansion of the tumor resulting in atrophy, but no invasive growth was observed.

Myxoma. The section was gray and white, lobulated, and hard in texture

Myxoma. Tumor tissue contained large amounts of mucus, with clear boundary and schistosome eggs in the capsule
The diagnosis of hepatic myxoma is generally of no difficulties; however, attention should be paid to differential diagnosis from other tumors with local mucinous degeneration. For an instance, mesenchymal hamartoma of the liver can have mucinous degeneration, but the mucus contains bile ducts and liver tissue, while myxoma contains homogeneous mucus mostly with single component. Hepatic myxoma is a benign mesenchymal tumor, which can be treated by surgical resection with a good prognosis.
6.6.4 Chondroma
Only one case of chondroma has been found in the current literature, which was reported by Fried in 1992. The patient was a 44-year-old female manifesting dull pain and oppression symptoms in the liver zone. She was followed up by CT imaging for 6 years, after when a surgical resection was conducted for the tumor which increased significantly in volume. Pathological examination showed that hepatic tumor was 19 cm×15 cm×9.5 cm in size with a clear boundary and was a huge oval nodular mass with an envelope and tender in texture similar to a rubber. The cross section was yellow and myxoid, or gray hyaline and cartilage-like, with calcification or ossification, and a jelly area of cystic degeneration was found in the center which was 6 cm×4.5 cm in size. Microscopic observation showed that it contained lobulated well-differentiated hyaline cartilage, fibrous cartilage, or myxoid cartilage, with mature hyaline cartilage as the majority. The cartilage was separated into lobules by fibrous tissue, and tumor cells were without atypia or obvious mitotic figures. And it was difficult to find hypertrophic or binucleate cells. Tumors with peripheral foci of fibrosis surrounding the lobules are called fibrochondroma, and those with obvious calcification and ossification are called osteochondroma, while tumors with abundant myxoid interstitial tissue in the center or the peripheral regions of the cartilage lobules are known as myxoid chondroma. The tumors were separated from the hepatic parenchyma by fibrous tissue, and infiltration of chronic inflammatory cells in surrounding liver tissues with formation of focal lymphoid follicles could be observed. Immunohistochemistry: cartilaginous cells were positive for vimentin and S-100 and negative for CK and SMA. Analysis via flow cytometry showed no heteroploid tumor cells.
6.6.5 Langerhans Cell Histiocytosis
6.6.5.1 Pathogenesis and Mechanism
Langerhans cell histiocytosis (LCH) is caused by hyperplasia of histiocytes and dendritic cells in the mononuclear phagocytic system, and Langerhans cells (LCs) are equivalent to normal dendritic cells. The pathogenesis of LHC is unknown. Yousem et al. (2001) conducted human androgen receptor assay (HUMARA) of chromosome X polymorphism in CD1a-positive LCH cells by microdissection in women, finding that mono-organ (lung) LCH is non-clonal or polyclonal, while systemic LCH is monoclonal with potential tumorous properties. Recently, Badalian-Very et al. [176] discovered BRAF V600E mutations in LCH tissues, while mutations of TP53 and MET have also been found in individual cases, suggesting that LCH is a tumorous lesions and may be sensitive to inhibitors of the RAF pathway.
LCH can be either diffuse lesions involving multiple systems and organs or focal lesions involving a single system. And LCH accumulation and infiltration can be found to form masses in almost all organs of the body, and the predilection sites include the bone, skin, lymph nodes, and bone marrow, followed by the spleen, lung, endocrine organs, and gastrointestinal tract. The liver is often less involved and can be the only affected organ or as part of multiorgan diseases. LCH subtypes include three kinds of lesions:
-
1.
Solitary or multiple bone destruction LCH (eosinophilic granuloma ), confined to bone destruction
-
2.
Local disseminated LCH with bone destruction and ectosteal soft tissue invasion (Hand-Schuller-Christian disease), the triad signs of which include skull defects, diabetes insipidus (pituitary involvement), and exophthalmos
-
3.
Diffuse disseminated LCH involving multiple systems and organs (liver, spleen, lung, and bone marrow) (Letterer-Siwe disease)
-
4.
Congenital self-healing reticulohistiocytosis (Hashimoto-Pritzker disease), the children patients of the disease are with skin damage as early as at birth [177, 178]
Diffuse disseminated LCH mainly involves infants and young children, and the incidence of cases with liver involvement was 40–60%, while cases of adult hepatic solitary LCH have been occasionally reported. Thus, LCH can be divided into single system with single lesion type, single system with multifocal type, and multisystem with multifocal type. The latter is further divided into low-risk group (involvement of multiple organs and systems except the hematopoietic system, liver, spleen, and lung) and high-risk group (involvement of multiple organs and systems including hematopoietic system, liver, spleen, and lung). The mechanism of the damage caused by hepatic LCH is associated with the invasion and destruction of bile ducts by LC, leading to damage and disappearance of bile ducts and further peribiliary fibrosis, inducing secondary sclerosing cholangitis changes, during which the chronic injury eventually evolved into biliary cirrhosis.
6.6.5.2 Clinical Features
Hepatic LCH is a visceral LCH, and the patients are predominantly infants and young children, affecting few adults and the elderly rarely. Shi et al. [179] reported 13 cases of diffuse LCH involving the liver and other organs and tissues, including seven males and six females, aged 13–52 months with an average of 28.9 months old. Kaplan et al. [184] reported one group of nine hepatic LCH cases, with a male to female ratio of 1:2 and a median age of 18 months old (ranged 7 days to 62 years old), among which five cases were solitary hepatic LCH and the remaining four cases were accompanied with involvement of the lymph nodes, skin, lung, thymus, spleen, pancreas, bone, and heart. Mulitifocal (multiple) LCH is more common in children, while monofocal (solitary) LCH can occur at any age. Adult patients with LCH can also be complicated by diseases, such as breast cancer, lymphoma , or leukemia. The clinical symptoms mainly relate to the number of organs involved, and the most common manifestations in pediatric patients are multisystem LCH, fever, and poor growth. Patients of hepatic LCH have normal extrahepatic bile ducts, and their intrahepatic bile ducts show segmental dilatation with stones, similar to the characteristics of sclerosing cholangitis. The patients usually manifest as chronic cholestasis, portal hypertension, ascites, hepatosplenomegaly, and abnormal liver function, including elevated serum alkaline phosphatase and γ-glutamyl transpeptidase in most cases. Braier et al. [180] reported 182 cases of children with LCH, including 36 cases with liver damage, among which 12 patients had cholestasis. Imaging examination primarily shows multiple liver nodular and beaded lesions distributed along the portal vein system with cholangitis. In 2009, the International Association of Tissue and Cells divided LCH into two groups: ① low risk group, patients older than 2 years without damage on the liver, lung, and bone, and ② high risk groups, with damage on multiple organs including the liver, lung, and bone.
6.6.5.3 Gross Features
The lesions are multiple nodular or intranodular cystic, ranging in diameter from 2 mm microgranulomas to 10 cm solid masses, brown yellow, tenacious, with clear boundaries.
6.6.5.4 Microscopic Features
Histologically, LCH can be divided into four stages: proliferation, granuloma , xanthoma, and fibrosis, mainly in the following three kinds of pathological presentations:
-
1.
Formation of LC foci. Small granulomas or tumorlike lesions are of diagnostic value. Typical LCs are 10–12 μm in diameter, rich in eosinophilic cytoplasm with phagocytosis, and the nuclei are often oval, lobulated, and horseshoe shaped or twisted with obvious nuclear grooves, fine granular chromatin, and visible nucleolus. In most cases, the LCs are surrounded by varying amounts of eosinophilic granulocytes, lymphocytes, neutrophils, plasma cells, and non-LC multinucleated giant cells, which can be mingled with fibroblasts and hematopoietic cells.
-
2.
LC infiltration into bile duct walls. LCs mainly invade the basement membrane of bile ducts, with infiltration of inflammatory cells, resulting in stripping of biliary epithelial cells from the basement membrane. Clinically, this can lead to chronic cholestasis, similar to the performance of primary sclerosing cholangitis. Residual small bile ducts can be observed in the LC clusters, or they are completely replaced by LC, and only the basement membrane of the bile duct can be seen, which is also considered to be the characteristic lesions of hepatic LCH. And a large number of LCs and infiltration of inflammatory cells can also be discovered in the lumen of large bile ducts, resulting in dilatation of the bile ducts.
-
3.
Peribiliary fibrosis. It is observed in most cases. On the basis of second pathological lesions, the large bile ducts are surrounded by a large amount of fibrous tissue with dilatation of the bile ducts, presenting the changes of secondary sclerosing cholangitis, which is the main reason for jaundice. Other secondary changes include hyperplasia of small bile ducts and fibrous tissue in the portal district, expansion of periportal area, an “onion skin” appearance around the bile ducts, cholestasis in liver cells, and formation of fat nodules around the portal veins in the liver parenchyma.
In 1987, the International Association of Tissue and Cells classified LCH into three grades:
-
Grade I: according to findings, the clinical and laboratory tests, X-ray changes of organs including bone and lung, and abnormal LCs on histological sections, a preliminary diagnosis can be made.
-
Grade II: immunohistochemical staining shows S-100 positive in LC.
-
Grade III: visible Birbeck bodies or positive CD1a immunohistochemistry staining under electron microscope.
6.6.5.5 Immunohistochemistry
Positive CD1a (OKT-6) and langerin (CD207) are of the greatest diagnostic significance (Fig. 6.95). In addition, positive vimentin, CD11, S-100, and CD14, negative lymphocyte markers, and partial positive CD68 and lysozyme suggest the histiocyte origin of LC rather than lymphocyte lines.

Hepatic Langerhans cell histiocytosis. Birbeck bodies in the cytoplasm (From Kaplan[182])
6.6.5.6 Electron Microscopic Observation
Poor development of LC can be found, and Birbeck bodies in the cytoplasm are of diagnostic value with specificity [182] (Fig. 6.95).
6.6.5.7 Differential Diagnosis
The basis pathological change of LCH is sclerosing cholangitis, while LC is not visible in all cases, which makes it difficult to differentiate from primary sclerosing cholangitis; however, the latter is rare in children. Thus, the correct diagnosis of LC can be made if phenomenon in the portal area of a child’s liver is discovered, including accumulation of histiocytes accompanied by infiltration of inflammatory cell, damage on bile ducts, the absence of small bile ducts, sclerosing cholangitis, cholestasis, confirmation of LC by immunohistochemical staining of S-100 and CD-1a, or visible LC in extrahepatic lesions. Moreover, IDC tumor is a progressive malignant tumor, which has long processed cytoplasm, decreased expression of CD-1a, and fewer Birbeck granules [181].
6.6.5.8 Treatment and Prognosis
LCH with hepatic involvement usually suggests a poor prognosis. However, the prognosis of patients varies greatly according to different ages, numbers, and severity of organs that are involved. LCH patients under 2 years old or over 65 years old with multiple organ involvement have a poorer prognosis. Generally, hepatic LCH is with a good prognosis, and the median follow-up period reaches 10 years and 6 months long. Yagita et al. (2001) reported a case of a 58-year-old patient who had hepatic and splenic LCH, including multiple lesions in the liver and a mass of 6 cm in diameter in the spleen. Excochleation of part of the hepatic lesions and splenectomy were conducted as well as CHOP chemotherapy, after which the volumes of the remaining hepatic lesions reduced, and the patients survived well 4 years later without recurrence, suggesting the benign nature of the LCH. Effective chemotherapy facilitates the reduction of the number and size of LCH lesions and long-term survival for the patients. For LCH patients with progressive sclerosing cholangitis, liver transplantation is an effective choice [183]. Hepatic LCH can be complicated by hepatocellular carcinoma in a few cases.
6.6.6 Spongiotic Pericytoma
In 2005, Kaiserling et al. [185] reported a case of a 35-year-old woman with a history of several years’ oral contraceptive, who received a surgical resection of a 6 cm×5 cm×3 cm spongiotic pericytoma. The cross section of the tumor was brownish red, with gray-white fibrous septa and a clear boundary with the surrounding tissue. Under the microscope, the spindle-shaped tumor cells were rich in translucent cytoplasm, with a vascular fibrous septa in the center, arranged in nests, in diffuse nodular distribution. Liver plates, hepatic lobule, and hepatic sinusoid remain in the tumor with no envelope. The immunohistochemical staining shows that the tumor cells express VI, CD34, CD105 (endoglin), CD99 (Mic1), CD56, and SMA, and the stroma cells around the tumor express collagens I, III, and V and fibronectin, while Syn and sGFAP are negative, and a small amount of CD105 (endoglin)-positive staining cells are found in the adjacent hepatic sinusoid, which are fibroblast-like but contain fat droplets under electron microscope. Kaiserling et al. suggested that the tumor was similar to cell tumor discovered in animal experiments, which expressed CD34 along with the hepatic sinusoidal endothelium, suggesting that the tumor derived from the lining cells of the hepatic sinusoid.
This is the only case report on human hepatic spongiotic pericytoma, and previous reports were on similar lesions in mice, rats, and fish induced by carcinogenic drugs on the liver [187, 188]. Karbe et al. [186] and Bannasch et al. [189] supported that astrocytoma is an independent tumor and that the tumor is the result of carcinogenic agents. However, Kerlin et al. [190] suggested that astrocytoma is not a true tumor. Though whether the tumor is benign or malignant has not been determined, it may have a good prognosis based on the evidence of low proliferation activity of the tumor cells and no recurrence or metastasis during the 12-month follow-up.
References
Rebouissou S, Bioulac-Sage P, Zucman-Rossi J. Molecular pathogenesis of focal nodular hyperplasia and hepatocellular adenoma. J Hepatol. 2008;48(1):163–70.
Van Aalten SM, Terkivatan T, De Man RA, et al. Diagnosis and treatment of hepatocellular adenoma in the Netherlands: similarities and differences. Dig Surg. 2010;27(1):61–7.
Socas L, Zumbado M, Perez-Luzardo O, et al. Hepatocellular adenomas associated with anabolic androgenic steroid abuse in bodybuilders: a report of two cases and a review of the literature. Br J Sports Med. 2005;39(5):e27.
Labrune P, Trioche P, Duvaltier I, et al. Hepatocellular adenomas in glycogen storage disease type I and III: a series of 43 patients and review of the literature. J Pediatr Gastroenterol Nutr. 1997;24(3):276–9.
Jeannot E, Mellottee L, Bioulac-Sage P, et al. Spectrum of HNF1A somatic mutations in hepatocellular adenoma differs from that in patients with MODY3 and suggests genotoxic damage. Diabetes. 2010;59(7):1836–44.
Bioulac-Sage P, Taouji S, Possenti L, et al. Hepatocellular adenoma subtypes: the impact of overweight and obesity. Liver Int. 2012;32(8):1217–21.
Chang CY, Hernandez-Prera JC, Roayaie S, et al. Changing epidemiology of hepatocellular adenoma in the United States: review of the literature. Int J Hepatol. 2013;2013:604860.
Liu HP, Cong WM. Hepatocellular adenoma: new understandings of molecular pathology and novel model of clinical diagnosis and therapy. Chin J Clin Hepatol. 2013;29(11):801–4.
Bioulac-Sage P, Laumonier H, Couchy G, et al. Hepatocellular adenoma management and phenotypic classification: the Bordeaux experience. Hepatology. 2009;50(2):481–9.
Grazioli L, Bondioni MP, Haradome H, et al. Hepatocellular adenoma and focal nodular hyperplasia: value of gadoxetic acid-enhanced MR imaging in differential diagnosis. Radiology. 2012;262(2):520–9.
Bioulac-Sage P, Rebouissou S, Thomas C, et al. Hepatocellular adenoma subtype classification using molecular markers and immunohistochemistry. Hepatology. 2007;46(3):740–8.
Zhang LH, Xu JJ. Molecular subtypes and clinical significance of Hepatocellular adenoma. Chin J Pathol. 2014;43(6):428–30.
Rebouissou S, Amessou M, Couchy G, et al. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature. 2009;457(7226):200–4.
Bioulac-Sage P, Balabaud C, Zucman-Rossi J. Subtype classification of hepatocellular adenoma. Dig Surg. 2010;27(1):39–45.
Pan J, Cong WM. Analysis on loss of heterozygosity of four tumor suppressor genes in hepatocellular adenoma. Chin J Clin Exp Pathol. 2003;19(5):481–3.
Fu HH, JIN GZ, Liu HP, et al. Analysis of microsatellite instability in hepatocellular adenoma cases with overweight and obesity. Chin J Cancer Prev Treat. 2013;20(20):1557–60.
Chen LL, JI Y, Xu JF, et al. Focal nodular hyperplasia of liver: a clinicopathologic study of 238 patients. Chin J Pathol, 2011, 40(1): 17–22.
Tanaka J, Baba N, Arii S, et al. Typical fibrolamellar hepatocellular carcinoma in Japanese patients: report of two cases. Surg Today. 1994;24(5):459–63.
Nault JC, Bioulac-Sage P, Zucman-Rossi J. Hepatocellular benign tumors-from molecular classification to personalized clinical care. Gastroenterology. 2013;144(5):888–902.
Flejou JF, Barge J, Menu Y, et al. Liver adenomatosis. An entity distinct from liver adenoma? Gastroenterology. 1985;89(5):1132–8.
De Kock I, Mortele KJ, Smet B, et al. Hepatic adenomatosis: MR imaging features. JBR-BTR. 2014;97(2):105–8.
Liu LG, Wu F, Wu JX, et al. Hepatocellular adenoma and liver adenomatosis: a report of 11 patients. Chin J Hepatobiliary Surg. 2012;18(3):166–8.
Greaves WO, Bhattacharya B. Hepatic adenomatosis. Arch Pathol Lab Med. 2008;132(12):1951–5.
Asran MK, Loyer EM, Kaur H, et al. Case 177: congenital absence of the portal vein with hepatic adenomatosis. Radiology. 2012;262(1):364–7.
Babaoglu K, Binnetoglu FK, Aydogan A, et al. Hepatic adenomatosis in a 7-year-old child treated earlier with a Fontan procedure. Pediatr Cardiol. 2010;31(6):861–4.
Iwen KA, Klein J, Hubold C, et al. Maturity-onset diabetes of the young and hepatic adenomatosis - characterisation of a new mutation. Exp Clin Endocrinol Diabetes. 2013;121(6):368–71.
Shen WF, Yang JM. Diagnosis and treatment of hepatic adenoma and liver adenomatosis. Chin J Hepatobiliary Surg. 2007;13(9):646–8.
An C, Park S, Choi YJ. Diffusion-weighted MRI in intrahepatic bile duct adenoma arising from the cirrhotic liver. Korean J Radiol. 2013;14(5):769–75.
Koga F, Tanaka H, Takamatsu S, et al. A case of very large intrahepatic bile duct adenoma followed for 7 years. World J Clin Oncol. 2012;3(4):63–6.
Wu WW, Gu M, Lu D. Cytopathologic, histopathologic, and immunohistochemical features of intrahepatic clear cell bile duct adenoma: a case report and review of the literature. Case Rep Pathol. 2014;2014:874826.
Johannesen EJ, Wu Z, Holly JS. Bile duct adenoma with oncocytic features. Case Rep Pathol. 2014;2014:282010.
Albores-Saavedra J, Hoang MP, Murakata LA, et al. Atypical bile duct adenoma, clear cell type: a previously undescribed tumor of the liver. Am J Surg Pathol. 2001;25(7):956–60.
Arena V, Arena E, Stigliano E, et al. Bile duct adenoma with oncocytic features. Histopathology. 2006;49(3):318–20.
Hastir D, Verset L, Demetter P. Intrahepatic bile duct adenoma with oncocytic features. Liver Int. 2013;33(2):273.
Bhathal PS, Hughes NR, Goodman ZD. The so-called bile duct adenoma is a peribiliary gland hamartoma. Am J Surg Pathol. 1996;20(7):858–64.
Sasaki M, Matsubara T, Kakuda Y, et al. Immunostaining for polycomb group protein EZH2 and senescent marker p16INK4a may be useful to differentiate cholangiolocellular carcinoma from ductular reaction and bile duct adenoma. Am J Surg Pathol. 2014;38(3):364–9.
Komuta M, Spee B, Vander Borght S, et al. Clinicopathological study on cholangiolocellular carcinoma suggesting hepatic progenitor cell origin. Hepatology. 2008;47(5):1544–56.
Nishihara Y, Aishima S, Hayashi A, et al. CD10+ fibroblasts are more involved in the progression of hilar/extrahepatic cholangiocarcinoma than of peripheral intrahepatic cholangiocarcinoma. Histopathology. 2009;55(4):423–31.
Tretiakova M, Antic T, Westerhoff M, et al. Diagnostic utility of CD10 in benign and malignant extrahepatic bile duct lesions. Am J Surg Pathol. 2012;36(1):101–8.
Haas S, Gutgemann I, Wolff M, et al. Intrahepatic clear cell cholangiocarcinoma: immunohistochemical aspects in a very rare type of cholangiocarcinoma. Am J Surg Pathol. 2007;31(6):902–6.
Toriyama E, Nanashima A, Hayashi H, et al. A case of intrahepatic clear cell cholangiocarcinoma. World J Gastroenterol. 2010;16(20):2571–6.
Nakanuma Y, Tsutsui A, Ren XS, et al. What are the precursor and early lesions of peripheral intrahepatic cholangiocarcinoma? Int J Hepatol. 2014;2014:805973.
Rafiq E, Alaradi O, Bawany M, et al. A combination of snare polypectomy and apc therapy for prolapsing common bile duct adenoma. J Interv Gastroenterol. 2012;2(4):193–5.
Munshi AG, Hassan MA. Common bile duct adenoma: case report and brief review of literature. Surg Laparosc Endosc Percutan Tech. 2010;20(6):e193–4.
Soares KC, Arnaoutakis DJ, Kamel I, et al. Cystic neoplasms of the liver: biliary cystadenoma and cystadenocarcinoma. J Am Coll Surg. 2014;218(1):119–28.
Yang ZZ, Li Y, Liu J, et al. Giant biliary cystadenoma complicated with polycystic liver: a case report. World J Gastroenterol. 2013;19(37):6310–4.
Abhishek S, Jino T, Sarin GZ, et al. An uncommon cause of ascites: spontaneous rupture of biliary cystadenoma. Australas Med J. 2014;7(1):6–10.
Yu YQ, Lou BH, Yan HC, et al. Intrahepatic biliary cystadenoma presenting with pleural effusion. Chin Med J. 2012;125(7):1355–7.
Sang X, Sun Y, Mao Y, et al. Hepatobiliary cystadenomas and cystadenocarcinomas: a report of 33 cases. Liver Int. 2011;31(9):1337–44.
Wang C, Miao R, Liu H, et al. Intrahepatic biliary cystadenoma and cystadenocarcinoma: an experience of 30 cases. Dig Liver Dis. 2012;44(5):426–31.
Rayapudi K, Schmitt T, Olyaee M. Filling defect on ERCP: Biliary Cystadenoma, a Rare Tumor. Case Rep Gastroenterol. 2013;7(1):7–13.
Zen Y, Pedica F, Patcha VR, et al. Mucinous cystic neoplasms of the liver: a clinicopathological study and comparison with intraductal papillary neoplasms of the bile duct. Mod Pathol. 2011;24(8):1079–89.
Abdul-Al HM, Makhlouf HR, Goodman ZD. Expression of estrogen and progesterone receptors and inhibin-alpha in hepatobiliary cystadenoma: an immunohistochemical study. Virchows Arch. 2007;450(6):691–7.
Daniels JA, Coad JE, Payne WD, et al. Biliary cystadenomas: hormone receptor expression and clinical management. Dig Dis Sci. 2006;51(3):623–8.
Ahanatha Pillai S, Velayutham V, Perumal S, et al. Biliary cystadenomas: a case for complete resection. HPB Surg. 2012;2012:501705.
Kazama S, Hiramatsu T, Kuriyama S, et al. Giant intrahepatic biliary cystadenoma in a male: a case report, immunohistopathological analysis, and review of the literature. Dig Dis Sci. 2005;50(7):1384–9.
Ratti F, Ferla F, Paganelli M, et al. Biliary cystadenoma: short- and long-term outcome after radical hepatic resection. Updat Surg. 2012;64(1):13–8.
Diaz De Liano A, Olivera E, Artieda C, et al. Intrahepatic mucinous biliary cystadenoma. Clin Transl Oncol. 2007;9(10):678–80.
Delis SG, Touloumis Z, Bakoyiannis A, et al. Intrahepatic biliary cystadenoma: a need for radical resection. Eur J Gastroenterol Hepatol. 2008;20(1):10–4.
Meng XF, Li J, Zhang WZ, et al. Intrahepatic biliary cystadenoma: experience with 10 consecutive cases at a single center. J South Med Univ. 2011;31(10):1733–6.
Sun ZP, Peng C, Jiang B, et al. Intrahepatic biliary cystadenoma: diagnosis and Treatment experience of 15 cases. J Clin Res. 2013;30(5):1030–2.
Shen H. Diagnoses and treatments of 11 cases of intrahepatic Bile Duct Cystadenoma. Zhejiang Pract Med. 2012;17(3):196–8.
Romagnoli R, Patrono D, Paraluppi G, et al. Liver transplantation for symptomatic centrohepatic biliary cystadenoma. Clin Res Hepatol Gastroenterol. 2011;35(5):408–13.
Yeh TS, Tseng JH, Chen TC, et al. Characterization of intrahepatic cholangiocarcinoma of the intraductal growth-type and its precursor lesions. Hepatology. 2005;42(3):657–64.
Lee SS, Kim MH, Lee SK, et al. Clinicopathologic review of 58 patients with biliary papillomatosis. Cancer. 2004;100(4):783–93.
Zhang XF, Qiu FB, He JC, et al. Clinical epidemiology of biliary papillomatosis in China during the past 32 years(1979~2011). Chin J Curr Adv Gen Surg. 2012;15(6):455–8.
Nakanuma Y, Sato Y, Ojima H, et al. Clinicopathological characterization of so-called “cholangiocarcinoma with intraductal papillary growth” with respect to “intraductal papillary neoplasm of bile duct (IPNB)”. Int J Clin Exp Pathol. 2014;7(6):3112–22.
White AD, Young AL, Verbeke C, et al. Biliary papillomatosis in three Caucasian patients in a Western centre. Eur J Surg Oncol. 2012;38(2):181–4.
Sasaki M, Matsubara T, Yoneda N, et al. Overexpression of enhancer of zeste homolog 2 and MUC1 may be related to malignant behaviour in intraductal papillary neoplasm of the bile duct. Histopathology. 2013;62(3):446–57.
Murakami S, Ajiki T, Hori Y, et al. Establishment of a novel cell line from intraductal papillary neoplasm of the bile duct. Anticancer Res. 2014;34(5):2203–9.
Rocha FG, Lee H, Katabi N, et al. Intraductal papillary neoplasm of the bile duct: a biliary equivalent to intraductal papillary mucinous neoplasm of the pancreas? Hepatology. 2012;56(4):1352–60.
Kim KM, Lee JK, Shin JU, et al. Clinicopathologic features of intraductal papillary neoplasm of the bile duct according to histologic subtype. Am J Gastroenterol. 2012;107(1):118–25.
Tsui WM, Loo KT, Chow LT, et al. Biliary adenofibroma. A heretofore unrecognized benign biliary tumor of the liver. Am J Surg Pathol. 1993;17(2):186–92.
Parada LA, Bardi G, Hallen M, et al. Monosomy 22 in a case of biliary adenofibroma. Cancer Genet Cytogenet. 1997;93(2):183–4.
Akin O, Coskun M. Biliary adenofibroma with malignant transformation and pulmonary metastases: CT findings. AJR Am J Roentgenol. 2002;179(1):280–1.
Varnholt H, Vauthey JN, Dal Cin P, et al. Biliary adenofibroma: a rare neoplasm of bile duct origin with an indolent behavior. Am J Surg Pathol. 2003;27(5):693–8.
Xu L, Wang FS, Chen L, et al. Biliary adenofibroma of the liver: report of a case. Chin J Gen Surg. 2009;11:925–925.
Gurrera A, Alaggio R, Leone G, et al. Biliary adenofibroma of the liver: report of a case and review of the literature. Pathol Res Int, 2010, 504584.
Fu XH, Chun KJ, Lu CD, et al. Hepatic cavernous hemangioma: an analysis of 172 cases. Chin J Pract Surg. 2009;9:756–8.
Mazereeuw-Hautier J, Hoeger PH, Benlahrech S, et al. Efficacy of propranolol in hepatic infantile hemangiomas with diffuse neonatal hemangiomatosis. J Pediatr. 2010;157(2):340–2.
Mo JQ, Dimashkieh HH, Bove KE. GLUT1 endothelial reactivity distinguishes hepatic infantile hemangioma from congenital hepatic vascular malformation with associated capillary proliferation. Hum Pathol. 2004;35(2):200–9.
Zhou W, Li A, Pan Z, et al. Selective hepatic vascular exclusion and Pringle maneuver: a comparative study in liver resection. Eur J Surg Oncol. 2008;34(1):49–54.
Mcgrath FP, Gibney RG, Morris DC, et al. Case report: multiple hepatic and pulmonary haemangioblastomas--a new manifestation of von Hippel-Lindau disease. Clin Radiol. 1992;45(1):37–9.
Hayasaka K, Tanaka Y, Satoh T, et al. Hepatic hemangioblastoma: an unusual presentation of von Hippel-Lindau disease. J Comput Assist Tomogr. 1999;23(4):565–6.
Rojiani AM, Owen DA, Berry K, et al. Hepatic hemangioblastoma. An unusual presentation in a patient with von Hippel-Lindau disease. Am J Surg Pathol. 1991;15(1):81–6.
Rapini N, Lidano R, Pietrosanti S, et al. De novo 13q13.3-21.31 deletion involving RB1 gene in a patient with hemangioendothelioma of the liver. Ital J Pediatr. 2014;40:5.
Riley MR, Garcia MG, Cox KL, et al. Hepatic infantile hemangioendothelioma with unusual manifestations. J Pediatr Gastroenterol Nutr. 2006;42(1):109–13.
Sondhi V, Kurkure PA, Vora T, et al. Successful management of multi-focal hepatic infantile hemangioendothelioma using TACE/surgery followed by maintenance metronomic therapy. BMJ Case Rep. 2012;2012:1–4.
Kochin IN, Miloh TA, Arnon R, et al. Benign liver masses and lesions in children: 53 cases over 12 years. Isr Med Assoc J. 2011;13(9):542–7.
Choi WJ, Jeong WK, Kim Y, et al. MR imaging of hepatic lymphangioma. Korean J Hepatol. 2012;18(1):101–4.
Ra SH, Bradley RF, Fishbein MC, et al. Recurrent hepatic lymphangiomatosis after orthotopic liver transplantation. Liver Transpl. 2007;13(11):1593–7.
Zhang YZ, Ye YS, Tian L, et al. Rare case of a solitary huge hepatic cystic lymphangioma. World J Clin Cases. 2013;1(4):152–4.
Datz C, Graziadei IW, Dietze O, et al. Massive progression of diffuse hepatic lymphangiomatosis after liver resection and rapid deterioration after liver transplantation. Am J Gastroenterol. 2001;96(4):1278–81.
Liu XW, Sun B, Guo TG. CT, MRI diagnosis of hepatic leiomyoma. Chin J Lab Diagn. 2011;5(15):2.
Liu Q, Liu J, Chen W, et al. Primary solitary fibrous tumors of liver: a case report and literature review. Diagn Pathol. 2013;8:195.
Fisher C. Atlas of soft tissue tumor pathology. New York/Heidelberg/Dordrecht/London: Springer; 2013.
Theise ND, Park YN, Curado MP, et al. Tumours of the liver and intrahepatic bile ducts. In: Bosman FT, Carneiro F, Hruban RH, et al., editors. WHO classification of tumours of the digestive system. Geneva: WHO Press; 2010. p. 195–261.
Beyer L, Delpero JR, Chetaille B, et al. Solitary fibrous tumor in the round ligament of the liver: a fortunate intraoperative discovery. Case Rep Oncol. 2012;5(1):187–94.
Yilmaz S, Kirimlioglu V, Ertas E, et al. Giant solitary fibrous tumor of the liver with metastasis to the skeletal system successfully treated with trisegmentectomy. Dig Dis Sci. 2000;45(1):168–74.
Wang H, Shen D, Hou Y. Malignant solitary tumor in a child: a case report and review of the literature. J Pediatr Surg. 2011;46(3):e5–9.
Munoz E, Prat A, Adamo B, et al. A rare case of malignant solitary fibrous tumor of the spinal cord. Spine (Phila Pa 1976). 2008;33(12):e397–9.
Sasaki H, Kurihara T, Katsuoka Y, et al. Distant metastasis from benign solitary fibrous tumor of the kidney. Case Rep Nephrol Urol. 2013;3(1):1–8.
Cong WM, Wu MC, Chen H, et al. Hepatic angiomyolipoma: a case report. Chin J Surg. 1992;30(10):618.
Ren N, Qin LX, Tang ZY, et al. Diagnosis and treatment of hepatic angiomyolipoma in 26 cases. World J Gastroenterol. 2003;9(8):1856–8.
Ji Y, Zhu X, Xu J, et al. Hepatic angiomyolipoma: a clinicopathologic study of 10 cases. Chin Med J. 2001;114(3):280–5.
You J, Xu W, Zhu JH. The diagnostic imaging and interventional therapy of hepatic angiomyolipoma associated with tuberous sclerosis. J Interven Radiology. 2001;10(6):333–5.
Wang JR, Qiu FB, Li ZC, et al. The epidemiological characteristics, diagnosis and treatment of hepatic angiomyolipoma(HAML) in China in the past 23 years. J Hepatopancreatobiliary Surg. 2012;24(3):183–7.
Wang JR, Qiu FB. Diagnosis and treatment of Hepatic Angiomyolipoma. Med Recapitulate. 2011;17(12):1826–8.
Zhu L, Hao YZ, Huang SL, et al. Ultrasonographic diagnosis of hepatic angiomyolipoma. Chin J Clin Oncol Rehabil. 2004;11(3):244–6.
Li SL, Qian JZ, Xu HM. Analysis of the imaging and clinicopathological features of hepatic angiomyolipoma. Chin J Gen Surg. 2011;20(7):696–9.
Yang B, Chen WH, Li QY, et al. Hepatic angiomyolipoma: dynamic computed tomography features and clinical correlation. World J Gastroenterol. 2009;15(27):3417–20.
Jeon TY, Kim SH, Lim HK, et al. Assessment of triple-phase CT findings for the differentiation of fat-deficient hepatic angiomyolipoma from hepatocellular carcinoma in non-cirrhotic liver. Eur J Radiol. 2010;73(3):601–6.
Zhang LP, Tang BH, Li LC, et al. CT features of epithelioid angiomyolipoma in liver: report of 2 cases and literature review. J Clin Radiology. 2012;31(12):1805–7.
Balci NC, Akinci A, Akun E, et al. Hepatic angiomyolipoma: demonstration by out of phase MRI. Clin Imaging. 2002;26(6):418–20.
Ji Y, Zhu XZ. Hepatic angiomyolipoma-a clinicopathologic study. Chin J Clin Exp Pathol. 2000;16(3):192–5.
Li M, Liu JS, Xu GR, et al. Clinicopathological Analysis for Hepatic Angioleiomyolipoma: a Report of Two Cases and Review of Literature. Chin J Clin Exp Pathol. 2012;28(9):1049–51.
Ji XL, Ji Y, Zhong DR, et al. Hepatic angiomyolipoma: a clinicopathological study of 21 cases. Chin J Diagn Pathol. 2001;8(5):267–9.
Tsui WM, Colombari R, Portmann BC, et al. Hepatic angiomyolipoma: a clinicopathologic study of 30 cases and delineation of unusual morphologic variants. Am J Surg Pathol. 1999;23(1):34–48.
Zhou Y, Chen F, Jiang W, et al. Hepatic epithelioid angiomyolipoma with an unusual pathologic appearance: expanding the morphologic spectrum. Int J Clin Exp Pathol. 2014;7(9):6364–9.
Nonomura A, Enomoto Y, Takeda M, et al. Angiomyolipoma of the liver: a reappraisal of morphological features and delineation of new characteristic histological features from the clinicopathological findings of 55 tumours in 47 patients. Histopathology. 2012;61(5):863–80.
Rao Q, Cheng L, Xia QY, et al. Cathepsin K expression in a wide spectrum of perivascular epithelioid cell neoplasms (PEComas): a clinicopathological study emphasizing extrarenal PEComas. Histopathology. 2013;62(4):642–50.
Makhlouf HR, Ishak KG, Shekar R, et al. Melanoma markers in angiomyolipoma of the liver and kidney: a comparative study. Arch Pathol Lab Med. 2002;126(1):49–55.
Lenci I, Angelico M, Tisone G, et al. Massive hepatic angiomyolipoma in a young woman with tuberous sclerosis complex: significant clinical improvement during tamoxifen treatment. J Hepatol. 2008;48(6):1026–9.
Gennatas C, Michalaki V, Kairi PV, et al. Successful treatment with the mTOR inhibitor everolimus in a patient with perivascular epithelioid cell tumor. World J Surg Oncol. 2012;10:181.
Higa F, Uchihara T, Haranaga S, et al. Malignant epithelioid angiomyolipoma in the kidney and liver of a patient with pulmonary lymphangioleiomyomatosis: lack of response to sirolimus. Intern Med. 2009;48(20):1821–5.
Deng YF, Lin Q, Zhang SH, et al. Malignant angiomyolipoma in the liver: a case report with pathological and molecular analysis. Pathol Res Pract. 2008;204(12):911–8.
Folpe AL, Mentzel T, Lehr HA, et al. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol. 2005;29(12):1558–75.
Brimo F, Robinson B, Guo C, et al. Renal epithelioid angiomyolipoma with atypia: a series of 40 cases with emphasis on clinicopathologic prognostic indicators of malignancy. Am J Surg Pathol. 2010;34(5):715–22.
Martin-Benitez G, Marti-Bonmati L, Barber C, et al. Hepatic lipomas and steatosis: an association beyond chance. Eur J Radiol. 2012;81(4):e491–4.
Moreno Gonzalez E, Seoane Gonzalez JB, Bercedo Martinez J, et al. Hepatic myelolipoma: new case and review of the literature. Hepato-Gastroenterology. 1991;38(1):60–3.
Allison KH, Mann GN, Norwood TH, et al. An unusual case of multiple giant myelolipomas: clinical and pathogenetic implications. Endocr Pathol. 2003;14(1):93–100.
Radhi J. Hepatic myelolipoma. J Gastrointestin Liver Dis. 2010;19(1):106–7.
Chen XX, Jiang XW, Huang W. Hepatic myelolipoma: a case report. Chin J Intern Med. 2010; 49(3):262–262.
Hayashi M, Takeshita A, Yamamoto K, et al. Primary hepatic benign schwannoma. World J Gastrointest Surg. 2012;4(3):73–8.
Kapoor S, Tevatia MS, Dattagupta S, et al. Primary hepatic nerve sheath tumor. Liver Int. 2005;25(2):458–9.
Kim YC, Park MS. Primary hepatic schwannoma mimicking malignancy on fluorine-18 2-fluoro-2-deoxy-D-glucose positron emission tomography-computed tomography. Hepatology. 2010;51(3):1080–1.
Zacharia TT, Jaramillo D, Poussaint TY, et al. MR imaging of abdominopelvic involvement in neurofibromatosis type 1: a review of 43 patients. Pediatr Radiol. 2005;35(3):317–22.
Malagari K, Drakopoulos S, Brountzos E, et al. Plexiform neurofibroma of the liver: findings on mr imaging, angiography, and CT portography. AJR Am J Roentgenol. 2001;176(2):493–5.
Delgado J, Jaramillo D, Ho-Fung V, et al. MRI features of plexiform neurofibromas involving the liver and pancreas in children with neurofibromatosis type 1. Clin Radiol. 2014;69(6):e280–4.
Imbert JP, Pilleul F, Valette PJ. Value of MRI in hepatic plexiform neurofibromatosis. Case report. Gastroenterol Clin Biol. 2002;26(8–9):791–3.
Robertson KA, Nalepa G, Yang FC, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1: a phase 2 trial. Lancet Oncol. 2012;13(12):1218–24.
Ucar C, Caliskan U, Toy H, et al. Hepatoblastoma in a child with neurofibromatosis type I. Pediatr Blood Cancer. 2007;49(3):357–9.
Andreu V, Elizalde I, Mallafre C, et al. Plexiform neurofibromatosis and angiosarcoma of the liver in von Recklinghausen disease. Am J Gastroenterol. 1997;92(7):1229–30.
Lederman SM, Martin EC, Laffey KT, et al. Hepatic neurofibromatosis, malignant schwannoma, and angiosarcoma in von Recklinghausen’s disease. Gastroenterology. 1987;92(1):234–9.
Kakitsubata Y, Kakitsubata S, Sonoda T, et al. Neurofibromatosis type 1 involving the liver: ultrasound and CT manifestations. Pediatr Radiol. 1994;24(1):66–7.
Levi Sandri GB, Ettorre GM, Vennarecci G. Hepatic angiomyolipoma and neurofibromatosis type 2: a novel association. Liver Int. 2014;34(9):1445.
Miller MB, Tonsgard JH, Soltani K. Late-onset neurofibromatosis in a liver transplant recipient. Int J Dermatol. 2000;39(5):376–9.
Yohay KH. The genetic and molecular pathogenesis of NF1 and NF2. Semin Pediatr Neurol. 2006;13(1):21–6.
Maruta H. Effective neurofibromatosis therapeutics blocking the oncogenic kinase PAK1. Drug Discov Ther. 2011;5(6):266–78.
Roman SA, Sosa JA. Functional paragangliomas presenting as primary liver tumors. South Med J. 2007;100(2):195–6.
Koh PS, Koong JK, Westerhout CJ, et al. Education and imaging. Hepatobiliary and pancreatic: A huge liver paraganglioma. J Gastroenterol Hepatol. 2013;28(7):1075.
Zhang L, Liao Q, Hu Y, et al. Paraganglioma of the pancreas: a potentially functional and malignant tumor. World J Surg Oncol. 2014;12(1):218.
Baba Y, Beppu T, Imai K, et al. A case of adrenal rest tumor of the liver: radiological imaging and immunohistochemical study of steroidogenic enzymes. Hepatol Res. 2008;38(11):1154–8.
Yan ML, Wang YD, Tian YF, et al. Adenocarcinoma arising from intrahepatic heterotopic pancreas: a case report and literature review. World J Gastroenterol. 2012;18(22):2881–4.
Guillou L, Nordback P, Gerber C, et al. Ductal adenocarcinoma arising in a heterotopic pancreas situated in a hiatal hernia. Arch Pathol Lab Med. 1994;118(5):568–71.
Naoe H, Iwasaki H, Kawasaki T, et al. Primary hepatic gastrinoma as an unusual manifestation of zollinger-ellison syndrome. Case Rep Gastroenterol. 2012;6(3):590–5.
Moriura S, Ikeda S, Hirai M, et al. Hepatic gastrinoma. Cancer. 1993;72(5):1547–50.
Diaz R, Aparicio J, Pous S, et al. Primary hepatic gastrinoma. Dig Dis Sci. 2003;48(8):1665–7.
Tsalis K, Vrakas G, Vradelis S, et al. Primary hepatic gastrinoma: report of a case and review of literature. World J Gastrointest Pathophysiol. 2011;2(2):26–30.
Ogawa S, Wada M, Fukushima M, et al. Case of primary hepatic gastrinoma: diagnostic usefulness of the selective arterial calcium injection test. Hepatol Res. 2014;45(7):823–6.
Ayub A, Zafar M, Abdulkareem A, et al. Primary hepatic vipoma. Am J Gastroenterol. 1993;88(6):958–61.
Hachicha I, Zayene A, Mnif Hachicha L, et al. Primary hepatic vipoma. Gastroenterol Clin Biol. 2003;27(5):551–4.
Ohwada S, Joshita T, Ishihara T, et al. Primary liver somatostatinoma. J Gastroenterol Hepatol. 2003;18(10):1218–9.
Morisawa Y, Tanaka A, Yamamoto T, et al. Primary hepatic somatostatinoma developed in a patient with von Recklinghausen’s disease. J Gastroenterol. 2006;41(4):389–91.
Do Cao C, Mekinian A, Ladsous M, et al. Hypercalcitonemia revealing a somatostatinoma. Ann Endocrinol (Paris). 2010;71(6):553–7.
Zhang ZY, Zhou GW, Shen C, et al. Liver transplantation revealing a case of metastatic pancreatic somatostatinoma. Chin J Surg. 2007;45(15):1075–6.
Rosenau J, Bahr MJ, Von Wasielewski R, et al. Ki67, E-cadherin, and p53 as prognostic indicators of long-term outcome after liver transplantation for metastatic neuroendocrine tumors. Transplantation. 2002;73(3):386–94.
Einarsson JI, Edwards CL, Zurawin RK. Immature ovarian teratoma in an adolescent: a case report and review of the literature. J Pediatr Adolesc Gynecol. 2004;17(3):187–9.
Karlo C, Leschka S, Dettmer M, et al. Hepatic teratoma and peritoneal gliomatosis: a case report. Cases J. 2009;2:9302.
Col C. Immature teratoma in both mediastinum and liver of a 21-Year-old female patient. Acta Med Austriaca. 2003;30(1):26–8.
Certo M, Franca M, Gomes M, et al. Liver teratoma. Acta Gastroenterol Belg. 2008;71(2):275–9.
Rout P, Rameshkumar K, Srikrishna NV. Hepatic mesothelioma. Indian J Gastroenterol. 1999;18(4):176–7.
Flemming P, Becker T, Klempnauer J, et al. Benign cystic mesothelioma of the liver. Am J Surg Pathol. 2002;26(11):1523–7.
Wang JB, Cao BK. One case of hepatic fibrous mesothelioma. Chin J Gen Surg. 2004;19(12):715–715.
Zhang JP, Ni JL. One case of large hepatic myxoma. Chin J Hepatobiliary Surg. 2007;9(13):610–1.
Badalian-Very G, Vergilio JA, Degar BA, et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood. 2010;116(11):1919–23.
Wu ZS, Li DH, Kong LB, et al. One case of liver Langerhans cell histiocytosis. Chin J Hepatobiliary Surg. 2012;18(2):138–138.
Hu X, Dong A, Lv S, et al. F-18 FDG PET/CT imaging of solitary liver Langerhans cell histiocytosis: preliminary findings. Ann Nucl Med, 2012, 2012.
Shi Y, Qiao Z, Xia C, et al. Hepatic involvement of Langerhans cell histiocytosis in children–imaging findings of computed tomography, magnetic resonance imaging and magnetic resonance cholangiopancreatography. Pediatr Radiol. 2014;44(6):713–8.
Braier J, Ciocca M, Latella A, et al. Cholestasis, sclerosing cholangitis, and liver transplantation in Langerhans cell Histiocytosis. Med Pediatr Oncol. 2002;38(3):178–82.
Ma J, Jiang Y, Chen X, et al. Langerhans cell histiocytosis misdiagnosed as liver cancer and pituitary tumor in an adult: a case report and brief review of the literature. Oncol Lett. 2014;7(5):1602–4.
Valladeau J, Ravel O, Dezutter-Dambuyant C, et al. Langerin, a novel C-type lectin specific to Langerhans cells, is an endocytic receptor that induces the formation of Birbeck granules. Immunity. 2000;12(1):71–81.
Lee RJ, Leung C, Lim EJ, et al. Liver transplantation in an adult with sclerosing cholangitis due to multisystem Langerhans cell histiocytosis. Am J Transplant. 2011;11(8):1755–6.
Kaplan KJ, Goodman ZD, Ishak KG. Liver involvement in Langerhans’ cell histiocytosis: a study of nine cases. Mod Pathol. 1999;12(4):370–8.
Kaiserling E, Muller H. Neoplasm of hepatic stellate cells (spongiotic pericytoma): a new tumor entity in human liver. Pathol Res Pract. 2005;201(11):733–43.
Karbe E, Kerlin RL. Cystic degeneration/Spongiosis hepatis in rats. Toxicol Pathol. 2002;30(2):216–27.
Couch JA. Spongiosis hepatis: chemical induction, pathogenesis, and possible neoplastic fate in a teleost fish model. Toxicol Pathol. 1991;19(3):237–50.
Stroebel P, Mayer F, Zerban H, et al. Spongiotic pericytoma: a benign neoplasm deriving from the perisinusoidal (Ito) cells in rat liver. Am J Pathol. 1995;146(4):903–13.
Bannasch P. Comments on R. Karbe and R. L. Kerlin (2002). Cystic degeneration/spongiosis hepatis (Toxicol Pathol 30 (2), 216–227). Toxicol Pathol, 2003, 31(5): 566–570.
Kerlin RL, Karbe E. Response to comments on E. Karbe and R. L. Kerlin (2002). Cystic degeneration/spongiosis hepatis (Toxicol Pathol 30(2): 216–227). Toxicol Pathol, 2004; 32(2): 271.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Nature Singapore Pte Ltd. and People's Medical Publishing House
About this chapter
Cite this chapter
Cong, WM. et al. (2017). Benign Tumors of the Liver and Intrahepatic Bile Duct. In: Cong, WM. (eds) Surgical Pathology of Hepatobiliary Tumors. Springer, Singapore. https://doi.org/10.1007/978-981-10-3536-4_6
Download citation
DOI: https://doi.org/10.1007/978-981-10-3536-4_6
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-3535-7
Online ISBN: 978-981-10-3536-4
eBook Packages: MedicineMedicine (R0)