
Abstract
Chronic administration of aluminium has been proposed as an environmental factor that may affect some pathological changes related to neurotoxicity and Alzheimer’s disease (AD). The abnormal generation and deposition of β-amyloid (Aβ) in senile plaques are hallmark features in the brains of AD patients. Furthermore, Aβ is generated by the sequential cleavage of the amyloid precursor protein (APP) via the APP cleaving enzyme (α-secretase, or β-secretase) and γ-secretase. In the present study, we investigated the modulation of Aβ deposition and neurotoxicity in aluminium-maltolate-treated (0, 15, 30, 45 mmol/kg body weight via intraperitoneal injection) in experimental rats. We measured Aβ1–40 and Aβ1–42 in the cortex and hippocampus in rat brains using ELISA. Subtypes of α-secretase, β-secretase, and γ-secretase, including ADAM9, ADAM10, ADAM17 (TACE), BACE1, presenilin 1 (PS1) and nicastrin (NCT), were determined using western blotting analyses. These results indicated that aluminium-maltolate induced an AD-like behavioural deficit in rats at 30 and 45 mmol/kg body weight. Moreover, the Aβ1–42 content increased significantly, both in the cortex and hippocampus, although no changes were observed in Aβ1–40. Furthermore, ADAM9, ADAM10, and ADAM17 decreased significantly; in contrast, BACE1, PS1, and NCT showed significant increase. Taken together, these results suggest that the changes in secretases may correlate to the abnormal deposition of Aβ by aluminium in rat brains.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is a chronic neurodegenerative disorder characterised by the progressive loss of memory and cognitive ability [1]. The pathogenesis of AD involves a key event of changes in the morphology of the amyloid-β (Aβ) peptide, from its soluble monomeric form into fibrillated aggregates in the brain [2]. The excessive production and accumulation of Aβ plays a critical role in the pathogenesis of AD at early stages and may represent a common pathway to AD induced by various factors [3–5] Aluminium is a common chronic neurotoxin, and considerable evidence supports the possibility that it is a possible risk factor for AD [3, 6–10]. It is also known that as an element, aluminium accelerates Aβ generation and aggregation, induces structural changes of Aβ, and increases the formation of Aβ oligomers [11–13].
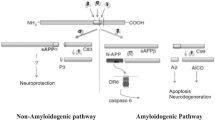
Amyloid-β, the primary component of amyloid plaques, is produced from amyloid precursor protein (APP) via a series of protease cleavages, including β- and γ-secretases [3]. APP is a transmembrane glycoprotein consisting of a large extracellular domain, a short transmembrane domain, and a cytoplasmic tail. β-secretase cleaves APP between amino acids 671 and 672 and produces sAPPβ and C99, while γ-secretase cleaves C99 in at amino acids 711–714 [14]. The key step in the generation of Aβ is the cleavage of APP by β-secretases [beta-site APP-cleaving enzyme 1 (BACE1)] [3]. Furthermore, APP is also cleaved by α-secretases within the Aβ region and produces sAPPα and C83, which preclude the formation of Aβ [15, 16]. In addition, α- secretase appears to compete with β-secretase for the initial cleavage of APP but has opposite effects on Aβ generation [17]. Thus the expression and activity of both of these enzymes may affect the level of Aβ.
More than 20 years ago, Esch [18] described, for the first time, that APP is cleaved within the Aβ sequence by an unknown protease, which was called secretase and is now known as α-secretase. The initial protease α-secretase was shown to have characteristics of a metalloprotease [19], and different metalloproteases were suggested as potential constitutive a-secretases. The best studied α-secretases were members of the ADAM (a disintegrin and metalloprotease) family, in particular ADAM9, ADAM10 and ADAM17 [20], which together constitute α-secretase activity and mediate non-amyloidogenic α-processing of APP [21].
BACE1 is a type 1 transmembrane aspartic protease that is related to the pepsin and retroviral aspartic protease families. BACE1 activity has a low optimum pH, and the enzyme is predominantly localised in acidic intracellular compartments. Soon after the discovery of BACE1, a homologous protein, BACE2, was identified. BACE1 and BACE2 share 64 % amino acid sequence homology and thus raise the possibility that BACE2 is also a β-secretase. However, unlike BACE1, which is highly expressed in neurons of the brain, BACE2 is expressed at low levels in the brain and does not exhibit the same cleavage specificity for APP as BACE1, thus indicating that it is a poor β-secretase candidate.
γ-secretase is a multisubunit intramembranous protein complex consisting of at least four proteins; presenilin (PS), nicastrin (NCT), anterior pharynx defective-1 (Aph-1) and presenilin enhancer-2 (Pen-2) [22]. Presenilin is a central, catalytic component of the γ-secretase complex, which induces intramembrane cleavage of various protein substrates [23]. Nicastrin is a large protein, which contains a single transmembrane domain. NCT is itself not catalytically active but instead promotes the maturation and proper trafficking of other components of the γ-secretase complex. PEN-2, which contains 101 amino acids, is the smallest subunit of the γ-secretase complex and shares no significant domain homology with any other known protein family [24]. APH-1 protein has seven transmembrane domains and potentially acts as a scaffold for complex formation. In addition, APH-1 has been shown to contribute to the proper assembly of the complex. Furthermore, γ-secretase is the final protease in the course of Aβ generation, and its amount and activity can affect the generation and aggregation of Aβ.
However, whether α -, β-, γ-secretases are all involved in Al-induced elevation and aggregation of Aβ is still unknown. Because the hippocampus and cerebral cortex in humans and rodents can accumulate Aβ, they are considered to be AD vulnerable regions [25]. Thus, in the present study, changes in Aβ1–40 and Aβ1–42 in the hippocampus and cerebral cortex in aluminium-maltolate treated neurotoxic rats were shown. We also analysed the contents of amyloidogenic-secretase BACE1; non-amyloidogenic secretases, ADAM9, ADAM10 and ADAM17; presenilin1; and NCT in brain extracts in response to aluminium-maltolate exposure.
Materials and Methods
Materials
AlCl3·6H2O (purity 99 %) and maltolate (purity 99 %) were purchased from Sigma-Aldrich Chemical Co. (Steinheim, Germany). Rat Aβ1–40 and Aβ1–42 ELISA kits were obtained from Uscn Life Science, Inc. (Wuhan, China). The BCA protein quantitative kit, IgG (H+L), ECL western blot Kit, and anti-β-actin antibody were obtained from Beijing CoWin Bioscience Co. Ltd. (Beijing, China). The anti-ADAM9 antibody was purchased from Signaling Technology. Inc. (Boston, USA), and the anti ADAM17 antibody was purchased from Abcam Inc. (Cambridge, England). The anti-ADAM10, BACE1, PS1 and NCT antibodies were obtained from Epitomics, Inc. (California, USA). All chemicals were of analytical grade unless otherwise indicated.
Al(mal)3 Preparation
Al(mal)3 was prepared according to previously described methods [26]. AlCl3·6H2O was dissolved in distilled water to a final concentration of 90 mmol/L, and maltolate was dissolved in phosphate-buffered saline (PBS) to a final concentration of 270 mmol/L. Two solutions were then mixed in equal volumes, and the pH was adjusted to 7.4 with 1 mol/L NaOH. The Al(mal)3 solution used in the animal experiments was freshly prepared for each experiment, and all solutions were sterile filtered using 0.22 μm syringe filters immediately following preparation.
Animals
Forty male adult Sprague-Dawley (SD) rats (200–220 g) were supplied by the Experimental Animal Center of Shanxi Medical University (Taiyuan, China). The rats were housed with free access to food and water in a room with an ambient temperature of 22 ± 2 °C and a 12:12 h light/dark cycle. All experiments were performed strictly according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animals were induced using aluminium-maltolate. The animals were randomly assigned to four groups: control groups with saline treatment and Al(mal)3 groups with 15 mmol/kg BW, 30 mmol/kg BW, or 45 mmol/kg BW. Saline and Al(mal)3 were administered to the rats via intraperitoneal injections. Administration was performed daily for 2 months and at the same time every day.
Morris Water Maze
The Morris water maze consisted of a black pool (200 cm in diameter, 75 cm high, and 45 cm above floor level) filled with water, and a black platform (10 cm in diameter, 50 cm high) was submerged 1 cm below the water surface and placed in the centre of the Southwest quadrant. The four starting locations were labelled North (N), East (E), South (S), and West (W) at an equal distance from the rim. The water was maintained at 22 ± 2 °C, and the platform was placed in either one of four virtual quadrants at 20 cm from the sidewall. The movements of the rats were recorded using a video camera connected to a computer. Data were analysed using a tracking program (version 1.0.0.3, Pharmaceutical Institute of the Academy of Medical Science of China, Beijing, China). Over four consecutive days at 7 a.m., the rats were trained four times per day and given a maximum of 120 s to locate the hidden platform. Once the platform was found, the rats were allowed to stay on the platform for 30 s. Rats that failed to locate the platform were then placed on it. Each rat was gently placed into the water with their nose facing the wall at one of the starting points. All of the animals were tested in sequence, and the time interval between two trials was approximately 1–1.5 h. The animals were placed in the same starting points on each day. The escape latency was averaged from four trials per day. The platform was removed from the pool on the 5th day, and the average frequency in finding the location of the platform in all rats was recorded in 120 s.
Sample Preparation
After the Morris water maze test, the rats were anaesthetised, perfused and sacrificed. The brains were rapidly removed from the skulls and dissected into different regions. All subsequent operations were performed on ice and the samples were frozen in cryogenic refrigerators at −80 °C for further analysis. Rat cortical and hippocampal tissues were homogenised and solubilised in lysis buffer (Beijing CoWin Bioscience Co. Ltd, Beijing, China). Protein concentrations were determined using the BCA Kit (Beijing CoWin Bioscience Co. Ltd, Beijing, China).
Detection for Aluminium Levels in the Rat Brain
The cortical (Co) and hippocampal (Hi) tissues of the rats were digested using a microwave. Detection for aluminium levels in rat brains were performed using graphite furnace atomic absorption spectrometry (gfaas).
ELISA for Aβ1–40, and Aβ1–42 Proteins
All experimental procedures were performed according to the manufacturer’s instructions.
Western Blotting Analyses for APP, ADAM9, ADAM10, ADAM17, BACE1, PS1 and NCT
Proteins (30 µg) were separated using 10 % sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and were then transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with antibodies directed against the following proteins: APP (1:1,000); ADAM9 (1:1,000), ADAM10 (1:1,000); ADAM17 (1:2,000); BACE1 (1:2,000); PS1 (1:1,000); and NCT (1:1,000). Immunoreactive proteins were visualised by exposure to X-ray film. The protein bands were quantified using image scanning, and the optical density was measured using computer software (Image lab 3.0, Bio-Rad Laboratories, Inc., California, USA). The data were then corrected by background subtraction and normalised against β-actin as an internal control.
Data Analyses
Statistical analysis of the data was performed using SPSS 17.0. All results were expressed as the mean ± SEM. Behavioural tests were analysed using repeated measure and one-way ANOVA. The level of proteins was analysed using one-way ANOVA, followed by the post hoc Dunnet test. The correlation between the expression of proteins and Aβ was assessed using Pearson correlation analyses. The criterion for statistical significance was established at P < 0.05.
Results
Behavioural Results
None of the tested mice showed obvious health problems (e.g., weight loss, cataracts, or toxicity reaction). During the 4-day acquisition phase of the Morris water maze test, the escape latency of all rats in the platform quadrant gradually improved, and the escape latency of Al(mal)3-treated rats was longer compared to the control rats (F(3,39) = 4.887, P < 0.01, Fig. 1(1)). Consistent with the results of the escape latency, the average frequency in finding the platform within 120 s in normal rats was markedly higher compared to Al(mal)3 rats on the 5th day (F(3,39) = 9.575, P < 0.01, Fig. 1(2)). The results of the Morris water maze test demonstrated a longer escape latency and reduced frequency in finding the platform in the Al(mal)3 rats, indicating that Al(mal)3 affected learning and memory.

SD rats were treated with aluminium-maltolate at 0, 15, 30, and 45 mmol/kg BW, i.p. The Morris water maze test was applied after treatment. Repeated measure analysis; one-way ANOVA, n = 10 for each group; error bars indicate the SEM. *P < 0.05 indicates the significant difference compared to control
Aluminium Levels in Rat Brains
As shown in Fig. 2, the levels of aluminium increased significantly in both Co and Hi compared to control expression (F(3,39) = 82.475, P < 0.01 for Co, F(3,39) = 94.065, P < 0.01 for Hi).

Aluminium levels in the rat hippocampus (Hippo) and cortex (CTX) were measured after aluminium-maltolate treatment using gfaas. Each experiment consisted of three parallel samples. One-way ANOVA, n = 10 for each group; error bars indicate the SEM. *P < 0.05 indicates the significant difference compared to control
Aβ1–40 and Aβ1–42 Protein Levels
As shown in Fig. 3, there was no significant difference in the Aβ1–40 content between the Co and Hi of the Al(mal)3 -treated rats and control rats (F(3,39) = 0.880, P > 0.05 for Co, F(3,39) = 1.590, P > 0.05 for Hi, Fig. 3(1)). In contrast, a significant increase in Aβ1–42 expression was observed in both Co and Hi (F(3,39) = 6.442, P < 0.01 for Co, F(3,39) = 29.908, P < 0.01 for Hi, Fig. 3(2)).

Aβ content in the rat Hippo and CTX were measured after aluminium-maltolate treatment using ELISA. Each experiment consisted of three parallel samples. One-way ANOVA, n = 10 for each group; error bars indicate the SEM. *P < 0.05 indicates a significant difference compared to control
APP Protein Expression
The expression of APP increased in both Co and Hi compared to control expression (F(3,39) = 81.325, P < 0.05 for Co, F(3,39) = 95.600, P < 0.05 for Hi, Fig. 4(1),(2),(3)). Statistical analyses showed that the expression of APP did not correlate with the content of Aβ1–42 (R = 0.294, P > 0.05 for Co; R = 0.353, P > 0.05 for Hi).

Proteins of Aβ metabolism-related molecules from the rat Hippo and CTX were measured after aluminium-maltolate treatment using western blotting analyses. β-actin was used as the loading control. All experiments were performed at least in triplicate with independent experiments. One-way ANOVA, n = 10 for each group; error bars indicate the SEM. *P < 0.05 indicates a significant difference compared to control
ADAM9, ADAM10 and ADAM17 Protein Expression
As shown in Fig. 5, the expression of ADAM9 decreased in the Co of Al(mal)3 -treated rats but was not significant compared to the control group (F(3,39) = 0.609, P > 0.05, Fig. 5(1),(3)); it did not correlate with Aβ1–42 (R = −0.134, P > 0.05) and decreased significantly in the Hi (F(3,39) = 11.368, P < 0.01, Fig. 5(2),(3)). Statistical analysis showed that the expression of ADAM9 correlated with the content of Aβ1–42 (R = −0.761, P < 0.01,). Furthermore, ADAM10 was significantly reduced in both tissues (F(3,39) = 67.177, P < 0.01 for Co, F(3,39) = 81.093, P < 0.01 for Hi, Fig. 5(1),(2),(4)) Statistical analysis also revealed that the expression of ADAM10 correlated with the content of Aβ1–42 (R = −0.708, P < 0.01 for Co; R = −0.897, P < 0.01 for Hi). ADAM17 was significantly reduced in both tissues ((F(3,39) = 142.223, P < 0.01 for Co,F(2,39) = 5.119, P < 0.01 for Hi, Fig. 5(1),(2),(5)). Statistical analysis showed that the expression of ADAM17 correlated with the level of Aβ1–42 (R = −0.634, P < 0.01 for Co; R = −0.718, P < 0.01 for Hi).
BACE1 Protein Expression
The expression of BACE1 increased significantly in both Co and Hi compared to control expression ((F(3,39) = 62.200, P < 0.01 for Co, F(3,39) = 114.318, P < 0.05 for Hi, Fig. 6(1),(2),(3)). Statistical analysis showed that the expression of BACE1 correlated with the content of Aβ1–42 (R = 0.608, P < 0.01 for Co; R = 0.804, P < 0.01 for Hi).
PS1 and NCT Protein Levels
The protein expression of PS1 was significantly increased in both tissues (F(3,39) = 79.720, P < 0.01 for Co, F(3,39) = 30.614, P < 0.01 for Hi, see Fig. 7(1),(2),(3)), and was significantly correlated with Aβ1–42 (R = 0.744,P < 0.01 for Co; R = 0.757, P < 0.01 for Hi). However, the expression of NCT was significantly increased in both tissues (F(3,39) = 26.147, P < 0.01 for Co, F(3,39) = 241.001, P < 0.05 for Hi, see Fig. 7(1),(2),(4)). Statistical analysis showed that the expression of NCT correlated significantly with the content of Aβ1–42 (R = 0.622, P < 0.01 for Hi; R = 0.859, P < 0.01 for Co).
Discussion
Epidemiological studies have revealed an association between chronic exposure to aluminium in drinking water and AD risk [27, 28]. Aluminium can reach and accumulate in nearly every organ in the human body, and the central nervous system is a specific target of deleterious effects [29]. Various mechanisms have been proposed to underlie aluminium-induced neurotoxicity, such as oxidative stress [30], alterations of the phosphorylation levels of neurofilaments [31], among other mechanisms. However, the contribution of aluminium to AD still requires further investigation. During the development of AD, extracellular accumulation of Aβ in the brain is regarded as an early and main event that occurs when massive cell death is not observed [31]. Importantly, aluminium has been found to enhance the production and aggregation of Aβ both in in vitro and in vivo studies [11, 32]; however, its underlying mechanism remains unknown.
The present study revealed that aluminium-maltolate could enter the brain in rats and damage learning and memory functions as well as enhance the Aβ1–42 content in the cortex and hippocampus. Moreover, aluminium-maltolate induced Aβ1–42 increase in the hippocampus is more profound compared to the cortex (Fig. 4). Furthermore, evidence showed that aluminium may be involved in the deposition of Aβ peptides, which causes severe damage to brain tissues [33, 34]. Aβ exists in a dynamic equilibrium of soluble monomeric, oligomeric, protofibrillar, and fibrillar forms, and the levels of which reflect a balance between their production and removal from the brain. Aβ1–42 is relatively insoluble in the interstitial fluid and is prone to parenchymal deposition, whereas Aβ1–40 is more soluble and less prone to parenchymal deposition. In our study, aluminium increased Aβ1–42 compared to Aβ1–40 in rat brains (Fig. 4). On the basis, the possible reason which aluminium increased Aβ1–42 in rat brains, rather than Aβ1–40 was that Aβ1–40 is more soluble and less prone to parenchymal increasing.
An increasing number of reports has shown that the amount of Aβ in the brain is correlated to Aβ generation [35]. Thus, it was suggested that an alteration in the expression of metabolism-associated molecules of Aβ might be developed into early markers of aluminium neurotoxicity. In our study, aluminium increased the expression of APP, but these changes did not correlate with the content of Aβ1–42. In addition, the present study showed that the expression of enzymes that participate in Aβ metabolism obviously changed: (1) ADAM9, ADAM10 and ADAM17 were decreased; (2) BACE1 was increased; and (3) PS1 and NCT were increased. In addition, they correlated with the content of Aβ1–42 in rat brains. This finding suggested that, in addition to Aβ, Aβ-pathway-related molecules, such as ADAM, BACE1, PS1 and NCT, could also be developed into critical biological markers for aluminium neurotoxicity.
Evidence showed that the protease ADAM10 but not ADAM9 or ADAM 17 was essential for α-secretase cleavage. Moreover, ADAM9 and ADAM17 were not able to compensate for ADAM10 in constitutive α-secretase cleavage [17]. Furthermore, ADAM10 showed coordinated expression with APP in the human brain [36]. In a subset of ADAM10-deficient fibroblasts, APP α-secretase cleavage was altered to a variable degree [17]. In our study, the decrease in ADAM9, ADAM10 and ADAM17 were obvious in the hippocampus compared to the cortex, and the decrease in ADAM10 was most profound in both the cortex and hippocampus. This appearance was similar to Aβ1–42. Taken together, these results suggested that the damage induced by aluminium on the hippocampus was more serious compared to the cortex. Furthermore, the decrease in ADAM9, 10, and 17 played a role in the increase in Aβ1–42 induced by aluminium. In the hippocampus and cortex of AD patients, ADAM17 was localised to senile plaques [37]. Evidence has shown that although ADAM17 is capable of cleaving APP, it is likely that additional members of the ADAM family mediate the endogenous constitutive and regulated α-secretase cleavage of APP in HEK293 cells [16]. In the present study, the decrease in ADAM17 may be one of the enzymes that regulate α-secretase cleavage of APP. Although the involvement of ADAM9 is less convincing, emerging evidence has demonstrated that there is most likely not a single a-secretase activity but rather several enzymes activities that act together to cleave APP at the α-secretase site. The decrease in ADAM9 was not profound compared to ADAM10 and ADAM17. We proposed that the role of ADAM9 is still unclear or that there may be a functional overlap or redundancy with other members of the ADAMs family in aluminium-treated rats.
BACE1 cleavage is a pre-requisite for Aβ formation and is the initiating step in Aβ generation. BACE1 levels are elevated in both AD experimental models and in the AD patient brain [38–40]. Moreover, BACE1 activity is correlated with brain Aβ production in the frontal cortex [41, 42]. Intraventricular Al-maltol injection in the aged New Zealand white rabbit induced behavioural, biochemical and neuropathological changes, which were similar to what occurs in clinical AD, including Aβ deposition close to the cerebral cortex and hippocampus and increased β-secretase activity in the hippocampus [43]. In addition, there was a marked increase in β-secretase and amyloid-β induced by AlCl3 [13]. Differentiated neuronal SH-SY5Y cells exposed to AlCl3 and Aβ demonstrated slightly reduced BACE1 levels after 1 h and significantly increased BACE1 levels after 3 h of exposure. Moreover, BACE2 levels were increased at both times. The mRNA levels of both genes were downregulated with longer time periods of exposure (6, 12, and 24 h). Taken together, these findings indicated that aluminium toxicity is correlated with changes in both BACE1 and BACE2 expression levels [3]. In our study, BACE1 was detected and changes in BACE1 showed a positive correlation with changes in brain Aβ42. Consistent with other studies, our data suggested that the increase in BACE1 expression might be a key factor of aluminium-induced Aβ1–42 content in the rat brain.
γ-secretase, an intramembrane aspartyl protease, is critically involved in AD via proteolysis of the APP, which generates the pathogenic and amyloid plaque-forming Aβ1–42 peptide [44]. However, it is not completely clear how the different subunits of γ-secretase contribute to substrate docking and cleaving. It appears that the major obstacle lies in the lack of structural information related to the entire complex, which consists of four subunits [23]. PS was thought to be a central, catalytic moiety of the γ-secretase complex. Some studies found that PS1 was able to cleave substrates in the absence of NCT, APH1, and PEN-2 in an activity test performed in liposomes. However, NCT has been proposed to participate in the recruitment of Notch1 and APP substrates to γ-secretase [45, 46]. NCT was shown to co-localise with PS1 in murine lysosomes. Furthermore, γ-secretase activity against APP could also be detected in lysosomal membranes [47]. Moreover, γ-secretase was thought to be important for PS stabilisation and trafficking [48–50]. In addition, further evidence revealed that there was a key role for NCT in modulating Aβ production and amyloid plaque formation in vivo [51]. In the present study, we investigated the levels of PS1 and NCT, which are different γ-secretase components, in the cortex and hippocampus of rat brains. Both PS1 and NCT increased and correlated well with Aβ1–42 levels. Our study indicated that PS1 and NCT played a role during the deposition of Aβ1–42 induced by aluminium-maltolate.
In conclusion, our findings suggested that aluminium-maltolate induced the depression of learning and memory in rats in vivo and generated a high content of Aβ1–42 in rat brains. Furthermore, β-secretase increased significantly, and different subtypes or components of α-secretase (particularly ADAM10 and ADAM17) significantly decreased. Thus, APP cleavage by BACE1 was increased, and APP cleavage by α-secretase was decreased. Consistent with these findings, C99 cleavage by increased subtype components of γ-secretase was increased in the aluminium-treated groups, and Aβ1–42 was produced and increased in the cortex and hippocampus of aluminium-treated animals. All of these factors may be developed into useful markers to detect the early neurotoxic effect of aluminium and can also be regarded as targets for a therapeutic strategy against the neurotoxicity of aluminium. Furthermore, some of these factors may be developed into valid targets to treat AD due to aluminium neurotoxicity.
References
Wu G, Sankaranarayanan S, Hsieh SH, Simon AJ, Savage MJ (2011) Decrease in brain soluble amyloid precursor protein beta (sAPPbeta) in Alzheimer’s disease cortex. J Neurosci Res 89:822–832
Jiang T, Zhi XL, Zhang YH, Pan LF, Zhou P (2012) Inhibitory effect of curcumin on the Al(III)-induced Abeta(4)(2) aggregation and neurotoxicity in vitro. Biochim Biophys Acta 1822:1207–1215
Castorina A, Tiralongo A, Giunta S, Carnazza ML, Scapagnini G, D’Agata V (2010) Early effects of aluminum chloride on beta-secretase mRNA expression in a neuronal model of beta-amyloid toxicity. Cell Biol Toxicol 26:367–377
Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH (2005) Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci 8:79–84
Banks WA, Niehoff ML, Drago D, Zatta P (2006) Aluminum complexing enhances amyloid beta protein penetration of blood-brain barrier. Brain Res 1116:215–221
Walton JR (2013) Aluminum involvement in the progression of Alzheimer’s disease. J Alzheimers Dis (JAD) 35:7–43
Brenner S (2013) Aluminum may mediate Alzheimer’s disease through liver toxicity, with aberrant hepatic synthesis of ceruloplasmin and ATPase7B, the resultant excess free copper causing brain oxidation, beta-amyloid aggregation and Alzheimer disease. Med Hypotheses 80:326–327
Walton JR (2012) Cognitive deterioration and associated pathology induced by chronic low-level aluminum ingestion in a translational rat model provides an explanation of Alzheimer’s disease, tests for susceptibility and avenues for treatment. Int J Alzheimers Dis 2012:914947
Liang RF, Li WQ, Wang XH, Zhang HF, Wang H, Wang JX, Zhang Y, Wan MT, Pan BL, Niu Q (2012) Aluminium-maltolate-induced impairment of learning, memory and hippocampal long-term potentiation in rats. Ind Health 50:428–436
Ferreira PC, Piai Kde A, Takayanagui AM, Segura-Munoz SI (2008) Aluminum as a risk factor for Alzheimer’s disease. Rev Lat Am Enfermagem 16:151–157
Ricchelli F, Drago D, Filippi B, Tognon G, Zatta P (2005) Aluminum-triggered structural modifications and aggregation of beta-amyloids. Cell Mol Life Sci (CMLS) 62:1724–1733
Drago D, Folin M, Baiguera S, Tognon G, Ricchelli F, Zatta P (2007) Comparative effects of Abeta(1-42)-Al complex from rat and human amyloid on rat endothelial cell cultures. J Alzheimers Dis (JAD) 11:33–44
Zaky A, Mohammad B, Moftah M, Kandeel KM, Bassiouny AR (2013) Apurinic/apyrimidinic endonuclease 1 is a key modulator of aluminum-induced neuroinflammation. BMC Neurosci 14:26
Fiorelli T, Kirouac L, Padmanabhan J (2013) Altered processing of amyloid precursor protein in cells undergoing apoptosis. PLoS One 8:e57979
Deuss M, Reiss K, Hartmann D (2008) Part-time alpha-secretases: the functional biology of ADAM 9, 10 and 17. Curr Alzheimer Res 5:187–201
Allinson TM, Parkin ET, Turner AJ, Hooper NM (2003) ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res 74:342–352
Lichtenthaler SF (2011) Alpha-secretase in Alzheimer’s disease: molecular identity, regulation and therapeutic potential. J Neurochem 116:10–21
Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ (1990) Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science (New York, NY) 248:1122–1124
Roberts SB, Ripellino JA, Ingalls KM, Robakis NK, Felsenstein KM (1994) Non-amyloidogenic cleavage of the beta-amyloid precursor protein by an integral membrane metalloendopeptidase. J Biol Chem 269:3111–3116
Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F (1999) Constitutive and regulated alpha-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 96:3922–3927
Delarasse C, Auger R, Gonnord P, Fontaine B, Kanellopoulos JM (2011) The purinergic receptor P2X7 triggers alpha-secretase-dependent processing of the amyloid precursor protein. J Biol Chem 286:2596–2606
Frykman S, Hur JY, Franberg J, Aoki M, Winblad B, Nahalkova J, Behbahani H, Tjernberg LO (2010) Synaptic and endosomal localization of active gamma-secretase in rat brain. PLoS One 5:e8948
Smolarkiewicz M, Skrzypczak T, Wojtaszek P (2013) The very many faces of presenilins and the γ-secretase complex. Protoplasma 250:997–1011
Dries DR, Yu G (2008) Assembly, maturation, and trafficking of the gamma-secretase complex in Alzheimer’s disease. Curr Alzheimer Res 5:132–146
Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM (2005) Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol Aging 26:645–654
Langui D, Probst A, Anderton B, Brion JP, Ulrich J (1990) Aluminium-induced tangles in cultured rat neurones. Enhanced effect of aluminium by addition of maltol. Acta Neuropathol 80:649–655
Flaten TP (2001) Aluminium as a risk factor in Alzheimer’s disease, with emphasis on drinking water. Brain Res Bull 55:187–196
Martyn CN (1992) The epidemiology of Alzheimer’s disease in relation to aluminium. Ciba Found Symp Ciba 169:69–79 discussion 79–86
Chen TJ, Hung HS, Wang DC, Chen SS (2010) The protective effect of Rho-associated kinase inhibitor on aluminum-induced neurotoxicity in rat cortical neurons. Toxicol Sci 116:264–272
Bhalla P, Dhawan DK (2009) Protective role of lithium in ameliorating the aluminium-induced oxidative stress and histological changes in rat brain. Cell Mol Neurobiol 29:513–521
Kaur A, Joshi K, Minz RW, Gill KD (2006) Neurofilament phosphorylation and disruption: a possible mechanism of chronic aluminium toxicity in Wistar rats. Toxicology 219:1–10
Campbell A, Kumar A, La Rosa FG, Prasad KN, Bondy SC (2000) Aluminum increases levels of beta-amyloid and ubiquitin in neuroblastoma but not in glioma cells. Proc Soc Exp Biol Med (New York, NY) 223:397–402
Yumoto S, Kakimi S, Ohsaki A, Ishikawa A (2009) Demonstration of aluminum in amyloid fibers in the cores of senile plaques in the brains of patients with Alzheimer’s disease. J Inorg Biochem 103:1579–1584
Pratico D, Uryu K, Sung S, Tang S, Trojanowski JQ, Lee VM (2002) Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J 16:1138–1140
Sun ZZ, Chen ZB, Jiang H, Li LL, Li EG, Xu Y (2009) Alteration of Abeta metabolism-related molecules in predementia induced by AlCl3 and D-galactose. Age 31:277–284
Marcinkiewicz M, Seidah NG (2000) Coordinated expression of beta-amyloid precursor protein and the putative beta-secretase BACE and alpha-secretase ADAM10 in mouse and human brain. J Neurochem 75:2133–2143
Skovronsky DM, Fath S, Lee VM, Milla ME (2001) Neuronal localization of the TNFalpha converting enzyme (TACE) in brain tissue and its correlation to amyloid plaques. J Neurobiol 49:40–46
Harada H, Tamaoka A, Ishii K, Shoji S, Kametaka S, Kametani F, Saito Y, Murayama S (2006) Beta-site APP cleaving enzyme 1 (BACE1) is increased in remaining neurons in Alzheimer’s disease brains. Neurosci Res 54:24–29
Zhao J, Fu Y, Yasvoina M, Shao P, Hitt B, O’Connor T, Logan S, Maus E, Citron M, Berry R, Binder L, Vassar R (2007) Beta-site amyloid precursor protein cleaving enzyme 1 levels become elevated in neurons around amyloid plaques: implications for Alzheimer’s disease pathogenesis. J Neurosci 27:3639–3649
Tamagno E, Parola M, Bardini P, Piccini A, Borghi R, Guglielmotto M, Santoro G, Davit A, Danni O, Smith MA, Perry G, Tabaton M (2005) Beta-site APP cleaving enzyme up-regulation induced by 4-hydroxynonenal is mediated by stress-activated protein kinases pathways. J Neurochem 92:628–636
Li Q, Sudhof TC (2004) Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem 279:10542–10550
Gustaw KA, Garrett MR, Lee HG, Castellani RJ, Zagorski MG, Prakasam A, Siedlak SL, Zhu X, Perry G, Petersen RB, Friedland RP, Smith MA (2008) Antigen-antibody dissociation in Alzheimer disease: a novel approach to diagnosis. J Neurochem 106:1350–1356
Panahi N, Mahmoudian M, Mortazavi P, Hashjin GS (2013) Effects of berberine on beta-secretase activity in a rabbit model of Alzheimer’s disease. Arch Med Sci (AMS) 9:146–150
Hass MR, Sato C, Kopan R, Zhao G (2009) Presenilin: RIP and beyond. Semin Cell Dev Biol 20:201–210
Chen F, Yu G, Arawaka S, Nishimura M, Kawarai T, Yu H, Tandon A, Supala A, Song YQ, Rogaeva E, Milman P, Sato C, Yu C, Janus C, Lee J, Song L, Zhang L, Fraser PE, St George-Hyslop PH (2001) Nicastrin binds to membrane-tethered Notch. Nat Cell Biol 3:751–754
Dries DR, Shah S, Han YH, Yu C, Yu S, Shearman MS, Yu G (2009) Glu-333 of nicastrin directly participates in gamma-secretase activity. J Biol Chem 284:29714–29724
Pasternak SH, Bagshaw RD, Guiral M, Zhang S, Ackerley CA, Pak BJ, Callahan JW, Mahuran DJ (2003) Presenilin-1, nicastrin, amyloid precursor protein, and gamma-secretase activity are co-localized in the lysosomal membrane. J Biol Chem 278:26687–26694
LaVoie MJ, Fraering PC, Ostaszewski BL, Ye W, Kimberly WT, Wolfe MS, Selkoe DJ (2003) Assembly of the gamma-secretase complex involves early formation of an intermediate subcomplex of Aph-1 and nicastrin. J Biol Chem 278:37213–37222
Zhang YW, Luo WJ, Wang H, Lin P, Vetrivel KS, Liao F, Li F, Wong PC, Farquhar MG, Thinakaran G, Xu H (2005) Nicastrin is critical for stability and trafficking but not association of other presenilin/gamma-secretase components. J Biol Chem 280:17020–17026
Mao G, Cui MZ, Li T, Jin Y, Xu X (2012) Pen-2 is dispensable for endoproteolysis of presenilin 1, and nicastrin-Aph subcomplex is important for both gamma-secretase assembly and substrate recruitment. J Neurochem 123:837–844
Sesele K, Thanopoulou K, Paouri E, Tsefou E, Klinakis A, Georgopoulos S (2013) Conditional inactivation of nicastrin restricts amyloid deposition in an Alzheimer’s disease mouse model. Aging cell 12:1032–1040
Acknowledgments
This work was supported by the National Nature Science Foundation of China (81302410).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Wang, L., Hu, J., Zhao, Y. et al. Effects of Aluminium on β-Amyloid (1–42) and Secretases (APP-Cleaving Enzymes) in Rat Brain. Neurochem Res 39, 1338–1345 (2014). https://doi.org/10.1007/s11064-014-1317-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-014-1317-z