
Abstract
The link between the Anrep effect -the increase in cardiac contractility that develops 10–15 min following myocardial stretch- and myocardial hypertrophy and failure was not appreciated until we proposed it in the 2005 edition of the book “Mechanosensitivity in Cells and Tissues”. In this new version of the chapter we will present the updated experimental evidence that led us to propose the autocrine/paracrine mechanism underlying the Anrep effect, as well as its resemblance to signals that have been described for cardiac hypertrophy development and heart failure. Interesting novel data supporting a crucial role for stretch-induced mineralocorticoid receptor activation, EGFR transactivation and increased mitochondrial production of reactive oxygen species leading to NHE-1 stimulation will be thoroughly described. A clear understanding of the early triggering mechanisms that stretch imposes to the myocardium will allow us to design novel weapons to win the battle against cardiac hypertrophy and failure, a major disease spread worldwide.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
- Stretch
- Sodium–hydrogen exchange
- Slow force response
- Hypertrophy
- Angiotensin
- Endothelin
- Aldosterone
- Epidermal growth factor
- Oxidative stress
12.1 Introduction
The link between the Anrep effect and myocardial hypertrophy and failure, although obvious—since this effect is the result of myocardial strain- was not appreciated until we proposed it in the 2005 edition of the book “Mechanosensitivity in Cells and Tissues”. Previous experiments by Izumo and Sadoshima (Sadoshima et al. 1993), Ito et al. (1993) and Yamazaki et al. (1996) performed in neonatal cardiomyocytes showed that mechanical stretch induces the release of preformed angiotensin II (A2) to the surrounding media, yet these findings were never linked to the Anrep effect, a phenomenon originally described in the open chest dog model (von Anrep 1912). Furthermore, Ito et al. (1993) showed that in stretched-conditioned medium where A2 was released, the hypertrophic signals were abolished by interfering with the endothelin (ET) action, finding that is in agreement with our own results demonstrating that stretching adult myocardium releases A2 (or activates the AT1 receptor by deformation) triggering the release and/or formation of ET (for review see (Cingolani et al. 2011a)).
12.2 The Anrep Effect
Although the contractile performance of the heart is under continuous neurohormonal and electrophysiological influence, the heart possesses intrinsic mechanisms, adapting to different hemodynamic conditions by changing its cardiac output. An increase in ventricular end diastolic volume, induced by increasing aortic resistance to ejection or venous return, leads to a more powerful contraction. This occurs immediately and is the well-known Frank-Starling mechanism that allows the heart to increase its output after an increase in venous return or to eject the same stroke volume against a greater afterload. However, over the next 10 or 15 min after the sudden stretch, there is a further increase in myocardial performance and the end diastolic volume returns towards its original value. The time constant of this phenomenon will depend on several factors such as species differences, temperature, coronary blood flow, etc. In 1912, von Anrep showed that after clamping the ascending aorta in a dog (acutely decreasing outflow and increasing intraventricular pressures), its heart initially dilated. This was followed by a progressive decline in heart volumes towards initial values over the next minutes (von Anrep 1912). Von Anrep interpreted these findings as secondary to a positive inotropic effect mediated by the release of cathecolamines by the adrenal glands, which were receiving low blood flow. In 1959 Rosenblueth et al. (1959) called attention to the fact that both, an increase in heart rate (Bowditch effect) and in afterload augmented the contractility in the isolated canine right ventricle, “the two staircase phenomenon”. Sarnoff, in 1960, coined the term “homeometric autoregulation” to define the decrease in left ventricular end diastolic volume after the initial increase in diastolic volume that occurs after an increase in afterload (Sarnoff et al. 1960). Since both reports (Rosenblueth et al. 1959; Sarnoff et al. 1960) were based on experiments performed in isolated hearts, the possibility of a positive inotropic effect due to the release of cathecolamines by the adrenal glands was ruled out.
In 1973, Parmley and Chuck reproduced this phenomenon in isolated strips of ventricular myocardium (Parmley and Chuck 1973). They showed that if the length of the muscle was increased, there were corresponding rapid and slow increases in developed force. The rapid change in force is thought to be the basis of the Frank-Starling mechanism and occurs secondary to an increase in myofilament Ca2 + sensitivity (Hofmann and Fuchs 1988). The slow force response (SFR) after a change in length is due to a progressive increase in the Ca2 + transient, as demonstrated by Allen and Kurihara in 1982 (Allen and Kurihara 1982) and later on confirmed by other authors, including us (Kentish and Wrzosek 1998; Alvarez et al. 1999). Parmley and Chuck also ruled out the possible role played by cathecolamines at the nerve endings in the development of the SFR, since the response was also present in isolated muscles from reserpinized animals (Parmley and Chuck 1973).
Although the cellular and molecular bases of the Frank-Starling mechanism (or “heterometric autoregulation”) are well-known and involve mainly an increase in the response of cardiac myofilaments to calcium (Hofmann and Fuchs 1988), the mechanism of the Anrep effect (homeometric autoregulation) is less understood. It is accepted that the increase in cardiac contractility that develops during the 10–15 min following muscle stretch can be quantitatively explained by a progressive increase in calcium transients (Allen and Kurihara 1982; Kentish and Wrzosek 1998; Alvarez et al. 1999). However, the source for this increase in calcium was less understood. It could not be explained by a hyperactive sarcoplasmic reticulum (SR) (Kentish and Wrzosek 1998) nor by an increased transarcolemmal calcium current (Hongo et al. 1996). The mechanism leading to the increase in the calcium transient was clarified by experiments performed in our laboratory that demonstrated a link between calcium influx and an autocrine/paracrine response to muscle stretch (Cingolani et al. 1998, 2003a; Alvarez et al. 1999; Perez et al. 2001, 2011a; Caldiz et al. 2007; Villa-Abrille et al. 2010).
12.3 The Autocrine/Paracrine Loop Triggered by Myocardial Stretch
As stated before, the stretch of cardiac muscle increases developed force in two phases. The first phase, which occurs rapidly, is generally attributed to enhanced myofilament responsiveness to calcium and is probably not affected by the autocrine/paracrine mechanism. The second phase (SFR) occurs gradually and is due to an increase in the calcium transient amplitude as a result of the autocrine/paracrine mechanism. The SFR was proposed to be the in vitro equivalent of the Anrep phenomenon and its genesis was unknown until we proposed that Na+/H+ exchanger (NHE-1) activation was the main step in the autocrine/paracrine mechanism leading to the increase in contractility by increasing intracellular sodium and calcium (Alvarez et al. 1999).
Most intracellular pathways leading to cardiac hypertrophy and failure are triggered by increases in intracellular calcium levels. Actually, the rise in cardiac muscle calcium causing the SFR or Anrep effect occurs as fast as 10–15 min after stretch. It is surprising that most investigators working in the field of excitation-contraction coupling and cardiac mechanics have not established a link between the Anrep effect and cardiac hypertrophy and failure. Interestingly, while several years ago we proposed the crucial role of the NHE-1 in the SFR development, more recently, elegant experiments by Wakabayashi's group demonstrated that NHE-1 activation is sufficient to generate calcium signals causing cardiac hypertrophy and failure (Nakamura et al. 2008).
An attractive idea, albeit speculative, will be that the fate of the myocardium may be determined during the first few minutes after stretch (i.e. it is possible that a pharmacological intervention that prevents the development of the Anrep effect might blunt the subsequent hypertrophy and failure). Approximately 23 million people are affected with heart failure, and 2 million new cases of heart failure are diagnosed each year worldwide. All the basic studies mentioned in this chapter need to be considered when designing new therapeutic strategies in the treatment of cardiac hypertrophy and failure. A clear understanding of the early triggering mechanisms that stretch imposes to the myocardium will allow us to design novel weapons to win the battle against this major disease.
In the next sections, we will present the experimental evidence that led us to propose the autocrine/paracrine mechanism underlying the SFR, as well as its resemblance to signals that have been described for cardiac hypertrophy development and heart failure.
12.4 Recent Advances in the Anrep Effect and Myocardial Hypertrophy and Failure
Figure 12.1 (left panel) depicts the state of knowledge on this subject when we wrote the chapter in the 2005 edition of the present book. In summary, the chain of events hypothesized at that time comprised the following: (1) AT1 receptor activation, (2) release/formation of ET, (3) NHE-1 hyperactivity, (4) increase in intracellular Na+ concentration, and (5) increase in Ca2 + transient amplitude through the Na+/Ca2 + exchanger (NCX).

State of knowledge of the chain of events triggered by myocardial stretch at 2005 (left panel) and the updated sequence at 2011 (right panel)
On the right panel of Fig. 12.1 we present the recent advances in this particular field, stressing the idea that myocardial hypertrophy and failure begins with cardiac strain-triggered intracellular pathways that are in part common to hypertrophy and failure development and the mechanical counterpart, the so called SFR. Our updated proposal is that the chain of events triggered by myocardial stretch is as follows: (1) release of A2, (2) release/formation of ET, (3) MR activation, (4) transactivation of the EGFR, (5) NADPH oxidase activation, (6) mitochondrial reactive oxygen species (ROS) production, (7) activation of redox-sensitive kinases, (8) NHE-1 hyperactivity, (9) increase in intracellular Na+ concentration, and (10) increase in Ca2 + transient amplitude through the NCX.
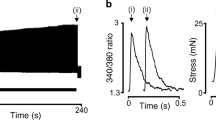
The left panel of Fig. 12.2 shows the typical behavior of a control papillary muscle before and after stretch. The first increase in force occurs immediately after stretch without changes in the Ca2 + transient and is attributed to the Frank-Starling mechanism. The SFR is the mechanical result of a chain of intracellular signals triggered by the stretch that ends with an increase in the Ca2 + transient. The pharmacological interventions that abolished the SFR or Anrep effect are summarized in the right panel of Fig. 12.2. Note the relationships between these interventions and the therapeutic treatments used to regress myocardial hypertrophy or to treat heart failure.

Left: Contractile response to stretch of an isolated papillary muscle. The first increase in force (from “a” to “b”, top) occurs without changes in the Ca2 + transient (“a” to “b”, bottom) while the SFR (from “b” to “c”, top) is due to an increase in the Ca2+ transient (“b” to “c”, bottom). Right: Pharmacological interventions proved to inhibit the SFR. (Modified with permission from Cingolani et al. (2001))
During the 5–6 years after the 2005 chapter was written, the followings steps were added to the sequence of the SFR generation:
12.4.1 The Critical Role of Mitochondrial ROS in the Activation of Redox Sensitive Kinases Leading to the Anrep Effect
The participation of ROS as intracellular signalling markers of A2/ET-1 in the myocardium is a well accepted fact (Sugden and Clerk 2006). In fact, we have demonstrated that a low dose of A2 (1 nmol/L) increases sarcomere shortening of isolated cat cardiomyocytes through an autocrine crosstalk with endogenous ET-1 (Cingolani et al. 2006), being this effect dependent on ROS production. Both peptides, A2 and ET-1, are well known activators of the NADPH oxidase (Giordano 2005; Kimura et al. 2005b) and through this action it has been reported a phenomenon called “ROS-induced ROS-release”, by which a small amount of ROS triggers a greater ROS production from the mitochondria (Zorov et al. 2000; Brandes 2005; Kimura et al. 2005a). The fact that ROS were implicated in myocardial strain-triggered hypertrophy (Pimentel et al. 2006), lead us to explore the possibility that the ROS-induced ROS-release mechanism would underlie the SFR. Figure 12.3a shows that stretch -in addition to its mechanical effect- induces an increase in intracellular ROS formation of approximately 30 % above baseline levels. Furthermore, scavenging ROS with N-(2-mercaptopropionyl)-glycine (MPG) or EUK8 inhibited both stretch-induced increase in ROS (Fig. 12.3a) and the SFR (Fig. 12.3b). We also found that ROS scavenging inhibited the increase in [Na+]i that occurs in response to stretch (Fig. 12.3c).

Myocardial stretch induced an intracellular ROS increase of ~30 % above the baseline levels that was cancelled by the ROS scavengers MPG and EUK8 (Panel a). MPG and EUK8 also cancelled the slow force response (expressed as percent of initial rapid phase) (Panel b). Furthermore, ROS scavenging also blunted stretch-induced increase in (Na+)i (Panel c). Insets show original raw data. * indicates P < 0.05 control vs. MPG and EUK8. DF = developed force. (Modified with permission from Caldiz et al. (2007))
These results allow us to hypothesize that activation of NAPDH oxidase after stretch would produce a small amount of O2 −, which may open the ATP-sensitive mitochondrial potassium (mKATP) channels and produce a larger amount of O2 − enough to generate the SFR. Experimental evidence supports these assumptions since the SFR was abolished after NADPH oxidase inhibition (apocynin or diphenyleneiodonium chloride, DPI) or after blockade of mKATP channels (5-hydroxydecanoate, 5HD, or glibenclamide) (Fig. 12.4a). Furthermore, the NHE-1-induced increase in [Na+]i underlying the SFR was also abolished by these interventions (Fig. 12.4b).

NADPH oxidase inhibition by apocynin (Apo) or diphenyleneiodonium chloride (DPI) as well as mKATP channels blockade with 5-hydroxydecanoate (5HD) or glybenclamide (Gly) abolished slow force response (expressed as percent of initial rapid phase) (Panel a). All these interventions also cancelled NHE-1-mediated increase in [Na+]i that accompanied the slow force response (Panel b). Insets show original raw data. * indicates P < 0.05 control vs. all other groups. DF = developed force. (Modified with permission from Caldiz et al. (2007))
In this context, it appears reasonable to speculate that stretch-mediated mitochondrial ROS production leads to phosphorylation and activation of the NHE-1. Actually, ROS-mediated activation of NHE-1 has been reported to be due to redox sensitive kinase-mediated phosphorylation of the exchanger cytosolic tail, being MEK, ERK1/2 and p90rsk kinases the favourite candidates (Rothstein et al. 2002). In this regard: (1) RAS-dependent activation of these kinases has been reported after stretch in neonatal cardiomyocytes (Pimentel et al. 2006); (2) we have detected significant increases in ERK1/2 and p90rsk phosphorylation after stretch (Fig. 12.5) that were abolished with 1 mmol/L losartan (Fig. 12.5); and (3) inhibition of MEK (a kinase upstream ERK1/2) also blunted the SFR (Fig. 12.5).

a Myocardial stretch significantly increased ERK1/2 and p90rsk phosphorylation, effect that was cancelled by losartan (Los). b Inhibition of MEK (a kinase upstream ERK1/2 and downstream RAS) by PD98059 cancelled the SFR (expressed as percent of the initial rapid phase). DF = developed force. * indicates P < 0.05 vs. non-stretched control (cont);† indicates P < 0.05 control vs. PD98059. (Modified with permission from Caldiz et al. (2007))
12.4.2 The Role of Epidermal Growth Factor Receptor (EGFR) in the SFR Development
It has been recently established that transactivation of the EGFR is the primary mechanism underlying G-protein coupled receptor (GPCR) agonist activation of ERK1/2 and its downstream intracellular pathways (Lemarie et al. 2008). Furthermore, myocardial stretch (Anderson et al. 2004; Duquesnes et al. 2009), myocardial hypertrophy (Kagiyama et al. 2002), and ET-1 signaling (Asakura et al. 2002) have been shown to involve EGFR transactivation. The possibility that this receptor was playing a role in the chain of events following myocardial stretch was examined. To this aim we explored whether inhibiting EGFR transactivation would impact on the SFR. Several mediators are known to be involved in the transactivation process, but the precise communication between GPCR and EGFR remains not entirely understood (Wetzker and Bohmer 2003). One proposed mediator is Src tyrosine kinase (Wetzker and Bohmer 2003); thus, as a first step in probing our hypothesis, we inhibited Src kinase with the specific tyrosine kinase inhibitor PP1. Figure 12.6b shows that PP1 (1 mmol/l) completely abolished the SFR. Another proposed mediator (Krieg et al. 2004; Szokodi et al. 2008) of EGFR transactivation is heparin-binding EGF (HB-EGF). HB-EGF is generated through extracellular proteolytic cleavage of proHB-EGF by the action of a matrix metalloproteinase (MMP). To test the contribution of this signaling pathway to the SFR, we inhibited MMP with MMP inhibitor III (MMPI), which specifically targeted MMPs 1, 2, 3, 7 and 13. MMPI (3 mmol/l) did not completely eliminate the SFR, but significantly reduced its magnitude by < 60 % (Fig. 12.6c) providing further support to the notion that EGFR transactivation was required for a fully developed SFR. Finally, we specifically inhibited the EGFR with AG1478 (1 mmol/l), which is known to prevent receptor phosphorylation, and consequently its activation. Under these conditions, the SFR was completely abolished (Fig. 12.6d). Thus, these three interventions that interfered with the mechanism of EGFR transactivation significantly decreased the SFR confirming that EGFR transactivation plays an essential role in the development of the SFR in cat myocardium.

SFR and EGFR transactivation. (a) a typical force record from a cat papillary muscle subjected to an increase in length from 92 to 98 % of Lmax; the biphasic response to stretch can be seen (vertical dotted lines indicate stretching interval). (b–d), same as a but from muscles pretreated with matrix metalloproteinase inhibitor (MMPI, b), the Src kinase inhibitor PP1 (c) or the EGFR blocker AG1478 (d), interventions that cancel EGFR transactivation. As can be seen, all these pharmacological interventions prevented the development of the SFR to stretch. (e), the averaged results obtained under the different experimental conditions expressed as a percentage of the initial rapid phase. ∗P < 0.05 control curve vs. others (2-way ANOVA). §P < 0.05 vs. initial rapid phase (for the sake of clarity, significance is indicated only for 15 min of stretch. (Modified with permission from Villa-Abrille et al. (2010))
12.4.3 Activation of Redox Sensitive Kinases and NHE Phosphorylation after Myocardial Stretch. Role of the EGFR Transactivation
We (Cingolani et al. 2005; Caldiz et al. 2007) and others (Zhang et al. 2009) previously showed that the SFR depends on the activation of NHE-1, which is a target of the redox sensitive kinases, ERK1/2. Others showed that ROS stimulated NHE-1 through MAPK (Rothstein et al. 2002; Haworth et al. 2003; Fliegel and Karmazyn 2004; Akram et al. 2006), and we recently proposed that stretch induced the mitochondrial production of ROS (Caldiz et al. 2007). We detected a significant increase in ERK1/2 phosphorylation after myocardial stretch. This effect was cancelled by pre-treatment with either AG1478 or PP1 (Fig. 12.7), two inhibitors of EGFR transactivation that also blocked the mechanical response. These findings showed that the prevention of EGFR activation is able to cancel both the increase in ERK1/2 phosphorylation and the mechanical response to stretch.

ERK1/2 phosphorylation after stretch. Myocardial stretch (S) significantly increased ERK1/2 phosphorylation. This effect was blunted either by EGFR blockade with AG1478 (AG) (a) or by Src kinase inhibition with PP1 (b), demonstrating that EGFR transactivation after stretch is necessary for ERK1/2 phosphorylation. AG1478 and PP1 alone did not modify basal ERK1/2 phosphorylation (92 ± 4 %, n = 4, and 107 ± 8 %, n = 4, of control respectively). ∗P < 0.05 vs. non-stretched control (control). (Modified with permission from Villa-Abrille et al. (2010))
Additionally, we estimated the levels of NHE-1 phosphorylation at Ser703 with a phospho-Ser 14-3-3 binding motif antibody. Figure 12.8 shows that phosphorylation at the 14-3-3 binding motif was increased after myocardial stretch, and this increase was prevented with AG1478. Our results support the concept that GPCR induced-EGFR transactivation plays a role in the chain of events that lead to NHE-1 phosphorylation and SFR development.

Stretch-induced NHE-1 phosphorylation. Myocardial stretch significantly increased NHE-1 phosphorylation at Ser703 estimated by a phospho-Ser 14-3-3 binding motif antibody. This effect was cancelled when the EGFR was blocked by AG1478 (AG). These results support a role of the EGFR transactivation in the chain of events leading to NHE-1 phosphorylation and SFR development. AG1478 alone did not modify basal NHE-1 phosphorylation (93 ± 4 % of control, n = 4). ∗P < 0.05 vs. control. (Modified with permission from Villa-Abrille et al. (2010))
Recently, it has been demonstrated that the Anrep effect was absent in a transgenic mouse lacking thrombospondin-4 (Cingolani et al. 2011b), a matricellular protein that is normally expressed at modest levels in the heart under normal conditions, but has been shown to be elevated in animals as well as humans with heart failure. Interestingly, the thrombospondin-4 molecule carries an EGF-like repeat, and as mentioned before, mice not expressing this protein not only had the Anrep effect blunted, but also failed to phosphorylate ERK1/2, as controls did. Surprisingly, these mice showed a phenotype of dilated cardiomyopathy after their aortas were banded, suggesting that a complex- not yet completely understood- cross-talk between the extracellular matrix and myocytes takes place after stretch. Further studies will continue to address the mechanistic role these matricellular proteins have in the development of heart failure.
12.4.4 Activation of the MR as a Consequence of Muscle Stretch
The link between A2 or its AT1 receptor and the mineralocorticoid receptor (MR) is an accepted fact (Lemarie et al. 2008, 2009; Grossmann and Gekle 2009). Although still somewhat controversial, aldosterone (ALD), which is known to be regulated by A2, appears to be synthesized and/or released by cardiac muscle (Gomez-Sanchez et al. 2004; Chai and Danser 2006); (Silvestre et al. 1998, 1999; Takeda et al. 2000). We have recently hypothesized that if a crosstalk between A2 and the MR occurs during ROS production, and at the same time A2 and ROS are crucial for SFR development, therefore MR inhibition would blunt the SFR. In this section we will present evidence that MR activation is involved in the signalling pathway leading to the Anrep effect.
Figure 12.9 shows that MR activation is necessary to promote ROS formation by a physiological concentration of A2 (1 nmol/L), since the increase in superoxide anion formation of ~50 % was suppressed after blocking MR with spironolactone or eplerenone. This effect was also suppressed by blocking AT1, ET1 (type A) receptor or EGFRs, by inhibiting NADPH oxidase, or by targeting mitochondria; and it was unaffected by glucocorticoid receptor inhibition.

Superoxide anion production induced by angiotensin II. a MR blockade with spirolactone (Sp, 10 mmol/L) or eplerenone (Ep, 10 mmol/L) abrogated the effect of 1 nmol/L A2 on the basal rate of O2˙ production. This effect was also blunted by the AT1 and ETA receptor antagonists losartan (Los, 1 mmol/L) and BQ123 (BQ, 10 mmol/L), respectively, and by NADPH oxidase inhibition with apocynin (Apo, 300 mmol/L). b A2-induced O2˙ formation was also blunted by targeting mitochondria with 5HD (100 mmol/L), glibenclamide (Gli, 50 mmol/L), or rotenone (Rot, 10 mmol/L), and by preventing EGFR activation either by EGFR blockade with AG1478 (AG, 1 mmol/L) or by inhibiting the metalloproteinase involved in EGFR transactivation with MMPI (3 mmol/L). Glucocorticoid receptor inhibition with Ru-486 (10 mmol/L) did not influence the effect of A2. *P < 0.05 vs. basal O2˙ production. (Modified with permission from Caldiz et al. (2011))
All interventions except AT1 receptor blockade blunted the increase in superoxide anion promoted by an equipotent dose of ET-1 (1 nmol/L), confirming that ET receptor activation is downstream of AT1 receptor (not shown, (Caldiz et al. 2011)). Similarly, an increase in superoxide anion promoted by an equipotent dose of ALD (10 nmol/L) was blocked by spironolactone or eplerenone, by preventing EGFR transactivation, but not after inhibiting glucocorticoid receptors or protein synthesis, suggesting a non-genomic MR effect (Fig. 12.10a). Combination of ALD and ET-1 did not further increase superoxide anion formation (Fig. 12.10b). ALD increased phosphorylation of the redox-sensitive kinases ERK1/2, p90RSK, and the NHE-1, effects that were eliminated by eplerenone or by preventing EGFR transactivation (not shown, see ref. (Caldiz et al. 2011)).

Superoxide anion production induced by aldosterone. a The effect of ALD at a concentration (10 nmol/L) that mimicked the effect of A2 and ET on the basal rate of O2˙ production was suppressed by spirolactone (Sp) and eplerenone (Ep), but not by the glucocorticoid receptor inhibitor Ru-486 or by preventing protein synthesis with cycloheximide (CicHex, 7 mmol/L). This demonstrates that MR activation has nongenomic consequences and excludes the possibility of glucocorticoid receptor activation. On the other hand, as shown for A2 and ET, the ALD-mediated increase in ROS formation was prevented by NADPH oxidase inhibition (Apo) and by preventing EGFR activation (AG and MMPI). This suggests that transactivation occurs in the direction of activated MR to EGFR, and that metalloproteinase activation downstream of MR is crucial for EGFR transactivation. b The combination of ALD and ET did not promote any further increase in O2˙ production. Under this condition, mitochondrial O2˙ production was abrogated by spironolactone (Sp), but unaffected by ETA receptor blockade with BQ123 (BQ), indicating that the only possible sequence of events is from ETA to MR. *P < 0.05 vs. basal O2˙ production. (Modified with permission from Caldiz et al. (2011))
Finally, the SFR was suppressed by MR blockade, by preventing EGFR transactivation or by scavenging ROS, but it was unaffected by glucocorticoid receptor blockade or protein synthesis inhibition as shown in Fig. 12.11. These results clearly suggest that MR activation is a necessary step in stretch-triggered mitochondrial ROS that mediates the activation of redox-sensitive kinases upstream NHE-1, leading to de Anrep effect.

SFR and MR activation. a Typical force record from rat papillary muscle subjected to an increase in length from 92 to 98 % of Lmax. The biphasic force response to stretch can be observed. b Same as (a) but from a muscle pre-treated with the MR blocker eplerenone, demonstrating that prevention of MR activation after stretch eliminated the SFR. c Averaged results of the SFR expressed as percentages of the initial rapid phase. MR blockade, not only by eplerenone but also by spironolactone, completely suppressed the SFR. However, the SFR was unaffected by the glucocorticoid receptor inhibitor Ru-486 or the protein synthesis inhibitor cycloheximide. d As reported previously in cat myocardium (Villa-Abrille et al. 2010) the SFR required EGFR transactivation, since it was blunted either by direct EGFR inhibition (AG1478) or by blocking transactivation with MMPI. Furthermore, the SFR was suppressed by the ROS scavenger MPG, supporting the notion that ROS formation is a key factor in the chain of events leading to the Anrep effect. (Modified with permission from Caldiz et al. (2011))
12.5 Direct Measurements of NHE-1 Stimulation by Aldosterone: Transactivation of the EGFR
Fujisawa et al. (2003) demonstrated that mineralocorticoid/salt-induced rat cardiac fibrosis and hypertrophy was prevented by the selective NHE-1 blocker cariporide. It has also been reported that ALD upregulates the expression and function of NHE-1 (Ebata et al. 1999; Karmazyn et al. 2003; Barbato et al. 2004; Matsui et al. 2007) and that selective blockade of this transporter prevents and/or reverts left ventricular hypertrophy in various animal models (Cingolani and Ennis 2007). According to these data and in agreement with our previous results on the SFR described above, we have recently shown that ALD increases NHE-1 activity in rat ventricular myocytes through a non-genomic pathway (Fig. 12.12a, b) (De Giusti et al. 2011).

Aldosterone induced activation of the NHE-1 and its blockade by inhibiting the EGFR. Panel a: representative traces of pHi during the application of two consecutive ammonium pulses (20 mmol/L NH4Cl), in the absence (first pulse) and presence of 10 nmol/L ALD (aldo, second pulse). ALD was applied 10 min before the second pulse. Panel b: average proton efflux JH, carried by the NHE-1, before (first pulses, closed circles, n = 5) and after application of 10 nmol/L ALD (second pulses, open circles, n = 5). JH is significantly enhanced by ALD. *P < 0.05 vs. control. Panel c: representative traces of pHi during the application of two consecutive ammonium pulses (20 mmol/L NH4Cl), in the absence (first pulse) and presence of 10 nmol/L ALD (second pulse). The EGFR blocker AG1478 (AG, 1 μmol/L) was applied 10 min before the first pulse and maintained throughout the experiment. ALD was applied 10 min before the second pulse. Panel d: average proton efflux JH, carried by the NHE-1, before (first pulses, open circles, n = 4) and after application of 10 nmol/L ALD (second pulses, closed circles, n = 4) in the continuous presence of 1 μmol/L AG1478. The transactivation of the EGFR by ALD leads to the activation of the NHE-1. (Modified with permission from De Giusti et al. (2011))
As commented above, EGFR activation represents one of the signaling pathways involving ALD (Grossmann and Gekle 2007; Grossmann et al. 2007). It has been shown that the MR blocker spironolactone reduces the EGFR mRNA synthesis after cerebral ischemia (Dorrance et al. 2001). Accordingly, Grossmann et al. (2007) reported that MR activation by ALD enhanced EGFR expression via an interaction with the EGFR promoter of vascular smooth muscle. In addition to these genomic effects, non-genomic actions of ALD involving EGFR transactivation have also been reported (Grossmann and Gekle 2008, 2009). Consistent with this evidence, we have recently shown that ALD enhances NHE-1 activity via transactivation of EGFR (Fig. 12.12c, d) (De Giusti et al. 2011). The stimulatory effect of this hormone on NHE-1 was blocked by the inhibitor of the Src-kinase PP1 and the blocker of metalloproteinases MMPI (De Giusti et al. 2011).
As commented above, these proteases release HB-EGF from its precursor, proHB-EGF. Figure 12.13 depicts that activation of EGFR by HB-EGF increases the production of intracellular ROS and triggers the ERK1/2 pathway, which phosphorylates p90RSK (De Giusti et al. 2011).

Signaling cascade involved in the aldosterone-induced NHE-1 stimulation in rat cardiomyocytes. The activation of the MR by ALD (Aldo) (1) leads to EGFR transactivation. This mechanism is mediated by the activation of the src-kinase (2) and the metalloproteinases. HB-EGF is released from the cell surface following shedding of the extracellular domain (ectodomain shedding) by this zinc-dependent metalloproteinases (3). HB-EGF binds to the EGFR and increases the production of superoxide anion (O2−) (4). These reactive oxygen species (ROS) stimulates the redox-sensitive kinase ERK1/2 (5), which phosphorylates the kinase p90RSK. This kinase phosphorylates the NHE-1 (6), stimulating its activity. Eplerenone (eple) and spironolactone (spiro) (MR blockers), PP1 (src-kinase inhibitor), MMPI (metalloproteinases blocker) and MPG (ROS scavenger) were employed to investigate pathways 1–4 (Ref. (De Giusti et al. 2011)). The inhibitor of the EGFR kinase AG1478 (AG) was used to evaluate the transactivation of this receptor by aldosterone. (Modified with permission from De Giusti et al. (2011))
This kinase, in turn, phosphorylates Serine703 of the NHE-1, leading to the activation of the transporter. As noted in Fig. 12.13, at least a fraction of the total amount of MR appears to be linked to the sarcolemma, likely co-localized to the EGFR (Grossmann et al. 2010) and/or associated to caveolin-1 (Krug et al. 2011). This data would explain the binding of ALD to the sarcolemmal fraction reported by Le Moellic et al. (2004). In addition, non genomic effects of ALD altering stimulation of a GPCR (GPR30) has been recently reported in vascular smooth muscle and endothelial cells (Gros et al. 2011).
12.6 Molecular Approach Targeting the Anrep Effect: Silencing NHE-1 Expression by Interference RNA
Gene silencing by RNA interference is a natural process occurring in cells by which a specific mRNA is degraded and therefore the expression of the encoded protein prevented. This mechanism is mediated by a double-stranded RNA (dsRNA) of approximately 18–23 nucleotides of length present inside the cell, known as siRNA (small interfering RNA) (Mello and Conte 2004; Kim and Rossi 2007). Briefly, through a multiple step pathway, one of the RNA strands of the siRNA is matched to a complementary mRNA which in turn is cleaved and ultimately degraded. In the research arena, in vivo or in vitro delivery of siRNA molecules to cells provides a powerful tool to specifically silence a single type of protein (Akhtar and Benter 2007; Kim and Rossi 2007). This technology has several advantages: (1) It is highly specific and can differentiate between members of the same family and even between isoforms of the same protein; (2) It has a lasting effect whose extension varies according to the strategy used to deliver the siRNA or modifications of the siRNA molecule; (3) It can be reversed; (4) It is relatively easy to obtain a siRNA. Moreover, siRNA technology has the potential to be used in the therapeutic field, to validate a protein as a suitable target whose inhibition would mediate the cure or alleviation of a disease. After finding the target protein, a synthetic drug could be designed to treat the disease. However, the RNA interference methodology also allows consideration of the siRNA molecule itself as a possible therapeutic tool (Kim and Rossi 2007). Delivery of siRNA molecules into the cell portends several challenges, starting with penetration of the plasma membrane, stability of siRNA inside and outside the cell, toxicity, and triggering of immune responses (Akhtar and Benter 2007; Kim and Rossi 2007; Manjunath et al. 2009).
Classic pharmacological techniques to inhibit a desired protein in vivo have several disadvantages compared to interference RNA: (1) The drug distributes broad-wide in the organism, condition that may affect undesired targets or generate side effects; (2) It is very difficult to make a drug that can differentiate between members of the family or isoforms; (3) Drug concentration change along time therefore requiring frequent administration.
Pharmacological inhibition of NHE-1 was beneficial in different experimental models of cardiac pathologies (Ennis et al. 1998, 2003; Karmazyn 1999; Avkiran and Marber 2002; Camilion de Hurtado et al. 2002; Engelhardt et al. 2002; Cingolani et al. 2003b). However, clinical trials with NHE-1 inhibitors like GUARDIAN (Theroux et al. 2000), ESCAMI (Zeymer et al. 2001) and EXPEDITION (Mentzer et al. 2008) failed to provide such benefits, and one of these studies was suspended due to undesired cerebrovascular side effects (Mentzer et al. 2008). Although the mechanism for this negative effect is still not clear, it could be related to blockade of NHE-1 activity in the brain where NHE-1 function seems to be essential, since animals lacking NHE-1 showed aberrant phenotype that included ataxia and epileptic-like seizures (Cox et al. 1997; Bell et al. 1999). It is possible then, that pharmacological inhibition of NHE-1 affects the exchanger in tissues other than the myocardium where its role is crucial or, even inhibits an unrelated protein required for normal function (Villafuerte et al. 2007). Activation of the RNA interference pathway appeared to be a suitable method to specifically block the NHE-1 because it spares the other nine members of the protein family. In our laboratory, we designed two different approaches to target the NHE-1 protein exclusively in the heart: (a) the delivery of naked siRNA, and (b) the delivery of siRNA using the lentivirus backbone. The use of viral vectors for delivering siRNA inside the cells facilitates its entry and provides a long lasting inhibition of a protein. Disadvantages of this strategy are potential changes of the gene expression after integration of viral DNA in the host genome and generation of immune responses. The use of naked siRNA has the advantage of preventing a massive immune response, but carries the difficult task of crossing the plasma membranes, allowing optimal propagation, mainly due to its size and electrostatic charge. Furthermore, it is more susceptible to be degraded by nucleases producing a less lasting silencing effect.
We have demonstrated that after a week of a single injection of naked siRNANHE-1 to the left ventricle, an extensive reduction of the NHE-1 expression and function took place (Morgan et al. 2011) (Fig. 12.14).

NHE-1 expression reduction distally from the injection site. a Mice were injected once in the apex of the left ventricle with naked siRNANHE-1 or siRNAscramble (siRNASCR) as shown in the heart scheme, and sacrificed after 72 h. b Left ventricle was divided into three parts (apex, middle, and base) to evaluate NHE-1 expression. c Representative immunoblots of lysates of the different fractions of the left ventricle. d Average expression of protein, quantified by densitometry and normalized to the amount of GAPDH (n = 7, *P < 0.05 vs. siRNASCR). (Modified with permission from Morgan et al. (2011))
These results, suggests the ability of siRNA molecules to spread through the myocardium and reduce NHE-1 expression and activity faraway from the injection site. In agreement with our findings, Kizana et al. (2009) demonstrated that siRNA molecules can move through cultured neighbour neonatal rat ventricle myocytes when coupled by gap junctions. Accordingly, it was found in different cultured cells that siRNA molecules can travel from one cell to another through gap junctions (Valiunas et al. 2005; Wolvetang et al. 2007) if connexin 43 (the predominant connexin isoform in adult hearts (Dhein 1998) is expressed (Valiunas et al. 2005). This local injection of siRNA in the left ventricle did not produce any effect on other organs, which allowed concluding that the procedure was successful in limiting the effects of siRNA to the heart (Morgan et al. 2011).
We have also recently incorporated the siRNA sequence able to mediate specific NHE-1 knockdown into a lentiviral vector (l-shNHE1) and injected into the left ventricular wall of Wistar rats (Perez et al. 2011). A separated group of rats injected with a vector expressing a non-silencing sequence (scramble) served as control. Confocal microscopy analysis of heart tissue revealed spreading of l-shNHE1 (DsRed tagged) from the sites of injection throughout the myocardium. Hearts with l-shNHE1 showed reduced NHE-1 protein expression (44 ± 8 % of controls, n = 4, P < 0.05) that correlated with depressed pHi recovery after acidosis and abolishment of the SFR (Fig. 12.15), despite preserved ERK1/2 activation (in % of control: stretch 241 ± 10 n = 5; stretch l-shNHE1 285 ± 36 n = 6). These data provide unequivocal support to our proposal that NHE-1 activation is crucial to the Anrep effect.

Functional activity of the NHE-1 was evaluated by the recovery of pHi after an acidic load (NH4+ prepulse) in isolated papillary muscles where the SFR was also tested. The original pHi recordings (left) as well as the averaged initial H+ fluxes (JH+, middle) clearly demonstrate the significant depression of NHE-1 activity in l-shNHE1 injected muscles compared to scramble. Furthermore, silencing the NHE-1 blunted the SFR (right). *P < 0.05 vs. scramble
12.7 Potential Link Between NHE-1 Activation and Cardiac Hypertrophy and Failure
Cardiac hypertrophy is known to be one of the main cardiovascular risk factors and a poor prognostic sign associated with nearly all forms of heart failure (Koren et al. 1991; Lloyd-Jones et al. 2002). Most intracellular pathways leading to pathological cardiac hypertrophy and failure converge at the increase in intracellular calcium levels and downstream activation of the calcineurin-dependent transcriptional pathway. The rise in calcium may occur through different mechanisms. One of them is an increase in intracellular Na+ resulting from enhanced function of the NHE-1, which drives the NCX to increase cytosolic calcium. As stated before, after cardiac muscle is stretched, an autocrine/paracrine chain of steps occur in which AT1 receptor activation is an early event (Sadoshima et al. 1993). This pathway also involves NADPH oxidase-dependent mitochondrial reactive oxygen species release, which itself activates the NHE-1 redox-sensitive kinasep90RSK, among others.
Enhanced NHE-1 activity as a possible mechanism involved in cardiac hypertrophy and failure was previously reported in the hypertrophic myocardium of adult spontaneously hypertensive rats (SHR) (Perez et al. 1995), in human ventricular myocytes from hearts with chronic end-stage heart failure (Yokoyama et al. 2000), in a pressure-volume overload model of cardiac hypertrophy and failure in rabbits (Baartscheer et al. 2008), in the hypertrophied heart of a type 2 diabetic rat model (Darmellah et al. 2007) and in neonatal rats (Dulce et al. 2006).
Interestingly, Nakamura et al. (2008) have recently demonstrated in vitro that NHE-1 hyperactivity is sufficient to generate calcium signals required for cardiac hypertrophy to take place. Although in vivo physiological data supporting the involvement of this mechanism in the transition to chronic cardiac hypertrophy and its consequences is scant, Baartscheer et al. (2005) have shown in elegant experiments that long-term NHE-1 inhibition with cariporide in rabbits with combined pressure and volume overload cardiac hypertrophy and failure attenuated hypertrophy and decreased the previously augmented diastolic calcium without significant alteration of systolic calcium (Fig. 12.16).

Prevention of cardiac hypertrophy and normalization of the previously augmented diastolic Ca2+ observed in rabbits with cardiac hypertrophy and failure (caused by combined pressure and volume overload) treated during 3 months with the NHE-1 inhibitor cariporide. Action potentials (top) and Ca2+ transients (bottom) in isolated myocytes from control (Ctrl), control plus cariporide (Ctrl-car), heart failure (HF), and heart failure plus cariporide (HF-car) groups. (Modified with permission from Baartscheer et al. (2005))
An increased activity of calcineurin in the myocardium of the SHR, and its suppression by the antihypertrophic treatment has been reported previously (Zou et al. 2002; Ennis et al. 2007). Similarly, in the hypertrophied myocardium of rats with salt-sensitive hypertension, an increase in the activity of calcineurin and its prevention by treatment with an AT1 blocker has been reported (Nagata et al. 2002). We were the first to report that the regression of cardiac hypertrophy caused by NHE-1 inhibition was accompanied by normalization of the activity of the calcineurin pathway and preservation or even improvement of cardiac function. (Ennis et al. 2007). NHE-1 inhibition by decreasing [Na+]i diminishes calcium either by decreasing calcium entry (reverse mode) or by increasing calcium efflux (forward mode) through the NCX. At first glance, it may appear difficult to understand how a decrease in cytosolic calcium induced by NHE-1 inhibition can improve myocardial contractility in the long term. However, the preservation of cardiac function after regression of cardiac hypertrophy seems not to be unique to the regression of cardiac hypertrophy induced by NHE-1 inhibition (Esposito et al. 2002). In the myocardium, intracellular calcium is compartmentalized in such way that the contractile pool is different from the pool that regulates reactive signaling. In agreement with this, it has been suggested that calcineurin, as well as CaMKII are preferentially activated by specific sub-cellular calcium pools (Frey et al. 2004; Wu et al. 2010). Therefore we can speculate that the decrease in diastolic calcium might be sensed by the calcium calmodulin-calcineurin pathway, but not by the contractile machinery. Moreover, a negative inotropic effect of calcineurin through different mechanisms has been described (Sah et al. 2002; Li et al. 2003), and a positive inotropic effect could therefore be expected with the phosphatase deactivation.
In our scheme, stretch-triggered NHE-1 activation is the main step leading to cardiac hypertrophy and failure. The experiments that originally induced us to consider the activation of NHE-1 after myocardial stretch were performed in the absence of bicarbonate in the medium, where the only active pHi regulating mechanism was this exchanger (Cingolani et al. 1998). Therefore, the increase in pHi served as a “marker” for NHE-1 activation by A2-ET. This activation was protein kinase C-dependent since chelerythrine prevented it (Cingolani et al. 1998). It is interesting to emphasize that the NHE-1 exchanges one intracellular H+ for one extracellular Na+, therefore, its activation would be followed by an increase in both [Na+]i and pHi. However, during our experiments it became evident that the increase in pHi occurred only in the absence of bicarbonate in the medium. In contrast, when bicarbonate was present in the media, the simultaneous activation by the stretch of the NHE-1 and the Na+ independent Cl−/HCO3 − exchanger (AE) precluded significant changes in pHi but not in [Na+]i (Fig. 12.17). Regarding the intracellular signals leading to activation of NHE-1 by ET, they are not fully understood. If we consider that part of the positive inotropic effect of ET-1 is the result of endogenously generated ROS (Sand et al. 2003) and that ROS, through MAPK pathways, phosphorylate the cytosolic tail of the NHE-1 increasing its activity (Rothstein et al. 2002), we could suggest that ROS may be involved in the activation of the NHE-1 after stretch.

Representative experiments showing that in the presence of bicarbonate, NHE-1 activation by stretch causes elevation of [Na+]i (assessed by SBFI 340/380 fluorescence ratio) but not of pHi, due to the simultaneous activation of the AE. (Modified with permission from Cingolani et al. (2003))
As pointed out before, the increase in [Na+]i induced by NHE-1 activation is the most important step in the chain of events leading to its mechanical counterpart, the SFR, and perhaps portending implications in the mechanism(s) that lead to myocardial hypertrophy and failure (Gray et al. 2001). In connection with this, we have demonstrated that exogenous applied A2 stimulates AE activity through endogenous ET (Camilión de Hurtado et al. 2000). Therefore, under physiological conditions, stretch will be followed by a sequential release of A2 and ET leading to the simultaneous activation of NHE and AE. The rise in pHi induced by NHE-1 activation might be prevented by AE, but this is not the case for the increase in [Na+]i, due to its Na+ independency. This increase in [Na+]i will trigger an increase in calcium influx (and consequently the calcium transient) by reducing the NCX forward mode and/or favoring its reverse mode of operation. Instead, when the activation of the AE is prevented by a functional antibody, an increase in [Na+]i takes place (Fig. 12.18a) together with an increase in pHi. The mechanical counterpart may therefore result from the increase in calcium transient and also from the increase in myofilament calcium responsiveness due to cytosolic alkalization (Fabiato and Fabiato 1978; Mattiazzi et al. 1979; Orchard and Kentish 1990). In agreement with the latter, Fig. 12.18b, c shows that in the presence of the AE antibody, the stretch of a cat papillary muscle produces a greater SFR.

When the AE activation is prevented by a functional antibody (antiAE3Loop III) an increase in pHi takes place (panel a), Thus, the SFR in this condition results from the increase in the calcium transient plus an increase in myofilament calcium responsiveness due to cytosolic alkalization and, therefore, the SFR to stretch is greater (compare panels b and c). Modified with permission from Cingolani et al. (2003a)
It may be argued that the Na+/K+ATPase should prevent the increase in [Na+]i elicited by NHE-1 hyperactivity, however, Bers et al. (2003) have shown that the changes in [Na+]i necessary to alter the Na+ pump activity should be greater than those detected by us after the stretch. Furthermore, we should consider that an enhanced activity of the pump would be probably detected due to the increase in [Na+]i after stretch, but not enough to normalize the [Na+]i. In other words, the rise in [Na+]i detected during the SFR should be higher if the pump was inhibited. We may also speculate that, similarly to the Na+ pump lag hypothesis for the force-frequency relationship, the greater Na+ entry is balanced by an increased Na+ pump activity, but only at the cost of elevated [Na+]i and hence increased calcium entry. In contrast, the changes in [Na+]i detected after stretch may suffice to alter the activity of the NCX, specially if the NHE and the NCX are co-localized (Petrecca et al. 1999; Brette et al. 2002). Accordingly, we recently showed that the positive inotropic effect of exogenous A2 or ET-1 is accompanied by a cariporide-sensitive increase in [Na+]i (Fig. 12.19) (Perez et al. 2003). Additionally, the fact that ET receptors blockade with TAK044 canceled the A2-induced rise in [Na+]i reinforces the role of ET as mediator of A2 effects (Perez et al. 2003).

The positive inotropic effect of exogenous Ang II or ET-1 is accompanied by a cariporide-sensitive rise in [Na+]i. Panel a: Typical experiment and averaged results of the effect of a low dose of A2 on pHi. Panel b: Time course of [Na+]i changes induced by A2 and averaged values after 30 min, in control and after NHE-1 inhibition with cariporide (HOE642) or ET receptors blockade (TAK044). Panels c and d: Same as A and B respectively, but after addition of an equipotent dose of exogenous ET-1. Note that despite the lack of pHi change, there is an A2-induced NHE-1 stimulation detected by the increase in [Na+]i that requires available ET receptors and that this effect can be mimicked by an equipotent dose of exogenous ET-1. *P < 0.05 vs pre-peptide control value. (Modified with permission from Perez et al. (2003))
Although the role of NHE-1 activation early after stretch leading to the SFR development (and possibly to cardiac hypertrophy and failure) has been detected in different species including cat (Perez et al. 2001; Caldiz et al. 2007), human (von Lewinski et al. 2004), rabbit (Luers et al. 2005), and rat (Alvarez et al. 1999; Calaghan and White 2004) myocardium, involvement of stretch-operated channels in this response was recently proposed in mouse ventricular muscle by Ward et al. (2008), who showed that canonical transient receptor-operated channels (TRPC) are sensitive to stretch in mice myocardium. Furthermore, Takahashi et al. (2007) showed TRPC sensitivity to A2 in human coronary artery smooth muscle cells. Interestingly, it was proposed that TRPC channels were necessary mediators of pathological cardiac hypertrophy in mice, in part through calcineurin-NFAT signaling (Wu et al. 2010), a pathway that we showed to be sensitive to NHE-1 inhibition in rats (Ennis et al. 2007). This discrepancy may be explained by two alternative hypotheses: (1) the TRPC channels were involved in one or some of the steps in the chain of events described previously; i.e., to induce A2 release after stretch or (2) by species differences.
Regarding whether some early intracellular signals triggered by the autocrine/paracrine mechanism, (i.e; NHE-1 activation) persists over time, we recently explore this in a mouse model of cardiac hypertrophy and failure by transverse aortic constriction (TAC). After 7 weeks of TAC, cardiac hypertrophy and decreased myocardial performance was detected, along with enhanced oxidative stress, as well as increased activity of redox-sensitive p90RSK kinase and NHE-1 phosphorylation. Selective AT1 receptors blockade with losartan prevented p90RSK and NHE-1 activation and decreased hypertrophy development, preserving contractility in spite of a higher workload (Cingolani et al. 2010, 2011c). It is important to highlight that losartan treatment did not restore wall thickness to control values (it remained ~24 % higher than controls), but certainly reduced it to levels “necessary” to counteract for the increase in pressure induced by the aortic constriction. In other words, an excessive cardiac hypertrophy was eliminated. Interestingly, in spite of the increase in wall stress seen with losartan in the present study, the reduction in cardiac hypertrophy was accompanied by an increased cardiac performance. These findings suggest that the degree of cardiac hypertrophy prevented by losartan was maladaptive or “inappropriate”, a concept previously coined by others (Mureddu et al. 2009). In this regard, it seems that pressure overload may trigger multiple intracellular signaling pathways in addition to enhanced AT1 receptor stimulation. Whereas some of these may be deleterious, others may benefit the heart allowing it to adapt to different stressors. The hypothetical proposal to explain these striking findings is schematized in Fig. 12.20.

Schematic representation of the proposed signaling pathway involved in the prevention of cardiac hypertrophy by AT1 receptors blockade. In our scheme, the AT1 receptors-sensitive part of the TAC-induced CH is maladaptive and related to redox-sensitive p90RSK activation, NHE-1 phosphorylation/activation, increase in intracellular Na+ and the consequent increase in intracellular calcium through the NCX. The increased calcium concentration would then activate the calcineurin-NFAT signaling pathway responsible for triggering an abnormal cardiac growth. On the other hand, the same mechanical stimulus (stretch of cardiac muscle) may trigger other prohypertrophic signals intended to compensate for the increased wall stress (“adaptive hypertrophy”) (Catalucci et al. 2008). The reason for an improvement in cardiac performance accompanying the regression in cardiac hypertrophy due to AT1 receptors blockade is not apparent to us at present. However, we could speculate about cancellation of the negative inotropic effect assigned to calcineurin phosphatase activation (Sah et al. 2002; Li et al. 2003). (Modified with permission from Cingolani et al. (2011c))
12.7.1 Hypertrophic Signals Triggered by NHE-1
The possible link between the SFR and myocardial hypertrophy and failure is supported by the fact that an enhanced activity of NHE-1 is detected in several models of cardiac hypertrophy (Kusumoto et al. 2001; Engelhardt et al. 2002). In the hypertrophied myocardium of SHR (Wang et al. 2003), an increased activity of NHE-1 has been detected (Perez et al. 1995; Schussheim and Radda 1995) due to a kinase-dependent posttranslational phosphorylation of its cytosolic tail (Siczkowski et al. 1995; Ennis et al. 1998). The regression of myocardial hypertrophy produced by several pharmacological interventions was accompanied by normalization of the NHE-1 activity (Ennis et al. 1998; Alvarez et al. 2002). Moreover, chronic treatment of SHR rats with NHE-1 inhibitors caused load-independent regression of cardiomyocyte hypertrophy and fibrosis (Camilion de Hurtado et al. 2002; Cingolani et al. 2003b), although the latter effect took longer than the regression of myocyte size (Cingolani et al. 2003b), possibly as a reflection of the slower turnover rate of collagen metabolism (Weber and Brilla 1991).
Based on our previous results in adult multicellular cardiac preparations, hypertensive cardiac hypertrophy and failure are caused by an autocrine/paracrine chain of events triggered by myocardial stretch that begins with the activation of the AT1 receptors followed by the release/formation of endothelin-1 (ET-1), MR activation, EGFR transactivation and stimulation of the NHE-1 (Cingolani et al. 2005; Villa-Abrille et al. 2010; Caldiz et al. 2011).). The increased production of ROS that results from A2/ET-1 stimulation of the NADPH oxidase may be responsible for ERK1/2-p90RSK activation and NHE-1 stimulation (Caldiz et al. 2007; Garciarena et al. 2008). NHE-1 hyperactivity leads to an increase in intracellular Na + concentration that promotes cytoplasmic calcium overload through the NCX (Cingolani et al. 2005). Calcium is widely recognized as one of the main pro-hypertrophic intracellular signal. It activates several intracellular pathways like calcineurin, nuclear factor of activated T cells (NFAT), calcium/calmodulin-dependent kinase II, protein kinase C and possibly other intracellular signaling pathways. Calcineurin is a prohypertrophic serine-threonine protein phosphatase that is activated in response to sustained elevations of intracellular levels of calcium. Once activated, calcineurin directly dephosphorylates NFATs within the cytoplasm and promotes their translocation into the nucleus to induce the transcription of several genes. In SHR, Ennis et al. (2007) described that NHE-1 blockade regressed cardiac hypertrophy, decreased myocardial BNP, calcineurin Ab and nuclear NFAT expression. Additionally, they demonstrated by echocardiography, a reduction in left ventricular wall thickness without changes in cavity dimensions or a significant decrease in blood pressure (Ennis et al. 2007).
Emerging evidence indicates that NHE-1 can be activated by ROS (Sabri et al. 1998; Snabaitis et al. 2002). Less well explored is the possibility that the increased ROS production, in addition to its role played upstream to NHE-1, may be induced by a rise in [Na+]i secondary to NHE-1 hyperactivity. Javadov et al. (2005, 2006) showed in rats with myocardial infarction that NHE-1 inhibition was able to prevent cardiac hypertrophy and decreased the vulnerability of mitochondria to calcium. In addition, they attributed the anti-hypertrophic effect of NHE-1 inhibition to the decreased generation of mitochondrial-derived ROS. In relation to this, we recently reported that the cardiac superoxide production induced by A2 was reduced under NHE-1 inhibition (Javadov et al. 2006; Garciarena et al. 2008). Moreover, the decrease in infarct size and level of tissue lipoperoxidation, induced by ROS scavengers administered during the reperfusion, can be mimicked by specific blockade of NHE-1 (Fantinelli et al. 2006). Therefore, NHE-1 inhibition may exert its beneficial effects by decreasing [Na+]i and/or ROS production. Both [Na+]i and ROS target the NCX to modify its activity, and therefore target calcium either at the bulk of the cytosol, or to more restricted spaces.
12.8 Conclusion and Perspectives. Possible Applications in the Clinical Arena
The RALES trial in 1999, the EPHESUS in 2003 and the EMPHASIS-HF in 2010, called attention to the beneficial effects of ALD antagonism in the treatment of heart failure. Cardiovascular disease and specially heart failure is one of the most important health problems in the world. Cardiac hypertrophy is known to be the main entrance door to the failing heart. As described above, cardiac hypertrophy and failure are triggered by intracellular signals that occur following myocardial stretch. Surprisingly, investigators working in the area of cardiac mechanics did not often extrapolate their early findings seen after stretch to the development of cardiac hypertrophy and/or failure. The reason for this could be that time frames in which these two phenomena occur are quite different. However, the long journey toward myocardial hypertrophy and failure begins with one step, and this first step may well be the autocrine/paracrine intracellular signaling pathway triggered by myocardial stretch as was proposed by Izumo and Sadoshima in neonatal cardiac myocytes (Sadoshima et al. 1993) and by us (Cingolani et al. 1998; Alvarez et al. 1999; Perez et al. 2001; Caldiz et al. 2007; Villa-Abrille et al. 2010) in adult multicellular preparations.
Current treatment against cardiac failure is mainly based on inhibition of hormones (A2, ALD, catecholamines). Despite the term “ALD inhibition” has been widespread used, this is often misleading and should be replaced by MR antagonism, mainly because ALD is not the only agonist binding to and activating MR (Mihailidou et al. 2009). Although several studies have demonstrated the important benefits of MR antagonists in heart failure, their clinical use remains lower than expected and the exact mechanism of the beneficial effect is still unknown.
In 1990 Swedberg et al. (1990) established a relationship between plasma levels of ALD and mortality in patients with heart failure. This finding called attention to the possibility of predicting indexes of morbidity and mortality in patients suffering from this disease with excessive plasma levels of ALD. On the other hand, Karl Weber's laboratory carried out several investigations demonstrating that ALD itself is able to increase myocardial fibrosis independently of blood pressure level (for review see (Gandhi et al. 2011)). Furthermore, it was also shown that spironolactone, an MR blocker with diuretic properties developed by Searle laboratories, abolished these effects.
Contemporarily, several research labs reported that MR are not only expressed in classical ALD target tissues, but also in many others, including smooth and cardiac muscle.
The most potent stimulator of ALD synthesis is A2. Consequently, interfering with A2 actions should decrease systemic ALD levels. However, despite complete vascular angiotensin converting enzyme inhibition plasma ALD levels were elevated in patients with heart failure (Jorde et al. 2002). Even the combination of angiotensin converting enzyme inhibition and A2 antagonism only transiently reduces ALD plasma levels in patients with heart failure (McKelvie et al. 1999) suggesting A2 independent ALD production. This phenomenon known as ALD escape and whose underlying mechanism has not been completely clarified yet constitutes a strong proof to directly inhibit MR activation on top of angiotensin converting enzyme inhibition or AT1 blockers in the treatment of heart failure.
Among MR inhibitors, spironolactone was the first marketed compound in the early 1960s, and although proved to be clinically useful, it also showed tolerability problems due to painful gynecomastia or menstrual disturbances due to its androgenic and progesteronergic effects. Nevertheless, it was the only compound approved to be used in the RALES in 1663 patients with severe heart failure (Class III-IV NYHA). The trial was discontinued after a mean follow-up period of 24 months, because interim analysis determined that spironolactone reduced the risk of death by 30 %. Later on, more specific compounds that inhibit MR were developed, and after several years of delay Searle patented eplerenone in 1984. Eplerenone was tested in a clinical study called EPHESUS performed on 6642 patients with acute myocardial infarction complicated with left ventricular systolic dysfunction (ejection fraction less than 40 %). Treatment started 3–14 days after myocardial infarction and was maintained during 16 months. The results were positive, favoring the active treatment arm, and the main difference with RALES was that most of the patients in EPHESUS were receiving beta blockers (75 % vs. 11 % in RALES). All cause mortality decreased by ~15 % and sudden cardiac death by ~21 %. Interestingly, a post hoc analysis of the EPHESUS (Pitt et al. 2005) showed a reduction of all cause mortality by ~31 % as early as 30 days after eplerenone treatment. One important fact to emphasize, specially after the widely spread concept that high levels of ALD characterizes heart failure, is that plasma levels of ALD and Na+ were in the normal range in both RALES and EPHESUS trials at randomization, and the beneficial effects were seen early, probably before the “ALD escape” took place. These findings may suggest that: (1) MRs are activated by ligands other than ALD, or (2) cytosolic MRs are activated by increased intracellular levels of ALD independently of its plasma levels (Silvestre et al. 1998).
In contrast with the two mentioned clinical trials, the recently published EMPHASIS (Zannad et al. 2010, 2011) was carried out on patients with less severe heart failure. This study enrolled 2737 patients with heart failure class II and III of the NYHA and left ventricular ejection fraction of no more than 35 %. The trial was stopped prematurely according to their rules, after a median follow-up of 21 months, due to the excess of benefit in reducing the risk of cardiovascular death or hospitalization for heart failure, obtained by anti-aldosteronic therapy with eplerenone, which was then extended to both arms of the trail. Another clinical trial which is currently running is the TOPCAT. This study will test the effects of MR inhibition in patients suffering from heart failure with left ventricular ejection fraction of at least 45 %. TOPCAT will probably end in 2013.
Finally, ALBATROS is a clinical trial designed to asses the potential superiority of MR inhibition early after myocardial infarction. The study will evaluate the intravenous bolus of potassium camreonate followed by a daily dose of 25 mg of spironolactone for 6 months on top of standard therapy in 1600 patients with myocardial infarction.
Although clinical evidence undoubtedly showed beneficial effects of treating heart failure patients with MR blockers, the mechanisms by which MR antagonism provide cardiovascular protection are not completely understood. In this regard, our own results assigning a crucial role for MR activation as an early hypertrophic signal triggered by myocardial stretch (presented before in this chapter) encouraged us to suggest that prevention of oxidative stress and NHE-1 activation should be considered as a potential key factor for the salutary effects of ALD antagonism in humans.
References
Akhtar S, Benter IF (2007) Nonviral delivery of synthetic siRNAs in vivo. J Clin Invest 117:3623–3632
Akram S, Teong HF, Fliegel L, Pervaiz S, Clement MV (2006) Reactive oxygen species-mediated regulation of the Na+-H+ exchanger 1 gene expression connects intracellular redox status with cells' sensitivity to death triggers. Cell Death Differ 13:628–641
Alvarez BV, Perez NG, Ennis IL, Camilion de Hurtado MC, Cingolani HE (1999) Mechanisms underlying the increase in force and Ca2+ transient that follow stretch of cardiac muscle: a possible explanation of the Anrep effect. Circ Res 85:716–722
Alvarez BV, Ennis IL, De Hurtado MC, Cingolani HE (2002) Effects of antihypertensive therapy on cardiac sodium/hydrogen ion exchanger activity and hypertrophy in spontaneously hypertensive rats. Can J Cardiol 18:667–672
Allen DG, Kurihara S (1982) The effects of muscle length on intracellular calcium transients in mammalian cardiac muscle. J Physiol 327:79–94
Anderson HD, Wang F, Gardner DG (2004) Role of the epidermal growth factor receptor in signaling strain-dependent activation of the brain natriuretic peptide gene. J Biol Chem 279:9287–9297
Asakura M, Kitakaze M, Takashima S, Liao Y, Ishikura F, Yoshinaka T, Ohmoto H, Node K, Yoshino K, Ishiguro H, Asanuma H, Sanada S, Matsumura Y, Takeda H, Beppu S, Tada M, Hori M, Higashiyama S (2002) Cardiac hypertrophy is inhibited by antagonism of ADAM12 processing of HB-EGF: metalloproteinase inhibitors as a new therapy. Nat med 8:35–40
Avkiran M, Marber MS (2002) Na+/H+ exchange inhibitors for cardioprotective therapy: progress, problems and prospects. J Am Col Cardiol 39:747–753
Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Opthof T, Fiolet JW (2005) Chronic inhibition of Na+/H+-exchanger attenuates cardiac hypertrophy and prevents cellular remodeling in heart failure. Cardiovasc Res 65:83–92
Baartscheer A, Hardziyenka M, Schumacher CA, Belterman CN, van Borren MM, Verkerk AO, Coronel R, Fiolet JW (2008) Chronic inhibition of the Na+/H+ – exchanger causes regression of hypertrophy, heart failure, and ionic and electrophysiological remodelling. Br J Pharmacol 154:1266–1275
Barbato JC, Rashid S, Mulrow PJ, Shapiro JI, Franco-Saenz R (2004) Mechanisms for aldosterone and spironolactone-induced positive inotropic actions in the rat heart. Hypertension 44:751–757
Bell SM, Schreiner CM, Schultheis PJ, Miller ML, Evans RL, Vorhees CV, Shull GE, Scott WJ (1999) Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am J Physiol 276:C788–C795
Bers DM, Barry WH, Despa S (2003) Intracellular Na+ regulation in cardiac myocytes. Cardiovasc Res 57:897–912
Brandes RP (2005) Triggering mitochondrial radical release: a new function for NADPH oxidases. Hypertension 45:847–848
Brette F, Komukai K, Orchard CH (2002) Validation of formamide as a detubulation agent in isolated rat cardiac cells. Am J Physiol 283:H1720–H1728
Calaghan S, White E (2004) Activation of Na+-H+ exchange and stretch-activated channels underlies the slow inotropic response to stretch in myocytes and muscle from the rat heart. J Physiol 559:205–214
Caldiz CI, Garciarena CD, Dulce RA, Novaretto LP, Yeves AM, Ennis IL, Cingolani HE, Chiappe de Cingolani G, Perez NG (2007) Mitochondrial reactive oxygen species activate the slow force response to stretch in feline myocardium. J Physiol 584:895–905
Caldiz CI, Diaz RG, Nolly MB, Chiappe de Cingolani GE, Ennis IL, Cingolani HE, Perez NG (2011) Mineralocorticoid receptor activation is crucial in the signalling pathway leading to the Anrep effect. J Physiol 589:6051–6061
Camilión de Hurtado MC, Alvarez BV, Ennis IL, Cingolani HE (2000) Stimulation of myocardial Na+ -independent Cl−HCO3− exchanger by angiotensin II is mediated by endogenous endothelin. Circ Res 86:622–627
Camilión de Hurtado MC, Portiansky EL, Perez NG, Rebolledo OR, Cingolani HE (2002) Regression of cardiomyocyte hypertrophy in SHR following chronic inhibition of the Na+/H+ exchanger. Cardiovasc Res 53:862–868
Catalucci D, Latronico MV, Ellingsen O, Condorelli G (2008) Physiological myocardial hypertrophy: how and why? Front Biosci 13:312–324
Cingolani HE, Ennis IL (2007) Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 115:1090–1100
Cingolani HE, Alvarez BV, Ennis IL, Camilion de Hurtado MC (1998) Stretch-induced alkalinization of feline papillary muscle: an autocrine-paracrine system. Circ Res 83:775–780
Cingolani HE, Pérez NG, Camilión de Hurtado MC (2001) An autocrine/paracrine mechanism triggered by myocardial stretch induces changes in contractility. NIPS 16:88–91
Cingolani HE, Chiappe GE, Ennis IL, Morgan PG, Alvarez BV, Casey JR, Dulce RA, Perez NG, Camilion de Hurtado MC (2003a) Influence of Na+ -independent Cl−HCO3− exchange on the slow force response to myocardial stretch. Circ Res 93:1082–1088
Cingolani HE, Rebolledo OR, Portiansky EL, Perez NG, Camilion de Hurtado MC (2003b) Regression of hypertensive myocardial fibrosis by Na(+)/H(+) exchange inhibition. Hypertension 41:373–377
Cingolani HE, Perez NG, Aiello EA, de Hurtado MC (2005) Intracellular signaling following myocardial stretch: an autocrine/paracrine loop. Regul Pept 128:211–220
Cingolani HE, Villa-Abrille MC, Cornelli M, Nolly A, Ennis IL, Garciarena C, Suburo AM, Torbidoni V, Correa MV, Camilionde Hurtado MC, Aiello EA (2006) The positive inotropic effect of angiotensin II: role of endothelin-1 and reactive oxygen species. Hypertension 47:727–734
Cingolani OH, Perez NG, Mosca SM, Schinella GR, Console GM, Ennis IL, Escudero EM, Cingolani HE (2010) AT1 receptor blockade with losartan prevents Maladaptive hypertrophy in pressure overload by inhibiting ROS release. Hypertension 56:e119 (Abstract)
Cingolani HE, Ennis IL, Aiello EA, Perez NG (2011a) Role of autocrine/paracrine mechanisms in response to myocardial strain. Pflugers Arch 426(1):29–38
Cingolani OH, Kirk JA, Seo K, Koitabashi N, Lee DI, Ramirez-Correa G, Bedja D, Barth AS, Moens AL, Kass DA (2011b) Thrombospondin-4 is required for stretch-mediated contractility augmentation in cardiac muscle. Circ Res 109:1410–1414
Cingolani OH, Perez NG, Ennis IL, Alvarez MC, Mosca SM, Schinella GR, Escudero EM, Console G, Cingolani HE (2011c) In vivo key role of reactive oxygen species and NHE-1 activation in determining excessive cardiac hypertrophy. Pflugers Arch 462:733–743
Cox GA, Lutz CM, Yang CL, Biemesderfer D, Bronson RT, Fu A, Aronson PS, Noebels JL, Frankel WN (1997) Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 91:139–148
Chai W, Danser AH (2006) Why are mineralocorticoid receptor antagonists cardioprotective? N-S Arch Pharmacol 374:153–162
Darmellah A, Baetz D, Prunier F, Tamareille S, Rucker-Martin C, Feuvray D (2007) Enhanced activity of the myocardial Na+/H+ exchanger contributes to left ventricular hypertrophy in the Goto-Kakizaki rat model of type 2 diabetes: critical role of Akt. Diabetologia 50:1335–1344
De Giusti VC, Nolly MB, Yeves AM, Caldiz CI, Villa-Abrille MC, Chiappe de Cingolani G, Ennis IL, Cingolani HE, Aiello EA (2011) Aldosterone stimulates the cardiac Na+/H+ exchanger via transactivation of the epidermal growth factor receptor. Hypertension 58:912–919
Dhein S (1998) Gap junction channels in the cardiovascular system: pharmacological and physiological modulation. Trends Pharmacol Sci 19:229–241
Dorrance AM, Osborn HL, Grekin R, Webb RC (2001) Spironolactone reduces cerebral infarct size and EGF-receptor mRNA in stroke-prone rats. Am J Physiol Regul Integr Comp Physiol 281:R944–R950
Dulce RA, Hurtado C, Ennis IL, Garciarena CD, Alvarez MC, Caldiz C, Pierce GN, Portiansky EL, Chiappe de Cingolani GE, Camilion de Hurtado MC (2006) Endothelin-1 induced hypertrophic effect in neonatal rat cardiomyocytes: involvement of Na+/H+ and Na+/Ca2+ exchangers. J Mol Cell Cardiol 41:807–815
Duquesnes N, Vincent F, Morel E, Lezoualc'h F, Crozatier B (2009) The EGF receptor activates ERK but not JNK Ras-dependently in basal conditions but ERK and JNK activation pathways are predominantly Ras-independent during cardiomyocyte stretch. Int J Biochem Cell Biol 41:1173–1181
Ebata S, Muto S, Okada K, Nemoto J, Amemiya M, Saito T, Asano Y (1999) Aldosterone activates Na+/H+ exchange in vascular smooth muscle cells by nongenomic and genomic mechanisms. Kidney Int 56:1400–1412
Engelhardt S, Hein L, Keller U, Klambt K, Lohse MJ (2002) Inhibition of Na+-H+ exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)-adrenergic receptor transgenic mice. Circ Res 90:814–819
Ennis IL, Alvarez BV, Camilion de Hurtado MC, Cingolani HE (1998) Enalapril induces regression of cardiac hypertrophy and normalization of pHi regulatory mechanisms. Hypertension 31:961–967
Ennis IL, Escudero EM, Console GM, Camihort G, Dumm CG, Seidler RW, Camilion de Hurtado MC, Cingolani HE (2003) Regression of isoproterenol-induced cardiac hypertrophy by Na+/H+ exchanger inhibition. Hypertension 41:1324–1329
Ennis IL, Garciarena CD, Escudero EM, Perez NG, Dulce RA, Camilion de Hurtado MC, Cingolani HE (2007) Normalization of the calcineurin pathway underlies the regression of hypertensive hypertrophy induced by Na+/H+exchanger-1 (NHE-1) inhibition. Can J Physiol Pharm 85:301–310
Esposito G, Rapacciuolo A, Naga Prasad SV, Takaoka H, Thomas SA, Koch WJ, Rockman HA (2002) Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation 105(1):85–92
Fabiato A, Fabiato F (1978) Effects of pH on the myofilaments and the sarcoplasmic reticulum of skinned cells from cardiace and skeletal muscles. J Physiol 276:233–255
Fantinelli JC, Cingolani HE, Mosca SM (2006) Na+/H+ exchanger inhibition at the onset of reperfusion decreases myocardial infarct size: role of reactive oxygen species. Cardiovasc Pathol 15:179–184
Fliegel L, Karmazyn M (2004) The cardiac Na-H exchanger: a key downstream mediator for the cellular hypertrophic effects of paracrine, autocrine and hormonal factors. Biochem Cell Biol 82:626–635
Frey N, Barrientos T, Shelton JM, Frank D, Rütten H, Gehring D, Kuhn C, Lutz M, Rothermel B, Bassel-Duby R, Richardson JA, Katus HA, Hill JA, Olson EN (2004) Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med 10(12):1336–1343
Fujisawa G, Okada K, Muto S, Fujita N, Itabashi N, Kusano E, Ishibashi S (2003) Na/H exchange isoform 1 is involved in mineralocorticoid/salt-induced cardiac injury. Hypertension 41:493–498
Gandhi MS, Kamalov G, Shahbaz AU, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT (2011) Cellular and molecular pathways to myocardial necrosis and replacement fibrosis. Heart Failure Rev 16:23–34
Garciarena CD, Caldiz CI, Correa MV, Schinella GR, Mosca SM, Chiappe de Cingolani GE, Cingolani HE, Ennis IL (2008) Na+/H+ exchanger-1 inhibitors decrease myocardial superoxide production via direct mitochondrial action. J Appl Physiol 105:1706–1713
Giordano FJ (2005) Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest 115:500–508
Gomez-Sanchez EP, Ahmad N, Romero DG, Gomez-Sanchez CE (2004) Origin of aldosterone in the rat heart. Endocrinology 145:4796–4802
Gray RP, McIntyre H, Sheridan DS, Fry CH (2001) Intracellular sodium and contractile function in hypertrophied human and guinea-pig myocardium. Pflugers Arch 442:117–123
Gros R, Ding Q, Sklar LA, Prossnitz EE, Arterburn JB, Chorazyczewski J, Feldman RD (2011) GPR30 expression is required for the mineralocorticoid receptor-independent rapid vascular effects of aldosterone. Hypertension 57:442–451
Grossmann C, Gekle M (2007) Non-classical actions of the mineralocorticoid receptor: misuse of EGF receptors? Mol Cell Endocrinol 277:6–12
Grossmann C, Gekle M (2008) Nongenotropic aldosterone effects and the EGFR: interaction and biological relevance. Steroids 73:973–978
Grossmann C, Gekle M (2009) New aspects of rapid aldosterone signaling. Mol Cell Endocrinol 308:53–62
Grossmann C, Krug AW, Freudinger R, Mildenberger S, Voelker K, Gekle M (2007) Aldosterone-induced EGFR expression: interaction between the human mineralocorticoid receptor and the human EGFR promoter. Am J Physiol Endocrinol Metab 292:E1790–E1800
Grossmann C, Husse B, Mildenberger S, Schreier B, Schuman K, Gekle M (2010) Colocalization of mineralocorticoid and EGF receptor at the plasma membrane. Biochim Biophys Acta 1803:584–590
Haworth RS, McCann C, Snabaitis AK, Roberts NA, Avkiran M (2003) Stimulation of the plasma membrane Na+/H+ exchanger NHE1 by sustained intracellular acidosis. Evidence for a novel mechanism mediated by the ERK pathway. J Biol Chem 278:31676–31684
Hofmann PA, Fuchs F (1988) Bound calcium and force development in skinned cardiac muscle bundles: effect of sarcomere length. J Mol Cell Cardiol 20:667–677
Hongo K, White E, Le Guennec JY, Orchard CH (1996) Changes in [Ca2+]i, [Na+]i and Ca2+ current in isolated rat ventricular myocytes following an increase in cell length. J Physiol 491(Pt 3):609–619
Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A, Nogami A, Murumo F, Hiroe M (1993) Endothelin-1 is an autocrine/paracrine factor in the mechanism of angiotensin II-induced hypertrophy in cultured rat cardiomyocytes. J Clin Invest 92:398–403
Javadov S, Huang C, Kirshenbaum L, Karmazyn M (2005) NHE-1 inhibition improves impaired mitochondrial permeability transition and respiratory function during postinfarction remodelling in the rat. J Mol Cell Cardiol 38:135–143
Javadov S, Baetz D, Rajapurohitam V, Zeidan A, Kirshenbaum LA, Karmazyn M (2006) Antihypertrophic effect of Na+/H+ exchanger isoform 1 inhibition is mediated by reduced mitogen-activated protein kinase activation secondary to improved mitochondrial integrity and decreased generation of mitochondrial-derived reactive oxygen species. J Pharmacol Exp Ther 317:1036–1043
Jorde UP, Vittorio T, Katz SD, Colombo PC, Latif F, Le Jemtel TH (2002) Elevated plasma aldosterone levels despite complete inhibition of the vascular angiotensin-converting enzyme in chronic heart failure. Circulation 106:1055–1057
Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, Phillips MI (2002) Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation 106:909–912
Karmazyn M (1999) The role of the myocardial sodium-hydrogen exchanger in mediating ischemic and reperfusion injury. From amiloride to cariporide. Ann N Y Acad Sci 874:326–334
Karmazyn M, Liu Q, Gan XT, Brix BJ, Fliegel L (2003) Aldosterone increases NHE-1 expression and induces NHE-1-dependent hypertrophy in neonatal rat ventricular myocytes. Hypertension 42:1171–1176
Kentish JC, Wrzosek A (1998) Changes in force and cytosolic Ca2+ concentration after length changes in isolated rat ventricular trabeculae. J Physiol 506(Pt 2):431–444
Kim DH, Rossi JJ (2007) Strategies for silencing human disease using RNA interference. Nat Rev 8:173–184
Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Abe Y (2005a) Mitochondria-derived reactive oxygen species and vascular MAP kinases: comparison of angiotensin II and diazoxide. Hypertension 45:438–444
Kimura S, Zhang GX, Nishiyama A, Shokoji T, Yao L, Fan YY, Rahman M, Suzuki T, Maeta H, Abe Y (2005b) Role of NAD(P)H oxidase- and mitochondria-derived reactive oxygen species in cardioprotection of ischemic reperfusion injury by angiotensin II. Hypertension 45:860–866
Kizana E, Cingolani E, Marban E (2009) Non-cell-autonomous effects of vector-expressed regulatory RNAs in mammalian heart cells. Gene Ther 16:1163–1168
Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH (1991) Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 114:345–352
Krieg T, Cui L, Qin Q, Cohen MV, Downey JM (2004) Mitochondrial ROS generation following acetylcholine-induced EGF receptor transactivation requires metalloproteinase cleavage of proHB-EGF. J Mol Cell Cardiol 36:435–443
Krug AW, Pojoga LH, Williams GH, Adler GK (2011) Cell membrane-associated mineralocorticoid receptors? New evidence. Hypertension 57:1019–1025
Kusumoto K, Haist JV, Karmazyn M (2001) Na+/H+ exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am J Physiol 280:H738–H745
Le Moellic C, Ouvrard-Pascaud A, Capurro C, Cluzeaud F, Fay M, Jaisser F, Farman N, Blot-Chabaud M (2004) Early nongenomic events in aldosterone action in renal collecting duct cells: PKCalpha activation, mineralocorticoid receptor phosphorylation, and cross-talk with the genomic response. J Am Soc Nephrol 15:1145–1160
Lemarie CA, Paradis P, Schiffrin EL (2008) New insights on signaling cascades induced by cross-talk between angiotensin II and aldosterone. J Mol Med 86:673–678
Lemarie CA, Simeone SM, Nikonova A, Ebrahimian T, Deschenes ME, Coffman TM, Paradis P, Schiffrin EL (2009) Aldosterone-induced activation of signaling pathways requires activity of angiotensin type 1a receptors. Circ Res 105:852–859
Li J, Yatani A, Kim SJ, Takagi G, Irie K, Zhang Q, Karoor V, Hong C, Yang G, Sadoshima J, Depre C, Vatner DE, West MJ, Vatner SF (2003) Neurally-mediated increase in calcineurin activity regulates cardiac contractile function in absence of hypertrophy. Cardiovasc Res 59:649–657
Luers C, Fialka F, Elgner A, Zhu D, Kockskamper J, von Lewinski D, Pieske B (2005) Stretch-dependent modulation of [Na+]i, [Ca2+]i, and pHi in rabbit myocardium – a mechanism for the slow force response. Cardiovasc Res 68:454–463
Lloyd-Jones DM, Larson MG, Leip EP, Beiser A, D'Agostino RB, Kannel WB, Murabito JM, Vasan RS, Benjamin EJ, Levy D (2002) Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation 106:3068–3072
Manjunath N, Wu H, Subramanya S, Shankar P (2009) Lentiviral delivery of short hairpin RNAs. Adv Drug Deliver Rev 61:732–745
Matsui S, Satoh H, Kawashima H, Nagasaka S, Niu CF, Urushida T, Katoh H, Watanabe Y, Hayashi H (2007) Non-genomic effects of aldosterone on intracellular ion regulation and cell volume in rat ventricular myocytes. Can J Physiol Pharm 85:264–273
Mattiazzi AR, Cingolani HE, de Castuma ES (1979) Relationship between calcium and hydrogen ions in heart muscle. Am J Physiol 237:H497–H503
McKelvie RS, Yusuf S, Pericak D, Avezum A, Burns RJ, Probstfield J, Tsuyuki RT, White M, Rouleau J, Latini R, Maggioni A, Young J, Pogue J (1999) Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 100:1056–1064
Mello CC, Conte D Jr (2004) Revealing the world of RNA interference. Nature 431:338–342
Mentzer RM Jr, Bartels C, Bolli R, Boyce S, Buckberg GD, Chaitman B, Haverich A, Knight J, Menasche P, Myers ML, Nicolau J, Simoons M, Thulin L, Weisel RD (2008) Sodium-hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg 85:1261–1270
Mihailidou AS, Loan Le TY, Mardini M, Funder JW (2009) Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension 54:1306–1312
Morgan PE, Correa MV, Ennis IL, Diez AA, Perez NG, Cingolani HE (2011) Silencing of sodium/hydrogen exchanger in the heart by direct injection of naked siRNA. J Appl Physiol 111(2):566–572
Mureddu GF, Cioffi G, Stefenelli C, Boccanelli A, de Simone G (2009) Compensatory or inappropriate left ventricular mass in different models of left ventricular pressure overload: comparison between patients with aortic stenosis and arterial hypertension. J Hypertens 27:642–649
Nagata K, Somura F, Obata K, Odashima M, Izawa H, Ichihara S, Nagasaka T, Iwase M, Yamada Y, Nakashima N, Yokota M (2002) AT1 receptor blockade reduces cardiac calcineurin activity in hypertensive rats. Hypertension 40(2):168–174
Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S (2008) Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res 103:891–899
Orchard CH, Kentish JC (1990) Effects of changes of pH on the contractile function of cardiac muscle. Am J Physiol 258:C967–C981
Parmley WW, Chuck L (1973) Length-dependent changes in myocardial contractile state. Am J Physiol 224:1195–1199
Perez NG, Alvarez BV, Camilion de Hurtado MC, Cingolani HE (1995) pHi regulation in myocardium of the spontaneously hypertensive rat. Compensated enhanced activity of the Na+-H+ exchanger. Circ Res 77:1192–1200
Perez NG, de Hurtado MC, Cingolani HE (2001) Reverse mode of the Na+–Ca2+ exchange after myocardial stretch: underlying mechanism of the slow force response. Circ Res 88:376–382
Perez NG, Villa-Abrille MC, Aiello EA, Dulce RA, Cingolani HE, Camilion de Hurtado MC (2003) A low dose of angiotensin II increases inotropism through activation of reverse Na(+)/Ca(2+) exchange by endothelin release. Cardiovasc Res 60:589–597
Perez NG, Nolly MB, Roldan MC, Villa-Abrille MC, Cingolani E, Portiansky EL, Alvarez BV, Ennis IL, Cingolani HE (2011) Silencing of NHE-1 blunts the slow force response to myocardial stretch. J Appl Physiol 111:874–880
Perez NG, Nolly MB, Roldan MC, Villa-Abrille MC, Cingolani E, Portiansky EL, Alvarez BV, Ennis IL, Cingolani HE (2011b) Silencing of NHE-1 blunts the slow force response to myocardial stretch. J Appl Physiol
Petrecca K, Atanasiu R, Grinstein S, Orlowski J, Shrier A (1999) Subcellular localization of the Na+/H+ exchanger NHE1 in rat myocardium. Am J Physiol 276:H709–H717
Pimentel DR, Adachi T, Ido Y, Heibeck T, Jiang B, Lee Y, Melendez JA, Cohen RA, Colucci WS (2006) Strain-stimulated hypertrophy in cardiac myocytes is mediated by reactive oxygen species-dependent Ras S-glutathiolation. J Mol Cell Cardiol 41:613–622
Pitt B, White H, Nicolau J, Martinez F, Gheorghiade M, Aschermann M, van Veldhuisen DJ, Zannad F, Krum H, Mukherjee R, Vincent J (2005) Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 46:425–431
Rosenblueth A, Alanis J, Lopez E, Rubio R (1959) The adaptation of ventricular muscle to different circulatory conditions. Arch Int Physiol Biochim 67:358–373
Rothstein EC, Byron KL, Reed RE, Fliegel L, Lucchesi PA (2002) H(2)O(2)-induced Ca2+ overload in NRVM involves ERK1/2 MAP kinases: role for an NHE-1-dependent pathway. Am J Physiol 283:H598–H605
Sabri A, Byron KL, Samarel AM, Bell J, Lucchesi PA (1998) Hydrogen peroxide activates mitogen-activated protein kinases and Na+–H+ exchange in neonatal rat cardiac myocytes. Circ Res 82:1053–1062
Sadoshima J, Xu Y, Slayter HS, Izumo S (1993) Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 75:977–984
Sah R, Oudit GY, Nguyen TT, Lim HW, Wickenden AD, Wilson GJ, Molkentin JD, Backx PH (2002) Inhibition of calcineurin and sarcolemmal Ca2+ influx protects cardiac morphology and ventricular function in K(v)4.2 N transgenic mice. Circulation 105:1850–1856
Sand C, Peters SL, Pfaffendorf M, van Zwieten PA (2003) The influence of endogenously generated reactive oxygen species on the inotropic and chronotropic effects of adrenoceptor and ET-receptor stimulation. N-S Arch Pharmacol 367:635–639
Sarnoff SJ, Mitchell JH, Gilmore JP, Remensnyder JP (1960) Homeometric autoregulation in the heart. Circ Res 8:1077–1091
Schussheim AE, Radda GK (1995) Altered Na+-H+ - exchange activity in the spontaneously hypertensive perfused rat heart. J Mol Cell Cardiol 27:1475–1481
Siczkowski M, Davies JE, Ng LL (1995) Na+-H+ exchanger isoform 1 phosphorylation in normal Wistar-Kyoto and spontaneously hypertensive rats. Circ Res 76:825–831
Silvestre JS, Robert V, Heymes C, Aupetit-Faisant B, Mouas C, Moalic JM, Swynghedauw B, Delcayre C (1998) Myocardial production of aldosterone and corticosterone in the rat. Physiological regulation. J Biol Chem 273:4883–4891
Silvestre JS, Heymes C, Oubenaissa A, Robert V, Aupetit-Faisant B, Carayon A, Swynghedauw B, Delcayre C (1999) Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation 99:2694–2701
Snabaitis AK, Hearse DJ, Avkiran M (2002) Regulation of sarcolemmal Na + /H + exchange by hydrogen peroxide in adult rat ventricular myocytes. Cardiovasc Res 53:470–480
Sugden PH, Clerk A (2006) Oxidative stress and growth-regulating intracellular signaling pathways in cardiac myocytes. Antioxidants and redox signaling 8:2111–2124
Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L (1990) Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 82:1730–1736
Szokodi I, Kerkela R, Kubin AM, Sarman B, Pikkarainen S, Konyi A, Horvath IG, Papp L, Toth M, Skoumal R, Ruskoaho H (2008) Functionally opposing roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the regulation of cardiac contractility. Circulation 118:1651–1658
Takahashi Y, Watanabe H, Murakami M, Ohba T, Radovanovic M, Ono K, Iijima T, Ito H (2007) Involvement of transient receptor potential canonical 1 (TRPC1) in angiotensin II-induced vascular smooth muscle cell hypertrophy. Atherosclerosis 195:287–296
Takeda Y, Yoneda T, Demura M, Miyamori I and Mabuchi H (2000) Cardiac aldosterone production in genetically hypertensive rats. Hypertension 36:495–500
Theroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, Tognoni G, White HD, Willerson JT, Jessel A (2000) Inhibition of the sodium-hydrogen exchanger with cariporide to prevent myocardial infarction in high-risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation 102:3032–3038
Valiunas V, Polosina YY, Miller H, Potapova IA, Valiuniene L, Doronin S, Mathias RT, Robinson RB, Rosen MR, Cohen IS, Brink PR (2005) Connexin-specific cell-to-cell transfer of short interfering RNA by gap junctions. J Physiol 568:459–468
Villa-Abrille MC, Caldiz CI, Ennis IL, Nolly MB, Casarini MJ, Chiappe de Cingolani GE, Cingolani HE, Perez NG (2010) The Anrep effect requires transactivation of the epidermal growth factor receptor. J Physiol 588:1579–1590
Villafuerte FC, Swietach P, Vaughan-Jones RD (2007) Common inhibitors of membrane H+-transport also inhibit carbonic anhydrase The FASEB Journal 21. (Abstract)
von Anrep G (1912) On the part played by the suprarenals in the normal vascular reactions of the body. J Physiol 45:307–317
von Lewinski D, Stumme B, Fialka F, Luers C, Pieske B (2004) Functional relevance of the stretch-dependent slow force response in failing human myocardium. Circ Res 94:1392–1398
Wang Y, Meyer JW, Ashraf M, Shull GE (2003) Mice with a null mutation in the NHE1 Na+-H+ exchanger are resistant to cardiac ischemia-reperfusion injury. Circ Res 93:776–782
Ward ML, Williams IA, Chu Y, Cooper PJ, Ju YK, Allen DG (2008) Stretch-activated channels in the heart: contributions to length-dependence and to cardiomyopathy. Prog Biophys Mol Biol 97:232–249
Weber KT, Brilla CG (1991) Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation 83:1849–1865
Wetzker R, Bohmer FD (2003) Transactivation joins multiple tracks to the ERK/MAPK cascade. Nat Rev Mol Cell Biol 4:651–657
Wolvetang EJ, Pera MF, Zuckerman KS (2007) Gap junction mediated transport of shRNA between human embryonic stem cells. Biochem Biophys Res Commun 363:610–615
Wu X, Eder P, Chang B, Molkentin JD (2010) TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci USA 107:7000–7005
Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiroi Y, Mizuno T, Maemura K, Kurihara H, Aikawa R, Takano H, Yazaki Y (1996) Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem 271:3221–3228
Yokoyama H, Gunasegaram S, Harding SE, Avkiran M (2000) Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J Am Coll Cardiol 36:534–540
Zannad F, McMurray JJ, Drexler H, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pitt B (2010) Rationale and design of the Eplerenone in Mild Patients Hospitalization And SurvIval Study in Heart Failure (EMPHASIS-HF). Eur J Heart Fail 12:617–622
Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B (2011) Eplerenone in patients with systolic heart failure and mild symptoms. New Engl J Med 364:11–21
Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, Linssen G, Tebbe U, Schroder R, Tiemann R, Machnig T, Neuhaus KL (2001) The Na+/H+ exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol 38:1644–1650
Zhang YH, Dingle L, Hall R, Casadei B (2009) The role of nitric oxide and reactive oxygen species in the positive inotropic response to mechanical stretch in the mammalian myocardium. Biochim Biophys Acta 1787:811–817
Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ (2000) Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med 192:1001–1014
Zou Y, Yamazaki T, Nakagawa K, Yamada H, Iriguchi N, Toko H, Takano H, Akazawa H, Nagai R, Komuro I (2002) Continuous blockade of L-type Ca2+ channels suppresses activation of calcineurin and development of cardiac hypertrophy in spontaneously hypertensive rats. Hypertens Res 25:117–124
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2012 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Cingolani, H.E. et al. (2012). Early Activation of Intracellular Signals after Myocardial Stretch: Anrep Effect, Myocardial Hypertrophy and Heart Failure. In: Kamkin, A., Lozinsky, I. (eds) Mechanically Gated Channels and their Regulation. Mechanosensitivity in Cells and Tissues, vol 6. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-5073-9_12
Download citation
DOI: https://doi.org/10.1007/978-94-007-5073-9_12
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-007-5072-2
Online ISBN: 978-94-007-5073-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)