
Abstract
Fibrosis is a fundamental component of the adverse structural remodeling of myocardium present in the failing heart. Replacement fibrosis appears at sites of previous cardiomyocyte necrosis to preserve the structural integrity of the myocardium, but not without adverse functional consequences. The extensive nature of this microscopic scarring suggests cardiomyocyte necrosis is widespread and the loss of these contractile elements, combined with fibrous tissue deposition in the form of a stiff in-series and in-parallel elastic elements, contributes to the progressive failure of this normally efficient muscular pump. Cellular and molecular studies into the signal-transducer-effector pathway involved in cardiomyocyte necrosis have identified the crucial pathogenic role of intracellular Ca2+ overloading and subsequent induction of oxidative stress, predominantly confined within its mitochondria, to be followed by the opening of the mitochondrial permeability transition pore that leads to the destruction of these organelles and cells. It is now further recognized that Ca2+ overloading of cardiac myocytes and mitochondria serves as a prooxidant and which is counterbalanced by an intrinsically coupled Zn2+ entry serving as antioxidant. The prospect of raising antioxidant defenses by increasing intracellular Zn2+ with adjuvant nutriceuticals can, therefore, be preferentially exploited to uncouple this intrinsically coupled Ca2+–Zn2+ dyshomeostasis. Hence, novel yet simple cardioprotective strategies may be at hand that deserve to be further explored.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
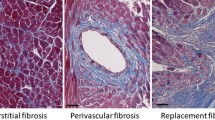
Fibrosis, including microscopic scarring, is a fundamental component of the adverse structural remodeling found in the myocardium of the failing human heart [1, 2]. Scarring, a morphologic footprint of earlier cardiomyocyte necrosis, serves to replace lost contractile cells and thereby plays a vital role in preserving myocardial structure and function. The extensive distribution of this replacement fibrosis suggests a widespread and ongoing necrosis of cardiomyocytes. Apoptosis also occurs in the failing heart, but to a lesser extent, often involving such noncardiomyocytes as macrophages and endothelial cells [3]. Furthermore, programmed cell death begets neither inflammatory cells nor fibroblast responses. As a consequence, fibrous tissue does not appear at the site of lost myocytes and, therefore, apoptosis has been referred to as a sterile form of cell death [4, 5]. The cumulative loss of contractile elements, together with the deposition of fibrous tissue, stiff in-series and in-parallel elastic elements composed primarily of type I fibrillar collagen having the tensile strength of steel, each contributes to the progressive failure of this previously efficient muscular pump during systolic and/or diastolic phases of the cardiac cycle [6].
Elevations in serum troponins, which do not stem from an acute coronary event or significant renal dysfunction, that compromises urinary troponin excretion have been reported in patients hospitalized because of the symptoms and signs of congestive heart failure (CHF) [7–15]. This would be consistent with an ongoing necrotic loss of cardiomyocytes. Coronary microembolization with microinfarcts could be held responsible for the rise in serum troponins [16]. We speculate neurohormonal activation could be another plausible mechanism. In either case, these serum-derived biomarkers of cardiomyocyte necrosis are associated with increased in-hospital and overall cardiac morbidity and mortality. Insights into cellular and molecular pathways involved in ongoing cardiomyocyte necrosis are quintessential in developing novel cardioprotective strategies that would salvage them and prevent myocardial fibrosis.
The failing heart and congestive heart failure syndrome
The heart fails as the contractility of this muscular pump declines and it becomes unable to propel sufficient amounts of blood and nutrients into the circulation for their delivery to metabolizing tissues at a rate commensurate with their prevailing metabolic demands. Likewise, the heart also fails when its ability to receive blood during diastolic filling is compromised by abnormalities in myocardial relaxation and/or tissue stiffness, and consequently atrial and ventricular filling pressures are inappropriately elevated and the displacement of blood is suboptimal due to the inadequately stretched ventricle. Systemic blood flow can, therefore, be impaired by ventricular dysfunction in either systole or diastole. In turn, renal perfusion is correspondingly reduced setting the primary prerequisite for the appearance of CHF.
In order to sustain a steady-state filtration fraction at equilibrium over a wide range of conditions where the intake of water is either plentiful or limited, renal blood flow normally constitutes 20% of the cardiac output. However, when renal perfusion is reduced, juxtaglomerular cells elaborate renin leading to an ensuing activation of the renin–angiotensin–aldosterone system (RAAS). A concomitant activation of the hypothalamic–pituitary–adrenal (HPA) axis stimulates the adrenergic nervous system (ANS) and release of catecholamines. The HPA axis also contributes to adrenocorticotropin (ACTH)-mediated release of glucocorticoids and mineralocorticoids by the adrenal glands. Activation of the RAAS and ANS normally involves short-term, physiologic homeostatic responses to reduced dietary Na+ intake or intravascular volume contraction. In the patient with heart failure, however, sustained homeostatic neurohormonal responses beget dyshomeostasis, which over time is expressed as an inappropriate (relative to dietary Na+ and intravascular volume) salt-avid state. The accompanying retention of salt and water, and subsequent expansion of intra- and later extravascular volumes, leads to the symptoms and signs of the clinical syndrome CHF. CHF occurs when these homeostatic responses are inappropriate and persistent. Treatment of the decompensated, often-hospitalized, patient with CHF will restore euvolemia and ameliorate their signs and symptoms returning them to a state of compensation. Episodes of neurohormonal activation that beget CHF disrupt the stable state of compensation and as such represent an acute stressor state, which proves integral to the pathologic remodeling of myocardium. Less well recognized are adverse consequences of effector hormones of the RAAS and ANS that extend beyond salt and water retention to involve a dyshomeostasis of several cations, including K+, Ca2+, Mg2+, and Zn2+, which have been shown to adversely influence intracellular homeostasis and survival of cardiomyocytes [17–21].
Acute stressor state
Bodily injury, such as accompanies burns, head trauma, subarachnoid hemorrhage, or acute myocardial infarction, represents a hyperadrenergic acute stressor state with marked elevations in circulating catecholamines. Acting through β2 receptors, epinephrine and norepinephrine promote the translocation of K+ from blood into the intracellular compartment of various cells, including cardiac myocytes and peripheral blood mononuclear cells (PBMC), leading to hypokalemia [22–24]. Likewise, plasma-ionized hypocalcemia and hypomagnesemia, and hypozincemia are common findings at the time of or soon after admission in patients hospitalized in the medical/surgical intensive care unit or the coronary care unit [25, 26]. The magnitude to which their respective plasma concentrations fall correlates well with the severity of injury, and therefore they may have predictive roles as relevant clinical biomarkers of poor prognosis. Necrosis of cardiomyocytes, with an elevation in serum troponins (albeit modest vis-à-vis myocardial infarction), occurs in response to the marked rise in circulating catecholamines and which can be ascertained postmortem from the morphologic evidence of contraction band necrosis and microscopic scarring of the right and left heart.
Acute stressor state: an animal model
The cellular and molecular pathways leading to this adverse myocardial remodeling during the systemic hyperadrenergic state have been investigated in animal models. In rodents, the administration of a single dose of a catecholamine leads to acute intracellular Ca2+ overloading of cardiac myocytes and mitochondria, as well as the concomitant generation of reactive oxygen and nitrogen species that overwhelm antioxidant defenses [27]. Cardiomyocyte necrosis, for example, occurs within hours of isoproterenol infusion and can be prevented by cotreatment with either a β1 adrenergic receptor antagonist or Ca2+ channel blocker [20, 21, 28]. The induction of oxidative stress that accompanies consequent excessive intracellular Ca2+ accumulation (EICA) begins in mitochondria with the necrotic cell death pathway initiated by the opening of the mitochondrial permeability transition pore (mPTP) regulated by cyclophilin D [21, 27, 29, 30]. Cyclophilin D, a prolyl isomerase located within the matrix of mitochondria, is required for mediating EICA- and oxidative stress–induced cardiomyocyte necrosis (vis-à-vis apoptosis). Its pathophysiologic significance has been underscored by cyclophilin D null mice which are protected from cardiomyocyte necrosis due to ischemia/reperfusion (I/R) injury, including Ca2+ overloading and oxidative damage. Contrariwise, cyclophilin D-overexpressing mice have spontaneous cardiomyocyte necrosis with mitochondrial swelling [31, 32]. Inhibitors of mPTP opening of cyclophilin D have each been proven cardioprotective in the isoproterenol-induced experimental model, as well as in I/R-induced injury, another example of acute EICA [33–35].
Chronic stressor state
Persistent neurohormonal activation involving the RAAS and ANS occurs with protracted CHF, especially in newly diagnosed and previously untreated patients or those who have discontinued taking their medications, or in whom the dosage of pharmacologic agents interfering with these effector hormones is suboptimal. A dyshomeostasis of divalent cations is frequently found in patients hospitalized with decompensated biventricular failure due to a dilated (idiopathic) cardiomyopathy [36–45]. These include plasma-ionized hypocalcemia and hypomagnesemia that account for secondary hyperparathyroidism (SHPT) and elevated plasma parathyroid hormone (PTH) levels, together with hypovitaminosis D that further compromises Ca2+ balance, hypozincemia, and hyposelenemia. Systemic evidence of oxidative stress, expressed as elevated plasma malondialdehyde and 8-isoprostane, has also been reported in patients with CHF [46–48].
Chronic stressor state: an experimental model
Eight-week-old male Sprague–Dawley rats are uninephrectomized, followed by the implantation of an osmotic minipump containing aldosterone. Infusion of aldosterone (0.75 μg/h) raises their plasma levels to those found in CHF which are inappropriate relative to dietary Na+ intake. Drinking water is fortified with 1% NaCl and 0.4% KCl to prevent hypokalemia. At week 1 of aldosterone/salt treatment (ALDOST), animals are clinically healthy and the myocardium appears normal by light microscopy. This preclinical stage gives way to a clinical stage with anorexia and a failure to gain weight at week 2 and beyond. Cardiac pathology first appears at week 4. This pathologic stage features microscopic scarring scattered throughout both the right and left atria and ventricles [49]. A perivascular/interstitial fibrosis involving the coronary, renal, and mesenteric circulations is also found. This topic has been reviewed extensively elsewhere [50, 51].
A series of studies have addressed the relevance of hemodynamic factors that could potentially contribute to cardiac fibrosis and have concluded those are not directly involved (reviewed in [52]). This viewpoint was primarily based on: (a) the presence of fibrosis in nonpressure-overloaded right atria and ventricle; (b) the absence of fibrosis when the LV pressure overload is created by infrarenal aortic banding without subsequent RAAS activation or when treatment is based on aldosterone together with a low-Na+ diet, or when 1% NaCl alone is given; (c) the prevention of fibrosis with either a small (nondepressor) or large (depressor) dose of spironolactone, which respectively fails to or does prevent hypertension; (d) an intracerebroventricular infusion of a mineralocorticoid receptor antagonist that prevents hypertension, but not fibrosis [53]; and (e) when a cardiospecific upregulation of aldosterone synthase accounts for increased tissue levels of aldosterone unaccompanied by cardiac fibrosis [54]. Thus, the evidence gathered to date indicates the adverse remodeling of myocardium during ALDOST is both (a) independent of hypertension and (b) unrelated to plasma or tissue-derived aldosterone per se, but some other circulating factor that accompanies aldosteronism (vide infra).
Cellular and molecular pathways leading to cardiomyocyte necrosis
Pathways accounting for cardiomyocyte necrosis and subsequent scarring of myocardium found at 4 weeks ALDOST have been examined and the identity and pathogenic role of circulating factors elucidated.
Oxidative stress
Evidence of oxidative stress in the myocardium is found during chronic mineralocorticoidism [55–59]. This includes (a) the presence of 3-nitrotyrosine, a byproduct of the reaction involving superoxide and nitric oxide; (b) an activation of the gp91phox subunit of NADPH oxidase found in inflammatory cells invading the injured myocardium that contributes to superoxide generation; (c) upregulated redox-sensitive nuclear transcription factor (NF)-κB and a proinflammatory gene cascade it regulates that includes intercellular adhesion molecule (ICAM)-1, monocyte chemoattractant protein (MCP)-1, and tumor necrosis factor (TNF)-alpha; and (d) increased tissue levels of 8-isoprostane and malondialdehyde, biomarkers of lipid peroxidation [58, 59]. There is also considerable evidence of oxidative stress in blood and urine consistent with the systemic nature of an altered redox state during chronic aldosteronism.
Intracellular Ca2+ overloading
Our hypothesis for the induction of oxidative stress during ALDOST would draw upon Albrecht Fleckenstein’s original concept that intracellular Ca2+ overloading of the heart is an integral and adverse pathophysiologic feature leading to myocardial necrosis [60]. In rats receiving 1 and 4 weeks ALDOST, we monitored intracellular Ca2+ levels in several tissues that included the heart and PBMC. We found increased Ca2+ levels in the myocardium and PBMC during preclinical, clinical, and pathologic stages, accompanied by biomarker evidence of oxidative stress that included increased levels of malondialdehyde and 8-isoprostane in the heart and increased H2O2 production by PBMC [59, 61–63].
Calcium and magnesium dyshomeostasis
Why intracellular Ca2+ overloading occurred during ALDOST was next addressed. Elevations in dietary Na+, albeit modest but inappropriate during ALDOST, are accompanied by increased tubular Na+ and, in turn, elevated urinary concentrations of Ca2+ and Mg2+. Aldosterone promotes epithelial cell Na+ channel-mediated Na+ reabsorption without influencing Ca2+ and Mg2+ excretion, which in turn accounts for the marked urinary losses of Ca2+ and Mg2+ [61]. A similar scenario unfolds in the Na+ channels of the colon’s epithelial cells that represent another site of high-density aldosterone receptor binding. The fecal excretion of Ca2+ and Mg2+, in fact, is many-fold greater than their urinary losses [61].
Secondary hyperparathyroidism (SHPT)
Metabolic studies accounted for the marked increase in urinary and fecal excretion of Ca2+ and Mg2+ during ALDOST, which leads to plasma-ionized hypocalcemia and hypomagnesemia. The calcium-sensing receptor of the parathyroid glands, in turn, responds to hypocalcemia with increased secretion of PTH. Accordingly, plasma PTH levels rise [61] and SHPT is evidenced by marked and progressive reductions in bone mineral density and bone strength [64]. We therefore hypothesized the intracellular Ca2+ overloading and induction of oxidative stress that accompanies ALDOST leading to cardiomyocyte necrosis, and scarring is mediated by the calcitropic hormone, PTH and not aldosterone (see Fig. 1). It represents an example of the Ca2+ paradox associated with SHPT as characterized by Fujita and Palmieri [65]. Furthermore, PTH-mediated intracellular Ca2+ overloading is coupled to an induction of oxidative stress in diverse tissues that includes cardiomyocytes and their mitochondria, as well as PBMC. The generation of reactive oxygen (ROS) and nitrogen (RNS) species appear to overwhelm their rate of detoxification by the cumulative capacity of antioxidant defenses. In mitochondria, Ca2+ overloading and oxidative stress lead to a nonphysiologic opening of the mPTP, with the ensuing osmotic-based structural and functional degeneration of these organelles that triggers the downhill final common cell death pathway leading to cardiomyocyte necrosis and subsequent replacement fibrosis [66].

The appearance of secondary hyperparathyroidism (SHPT) during aldosterone/salt treatment (ALDOST) is associated with increased excretory losses of Ca2+ and Mg2+, and the consequent appearance of plasma-ionized hypocalcemia and hypomagnesemia. Elevations in parathyroid hormone (PTH) seek to restore extracellular homeostasis of these divalent cations through their resorption from bone, absorption from gut, and reabsorption by kidney promoted by the steroid hormone 1,25(OH)2D3. Paradoxically, PTH elaboration is responsible for intracellular Ca2+ overloading and the induction of oxi-/nitrosative stress, which leads to cardiomyocyte necrosis and consequent replacement fibrosis, or scarring. Adapted from Alsafwah et al. [115]
A series of site-directed, sequential pharmacologic interventions targeted along the cellular–molecular cascades to block downstream events leading to cardiomyocyte necrosis and myocardial scarring were conducted. They collectively validated our hypothesis regarding the pathologic sequelae of events leading to this structural remodeling of myocardium in rats with chronic aldosteronism. These interventions included (a) cotreatment with spironolactone, an aldosterone receptor antagonist that attenuated the enhanced urinary and fecal losses of these cations to prevent hypocalcemia and hypomagnesemia and thereby ensuing SHPT [61]; (b) cotreatment with a Ca2+- and Mg2+-supplemented diet, together with vitamin D3 to enhance Ca2+ absorption, which prevented hypocalcemia and SHPT [67]; (c) parathyroidectomy, performed prior to starting ALDOST, to prevent SHPT [68]; (d) cotreatment with cinacalcet, a calcimimetic that resets the threshold of the parathyroid glands’ Ca2+-sensing receptor to prevent SHPT despite modest hypocalcemia [69]; (e)cotreatment with amlodipine, a Ca2+ channel blocker, which prevents intracellular Ca2+ overloading [62]; and finally (f) cotreatment with N-acetylcysteine, an antioxidant that abrogated oxidative stress [58].
Thus, taken together, the multitude of evidence gathered to date congruently supports the mechanism of PTH-mediated intracellular Ca2+ overloading that leads to the induction of oxidative stress during aldosteronism where ROS and RNS, primarily derived from mitochondria, overwhelm cellular antioxidant defenses. This scenario anticipates whether the overall consequence of an excessive generation of prooxidants or cumulative endogenous antioxidant defenses in combating ROS and RNS had been compromised. In this context, we next addressed plausible association of Zn2+ dyshomeostasis during ALDOST given its importance to these endogenous defenses, including Cu/Zn-superoxide dismutase (SOD).
Zinc dyshomeostasis
Chronic inappropriate excess of aldosterone is accompanied by increased urinary and fecal excretory Zn2+ losses, hypozincemia, and a fall in plasma Cu/Zn-SOD activity [70]. The hyperzincuria seen with ALDOST is related to urinary acidification, which contributes to the consequent metabolic alkalosis of aldosteronism [59]. Also contributory to hypozincemia is a coordinated selective translocation of Zn2+ to the sites of tissue injury, facilitated by corresponding upregulation of a Zn2+-binding protein, metallothionein (MT)-1 in targeted tissues [59, 70].
We also used a 65Zn tracer to systematically monitor Zn2+ kinetics during 1 and 4 weeks of ALDOST. A simultaneous fall in plasma 65Zn and a selective accumulation of 65Zn was found at sites of injury that included its translocation to recently incised skin at week 1 used for osmotic minipump implantation, as well as the injured heart and kidneys at week 4. This intracellular trafficking of 65Zn to injured tissues was facilitated by the upregulation of MT-1 [71]. However, at week 4, there was a decline in 65Zn in healed skin and bone, which serve as Zn2+ reservoirs and participate in resolving acute hypozincemia. Thus, the preferential translocation of circulating Zn2+ to injured tissues contributes to hypozincemia found with ALDOST, where increased tissue Zn2+ is essential in wound healing at these sites [72]. Since dyshomeostasis of Zn2+ proved to be another integral feature of aldosteronism, it became crucial to investigate whether the rise in myocardial tissue Zn2+ involved both its cardiac myocytes and mitochondria.
Zinc and antioxidant defenses
Cardiac myocytes and mitochondria were harvested from rats with 4 weeks of ALDOST, involving the pathologic stage, as well as from experimental controls. We found increased cytosolic free [Zn2+]i in cardiac myocytes and total Zn2+ concentration in mitochondria [59]. The rise in cardiomyocyte Zn2+ was facilitated by the increased expression of membranous Zn2+ transporters. Increased [Zn2+]i serves to augment the antioxidant defenses of cardiomyocytes, including their upregulation of MT-1 and activation of metal-responsive transcription factor (MTF)-1, which encodes genes related to various antioxidant defenses, such as Cu/Zn-SOD, MT-1, and glutathione synthase. Thus, intracellular Zn2+ loading in chronic aldosteronism is contemporaneous with intracellular Ca2+ overloading and relevant biomarkers of oxidative stress [73]. Pathophysiologically and in terms of innate redox states, Zn2+ serves as antioxidant and Ca2+ as prooxidant in our experimental model. This concept offers the prospect of exploiting Zn2+ supplementation as a novel therapeutic strategy to uncouple the intrinsically coupled Ca2+ and Zn2+ dyshomeostasis in favor of increasing [Zn2+]i, thus enhancing the overall endogenous antioxidant defense capacity and attenuating adverse myocardial remodeling.
The efficacy of a Zn2+ supplement in augmenting intracellular [Zn2+]i, and thereby antioxidant defenses, in rats receiving ALDOST was examined using ZnSO4. Cotreatment of ALDOST rats with ZnSO4 prevented hypozincemia and a fall in plasma Cu/Zn-SOD activity, while significantly increasing cardiomyocyte cytosolic [Zn2+]i. It attenuated biomarkers of oxidative stress, such as cardiac 8-isoprostane, and microscopic scarring [59]. Thus, increased tissue Zn2+ in the heart serves as an antioxidant, and intracellular Ca2+ overloading as prooxidant, with cardiomyocyte necrosis that highlights the intrinsic codependency of these two biologically essential and dynamic divalent cations (vide infra). Others have also reported a Zn2+ supplement to be cardioprotective in mice with streptozocin-induced diabetic cardiomyopathy, in rat hearts with ischemia/reperfusion injury or following isoproterenol administration [28, 33, 59, 74].
Coupled Ca2+ and Zn2+ dyshomeostasis
The dyshomeostasis of extra- and intracellular Ca2+ and Zn2+ that accompanies ALDOST contributes to a deleterious but reversible dysequilibrium between pro- and antioxidants. We hypothesized that intrinsic coupling of intracellular Ca2+ and Zn2+ dyshomeostasis regulates the redox state of cardiac myocytes and mitochondria. To test our hypothesis, we monitored each of these two cations using relevant fluorescent tags and fluorescence microscopy in cardiac myocytes and mitochondria harvested from rats receiving 4 weeks ALDOST alone, or in combination with spironolactone or amlodipine cotreatment. Compared to untreated, age-/sex-matched controls, we found (see Fig. 2) increased cardiomyocyte cytosolic free [Ca2+]i and [Zn2+]i, together with increased mitochondrial [Ca2+]m and [Zn2+]m, each of which could be prevented by spironolactone and attenuated by amlodipine cotreatment [73]. These iterations in divalent cation composition were accompanied by increased levels of 3-nitrotyrosine and 4-hydroxy-2-nonenal in cardiomyocytes, together with increased H2O2 production, malondialdehyde and oxidized glutathione in mitochondria that were coincident with increased activities of Cu/Zn-SOD and glutathione peroxidase (GSH-Px) [59, 66, 73]. Furthermore, alterations in intracellular [Zn2+]i were accompanied by the contemporaneous upregulation of MT-1, a Zn2+ importer and exporter (Zip1 and ZnT-1, respectively) and MTF-1.

Our current understanding of the pathways involving intrinsically coupled dyshomeostasis of Ca2+ and Zn2+ found in ALDOST. Increased excretory losses of these divalent cations lead to hypocalcemia and hypozincemia. Consequent secondary hyperparathyroidism with persistent elevations in circulating parathyroid hormone (PTH) are accompanied by uncontrolled Ca2+ entry via L-type Ca2+ channels (LTCC) to saturate intracellular binding and storage sites, and ultimately to intracellular [Ca2+]i overloading and excessive Ca2+ sequestration within mitochondria. An induction of oxidative stress and generation of reactive oxygen species (ROS) ensue involving mitochondria. The rise in [Zn2+]i and [Zn2+]m involves increased Zn2+ entry via LTCC to a minor extent, while the majority of [Zn2+]i is regulated by membrane-bound Zn transporters, including importers and exporters, Zip1 and ZnT-1, respectively, and its binding to metallothionein (MT)-1 to minimize cytotoxicity. Reprinted from Kamalov et al. [66]
Thus, in cardiac myocytes and mitochondria, an intrinsically coupled dyshomeostasis of intracellular Ca2+ and Zn2+ serves to regulate the redox state via induction of oxidative stress and generation of antioxidant defenses, respectively. These findings underscore the clinical relevance of both pharma- and nutriceutical strategies that can uncouple the coupled dyshomeostasis of these biologically essential cations and modulate them in favor of sustained antioxidant defenses. The coupled Ca2+ and Zn2+ dyshomeostasis seen in aldosteronism resembles the Ca2+ overloading and oxidative stress that exists in the hearts of hamsters with hereditary muscular dystrophy which is also accompanied by increased tissue Zn2+ [75–79]. This divalent cation dyshomeostasis seen in muscular dystrophy could be prevented by parathyroidectomy or a Ca2+ channel blocker. Furthermore, our findings with ALDOST resemble the protective role of increased [Zn2+]i induced by a Zn2+ supplement or Zn2+ ionophore, when intracellular [Ca2+]i overloading of the heart is present [80].
The temporal response to coupled Ca2+ and Zn2+ dyshomeostasis
Intracellular [Ca2+]i overloading, coupled with the induction of oxidative stress, is present at 4 weeks ALDOST. This prooxidant reaction in cardiac myocytes and mitochondria accounts for necrotic cell death and subsequent myocardial scarring. The rise in [Ca2+]i, a prooxidant, is intrinsically linked to increased [Zn2+]i serving as antioxidant. We addressed the temporal responses in coupled Ca2+ and Zn2+ dyshomeostasis, reflecting the prooxidant/antioxidant equilibrium, by examining preclinical and pathologic stages of ALDOST and by observing whether endogenous antioxidant defenses were ultimately overwhelmed accounting for the delay in cardiac remodeling. Responses in cardiomyocyte free [Ca2+]i and [Zn2+]i and mitochondrial total [Ca2+]m and [Zn2+]m, together with biomarkers of oxidative stress and antioxidant defenses, during 1 and 4 weeks ALDOST were monitored and compared. At week 1 and compared to controls, we found (i) elevations in [Ca2+]i and [Ca2+]m to be coupled with [Zn2+]i and [Zn2+]m; (ii) increased mitochondrial H2O2 production, cardiomyocyte xanthine oxidase activity, and cardiac and mitochondrial 8-isoprostane levels, counterbalanced by increased activity of antioxidant proteins, enzymes, and the nonenzymatic antioxidants that can be considered as cumulative antioxidant capacity. Some of these enzymes and proteins (e.g., metallothionein-1, Cu/Zn-superoxide, glutathione synthase) are regulated by MTF-1; and (iii) although these augmented antioxidant defenses were sustained at week 4, overall they fell short in combating the persistent intracellular Ca2+ overloading together with a marked rise in cardiac tissue 8-isoprostane and mPTP opening.
Thus, the intrinsically coupled Ca2+ and Zn2+ dyshomeostasis occurs early during ALDOST in cardiac myocytes and mitochondria that regulate redox equilibrium until week 4, when ongoing intracellular Ca2+ overloading and the accelerated rate of prooxidant generation overwhelm their rate of detoxification by antioxidant defenses. These observations support our contention that intracellular [Ca2+]i overloading accounts for the induction of oxidative stress that leads to necrotic cell death and consequent replacement fibrosis or myocardial scarring.
Uncoupling the coupled dyshomeostasis of Ca2+ and Zn2+
The prooxidant response to Ca2+ overloading in cardiac myocytes and mitochondria has been shown to be intrinsically coupled to simultaneous increased Zn2+ entry serving as an antioxidant [73]. Later, we investigated whether Ca2+ and Zn2+ dyshomeostasis and prooxidant/antioxidant dysequilibrium seen at 4 weeks, the pathologic stage of ALDOST, could be uncoupled in favor of antioxidants, using cotreatment with a ZnSO4 supplement, pyrrolidine dithiocarbamate (PDTC), a Zn2+ ionophore, or ZnSO4 in combination with amlodipine. Responses in cardiomyocyte free [Ca2+]i and [Zn2+]i, together with biomarkers of oxidative stress in cardiac myocytes and mitochondria, were monitored and statistically contrasted. At week 4 ALDOST and compared to controls, we found (i) an elevation in [Ca2+]i was coupled with [Zn2+]i and (ii) increased mitochondrial H2O2 production, and increased mitochondrial and cardiac 8-isoprostane levels. Cotreatment with the ZnSO4 supplement alone, PDTC alone, or ZnSO4+ amlodipine augmented the rise in cardiomyocyte [Zn2+]i beyond that seen with ALDOST alone, while attenuating the rise in [Ca2+]i which together served to reduce oxidative stress. Furthermore, ZnSO4, PDTC, and ZnSO4 + amlodipine were cardioprotective attenuating myocardial fibrosis [58, 59, 80].
Thus, the intrinsically coupled dyshomeostasis of intracellular Ca2+ and Zn2+ found in cardiac myocytes and mitochondria during 4 weeks ALDOST could be uncoupled in favor of antioxidant defenses by selectively increasing free [Zn2+]i and/or reducing [Ca2+]i using cotreatment with ZnSO4 or PDTC alone or ZnSO4+ amlodipine in combination. Each intervention proved to be cardioprotective at varying degrees. These cumulative salutary observations raise the therapeutic prospect that nutriceuticals capable of influencing extra- and intracellular Ca2+ and Zn2+ balance could prevent cardiac myocyte necrosis and myocardial scarring.
Translational research: divalent cation dyshomeostasis in human CHF
The secondary aldosteronism of CHF is accompanied by ionized hypocalcemia with SHPT [36–38, 45, 81]. As noted earlier, dyshomeostasis of divalent cations frequently occurs in patients hospitalized with decompensated biventricular failure due to a dilated (idiopathic) cardiomyopathy. Elevated serum PTH with SHPT is an established feature of primary and secondary aldosteronism [82]. Increased PTH levels found in patients with primary aldosteronism can be reduced by either an aldosterone receptor antagonist, spironolactone, or by adrenal surgery which also corrects ionized hypocalcemia [82]. SHPT is especially prevalent in African-Americans (AA) with protracted decompensated biventricular failure, where chronic elevations in plasma aldosterone account for symptoms and signs of CHF [38]. This is also related to the prevalence of hypovitaminosis D in AA with CHF [38]. The increased melanin content of darker skin in AA serves as a natural sunscreen. Accordingly, the prevalence of hypovitaminosis D, often of marked severity, compromises Ca2+ homeostasis predisposing AA patients to hypocalcemia and consequent SHPT [38, 83, 84]. Hypomagnesemia is another common clinical feature of aldosteronism, which too is corrected by spironolactone or adrenal surgery [85–87]. In addition to the ionized hypocalcemia and hypomagnesemia that accompany increased urinary and fecal losses of these divalent cations, other studies have now identified a concomitant dyshomeostasis of Zn2+ with hypozincemia [88, 89]. Urinary Zn2+ excretion is increased in response to angiotensin-converting enzyme inhibitor or angiotensin receptor antagonist [90, 91].
We documented for the first time serum Zn2+ and Se2+ levels to be reduced in our AA patients followed here in Memphis [36, 37]. This included those with decompensated failure and compensated failure, as well as with heart disease but not heart failure. Reasons for the deficiency of these divalent cations, including their reduced dietary intake, are under investigation. Tennessee soil is not deficient in these minerals. Se is an essential trace mineral and cofactor of antioxidant selenoenzymes, such as GSH-Px and thioredoxin reductase, that promote optimal antioxidant/oxidant balance [92]. Furthermore, a Se-deficient diet is associated with a diminished activation potential of NFκB and downregulation of Se-GSH-Px activity, which detoxifies peroxides and hydroperoxides in such diverse tissues as kidney, cardiovasculature, and red cells [93–98], which is readily reversible when dietary Se is adequately fortified [96, 97, 99]. Monitoring serum Se, Se-dependent enzyme activities, and Se-GSH-Px mRNA expression are clinically useful in addressing optimal Se supplementation [100, 101].
There is an appearance of a dilated cardiomyopathy in greater abundance in general populations where dietary Se2+ deficiencies are found, such as in the Se-poor soil of the Keysan Province of China, or when parenteral nutrition is deficient in Zn and/or Se, [102–104]. It has been suggested that the myopathic process in these scenarios is more closely related to altered immunocompetence and predisposition to viral pathogens than a direct effect of the specific trace mineral deficiencies on the myopathic heart. However, this is arguable. A redistribution of Se from the vascular compartment into tissues that may occur in chronic illness has also been suggested as contributory to the genesis of a dilated (idiopathic) cardiomyopathy [105–109].
Therefore, the metabolic–hormonal profile of CHF with aldosteronism depicts a concerted and contemporaneous dyshomeostasis of multiple nutrients that include Ca2+, Mg2+, Zn2+, and Se2+. Vitamin D deficiency is another crucial and common confounding variable. Other factors which may relate to compromised Ca2+ stores and contribute to the appearance of SHPT in AA with CHF include reduced dietary Ca2+ intake because of lactose intolerance and an active avoidance of dairy products rich in Ca2+ [110] and a preference for a high Na+ diet that enhances urinary Ca2+ excretion. A high-salt diet and consequential calciuria is well known for predisposing a patient to ionized hypocalcemia and SHPT with a resorption of bone which is invoked to restore extracellular Ca2+ homeostasis. Over time, osteopenia and osteoporosis accompany the calciuria of long-term dietary Na+ excess further predisposing them to atraumatic bone fractures [111, 112]. The risk of such fractures is now shown to be increased in patients with heart failure and appears to be preventable by spironolactone together with today’s standard of care [113, 114].
Summary
Previous myocardial infarction, hypertensive heart disease, or a dilated (idiopathic) cardiomyopathy may each contribute to the heart’s failure as a muscular pump that is perpetuated by a sporadic and progressive necrosis of cardiomyocytes, replaced by fibrous tissue, and promoted by inappropriate neurohormonal activation and effector hormones of the RAAS and ANS. The hyperadrenergic, acute stressor state in CHF is accompanied by a translocation of K+, Ca2+, Mg2+, and Zn2+ from the vascular to intracellular compartment. In the case of cardiac myocytes and mitochondria, intracellular Ca2+ overload and the induction of oxidative stress leads to mPTP opening, organellar degeneration, cardiomyocyte necrosis with myocardial fibrosis and subsequent scarring. A similar signal-transducer-effector pathway is at play with the chronic stressor state of aldosteronism where inappropriate elevations in plasma aldosterone, together with the heightened excretory losses of Ca2+ and Mg2+, lead to resultant hypocalcemia and hypomagnesemia and each contributes to the appearance of SHPT. Elevations in plasma PTH seek to restore extracellular homeostasis of these divalent cations through their resorption from bone. At the same time, PTH-mediated intracellular Ca2+ overloading becomes highly prevalent in affected cardiac myocytes and mitochondria. This Ca2+ paradox accounts for an induction of oxidative stress, leading to cardiomyocyte necrosis and subsequent reparative fibrosis. Fibrosis contributes to the adverse structural remodeling of the right and left heart with its attending pathologic influences on myocardial stiffness and contractility while it also serves as substrate for re-entrant arrhythmias. An intrinsically coupled dyshomeostasis of Zn2+ and Se2+ is also seen, which compromises antioxidant defenses.
Hence, several pathways are involved in the necrosis of cardiomyocytes and appearance of cardiac fibrosis. Included among these are catecholamine- and PTH-mediated intracellular Ca2+ overloading and induction of oxidative stress where the latter has been characterized as a Ca2+ paradox [65]. Another pathway relates to impaired antioxidant defenses that accompany Zn2+ dyshomeostasis with hypozincemia and Se2+ deficiency with hyposelenemia. The dyshomeostasis of intracellular Ca2+ and Zn2+ in cardiac myocytes and mitochondria is indeed intrinsically coupled where Ca2+ serves as prooxidant and Zn2+ as antioxidant [66, 73].
Conclusions
In unraveling these intricate cellular and molecular pathways leading to myocardial necrosis and fibrosis, it raises the prospect for nutriceutical uncoupling of Ca2+ and Zn2+ dyshomeostasis in favor of antioxidant defenses by raising [Zn2+]i through Zn2+ supplementation alone, or in combination with a Ca2+ supplement or Ca2+ channel blocker. In addressing the importance of a simultaneous dyshomeostasis involving multiple macro- and micronutrients, particularly in African-Americans with CHF, it is plausible to prevent the adverse structural remodeling of myocardium, which inevitably contributes to increased cardiovascular risk. The time is propitious to develop novel strategies that combine current pharmacologic interventions with inexpensive nutriceutical adjuvants to achieve the greatest therapeutic potential in CHF, which is fast becoming the most serious burden to health care cost containment in today’s industrialized world.
Abbreviations
- ACTH:
-
Adrenocorticotropin hormone
- ALDOST:
-
Aldosterone/salt treatment
- ANS:
-
Adrenergic nervous system
- CHF:
-
Congestive heart failure
- EICA:
-
Excessive intracellular Ca2+ accumulation
- GSH-Px:
-
Glutathione peroxidase
- HPA:
-
Hypothalamic–pituitary–adrenal
- I/R:
-
Ischemia/reperfusion
- ICAM:
-
Intercellular adhesion molecule
- MCP:
-
Monocyte chemoattractant protein
- mPTP:
-
Mitochondrial permeability transition pore
- MT-1:
-
Metallothionein-1
- MTF-1:
-
Metal-responsive transcription factor-1
- NFκB:
-
Nuclear transcription factor-κB
- PBMC:
-
Peripheral blood mononuclear cells
- PDTC:
-
Pyrrolidine dithiocarbamate
- PTH:
-
Parathyroid hormone
- RAAS:
-
Renin–angiotensin–aldosterone system
- RNS:
-
Reactive nitrogen species
- ROS:
-
Reactive oxygen species
- SHPT:
-
Secondary hyperparathyroidism
- SOD:
-
Superoxide dismutase
- TNF:
-
Tumor necrosis factor
References
Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P (1994) Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation 89:151–163
Cotran RS, Kumar V, Robbins SL (1989) The heart. In: Cotran RS, Kumar V, Robbins SL (eds) Robbins pathologic basis of disease, 4th edn. W B Saunders, Philadelphia, pp 597–656
Park M, Shen YT, Gaussin V, Heyndrickx GR, Bartunek J, Resuello RR, Natividad FF, Kitsis RN, Vatner DE, Vatner SF (2009) Apoptosis predominates in nonmyocytes in heart failure. Am J Physiol Heart Circ Physiol 297:H785–H791
Li H, Ambade A, Re F (2009) Cutting edge: necrosis activates the NLRP3 inflammasome. J Immunol 183:1528–1532
Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, Apte RN (2010) Differential release of chromatin-bound IL-1α discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci U S A 107:2574–2579
Weber KT (1989) Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 13:1637–1652
Ishii J, Nomura M, Nakamura Y, Naruse H, Mori Y, Ishikawa T, Ando T, Kurokawa H, Kondo T, Nagamura Y, Ezaki K, Hishida H (2002) Risk stratification using a combination of cardiac troponin T and brain natriuretic peptide in patients hospitalized for worsening chronic heart failure. Am J Cardiol 89:691–695
Kuwabara Y, Sato Y, Miyamoto T, Taniguchi R, Matsuoka T, Isoda K, Yamane K, Nishi K, Fujiwara H, Takatsu Y (2007) Persistently increased serum concentrations of cardiac troponin in patients with acutely decompensated heart failure are predictive of adverse outcomes. Circ J 71:1047–1051
Peacock WF IV, De Marco T, Fonarow GC, Diercks D, Wynne J, Apple FS, Wu AH (2008) Cardiac troponin and outcome in acute heart failure. N Engl J Med 358:2117–2126
Zairis MN, Tsiaousis GZ, Georgilas AT, Makrygiannis SS, Adamopoulou EN, Handanis SM, Batika PC, Prekates AA, Velissaris D, Kouris NT, Mytas DZ, Babalis DK, Karidis KS, Foussas SG (2009, Jan 19) Multimarker strategy for the prediction of 31 days cardiac death in patients with acutely decompensated chronic heart failure. Int J Cardiol [Epub ahead of print]
Löwbeer C, Gustafsson SA, Seeberger A, Bouvier F, Hulting J (2004) Serum cardiac troponin T in patients hospitalized with heart failure is associated with left ventricular hypertrophy and systolic dysfunction. Scand J Clin Lab Invest 64:667–676
Sukova J, Ostadal P, Widimsky P (2007) Profile of patients with acute heart failure and elevated troponin I levels. Exp Clin Cardiol 12:153–156
Ilva T, Lassus J, Siirilä-Waris K, Melin J, Peuhkurinen K, Pulkki K, Nieminen MS, Mustonen H, Porela P, Harjola VP (2008) Clinical significance of cardiac troponins I and T in acute heart failure. Eur J Heart Fail 10:772–779
Sato Y, Nishi K, Taniguchi R, Miyamoto T, Fukuhara R, Yamane K, Saijyo S, Tanada Y, Yamamoto E, Goto T, Takahashi N, Fujiwara H, Takatsu Y (2009) In patients with heart failure and non-ischemic heart disease, cardiac troponin T is a reliable predictor of long-term echocardiographic changes and adverse cardiac events. J Cardiol 54:221–230
Miller WL, Hartman KA, Burritt MF, Grill DE, Jaffe AS (2009) Profiles of serial changes in cardiac troponin T concentrations and outcome in ambulatory patients with chronic heart failure. J Am Coll Cardiol 54:1715–1721
Heusch G, Kleinbongard P, Böse D, Levkau B, Haude M, Schulz R, Erbel R (2009) Coronary microembolization: from bedside to bench and back to bedside. Circulation 120:1822–1836
Chen J, Mehta JL (2006) Interaction of oxidized low-density lipoprotein and the renin-angiotensin system in coronary artery disease. Curr Hypertens Rep 8:139–143
Delafontaine P, Akao M (2006) Angiotensin II as candidate of cardiac cachexia. Curr Opin Clin Nutr Metab Care 9:220–224
Tan LB, Jalil JE, Pick R, Janicki JS, Weber KT (1991) Cardiac myocyte necrosis induced by angiotensin II. Circ Res 69:1185–1195
Benjamin IJ, Jalil JE, Tan LB, Cho K, Weber KT, Clark WA (1989) Isoproterenol-induced myocardial fibrosis in relation to myocyte necrosis. Circ Res 65:657–670
Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, Jaleel N, Chua BH, Hewett TE, Robbins J, Houser SR, Molkentin JD (2007) Ca2+- and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest 117:2431–2444
Brown MJ, Brown DC, Murphy MB (1983) Hypokalemia from beta2-receptor stimulation by circulating epinephrine. N Engl J Med 309:1414–1419
Reid JL, Whyte KF, Struthers AD (1986) Epinephrine-induced hypokalemia: the role of beta adrenoceptors. Am J Cardiol 57:23F–27F
Hansen O, Johansson BW, Nilsson-Ehle P (1990) Metabolic, electrocardiographic, and hemodynamic responses to increased circulating adrenaline: effects of selective and nonselective beta adrenoceptor blockade. Angiology 41:175–188
Carlstedt F, Lind L, Wide L, Lindahl B, Hänni A, Rastad J, Ljunghall S (1997) Serum levels of parathyroid hormone are related to the mortality and severity of illness in patients in the emergency department. Eur J Clin Invest 27:977–981
Carlstedt F, Lind L, Rastad J, Stjernstrom H, Wide L, Ljunghall S (1998) Parathyroid hormone and ionized calcium levels are related to the severity of illness and survival in critically ill patients. Eur J Clin Invest 28:898–903
Javadov S, Karmazyn M, Escobales N (2009) Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharmacol Exp Ther 330:670–678
Chvapil M, Owen JA (1977) Effect of zinc on acute and chronic isoproterenol induced heart injury. J Mol Cell Cardiol 9:151–159
Chanoit G, Lee S, Xi J, Zhu M, McIntosh RA, Mueller RA, Norfleet EA, Xu Z (2008) Exogenous zinc protects cardiac cells from reperfusion injury by targeting mitochondrial permeability transition pore through inactivation of glycogen synthase kinase-3β. Am J Physiol Heart Circ Physiol 295:H1227–H1233
Halestrap AP (2006) Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem Soc Trans 34(Pt 2):232–237
Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y (2005) Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 434:652–658
Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434:658–662
Karagulova G, Yue Y, Moreyra A, Boutjdir M, Korichneva I (2007) Protective role of intracellular zinc in myocardial ischemia/reperfusion is associated with preservation of protein kinase C isoforms. J Pharmacol Exp Ther 321:517–525
Heusch G, Boengler K, Schulz R (2010) Inhibition of mitochondrial permeability transition pore opening: the holy grail of cardioprotection. Basic Res Cardiol 105:151–154
Tsujimoto Y, Shimizu S (2007) Role of the mitochondrial membrane permeability transition in cell death. Apoptosis 12:835–840
LaGuardia SP, Dockery BK, Bhattacharya SK, Nelson MD, Carbone LD, Weber KT (2006) Secondary hyperparathyroidism and hypovitaminosis D in African–Americans with decompensated heart failure. Am J Med Sci 332:112–118
Arroyo M, LaGuardia SP, Bhattacharya SK, Nelson MD, Johnson PL, Carbone LD, Newman KP, Weber KT (2006) Micronutrients in African–Americans with decompensated and compensated heart failure. Transl Res 148:301–308
Alsafwah S, LaGuardia SP, Nelson MD, Battin DL, Newman KP, Carbone LD, Weber KT (2008) Hypovitaminosis D in African Americans residing in Memphis, Tennessee with and without heart failure. Am J Med Sci 335:292–297
Oster O (1993) Trace element concentrations (Cu, Zn, Fe) in sera from patients with dilated cardiomyopathy. Clin Chim Acta 214:209–218
Ripa S, Ripa R, Giustiniani S (1998) Are failured cardiomyopathies a zinc-deficit related disease? A study on Zn and Cu in patients with chronic failured dilated and hypertrophic cardiomyopathies. Minerva Med 89:397–403
Topuzoglu G, Erbay AR, Karul AB, Yensel N (2003) Concentrations of copper, zinc, and magnesium in sera from patients with idiopathic dilated cardiomyopathy. Biol Trace Elem Res 95:11–17
Salehifar E, Shokrzadeh M, Ghaemian A, Aliakbari S, Saeedi Saravi SS (2008) The study of Cu and Zn serum levels in idiopathic dilated cardiomyopathy (IDCMP) patients and its comparison with healthy volunteers. Biol Trace Elem Res 125:97–108
Cénac A, Simonoff M, Djibo A (1996) Nutritional status and plasma trace elements in peripartum cardiomyopathy. A comparative study in Niger. J Cardiovasc Risk 3:483–487
Lee AH, Mull RL, Keenan GF, Callegari PE, Dalinka MK, Eisen HJ, Mancini DM, DiSesa VJ, Attie MF (1994) Osteoporosis and bone morbidity in cardiac transplant recipients. Am J Med 96:35–41
Shane E, Mancini D, Aaronson K, Silverberg SJ, Seibel MJ, Addesso V, McMahon DJ (1997) Bone mass, vitamin D deficiency, and hyperparathyroidism in congestive heart failure. Am J Med 103:197–207
Díaz-Vélez CR, García-Castiñeiras S, Mendoza-Ramos E, Hernández-López E (1996) Increased malondialdehyde in peripheral blood of patients with congestive heart failure. Am Heart J 131:146–152
Keith M, Geranmayegan A, Sole MJ, Kurian R, Robinson A, Omran AS, Jeejeebhoy KN (1998) Increased oxidative stress in patients with congestive heart failure. J Am Coll Cardiol 31:1352–1356
Campolo J, De Maria R, Caruso R, Accinni R, Turazza F, Parolini M, Roubina E, De Chiara B, Cighetti G, Frigerio M, Vitali E, Parodi O (2007) Blood glutathione as independent marker of lipid peroxidation in heart failure. Int J Cardiol 117:45–50
Sun Y, Ramires FJA, Weber KT (1997) Fibrosis of atria and great vessels in response to angiotensin II or aldosterone infusion. Cardiovasc Res 35:138–147
Sun Y, Zhang J, Zhang JQ, Ramires FJA (2000) Local angiotensin II and transforming growth factor-β1 in renal fibrosis of rats. Hypertension 35:1078–1084
Schiffrin EL (1995) Endothelin: potential role in hypertension and vascular hypertrophy. Hypertension 25:1135–1143
Weber KT (2001) Aldosterone in congestive heart failure. N Engl J Med 345:1689–1697
Young M, Fullerton M, Dilley R, Funder J (1994) Mineralocorticoids, hypertension, and cardiac fibrosis. J Clin Invest 93:2578–2583
Garnier A, Bendall JK, Fuchs S, Escoubet B, Rochais F, Hoerter J, Nehme J, Ambroisine ML, De Angelis N, Morineau G, d’Estienne P, Fischmeister R, Heymes C, Pinet F, Delcayre C (2004) Cardiac specific increase in aldosterone production induces coronary dysfunction in aldosterone synthase-transgenic mice. Circulation 110:1819–1825
Somers MJ, Mavromatis K, Galis ZS, Harrison DG (2000) Vascular superoxide production and vasomotor function in hypertension induced by deoxycorticosterone acetate-salt. Circulation 101:1722–1728
Pu Q, Neves MF, Virdis A, Touyz RM, Schiffrin EL (2003) Endothelin antagonism on aldosterone-induced oxidative stress and vascular remodeling. Hypertension 42:49–55
Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL (2002) Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension 40:504–510
Sun Y, Zhang J, Lu L, Chen SS, Quinn MT, Weber KT (2002) Aldosterone-induced inflammation in the rat heart. Role of oxidative stress. Am J Pathol 161:1773–1781
Gandhi MS, Deshmukh PA, Kamalov G, Zhao T, Zhao W, Whaley JT, Tichy JR, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT (2008) Causes and consequences of zinc dyshomeostasis in rats with chronic aldosteronism. J Cardiovasc Pharmacol 52:245–252
Fleckenstein A, Frey M, Fleckenstein-Grun G (1983) Consequences of uncontrolled calcium entry and its prevention with calcium antagonists. Eur Heart J 4(Suppl H):43–50
Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, Smith RA, Gerling IC, Weber KT (2005) Hyperparathyroidism and the calcium paradox of aldosteronism. Circulation 111:871–878
Ahokas RA, Sun Y, Bhattacharya SK, Gerling IC, Weber KT (2005) Aldosteronism and a proinflammatory vascular phenotype. Role of Mg2+, Ca2+ and H2O2 in peripheral blood mononuclear cells. Circulation 111:51–57
Ahokas RA, Warrington KJ, Gerling IC, Sun Y, Wodi LA, Herring PA, Lu L, Bhattacharya SK, Postlethwaite AE, Weber KT (2003) Aldosteronism and peripheral blood mononuclear cell activation. A neuroendocrine-immune interface. Circ Res 93:e124–e135
Chhokar VS, Sun Y, Bhattacharya SK, Ahokas RA, Myers LK, Xing Z, Smith RA, Gerling IC, Weber KT (2004) Loss of bone minerals and strength in rats with aldosteronism. Am J Physiol Heart Circ Physiol 287:H2023–H2026
Fujita T, Palmieri GM (2000) Calcium paradox disease: calcium deficiency prompting secondary hyperparathyroidism and cellular calcium overload. J Bone Miner Metab 18:109–125
Kamalov G, Ahokas RA, Zhao W, Johnson PL, Shahbaz AU, Bhattacharya SK, Sun Y, Gerling IC, Weber KT (2010) Temporal responses to intrinsically coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria during aldosteronism. Am J Physiol Heart Circ Physiol 298:H385–H394
Goodwin KD, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT (2006) Preventing oxidative stress in rats with aldosteronism by calcitriol and dietary calcium and magnesium supplements. Am J Med Sci 332:73–78
Vidal A, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Weber KT (2006) Calcium paradox of aldosteronism and the role of the parathyroid glands. Am J Physiol Heart Circ Physiol 290:H286–H294
Selektor Y, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT (2008) Cinacalcet and the prevention of secondary hyperparathyroidism in rats with aldosteronism. Am J Med Sci 335:105–110
Thomas M, Vidal A, Bhattacharya SK, Ahokas RA, Sun Y, Gerling IC, Weber KT (2007) Zinc dyshomeostasis in rats with aldosteronism. Response to spironolactone. Am J Physiol Heart Circ Physiol 293:H2361–H2366
Selektor Y, Parker RB, Sun Y, Zhao W, Bhattacharya SK, Weber KT (2008) Tissue 65Zinc translocation in a rat model of chronic aldosteronism. J Cardiovasc Pharmacol 51:359–364
Aureli L, Gioia M, Cerbara I, Monaco S, Fasciglione GF, Marini S, Ascenzi P, Topai A, Coletta M (2008) Structural bases for substrate and inhibitor recognition by matrix metalloproteinases. Curr Med Chem 15:2192–2222
Kamalov G, Deshmukh PA, Baburyan NY, Gandhi MS, Johnson PL, Ahokas RA, Bhattacharya SK, Sun Y, Gerling IC, Weber KT (2009) Coupled calcium and zinc dyshomeostasis and oxidative stress in cardiac myocytes and mitochondria of rats with chronic aldosteronism. J Cardiovasc Pharmacol 53:414–423
Wang J, Song Y, Elsherif L, Song Z, Zhou G, Prabhu SD, Saari JT, Cai L (2006) Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation 113:544–554
Lossnitzer K, Janke J, Hein B, Stauch M, Fleckenstein A (1975) Disturbed myocardial calcium metabolism: a possible pathogenetic factor in the hereditary cardiomyopathy of the Syrian hamster. Recent Adv Stud Cardiac Struct Metab 6:207–217
Bhattacharya SK, Johnson PL, Thakar JH (1993) Reversal of impaired oxidative phosphorylation and calcium overloading in the in vitro cardiac mitochondria of CHF-146 dystrophic hamsters with hereditary muscular dystrophy. J Neurol Sci 120:180–186
Palmieri GM, Nutting DF, Bhattacharya SK, Bertorini TE, Williams JC (1981) Parathyroid ablation in dystrophic hamsters. Effects on Ca content and histology of heart, diaphragm, and rectus femoris. J Clin Invest 68:646–654
Bhattacharya SK, Palmieri GM, Bertorini TE, Nutting DF (1982) The effects of diltiazem in dystrophic hamsters. Muscle Nerve 5:73–78
Crawford AJ, Bhattacharya SK (1987) Excessive intracellular zinc accumulation in cardiac and skeletal muscles of dystrophic hamsters. Exp Neurol 95:265–276
Kamalov G, Ahokas RA, Zhao W, Zhao T, Shahbaz AU, Johnson PL, Bhattacharya SK, Sun Y, Gerling IC, Weber KT (2010) Uncoupling the coupled calcium and zinc dyshomeostasis in cardiac myocytes and mitochondria seen in aldosteronism. J Cardiovasc Pharmacol 55:248–254
Khouzam RN, Dishmon DA, Farah V, Flax SD, Carbone LD, Weber KT (2006) Secondary hyperparathyroidism in patients with untreated and treated congestive heart failure. Am J Med Sci 331:30–34
Rossi E, Sani C, Perazzoli F, Casoli MC, Negro A, Dotti C (1995) Alterations of calcium metabolism and of parathyroid function in primary aldosteronism, and their reversal by spironolactone or by surgical removal of aldosterone-producing adenomas. Am J Hypertens 8:884–893
Bell NH, Greene A, Epstein S, Oexmann MJ, Shaw S, Shary J (1985) Evidence for alteration of the vitamin D-endocrine system in blacks. J Clin Invest 76:470–473
Sawaya BP, Monier-Faugere MC, Ratanapanichkich P, Butros R, Wedlund PJ, Fanti P (2002) Racial differences in parathyroid hormone levels in patients with secondary hyperparathyroidism. Clin Nephrol 57:51–55
Horton R, Biglieri EG (1962) Effect of aldosterone on the metabolism of magnesium. J Clin Endocrinol Metab 22:1187–1192
Conn JW (1963) Aldosteronism in man. Some clinical and climatological aspects. Part I. JAMA 183:775–781
Conn JW (1963) Aldosteronism in man. Some clinical and climatological aspects. Part II. JAMA 183:871–878
Garcia Zozaya JL, Padilla Viloria M (1997) Alterations of calcium, magnesium, and zinc in essential hypertension: their relation to the renin-angiotensin-aldosterone system [Spanish]. Invest Clin 38(Suppl 2):27–40
Tubek S (2005) Zinc content in lymphocytes and the activity of zinc ion efflux from lymphocytes in primary arterial hypertension. Biol Trace Elem Res 107:89–99
Golik A, Modai D, Averbukh Z, Sheffy M, Shamis A, Cohen N, Shaked U, Dolev E (1990) Zinc metabolism in patients treated with captopril versus enalapril. Metabolism 39:665–667
Golik A, Zaidenstein R, Dishi V, Blatt A, Cohen N, Cotter G, Berman S, Weissgarten J (1998) Effects of captopril and enalapril on zinc metabolism in hypertensive patients. J Am Coll Nutr 17:75–78
Prabhu KS, Zamamiri-Davis F, Stewart JB, Thompson JT, Sordillo LM, Reddy CC (2002) Selenium deficiency increases the expression of inducible nitric oxide synthase in RAW 264.7 macrophages: role of nuclear factor-κB in up-regulation. Biochem J 366(Pt 1):203–209
Reddi AS, Bollineni JS (2001) Selenium-deficient diet induces renal oxidative stress and injury via TGF-beta1 in normal and diabetic rats. Kidney Int 59:1342–1353
Moreno-Reyes R, Egrise D, Neve J, Pasteels JL, Schoutens A (2001) Selenium deficiency-induced growth retardation is associated with an impaired bone metabolism and osteopenia. J Bone Miner Res 16:1556–1563
Venardos K, Harrison G, Headrick J, Perkins A (2004) Effects of dietary selenium on glutathione peroxidase and thioredoxin reductase activity and recovery from cardiac ischemia-reperfusion. J Trace Elem Med Biol 18:81–88
Wu Q, Huang K, Xu H (2003) Effects of long-term selenium deficiency on glutathione peroxidase and thioredoxin reductase activities and expressions in rat aorta. J Inorg Biochem 94:301–306
Wu Q, Huang K (2004) Effect of long-term Se deficiency on the antioxidant capacities of rat vascular tissue. Biol Trace Elem Res 98:73–84
Faure P, Ramon O, Favier A, Halimi S (2004) Selenium supplementation decreases nuclear factor-kappa B activity in peripheral blood mononuclear cells from type 2 diabetic patients. Eur J Clin Invest 34:475–481
Quercia RA, Korn S, O’Neill D, Dougherty JE, Ludwig M, Schweizer R, Sigman R (1984) Selenium deficiency and fatal cardiomyopathy in a patient receiving long-term home parenteral nutrition. Clin Pharm 3:531–535
Hatanaka N, Nakaden H, Yamamoto Y, Matsuo S, Fujikawa T, Matsusue S (2000) Selenium kinetics and changes in glutathione peroxidase activities in patients receiving long-term parenteral nutrition and effects of supplementation with selenite. Nutrition 16:22–26
Weiss SL, Evenson JK, Thompson KM, Sunde RA (1996) The selenium requirement for glutathione peroxidase mRNA level is half of the selenium requirement for glutathione peroxidase activity in female rats. J Nutr 126:2260–2267
Xia YM, Hill KE, Burk RF (1989) Biochemical studies of a selenium-deficient population in China: measurement of selenium, glutathione peroxidase and other oxidant defense indices in blood. J Nutr 119:1318–1326
van Rij AM, Thomson CD, McKenzie JM, Robinson MF (1979) Selenium deficiency in total parenteral nutrition. Am J Clin Nutr 32:2076–2085
Reeves WC, Marcuard SP, Willis SE, Movahed A (1989) Reversible cardiomyopathy due to selenium deficiency. JPEN J Parenter Enteral Nutr 13:663–665
Sullivan JF, Blotcky AJ, Jetton MM, Hahn HK, Burch RE (1979) Serum levels of selenium, calcium, copper magnesium, manganese and zinc in various human diseases. J Nutr 109:1432–1437
Koch J, Neal EA, Schlott MJ, Garcia-Shelton YL, Chan MF, Weaver KE, Cello JP (1996) Zinc levels and infections in hospitalized patients with AIDS. Nutrition 12:515–518
van Lettow M, Harries AD, Kumwenda JJ, Zijlstra EE, Clark TD, Taha TE, Semba RD (2004) Micronutrient malnutrition and wasting in adults with pulmonary tuberculosis with and without HIV co-infection in Malawi. BMC Infect Dis 4:61
Dworkin BM, Rosenthal WS, Wormser GP, Weiss L, Nunez M, Joline C, Herp A (1988) Abnormalities of blood selenium and glutathione peroxidase activity in patients with acquired immunodeficiency syndrome and aids-related complex. Biol Trace Elem Res 15:167–177
Kavanaugh-McHugh AL, Ruff A, Perlman E, Hutton N, Modlin J, Rowe S (1991) Selenium deficiency and cardiomyopathy in acquired immunodeficiency syndrome. JPEN J Parenter Enteral Nutr 15:347–349
Jarvis JK, Miller GD (2002) Overcoming the barrier of lactose intolerance to reduce health disparities. J Natl Med Assoc 94:55–66
Cohen AJ, Roe FJ (2000) Review of risk factors for osteoporosis with particular reference to a possible aetiological role of dietary salt. Food Chem Toxicol 38:237–253
Teucher B, Dainty JR, Spinks CA, Majsak-Newman G, Berry DJ, Hoogewerff JA, Foxall RJ, Jakobsen J, Cashman KD, Flynn A, Fairweather-Tait SJ (2008) Sodium and bone health: impact of moderately high and low salt intakes on calcium metabolism in postmenopausal women. J Bone Miner Res 23:1477–1485
van Diepen S, Majumdar SR, Bakal JA, McAlister FA, Ezekowitz JA (2008) Heart failure is a risk factor for orthopedic fracture: a population-based analysis of 16, 294 patients. Circulation 118:1946–1952
Carbone LD, Cross JD, Raza SH, Bush AJ, Sepanski RJ, Dhawan S, Khan BQ, Gupta M, Ahmad K, Khouzam RN, Dishmon DA, Nesheiwat JP, Hajjar MA, Chishti WA, Nasser W, Khan M, Womack CR, Cho T, Haskin AR, Weber KT (2008) Fracture risk in men with congestive heart failure. Risk reduction with spironolactone. J Am Coll Cardiol 52:135–138
Alsafwah S, LaGuardia SP, Arroyo M, Dockery BK, Bhattacharya SK, Ahokas RA, Newman KP (2007) Congestive heart failure is a systemic illness. A role for minerals and micronutrients. Clin Med Res 5:238–243
Acknowledgments
This work was supported, in part, by NIH grants R01-HL73043 and R01-HL90867 (KTW). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. Authors have no conflicts of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gandhi, M.S., Kamalov, G., Shahbaz, A.U. et al. Cellular and molecular pathways to myocardial necrosis and replacement fibrosis. Heart Fail Rev 16, 23–34 (2011). https://doi.org/10.1007/s10741-010-9169-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-010-9169-3