
Abstract
Increasing evidence suggests that disruption of metal homeostasis contributes to the pathogenesis of various neurodegenerative diseases, including Alzheimer’s disease, Lewy body diseases, vascular dementia, and prion diseases. Conformational changes of disease-related proteins such as ß-amyloid protein, α-synuclein, and prion proteins are well-established contributors to the synaptotoxicity, neurotoxicity, and pathogenesis of these diseases. Recent studies have revealed that these proteins are metalloproteins that coexist in synapses and play significant roles in the maintenance of metal homeostasis in synapses. Trace elements such as zinc (Zn), iron (Fe), copper (Cu), and aluminum (Al) bind to these proteins, thereby influencing their conformations and functions. Additionally, these metals have common binding sites; binding of metals to proteins is nonspecific. Therefore, metal-metal interactions at synapses contribute to the neurodegenerative processes. We present a current review of the role of trace elements in the functions and toxicity of disease-related proteins, as well as in the pathogenesis of neurodegenerative diseases. Possible therapeutic approaches related to metal homeostasis are discussed.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
1 Introduction
The brain is a unique organ, representing only 2 % of the typical human body weight, but consuming 20 % of the total oxygen used by the body. The disproportionate oxygen metabolism of the brain contributes to increased susceptibility to oxidative stress. The brain is primary composed of two cell types: neurons and glial cells. Neurons generally do not regenerate and are maintained for the individual’s entire life span. Therefore, neurons are susceptible to accumulation of toxic substances such as heavy metals. The blood-brain barrier (BBB), which is composed of tight junctions between endothelial cells, is one mechanism to prevent brain exposure to toxic substances. However, this barrier system is not perfect, and when the brain experiences long durations of toxic substance exposure, the substances may accumulate and result in adverse effects such as Minamata disease.
Essential trace elements including iron (Fe), zinc (Zn), and copper (Cu) exist in the brain and have crucial roles in normal brain functions, such as myelination, neurotransmitter synthesis, and neural information processing. Deficiency of these metals produces severe adverse effects on central nervous system functions, especially learning and memory. Moreover, growing evidence suggests that dyshomeostasis of metals, either excess or deficiency, is implicated in the pathogenesis of various neurodegenerative diseases including Alzheimer’s disease (AD), prion diseases, Lewy body diseases (Parkinson’s disease, dementia with Lewy bodies (PD/DLB), etc.), and vascular dementia [1, 2]. All of these diseases, except for vascular dementia, include abnormal deposition of disease-related proteins in the brain. The disease-associated proteins, termed amyloidogenic proteins, are ß-amyloid protein (AßP) in AD, prion protein in prion diseases, and α-synuclein in Lewy body diseases. Although the primary sequences are identical to typical proteins, the disease-associated proteins form fibril-like structures of oligomers with the ß-pleated sheet structures (amyloid fibrils) and are associated with neurotoxicity. Since the conformational changes to these amyloidogenic proteins are central to the pathogenesis, a new category, termed “conformational diseases” (protein-misfolding diseases), has proposed [3]. Metals are of particular interest as factors that influence conformational changes to the amyloidogenic proteins [4]. Recent studies have demonstrated that these amyloidogenic proteins or their precursor proteins (such as amyloid precursor protein; APP) possess the binding ability to metals and thereby participate in regulating metal homeostasis. Furthermore, metals and most of these amyloidogenic proteins are co-localized at the synapses which are the crucial node of neural networks. Table 14.1 summarizes the structure and properties of theses amyloidogenic proteins.
In the current neurometallomics study, we investigated the molecular mechanisms of neurodegenerative diseases, focusing on metal-protein interactions and metal-metal interactions at the synapse. Here, we describe the characteristics of three essential elements (Fe, Zn, Cu) and one toxic element (Al) in the brain, and thereafter, we review the functions and neurotoxicity of metals as well as the implications for neurodegenerative disease pathogenesis, based on recent studies from our lab and those by other authors.
2 Properties of Metals in the Brain
2.1 Iron (Fe) in the Brain
Considerable amounts of trace elements such as Fe, Zn, and Cu exist in the brain, in addition to ubiquitous elements such as sodium (Na), potassium (K), calcium (Ca), and magnesium (Mg). The distribution of each metal differs across brain regions [5]. Among these trace elements, Fe is the most abundant in the brain and in the whole body. Fe is essential for numerous biological functions as an enzyme cofactor for metabolic processes such as the oxygen transport, oxidative phosphorylation, and energy transfer. Fe has critical roles in specialized brain functions such as the synthesis of dopaminergic neurotransmitters and the myelination. Therefore, Fe deficiency impairs learning abilities, especially in children or infants, and it produces impaired working ability or learning ability also in adults [6]. However, excess Fe can generate reactive oxygen species (ROS) that damage DNA, proteins, and lipids and can therefore be toxic to neurons [7]. Fe exists in two different forms: ferrous iron (Fe2+) and ferric iron (Fe3+). In general, oxidized Fe3+ is insoluble and exists extracellularly, whereas reduced Fe2+ is soluble and intracellularly located. Orally administrated Fe is primary absorbed from the gastrointestinal pathway by divalent metal transporter-1 (DMT-1) as Fe2+ [8]. Once it enters the circulation, Fe2+ ions are oxidized to Fe3+ by ferroxidases such as ferritin or ceruloplasmin. It binds with transferrin, an iron binding protein that binds two Fe3+ions. Transferrin-bound iron (Fe3+) crosses BBB via transferrin receptors and enters into the cells. Finally, Fe3+ is reduced to Fe2+ by ferrireductase and function as a cofactor for neuronal enzymes such as tyrosine hydroxylase, which is necessary for the dopamine synthesis. Thus, Fe levels as well as the ratio between Fe2+ and Fe3+ are strictly regulated in normal brains. Two amyloidogenic proteins, prion protein and α-synuclein, reportedly possess ferrireductase activity [9, 10]. Ferroportin, a transmembrane protein, controls Fe export from intracellular to extracellular compartments. Recent findings suggest that APP, a precursor protein of Alzheimer’s AßP, binds to ferroportin and regulates Fe efflux [11].
The iron-responsive element/iron-regulatory protein (IRE/IRP) network also regulates Fe levels [12]. The mRNA encoding Fe-binding proteins such as ferritin or transferrin possess IRE domains in their mRNA. This system regulates production of Fe-binding proteins and prevents the formation of free Fe2+ and toxic free radicals. In Fe-deficient conditions, IRP binds to the IREs and inhibits their expression. As the concentration of free Fe2+ increases, binding of Fe to IRP and the expression of transferrin is downregulated, whereas that of ferritin is upregulated, thereby decreasing the amount of free Fe2+. The expression of two amyloid-related proteins, APP and α-synuclein, is also controlled by Fe [13] (see Sects. 14.3, 14.4, and 14.5).
2.2 Zinc (Zn) in the Brain
Zn is the second most abundant element in the brain. Zn is essential for most organisms and has important roles in various physiological functions such as mitotic cell division, immune system functioning, synthesis of proteins and DNA, and as a cofactor to more than 300 enzymes or metalloproteins. Recent studies have revealed that Zn signaling plays crucial roles as a second messenger in various human biological systems [14]. Zn deficiency in children results in dwarfism, delayed mental and physical development, immune dysfunction, and learning disabilities [15]. Zn deficiency also produces learning disorders, taste disorders, and odor disorders also in adults [16].
The human body contains approximately 2g of Zn, primary located in the testes, muscle, liver, and brain tissues. In the brain, Zn occurs at the highest concentrations in the hippocampus, amygdala, cerebral cortex, thalamus, and olfactory cortex [17]. The total Zn content of the hippocampus is estimated to be 70–90 mg/kg (dry weight). Although some Zn in the brain binds firmly to metalloproteins or enzymes, a substantial fraction (approximately 10 % or more) either forms free Zn ions (Zn2+) or is loosely bound and is histochemically detectable by staining using chelating reagents. This chelatable Zn is stored in the presynaptic vesicles of excitatory glutamatergic neurons and is secreted from these vesicles into synaptic clefts, together with glutamate, during neuronal excitation. The concentration of this secreted Zn is estimated to be 1–100 μM [18], although this value is still controversial. Secreted Zn modulates overall brain excitability by binding with N-methyl-D-aspartate (NMDA)-type glutamate receptors, GABA receptors, and glycine receptors. Recent studies have suggested that the secreted Zn2+ is critical to information processing, synaptic plasticity, learning, and memory [19, 20]. Indeed, Zn2+ in the hippocampus is essential for the induction of long-term potentiation (LTP), a form of synaptic information storage that has become a well-known paradigm for the mechanisms underlying memory formation [21].
There are two factors in the maintenance of Zn homeostasis: metallothioneins and Zn transporters. Metallothioneins are ubiquitous metal-binding proteins with 68 amino acids, which bind seven metal atoms (such as Zn, Cu, Cd, etc.) via its 20 cysteine residues. There are three types of metallothioneins: MT-1, MT-2, and MT-3. MT-1 and MT-2 are ubiquitously expressed throughout the whole body, whereas MT-3 is primary localized in the central nervous system [22]. Uchida et al. found that neuronal growth inhibitory factor (GIF) which inhibits neurite extensions and prevents neuronal death was decreased in the brains of AD patients and determined that GIF is equivalent to MT-3 [23]. Therefore, MT-3 (GIF) is implicated in AD-associated neuronal death [24].
Zn transporters also control Zn homeostasis by facilitating Zn influx when it is deficient and efflux when it is present in excess [25]. There are two types of mammalian Zn transporters: ZnT transporters and Zrt-, Irt-like protein (ZIP) transporters. ZnT transporters are involved with the gene family for solute carrier (SLC30) and decrease intracellular Zn via facilitation of Zn efflux from cells. There are 14 types of ZnT transporters in mammals, including ZnT-1 and ZnT-3, which are co-localized with chelatable Zn in the brain. ZnT-1 is a membrane protein with six transmembrane domains and is widely distributed in mammalian cells. ZnT-1 has a pivotal role in Zn efflux and in protection from excess Zn. The expression of ZnT-1 is induced after transient global ischemia and decreases following dietary Zn deficiency [26]. ZnT-3 is localized to the membranes of presynaptic vesicles, transports Zn into synaptic vesicles, and maintains high Zn concentrations in the vesicles. Although the physiological roles of ZnT-3 have not fully elucidated, ZnT-3 knockout mice have depleted synaptic Zn and impaired memory formation [27].
ZIP transporters are another type of Zn transporter encoded by SLC39 genes. ZIP transporters increase cytosolic Zn by promoting transport from extracellular to intracellular compartments. Fourteen ZIP genes have been identified in mammal, and the ZIP transporters are localized to cell membranes or in the membranes of the Golgi apparatus or endoplasmic reticulum (ER). These transporters control Zn influx into subcellular organs, and Zn transporter mutations produce severe diseases such as Ehlers-Danlos syndrome [28].
2.3 Cu (Copper) in the Brain
Cu is essential for brain functions and is a cofactor for numerous enzymes, such as cytochrome C, superoxide dismutase, lysyl oxidase, and tyrosinase. Cu is involved in Fe homeostasis as a component of ceruloplasmin and has neuroprotective activity as a component of Cu/Zn superoxide dismutase, an endogenous antioxidant. Cu deficiency results in adverse effects on myelination. Cu is a redox-active metal and exists both as oxidized Cu2+ and reduced Cu+. Thus, excess free Cu is toxic because it produces ROS and binds with the thiol groups of functional proteins. The Cu transporters, ATP7A and ATP7B, transport Cu in ATP dependently. Another Cu transporter, Ctr1, is also involved in neuronal Cu uptake. Mutations of these transporters are linked with neurodegenerative diseases such as Wilson disease and Menkes disease [29]. Recent studies suggest that Cu has modulatory effects on neuronal information processes. Intracellular Cu accumulates in synaptic vesicles and is then released into the synaptic clefts during neuronal excitation, similarly to Zn [30]. Its concentration is estimated to be approximately 15 μM. Although the physiological roles of the released Cu are still controversial, Cu reportedly blocks glutamate receptors and modulates neuronal spontaneous activity [31].
2.4 Aluminum (Al) in the Brain
Given that Al is implicated in many neurodegenerative diseases due to its peculiar chemical characteristics, it can be considered a neurotoxic metal. Al is the third most abundant element in the earth’s crust. Despite its widespread distribution throughout the environment, Al is not essential to living organisms. In contrast, Al reportedly inhibits more than 300 essential enzymatic functions. Al is suspected to contribute to various neurodegenerative diseases including Alzheimer’s disease (AD) [1, 32], amyotrophic lateral sclerosis (ALS) and Parkinsonism dementia (PD) in the Kii Peninsula and Guam [33], dialysis encephalopathy [34], and Gulf War syndrome [35].
Al exhibits only one oxidation state, Al3+. In acidic solutions with pH < 4, Al3+ exists as a soluble octahedral hexahydrate Al(H2O)6 3+ (Al3+). As the pH increases, its solubility decreases and Al(OH)3 precipitates at neutral pH. Al3+ has affinity for negatively charged, oxygen-donor ligands. Inorganic and organic phosphates, carboxylate, and deprotonated hydroxyl groups form strong bonds with Al3+. Owing to these chemical characteristics, Al3+ binds to the phosphate groups of DNA and RNA, affecting DNA topology and influencing the expression of various genes essential for brain functions. Al3+ also binds to the phosphate groups of nucleoside di- and triphosphates, such as ATP, and can thus influence energy metabolism. Al also inhibits the functions of various protein kinases and phosphatases. Furthermore, Al3+ has a very low ligand-exchange rate compared to other metals. For example, the ligand-exchange rate of Mg2+ is 105 times faster than that of Al3+, and therefore, Al3+ inhibits enzymes with Mg2+ cofactors. Al3+ also inhibits biological processes involving rapid Ca2+ exchange: the exchange rate for Al3+ is 108 times slower than that of Ca2+. Therefore, Al cannot participate in Ca2+- or Mg2+-related enzymatic reactions and has an extended half-life in the body.
The strong positive charges and a relatively small ionic radius of Al3+ in comparison to other metal ions such as Ca2+, Zn2+, and Na+ facilitate Al3+ binding firmly to metal-binding amino acids (histidine, His; tyrosine, Tyr; arginine, Arg; etc.) or phosphorylated amino acids and thereby influences their conformations. Al is a well-known protein cross-linker and is therefore used as a leather-tanning agent.
Additionally, Al3+ has characteristics similar to Fe and binds to Fe-binding proteins such as transferrin and ferritin. Al3+ also stimulates Fe-induced membrane lipid peroxidation and causes oxidative damage in vitro and in vivo, although Al3+ does not directly affect peroxidation [36].
Overall, Al influences various processes induced by Ca, Mg, Fe, and other elements. Metal-metal interaction by Al will be discussed later in this chapter.
3 Alzheimer’ s Disease (AD) and Metals
3.1 Amyloid Hypothesis
AD is a severe senile type of dementia that affects a large proportion of the elderly population worldwide. In Japan, there were more than 4,000,000 patients with senile dementia in 2013, and the number is increasing annually. The number of patients with mild cognitive incidence (MCI), which is the precursor stage of senile dementia, is also estimated to be more than 4,000,000. AD accounts for approximately half of patients with senile dementia.
AD is characterized by profound memory loss and the inability to form new memories. The pathological hallmarks of AD are the presence of numerous extracellular deposits, including senile plaques and neurofibrillary tangles (NFTs), and the selective loss of synapses and neurons in the hippocampal and cerebral regions [37]. Indeed, there is a strong correlation between the decrease in the number of synapses and the severity of memory impairment [38]. The major components of NFTs and senile plaques are phosphorylated tau protein and AßP, respectively.
Numerous biochemical, cell biological, and genetic studies support the idea termed “amyloid cascade hypothesis,” which proposes that AßP accumulation and the consequent neurodegeneration are central to AD, although this hypothesis requires further investigation [39, 40]. Recent studies of the identified AßP species have indicated that the oligomerization of AßP and its conformational changes are critical to the neurodegeneration process.
AßP is a small peptide of 39–43 amino acid residues, which results from cleavage of a large precursor protein (APP; amyloid precursor protein) at the N-terminus by the ß-APP cleaving enzyme (BACE) and the intramembrane cleavage of its C-terminus by γ-secretase (Fig. 14.1). Genetic studies of early-onset cases of familial AD have indicated that APP mutations and AßP metabolism are associated with AD [41]. Presenilins are considered to be one of γ -secretases, and their mutations also account for the majority of cases of early-onset familial AD [42].

Structure of AßP and its secretion from APP
AßP is secreted by the cleavage of the APP N-terminus by ß-secretase (BACE), followed by intramembrane cleavage of the C-terminus by γ-secretase. APP also binds to Cu or Zn. Human AßP and rodent AßP differ by three amino acids (Arg5, Tyr10, and His13). The expression of APP is regulated by Fe and Al
Yankner et al. reported that the first 40 amino acid residues of AßP (AßP (1–40)) causes the death of cultured rat hippocampal neurons or neurodegeneration in the brains of experimental animals [43]. AßP is a hydrophobic peptide with an intrinsic tendency to self-assemble and form SDS-stable oligomers in aqueous solutions. The monomeric form of AßP has a randomly coiled structure and is also less toxic. Oligomeric AßPs have ß-pleated sheet structures and form insoluble aggregates, termed amyloid fibrils. The neurotoxicity of AßP (1–40) peptides is enhanced by the process of “aging” (aggregation under incubation at 37 °C for several days), compared to freshly prepared AßP(1–40), and correlates with its ß-sheet contents [44]. Jarrett and Lansbury demonstrated that AßP formed oligomers via a nucleation-dependent process and that AßP (1-42) forms seeds for the aggregation of AßP (1-40) [45]. These results suggest that the ratio of AßP (1-40) and AßP (1-42) is critical to the pathogenesis. The point mutations of APP are located near the γ-secretase cleavage site of AßP (1-42) and influence the ratio of AßP (1-40) and AßP (1-42). Other mutations in presenilin genes influence the ratio of AßP (1-40) and AßP (1-42) by increasing production of AßP (1-42) in transfected cell lines.
3.2 Interaction Between Metals and AßP
AßP is secreted in the cerebrospinal fluid (CSF) of young individuals as well as in that of elderly individuals and non-dementia individuals [46]. Therefore, factors that accelerate or inhibit oligomerization may essentially contribute to the pathogenesis of AD. Several factors, such as the concentration of peptides, pH, composition of solvents, and temperature, influence the oligomerization processes. Oxidations, mutations, and racemization of AßP enhance its oligomerization. Additionally, substances including cholesterol or its oxidation products, apolipoprotein E, transthyretin, rifampicin, curcumin, aspirin, and ß-sheet breaker peptide, inhibit AßP oligomerization in vitro (Fig. 14.2) [47].

Oligomerization of AßP. AßP monomers exhibit random or α-helix structures. However, under aging conditions or in the presence of acceleratory factors, AßP self-aggregates and forms several types of oligomers (SDS-soluble oligomers, ADDLS, globulomers, protofibrils, etc.) before finally forming insoluble aggregates (amyloid fibrils). Oligomeric soluble Aßs are toxic, although the monomeric and fibril aggregates are relatively nontoxic
It is well established that rodent AßP exhibits a reduced tendency for oligomerization compared to human AßP in vitro, and AßP accumulation is rarely observed in the brains of rodents (rats or mice) compared to primates (humans or monkeys) [48]. As shown in Fig. 14.1, the amino acid sequences of human and rodent AßP are similar, but rodent AßP differs at three amino acids (Arg5, Tyr10, and His13) compared to primate AßP. Considering that all of these amino acids are able to bind metals and metals are important determinants for the conformation of proteins, it is possible that trace elements such as Al, Zn, Cu, and Fe may play central roles in the accumulation of AßP in the human brain. Al is of particular interest since epidemiological Al is considered to be a risk factor of AD [49]. Al3+ has strong positive charges and a relatively small ionic radius in comparison with the other metal ions. Thus, Al3+ firmly binds to metal-binding amino acids and induces conformational changes to proteins. Exley et al. first demonstrated that Al induces a conformational change in AßP (1-40), using circular dichroism (CD) spectroscopy [50]. Al also promotes aggregation of 125I-labeled AßP (1-40), with similar findings as for Fe and Zn [51]. We have developed a system for investigating AßP polymerization that involves immunoblotting and precipitation. Using this system, we have demonstrated that Al enhances oligomerization of AßP (1-40) and forms SDS-stable oligomers in vitro [52, 53]. The aggregated AßP (1-40) is redissolved by adding an Al chelator, deferoxamine (DFO). The oligomerization induced by Al is more marked than that induced by other metals, including Zn, Fe, Cu, and Cd. Furthermore, we have demonstrated that Al-aggregated AßPs bind tightly to the surface of cultured neurons and form fibrillar deposits, meanwhile, Zn-aggregated AßPs are rarely observed on the surface of cultured neurons. Furthermore, Al inhibits AßP degradation resulting from conformational changes and enhances AßP accumulation.
Other trace metals such as Zn and Cu enhance AßP oligomerization. Bush et al. have demonstrated that Zn2+, at a concentration similar to that in the CSF, produced AßP aggregation [54]. They reported that Cu2+ also binds to AßP, induces its aggregation, and increases ROS levels [55].
Despite all oligomers possess ß-pleated sheet structure, not all oligomers are equally neurotoxic. Recent studies using size-exclusion chromatography, gel electrophoresis, and atomic force microscopy revealed that several stable types of oligomers have been reported: naturally occurring soluble oligomers (dimmers or trimers), AßP-derived diffusible ligands (ADDLs), AßP globulomers, and protofibrils [4]. Hartley et al. separated aggregated AßP(1–40) into low-molecular-weight (mainly monomers), protofibrillar, and fibril fractions using size-exclusion chromatography and found that the protofibrillar fraction produced robust changes in the electrical activity of cultured neurons and was neurotoxic, but fibrils did not [56]. Walsh et al. found that SDS-stable oligomers exist in conditioned medium with cultured cells exogenously expressing the human APP gene [57]. The natural AßP oligomers obtained from the CSF of AD patients induced the loss of dendritic spines and synapses and blocked LTP [58].
The characteristics (size or shape) of AßP oligomers formed in the presence of Al, Zn Cu, and Fe are revealed to be identical by morphological analysis using atomic force microscopy [59]. Additionally, Sharma et al. revealed that Zn-aggregated AßPs are less toxic than Cu-aggregated AßP [60]. Meanwhile, Al-aggregated AßPs reportedly caused cytoskeletal changes, mitochondrial dysfunction, and increased production of ROS [61]. Bolognin et al. investigated aggregation and toxicity of AßP induced by Al, Cu, Fe, and Zn, finding that Al-aggregated AßPs induce overproduction of APP and tau, but AßP oligomers formed in the presence of other metals did not [62]. Everett et al. reported that AßP has the catalytic activity to reduce Fe3+ to redox-active Fe2+ and found that Al enhanced the reductive activity of AßP [63].
Metals can also participate in AßP-induced neurodegeneration pathways. Our previous studies, as well as numerous other studies, have demonstrated that AßPs are directly incorporated onto the surface of the cellular membrane and create unregulated pore-like channels that have cytotoxic effects [64–66]. These “amyloid channels” are giant multilevel pores that can facilitate the transport of large amounts of Ca2+ [67]. Zn binds to His residues, which are exposed to the internal site of the amyloid pore and inhibit Ca2+ influx [68, 69]. We found that AßP induced abnormal increases in [Ca2+]i using a high-resolution multi-site video imaging system in conjunction with a Ca2+-sensitive fluorescent dye (fura-2) [70]. Numerous studies, including those from our lab, have revealed that prion protein fragment peptide or α-synuclein also form amyloid channels on membranes [65, 66]. Zn can regulate Ca influx via these amyloid channels.
3.3 Interactions Between Metals and APP or Presenilin
Despite the wide distribution of APP in the brain, its physiological roles have not been elucidated. APP has distinct binding domains for Cu, Zn, and Fe [71]. Wong et al. found that APP does not possess ferroxidase activity, but binds to the Fe transporter ferroportin and enhances Fe efflux [11]. Furthermore, APP mRNA contains an IRE domain as well as ferritin, and its expression is regulated by Fe, as described in Fig. 14.1 [72]. Therefore, APP regulates Fe homeostasis, and Fe controls APP expression. There are other important findings implicating Fe homeostasis in the AD pathogenesis. Fe-related genes, such as transferrin C2 or the hemochromatosis gene, are risk factors for AD [73]. Imagawa et al. reported that Fe supplementation is effective for recovery of cognitive functions in patients with AD [74].
APP has two Cu-binding domains in its N-terminal and possesses the ability to reduce Cu2+ to Cu+ [75]. Cu induces the dimerization of APP, AßP production, and trafficking of APP from the ER to neurites [76]. Cu also influences APP processing and the expression of APP. These results suggest that APP has crucial roles in the maintenance of metal homeostasis, particularly Fe homeostasis. When this homeostasis is disrupted, increased Fe causes ROS production or increased APP causes AßP production, both initiate the degenerative processes. Indeed, APP knockout mice exhibited increased Cu levels in the brain [77]. Cicotosto et al. reported that knockout of APP or its analogue APLP2 in mice resulted in changes in the distribution of Cu, Zn, Fe, and Ca [78]. Decreased levels of ferroportin and accumulation of Fe in the brains of AD patients have been reported [79].
It is very possible that Al influences Fe homeostasis. As noted previously, Al has characteristics similar to Fe and binds to Fe-binding proteins, including ferritin and transferrin, or to Fe chelators such as DFO. Al3+ also binds to IRP [80, 81] and thus affects the expression of various Fe-regulated genes containing IREs, thereby causing elevation in the Fe concentration. Additionally, Al reportedly enhances the expression of APP [82–85]. Furthermore, Al influences the uptake of Fe into cultured neurons or glial cells [86].
Presenilins, one of the γ-secretases, and their mutations account for most cases of early-onset AD. Presenilins mainly exist in the ER and they are implicated in Ca homeostasis [87, 88]. Recent studies demonstrate that presenilins are also metal-binding proteins and participate in the neuronal uptake of Zn and Cu [89]. Presenilins also promote the uptake of dietary Cu [90].
4 Prion Disease and Metals
4.1 Overview
Prion diseases are fatal neurodegenerative diseases that include scrapie in sheep, bovine spongiform encephalopathy (BSE) in cattle, as well as Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS), and kuru disease in humans [91]. The common pathological hallmarks are the spongiform degeneration of neurons and glial cells, and the accumulation of amyloidogenic prion protein (PrP) in the brain of the infected animals or person. Prion diseases are also called transmissible spongiform encephalopathies, since their characteristic infections can be initiated by the administration of pathogenic tissue. Although the molecular pathogenesis and transmission mechanisms of prion diseases are still controversial, it is widely accepted that the conformational conversion of normal cellular prion protein (PrPC) to an abnormal scrapie-type isoform (PrPSc) is related to the transmissible characteristics of prion diseases. PrPC is a 30–35 kDa cell surface glycoprotein anchored at the plasma membrane by a glycosylphosphatidylinositol (GPI) domain and is ubiquitously expressed throughout the entire body, particularly in the brain. Both PrPC and PrPSc have the same characteristics in terms of chemical modifications as well as the same primary sequence. However, PrPC differs from PrPSc in that PrPSc is resistant to protease digestion, has high ß-sheet secondary structure, and has a propensity to form insoluble amyloid fibrils. When the misfolded PrPSc enters the body, for example, by the administration of contaminated food, the protease-resistant PrPSc can then aggregate. Next, fibril formation induces other PrPC molecules in the brain to misfold and aggregate.
4.2 Interactions Between Metals and PrP
The physiological roles of PrPC are still controversial; however, increasing evidence suggests that PrPC is a metalloprotein involved in metal homeostasis [92]. PrPC contains 208 amino acid residues and possesses a highly conserved octa-repeat domain composed of multiple tandem copies of the eight-residue sequence PHGGGWGQ in its N-terminal (Fig. 14.3). In 1997, Brown et al. reported decreased levels of Cu in the brains of PrP-knockout mice compared to wild-type mice. The activity of Cu-dependent enzymes was also reduced in PrP-null mice [93]. Jackson et al. reported that PrPC binds four Cu atoms at its octa-repeat domain and binds two additional Cu atoms to two other histidine (His) residues, His96 and His111 [94]. They also demonstrated that other metal ions including Zn2+, Mn2+, and Ni2+ can bind to these sites, although with lower affinity than Cu2+. PrPC reportedly transports Cu from the extracellular space to the intracellular space via endocytosis and regulates the intracellular concentration of Cu. PrPC possesses or modulates Cu/Zn superoxide dismutase (Cu/Zn SOD) activity in the brain and has protective roles against oxidative stress [95]. Recent studies have suggested that PrPC regulates the function of the NMDA-type glutamate receptor in a Cu-dependent manner [96]. In contrast, Cu2+ influences the gene expression and cellular trafficking of PrP [97]. It is also possible that the depletion of PrPC and a subsequent increase in Cu could cause oxidative damage to neurons [98]. Furthermore, PrP-deficient neurons exhibit lower glutathione reductase activity and increased susceptibility to damage by hydrogen peroxide [99].

The structure of the prion protein and its metal -binding properties
Zn2+ has the next highest affinity for binding with PrP, after Cu2+. As discussed previously, Zn accumulates in synaptic vesicles, and a considerable amount of Zn (~100 μM) is released with glutamate during neuronal excitation. Since the concentration of Zn in the brain is much higher than the concentration of Cu (~15 μM), it is possible that Zn2+ influences PrPC binding to Cu [100]. Moreover, recent bioinformatics analysis has revealed the evolutionary similarities between prion genes and genes encoding the ZIP transporters [101]. Of 14 ZIP transporters, sequence similarities have been reported between PrPC and N-terminal ectodomains of ZIP5, ZIP6, and ZIP10. Furthermore, PrPC co-localizes with ZIP5 in neuroblastoma cells [102]. Watt et al. found that PrPC facilitates the uptake of Zn into neuronal cells and that the effect is mediated by α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors [103]. These authors hypothesized that PrPC functions as a Zn sensor, detecting extracellular levels up to a particular threshold. Indeed, PrPC reportedly attenuates Zn-induced neurotoxicity.
In addition to Cu and Zn, other metals such as Fe and Mn are associated with PrP. Singh et al. suggested that PrPC functions as a ferrireductase that is responsible for reducing Fe3+ to bioavailable Fe2+ and modulating the cellular uptake of Fe [104]. PrP-knockout mice exhibit altered Fe metabolism [105]. Decreased levels of ferroxidase activity and transferrin have been observed in the cerebrospinal fluid of patients with CJD [106].
Furthermore, several studies have suggested that Mn may facilitate prion diseases. Johnson et al. investigated the levels of trace elements in prion-infected hamster brains using X-ray photoelectron emission microscopy with synchrotron radiation and found both reduced Cu and increased Mn in plaques composed of prion proteins [107]. Thackray et al. reported that PrPC loses its SOD-like activity when Cu is replaced with Mn [108]. Furthermore, Mn enhances the stability of PrP in soils and increases its infectivity [109]. The risk of contracting prion disease in elk, termed “chronic wasting disease,” has been associated with Mg deficiency and increased levels of Mn [110]. A recent epidemiological survey also suggested a relationship between the pathogenesis of CJD and Mn imbalance [111].
4.3 Toxicity of PrP Fragments and Metals
The neurodegenerative mechanisms of prion diseases include three possibilities: the “loss of normal protective functions of PrPC,” the “gain of toxic functions of PrPSc,” or a combination of both. PrPC is thought to regulate Cu homeostasis and to have antioxidant and cytoprotective effects against neurotoxicity induced by Cu2+ or free radicals. We have demonstrated that PrP fragment with octa-repeat domain attenuated Cu-induced neurotoxicity [112]. Thus, PrPC depletion may initiate various neurodegenerative processes.
Meanwhile, PrPSc produces synaptic impairment and apoptosis in neurons and astrocytes in vitro and in vivo. To investigate the mechanisms of PrPSc neurotoxicity, researchers (including our lab) have used synthetic fragment peptides of PrP (PrP106–126), because methodological difficulties occur when attempting to use a whole prion protein due to its strongly infectious characteristics. PrP106–126 has been used as a model peptide of PrPSc, as it coincides with the proposed sequence for ß-sheet structures, forms aggregates of ß-sheet structures to produce amyloid fibrils that share several characteristics with PrPSc, and causes the apoptotic death of cultured neurons or glial cells [113]. Additionally, PrP106–126 has the ability to bind to metals including Cu2+ and Zn2+ [114].
We observed that PrP106–126 forms ß-sheet structures during the “aging” process (incubation at 37 °C for several days) using a thioflavin T (ThT) fluorescence assay, far-UV circular dichroism (CD) spectroscopy, and atomic force microscopy (AFM) imaging [112]. Furthermore, aged PrP106–126 produced significant neurotoxicity in primary cultured rat hippocampal neurons. These characteristics are quite similar to that of AßP. We have added various trace elements to solutions of PrP106–126 during the aging processes and evaluated the resulting conformational changes and neurotoxicity. The presence of either Zn2+ or Cu2+ during the aging process significantly attenuated PrP106–126 neurotoxicity, whereas the presence of Al3+, Fe2+, and Fe3+ did not produce significant changes. Additionally, we investigated the effects of these metal ions on the ß-sheet formation of PrP106–126. We have utilized the changes in fluorescence of ThT, which binds with pleated ß-sheet structures, to observe the oligomerization of PrP106–126. The ThT fluorescence for solutions of aged PrP106–126 increased compared with freshly dissolved PrP106–126 solutions. The ThT fluorescence of PrP106–126 incubated with Zn2+, Fe2+, or Fe3+ was significantly decreased compared with aged PrP106–126 alone. In particular, the addition of Cu2+ dramatically decreased ThT fluorescence levels similar to fresh PrP106–126. Aged PrP106–126 forms amyloid fibrils with a distinctively straight and long morphology on mica plates as observed by AFM, although no fiber-like structures could be observed in freshly prepared PrP106–126. However, PrP106–126 aged with Cu or Zn exhibited different morphological features compared to aged PrP106–126 alone. Although the CD spectra of aged PrP106–126 exhibited ß-sheet structures, the CD spectra of PrP106–126 aged with Cu2+ exhibited random coil-like structures. Our results suggest that Cu and Zn influenced the ß-sheet formation of PrP106–126 and thereafter attenuated its neurotoxicity. These findings are consistent with other studies that Cu inhibits ß-sheet formation by PrP111–126 [115]. Thakur et al. investigated the conformational changes of full-length PrP by NMR and reported that Cu did not produce oligomerization of PrP at physiological temperatures and that Cu may act as an attenuator in prion diseases [116]. Although it is widely accepted that Cu enhances the aggregation of other amyloidogenic proteins such as AßP and α-synuclein, the effects of Cu on the conformational changes to amyloidogenic proteins may be complex and may depend on peptide structures [117].
5 Lewy Body Diseases and Metals
Lewy body disease is a category that includes Parkinson’s disease (PD), dementia with Lewy body (DLB), and multiple system atrophy (MSA). These diseases commonly exhibit abnormal cellular inclusions, termed Lewy bodies, which is the accumulation of α-synuclein, and therefore are termed “synucleopathy” [118]. DLB includes approximately 25 % senile dementia and has common pathological changes with AD, such as the deposition of senile plaques and tau protein. Moreover, the fragment peptide of α-synuclein, non-amyloid component (NAC), is co-accumulated with AßP in Alzheimer’s senile plaques. The oligomerization and fibrillation of α-synuclein have been implicated in the formation of Lewy bodies and the etiology of Lewy body diseases. It is demonstrated that α-synuclein is a 140-amino acid protein, abundantly present in the brain and particularly located in the nucleus and in the presynaptic terminals.
Involvement of metals in Lewy body diseases and α-synuclein has been extensively researched, since Mn toxicity exhibits Parkinson-like symptoms [119] and Fe-rich regions such as the substantia nigra are particularly vulnerable in Parkinson’s diseases [104]. Accumulation of Fe and Al has been reported in Lewy bodies of Parkinson patients. Furthermore, α-synuclein aggregation is accelerated by Cu and Al in vitro. Uversky et al. found the metals, such as Al and Mn, enhance the α-synuclein oligomerization [120]. Cu binds to α-synuclein in its N-terminal domain and C-terminal domain [121].
Increasing evidence suggests that α-synuclein primary exists in the presynaptic terminals of dopaminergic neurons and controls dopamine release, synaptic functions, and synaptic plasticity. Additionally, α-synuclein reportedly binds Fe and functions as a ferrireductase, which reduces Fe3+ to bioavailable Fe2+ [9]. Furthermore, α-synuclein mRNA has an IRE domain as well as APP or ferritin, and Fe regulates its expression [122]. In the postmortem brains of patients with Parkinson patient, Fe levels as well as the ratio of Fe2+ to Fe3+ were changed [123].
6 Vascular Dementia and Metals
Vascular dementia (VD) is a degenerative cerebrovascular disease that accounts for approximately one third of senile dementia cases. Risk factors include aging, sex (male), diabetes, and high blood pressure. The most common type of VD is caused by a series of small strokes or ischemia [124]. Following transient global ischemia or stroke, the interruption of blood flow and the resulting oxygen-glucose deprivation induce long-lasting membrane depolarization and result in excessive glutamate release into synaptic clefts. The excess glutamate then overstimulates receptors including NMDA-type receptors, AMPA-type receptors, and kainate-type receptors. Finally, Ca2+ dyshomeostasis (the entry of large quantities of Ca2+ to glutamate-responsive neurons) triggers delayed death of vulnerable populations of neurons, such as pyramidal neurons in the hippocampus, which is associated with learning and memory. Development of an infarct and the subsequent cognitive dysfunction produce the pathogenesis of VD. Approximately 30 % of patients with stroke experience show symptoms of dementia within 3 months of the initial stroke.
Increasing evidence suggests that Zn is central to ischemia-induced neuronal death and the pathogenesis of VD [125]. In ischemic conditions, as much as 300 μM Zn is co-released with glutamate into the synaptic clefts following membrane depolarization [126] and exhibits apoptotic cell death. Furthermore, the chelatable Zn moved from presynaptic terminals into cell bodies of degenerated neurons [127]. The increase in intracellular Zn2+ levels ([Zn2+]i), namely, Zn translocation, occurs in vulnerable neurons in the CA1 or CA3 regions of the hippocampus after transient global ischemia. Administration of calcium EDTA (Ca EDTA), a membrane-impermeable Zn chelator, blocked Zn translocation, protected hippocampal neurons after transient global ischemia, and reduced the infarct volume [128]. At least three major routes of Zn2+ entry were identified: voltage-gated Ca2+ channels (VGLC), NMDA-type glutamate receptors, and AMPA/kainate-type glutamate receptors [129]. Although NMDA-type glutamate receptors are present in most neurons, the permeability of Zn2+ and Ca2+ through AMPA/kainate channels is greater for NMDA receptor.
In the normal physiological condition, hippocampal neurons typically express AMPA receptors with GluR2 subunits, which are poorly permeable to divalent cations including Ca2+ and Zn2+. However, following ischemia, there is an acute reduction in GluR2 subunit expression, and neurons possess specific types of AMPA receptors with channels that are directly Ca2+-permeable (Ca-AMPA/kainate channels; Ca-A/K-R). The appearance of Ca-AMPA/kainate channels result in increased permeability of Ca2+, thereby enhancing toxicity. Therefore, the expression of Zn2+-permeable Ca-AMPA/kainate channels and the entry of Ca2+ and/or Zn2+ through the channels are mediators of the delayed neuronal death that follows ischemia. Considering that Ca EDTA, a Zn chelator, attenuates ischemia-induced downregulation of the GluR2 gene [128], Zn is also implicated in the transcriptional regulation of Ca-AMPA/kainate channels.
We investigated the molecular mechanism of Zn-induced neuronal death using GT1-7 cells (immortalized hypothalamic neurons), which are much more sensitive to Zn than other neuronal cells [130, 131]. GT1-7 cells possess neuronal characteristics such as neurite extensions, secretion of gonadotropin-releasing hormone (GnRH), and expression of neuron-specific proteins or receptors including microtubule-associated protein 2 (MAP2), tau protein, neurofilament, synaptophysin, GABAA receptors, dopamine receptors, and L-type Ca2+ channels [132]. We demonstrated that the ER stress pathway, the mitochondrial energy pathway, and the disruption of Ca homeostasis contribute to Zn-induced apoptosis. Screening for substances that are protective against Zn neurotoxicity yielded findings that carnosine (ß-alanyl histidine) attenuates Zn-induced neuronal death [133].
7 Hypothesis: Crosstalk of Metals and Amyloidogenic Proteins at the Synapse
The evidence indicates that the amyloidogenic proteins (or their fragment peptides), including APP (AβP), prion protein (PrP106-126), and α-synuclein (NAC), share similarities such as oligomerization with β-pleated sheet structures, neurotoxicity, and the ability to binding metals, as shown in Table 14.1. Furthermore, recent findings demonstrate that these proteins are co-localized at the synapse. APP occurs primarily in the presynaptic membrane, PrPC occurs in postsynaptic membranes, and α-synuclein occurs primarily in the presynaptic cytosol and to a lesser extent in membranes. Additionally, presenilins are predominantly located in the ER and also occur in the presynaptic and postsynaptic membranes.
The synapse is a local site of communications between neurons. It is small, but is the crucial node of brain neural networks. Neurotransmitters and metals (Zn or Cu) are co-released from synaptic vesicles in the presynaptic terminals to the synaptic clefts and bind to receptors in the postsynaptic domains (PSDs). Synapses are vulnerable regions for these neurodegenerative diseases, since synaptic plasticity is essential to memory formation. The synaptic cleft is conceptualized as a cylinder with 20 nm height and 200 nm radius [134]. Thus, the distance between presynaptic terminals and the postsynaptic membranes (~20 nm) may be small enough for proteins at the presynaptic terminal to interact with proteins in the postsynaptic terminal. Considering the small volume of synaptic clefts, it is plausible that neurotransmitters or metals are concentrated at synaptic clefts and their levels may be much higher than the level in the extracellular fluid. The concentration of glutamate in the synaptic cleft is estimated to reach a few millimolar range after 1 ms of neuronal depolarization, and the Zn concentration after depolarization is estimated to be 1–100 μM. Thus, amyloidogenic proteins must certainly interact with other proteins in a considerable amount of Zn and Cu.
Considering the combined evidence, we hypothesize that metal imbalance at the synapse contributes to the pathogenesis of neurodegenerative diseases. Under typical physiological conditions (Fig. 14.4), Zn can be released with glutamate and can bind with NMDA-type glutamate receptors or other receptors, inhibiting overall brain excitability. Secreted Zn can diffuse across the synaptic cleft, spill over to neighboring synapses, and influence the activity of neighboring synapses dose-dependently. Since glutamate produces excitation and Zn produced inhibition, differing concentrations of glutamate and Zn in the adjacent synapses create precise modulation of neuronal activity. Therefore, it is possible that Zn may have neuromodulator roles, transmitting the spatiotemporal information of neuronal activity. This may enable “lateral inhibition,” based on signaling contrast, and may be based on synaptic plasticity [135]. Synaptic Zn enters the postsynaptic neurons through Ca2+ channels and NMDA channels and regulates functions of various channels and receptors. Recent evidence suggests that the ZnT-1 transporter, which enhances Zn efflux to the extracellular compartment, is localized in postsynaptic membranes [136]. The ZnT-1 transporter binds with NMDA-type glutamate receptors and regulates the activity. In comparison, PrPC, an analogue of ZIP zinc transporters, localizes in postsynaptic membranes binding with AMPA-type glutamate receptor, which facilitate Zn influx. Thus, it is likely that synaptic Zn levels are controlled by both ZnT-1 and PrPC. MT-3 (GIF) secreted from neurons or glia may also regulate Zn homeostasis at synapses. Another contributor to Zn homeostasis is carnosine (CAR), an endogenous antioxidant and anti-cross-linking peptide, which is synthesized in glial cells [137].

Crosstalk between trace elements and amyloidogenic proteins at the synapse under normal condition. Zn and glutamate accumulate in synaptic vesicles and are released into synaptic clefts during neuronal excitation. Zn2+ regulates Ca2+ influx through glutamate receptors (NMDA-R, Ca-A/K), modulates neuronal information, and is implicated in the maintenance of synaptic plasticity and memory formation, similarly to Ca2+. Zn has important roles in information processing in the targeted neuron and also in neighboring neurons. APP and α-synuclein exist in the presynaptic domain, and PrPC is localized to the postsynaptic domain. These proteins are closely associated with the synaptic cleft and Zn and have cytoprotective roles via the regulation of metal homeostasis and protection from free radicals. PrPC binds to AMPA-type glutamate receptors and regulates Zn2+ levels similarly to ZIP Zn transporters. Additionally, the ZnT-1 Zn transporter is localized to postsynaptic membranes that express NMDA-type glutamate receptors and regulates Zn homeostasis. Moreover, PrPC has SOD activity and also regulates Cu2+ levels, which influence APP sequencing. APP converts Cu2+ to Cu+ and regulates Cu at the synapse. PrPC and α-synuclein have ferrireductase activity, which converts Fe3+ to Fe2+. APP binds to ferroportin and thereby regulates Fe2+ efflux. MT-3 and carnosine are released from glial cells, into synaptic clefts, and are also implicated in regulation of excess Zn. ZnT-1 zinc transporter 1, AMPA-R AMPA-type glutamate receptor, NMDA-R NMDA-type glutamate receptor, MT-3 metallothionein 3
Cu is also secreted at synaptic clefts following neuronal excitation. PrPC binds to Cu at its N-terminal domain and regulates synaptic Cu levels. It is also possible that PrPC provides Cu to APP or to NMDA-type glutamate receptor, thereby influencing the production of AβP or the neuronal excitability. APP also regulates Cu levels by reducing Cu2+ to Cu+, and both APP and PrPC reportedly attenuate Cu-induced toxicity.
Furthermore, APP controls Fe homeostasis by binding with ferroportin and promotes Fe efflux. In contrast, both PrPC and α-synuclein possess ferrireductase activity, regulate the Fe2+/Fe3+ ratio in synapses, and thereby control neurotransmitter synthesis.
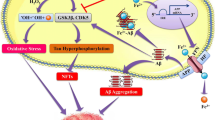
We have demonstrated the complex and subtle interactions between metals and amyloidogenic proteins at the synapse. When the homeostasis of metals is disrupted, synapses and neuron degradations occur, which contribute to the pathogenesis of neurodegenerative diseases (Fig. 14.5).

Disruption of metal homeostasis and the pathogenesis of neurodegenerative diseases. When toxic metals such as Al cross the blood-brain barrier, Al binds various Fe-binding proteins and influences Fe homeostasis, which initiates formation of free radicals and upregulation of APP and results in overproduction of AßP. Al also causes the oligomerization and accumulation of AßP. These adverse effects may enhance the synaptotoxicity and neurotoxicity of AβP and produce AD pathogenesis. Oligomerized AβPs form pore-like structures (AβP channels) in membranes, cause Ca dyshomeostasis, and initiate apoptotic neuronal death. Conversely, Zn inhibits amyloid channels and has neuroprotective properties. However, excess Zn resulting from overexcitation during ischemia also disrupts metal homeostasis at the synapse, initiates neurodegeneration, and contributes to the pathogenesis of vascular dementia. When exogenous PrPSc enters into the brain, it stimulates conversion of endogenous PrPC and results in PrPC depletion and PrPSc accumulation. The loss of neuroprotective functions by PrPC produces oxidative stress, enhances AßP neurotoxicity, and results in neuronal death. Accumulated PrPSc forms Ca2+-permeable channels (PrP channel) in membranes and disrupts Ca homeostasis
Interactions between one metal and other metals may disrupt metal homeostasis. Varying types of metal ions usually share binding sites, although their binding constants differ. In particular, Cu2+ and Zn2+share similar chemical characteristics and interact with each other. Similarly, Al3+ and Fe3+ share similar characteristics and interact with each other. Thus, the disruption of metal homeostasis may occur in the presence of toxic metals such as Al or the excess of essential metals such as Zn, Cu, or Fe.
In case of the pathogenesis of AD, Al enters into the brain. Al binds to Fe-binding proteins, inhibits IRP/IRE pathway as described previously, and influences the expression of Fe-binding proteins in the brain, including APP, α-synuclein, and ferritin, thereby increasing Fe levels. Overexpression of APP induced by Al results in accumulation of AβP. Al and other metals also accelerate AβP oligomerization and enhance the neurotoxicity. AβP oligomers may form amyloid channels on synaptic membranes, produce Ca dyshomeostasis, initiating synaptotoxicity and neurotoxicity, and produce the pathogenesis of AD. Zn inhibits AβP amyloid channel formations and neurotoxicity [69]. Zn also infounces Al-induced neurotoxicity [138]. In this context, Zn may have two faces in the pathogenesis of AD.
In case of the pathogenesis of prion diseases, pathogenetic PrPSc enters the brain primary by food contamination, triggers conformational changes to PrPC, and causes conversion of PrPC to PrPSc. PrPC is neuroprotective, since it has antioxidant activity and attenuates AßP neurotoxicity. Loss of neuroprotective PrPC may disrupt metal homeostasis. Increased Cu at the synapse produces the oxidative stress and influences the APP expression and processing. Thus, the depletion of normal PrPC results in oxidative stress, enhances the neurotoxicity of AßP, and results in apoptotic death of neurons and glia. Additionally, accumulated PrPSc may form channels (PrP channel) in the membrane and thereby disrupt Ca homeostasis as well as AßP. Cu2+ and Zn2+ inhibit the conformational conversion from PrPC to PrPSc and also attenuate PrPSc-induced Ca dyshomeostasis and neurotoxicity. However, Mn may enhance PrPSc toxicity by influencing the conversion of PrPC.
In the case of total ischemia, excess Zn is released with glutamate, which produces disruption of Ca homeostasis, triggers mitochondrial energy depletion and ER stress, and contributes to neurodegeneration. Zn also enhances the expressions of Ca-permeable AMPA-type glutamate receptors and enhances Ca dyshomeostasis. Considering that vascular degeneration may be linked with the early-stage AD, Zn neurotoxicity may also be associated with AD.
8 Metal-Related Treatments for Neurodegenerative Diseases
Several studies have investigated metal chelators as potential treatment for neurodegenerative diseases. Clioquinol (5-chloro-7-iodo-8-quinolinol) and its derivatives have been assessed in therapeutic trials for AD. Clioquinol (quinoform), a chelator of Cu2+ or Zn2+, inhibits oligomerization of AßP and attenuates the accumulation of amyloid in the brains of experimental animals [139]. Clinical trials using its analogue, PBT2, are under investigation [140]. Deferoxamine (DFO), a chelator of Al and Fe, attenuates the decline of daily living skills in AD patients [141]. Intranasal application of DFO reportedly attenuates memory loss and amyloid deposition [142]. Silicates, which couple with Al and reduce its toxicity, are also candidates for chelation therapy in AD [143].
Clioquinol treatment also had beneficial effects on scrapie-induced memory impairment [144]. It was also revealed that D-(-)-penicillamine, a Cu2+-specific chelator, attenuated the pathogenesis of prion diseases in vivo [145].
Our screen for substances that protect against PrP106–126-induced neurotoxicity revealed that carnosine may be a candidate treatment modality for prion diseases [112]. Carnosine is a water-soluble dipeptide contained in mammalian muscles and in the brain, particularly in the olfactory bulbs [137]. Carnosine has antioxidant and anti-glycation properties as well as the ability to bind to metals. Additionally, carnosine has anti-cross-linking properties, inhibits the oligomerization of AßP, and attenuates neurodegeneration in a mouse model of AD [146]. We previously reported that carnosine inhibited Zn2+-induced neuronal death after ischemia [2, 133]. Considering these beneficial characteristics, carnosine may have neuroprotective functions in the brain. Based on these findings, we have published a patent for carnosine as a possible basis for drugs designed to treat vascular dementia [147].
9 Conclusions
We have demonstrated the complex and subtle interactions between metals and amyloidogenic proteins at the synapse. This crosstalk is essential for normal brain functions, and therefore, the disruption of metal homeostasis may contribute to the conformational changes of amyloidogenic proteins and to the pathogenesis of neurodegenerative diseases. Our findings suggest that metal dyshomeostasis may be one of the common mechanisms for this pathogenesis and may elucidate the enigmatic roles of trace elements in neurodegenerative diseases and preventive drug development. In conclusion, our working hypothesis may contribute to enhanced understanding of the role of metals in the neurodegenerative diseases. Further research regarding “neurometallomics” is necessary, particularly in relation to the molecular mechanisms of synaptic degeneration and the quantitative analysis of neurometals.
References
Kawahara M, Kato-Negishi M (2011) Link between aluminum and the pathogenesis of Alzheimer’s disease: the integration of the aluminum and amyloid cascade hypotheses. Int J Alzheimer Dis 2011:276393
Kawahara M, Mizuno D, Koyama H, Konoha K, Ohkawara S, Sadakane Y (2013) Disruption of zinc homeostasis and the pathogenesis of senile dementia. Metallomics 6:209–219
Carrell RW, Lomas DA (1997) Conformational disease. Lancet 350:134–138
Kawahara M (2010) Role of calcium dyshomeostasis via amyloid channels in the pathogenesis of Alzheimer’s disease. Curr Pharm Des 16:2779–2789
Becker JSS, Matusch A, Palm C, Salber D, Morton K, Becker S (2010) Bioimaging of metals in brain tissue by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) and metallomics. Metallomics 2:104–111
Youdim MB (2008) Brain iron deficiency and excess; cognitive impairment and neurodegeneration with involvement of striatum and hippocampus. Neurotox Res 14:45–56
Crichton RR, Wilmet S, Legssyer R, Ward RJ (2002) Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J Inorg Biochem 91:9–18
Conrad ME, Umbreit JN (2000) Iron absorption and transport-an update. Am J Hematol 64:287–298
Davies P, Moualla D, Brown DR (2011) Alpha-synuclein is a cellular ferrireductase. PLoS One 6, e15814. doi:10.1371/journal.pone.0015814
Singh A, Haldar S, Horback K, Tom C, Zhou L, Meyerson H, Singh N (2013) Prion protein regulates iron transport by functioning as a ferrireductase. J Alzheimers Dis 35:541–552
Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA (2014) β-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 9:e114174. doi: 10.1371/journal.pone.0114174. eCollection 2014
Muckenthaler MU, Galy B, Hentze MW (2008) Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu Rev Nutr 28:197–213
Rogers JT, Bush AI, Cho HH, Smith DH, Thomson AM, Friedlich AL, Lahiri DK, Leedman PJ, Huang X, Cahill CM (2008) Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: riboregulation against neural oxidative damage in Alzheimer’s disease. Biochem Soc Trans 36:1282–1287
Fukada T, Yamasaki S, Nishida K, Murakami M, Hirano T (2011) Zinc homeostasis and signaling in health and diseases: zinc signaling. J Biol Inorg Chem 16:1123–1134
Sandstead HH (2012) Subclinical zinc deficiency impairs human brain function. J Trace Elem Med Biol 26:70–73
Takeda A, Tamano H (2009) Insight into zinc signaling from dietary zinc deficiency. Brain Res Rev 62:33–44
Frederickson CJ, Suh SW, Silva D, Frederickson CJ, Thompson RB (2000) Importance of zinc in the central nervous system: the zinc-containing neuron. J Nutr 130:1471S–1483S
Vogt K, Mellor J, Tong G, Nicoll R (2000) The actions of synaptically released zinc at hippocampal mossy fiber synapses. Neuron 26:187–196
Takeda A, Fujii H, Minamino T, Tamano H (2014) Intracellular Zn(2+) signaling in cognition. J Neurosci Res 92:819–824
Ueno S, Tsukamoto M, Hirano T, Kikuchi K, Yamada MK, Nishiyama N, Nagano T, Matsuki N, Ikegaya Y (2002) Mossy fiber Zn2+ spillover modulates heterosynaptic N-methyl-D-aspartate receptor activity in hippocampal CA3 circuits. J Cell Biol 158:215–220
Pan E, Zhang XA, Huang Z, Krezel A, Zhao M, Tinberg CE, Lippard SJ, McNamara JO (2011) Vesicular zinc promotes presynaptic and inhibits postsynaptic long-term potentiation of mossy fiber-CA3 synapse. Neuron 71:1116–1126
Bolognin S, Cozzi B, Zambenedetti P, Zatta P (2014) Metallothioneins and the central nervous system: from a deregulation in neurodegenerative diseases to the development of new therapeutic approaches. J Alzheimers Dis 41:29–42
Uchida Y, Takio K, Titani K, Ihara Y, Tomonaga M (1991) The growth inhibitory factor that is deficient in the Alzheimer’s disease brain is a 68 amino acid metallothionein-like protein. Neuron 7:337–347
Uchida Y (1994) Growth-inhibitory factor, metallothionein-like protein, and neurodegenerative diseases. Biol Signals 3:211–215
Fukada T, Kambe T (2011) Molecular and genetic features of zinc transporters in physiology and pathogenesis. Metallomics 3:662–674
Tsuda M, Imaizumi K, Katayama T, Kitagawa K, Wanaka A, Tohyama M, Takagi T (1997) Expression of zinc transporter gene, ZnT-1, is induced after transient forebrain ischemia in the gerbil. J Neurosci 17:6678–6684
Sindreu C, Palmiter RD, Storm DR (2011) Zinc transporter ZnT-3 regulates presynaptic Erk1/2 signaling and hippocampus-dependent memory. Proc Natl Acad Sci U S A 108:3366–3370
Bin BH, Fukada T, Hosaka T, Yamasaki S, Ohashi W, Hojyo S, Miyai T, Nishida K, Yokoyama S, Hirano T (2011) Biochemical characterization of human ZIP13 protein: a homo-dimerized zinc transporter involved in the spondylocheiro dysplastic Ehlers-Danlos syndrome. J Biol Chem 286:40255–40265
Kodama H, Fujisawa C, Bhadhprasit W (2010) Pathology, clinical features and treatments of congenital copper metabolic disorders–focus on neurologic aspects. Brain Dev 33:243–251
D’Ambrosi N, Rossi L (2015) Copper at synapse: release, binding and modulation of neurotransmission. Neurochem Int 90:36–45
Dodani SC, Firl A, Chan J, Nam CI, Aron AT, Onak CS, Ramos-Torres KM, Paek J, Webster CM, Feller MB, Chang CJ (2014) Copper is an endogenous modulator of neural circuit spontaneous activity. Proc Natl Acad Sci U S A 111:16280–16285
Exley C (2005) The aluminium-amyloid cascade hypothesis and Alzheimer’s disease. Subcell Biochem 38:225–234
Yase Y, Yoshida S, Kihira T, Wakayama I, Komoto J (2001) Kii ALS dementia. Neuropathology 21:105–109
Alfrey AC, LeGendre GR, Kaehny WD (1976) The dialysis encephalopathy syndrome: possible aluminium intoxication. New Engl J Med 294:184–188
Petrik MS, Wong MC, Tabata RC, Garry RF, Shaw CA (2007) Aluminum adjuvant linked to Gulf War illness induces motor neuron death in mice. Neruomol Med 9:83–92
Oteiza PI (1994) A mechanism for the stimulatory effect of aluminum on iron-induced lipid peroxidation. Arch Biochem Biophys 308:374–379
Selkoe DJ (1991) The molecular pathology of Alzheimer’s disease. Neuron 6:487–498
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572–580
Hardy JA, Higgins GA (1992) Alzheimer’s disease: the amyloid cascade hypothesis. Science 256:184–185
Wirths O, Multhaup G, Bayer TA (2004) A modified ß-amyloid hypothesis: intraneuronal accumulation of the beta-amyloid peptide–the first step of a fatal cascade. J Neurochem 91:513–520
Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L et al (1991) Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature 349:704–706
Selkoe DJ, Wolfe MS (2007) Presenilin: running with scissors in the membrane. Cell 131:215–221
Yankner BA, Duffy LK, Kirschner DA (1990) Neurotropic and neurotoxic effects of amyloid ß protein: reversal by tachykinin neuropeptides. Nature 250:279–282
Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW (1993) Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 13:1676–1687
Jarrett JT, Lansbury PT Jr (1993) Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell 73:1055–1058
Fukuyama R, Mizuno T, Mori S, Nakajima K, Fushiki S, Yanagisawa K (2000) Age-dependent change in the levels of Aβ40 and Aβ42 in cerebrospinal fluid from control subjects, and a decrease in the ratio of Aβ42 to Aβ40 level in cerebrospinal fluid from Alzheimer’s disease patients. Eur Neurol 43:155–160
Kawahara M, Negishi-Kato M, Sadakane Y (2009) Calcium dyshomeostasis and neurotoxicity of Alzheimer’s beta-amyloid protein. Expert Rev Neurother 9:681–693
Dyrks T, Dyrks E, Masters CL, Beyreuther K (1993) Amyloidogenicity of rodent and human ß A4 sequences. FEBS Lett 324:231–236
Flaten TP (2001) Aluminium as a risk factor in Alzheimer’s disease, with emphasis on drinking water. Brain Res Bull 55:187–196
Exley C, Price NC, Kelly SM, Birchall JD (1993) An interaction of β-amyloid with aluminium in vitro. FEBS Lett 324:293–295
Mantyh PW, Ghilardi JR, Rogers S, DeMaster E, Allen CJ, Stimson ER, Maggio JE (1993) Aluminum, iron, and zinc ions promote aggregation of physiological concentrations of beta-amyloid peptide. J Neurochem 61:1171–1174
Kawahara M, Muramoto K, Kobayashi K, Mori H, Kuroda Y (1994) Aluminum promotes the aggregation of Alzheimer’s β-amyloid protein in vitro. Biochem Biophys Res Commun 198:531–535
Kawahara M, Kato M, Kuroda Y (2001) Effects of aluminum on the neurotoxicity of primary cultured neurons and on the aggregation of ß-amyloid protein. Brain Res Bull 55:211–217
Bush AI, Pettingell WH, Multhaup G, d Paradis M, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE (1994) Rapid induction of Alzheimer Aß amyloid formation by zinc. Science 265:1464–1467
Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, Hartshorn MA, Tanzi RE, Bush AI (1998) Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis. J Biol Chem 273:12817–12826
Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ (1999) Protofibrillar intermediates of amyloid ß-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19:8876–8884
Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ (2000) The oligomerization of amyloid ß-protein begins intracellularly in cells derived from human brain. Biochemistry 39:10831–10839
Selkoe DJ (2008) Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav Brain Res 192:106–113
Chen WT, Liao YH, Yu HM, Cheng IH, Chen YR (2011) Distinct effects of Zn2+, Cu2+, Fe3+, and Al3+ on amyloid-beta stability, oligomerization, and aggregation: amyloid-beta destabilization promotes annular protofibril formation. J Biol Chem 286:9646–9656
Sharma AK, Pavlova ST, Kim J, Kim J, Mirica LM (2013) The effect of Cu(2+) and Zn(2+) on the Aβ42 peptide aggregation and cellular toxicity. Metallomics 5:1529–1536
Drago D, Cavaliere A, Mascetra N, Ciavardelli D, di Ilio C, Zatta P, Sensi SL (2008) Aluminum modulates effects of beta amyloid(1-42) on neuronal calcium homeostasis and mitochondria functioning and is altered in a triple transgenic mouse model of Alzheimer’s disease. Rejuvenation Res 11:861–871
Bolognin S, Zatta P, Lorenzetto E, Valenti MT, Buffelli M (2013) β-Amyloid-aluminum complex alters cytoskeletal stability and increases ROS production in cortical neurons. Neurochem Int 62:566–574
Everett J, Céspedes E, Shelford LR, Exley C, Collingwood JF, Dobson J, van der Laan G, Jenkins CA, Arenholz E, Telling ND (2014) Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide β-amyloid (1-42). J R Soc Interface 11:20140165. doi:10.1098/rsif.2014.0165
Arispe N, Diaz JC, Simakova O (2007) Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim Biophys Acta 1768:1952–1965
Kawahara M, Ohtsuka I, Yokoyama S, Kato-Negishi M, Sadakane Y (2011) Membrane incorporation, channel formation, and disruption of calcium homeostasis by Alzheimer’s ß-amyloid protein. Int J Alzheimer Dis:304583
Lal R, Lin H, Quist AP (2007) Amyloid beta ion channel: 3D structure and relevance to amyloid channel paradigm. Biochim Biophys Acta 1768:1966–1975
Arispe N, Rojas E, Pollard HB (1993) Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: blockade by tromethamine and aluminum. Proc Natl Acad Sci U S A 90:567–571
Arispe N, Pollard HB, Rojas E (1996) Zn2+ interaction with Alzheimer amyloid beta protein calcium channels. Proc Natl Acad Sci U S A 93:1710–1715
Kawahara M, Arispe N, Kuroda Y, Rojas E (1997) Alzheimer’s disease amyloid ß-protein forms Zn2+-sensitive, cation-selective channels across excised membrane patches from hypothalamic neurons. Biophys J 73:67–75
Kawahara M, Arispe N, Kuroda Y, Rojas E (2000) Alzheimer’s ß-amyloid, human islet amylin and prion protein fragment evoke intracellular free-calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell-line. J Biol Chem 275:14077–14083
Ayton S, Lei P, Bush AI (2013) Metallostasis in Alzheimer’s disease. Free Radic Biol Med 62:76–89
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, Greig NH, Lahiri DK, Tanzi RE, Bush AI, Giordano T, Gullans SR (2002) An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J Biol Chem 277:45518–45528
Namekata K, Imagawa M, Terashi A, Ohta S, Oyama F, Ihara Y (1997) Association of transferrin C2 allele with late-onset Alzheimer’s disease. Hum Genet 101:126–129
Imagawa M, Naruse S, Tsuji S, Fujioka A, Yamaguchi H (1992) Coenzyme Q10, iron, and vitamine B6 in genetically-confirmed Alzheimer’s disease. Lancet 340:671
White AR, Multhaup G, Maher F, Bellingham S, Camakaris J, Zheng H, Bush AI, Beyreuther K, Masters CL, Cappai R (1999) The Alzheimer’s disease amyloid precursor protein modulates copper-induced toxicity and oxidative stress in primary neuronal cultures. J Neurosci 19:9170–9179
Baumkötter F, Schmidt N, Vargas C, Schilling S, Weber R, Wagner K, Fiedler S, Klug W, Radzimanowski J, Nickolaus S, Keller S, Eggert S, Wild K, Kins S (2014) Amyloid precursor protein dimerization and synaptogenic function depend on copper binding to the growth factor-like domain. J Neurosci 34:11159–11172
White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R (1999) Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res 842:439–444
Ciccotosto GD, James SA, Altissimo M, Paterson D, Vogt S, Lai B, de Jonge MD, Howard DL, Bush AI, Cappai R (2014) Quantitation and localization of intracellular redox active metals by X-ray fluorescence microscopy in cortical neurons derived from APP and APLP2 knockout tissue. Metallomics 6:1894–1904
Raha AA, Vaishnav RA, Friedland RP, Bomford A, Raha-Chowdhury R (2013) The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun 1:55. doi:10.1186/2051-5960-1-55
Oshiro S, Kawahara M, Shirao M, Muramoto K, Kobayashi K, Ishige R, Nozawa K, Hori M, Yung C, Kitajima S, Kuroda Y (1998) Aluminum taken up by transferrin independent iron uptake affects the iron metabolism in rat cortical cells. J Biochem 123:42–46
Yamanaka K, Minato N, Iwai K (1999) Stabilization of iron regulatory protein 2, IRP2, by aluminum. FEBS Lett 462:216–220
Li XB, Zhang ZY, Yin LH, Schluesener HJ (2012) The profile of β-amyloid precursor protein expression of rats induced by aluminum. Environ Toxicol Pharmacol 33:135–140
Liang RF, Li WQ, Wang H, Wang JX, Niu Q (2013) Impact of sub-chronic aluminium-maltolate exposure on catabolism of amyloid precursor protein in rats. Biomed Environ Sci 26:445–452
Walton JR, Wang MX (2009) APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J Inorg Biochem 103:1548–1554
Wang L, Hu J, Zhao Y, Lu X, Zhang Q, Niu Q (2014) Effects of aluminium on β-amyloid (1-42) and secretases (APP-cleaving enzymes) in rat brain. Neurochem Res 39:1338–1345
Oshiro S, Kawahara M, Kuroda Y, Zhang C, Cai Y, Kitajima S, Shirao M (2000) Glial cells contribute more to iron and aluminum accumulation but are more resistant to oxidative stress than neuronal cells. Biochim Biophys Acta 1502:405–414
Green KN, LaFerla FM (2008) Linking calcium to Aß and Alzheimer’s disease. Neuron 59:190–42008
Mattson MP (2010) ER calcium and Alzheimer’s disease: in a state of flux. Sci Signal 3:pe10. doi:10.1126/scisignal.3114pe10
Greenough MA, Volitakis I, Li QX, Laughton K, Evin G, Ho M, Dalziel AH, Camakaris J, Bush AI (2011) Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J Biol Chem 286:9776–9786
Southon A, Greenough MA, Ganio G, Bush AI, Burke R, Camakaris J (2013) Presenilin promotes dietary copper uptake. PLoS One 8, e62811
Prusiner SB (1997) Prion diseases and the BSE crisis. Science 278:245–251
Mizuno D, Koyama H, Ohkawara S, Sadakane Y, Kawahara M (2014) Involvement of trace elements in the pathogenesis of prion diseases. Curr Pharam Biotech 15:1049–1057
Brown DR, Qin K, Herms JW, Madlung A, Manson J, Strome R, Fraser PE, Kruck T, von Bohlen A, Schulz-Schaeffer W, Giese A, Westaway D, Kretzschmar H (1997) The cellular prion protein binds copper in vivo. Nature 390:684–687
Jackson GS, Murray I, Hosszu LL, Gibbs N, Waltho JP, Clarke AR, Collinge J (2001) Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci U S A 98:8531–8535
Brown DR (2009) Brain proteins that mind metals: a neurodegenerative perspective. Dalton Trans 21:4069–4076
Gasperini L, Meneghetti E, Pastore B, Benetti F, Legname G (2015) Prion protein and copper cooperatively protect neurons by modulating NMDA receptor through S-nitrosylation. Antioxid Redox Signal 22:772–784
Alfaidy N, Chauvet S, Donadio-Andrei S, Salomon A, Saoudi Y, Richaud P, Aude-Garcia C, Hoffmann P, Andrieux A, Moulis JM, Feige JJ, Benharouga M (2013) Prion protein expression and functional importance in developmental angiogenesis: role in oxidative stress and copper homeostasis. Antioxid Redox Signal 18:400–411
Rotilio G1, Carrì MT, Rossi L, Ciriolo MR (2000) Copper-dependent oxidative stress and neurodegeneration. IUBMB Life 50:309–314
White AR, Collins SJ, Maher F, Jobling MF, Stewart LR, Thyer JM, Beyreuther K, Masters CL, Cappai R (1999) Prion protein-deficient neurons reveal lower glutathione reductase activity and increased susceptibility to hydrogen peroxide toxicity. Am J Pathol 155:1723–1730
Stellato F, Spevacek A, Proux O, Minicozzi V, Millhauser G, Morante S (2011) Zinc modulates copper coordination mode in prion protein octa-repeat subdomains. Eur Biophys J 40:1259–1270
Schmitt-Ulms G, Ehsani S, Watts JC, Westaway D, Wille H (2009) Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS One 4, e7208
Pocanschi CL, Ehsani S, Mehrabian M, Wille H, Reginold W, Trimble WS, Wang H, Yee A, Arrowsmith CH, Bozóky Z, Kay LE, Forman-Kay JD, Rini JM, Schmitt-Ulms G (2013) The ZIP5 ectodomain co-localizes with PrP and may acquire a PrP-like fold that assembles into a dimer. PLoS One 8, e72446
Watt NT, Griffiths HH, Hooper NM (2013) Neuronal zinc regulation and the prion protein. Prion 7:203–208
Singh N, Haldar S, Tripathi AK, McElwee MK, Horback K, Beserra A (2014) Iron in neurodegenerative disorders of protein misfolding: a case of prion disorders and Parkinson’s disease. Antioxid Redox Signal 21:471–484
Singh A, Kong Q, Luo X, Petersen RB, Meyerson H, Singh N (2009) Prion protein (PrP) knock-out mice show altered iron metabolism: a functional role for PrP in iron uptake and transport. PLoS One 4, e6115
Haldar S, Beveridge J, Wong J, Singh A, Galimberti D, Borroni B, Zhu X, Blevins J, Greenlee J, Perry G, Mukhopadhyay CK, Schmotzer C, Singh N (2013) A low-molecular-weight ferroxidase is increased in the CSF of sCJD cases: CSF ferroxidase and transferrin as diagnostic biomarkers for sCJD. Antioxid Redox Signal 19:1662–1675
Johnson CJ, Gilbert PU, Abrecht M, Baldwin KL, Russell RE, Pedersen JA, Aiken JM, McKenzie D (2013) Low copper and high manganese levels in prion protein plaques. Viruses 5:654–662
Thackray AM, Madec JY, Wong E, Morgan-Warren R, Brown DR, Baron T, Bujdoso R (2003) Detection of bovine spongiform encephalopathy, ovine scrapie prion-related protein (PrPSc) and normal PrPc by monoclonal antibodies raised to copper-refolded prion protein. Biochem J 370:81–90
Davies P, Brown DR (2009) Manganese enhances prion protein survival in model soils and increases prion infectivity to cells. PLoS One 4:e7518
White SN, O’Rourke KI, Gidlewski T, VerCauteren KC, Mousel MR, Phillips GE, Spraker TR (2010) Increased risk of chronic wasting disease in Rocky Mountain elk associated with decreased magnesium and increased manganese in brain tissue. Can J Vet Res 74:50–53
Slivarichová D, Mitrová E, Ursínyová M, Uhnáková I, Koscová S, Wsólová L (2011) Geographic accumulation of Creutzfeldt-Jakob disease in Slovakia–environmental metal imbalance as a possible cofactor. Cent Eur J Public Health 19:158–164
Kawahara M, Koyama H, Nagata T, Sadakane Y (2011) Zinc, copper, and carnosine attenuate neurotoxicity of prion fragment PrP106-126. Metallomics 3:726–734
Bonetto V, Massignan T, Chiesa R, Morbin M, Mazzoleni G, Diomede L, Angeretti N, Colombo L, Forloni G, Tagliavini F, Salmona M (2002) Synthetic miniprion PrP106. J Biol Chem 277:31327–31334
Grasso D, Milardi D, La Rosa C, Rizzarelli E (2004) The different role of Cu++ and Zn++ ions in affecting the interaction of prion peptide PrP106-126 with model membranes. Chem Commun (Camb) 21:246–247
Inayathullah M, Satheeshkumar KS, Malkovskiy AV, Carre AL, Sivanesan S, Hardesty JO, Rajadas J (2013) Solvent microenvironments and copper binding alters the conformation and toxicity of a prion fragment. PLoS One 8, e85160
Thakur AK, Srivastava AK, Srinivas V, Chary KV, Rao CM (2011) Copper alters aggregation behavior of prion protein and induces novel interactions between its N- and C-terminal regions. J Biol Chem 286:38533–38545
Ward B, Walker K, Exley C (2008) Copper(II) inhibits the formation of amylin amyloid in vitro. J Inorg Biochem 102:371–375
Dettmer U, Selkoe D, Bartels T (2015) New insights into cellular α-synuclein homeostasis in health and disease. Curr Opin Neurobiol 36:15–22
Chen P, Chakraborty S, Mukhopadhyay S, Lee E, Paoliello MM, Bowman AB, Aschner M (2015) Manganese homeostasis in the nervous system. J Neurochem 134:601–610
Uversky VN, Li J, Fink AL (2001) Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J Biol Chem 276:44284–44296
Tavassoly O, Nokhrin S, Dmitriev OY, Lee JS (2014) Cu(II) and dopamine bind to α-synuclein and cause large conformational changes. FEBS J 281:2738–2753
Cahill CM, Lahiri DK, Huang X, Rogers JT (2009) Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim Biophys Acta 1790:615–628
Kienzl E, Jellinger K, Stachelberger H, Linert W (1999) Iron as catalyst for oxidative stress in the pathogenesis of Parkinson’s disease? Life Sci 65:1973–1976
Lee J, Grabb MC, Zipfel GJ, Choi DW (2000) Brain tissue responses to ischemia. J Clin Invest 106:723–731
Weiss JH, Sensi SL, Koh JY (2000) Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci 21:395–401
Frederickson CJ, Klitenick MA, Manton WI, Kirkpatrick JB (1983) Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain Res 27:335–339
Koh JY, Suh SW, Gwag BJ, He YY, Hsu CY, Choi DW (1996) The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 272:1013–1016
Calderone A1, Jover T, Mashiko T, Noh KM, Tanaka H, Bennett MV, Zukin RS (2004) Late calcium EDTA rescues hippocampal CA1 neurons from global ischemia-induced death. J Neurosci 24:9903–9913
Weiss JH, Sensi SL (2000) Ca2+-Zn2+ permeable AMPA or kainate receptors: possible key factors in selective neurodegeneration. Trends Neurosci 23:365–371
Kawahara M, Kato-Negishi M, Kuroda Y (2002) Pyruvate blocks zinc-induced neurotoxicity in immortalized hypothalamic neurons. Cell Mol Neurobiol 22:87–93
Koyama H, Konoha K, Sadakane Y, Ohkawara S, Kawahara M (2012) Zinc neurotoxicity and the pathogenesis of vascular-type dementia: involvement of calcium dyshomeostasis and carnosine. J Clin Toxicol S3-002. doi:10.4172/2161-0495.S3-002
Mellon PL, Windle JJ, Goldsmith PC, Padula CA, Roberts JL, Weiner RI (1990) Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron 5:1–10
Mizuno D, Kawahara M (2014) Carnosine: a possible drug for vascular dementia. J Vasc Med Surg 2:3. doi.org/10.4172/2329-6925.1000146
Schikorski T, Stevens CF (1997) Quantitative ultrastructural analysis of hippocampal excitatory synapses. J Neurosci 17:5858–5867
Wang X, Sommer FT, Hirsch JA (2011) Inhibitory circuits for visual processing in thalamus. Curr Opin Neurobiol 21:726–733
Mellone M, Pelucchi S, Alberti L, Genazzani AA, Di Luca M, Gardoni F (2015) Zinc transporter-1: a novel NMDA receptor-binding protein at the postsynaptic density. J Neurochem 132:159–168
Hipkiss AR (2009) Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res 57:87–154
Singla N, Dhawan DK (2014) Zinc modulates aluminium-induced oxidative stress and cellular injury in rat brain. Metallomics 6:1941–1950
Zhang YH, Raymick J, Sarkar S, Lahiri DK, Ray B, Holtzman D, Dumas M, Schmued LC (2013) Efficacy and toxicity of clioquinol treatment and A-beta42 inoculation in the APP/PSI mouse model of Alzheimer’s disease. Curr Alzheimer Res 10:494–506
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, Ingelsson M, Masters CL, Tanzi RE, Cummings JL, Herd CM, Bush AI (2010) PBT2 rapidly improves cognition in Alzheimer’s disease: additional phase II analyses. J Alzheimers Dis 20:509–516
Crapper McLachlan DR, Dalton AJ, Kruck TP, Bell MY, Smith WL, Kalow W, Andrews DF (1991) Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337:1304–1308
Fine JM, Renner DB, Forsberg AC, Cameron RA, Galick BT, Le C, Conway PM, Stroebel BM, Frey WH, Hanson LR (2015) Intranasal deferoxamine engages multiple pathways to decrease memory loss in the APP/PS1 model of amyloid accumulation. Neurosci Lett 584:362–367. doi:10.1016/j.neulet.2014.11.013
Exley C (2007) Organosilicon therapy in Alzheimer’s disease? J Alzheimers Dis 11:301–302
Bareggi SR, Braida D, Pollera C, Bondiolotti G, Formentin E, Puricelli M, Poli G, Ponti W, Sala M (2009) Effects of clioquinol on memory impairment and the neurochemical modifications induced by scrapie infection in golden hamsters. Brain Res 1280:195–200
Sigurdsson EM, Brown DR, Alim MA, Scholtzova H, Carp R, Meeker HC, Prelli F, Frangione B, Wisniewski T (2003) Copper chelation delays the onset of prion disease. J Biol Chem 278:46199–46202
Corona C, Frazzini V, Silvestri E, Lattanzio R, La Sorda R, Piantelli M, Canzoniero LM, Ciavardelli D, Rizzarelli E, Sensi SL (2011) Effects of dietary supplementation of carnosine on mitochondrial dysfunction, amyloid pathology, and cognitive deficits in 3xTg-AD mice. PLoS One 6:e17971. doi:10.1371/journal.pone.0017971
Kawahara M, Konoha K, Nagata T, Sadakane Y (2007) Protective substances against zinc-induced neuronal death after ischemia: carnosine a target for drug of vascular type of dementia. Recent Patents CNS Drug Discov 2:145–149
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer Japan KK
About this chapter
Cite this chapter
Mizuno, D., Kawahara, M. (2017). Link Between Metal Homeostasis and Neurodegenerative Diseases: Crosstalk of Metals and Amyloidogenic Proteins at the Synapse. In: Ogra, Y., Hirata, T. (eds) Metallomics. Springer, Tokyo. https://doi.org/10.1007/978-4-431-56463-8_14
Download citation
DOI: https://doi.org/10.1007/978-4-431-56463-8_14
Published:
Publisher Name: Springer, Tokyo
Print ISBN: 978-4-431-56461-4
Online ISBN: 978-4-431-56463-8
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)