
Abstract
The precise mechanisms, how (1) cardiomyocytes sense information about mechanical load and cellular stretch and strain and how (2) downstream signal transduction turns from an initially adaptive to a maladaptive response are far from being fully elucidated. What seems increasingly clear is that one single “cardiac stretch sensor” likely doesn’t exist, but cardiac strain sensing is based on shared general molecular principles at various subcellular domains. A tremendous scientific effort has been spent over the past years, using novel evolving biophysical techniques as well as human and mouse genetics, combined with molecular and functional studies to gain more insight into these principles. These studies have analyzed multiple cellular and subcellular structures in cardiac myocytes (and fibroblasts) as possible signaling hubs for strain perception and altered biochemical processes. Together, these bona fide “cardiac stretch sensor” areas and complexes share common principles like involvement of specialized protein modules, containing domains serving as molecular springs and specific binding sites for protein-protein interactions or phosphorylation/dephosphorylation upon tension related unfolding. In this chapter, we aimed to summarize important findings that have led to a deeper understanding of cardiac mechanoperception and that have uncovered complex and precisely controlled signaling machineries, linked to mechanotransduction and mechanotransmission within various subcellular compartments. In detail, we discuss cardiac mechanotransduction and stretch sensing that can occur at the level of the sarcomeric Z-discs and M-bands, the intercalated disc, as well as at the sarcolemma, microtubule networks, and organelles like the mitochondria and the nucleus. These hubs typically involve specific structural and regulatory proteins, certain transcription factors and enzymes, as well as highly volatile biochemical compounds (e.g., reactive oxygen species and nitric oxide) and various ion channels. Moreover, cardiac mechanotransduction not only involves cardiomyocytes but also includes a complex interplay with cardiac fibroblasts and the extracellular matrix. Finally, we provide an outlook how altered mechanoperception and mechanotransduction modulate heart disease and how these findings could be therapeutically exploited in the future.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


1 Introduction
Increased myocardial distension, as a result of acute or chronic hemodynamic pressure overload, as it occurs in settings of persistent arterial hypertension and valvular or ischemic heart disease, is one of the most frequent challenges for the heart [1]. From a hemodynamic point of view, the short-term adaptation to increased cardiac load as an initial increase in systolic force generation has been described more than 100 years ago and is still known as “Frank-Starling mechanism” [2].
But since, the ability of cardiac tissue and in particular the cardiomyocyte itself, to respond and chronically adapt to various loading conditions throughout development and disease, crucial to maintain hemodynamic stability and prevent circulatory congestion or to compensate for increased wall stress, has also been extensively studied.
In particular, it has been analyzed how cardiomyocytes that are coupled chemically, electrically, and mechanically sense active contractile forces and passive tissue stiffness and how this affects signal transduction and gene regulation [2,3,4,5]. The rapid development of new biochemical, genetic, and imaging techniques has allowed us to gain even more insight in the innumerable and diverse molecular components and signaling processes that are involved herein. These intracellular and extracellular processes mostly include conformational protein alterations as initial events resulting in altered protein-protein interactions, differential protein expression, posttranslational modifications (i.e., phosphorylation), and changes in subcellular protein localization, as well as altered ion channel functions and generation of small volatile molecules like nitric oxide (NO) and reactive oxygen species (ROS) [3, 6,7,8,9,10,11,12]. Of note, the latter are mediators that are involved in almost all mechanotransduction processes and provide ultra-short-term mechano-electrical feedback and also enable mechanochemical transmission [6, 13].
Beyond these structural and signaling-based mechanisms, cardiomyocytes feature a complex network of calcium entry mechanisms and calcium storages, as well as a large number of mitochondria, responsible for synthesizing and delivering ATP molecules ensuring constant and proper sarcomeric contraction [2, 14,15,16]. Structural and regulatory components involved in these metabolic mechanisms are closely aligned near the cardiac sarcomere, forming important microdomains that are also able to rapidly adapt to increased energy/metabolic and calcium demands under pressure overload, hypertrophy, and heart failure and thus participate in muscular mechanoperception [11, 17].
Scientific evidence is constantly growing that there are distinct cellular pathways that mediate cardiomyocyte mechanotransduction under either pathological or physiological (i.e., exercise) conditions, involving mostly different intracellular signaling cascades [18]. To add another layer of complexity, these signaling pathways and involved components are believed to also change upon developmental and aging processes [4, 5, 19, 20]. In general cardiomyocyte hypertrophy, denoting initially an adaptive cardiac response to compensate for increased wall stress, results in a pathological maladaptive condition upon persistent activation, so there have to be common mechanisms (and potential future therapeutic targets) dividing physiological or adaptive cardiac hypertrophy signaling cascades from those driving intracellular and extracellular processes leading to increased cardiac morbidity and mortality [1, 5]. To precisely dissect these highly variable and complex mechanoperception and mechanotransmission mechanisms, signaling cascades will help to design future gene therapy-based or pharmacological strategies to improve patient outcomes.
2 The Z-Disc and Mechanotransduction
Multiple studies have identified the cardiac sarcomere with its complex assembly of myofilament proteins as a key structure for mechanotransduction [9, 21, 22]. In particular, the cardiac Z-disc as the lateral boundary within each sarcomere not only consists of multiple layers of α-actinin aligned in antiparallel organization [17] but also forms a complex protein network for sensing sarcomeric strain. The view of the cardiac Z-disc as a mere structural sarcomeric component has extensively changed over the years and is now believed to be the most important signaling nodal point for strain-related downstream signal transduction [23]. Beyond carrying myofilaments, the sarcomere connects to cardiac sarcolemma by cytoskeletal protein complexes that link in particular the Z-disc to the integrin and dystroglycan complexes at costameres. Indirectly, this interface can connect the Z-disc to components of the cardiac extracellular matrix (ECM) through components containing transmembrane domains [5, 8, 17, 24]. Among others, the intermediate filament desmin has also been shown to connect the Z-disc to the cardiomyocyte nucleus [25, 26], while other proteins like mink (minimal potassium channel subunit) or telethonin/TCAP are believed to tether cardiac t-tubules to the sarcomeric Z-disc [27, 28]. Taken together, this places the sarcomeric Z-disc within the center of multiple cardiomyocyte signaling microdomains.
Titin, muscle LIM protein (MLP), calsarcin 1, and filamin C are likely critical players in force transmission and sensing within the sarcomeric Z-disc [23]. But a constantly increasing number of Z-disc proteins (TCAP, LMCD1, ALP, ABLIM, Cypher/ZASP/Oracle, PICOT, FHL1/2, CEFIP, muscle-specific ankyrin repeat protein (MARP), nexilin, myopalladin, myotilin) have been recently discovered and shown to participate in complex signaling networks implicated in mechanotransduction, hypertrophy signaling, and cardiomyocyte contractile (dys)function as discussed below [5, 22, 23, 28].
2.1 Strain Sensing and Mechanotransduction at the Sarcomeric Z-Disc/I-Band
Titin is a giant protein that pervades the sarcomere from the sarcomeric Z-disc to the center of the sarcomere at the M-band [29, 30]. Titin contains multiple distinct protein domains that have been shown to serve as anchoring hubs for a variety of additional proteins that are all involved in myocardial stretch responses during cardiac hypertrophy and failure [29,30,31]. The overall cellular location along the sarcomeric filaments, as well as the unique structural properties of each titin molecule itself, makes titin an ideal candidate for sensing systolic or diastolic biomechanical forces acting on the sarcomere and those being generated by the sarcomere itself during active contraction [29, 32]. A large variety of functional roles for titin have been revealed, including the control of correct sarcomere assembly, sarcomere length and stability, the generation of stretch-adapted passive sarcomeric stiffness, and as signal transducer in response to pressure overload [16, 29,30,31, 33,34,35,36]. I-band titin contains unique domains and interdomain linkers that are believed to follow a “stepwise extension” model. Initially, upon moderate strain application, at the sarcomeric I-band, N2BA titin is elongated by straightening their Ig interdomain linkers [29, 37]. When this capacity is worn out upon further increase of strain forces, the PEVK-domain and finally the N-Bus domain further extend and unfold to expose phosphorylation and binding sites [30, 31, 34]. Taken together, titin and its unique structural and regulatory features are thus believed to represent the potential molecular fundament for the aforementioned Frank-Starling mechanism.
2.2 The Z-Disc/I-Band Titin/MLP/TCAP Complex: Stretch Sensing Within Sarcomeric Borders
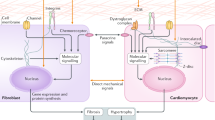
The muscle LIM domain protein (MLP) is specifically expressed in all striated muscle cells and has been implicated in a broad range of different cellular functions [5, 21, 28]. It contains two LIM domains and thereby interacts with multiple proteins, including α-actinin, titin, and TCAP at the Z-disc and other subcellular compartments including intercalated discs and even the nucleus [21]. Of note, MLP protein was also found at sarcomeric I-bands. At Z-discs, MLP-associated TCAP is hereby able to weave two titin filaments at the Z-disc [5]. This protein complex has been shown to be exceptionally resistant to biomechanical forces, due to multiple hydrogen bonds between TCAP and titin [5, 30]. Moreover MLP is believed to shuttle into the nucleus to promote nuclear signaling in muscle cells (possibly via GATA4 and SRF) [21]. MLP knockout mice display a severe dilated cardiomyopathy phenotype resulting in heart failure and premature death, whereas in vitro, MLP deficiency resulted in abrogation of hypertrophic signaling upon neurohumoral or strain-related activation [21, 23, 28]. The W4R-MLP polymorphism, found in human DCM, results in an altered MLP structure, incapable to interact with TCAP [38]. Knock-in mice with homozygous expression of the W4R-MLP show spontaneous cardiac hypertrophy without pressure overload possibly via alteration of elastic intrinsic properties of Z-disc titin and/or shuttling of mutant MLP to the nucleus [28, 38]. Yet, cardiac-specific overexpression of MLP does not protect against increased biomechanical stress due to pressure overload [39]. In addition to the aforementioned MLP functions, it is believed to recruit the phosphatase calcineurin to the Z-disc, a major signaling enzyme linked to pathological cardiac hypertrophy as discussed below [21] (Fig. 1).

Signaling molecules embedded in the sarcomeric Z-disc in context to the nuclear and cellular lamina. Schematic illustration of typical multiprotein complexes involved in cardiomyocyte mechanotransduction. At the sacromeric Z-disc, the calcineurin-NFAT pathway tethered via calsarcin 1 onto the sarcomere is shown. NFAT as well as sarcomeric MLP is believed to shuttle between the Z-disc and the nucleus. At the sarcomeric I-band, a titin-associated complex involving FHL1 and 1 and MAP-Kinases Erk1/2 is displayed. ERK1/2 is also believed to shuttle in the nucleus to promote hypertrophic gene expression via transcription factors like GATA4. GPCR G-protein-coupled receptor, At1R angiotensin II type 1 receptor, SR sarcoplasmic reticulum, LINC linker of nucleoskeleton and cytoskeleton
Myopodin, also known as Synaptopodin2, was initially described as actin-bundling protein enriched in the heart and skeletal muscle and is believed to be responsive to cellular stress induced by heating of myoblasts to 43 °C [40]. The fact that myopodin is also part of a Z-disc signaling complex containing alpha-actinin, calcineurin, Ca2+/calmodulin-dependent kinase II (CaMKII), muscle-specific A-kinase anchoring protein, and myomegalin suggests an important function in Z-disc’s associated signaling [41]. This is further supported by the finding that protein kinase A (PKA) or CaMKII-dependent myopodin phosphorylation mediates its binding to the chaperone 14-3-3 and subsequent nuclear import, whereas dephosphorylation of myopodin by calcineurin counteracts this process. Another relation of myopodin function to cellular tension was reported in mammalian A7r5 cells, where tension-induced unfolding of filamin disrupted a complex of myopodin, BAG3, and filamin which led to autophagosome formation and subsequent filamin degradation [42].
2.3 Calcineurin as Major Player in Cardiac Stress Signaling
The calcium/calmodulin-dependent phosphatase calcineurin (CaN) and its downstream targets, transcription factors of the nuclear factor of activated T cells (NFAT) family, play essential roles in cardiomyocyte signaling [10]. Calcineurin typically forms a heterodimer expressed as three different isoforms, namely, αCaN, βCaN, and γCaN, while the structure of each isoform shows similar features, containing a catalytic A chain (CnA) and calcium-binding regulatory B domain (CnB1) [43]. A direct link of calcineurin activity to mechanotransduction has initially been demonstrated by the fact that in vivo, transgenic overexpression of constitutively active calcineurin in mouse hearts leads to massive ventricular hypertrophy and subsequent heart failure [44]. In contrast, mice lacking βCaN show impaired ability to induce cardiac hypertrophy upon pressure overload [45]. Calcineurin is activated in response to increases in local calcium concentrations as stepwise process with an inactive, partially active, and fully activated state [43]. At basal cardiomyocyte calcium concentrations, calcineurin remains in its inactive state, in which the regulatory domain is folded onto the B chain binding helix, leaving CnB1 unbound. The partially active state occurs when loose calcium ions bind to CnB1 upon increasing concentrations, resulting in interaction of the N-terminal lobe of CnB1 to the B chain binding helix of CaN. The fully active state occurs when also calmodulin (a calcium-sensing protein) associates to the holoenzyme, causing calcineurin’s regulatory domain to correctly fold, thereby removing its autoinhibitory domain from the catalytic site [43]. To ensure tightly controlled calcineurin activity, many factors are likely involved, including the rate of rise and fall of calcium concentrations, association with calmodulin, and regional differences in availability of these factors in certain microdomains [46,47,48,49,50,51,52]. Within this global model, cardiomyocyte calcium entry is linked to dephosphorylation, whereas in contrast, calcium signaling is also coupled to phosphorylation through various calmodulin-modulated kinases (CAMKs) [10]. In this interplay, these processes offer a distinct coordination between calcium-induced calcium release and excitation contraction coupling and phosphorylation/dephosphorylation processes affecting signaling and gene regulation. While calcineurin is primarily located in the cytosol, various protein binding partners, like MLP, calsarcins, and A-kinase anchoring proteins, are believed to tether calcineurin to different subcellular signaling microdomains, like the t-tubule-Z-disc interface as well as the nuclear envelope. This localization in close proximity to voltage-activated L-type calcium channels (LTCC) and transient receptor potential canonical (TRPC) channels at t-tubule membranes and ryanodine receptors (RyR2) in the sarcoplasmic reticulum (SR) place calcineurin in an ideal location for sensing calcium fluxes upon short-term increases in local intracellular concentration [43,44,45,46,47,48,49]. The most studied substrates of calcineurin are the family of nuclear factors of activated T cells (NFATs). Desphosphorylation of these transcription factors by calcineurin in the cytosol results in exposure of a nuclear localization signal, resulting in NFAT translocation to the nucleus, thereby activating prohypertrophic gene expression programs [10, 43]. Interestingly, NFATs function as heterodimers in cooperation with other prohypertrophic transcription factors like MEF2 and GATA4, which can also be directly activated by calcineurin-mediated dephosphorylation [10]. Because of its crucial functions in maintaining cardiac homeostasis, calcineurin activity is tightly regulated by multiple endogenous mechanisms. The calcineurin inhibitor CABIN-1/CAIN interacts with calcineurin as well as acting directly as a co-repressor of the SRF-associated transcription factor MEF2. Since CABIN-1 is a large, multidomain protein, it is assumed to function as a calcineurin scaffold, facilitating interactions with other regulatory proteins [10]. Carabin is another endogenous calcineurin inhibiting protein showing reduced abundance in models of pressure overload and in human heart failure. Knockout of carabin in vivo results in exaggerated pressure overload-associated cardiac hypertrophy and heart failure, whereas cardiomyocyte-specific overexpression of carabin has shown to be protective [10]. The family of regulators of calcineurin (Rcan1, 2, and 3) consists of small proteins that can potently inhibit calcineurin activity through a unique C-terminal calcineurin binding domain. Of note, the expression of the cardiac Rcan1.4 isoform is regulated under almost exclusive calcineurin/NFAT transcriptional control and thus provides a direct feedback loop for calcineurin activity [10]. Other negative regulators of calcineurin activity at the sarcomeric Z-disc include calsarcin 1 and the protein PICOT (protein kinase C-interacting cousin of thioredoxin) [48, 53]. PICOT directly interacts and colocalizes with MLP at the Z-disc and thereby disturbs the MLP-calcineurin interaction, resulting in a concentration-dependent displacement of calcineurin from the Z-disc [5, 10, 23]. The calsarcin family of proteins includes three known members, all localizing specifically to the Z-disc. In this regard, calsarcin-1, representing the only isoform expressed in the adult heart, has been shown to be a negative regulator of calcineurin activity [5, 10, 23, 47].
2.4 N2B Titin Controlled Mechanosensing Hubs
An I-band localized subdomain of N2B (N2Bus) titin can act as a molecular spring element, able to unravel upon heavily increased sarcomeric strain and subsequent diastolic distension, thereby functioning as a diastolic length sensor (see above) [32, 36, 37]. At the N2Bus, titin interacts with 4½-LIM domain proteins, FHL1 and FHL2, that are believed to form a unique multiprotein complex with the kinases MAPK/ERK1/2, directly linking mechanical strain to hypertrophic signaling and growth. Load-dependent unfolding of the N2Bus spring-like subdomain releases activated MAPK/ERK2 and enables their active shuttling to the nucleus to promote hypertrophy-associated gene expression [5, 23, 34]. FHL2, which is known to show additional interactions in cardiac muscle, also connects the N2B-domain to integrins, thereby connecting titin to the integrin-associated mechanotransduction pathway at costameres. FHL protein-associated signalosomes gained more attention, since recently a previously uncharacterized protein named cardiac-enriched FHL2-interacting protein (CEFIP) was discovered. CEFIP is upregulated in cardiomyopathy and shows a heart- and skeletal muscle-specific expression profile. CEFIP also localizes to sarcomeric Z-discs where it interacts with FHL2, thereby enhancing calcineurin activity [46]. Beyond FHL2 regulatory processes, titin N2Bus can be phosphorylated by multiple cardiac kinases, including ERK1/2, PKA, and CamKII, possibly in order to provide local adaptation of this regulatory pathway by reducing titin/sarcomeric passive tension [5, 23, 34].
The muscle-specific intermediate filament component desmin not only connects nuclei and mitochondria but also intercalated discs to the Z-disc [54]. Mutations in desmin cause a variety of cardiac diseases like dilated cardiomyopathy, arrhythmogenic right ventricular cardiomyopathy (ARVC), and familial restrictive cardiomyopathy (often sharing the common feature of variable associated conduction system defects) as well as skeletal myopathies [54,55,56]. Due to its assumed unique elasticity, desmin is believed to sense cellular and protein complex deformations, which may trigger certain conformational changes in response to strain-related mechanical alterations, resulting in diverse signaling events as well as structural alterations (further discussed below) [5, 9, 23, 57].
The Z-disc protein filamin C (FLNC) is also recognized as a potential molecular strain sensor in cardiomyocytes. Moreover, filamin C can act as a structural crosslinker of actin rods at the sarcomeric Z-disc [22]. For all three filamins (filamin A, B, and C), a subcellular localization at either the sarcomeric Z-disc, the intercalated discs, cell membranes, and myotendinous junctions has been described. It is speculated that due to its unique structural characteristics, in particular filamin A and filamin C can also serve as a molecular interface for mechanotransduction [22]. As it has been assumed for other proteins, filamin molecules are able to straighten upon tension, thus changing affinities for protein-protein interactions and subsequent signaling [22]. This seems of special interest, since at Z-discs, filamin C interacts with various proteins implicated in mechanotransduction like calsarcins, myotilin, myopodin, and others [23], and binds to sarcolemma via integrin-1β and sarcoglycan-delta. Mutations in the filamin C gene have also been linked to a variety of cardiac diseases like dilated cardiomyopathy, arrhythmogenic right-ventricular cardiomyopathy (ARVC), familial restrictive cardiomyopathy, and hypertrophic cardiomyopathy (sharing some important features of desmin-related diseases like increased rates of associated cardiac conduction defects) [22].
3 Sensing of Mechanical Strain and Signal Transduction at the Center of the Sarcomere, the M-Band
Beyond its function as a structural anchor for thick and thin filaments within the center of the sarcomere, the M-band is a precisely organized nodal point in signaling and has been recognized as biomechanical strain sensor and regulator of sarcomeric force imbalances during active muscle contraction [58]. Additionally, it is involved in transfer of these biomechanical signals into altered signal transduction and has its role in hypertrophic signaling, cardiac protein turnover, and cardiomyocyte calcium handling. Like the Z-disc, the sarcomeric M-band is composed of a multiprotein structural and signaling hub with numerous interactions and associated pathways [29,30,31, 33, 34].
From a structural point of view, it has been largely accepted that regular M-band assembly is needed for proper packaging of the thick filaments with appropriate distances and overlap to the thin filaments at the onset of sarcomeric contraction (termed “M-bridges”) [58]. In this context, the M-band domain of titin plays a pivotal role besides the proteins myomesin 1 and myomesin 2 that have been shown to directly interact with M-band titin and also function as “M-bridges.” Myomesins are organized into antiparallel dimers to the central zone of myosins and to the C-terminal region of titin and thereby crosslink thick filaments [58].
As outlined above, due to its elastic I-band domains and its unique filament-like structure, titin is an ideal stretch sensing molecule and adaptable molecular spring that can integrate various biophysical signals about acute and chronically altered myofilament tension [30, 34]. The M-band domain of titin shows multiple interaction hubs linking the M-band to the protein turnover machinery (via Nbr1/p62/Murf2) [59]. Hence, upon inactivation in the skeletal muscle, titin kinase (TK)-mediated conformational change results in disruption of the Nbr1/p62/Murf2 complex and subsequent inhibition of SRF signaling [58]. Single-molecule analysis experiments by atomic force spectroscopy revealed that exposure of recombinant TK domain to stretch forces causes a conformational change required for ATP binding and access of the autoinhibitory tyrosine, which fully activates the kinase by removal of the carboxy-terminal autoinhibitory tail by yet unknown protein cofactors [29, 58].
Beyond that, M-band titin interacts with myospryn, which is known to also bind the cardiac phosphatase calcineurin, thereby indicating an SRF-independent sarcomeric hub at the M-band for prohypertrophic or anti-hypertrophic signaling [49]. Analyses in titin knockout mouse models revealed that specific depletion of M-band titin leads to atrophy and preserved cardiac function, whereas depletion of full-length titin leads to dilated cardiomyopathy and heart failure, while both phenotypes are associated with premature cardiac death before day 40 [35]. In line with these results, a mouse model with a conditional depletion of the M-band-associated titin kinase region developed dilated cardiomyopathy with altered sarcomeric architecture and dissociation of MURF1 [60]. A mouse model mimicking the human DCM related c.43628insAT mutation, as well as a mouse model expressing the A178D missense variant, develops a dilated cardiomyopathy phenotype that can be aggravated under neurohumoral stimulation via prohypertrophic factors [61].
But beyond titin’s functions, the overall M-band composition varies upon different developmental stages and under pathological conditions. Differential expression levels of mature myomesin and an alternatively spliced embryonic (“EH”-) myomesin isoform are thought to contribute to specific functional and structural features at different cardiac developmental stages. Alterations of M-band assembly with re-expression of EH-myomesins and “fuzzy”—less stiff—sarcomeres are believed to accompany and contribute to the progression of cardiomyopathy [58]. A similar phenomenon was described for titin itself that exists in two main co-expressed isoforms in the mammalian heart, which are differentially expressed in the developed and diseased heart (“titin switch” with exchange to N2BA (more compliant) from N2B (stiffer) or vice versa) [8, 16, 30]. Another novel component of the sarcomeric M-band is the leucine-rich repeat containing protein myomasp/LRRC39, which binds to the rod domain of MYH7 at the M-band, and that is downregulated upon pressure overload. Loss of myomasp in vivo results in M-band alterations and reduced contractility associated with impaired SRF signaling, supporting the notion that it is required for M-band integrity and associated stretch signaling [62].
4 Stretch Sensing Between Cardiomyocytes and the Extracellular Matrix via the Intercalated Discs and Costameres
The cardiac cytoskeleton can be broadly subdivided into intermediate filaments, microtubules, myofibrils (with the sarcomere as main contractile unit), and the intercalated discs that all are connected via multiple mechanisms involving multiprotein complexes [5, 8, 13, 17]. Myofibrils are also attached to the plasma membrane at adherens junctions (zonula, fascia, punctum adherens) as zonal subtype of intercalated discs (ID), whereas in turn the sarcomeric Z-disc is anchored to the plasma membrane at the costameres [63]. Additional specialized subtypes of cell-cell contacts at the ID are desmosomes and gap junctions as well as adherens junctions, all of which have been proposed to form a specific joint structure in the heart termed “area composita” [64, 65]. All these highly specified substructures are involved in cell-cell adhesion, cell connection to the extracellular matrix, bidirectional force transmission, and electrical coupling of cardiac muscle cells [13, 63, 66]. Importantly, the extracellular matrix is not only regarded as an environmental milieu but also organizes, connects to, and communicates with neighboring cardiomyocytes and fibroblasts, while absorbing and transmitting biomechanical forces through interaction with cytoskeletal networks [66, 67]. Their main components are laminin proteins and various collagen subtypes with different levels of molecular stiffness (collagen I stiffer than III) [68]. The main connection of the ECM to the cytoskeleton is achieved by transmembrane receptors from the integrin family that link the ECM with extracellular domains and also connect to macromolecular complexes containing kindling/paxilin, talin, vinculin, focal adhesion kinase, desmin, and VASP (vasodilator-stimulated phosphoprotein) with cytosolic domains [8]. Integrin signaling complexes hereby transduce mechanical forces outside-in and inside-out, a process that is tightly regulated by multiple signaling mechanisms [63, 67]. Beta1D integrin was found to be expressed at intercalated discs as well as costameres and binds to alpha 7B integrin, which forms a heterodimeric complex that can phosphorylate focal adhesion kinase (FAK) at Tyr 397. This signaling complex also includes scr/PI3K/Grb2, and has also been associated with mitogen-activated kinase (MAP-Kinase) signaling (e.g., MEK1/2 and ERK1/2-), directly linking ID-based mechanotransduction to hypertrophic signaling and cardiomyocyte growth [69,70,71,72]. In addition, the protein melusin has also been shown to interact with FAK at costameres to promote strain-related hypertrophic signaling. Melusin knockout mice fail to execute a hypertrophic response upon pressure overload and directly undergo cardiac failure, emphasizing melusin’s role as additional strain sensor [63, 67, 73, 74].
4.1 The Extracellular Matrix: More Than Just Collagens
As it has already been outlined, the ECM is not only a static environmental milieu interacting with cytoskeletal networks. Acute and chronic tissue biomechanical strain in the myocardium is associated with profound and dynamic changes in the composition of the ECM [75, 76]. Under basal conditions, quiescent cardiac fibroblasts are responsible for constant renewal of ECM proteins, but upon biomechanical stress, fibroblasts are able to proliferate and convert into myofibroblasts, contractile and matrix remodeling cells. This is one of the earliest effects upon cardiac pressure overload, thus leading to deposition of additional collagenous matrix and expansion of the interstitium. Together with ECM crosslinking, matrix deposition results in a profound increase in overall myocardial stiffness [75].
Within the extracellular matrix, cardiac pressure overload is associated with a local induction and activation of various collagenases (MMP-1, MMP-8, MMP-13), gelatinases (MMP-2, MMP-9), stromelysins/matrilysins, and also membrane-type MMPs. Within this context MMP induction may be mediated through activation of proinflammatory signaling pathways like TNF-α and IL-1β. Members of the MMPs family have been implicated in TGF-β activation and may cleave transmembrane receptors, such as integrins or syndecans in order to modify proinflammatory or fibrogenic cascades. MMPs may also act as intracellular and intercellular mediators, promoting degradation of contractile proteins in cardiomyocytes or modulating signal transduction responses in interstitial cells. In contrast to MMP activation, rather decreasing myocardial stiffness, matrix-preserving mediators like members of tissue inhibitors of metalloproteinases (TIMPs) control the deposition of structural ECM proteins, thus increasing myocardial stiffness as a counter regulatory process. Imbalances between those two regulatory systems might accompany the transition of compensated cardiac hypertrophy to decompensated heart failure during long-term cardiac remodeling [75,76,77].
Stress-induced fibroblast activation in the pressure-overloaded myocardium may also be indirect, involving paracrine factors and interaction via cardiomyocytes and various immune cells involving the release of fibrogenic growth factors. The most prominent cascade is activation of the renin-angiotensin-aldosterone system (RAAS) triggering broad inflammatory signaling and leading to downstream stimulation of TGF-β and other associated pathways [75, 78].
Besides those characterized local and systemic pathways, matricellular proteins play a pivotal role in the paracrine regulation of the pressure overload-associated stress response. Members of the matricellular protein family thereby serve as dynamic integrators of microenvironmental changes between ECM and any other myocardial cell type. Fibronectin deposition is believed to be crucially involved in myofibroblast transdifferentiation and has been implicated as an important mediator in subsequent cardiomyocyte hypertrophy. Increased myocardial load or strain triggers marked upregulation of various splice variants of fibronectin which has in turn been linked to TGF-β pathways promoting fibroblast to myofibroblast conversion [75].
Further members of the matricellular family are tenascin-C; tenascin-X; SPARC; thrombospondin-1 (TSP-1), TSP-2, and TSP-4; osteopontin; periostin; and the members of the CCN family. All protein members of this family share the common function that they are able to modulate fibroblast proliferation, survival, and activation and in myofibroblast conversion that they control certain subpopulations of resident and infiltrating immune cells, directly interfere with cardiomyocytes to control cell survival and apoptosis, and are involved in ECM crosslinking processes [75]. This inducible paracrine repertoire offers a tissue wide adaptive control connecting various cell types and the ECM, and also offers systemic regulatory functions. Whereas tenascin-C can act as modulator of macrophage phenotypes in pressure-overloaded hearts, osteopontin is present at very low levels under basal conditions but becomes rapidly induced upon pressure overload and may act by stimulating a fibrogenic program in cardiac fibroblasts. Beyond that, ECM fragments (so-called matrikines) generated through protease activation are able to induce proinflammatory cascades or activate pro-apoptotic pathways in cardiomyocytes upon pressure overload [76, 78, 79].
But not only structural and paracrine factors are involved in ECM-based mechanotransduction and mechanoperception. The Hippo pathway-associated transcriptional complex YAP/TAZ is believed to be involved in ECM-based mechanosensing since it plays a crucial role in adult cardiac fibroblast migration, proliferation, and differentiation. In adult murine heart’s post myocardial infarction, cells with nuclear YAP/TAZ localize at the infarct border zone, suggesting a prompt response of resident stromal cells to ischemia or increased biomechanical load.
4.2 Stretch Transmission at Costameric Integrin-Talin-Vinculin Clutches
The integrin interacting protein talin also seems to be of special interest as potential local molecular stretch sensor, since it can undergo conformational extension upon increased strain, unfolding a binding site for vinculin and thereby easing subsequent actin binding [70, 80, 81]. Talin has been mainly found at costamers but is also highly associated with intercalated discs. This integrin-talin-vinculin-actin-ECM-cell adhesion complex hereby re-forms within seconds, representing a dynamic transmission-like interface with constant engagement and disengagement reinforced by cyclic vinculin binding [80, 81]. Comparable to other mechanotransduction initial events, stepwise unfolding of talin domains represents a key feature, enabling stabilized vinculin actin adhesions under conditions of increased tissue tension [81, 82]. Beyond that, binding of talin to integrin per se activates integrin signaling through open conformation stabilization [69]. Of note, talin exists in two isoforms (talin 1 and 2), of which talin 2 represents the most prominent isoform in adult hearts. Upon pressure overload and in human cardiomyopathy, a re-expression of fetal-like talin 1 occurs, which is considered as rather maladaptive response [81]. Beyond that, talins also connect costameres to the sarcomeric Z-disc via interaction with gamma-actin and alpha-actinin [81, 82]. The intermediate filament protein desmin has also been shown to connect costameres to sarcomeric Z-discs and serves as strain-sensing molecule (as discussed above) and additional shock absorber in force transduction [54].
4.3 Stretch Signaling at Costameric Dystrophin Glycoprotein Complexes
Besides integrin-talin signaling and desmin, the cardiomyocyte dystrophin glycoprotein complex (DGC) is another important signaling hub for costameric mechanotransduction, also regarded as a “shock absorber” [24]. Known members of the core DGC complex are dystrophin, dystroglycan, and sarcoglycan-sarcospan subcomplexes as well as dystrobrevin and syntrophin [83]. DGC also associates with ILK/PINCH/Parvin and MLP [67]. Besides its role in absorption mechanical forces, it has been assumed that DGC protects costameres from fragmentation during strong contraction events as they can occur in skeletal but also cardiac muscle cells [67, 84]. DGC-associated hypertrophic signaling mainly involves MAPK and Rac1 [51]; additional signaling molecules include nitric oxide as highly volatile second messengers (see below) [67]. Dystrophinopathies are a group of specific clinical entities due to mutations in the dystrophin gene and include the Duchenne muscular dystrophy (DMD), Becker muscular dystrophy (BMD), and X-linked dilated cardiomyopathy (XLDCM) [85]. In all three subforms, the heart muscle can be affected to various degrees, depending on the precise genotype and the stage of the disease. Mutations in the dystrophin gene thereby result in altered protein structures, possibly affecting the function of the overall cardiac DCG, ultimately leading to regional replacement of myocardium by fibrotic tissue or fat [86, 87]. During cardiac progression of dystrophinopathies, left ventricular dysfunction and ventricular arrhythmias due to increased fibrosis can occur, ultimately leading to heart failure and sudden cardiac death in final stages [88]. Of note, in particular XLDCM represents a rapidly progressive myocardial disorder, starting in young male variant carriers as dilated cardiomyopathy, leading to death from refractory heart failure within 1–2 years after diagnosis [86]. Of further note, gene therapy for DMD has recently been successfully applied in large animals, paving the way for future treatment of patients affected with this severe disorder [89] (Fig. 2).

Signaling molecules and associated pathways at cardiac costameres. Schematic overview of multiple signaling principles connecting cardiac sarcomeres to the cardiomyocyte’s sarcolemma via costameres involving the integrin signaling complex and the dystrophin glycoprotein complex (DGC). Both signaling and sensing hubs directly connect intracellular stretch perception mechanisms to extracellular matrix components that are embedded in and also able to adapt to various loading conditions. Via sensing of ECM substrate tension and via adaptation of ECM stiffness mechanoperception, as well as via integration of the contraction-relaxation status of the sarcomere, cardiac mechanosensing is organized by inside-out and outside-in directed mechanisms. LTCC L-type calcium channel, TRPC transient receptor potential channels, SAC stretch-activated channels, NO nitric oxide
5 Force Sensing and Transmission at Cell-Cell Contacts in the Heart
Intercalated discs (ID) are also believed to be involved in cardiac mechanotransduction through conformational changes in associated proteins and in the sensing of stiffness of the ECM by various mechanisms [63]. While costameric stretch sensing mainly involves talin proteins, ID strain sensing is highly linked to the cadherin family of proteins on a molecular level [63, 90]. Cadherins bind through their cytoplasmic domain to β-catenin which in turn tethers cadherin to the actin cytoskeleton via α-catenin. Like in single talin molecules, catenins can undergo conformational straightening upon increased biomechanical load, exposing a vinculin binding domain and thereby facilitating vinculin binding [63]. Recruitment of vinculin and metavinculin to intercalated discs is believed to rather stiffen the whole ID complex, whereas loss of vinculin or metavinculin, as well as missense mutations in vinculin, have been linked to development and progression of dilated cardiomyopathy [63, 91]. Phosphorylation of vinculin protein at residue Tyr 822 occurs upon stretch and is recognized as another fine-tuning principle in ID-based vinculin signaling [63]. At intercalated discs, the protein N-RAP (nebulin-related-anchoring protein) is able to form another N-cadherin/integrin signaling complex by its interaction with talin, vinculin, MLP, and alpha-actinin and also crosslinks cadherin and integrin signaling. N-RAP is upregulated in MLP knockout mice and dilated cardiomyopathy [63, 92], while transgenic N-RAP overexpression in mice results in right ventricular cardiomyopathy [63], emphasizing its role in proper heart muscle function.
5.1 Stretch Signaling at Cardiac Desmosomes
The desmosome, another specific cell-cell contact structure, represents a multiprotein complex composed of transmembrane cadherin family members desmoglein (DSG2, which has been proposed to sense mechanical forces) or desmocollin, whose extracellular domain interacts with opposite facing cadherin molecules at neighboring cardiomyocytes [13, 93]. Their corresponding intracellular domain thereby interacts with intermediate filaments via the desmosomal proteins plakoglobin, plakophilin and desmoplakin [63]. As outlined, desmoglein is believed to be directly involved in desmosomal strain sensing, since targeted deletion of the extracellular domain of desmoglein in mice leads to a biventricular form of ARVC including biventricular dilatation and dysfunction as well as sudden cardiac death. At the ultrastructural level, mouse hearts showed profound enlargement of desmosomal intercellular gaps with destroyed desmosomal structure, which seemed to coincide with visible heart lesions at the macroscopical level. Moreover, multiple desmosomal proteins and associated cellular pathways are involved in the pathogenesis and architectural features of arrhythmogenic right ventricular cardiomyopathies and the risk of sudden cardiac death. Whether altered mechanoperception or mainly altered desmosomal architecture or conduction abnormalities and impairment of cell-cell connections are key features is still under constant scientific debate. To date adipogenic/fibrogenic gene expression and resulting ultrastructural changes are regarded as the main hallmark of ARVC.
6 Sarcolemmal Strain Sensing and Mechanoelectrical Feedback via Mechanosensitive Ion Channels
Besides elements of the cardiac contractile apparatus and the cell junction system, the sarcolemma, representing a global interface to neighboring cells and the extracellular matrix (ECM), and its embedded ion channel network seem to be an ideal subcellular structure for force sensing, conformational alterations, and downstream regulation of ion fluxes according to differential stages of membrane strain or increased cellular volumes [12, 15, 94]. The idea of mechanosensitive ion channels (MCS) has been largely analyzed in non-cardiomyocytes, but moreover, even every specific channel that is also expressed in cardiomyocytes has clearly proven its functional relevance in cardiovascular health and disease so far [12, 94]. In general, mechanosensitive ion channel signaling in muscle can be categorized into stretch-activated (SAC) and volume-activated (VAC) ion channel mechanisms [12, 15, 94]. But despite a potential lack of mechanistic understanding, since the clinical observation that a strong precordial fist thump, as well as mechanical thoracic compression, is eventually able to alter or restore cardiac electrical activity, the idea of a direct mechanoelectrical feedback without the need of ligands or second messengers has emerged [12] (Table 1).
Mechanistically, besides strain-dependent conformational changes, leading to enhanced open probabilities and increasing ion fluxes, additional modifications like phosphoinosite binding, phosphorylation, or altered protein-protein interactions have been described [94]. In terms of phosphoinosite-dependent sarcolemmal stretch signaling, the phosphatidylinositol 3-kinase (PI3K) pathway plays a pivotal role in cardiomyocytes [95]. In response to increased cardiomyocyte mechanical strain, the phosphoinosite-converting enzyme PI3K translocates to the plasma membrane to locally convert phosphatidylinositol (4,5)-bisphosphate (PIP2) into phosphatidylinositol (3,4,5)-trisphosphate (PIP3) [95]. In turn, PIP3 activates the phosphoinositide-dependent protein kinase-1 (PDK1) through its pleckstrin- homology (PH-) domain, and PDK1 subsequently phosphorylates and activates the serine/threonine-specific protein kinase Akt at threonine residue 308, which is further stabilized by additional phosphorylation of serine 473 by mammalian target or rapamycin (mTOR). Akt activation results in a variety of prohypertrophic and prosurvival signals, and is also believed to directly regulate the strain-dependent titin isoform switch [95]. PIP3 accumulation also subsequently recruits gelsolin to the plasma membrane, thereby negatively regulating its activity and pathological actin remodeling [95]. Finally, it has been recently shown that the protein kinase Akt together with the strain-sensitive channel polycystin-1 can stabilize sarcolemmal LTCC levels by phosphorylation-dependent prevention of proteosomal degradation [96]. This finding represents another important principle, directly and indirectly linking cardiomyocyte sarcolemmal stretch sensing to calcium-induced calcium release and excitation-contraction coupling.
6.1 Mechanosensitive Transient Receptor Potential Channels (Polycystins) in the Heart
Various transient receptor potential channels (TRPs), though initially and sometimes extensively studied in non-cardiac cells, are expressed in cardiomyocytes and are believed to play a pivotal role in mechanoelectrical feedback processes [94]. The subfamily of transient receptor potential polycystins (TRPPs), polycystin 1 and 2 (encoded by PKD1 and PKD2 genes), are involved in sensing mechanical forces and fluid shear stress, triggering multiple intracellular signaling pathways [97, 98]. The notion that cardiac specific polycystin-1 knockout mice develop spontaneous cardiomyopathy supports the concept that they are required to maintain normal cardiac function [99, 100]. Moreover, polycystin-1−/− mice displayed shorter action potential and shortening of QT intervals in surface ECGs, a phenotype that has also been linked to loss of function mutations in the LTCC coding CACNA1C gene or other mouse models of compromised LTCC function-associated cardiomyopathies [101]. TRPP2/polycystin-2 is expressed in cardiomyocyte endoplasmatic reticulum and has also been linked to sarcoplasmatic ryanodine receptor 2 (RYR2) function [97, 98].
6.2 Other Mechanosensitive Calcium Channels in the Heart
Besides TRPP channels, there are over 33 genes that encode TRP subunits, which form a superfamily of mammalian channels, further subclassified as TRP-C, TRP-V, TRP-M, TRP-P, TRP-ML, TRP-N, and TRP-A subfamilies [94]. Several members of the TRPC (“C” for canonical) family channels, especially TRPC1, TRPC3, and TRPC6, are considered to be mechanosensitive and important for accurate cardiomyocyte function and signaling [102, 103]. TRPC1 expression has been shown to increase in an induced rat heart hypertrophy model, while TRPC1−/− mice seem protected from cardiac hypertrophy, which underlines its crucial function in pathological hypertrophic signaling [104]. Most surprisingly, the functions of these multiple TRPC channels do not seem to be redundant in cardiomyocytes, potentially because they act through different regulatory mechanisms [105]. Mice with heart-specific overexpression of TRPC6 develop spontaneous cardiac hypertrophy and pathological remodeling, whereas TRPC6 deletion or inhibition has been shown to be cardioprotective [106, 107]. Of note, TRCPC6 channels are also partially regulated by PI3K-dependent exocytosis.
Since it has been shown that expression of TRP cation channel, subfamily C, member 3 (TRPC3) is increased in response to calcineurin signaling [52, 102], a potential link from TRPC6 function to calcineurin has also been analyzed. Interestingly, TRPC6 abundance was differentially upregulated in mouse hearts in response to pressure overload and, most strikingly, in failing human hearts. Two conserved NFAT consensus sites in the promoter of the TRPC6 gene have been discovered, representing a potential reciprocal activation circuit in which calcineurin activation results in increased TRPC6 expression [50, 108]. In turn, in vivo pathological cardiac overexpression of TRPC6 was associated with enhanced calcineurin signaling leading to pathologic cardiac growth and heart failure [50].
6.3 Mechanosensitive Potassium Channels in the Heart
Besides TRP channels, multiple types of mechanosensitive K+ channels are expressed in different parts of the heart [12, 94]. TREK-1 [(TWIK)-related K+ channel], also known as potassium channel subfamily K member 2, is expressed in both the atria and ventricles, and extensive research has shown that TREK-1 and its human homologue TRAAK (TWIK-related arachidonic acid stimulated K+ channel) are mechanosensitive [109,110,111,112]. TREK-1 channels are aligned in longitudinal stripes on the cardiomyocyte surface, which potentially facilitates bidirectional strain sensing [112]. TREK-1 channels that can easily be opened by either pipet suction or pressure in vitro have thereby been linked to arrhythmogenesis and the onset of atrial fibrillation as well as heart failure [12, 94]. The role for TREK-1 in arrhythmogenesis is further strengthened by the fact that several anti-arrhythmic drugs, including lidocaine, mexiletine, propafenone, dronedarone, and vernakalant, inhibit TREK-1 channel function. Compared to TREK-1, the human relevance of TRAAK is less well understood [12, 94].
Recently two newly discovered proteins, Piezo1 and Piezo2, which assemble as transmembrane trimers, were also proposed as bona fide stretch-activated channels with striking characteristics in sarcolemmal force sensing and potentially regulating calcium influx [94]. Piezo1 channels are upregulated in rodent heart failure but are downregulated and potentially delocalized in in vitro cardiomyocyte stretch models [113]. Being largely characterized in endothelial cells and shear stress scenarios, more research in hearts and cardiomyocytes is needed to fully clarify the role of Piezo channels in mechanotransduction in cardiac muscle health and disease.
7 The Microtubule Network in Cardiomyocyte Stretch Sensing: More Than Just a Skeleton
The view of the microtubule’s role in cardiac function and mechanotransduction has experienced a fundamental change along with the availability of more sophisticated observational and manipulative molecular techniques [114, 115]. Like with other molecules involved in cardiac strain sensing, direct observation of dynamic microtubule networks in beating myocytes also suggests a complex behavior with spring-like functions that is additionally fine-tuned by post-translational modifications [115]. Maybe most relevant, targeted detyrosination (dTyr) of microtubules is a result of enzymatic cleavage of a C-terminal tyrosine residue and is thereby able to facilitate specific interactions with intermediate filament proteins like desmin that link microtubules to the sarcomeric Z-disc or components of the nuclear envelope [26, 116, 117]. Beyond that, recent progress in elegant live imaging techniques has helped to visualize growing microtubule networks in cardiomyocytes. These experiments revealed that microtubules likely grow from Z-disc to Z-disc and even tend to pause, drift, or deviate on this central sarcomeric signaling hub. Strikingly, this growth behavior implicates local protein-microtubule interactions and possible modifications at the sarcomeric Z-disc in affecting microtubule architecture [115]. Modified microtubules are known to alter cardiomyocyte stiffness, resulting in overall increased myocyte viscoelasticity conditioned by elevated detyrosination [114]. Microtubules are also believed to modify cardiac contractility and cytoskeletal mechanosignaling through a variety of processes involving cellular organelles like mitochondria and the nucleus [7, 14, 25, 115]. Moreover, microtubules have been shown to form interactions with structural components and ion channels within the t-tubule system and sarcoplasmic reticulum, controlling ion channel membrane trafficking and indirectly EC coupling [114, 115]. Elevated levels of dTyr microtubules can be a result of elevated levels of reactive oxygen species (ROS) and levels of nitric oxide (NO) as they occur during increased mechanical stretch in cardiomyocytes [115]. dTyr microtubule abundance is also increased in human cardiomyopathy, and increasing detyrosinated micotubules herein correlated with declining systolic function [116]. Moreover, dTyr microtubules are believed to have in particular an increased affinity to bind the intermediate filament and strain-sensing molecule desmin [115]. In this setting, also proper positioning of organelles like mitochondria and the cardiomyocyte nucleus involves the cardiac actin network, desmin, and the microtubule system [26]. Alterations in all these structures (a hallmark of heart failure remodeling) also result in impaired cardiomyocyte mitochondrial metabolism and Ca2+ cycling as well as impaired nuclear function, integrity, and even possible DNA damage [25, 115, 116].
7.1 Mechanotransduction at the Cardiomyocyte’s Nuclear Lamina
The cardiomyocyte nucleus, representing the command center of cardiomyocyte gene regulation, is another important organelle involved in mechanoperception [25, 26, 118]. Lamins are the main protein components of the inner nuclear lamina and form stable filament webs inside the whole nucleus [119, 120]. Lamins are intermediate filament proteins divided into A-type lamins, derived from alternative splicing of the LMNA gene, and B-type lamins that are encoded by distinct LMNB1 and LMNB2 genes, respectively [121, 122]. Experimental evidence has demonstrated that lamin structures play pivotal roles in the maintenance of normal nuclear mechanics and cardiomyocyte mechanotransduction [119, 123]. Besides its nuclear functions, lamins affect the mechanical properties of the cytoplasm and the organization of cytoskeletal elements as well as regulation of the overall cell shape [119, 122, 124]. Hence, lamins are believed to interact with multiple proteins and thereby can regulate gene expression, chromatin homeostasis, and even nuclear positioning [124, 125]. Cells from lamin knockout mice show decreased association of desmin at the nuclear surface and severe alterations of actin-, vimentin-, and tubulin-based filament structures. A-type lamins, represented by lamins A and C, are developmentally regulated proteins found in high abundance in the skeletal and cardiac muscle. The crucial function of these proteins is emphasized by the facts that mice deficient in lamin A and C develop severe muscular dystrophy and die prematurely at the age of 6–8 weeks [124]. Moreover, human relevance is underlined by a group of diseases caused by mutation variants in the LMNA gene sequence [126, 127]. Laminopathies are usually classified into four groups, according to the number and the types of the affected tissues. The first group represents lamin myopathies affecting both the skeletal and the cardiac muscles [128]. Mutations in the lamin A gene are generally considered one of the most common mutations associated with this disease. LMNA gene defects are believed to account for almost one third of dilated cardiomyopathy cases accompanied by atrioventricular block [128]. Given the knowledge that cells expressing mutated A-type lamins display histological lobulations in the nuclear envelope, loss of peripheral heterochromatin, and anomalous nuclear pore complex distribution, two main models were hypothesized to explain the onset of laminopathies [25, 124]: according to the “structural model,” mutations in A-type lamins impair the nuclear resistance to mechanical stimuli, resulting in fragility, increased stress sensitivity, and possibly premature senescence [25, 124]. This model would explain why in particular striated muscle tissues, which are frequently exposed to mechanical strain, are mainly affected by “laminopathies.” The mechanistic role of lamins in cardiac mechanotransduction is also related to their unique filamental structure and biophysical behavior. Assembly of the nuclear lamin mesh starts with lamin dimer assembly and subsequent dimer aggregation head to tail, finally forming small polymer filaments [121].
Under increasing cardiomyocyte tension, integrins transmit a strain-dependent external impulse to the cytoskeleton, which then transfers the stimulus to the nuclear lamina structure. In turn, lamins are rearranged at the molecular level, leading to unfolding of lamin A immunoglobulin domains and/or alterations of flexible linkers L1, L12, and L3 in the protein structure, as well as alterations in the dimer head-tail interaction. Within these proposed “altered protein complex” models, mechanical strain straightens the lamin proteins as well as the overall lamin-nuclear-mesh potentially exposing protein-binding regions [128]. The global lamin-mesh rearrangement has been correlated with a higher nuclear localization of cardiac transcription factors (cFOS, RARG, JNK, SRF, MRTF, Yap1, and ERK), and their specific affinity to chromatin by exposing binding sites for these transcription factors to increase transcription of cytoskeletal components [119, 125, 128, 129]. Moreover, cardiomyocytes can modulate the nuclear biophysical properties by changing the phosphorylation level of the lamins, thereby affecting both the structural lamina conformation and stiffness. In this case increased force transmission results in hiding of the sites for phosphorylation. The inhibited phosphorylation activity increases the amount of lamins at the nuclear envelope with a consequent increase in lamina stiffness [130, 131].
7.2 Mechanotransduction by Protein Complexes Within the Nuclear Envelope
But besides the lamin web, there are other integral parts of the nuclear envelope that are involved in mechanotransduction like LINC complexes (LInker of Nucleoskeleton and Cytoskeleton) that contain different nesprins, emerin, and SUNs (named for SUN domain family members (Sad1p, Unc-84). LINC complexes can sense and regulate myocyte-wide strain transfer to the nucleus itself [25, 26, 132]. LINC complex disruptions have been analyzed and described in cells that typically are prone to experience high mechanical strain, such as myocytes and cardiomyocytes. Nesprins share the common capabilities of stretch sensing molecules, as they have been shown to unfold upon envelope strain, exposing binding sites that promote either dimerization or recruitment of additional binding proteins, facilitating complex stability and rigidity [25, 26]. In mice, deletion of nesprin 1 and 2 gene function results in cardiomyopathy and altered gene expression in response to myocyte pressure overload [133, 134]. Linking the nucleus to the cytoskeleton, microtubules not only can provide compressive forces on nuclei but also show multiple interactions to nesprin 1 and 2 [135, 136]. It is speculated that loss of desmin-nesprin-microtubule interactions or disruption of LINC in nuclear envelopes results in nuclear membrane infolding, driven by external forces applied through external microtubule-network compression [26]. Since correct assembly of LINC, desmin, and the microtubule network is required for accurate nuclear shape, the integrity, positioning, and homeostasis, nesprin-dependent regulation of LINC stability might represent another important cardiac mechanotransduction principle [25, 26, 134,135,136,137,138].
8 Mechanotransduction and Mechanosensitive Gene Induction
Extensive gene expression profiling in cardiomyocytes using cDNA microarrays or RNA sequencing has helped to gain more insight into strain-associated gene regulation and led to the discovery of cardiac mechanosensitive gene programs [139]. In vitro models of cardiomyocyte stretch as well as animal models of experimental pressure overload by transverse aortic constriction (TAC) have consistently demonstrated increased expression of natriuretic peptide genes and re-expression of a so-called fetal gene program. Beyond that Mt1 (encoding metallothionein 1) and various other genes encoding proteins involved in mitochondrial metabolism or the cytoskeleton are believed to be expressed or re-expressed in a strain-dependent manner in cardiomyocytes [140]. More cardiomyocyte genes with potentially specific mechanosensitive expression have been discovered like suppression of tumorigenicity-2 (ST2) and the TGF-beta superfamily member growth differentiation factor-15 (GDF15) [141], which subsequently have been developed as a clinical biomarker for myocardial infarction, hemodynamic cardiac load, or heart failure [142,143,144,145]. Myocardial gene expression can be regulated on the posttranscriptional level by a variety of mechanisms like certain microRNAs that are also believed to be differentially expressed in various loading conditions or that are involved in the regulation of mechanotransduction. MicroRNAs are short, non-coding RNAs that bind complementary mRNAs to control mRNA degradation or subsequent protein translation. In an effort to identify specific microRNAs regulated through myocyte stretch, we and others used microarrays under different experimental loading conditions in cardiomyocytes in vitro. These experiments identified miR-20a as being responsive to both stretch and simulated ischemia reperfusion and that overexpression of miR-20a was sufficient to protect cardiomyocytes from apoptosis [146].
9 Translational Perspectives
The increasing insight into molecular mechanisms of mechanoperception and transmission (mechanotransduction) is the potential basis for the development of new therapeutic approaches. Biomechanical stress sensors and downstream signaling pathways involving the above described sarcomere-associated molecules are essential for physiological cardiac function [2, 19]. Impairment or dysregulation of this complex network is directly associated with a variety of congenital and acquired cardiac diseases such as laminopathies, dystrophinopathies, desminopathies, arrhythmogenic right ventricular cardiomyopathy, familial restrictive cardiomyopathy (RCM), valvular and ischemic heart diseases, and dilated and/or hypertrophic cardiomyopathy (DCM, HCM) [27, 54, 56, 58, 63].
The causes for laminopathies, dystrophinopathies, and desminopathy are known mutations in specific genes encoding for proteins associated with sarcomeric proteins, namely, lamins (LMNA), dystrophin (DMD), and desmin (DES) [55, 56, 147,148,149]. In addition, dozens of mutations in genes encoding for different sarcomeric proteins involved in mechanoperception and translation of stretch-induced signals have been identified to contribute to different cardiomyopathies [38, 98, 150, 151]. Basic research using knockdown approaches in in vitro and in vivo models as well as OMICs studies analyzing patient data can help to identify even more mutations and polymorphisms as risks factors for cardiomyopathies (Fig. 3).


List of mostly accepted cardiomyopathy-associated genes in relation to their disease phenotypes. Genes/proteins that are believed to be directly or indirectly involved in cardiac stretch sensing and mechanotransduction are highlighted in red. Web access to comprehensive lists of cardiomyopathy-associated genes and gene variants is possible via https://www.ncbi.nlm.nih.gov/clinvar/?term=cardiomyopathy, https://www.cardiodb.org/acgv/, https://seidman.hms.harvard.edu/?page_id=1476
Besides congenital mutations, chronically increased biomechanical stress on the ventricular myocardium, which occurs as a result of pressure overload in hypertension and valvular but also after remodeling processes in ischemic heart diseases and dilated/hypertrophic cardiomyopathies, leads to alterations in signaling cascades triggered by mechanotransduction and subsequently accelerates disease progression to heart failure. A better understanding of the structural and regulatory role of sarcomere-associated proteins in mechanotransduction has already led to tests of new pharmaceutical approaches. In recent years especially ion channels/transporters have come into focus as targets to block detrimental signaling induced by biomechanical stress in the myocardium leading to cardiac remodeling and heart failure. Examples are cariporide treatment of patients with ischemic heart disease and in in vivo models of cardiac hypertrophy [152], rimeporide treatment of children with dystrophinopathy [153], and inhibition of K+ channels in models of atrial fibrillation [154, 155]. However, further research is essential, as the inhibition often lacks specificity and may increase the risk for side effects (e.g., strokes, as reported for the cariporide treatment [152]), and the molecular mechanisms are still not completely understood (e.g., association of anti-arrhythmic drugs with inhibition of TREK-113 on the one hand but correlation of impairment of TREK-1 with arrhythmogenesis on the other hand).
As described above, many other members of the network of cardiac mechanoperception and mechanotransduction are currently studied in in vitro and in vivo experiments with regard to their exact function, and further studies might demonstrate their potential in therapeutic approaches (e.g., titin, MLP, LMCD1, myomasp/LRRC39, melusin, polycystin, talin, TRPC1 and TRPC6, and nesprin).
In this context, not only the development of conventional pharmaceutics is of interest, but also the constantly growing field of gene therapies amplifies the potential targets and therapeutic approaches including a “personalized medicine” approach for genetic disease.
The most promising methods for gene therapy are CRISPR/Cas-based and vector, mostly adeno-associated virus (AAV), mediated approaches. In recent years more and more successful gene editing in vitro and in vivo has been performed, e.g., for removal of HIV proviral DNA sequences or host receptors to treat HIV infections, targeting the convertase PCSK9 which is associated with high blood lipid levels to treat cardiovascular diseases or to correct mutations causing sickle cell disease in stem cells [156]. Although gene editing in humans is ethically controversial, first human cells with induced disruption of genes associated with inhibition of anti-tumor responses and introduced tumor recognition genes have been used to treat cancer patients [157]. With regard to heart diseases, AAV9-mediated CRISPR/Cas9-based gene editing has been used to target specific genes in cardiomyocytes in animal models, too [156, 158]. Along these lines, a number of new animal models for, e.g., dystrophies, cardiovascular diseases, and dilated cardiomyopathies have been created for further research [156]. Furthermore, first in vivo corrections of mutations which cause heart diseases have been described [156, 159], e.g., an AAV9-Cas9-based approach to restore functional dystrophin levels in a canine model for Duchenne muscular dystrophy [89, 156]. The potential of gene therapies in general and for genetic and acquired cardiomyopathies in particular is immense, as beyond correcting mutations, possible targets for CRISPR/Cas approaches to treat cardiac diseases are discussed [159, 160]. In this context, proteins involved in mechanotransduction are promising targets and await further research. But beyond these potential promising possibilities of gene therapeutic approaches in genetic cardiomyopathies, there are numerous scientific, ethic, and economic limitations with respect to a broad bedside use in humans.
10 Conclusions
Cardiac and skeletal muscle cells bidirectionally sense and transmit (outside-in and inside-out) mechanical forces between the extracellular matrix, the contractile apparatus, and various organelles, and they consecutively respond via structural changes and altered signal transduction, a process known as cardiac mechanotransduction. Increases in mechanical load of cardiac myocytes lead to biochemical signals and induce cellular hypertrophy, an initially adaptive response that, through persistent strain exposure, ultimately leads to pathological hypertrophy, increased tissue fibrosis and stiffness, predisposition to various arrhythmias, and subsequent terminal heart failure. Over the past decade, increasing scientific evidence has emerged that sensing cardiac load involves not the “cardiac stretch sensor” but rather divergent, different, and multiple mechanisms involving a large variety of structural and regulatory proteins at distinct subcellular localizations. These recent scientific advances provide better mechanistic insight into the earliest manifestations of pathologic cardiac hypertrophy and heart failure. And beyond this, such progress also promises future possibilities of better therapeutic interventions for diseases still leading mortality and death rates in the Western world.
References
Gjesdal O, Bluemke DA, Lima JA (2011) Cardiac remodeling at the population level–risk factors, screening, and outcomes. Nat Rev Cardiol 8:673–685. https://doi.org/10.1038/nrcardio.2011.154
Neves JS et al (2015) Acute myocardial response to stretch: what we (don’t) know. Front Physiol 6:408. https://doi.org/10.3389/fphys.2015.00408
Garoffolo G, Pesce M (2019) Mechanotransduction in the cardiovascular system: from developmental origins to homeostasis and pathology. Cells 8. https://doi.org/10.3390/cells8121607
Kitmitto A, Baudoin F, Cartwright EJ (2019) Cardiomyocyte damage control in heart failure and the role of the sarcolemma. J Muscle Res Cell Motil 40:319–333. https://doi.org/10.1007/s10974-019-09539-5
Lyon RC, Zanella F, Omens JH, Sheikh F (2015) Mechanotransduction in cardiac hypertrophy and failure. Circ Res 116:1462–1476. https://doi.org/10.1161/CIRCRESAHA.116.304937
Boycott HE, Nguyen MN, Vrellaku B, Gehmlich K, Robinson P (2020) Nitric oxide and mechano-electrical transduction in cardiomyocytes. Front Physiol 11:606740. https://doi.org/10.3389/fphys.2020.606740
Chen-Izu Y, Izu LT (2017) Mechano-chemo-transduction in cardiac myocytes. J Physiol 595:3949–3958. https://doi.org/10.1113/JP273101
Gaetani R et al (2020) When stiffness matters: mechanosensing in heart development and disease. Front Cell Dev Biol 8:334. https://doi.org/10.3389/fcell.2020.00334
Haque ZK, Wang DZ (2017) How cardiomyocytes sense pathophysiological stresses for cardiac remodeling. Cell Mol Life Sci 74:983–1000. https://doi.org/10.1007/s00018-016-2373-0
Parra V, Rothermel BA (2017) Calcineurin signaling in the heart: the importance of time and place. J Mol Cell Cardiol 103:121–136. https://doi.org/10.1016/j.yjmcc.2016.12.006
Pasqualini FS, Nesmith AP, Horton RE, Sheehy SP, Parker KK (2016) Mechanotransduction and metabolism in cardiomyocyte microdomains. Biomed Res Int 2016:4081638. https://doi.org/10.1155/2016/4081638
Peyronnet R, Nerbonne JM, Kohl P (2016) Cardiac mechano-gated ion channels and arrhythmias. Circ Res 118:311–329. https://doi.org/10.1161/CIRCRESAHA.115.305043
Citi S (2019) The mechanobiology of tight junctions. Biophys Rev 11:783–793. https://doi.org/10.1007/s12551-019-00582-7
Cabassi A, Miragoli M (2017) Altered mitochondrial metabolism and mechanosensation in the failing heart: focus on intracellular calcium signaling. Int J Mol Sci 18. https://doi.org/10.3390/ijms18071487
Jones PP, MacQuaide N, Louch WE (2018) Dyadic plasticity in cardiomyocytes. Front Physiol 9:1773. https://doi.org/10.3389/fphys.2018.01773
Koser F, Loescher C, Linke WA (2019) Posttranslational modifications of titin from cardiac muscle: how, where, and what for? FEBS J 286:2240–2260. https://doi.org/10.1111/febs.14854
Gautel M (2011) The sarcomeric cytoskeleton: who picks up the strain? Curr Opin Cell Biol 23:39–46. https://doi.org/10.1016/j.ceb.2010.12.001
Nakamura M, Sadoshima J (2018) Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 15:387–407. https://doi.org/10.1038/s41569-018-0007-y
Takahashi K, Kakimoto Y, Toda K, Naruse K (2013) Mechanobiology in cardiac physiology and diseases. J Cell Mol Med 17:225–232. https://doi.org/10.1111/jcmm.12027
Ward M, Iskratsch T (2020) Mix and (mis-)match – the mechanosensing machinery in the changing environment of the developing, healthy adult and diseased heart. Biochim Biophys Acta Mol Cell Res 1867:118436. https://doi.org/10.1016/j.bbamcr.2019.01.017
Buyandelger B et al (2011) MLP (muscle LIM protein) as a stress sensor in the heart. Pflugers Arch 462:135–142. https://doi.org/10.1007/s00424-011-0961-2
Mao Z, Nakamura F (2020) Structure and function of filamin C in the muscle Z-disc. Int J Mol Sci 21. https://doi.org/10.3390/ijms21082696
Frank D, Frey N (2011) Cardiac Z-disc signaling network. J Biol Chem 286:9897–9904. https://doi.org/10.1074/jbc.R110.174268
Le S et al (2018) Dystrophin as a molecular shock absorber. ACS Nano 12:12140–12148. https://doi.org/10.1021/acsnano.8b05721
Graham DM, Burridge K (2016) Mechanotransduction and nuclear function. Curr Opin Cell Biol 40:98–105. https://doi.org/10.1016/j.ceb.2016.03.006
Heffler J et al (2020) A balance between intermediate filaments and microtubules maintains nuclear architecture in the cardiomyocyte. Circ Res 126:e10–e26. https://doi.org/10.1161/CIRCRESAHA.119.315582
Knoll R et al (2002) The cardiac mechanical stretch sensor machinery involves a Z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 111:943–955. https://doi.org/10.1016/s0092-8674(02)01226-6
Vafiadaki E, Arvanitis DA, Sanoudou D (2015) Muscle LIM protein: master regulator of cardiac and skeletal muscle functions. Gene 566:1–7. https://doi.org/10.1016/j.gene.2015.04.077
Herzog W (2018) The multiple roles of titin in muscle contraction and force production. Biophys Rev 10:1187–1199. https://doi.org/10.1007/s12551-017-0395-y
Linke WA (2008) Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res 77:637–648. https://doi.org/10.1016/j.cardiores.2007.03.029
Hamdani N, Herwig M, Linke WA (2017) Tampering with springs: phosphorylation of titin affecting the mechanical function of cardiomyocytes. Biophys Rev 9:225–237. https://doi.org/10.1007/s12551-017-0263-9
Swist S et al (2020) Maintenance of sarcomeric integrity in adult muscle cells crucially depends on Z-disc anchored titin. Nat Commun 11:4479. https://doi.org/10.1038/s41467-020-18131-2
Kruger M, Kotter S (2016) Titin, a central mediator for hypertrophic signaling, exercise-induced mechanosignaling and skeletal muscle remodeling. Front Physiol 7:76. https://doi.org/10.3389/fphys.2016.00076
Kruger M, Linke WA (2009) Titin-based mechanical signalling in normal and failing myocardium. J Mol Cell Cardiol 46:490–498. https://doi.org/10.1016/j.yjmcc.2009.01.004
Radke MH et al (2019) Deleting full length titin versus the titin M-band region leads to differential mechanosignaling and cardiac phenotypes. Circulation 139:1813–1827. https://doi.org/10.1161/CIRCULATIONAHA.118.037588
Tonino P et al (2017) The giant protein titin regulates the length of the striated muscle thick filament. Nat Commun 8:1041. https://doi.org/10.1038/s41467-017-01144-9
Granzier HL et al (2014) Deleting titin’s I-band/A-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci U S A 111:14589–14594. https://doi.org/10.1073/pnas.1411493111
Knoll R et al (2010) A common MLP (muscle LIM protein) variant is associated with cardiomyopathy. Circ Res 106:695–704. https://doi.org/10.1161/CIRCRESAHA.109.206243
Kuhn C et al (2012) Cardiac remodeling is not modulated by overexpression of muscle LIM protein (MLP). Basic Res Cardiol 107:262. https://doi.org/10.1007/s00395-012-0262-8
Weins A et al (2001) Differentiation- and stress-dependent nuclear cytoplasmic redistribution of myopodin, a novel actin-bundling protein. J Cell Biol 155:393–404. https://doi.org/10.1083/jcb.200012039
Faul C, Dhume A, Schecter AD, Mundel P (2007) Protein kinase A, Ca2+/calmodulin-dependent kinase II, and calcineurin regulate the intracellular trafficking of myopodin between the Z-disc and the nucleus of cardiac myocytes. Mol Cell Biol 27:8215–8227. https://doi.org/10.1128/MCB.00950-07
Ulbricht A et al (2013) Cellular mechanotransduction relies on tension-induced and chaperone-assisted autophagy. Curr Biol 23:430–435. https://doi.org/10.1016/j.cub.2013.01.064
Creamer TP (2020) Calcineurin. Cell Commun Signal 18:137. https://doi.org/10.1186/s12964-020-00636-4
Molkentin JD et al (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93:215–228. https://doi.org/10.1016/s0092-8674(00)81573-1
Bueno OF et al (2002) Impaired cardiac hypertrophic response in Calcineurin Abeta-deficient mice. Proc Natl Acad Sci U S A 99:4586–4591. https://doi.org/10.1073/pnas.072647999
Dierck F et al (2017) The novel cardiac z-disc protein CEFIP regulates cardiomyocyte hypertrophy by modulating calcineurin signaling. J Biol Chem 292:15180–15191. https://doi.org/10.1074/jbc.M117.786764
Frey N et al (2004) Mice lacking calsarcin-1 are sensitized to calcineurin signaling and show accelerated cardiomyopathy in response to pathological biomechanical stress. Nat Med 10:1336–1343. https://doi.org/10.1038/nm1132
Jeong D et al (2008) PICOT attenuates cardiac hypertrophy by disrupting calcineurin-NFAT signaling. Circ Res 102:711–719. https://doi.org/10.1161/CIRCRESAHA.107.165985
Kielbasa OM et al (2011) Myospryn is a calcineurin-interacting protein that negatively modulates slow-fiber-type transformation and skeletal muscle regeneration. FASEB J 25:2276–2286. https://doi.org/10.1096/fj.10-169219
Kuwahara K et al (2006) TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest 116:3114–3126. https://doi.org/10.1172/JCI27702
Nakamura A, Yoshida K, Takeda S, Dohi N, Ikeda S (2002) Progression of dystrophic features and activation of mitogen-activated protein kinases and calcineurin by physical exercise, in hearts of mdx mice. FEBS Lett 520:18–24. https://doi.org/10.1016/s0014-5793(02)02739-4
Nakayama H, Wilkin BJ, Bodi I, Molkentin JD (2006) Calcineurin-dependent cardiomyopathy is activated by TRPC in the adult mouse heart. FASEB J 20:1660–1670. https://doi.org/10.1096/fj.05-5560com
Cha H et al (2008) PICOT is a critical regulator of cardiac hypertrophy and cardiomyocyte contractility. J Mol Cell Cardiol 45:796–803. https://doi.org/10.1016/j.yjmcc.2008.09.124
McLendon PM, Robbins J (2011) Desmin-related cardiomyopathy: an unfolding story. Am J Physiol Heart Circ Physiol 301:H1220–H1228. https://doi.org/10.1152/ajpheart.00601.2011
Klauke B et al (2010) De novo desmin-mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum Mol Genet 19:4595–4607. https://doi.org/10.1093/hmg/ddq387
Goldfarb LG et al (1998) Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 19:402–403. https://doi.org/10.1038/1300
Kreplak L, Herrmann H, Aebi U (2008) Tensile properties of single desmin intermediate filaments. Biophys J 94:2790–2799. https://doi.org/10.1529/biophysj.107.119826
Lange S, Pinotsis N, Agarkova I, Ehler E (2020) The M-band: the underestimated part of the sarcomere. Biochim Biophys Acta Mol Cell Res 1867:118440. https://doi.org/10.1016/j.bbamcr.2019.02.003
Lange S et al (2005) The kinase domain of titin controls muscle gene expression and protein turnover. Science 308:1599–1603. https://doi.org/10.1126/science.1110463
Gotthardt M et al (2003) Conditional expression of mutant M-line titins results in cardiomyopathy with altered sarcomere structure. J Biol Chem 278:6059–6065. https://doi.org/10.1074/jbc.M211723200
Gramlich M et al (2009) Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J Mol Cell Cardiol 47:352–358. https://doi.org/10.1016/j.yjmcc.2009.04.014
Will RD et al (2010) Myomasp/LRRC39, a heart- and muscle-specific protein, is a novel component of the sarcomeric M-band and is involved in stretch sensing. Circ Res 107:1253–1264. https://doi.org/10.1161/CIRCRESAHA.110.222372
Pruna M, Ehler E (2020) The intercalated disc: a mechanosensing signalling node in cardiomyopathy. Biophys Rev 12:931–946. https://doi.org/10.1007/s12551-020-00737-x
Borrmann CM et al (2006) The area composita of adhering junctions connecting heart muscle cells of vertebrates. II. Colocalizations of desmosomal and fascia adherens molecules in the intercalated disk. Eur J Cell Biol 85:469–485. https://doi.org/10.1016/j.ejcb.2006.02.009
Franke WW, Borrmann CM, Grund C, Pieperhoff S (2006) The area composita of adhering junctions connecting heart muscle cells of vertebrates. I. Molecular definition in intercalated disks of cardiomyocytes by immunoelectron microscopy of desmosomal proteins. Eur J Cell Biol 85:69–82. https://doi.org/10.1016/j.ejcb.2005.11.003
McCain ML, Lee H, Aratyn-Schaus Y, Kleber AG, Parker KK (2012) Cooperative coupling of cell-matrix and cell-cell adhesions in cardiac muscle. Proc Natl Acad Sci U S A 109:9881–9886. https://doi.org/10.1073/pnas.1203007109
Sit B, Gutmann D, Iskratsch T (2019) Costameres, dense plaques and podosomes: the cell matrix adhesions in cardiovascular mechanosensing. J Muscle Res Cell Motil 40:197–209. https://doi.org/10.1007/s10974-019-09529-7
McDaniel DP et al (2007) The stiffness of collagen fibrils influences vascular smooth muscle cell phenotype. Biophys J 92:1759–1769. https://doi.org/10.1529/biophysj.106.089003
Anthis NJ et al (2009) The structure of an integrin/talin complex reveals the basis of inside-out signal transduction. EMBO J 28:3623–3632. https://doi.org/10.1038/emboj.2009.287
Gingras AR et al (2009) Structural determinants of integrin binding to the talin rod. J Biol Chem 284:8866–8876. https://doi.org/10.1074/jbc.M805937200
Israeli-Rosenberg S, Manso AM, Okada H, Ross RS (2014) Integrins and integrin-associated proteins in the cardiac myocyte. Circ Res 114:572–586. https://doi.org/10.1161/CIRCRESAHA.114.301275
Pham CG et al (2000) Striated muscle-specific beta(1D)-integrin and FAK are involved in cardiac myocyte hypertrophic response pathway. Am J Physiol Heart Circ Physiol 279:H2916–H2926. https://doi.org/10.1152/ajpheart.2000.279.6.H2916
Bauer MS et al (2019) Structural and mechanistic insights into mechanoactivation of focal adhesion kinase. Proc Natl Acad Sci U S A 116:6766–6774. https://doi.org/10.1073/pnas.1820567116
Brancaccio M et al (2003) Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat Med 9:68–75. https://doi.org/10.1038/nm805
Frangogiannis NG (2017) The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest 127:1600–1612. https://doi.org/10.1172/JCI87491
Frangogiannis NG (2019) The extracellular matrix in ischemic and nonischemic heart failure. Circ Res 125:117–146. https://doi.org/10.1161/CIRCRESAHA.119.311148
Chin IL, Hool L, Choi YS (2019) A review of in vitro platforms for understanding cardiomyocyte mechanobiology. Front Bioeng Biotechnol 7:133. https://doi.org/10.3389/fbioe.2019.00133
Takeda N, Manabe I (2011) Cellular interplay between cardiomyocytes and nonmyocytes in cardiac remodeling. Int J Inflam 2011:535241. https://doi.org/10.4061/2011/535241
Imanaka-Yoshida K, Aoki H (2014) Tenascin-C and mechanotransduction in the development and diseases of cardiovascular system. Front Physiol 5:283. https://doi.org/10.3389/fphys.2014.00283
del Rio A et al (2009) Stretching single talin rod molecules activates vinculin binding. Science 323:638–641. https://doi.org/10.1126/science.1162912
Goult BT, Yan J, Schwartz MA (2018) Talin as a mechanosensitive signaling hub. J Cell Biol 217:3776–3784. https://doi.org/10.1083/jcb.201808061
Manso AM et al (2017) Loss of mouse cardiomyocyte talin-1 and talin-2 leads to beta-1 integrin reduction, costameric instability, and dilated cardiomyopathy. Proc Natl Acad Sci U S A 114:E6250–E6259. https://doi.org/10.1073/pnas.1701416114
Lapidos KA, Kakkar R, McNally EM (2004) The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res 94:1023–1031. https://doi.org/10.1161/01.RES.0000126574.61061.25
Peter AK, Cheng H, Ross RS, Knowlton KU, Chen J (2011) The costamere bridges sarcomeres to the sarcolemma in striated muscle. Prog Pediatr Cardiol 31:83–88. https://doi.org/10.1016/j.ppedcard.2011.02.003
Fratter C et al (2020) EMQN best practice guidelines for genetic testing in dystrophinopathies. Eur J Hum Genet 28:1141–1159. https://doi.org/10.1038/s41431-020-0643-7
Finsterer J, Stollberger C (2003) The heart in human dystrophinopathies. Cardiology 99:1–19. https://doi.org/10.1159/000068446
Mavrogeni SI, Markousis-Mavrogenis G, Papavasiliou A, Papadopoulos G, Kolovou G (2018) Cardiac involvement in Duchenne muscular dystrophy and related dystrophinopathies. Methods Mol Biol 1687:31–42. https://doi.org/10.1007/978-1-4939-7374-3_3
Catanzaro JN et al (2020) Proarrhythmic manifestations of neuromuscular dystrophinopathies. Cardiol Rev. https://doi.org/10.1097/CRD.0000000000000305
Amoasii L et al (2018) Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science 362:86–91. https://doi.org/10.1126/science.aau1549
Chopra A, Tabdanov E, Patel H, Janmey PA, Kresh JY (2011) Cardiac myocyte remodeling mediated by N-cadherin-dependent mechanosensing. Am J Physiol Heart Circ Physiol 300:H1252–H1266. https://doi.org/10.1152/ajpheart.00515.2010
Kanoldt V et al (2020) Metavinculin modulates force transduction in cell adhesion sites. Nat Commun 11:6403. https://doi.org/10.1038/s41467-020-20125-z
Zhang JQ et al (2001) Ultrastructural and biochemical localization of N-RAP at the interface between myofibrils and intercalated disks in the mouse heart. Biochemistry 40:14898–14906. https://doi.org/10.1021/bi0107445
Baddam SR et al (2018) The desmosomal cadherin desmoglein-2 experiences mechanical tension as demonstrated by a FRET-based tension biosensor expressed in living cells. Cells 7. https://doi.org/10.3390/cells7070066
Teng J, Loukin S, Kung C (2014) Mechanosensitive ion channels in cardiovascular physiology. Exp Clin Cardiol 20:6550–6560
Krajnik A et al (2020) Phosphoinositide signaling and mechanotransduction in cardiovascular biology and disease. Front Cell Dev Biol 8:595849. https://doi.org/10.3389/fcell.2020.595849
Cordova-Casanova A et al (1865) Mechanical stretch increases L-type calcium channel stability in cardiomyocytes through a polycystin-1/AKT-dependent mechanism. Biochim Biophys Acta Mol Cell Res 289-296:2018. https://doi.org/10.1016/j.bbamcr.2017.11.001
Kuo IY et al (2014) The number and location of EF hand motifs dictates the calcium dependence of polycystin-2 function. FASEB J 28:2332–2346. https://doi.org/10.1096/fj.13-247106
Paavola J et al (2013) Polycystin-2 mutations lead to impaired calcium cycling in the heart and predispose to dilated cardiomyopathy. J Mol Cell Cardiol 58:199–208. https://doi.org/10.1016/j.yjmcc.2013.01.015
Balbo BE et al (2016) Cardiac dysfunction in Pkd1-deficient mice with phenotype rescue by galectin-3 knockout. Kidney Int 90:580–597. https://doi.org/10.1016/j.kint.2016.04.028
Boulter C et al (2001) Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc Natl Acad Sci U S A 98:12174–12179. https://doi.org/10.1073/pnas.211191098
Eden M et al (2016) Myoscape controls cardiac calcium cycling and contractility via regulation of L-type calcium channel surface expression. Nat Commun 7:11317. https://doi.org/10.1315/11317
Eder P, Molkentin JD (2011) TRPC channels as effectors of cardiac hypertrophy. Circ Res 108:265–272. https://doi.org/10.1161/CIRCRESAHA.110.225888
Wu X, Eder P, Chang B, Molkentin JD (2010) TRPC channels are necessary mediators of pathologic cardiac hypertrophy. Proc Natl Acad Sci U S A 107:7000–7005. https://doi.org/10.1073/pnas.1001825107
Seth M et al (2009) TRPC1 channels are critical for hypertrophic signaling in the heart. Circ Res 105:1023–1030. https://doi.org/10.1161/CIRCRESAHA.109.206581
Gottlieb P et al (2008) Revisiting TRPC1 and TRPC6 mechanosensitivity. Pflugers Arch 455:1097–1103. https://doi.org/10.1007/s00424-007-0359-3
Lin BL et al (2019) In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci U S A 116:10156–10161. https://doi.org/10.1073/pnas.1815354116
Xie J et al (2012) Cardioprotection by Klotho through downregulation of TRPC6 channels in the mouse heart. Nat Commun 3:1238. https://doi.org/10.1038/ncomms2240
Numaga-Tomita T, Nishida M (2020) TRPC channels in cardiac plasticity. Cells 9. https://doi.org/10.3390/cells9020454
Xian Tao L et al (2006) The stretch-activated potassium channel TREK-1 in rat cardiac ventricular muscle. Cardiovasc Res 69:86–97. https://doi.org/10.1016/j.cardiores.2005.08.018
Brohawn SG (2015) How ion channels sense mechanical force: insights from mechanosensitive K2P channels TRAAK, TREK1, and TREK2. Ann N Y Acad Sci 1352:20–32. https://doi.org/10.1111/nyas.12874
Brohawn SG, Campbell EB, MacKinnon R (2014) Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 516:126–130. https://doi.org/10.1038/nature14013
Brohawn SG, Su Z, MacKinnon R (2014) Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci U S A 111:3614–3619. https://doi.org/10.1073/pnas.1320768111
Wong TY et al (2018) Mechanical stretching simulates cardiac physiology and pathology through mechanosensor Piezo1. J Clin Med 7. https://doi.org/10.3390/jcm7110410
Caporizzo MA, Chen CY, Salomon AK, Margulies KB, Prosser BL (2018) Microtubules provide a viscoelastic resistance to myocyte motion. Biophys J 115:1796–1807. https://doi.org/10.1016/j.bpj.2018.09.019
Robison P, Prosser BL (2017) Microtubule mechanics in the working myocyte. J Physiol 595:3931–3937. https://doi.org/10.1113/JP273046
Chen CY et al (2018) Suppression of detyrosinated microtubules improves cardiomyocyte function in human heart failure. Nat Med 24:1225–1233. https://doi.org/10.1038/s41591-018-0046-2
Robison P et al (2016) Detyrosinated microtubules buckle and bear load in contracting cardiomyocytes. Science 352:aaf0659. https://doi.org/10.1126/science.aaf0659
Dahl KN, Ribeiro AJ, Lammerding J (2008) Nuclear shape, mechanics, and mechanotransduction. Circ Res 102:1307–1318. https://doi.org/10.1161/CIRCRESAHA.108.173989
Cho S et al (2019) Mechanosensing by the lamina protects against nuclear rupture, DNA damage, and cell-cycle arrest. Dev Cell 49:920–935 e925. https://doi.org/10.1016/j.devcel.2019.04.020
Dahl KN, Kahn SM, Wilson KL, Discher DE (2004) The nuclear envelope lamina network has elasticity and a compressibility limit suggestive of a molecular shock absorber. J Cell Sci 117:4779–4786. https://doi.org/10.1242/jcs.01357
Ahn J et al (2019) Structural basis for lamin assembly at the molecular level. Nat Commun 10:3757. https://doi.org/10.1038/s41467-019-11684-x
Makarov AA et al (2019) Lamin A molecular compression and sliding as mechanisms behind nucleoskeleton elasticity. Nat Commun 10:3056. https://doi.org/10.1038/s41467-019-11063-6
Davidson PM, Lammerding J (2014) Broken nuclei–lamins, nuclear mechanics, and disease. Trends Cell Biol 24:247–256. https://doi.org/10.1016/j.tcb.2013.11.004
Lammerding J et al (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113:370–378. https://doi.org/10.1172/JCI19670
Gonzalez JM, Navarro-Puche A, Casar B, Crespo P, Andres V (2008) Fast regulation of AP-1 activity through interaction of lamin A/C, ERK1/2, and c-Fos at the nuclear envelope. J Cell Biol 183:653–666. https://doi.org/10.1083/jcb.200805049
Tesson F et al (2014) Lamin A/C mutations in dilated cardiomyopathy. Cardiol J 21:331–342. https://doi.org/10.5603/CJ.a2014.0037
Lu JT, Muchir A, Nagy PL, Worman HJ (2011) LMNA cardiomyopathy: cell biology and genetics meet clinical medicine. Dis Model Mech 4:562–568. https://doi.org/10.1242/dmm.006346
Donnaloja F, Carnevali F, Jacchetti E, Raimondi MT (2020) Lamin A/C mechanotransduction in laminopathies. Cells 9. https://doi.org/10.3390/cells9051306
Dupont S et al (2011) Role of YAP/TAZ in mechanotransduction. Nature 474:179–183. https://doi.org/10.1038/nature10137
Buxboim A et al (2014) Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr Biol 24:1909–1917. https://doi.org/10.1016/j.cub.2014.07.001
Machowska M, Piekarowicz K, Rzepecki R (2015) Regulation of lamin properties and functions: does phosphorylation do it all? Open Biol 5. https://doi.org/10.1098/rsob.150094
Zhang X et al (2009) SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 64:173–187. https://doi.org/10.1016/j.neuron.2009.08.018
Banerjee I et al (2014) Targeted ablation of nesprin 1 and nesprin 2 from murine myocardium results in cardiomyopathy, altered nuclear morphology and inhibition of the biomechanical gene response. PLoS Genet 10:e1004114. https://doi.org/10.1371/journal.pgen.1004114
Chapman MA et al (2014) Disruption of both nesprin 1 and desmin results in nuclear anchorage defects and fibrosis in skeletal muscle. Hum Mol Genet 23:5879–5892. https://doi.org/10.1093/hmg/ddu310
Lu W et al (2012) Nesprin interchain associations control nuclear size. Cell Mol Life Sci 69:3493–3509. https://doi.org/10.1007/s00018-012-1034-1
Rajgor D, Mellad JA, Autore F, Zhang Q, Shanahan CM (2012) Multiple novel nesprin-1 and nesprin-2 variants act as versatile tissue-specific intracellular scaffolds. PLoS One 7:e40098. https://doi.org/10.1371/journal.pone.0040098
Lombardi ML et al (2011) The interaction between nesprins and sun proteins at the nuclear envelope is critical for force transmission between the nucleus and cytoskeleton. J Biol Chem 286:26743–26753. https://doi.org/10.1074/jbc.M111.233700
Driscoll TP, Cosgrove BD, Heo SJ, Shurden ZE, Mauck RL (2015) Cytoskeletal to nuclear strain transfer regulates YAP signaling in mesenchymal stem cells. Biophys J 108:2783–2793. https://doi.org/10.1016/j.bpj.2015.05.010
Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH (2019) Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol 16:361–378. https://doi.org/10.1038/s41569-019-0155-8
Frank D et al (2008) Gene expression pattern in biomechanically stretched cardiomyocytes: evidence for a stretch-specific gene program. Hypertension 51:309–318. https://doi.org/10.1161/HYPERTENSIONAHA.107.098046
Kempf T et al (2006) The transforming growth factor-beta superfamily member growth-differentiation factor-15 protects the heart from ischemia/reperfusion injury. Circ Res 98:351–360. https://doi.org/10.1161/01.RES.0000202805.73038.48
Eggers KM et al (2008) Growth-differentiation factor-15 for early risk stratification in patients with acute chest pain. Eur Heart J 29:2327–2335. https://doi.org/10.1093/eurheartj/ehn339
Kempf T et al (2007) Growth-differentiation factor-15 improves risk stratification in ST-segment elevation myocardial infarction. Eur Heart J 28:2858–2865. https://doi.org/10.1093/eurheartj/ehm465
Kempf T et al (2007) Prognostic utility of growth differentiation factor-15 in patients with chronic heart failure. J Am Coll Cardiol 50:1054–1060. https://doi.org/10.1016/j.jacc.2007.04.091
Kempf T, Wollert KC (2009) Growth-differentiation factor-15 in heart failure. Heart Fail Clin 5:537–547. https://doi.org/10.1016/j.hfc.2009.04.006
Frank D et al (2012) MicroRNA-20a inhibits stress-induced cardiomyocyte apoptosis involving its novel target Egln3/PHD3. J Mol Cell Cardiol 52:711–717. https://doi.org/10.1016/j.yjmcc.2011.12.001
Boriani G et al (2018) Cardiolaminopathies from bench to bedside: challenges in clinical decision-making with focus on arrhythmia-related outcomes. Nucleus 9:442–459. https://doi.org/10.1080/19491034.2018.1506680
Kubanek M et al (2020) Desminopathy: novel desmin variants, a new cardiac phenotype, and further evidence for secondary mitochondrial dysfunction. J Clin Med 9. https://doi.org/10.3390/jcm9040937
Peretto G et al (2018) Updated clinical overview on cardiac laminopathies: an electrical and mechanical disease. Nucleus 9:380–391. https://doi.org/10.1080/19491034.2018.1489195
Ahlberg G et al (2018) Rare truncating variants in the sarcomeric protein titin associate with familial and early-onset atrial fibrillation. Nat Commun 9:4316. https://doi.org/10.1038/s41467-018-06618-y
Brayson D, Shanahan CM (2017) Current insights into LMNA cardiomyopathies: existing models and missing LINCs. Nucleus 8:17–33. https://doi.org/10.1080/19491034.2016.1260798
Cingolani HE, Ennis IL (2007) Sodium-hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation 115:1090–1100. https://doi.org/10.1161/CIRCULATIONAHA.106.626929
Previtali SC et al (2020) Rimeporide as a fi rst-in-class NHE-1 inhibitor: results of a phase Ib trial in young patients with Duchenne Muscular Dystrophy. Pharmacol Res 159:104999. https://doi.org/10.1016/j.phrs.2020.104999
Wiedmann F et al (2020) The experimental TASK-1 potassium channel inhibitor A293 can be employed for rhythm control of persistent atrial fibrillation in a translational large animal model. Front Physiol 11:629421. https://doi.org/10.3389/fphys.2020.629421
Wiedmann F et al (2021) Mechanosensitive TREK-1 two-pore-domain potassium (K2P) channels in the cardiovascular system. Prog Biophys Mol Biol 159:126–135. https://doi.org/10.1016/j.pbiomolbio.2020.05.007
Zhang B (2021) CRISPR/Cas gene therapy. J Cell Physiol 236:2459–2481. https://doi.org/10.1002/jcp.30064
Stadtmauer EA et al (2020) CRISPR-engineered T cells in patients with refractory cancer. Science 367. https://doi.org/10.1126/science.aba7365
Carroll KJ et al (2016) A mouse model for adult cardiac-specific gene deletion with CRISPR/Cas9. Proc Natl Acad Sci U S A 113:338–343. https://doi.org/10.1073/pnas.1523918113
Landmesser U et al (2020) From traditional pharmacological towards nucleic acid-based therapies for cardiovascular diseases. Eur Heart J 41:3884–3899. https://doi.org/10.1093/eurheartj/ehaa229
Olivaes J, Bonamino MH, Markoski MM (2019) CRISPR/Cas 9 system for the treatment of dilated cardiomyopathy: a hypothesis related to function of a MAP kinase. Med Hypotheses 128:91–93. https://doi.org/10.1016/j.mehy.2019.05.013
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Ethics declarations
Declarations
-
Funding: This work was supported by the German Research Foundation (DFG) and the German Center for Cardiovascular Research (DZHK).
-
Availability of data and materials: Not applicable.
-
Authors Contributions: ME conceived and wrote the manuscript, NF supervised and reviewed the manuscript. DF and LK added the chapter “translational perspective and Fig. 2.”
-
Ethical approval: This article does not contain any studies with human participants by any of the authors. In all experiments involving animals and that have been conducted by the authors, ethical permit was granted by local committees of the University of Heidelberg (BW) and Kiel (SH).
-
Disclosure of interests: The authors declare that they have no conflicts of interest.
-
Consent for publication: All authors declare that they permit publication of this manuscript in its present form.
Rights and permissions
Copyright information
© 2023 The Author(s), under exclusive license to Springer Nature Switzerland AG
About this chapter

Cite this chapter
Eden, M., Kilian, L., Frank, D., Frey, N. (2023). Cardiac Mechanoperception and Mechanotransduction: Mechanisms of Stretch Sensing in Cardiomyocytes and Implications for Cardiomyopathy. In: Hecker, M., Duncker, D.J. (eds) Cardiac Mechanobiology in Physiology and Disease. Cardiac and Vascular Biology, vol 9. Springer, Cham. https://doi.org/10.1007/978-3-031-23965-6_1
Download citation
DOI: https://doi.org/10.1007/978-3-031-23965-6_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-23964-9
Online ISBN: 978-3-031-23965-6
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)