
Abstract
Microbes are integral part of our environment. They have enormous industrial and medicinal applications. Even they play a crucial role during digestion where they are present in the form of gut flora. Genomic sequences are a prerequisite for molecular taxonomic characterization of novel microbes, and traditional microbiology is dependent on clone cultures for DNA extraction of a specific microbe population. As a result vast varieties of species are missed since most of the microbes cannot be cultured in laboratory conditions. Metagenomics skips the requirement of culturing the microbes in lab as it studies genetic material which is directly taken from environmental samples.
Microorganisms are of great significance due to their applications in health, agriculture, and industry. Direct DNA sequencing of environmental samples has given opportunity to gather information about the microorganisms that were unexplored so far. Screening of useful bacteria that survive in different environmental conditions like heavily polluted soil, disease-affected tissues or cells, oil-contaminated water bodies, heavy metal-contaminated fields, etc. can be done easily by combining environmental and metagenomics approaches. The data obtained from environmental sample sequencing may be of great use in discovery of new drugs and antibiotics, new bacterial species, plant growth promoters, bioremediation as well as in many other industrial applications. The number of metagenomic studies has increased significantly in recent years, and it is believed that this trend will continue its pace due to huge applicability. This chapter also provides significant elaborations about methodology and tools, experimental design strategies, online resources, and databases applicable in metagenomic data analysis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


4.1 Introduction
During the evolution of life on earth, microbes have played an important role and have done much more for human beings for the sustenance and survival. As these microbes have adapted to the earth’s environment, they are found everywhere, viz., on earth, inside earth, in water, and in air. To understand their impact on global ecology, it is most important to understand their diversity and life. According to estimates, about 99% of the microbes are not culturable in pure culture. It acts as the major debacle in understanding the microbial genetics and community ecology. These microbial communities are responsible for biological activities carried out in all environments including the ocean (DeLong 2005), soils, and human-associated habitats (Ravel et al. 2011). Although metagenomics is quite a young and emerging field, it has helped in understanding the microbial diversity which was not possible by using traditional and classical methods of microbiology. Metagenomics has emerged as the most powerful and reliable technique for genome analysis of the entire community of microbes overruling the need to isolate and culture individual microbial species (Arrial et al. 2009). It has wide potential in discovering novel enzymes for industrial applications, antibiotics against many harmful microbes for curing diseases, and organisms for experimental purposes.
Major metagenomics themes are (a) marker metagenomics that surveys microbial community structure by targeting the highly conserved 16S rRNA gene; (b) functional metagenomics that takes the total environmental DNA, from which it infers the metabolic potential of the microbial community; and (c) identification of novel enzymes. Metagenomics uses two approaches: targeted metagenomics and shotgun sequencing. Targeted metagenomics is most commonly used to identify the phylogenetic diversity and the relative abundance in a given sample. This technique is mainly used to investigate the diversity of small subunit of rRNA (16S/18S rRNA) within a sample. It is often used to understand the impact of environmental contaminant that alters the microbial community structure. For conducting the study related to targeted metagenomics, the environmental DNA is extracted from the source, the particular gene of interest is amplified using PCR primers, and further these amplified results are sequenced using next-generation sequencing . Targeted metagenomics is useful in identifying the diversity of single gene of interest, but it is limited by the type of PCR primers used for the analysis (Shakya et al. 2013; Parada et al. 2016; Klindworth et al. 2013; Prosser 2015).
Similarly in shotgun metagenomics sequencing, the genomic complement of an environmental community is studied by using genome sequencing. Basically in this approach, the DNA is extracted from the environmental sample and fragmented to prepare sequencing libraries and further sequenced for the determination of total genomic content of that sample. Shotgun sequencing is often restricted by the depth of the sequencing.
Functional metagenomics has played a major role in understanding the role of microbial community in microbial ecology and global geochemical cycles. Furthermore it is a unique way to identify the novel enzymes from the environmental sample (Uchiyama and Miyazaki 2009). Therefore the functional metagenomics played major role in protein and nucleic acid database through addition of novel functional annotation. However major drawback of this technique includes a low hit rate of positive clones, low throughput, and time-consuming screening (Hosokawa et al. 2015).
Currently metagenomics is a powerful technique to have industrial applications in identification of novel biocatalysts , discovering novel antibiotics, and bioremediation . The application of metagenomics is increasing rapidly, and these are being listed below.
4.2 Application of Metagenomics and the Impact on Environmental Biotechnology
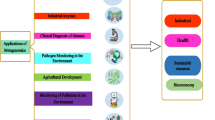
The new field of metagenomics is expected to bring fruitful result for the researchers working in the area of microbiology in mainly two ways: in first application it will provide knowledge about those bacteria which are still not cultivated so far (about 99% are uncultured in the pure culture). Secondly it will provide access to whole microbe community residing in variety of natural environment . Furthermore as we know that microbes are quite essential component of our life for the sustenance and these microbes play very crucial role in industries which are backbone of our present economy planet. Direct access to the genetic makeup of microbes of the whole ecosystem community will provide new basis for fundamental research and new tool for application in environment, agriculture , human health , bio-industry, etc. (Fig. 4.1).

Various aspects of applications of metagenomics (also known as environmental and community genomics) in different fields of biological science
4.2.1 Industrial Enzymes
There is an increasing demand of novel enzymes for industrial applications, and metagenomics is playing an important role in providing these biomolecules (Lorenz et al. 2002; Schloss and Handelsman 2003) specially enzymes that are used in wide range of applications (Kirk et al. 2002). These are required in minute amount to synthesize huge amount of key molecules that are used in producing active pharmaceuticals as these are the major building block of those products (Patel et al. 1994). There are many industrial enzymes which have a very wide application in industries and act as their backbone like cellulases, xylanases, lipases, amylases, etc.
Cellulases have attracted industrialists due to their wide application and crucial enzyme activities that are inherited in various forms within them such as endoglucanases (EC 3.2.1.4), exoglycosidase, and β-glucosidases (EC 3.2.1.21). Today cellulase is the third most widely used enzyme in industries (Wilson 2009). Cellulases are mainly used in animal feed and improving the digestibility. Furthermore de-inking of paper is another evolving application of this enzyme (Soni et al. 2008). Metagenomics has played a vital role in extracting cellulase from natural environments like compost soil, soil from cold region, rumen samples and many more. Even few workers have reported that cellulases are isolated from sugarcane soil and buffalo rumen (Alvarez et al. 2013; Duan et al. 2009).
Xylaneses are key enzymes that are widely used in degradation of xylan and are helpful in breaking of hemicellulose, regarded as essential component of cell wall. Xylaneses have wide spectrum of application in industries such as clarification of juices (Sharma 2012), detergents (Kumar et al. 2004), production of pharmacologically active polysaccharides for the antimicrobial agent use (Christakopoulos et al. 2001), antioxidants (Katapodis et al. 2003), and production of surfactants (Kashyap et al. 2014). Xylanases are produced by a wide range of microbes from different sources that have many application in industries. It is reported that xylaneses are present in insect gut that could be used for conversion of biomass into fermentable sugar which could be used for production of biofuels (Brennan et al. 2004; Lee et al. 2006; Jeong et al. 2012). This enzyme was reported in the saccharification of reed and could be used efficiently in the conversion of biomass to fermentable sugar for biofuel production (Wang et al. 2012).
Lipases are mainly triacylglycerol acylhydrolases that are actively involved in the conversion of triglycerides into diglycerides, monoglycerides, glycerols and fatty acids. Being resistant to varying environmental conditions like temperature, pH, organic solvent etc., they have great prospects in industries. It is widely found in many plant and animal sources and also reported in some microbes such as bacteria, fungus, and yeast, and these have varying application in oil industries, pharmaceutical industries, dairy industries etc. (Cardenas et al. 2001).
Amylases are mostly regarded as starch-degrading enzymes. They are quite abundant in plants, animals, and microbes. These have wide application in industries like food, fermentation, and pharma for hydrolysis of starch. AmyI3C6 commonly known as cold-adapted alpha amylase from the metagenomic libraries of cold and alkaline environment can be useful as it showed potent activity against two commercially known detergents. A novel amylase was isolated from a soil metagenome that showed 90% activity at low temperature which proved its potential for industrial exploitation (Sharma et al. 2010).
4.2.2 Bioactive Compounds and Antibiotics
Nowadays a major worldwide health-related problem involves treating infections which are resistant to antibiotics . These resistant microbes are able to cause severe mortality and impose a large budget on healthcare (Carlet et al. 2011). Earlier these antibiotics were used for treating human infection, but they became popular in agriculture and food industry as well as many other related sectors, thus finally imposing high impact on human health (Radhouani et al. 2014). In the current scenario, antibiotics are considered as the pillars of the modern medicine (Ball et al. 2013). This bacterial resistance against widely used common antibiotics has forced researchers to discover novel antibiotics against these microbial infectious diseases.
Today metagenomics is playing a very vital role in discovery of bioactive compounds and antibiotics . It is considered as an alternative way of isolating antibiotics from environmental samples as well as to trace the mechanism of bacterial gene resistance. The combined approach of metagenomics and next-generation sequencing has paved way for success in study of antimicrobial resistance and microbial genomes (Forsberg et al. 2012; McGarvey et al. 2012). Generally, the bacterial gene resistance is mainly developed due to the horizontal gene transfer or spontaneous mutation in target gene (Hassan et al. 2012). The transfer of antibiotic resistance gene involves the mobility of genetic material to other bacterial species or the same group (Thomas and Nielsen 2005).
Metagenomics is putting effort to sort out the drug resistance genes in microorganisms against various class of antibiotics. Its another application is identification of bioactive molecules having antimicrobial properties (MacNeil et al. 2001; Gillespie et al. 2002; Lim et al. 2005). Today, antibiotic resistance of microbes is an alarming worldwide problem and emerging as a major threat (Čivljak et al. 2014) as these microbes are developing resistance against many traditional antibiotics, and on the other hand, many researchers are discovering many novel antimicrobial compounds from different environmental sources including microorganisms, plants, and animals likewise (Roy et al. 2013; de Souza Candido et al. 2014). It is reported that uncultivated soil microbes have potential of novel biomolecules which could be very well exploited in any biotechnological application (Wilson and Piel 2013). In this way we can conclude that these soil microbes can be an alternative source of bioactive molecules. Various active biomolecules which are identified by metagenomic approach include teicoplanin, friulimicin, azinomycin, rapamycin, borregomycin, etc.
4.2.3 Bioremediation
The process to degrade and detoxify environmental contaminants through microbe-mediated process is known as bioremediation (Chakraborty et al. 2012). It involves removal of biological and anthropogenic contaminants through natural process, so it is considered as the most effective approach (Lovley 2003). Bioremediation approaches can be classified into three main classes, (a) natural attenuation, (b) biostimulation, and (c) bioaugmentation.
In natural attenuation native organisms are used for detoxifying contaminants through using natural process. This process is quite effective in terms of cost, and no need of altering additives is required for this. In biostimulation the rate of bioremediation is increased through using native organisms but needs to remove some environmental constraints. This approach required addition of some nutrients to achieve fast rate of bioremediation. Sometimes this approach failes to achieve their faster rate of bioremediation by using native organism due to their inability to degrade contaminant of concern. To overcome this problem, some nonnative organisms or enzymes are added to enhance the rate of bioremediation which is known as bioaugmentation. This approach is considered as most invasive as nonnative organism. In some cases bioaugmentation is considered as most convenient mean of remediation (Payne et al. 2011; Salanitro et al. 2000). The major drawback of bioaugmentation is that nonnative organism can’t survive under the condition found in the contaminated ecosystem.
In the present scenario, metagenomic approach is widely used for environmental monitoring and bioremediation. Metagenomics approaches that are often used for monitoring the environmental microbes are targeted metagenomics or shotgun metagenomics. Targeted metagenomics is widely exploited to study the phylogenetic diversity and relative abundance of a particular gene in the environment . This approach is used to study the diversity of the rRNA sequence in the sample (16S/18S rRNA). It is often used to study the impact of environmental contaminant in microbial community structure. The major advantage of targeted metagenomics is that it provides the information about microbial community present in the set of sample and change in microbial diversity before and after perturbation.
Likewise in the shotgun metagenomics, the total genomic complement of the environmental community is probed by using genome sequencing. In this approach, environmental DNA is extracted and fragmented to prepare genomic libraries and further sequenced to determine the total genomic content. Using this approach potential of a microbial community can be identified. Recently metatranscriptomics and metaproteomics are being widely used to apply over environmental system. In metatranscriptomics ribonucleic acid (RNA) is extracted from the sample and converted to complementary deoxyribonucleic acid (cDNA) in a similar function as in metagenomics. The metaproteomics approach does not involve the nucleic acid sequencing but high-resolution mass spectrometry combined with enzymatic digest of the proteins and liquid chromatography (Hettich et al. 2013). Metaproteomics provides an information about the kind of protein present inside the environmental sample including posttranslational modification in proteins that may impact their activity.
Many industries are responsible for increased level of hydrocarbons in the environment due to the incomplete combustion of fossil fuel. Generation of these anthropogenic compounds into the environment results into the accumulation of large amount of aromatic hydrocarbons which leads to contamination of ecosystem (Jacques et al. 2007). Microorganisms are involved in many biogeochemical cycles and have potential of degradation of hydrocarbons (Alexander 1994). Metagenomics can be helpful in degradation of aromatic compounds by screening and identifying suitable organisms in a metagenomic library obtained from oil source (Sierra-García et al. 2014). Many genes and their pathways were identified for the degradation of phenol and aromatic compound by using metagenomic approach (Silva et al. 2013). Some bacterial population having capacity for the degradation of polycyclic aromatic compound (PAH) were isolated from cold environment by identifying their functional target (Marcos et al. 2006).
As we know that oil spillage has badly affected many parts of the natural marine ecosystem (National Academy of Science 2005) due to increased anthropogenic activity (Hazen et al. 2016; Atlas and Hazen 2011). In this context Deepwater Horizon oil spill is considered as the worst marine oil spill in the USA and considered as major threat for marine ecosystem biology (King et al. 2015). The first application of metagenomics was to understand the mechanism behind the oil biodegradation in marine environment . The targeted metagenomics was applied to find out the microbial community in the surface water and reported as Cycloclasticus, Alteromonas, Halomonas, and Pseudoalteromonas (Redmond and Valentine 2012; Gutierrez et al. 2013). However they also reported that deep water is primarily composed of psychrophilic oil-degrading microbes related to Oceanospirillales, Colwellia, and Cycloclasticus (Hazen et al. 2010). Shotgun metagenomics approach was used for sample collected during Deepwater Horizon oil spill which revealed diverse group of genes responsible for chemotaxis and hydrocarbon degradation (Mason et al. 2012). The results of the single amplified genome showed genes involved in degradation of n-alkanes and cycloalkanes. Thus metagenomics sequencing approach helps in understanding the mechanism behind the oil degradation by microbial community in marine environment .
4.2.4 Applications in Agriculture
The productivity of agriculture is severely affected by presence of organic and inorganic anthropogenic pollutants that play a very significant role in abiotic stress. These kinds of abiotic stresses are responsible for reduction in crop yield. To improve the quality of such soil contaminated by anthropogenic pollutants, bioremediation is required. Microorganisms of soil metagenome are quite capable of producing biosurfactants which can remove many anthropogenic pollutants which may be either hydrocarbons or heavy metals (Sun et al. 2006). Biosurfactants are capable of removing hydrocarbons and heavy metals through the combination of soil washing and cleanup technology (Pacwa-Płociniczak et al. 2010; Liu et al. 2010a, b; Partovinia et al. 2010; Gottfried et al. 2010; Coppotelli et al. 2010; Kang et al. 2010). Some studies have revealed that biosurfactants isolated Lactobacillus pentosus had reduced the octane hydrocarbons from soil (Moldes et al. 2011). Some biosurfactant-producing species like Burkholderia isolated from oil-contaminated metagenome may act as a potential candidate for the reduction (bioremediation) of pesticides (Wattanaphon et al. 2008). Some studies have also revealed that biosurfactants are more efficient in removal of organic insoluble pollutant from soil than surfactants (Cameotra and Bollag 2003; Straube et al. 2003). The soil samples from such fields shall be subjected to metagenomics analysis, library preparation and subsequent analysis for identifying biosurfactant-producing microbes.
Besides application of biosurfactants for removal of many anthropogenic molecules which are either hydrocarbon or heavy metals, these may also be applicable in removal of plant pathogens due to their antimicrobial nature, thus promoting sustainable agriculture . Biosurfactants which are produced by rhizobacteria have antagonistic properties (Nihorimbere et al. 2011). For sustainable agriculture, biosurfactants and chemical surfactants are useful in controlling parasitism, antibiosis, competition, induced systemic resistance, and hypovirulence (Singh et al. 2007). In fact the application of surfactants in agriculture is mainly for enhancing the antagonistic activity of microbes and microbial products (Kim et al. 2004). Some studies have also revealed that these surfactants when applied in combination of certain fungus like Myrothecium verrucaria are found to be useful in the control of weed (Boyette et al. 2002).
Additionally, biosurfactants are also useful for inhibition of many phytopathogens. Biosurfactant isolated from Pseudomonas and Bacillus is reportedly used for the control of soft rot caused by Pectobacterium and Dickeya spp. and thus has been helpful in protection of economically valuable crops (Krzyzanowska et al. 2012). Many studies have reported that antipathogenic agents like rhamnolipids have the ability to kill zoospore of plant pathogens that are being resistant against many commercial pesticides (Sha et al. 2011, Kim et al. 2011). Some researchers have proposed that rhamnolipids also stimulate immunity in plants against various infectious agents (Vatsa et al. 2010). The lipopeptide biosurfactant of Bacillus origin was reported to inhibit growth of some phytopathogenic fungi like Fusarium spp., Aspergillus spp., and Bipolaris sorokiniana. Such biosurfactant of Bacillus origin can be very well exploited for their function as biocontrol agent (Velho et al. 2011). Surfactin isoform and this lipopeptide biosurfactant produced by Brevibacillus brevis strain HOB1 have reported potent antibacterial and antifungal properties which could be utilized for control of phytopathogens (Haddad 2008). Pseudomonas fluorescens biosurfactants are well reported for their antifungal property (Nielsen et al. 2002). Biosurfactants produced by the Pseudomonas fluorescens has potential in inhibition of certain fungal pathogens like Pythium ultimum (causes damping off and root rot of plants), Fusarium oxysporum (wilting in crop plants), and Phytophthora cryptogea (responsible for rotting of fruits and flowers) (Hultberg et al. 2008). Biosurfactants produced by Bacillus subtilis isolated from soil metagenome are found useful in the control of Colletotrichum gloeosporioides which is a causative agent of anthracnose on papaya leaves (Kim et al. 2010). A common plant pathogen Pseudomonas aeruginosa is found to be inhibited by the biosurfactants of staphylococcus of oil-contaminated soil metagenome (Eddouaouda et al. 2012). The abovementioned evidences support the claim that biosurfactants produced by many microbes could be very useful for control of various kinds of phytopathogens. Furthermore, these biosurfactants are emerging as an alternative source of commonly used pesticides and insecticides which are currently in agricultural practices. Metagenomics has great prospects in identifying many phytopathogens, plant growth-promoting microbes and biosurfactant-producing microbes as well.
4.2.5 Applications in Human Health
Human beings are always surrounded by microbes as they not only surface over them but also live within their body. The microbes which are residing inside the human flora are not fully characterized (less than 1%). Furthermore there are certain microbes in our environment which are causative agents of many infectious diseases. These infectious microbes are mainly characterized by laboratory-based surveillance and syndromic surveillance which are strictly relying on the non-laboratory data. Detecting these causative agents of infectious diseases is failed in approximately 40% gastroenteritis cases and 60% in encephalitis cases when conventional approach is used (Finkbeiner et al. 2008; Ambrose et al. 2011).
The Human Microbiome Project enabled the scientific community to know about the sophisticated sequencing technologies and association of microbiome toward human health and disease (Peterson et al. 2009). Metagenomics has the potential to detect both known and novel microorganisms using culture-independent sequencing and analysis of all nucleic acids taken from the sample. The whole genome sequences of the pathogens can be detected using the advance bioinformatics tools which further help in drawing inferences about antibiotic resistance, virulence and evolution.
In the present scenario, metagenomics is playing a very crucial role in investigating novel species and strains (Wan et al. 2013; Mokili et al. 2013; Xu et al. 2011), outbreaks (Loman et al. 2013; Greninger et al. 2010), and complex diseases (Wang et al. 2012; Cho and Blaser 2012). As with the advancement of the next-generation sequencing and its cost-effectiveness, it could become an essential approach in investigation of infectious diseases at very low abundance and can be performed from clinical samples (Seth-Smith et al. 2013) or from single cells (McLean et al. 2013). The metagenomics approaches which are used for the detection of these infectious or pathogenic agents include deep amplicon sequencing and shotgun sequencing.
In deep amplicon sequencing, certain gene families are reported in every known member species in a particular taxonomic group. It employs the amplification of certain taxonomic markers such as rRNA genes. By using next-generation sequencing, many different amplicons in a sample can be sequenced, and the resulting sequences are compared with the reference standard to identify the species/genus associated with each sequence. The deep amplicon sequencing is capable of identifying the novel microorganisms . In the case of bacterial deep amplicon sequencing, they use specific primers that are specific to the conserved genes such as 16S, rRNA, chaperonin-60 (Links et al. 2012), and RNA polymerase (rpoB) (Wu et al. 2011). Likewise in protozoan and fungal deep amplicon sequencing approach, they only target 18S rRNA gene regions (Leng et al. 2011; Sirohi et al. 2012; Iliev et al. 2012). Major advantage of the deep amplicon sequencing lies in an enhancement of the assay’s sensitivity for the microorganisms, with higher resolution. However the major drawback of this approach is the inaccurate estimation of the microbial community composition, which requires prior knowledge of pathogenic agent.
In shotgun metagenomics, all microbes are taken into account after sequencing all the nucleic acids extracted from a specimen. Extracted nucleic acids from the specimen are sequenced using next-generation approach, and their results are compared with their reference database. The database used in shotgun metagenomics are usually much larger than those used in deep amplicon sequencing and contain all the known sequences as compared to the set of sequence from a single gene family. The major advantage of shotgun metagenomics over deep amplicon sequencing is that it is less biased and generates data that better reflect the sample’s true population structure. Besides pathogen detection using shotgun metagenomics approach, it also has the potential to generate complete or nearly complete pathogen genome assemblies from the sample (Seth-Smith et al. 2013; McLean et al. 2013). These results provide an estimation of microbial phenotypes and microbial genotypes by determining the presence or absence of antimicrobial resistance and epidemic dynamics (Bertelli and Greub 2013).
Although metagenomics has immense potential to exploit genomics based information for identifying microbiomes that are relevant to the public health. Additionally it is of use in hospitals and healthcare facilities to identify unknown or novel pathogens as well as for characterization of normal and disease associated microbial communities. Through metagenomics approach, it became quite easier to identify the 78 species from the biofilm from the hospital sink with new bacterial phylum (McLean et al. 2013). Thus in the present scenario, metagenomics approach has proved itself as the most powerful tool for the detection of novel microorganisms .
4.2.6 Environmental Applications
Various kinds of microbes are living in our environments which are helpful in many ways. They play a very important role in decomposing dead material present in the environment and making it free from pollutants. There are certain microbes which are able to degrade oil whenever it spills over water surface. Many microbes also have the ability of cleaning the ground water. Here metagenomics may play very important role in identifying particular species which are concerned with water treatment purpose. Oil-consuming microbes that are present in sea are suitable examples of microbial bioremediation of water. Many other bacteria that are present in the soil have qualities of consuming heavy metals and may be helpful in reducing soil toxicity. Identification of these microbes is a major hurdle in further research and analysis in this regard. So this area is a hot cake for metagenomics and environmental scientists as well.
4.3 Methods and Tools
The steps involved in metagenomics analysis have been shown in the flowchart given in Fig. 4.2, and each part is explained in detail along with the tool used in particular methods. Figure 4.2 shows flowchart for experiment design, sampling, sample fractionation for obtaining DNA, that is further analyzed using different computational tools to find out solution to various research problems.

Flowchart of experimental and computational methods that are used to retrieve the genomics information which is further analyzed by different bioinformatics tools. This analysis helps in screening and identification of uncultured microbes that are directly taken from environmental samples
4.3.1 Experimental Design
Experimental design plays a major role in getting accurate, reliable, and high-quality data. Researchers working in the field of metagenomics need to focus on number of replication of data, cost-effectiveness for the sequencing, and accuracy of methods that are used to perform the metagenomics data analysis. In order to obtain accurate and qualitative results in the field of metagenomics, there should be minimum standards during experimental design. While designing the experiment, one must consider the biological and technical replicates, budget should be fixed for sequencing, best protocols should be searched for high yield and good quality of DNA, and sequencing platform should also be discussed. The place should be clearly defined in the terms of certain parameters, from where the sample has to be taken (Cooke et al. 2017).
4.3.2 Sampling
After the experimental design, sample is collected from different sources, i.e., soil, air, water, biopsy, plants, etc. which is known as sampling. The quality of data we obtained from metagenomics depends on sampling (Thomas et al. 2012). While describing the biodiversity, the sample should represent whole population (Wooley et al. 2010) and it should also represent habitat. While collecting the samples, one should know about the time (i.e., day, date, and year of sample collection), number of samples, and volume of samples needed to describe the environmental conditions. Strategy of sampling method and variability of experimental methods should be clear. For collection of representative sample, it is very important to know the amplitude of variation in habitat environment , for example, soil communities with varying soil types like clay, silt and sand particles, plant matter in various stages of decomposition, and variety of invertebrates. So, while sampling one must consider the scale i.e. size of habitat, biological variation, experimental variability, reproducibility, repository and singletons.(The New Science of Metagenomics).
4.3.3 Sample Fractionation
Sample fractionation is a process of lysing the cell to extract the genomic DNA. It is done for obtaining the genomic DNA from abundant as well as rare representative of each taxonomic groups possessing different thickness of cell wall and cell membrane. During sample fractionation or cell lysis, genomic DNA is also exposed to different types of nucleases. So, it’s very important to deactivate or inactivate the nucleases by adding strong denaturing agents to keep our genomic DNA safe (Virgin and Todd 2011; Claesson et al. 2012; Yatsunenko et al. 2012). Cell lysis can be performed by thermal, chemical, mechanical, and enzymatic methods (Felczykowska et al. 2015).
4.3.4 DNA Extraction
DNA extraction is a crucial step for analyzing the genome of unculturable microbe. So, it’s very important to select a qualitative and quantitative DNA extraction method for getting high yield and good quality of DNA (Felczykowska et al. 2015). The sample contains DNA in various packages like virus particles, eukaryotic DNA, and prokaryotic DNA including free DNA. This may be suspended in liquid, bound to solid, or trapped in the biofilm or tissue. So, extraction methods are selected on the basis of medium present and interest of population. Basically, there are two methods for extraction of DNA, i.e., direct method and indirect method. In the first method, cells are lysed within the sample, and then DNA is extracted, e.g., viruses, and later one includes separation of sample from noncellular material before lysis. The yield of DNA product is nearly 100 times lower in the indirect method of DNA extraction than direct, but the bacterial diversity of DNA recovered by indirect means was distinctly higher. (LaMontagne et al. 2002; Van et al. 1997; Ogram et al. 1987; Berry et al. 2003; Jacobsen and Rasmussen 1992).
4.3.5 DNA Sequencing
Generally, there are three types of sequencing methods, viz., amplicon sequencing, shotgun sequencing, and metagenomics sequencing. Amplicon sequencing is used for characterization of microbiota diversity and it is the most commonly used technique. It targets the small subunit of ribosomal RNA (16s) locus, which acts as marker which gives information about phylogeny and taxonomy (Pace et al. 1986; Hugenholtz and Pace 1996). This sequencing method is used to characterize a large range of microbial diversity in the human gut (Yatsunenko et al. 2012), Arabidopsis thaliana roots (Lundberg et al. 2012), ocean thermal vents (McCliment et al. 2006), hot springs (Bowen DeLeon et al. 2013), and Antarctic volcano mineral soils (Soo et al. 2009). Due to certain limitations of amplicon sequencing, shotgun sequencing came in the picture. Novel and highly diverged species were difficult to study using amplicon sequencing (Acinas et al. 2004).
Shotgun sequencing has capability to overcome the limitations of previous approach. This approach relies on extracting DNA from cells in community and fragmenting it into tiny parts (i.e., reads) that are used to align against the known genome and 16S rRNA. Hence, it provides opportunity to explore microbiota community with two aspects (Sharpton 2014). Shotgun sequencing has also limitation like large data handling, reads may not present in the whole genome, and sometimes two reads of the same gene don’t overlap (Schloss 2008; Sharpton et al. 2011). Advancement in shotgun sequencing enables it to answer the above-raised questions and has been used for identification of new viruses (Yozwiak et al. 2012) as well as characterization of uncultured bacteria (Wrighton et al. 2012). This advanced metagenomics sequencing has been used to characterize the microbes associated with roots (Bulgarelli et al. 2013; Vorholt 2012) and also used for identification of taxa that are associated with the human gut (Morgan et al. 2012).
4.3.6 Quality Control
The sequencing data obtained from NGS technology is first subjected to quality control studies. It is the process of sorting out and screening low-quality reads, which affect the downstream analysis (Zhou et al. 2014). The accuracy of microbial biodiversity can be improved by quality filtering (Handelsman 2004). There are several tools available for quality control as shown in Table 4.1.
4.3.7 Assembly
Assembly means reconstruction of genome from smaller fragment of DNA, i.e., reads obtained through sequencing (Reich et al. 1984). Basically, there are two types of assemblies, i.e., de novo assembly in which the genome is constructed from reads data and the second is comparative assembly which is used to reconstruct the genome using a closely related organism (Medvedev et al. 2007). For the de novo assembly, three algorithm-based strategies are used named as greedy (Pop and Salzberg 2008), overlap layout consensus (Myers 1995), and De Bruijn graph (Zerbino and Velvet 2008; Pevzner et al. 2004). Improved de novo assemblies have been generated with the help of a known reference genome to form a comparative assembly like OSLay (optimal syntenic layout of unfinished assemblies) (Richter et al. 2007), Projector 2 (Van et al. 2005) and ABACAS (algorithm-based automatic contiguation of assembled sequences) (Assefa et al. 2009).
4.3.8 Annotation
Functional annotation of metagenomics data obtained after the assembling of reads involves predicting the gene, biological function, gene pathway annotation, and metabolic pathway annotation. The tools used for different functional annotations are shown in Table 4.2.
4.4 Metagenomics Databases and Online Resources
There are many databases and online tools for analyzing and retrieving metagenomics data. Table 4.3 shows the name along with link of such databases/servers. The European Bioinformatics Institute (EBI) Metagenomics enables us to submit, analyze, visualize, and compare our data (Mitchell et al. 2015). MG-RAST is a metagenomics analysis server for annotation of sequence fragments, their phylogenetic classification, functional classification of samples, and comparison between multiple metagenomes. It also computes an initial metabolic reconstruction for the metagenome and allows comparison of metabolic reconstructions of metagenomes and genomes (Wilke et al. 2016). MEGAN (Huson et al. 2011) is a comprehensive toolbox for analyzing microbiome data. One can perform the different analytics using this tool like taxonomic analysis, functional analysis, etc. QIIME (Quantitative Insights Into Microbial Ecology) is a freely available bioinformatics tool for performing microbiome analysis from raw DNA sequencing data. One can perform demultiplexing and quality filtering, OTU (operational taxonomic unit) picking, taxonomic assignment, phylogenetic reconstruction, and diversity analyses and visualizations (Caporaso et al. 2010). Mothur is an open-source, expandable software to fill the bioinformatics needs of the microbial ecology community (Schloss et al. 2009). RDP (ribosomal database) provides quality-controlled, aligned, and annotated bacterial and archaeal 16S rRNA sequences, fungal 28S rRNA sequences, and a suite of analysis tools to the scientific community.
RDP is an online tool which is used to study the new fungal 28S rRNA sequence collection. RDP tools are now freely available in packages for users to incorporate in their local workflow (Cole et al. 2009). SILVA (from Latin silva) is an online freely accessible tool to check the quality of reads and aligned (16S/18S, small subunit ribosomal RNA) and large subunit (23S/28S, LSU) ribosomal RNA (rRNA) sequence data of bacteria, archaea, and eukarya (Quast et al. 2013). Real Time Metagenomics is an online freely available tool which performs annotation of metagenomes by relating the individual sequence reads with a database of known sequences and assigning a unique function to each read. They generated a novel approach to annotate metagenomes using unique k-mer oligopeptide sequences from 7 to 12 amino acids long (Edwards et al. 2012).
4.5 Bioinformatics-Based Data Analysis
Bioinformatics-based data analysis can be done using short reads and assembled contigs present in the short read archive (SRA) format (Fig. 4.3). The metagenomics SRA data is firstly treated to sort out high-quality reads or sequences. The pretreatment includes:
-
(a)
Removal of adapters and linkers
-
(b)
Removal of duplicate sequences (dereplication)
-
(c)
Quality assessment

Flowchart for analysis of data generated by different metagenomics experiments. The procedure involves use of several computational biology tools for retrieving functional information in terms of pathway, interaction network, and gene ontology hidden in the metagenomics data. GCNA (gene co-expression network analysis) and PPI (protein-protein interaction network) studies are useful for identification of interactors
Before pretreatment of data, quality of data is checked by checking base quality, GC content, sequence dereplication levels, and adapter content using FastQC. Quality control of metagenomics data is done by RSeQC (quality control of RNA-seq experiments) followed by RNA-SeQC (Wang et al. 2012; De Luca et al. 2012). Once data become clean, then it can be used for functional annotation. After pretreatment of data, assembling of reads is done for getting the functional contigs. Data size generated after sequencing can be reduced by metagenome assembly by using integrated computational approach (Howe et al. 2014).
Reference-based and de novo-based methods are used for assembling the reads. The previous one is used to align the short reads against the related genome, while the latter one is used to find out the novelty in genes against the similar reference genome. It requires a large memory and high computational methods. Once assembling is done, binning is performed. It is a computational process of clustering or assigning the contigs that may represent individual genome/taxon or closely related microbes. Homology-based tools are used to perform the binning, i.e., MetaPhlAn2, MetaPhyler, and CARMA (Segata et al. 2012; Liu et al. 2010a, b; Gerlach and Stoye 2011). Day by day, technology is improving which leads to reduction in sequencing cost; hence researchers can access the environmental metagenome, and bioinformatics tools can be integrated with metagenome data to produce useful results and findings (Albertsen et al. 2013).
Structural and functional annotation of microbial community can be done by using assembled reads and unassembled reads too. It is well proven that unassembled short reads contain original information that can explain about functional genes, metabolic profile, and quantitative composition of microbial taxa (Davit Bzhalava and Joakim Dillner 2013).
4.6 Conclusion
Metagenomics is a continuously increasing and developing field. Modern tools and techniques like bioinformatics, NGS technology, and data analysis methods are proving to be facilitators of the trending research field. Biological data is continuously increasing its size; hence researchers have golden opportunity to solve or retrieve the hidden information present in assembled or unassembled reads using modern analytical tools more efficiently. Direct DNA sequencing of environmental samples has given opportunity to gather information about the microorganisms that were unexplored so far. Screening of useful bacteria that survive in extreme environmental conditions, heavily polluted soil, disease-affected tissues or cells, oil-contaminated water bodies, heavy metal -contaminated fields, etc. can be done easily by combining environmental and metagenomics approaches. The data obtained from environmental sample sequencing may be of great use in discovery of new drugs and antibiotics , new bacterial species, plant growth promoters, bioremediation, as well as many other industrial applications. This article presents a detailed account of applications of metagenomics especially in the field of environmental biotechnology with special focus on methods and tools useful in sample collection, sequencing, and analyzing the metagenomics data.
References
Abarenkov K, Henrik Nilsson R, Larsson KH, Alexander IJ, Eberhardt U, Erland S et al (2010) The UNITE database for molecular identification of fungi: recent updates and future perspectives. New Phytol 186:281–285. https://doi.org/10.1111/j.1469-8137.2009.03160.x
Acinas SG, Marcelino LA, Klepac-Ceraj V, Polz MF (2004) Divergence and redundancy of 16S rRNA sequences in genomes with multiple Rrn operons. J Bacteriol 186:2629–2635. https://doi.org/10.1128/JB.186.9.2629-2635.2004
Albertsen M, Hugenholtz P, Skarshewski A, Nielsen KL, Tyson GW, Nielsen PH (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31(6):533–538. https://doi.org/10.1038/nbt.2579
Alexander M (1994) Biodegradation and bioremediation. Academic, San Diego. https://doi.org/10.12691/ijebb-2-2-1
Alvarez TM, Paiva JH, Ruiz DM, Cairo JPLF, Pereira IO, Paixão DAA, de Almeida RF, Tonoli CCC, Ruller R, Santos CR, Squina FM, Murakami MT (2013) Structure and function of a novel Cellulase 5 from sugarcane soil metagenome. PLoS 8(12):1–9. https://doi.org/10.1371/journal.pone.0083635
Ambrose HE, Granerod J, Clewley JP, Davies NWS, Keir G, Cunningham R, Zuckerman M, Mutton KJ, Ward KN, Ijaz S, Crowcroft NS, Brown DWG (2011) Diagnostic strategy used to establish etiologies of encephalitis in a prospective cohort of patients in England. J Clin Microbiol 49(10):3576–3583
Anupam R, Denial M, Debarati P, Suresh K, Ol F, Mandal SM (2013) Purification, biochemical characterization and self-assembled structure of a fengycin-like antifungal peptide from Bacillus thuringiensis strain SM1. Front Microbiol 4:332. https://doi.org/10.3389/fmicb.2013.00332
Arrial RT, Togawa RC, Brigido M (2009) Screening non-coding RNAs in transcriptomes from neglected species using PORTRAIT: case study of the pathogenic fungus Paracoccidioides brasiliensis. BMC Bioinform 10(1):239
Arumugam M, Harrington ED, Foerstner KU, Raes J, Bork P (2010) Smash community: a metagenomic annotation and analysis tool. Bioinformatics 26(23):2977–2978. https://doi.org/10.1093/bioinformatics/btq536
Assefa S, Keane TM, Otto TD, Newbold C, Berriman M (2009) ABACAS: algorithm-based automatic contiguation of assembled sequences. Bioinformatics 25(15):1968–1969. https://doi.org/10.1093/bioinformatics/btp347
Atlas RM, Hazen TC (2011) Oil biodegradation and bioremediation: a tale of the two worst spills in U.S. history. Environ Sci Technol 45(16):6709–6715
Ball AP, Bartlett JG, Craig WA, Drusano GL, Felmingham D, Garau JA, Klugman KP, Low DE, Mandell LA, Rubinstein E, Tillotson GS (2013) Future trends in antimicrobial chemotherapy: expert opinion on the 43 ICAAC. J Chemother 16(5):419–436
Berry AE, Chiocchini C, Selby T, Sosio M, Wellington EM (2003) Isolation of high molecular weight DNA from soil for cloning into BAC vectors. FEMS Microbiol Lett 223:15–20. https://doi.org/10.1016/S0378-1097(03)00248-9
Bertelli C, Greub G (2013) Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect 19(9):803–813
Bowen DLK, Gerlach R, Peyton BM, Fields MW (2013) Archaeal and bacterial communities in three alkaline hot springs in heart Lake Geyser Basin, Yellowstone National Park. Front Microbiol 4:330. https://doi.org/10.3389/fmicb.2013.00330
Boyette CD, Walker HL, Abbas HK (2002) Biological control of kudzu (Pueraria lobata) with an isolate of Myrothecium verrucaria. Biocontrol Sci Tech 12(1):75–82
Brennan Y, Callen WN, Christoffersen L, Dupree P, Goubet F, Healey S, Hernández M, Keller M, Li K, Palackal N, Sittenfeld A, Tamayo G, Wells S, Hazlewood GP, Mathur EJ, Short JM, Robertson DE, Steer BA (2004) Unusual microbial xylanases from insect guts. Appl Environ Microbiol 70(6):3609–3617. https://doi.org/10.1128/AEM.70.6.3609-3617.2004
Bulgarelli D, Schlaeppi K, Spaepen S, Ver Loren Van Themaat E, Schulze-Lefert P (2013) Structure and functions of the bacterial microbiota of plants. Annu Rev Plant Biol 64:807–838. https://doi.org/10.1146/annurev-arplant-050312-120106
Bzhalava D, Dillner J (2013) Bioinformatics for viral metagenomics. J Data Min Genomics Proteomics 4:3. https://doi.org/10.4172/2153-0602.1000134
Cameotra SS, Bollag JM (2003) Biosurfactant-enhanced bioremediation of polycyclic aromatic hydrocarbons. Crit Rev Environ Sci Technol 33(2):111–126. https://doi.org/10.1080/10643380390814505
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336. https://doi.org/10.1038/nmeth.f.303
Cardenas F, Emilio A, de Castro-Alvarez M-S, Jose-Maria S-M, Manuel V, Elson Steve W, Jose-Vicente S (2001) Screening and catalytic activity in organic synthesis of novel fungal and yeast lipases. J Mol Catal B Enzym 14:111–123. https://doi.org/10.1016/S1381-1177(00)00244-7
Chakraborty R, Wu CH, Hazen TC (2012) Systems biology approach to bioremediation. Curr Opin Biotechnol 23:483–490
Cho I, Blaser MJ (2012) The human microbiome: at the interface of health and disease. Nat Rev Genet 13:260–270
Christakopoulos P, Katapodis P, Kalogeris E, Kekos D, Macris BJ, Stamatis H, Skaltsa H (2001) Antimicrobial activity of acidic xylooligosaccharides produced by family 10 and 11 endoxylanases. Int J Biol Macromol 31(4–5):171–175. https://doi.org/10.1016/S0141-8130(02)00079-X
Čivljak R, Giannella M, Di Bella S, Petrosillo N (2014) Could chloramphenicol be used against ESKAPE pathogens? Are view of in vitro data in the literature from the 21st century. Expert Rev Anti-Infect Ther 12(2):249–264. https://doi.org/10.1586/14787210.2014.878647
Claesson MJ, Jeffery IB, Conde S, Power SE, O’Connor EM, Cusack S, Harris HMB, Coakley M, Lakshminarayanan B, O’Sullivan O, Fitzgerald GF, Deane J, O’Connor M, Harnedy N, O’Connor K, O’Mahony D, van Sinderen D, Wallace M, Brennan L, Stanton C, Marchesi JR, Fitzgerald AP, Shanahan F, Hill C, Ross RP, O’Toole PW (2012) Gut microbiota composition correlates with diet and health in the elderly. Nature 488(7410):178–184
Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ et al (2009) The ribosomal database project: improved alignments and new tools for rRNA analysis. Nucleic Acids Res 37:D141–D145. https://doi.org/10.1093/nar/gkn879
Cooke SJ, Birnie-Gauvin K, Lennox RJ, Taylor JJ, Rytwinski T, Rummer JL, Franklin CE, Bennett JR, Haddaway NR (2017) How experimental biology and ecology can support evidence-based decision-making in conservation: avoiding pitfalls and enabling application. Conserv Physiol 5(1):cox043
Coppotelli BM, Ibarrolaza A, Dias RL, Del Panno MT, Berthe-Corti L, Morelli IS (2010) Study of the degradation activity and the strategies to promote the bioavailability of phenanthrene by Sphingomonas paucimobilis strain 20006FA. Microb Ecol 59(2):266–276. https://doi.org/10.1007/s00248-009-9563-3
de Souza CE, e Silva Cardoso MH, Sousa DA, Viana JC, de Oliveira-Júnior NG, Miranda V, Franco OL (2014) The use of versatile plant antimicrobial peptides in agribusiness and human health. Peptides 55:65–78. https://doi.org/10.1016/j.peptides.2014.02.003
Delong EF (2005) Microbial community genomics in the ocean. Nat Rev Microbiol 3(6):459–469
Deluca DS, Levin JZ, Sivachenko A, Fennell T, Nazaire MD, Williams C, Reich M, Winckler W, Getz G (2012) RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28(11):1530–1532. https://doi.org/10.1093/bioinformatics/bts196
Desantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K et al (2006) Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 72:5069–5072. https://doi.org/10.1128/AEM.03006-05
Duan CJ, Xian L, Zhao GC, Feng Y, Pang H, Bai XL, Tang JL, Ma QS, Feng JX (2009) Isolation and partial characterization of novel genes encoding acidic cellulases from metagenomes of buffalo rumens. J Appl Microbiol 107(1):245–256. https://doi.org/10.1111/j.1365-2672.2009.04202.x
Eddouaouda K, Mnif S, Badis A, Younes SB, Cherif S, Ferhat S, Mhiri N, Chamkha M, Sayadi S (2012) Characterization of a novel biosurfactant produced by Staphylococcus sp. strain 1E with potential application on hydrocarbon bioremediation. J Basic Microbiol 52(4):408–418. https://doi.org/10.1002/jobm.201100268
Edwards et al (2012) Real time metagenomics: using k-mers to annotate metagenomes. Bioinformatics 28(24):3316–3317. https://doi.org/10.1093/bioinformatics/bts599
Felczykowska A, Krajewska A, Zielińska S, Łoś JM (2015) Sampling, metadata and DNA extraction important steps in metagenomic studies. Acta Biochim Pol 62(1):151–160. https://doi.org/10.18388/abp.2014_916
Finkbeiner SR, Allred AF, Tarr PI, Klein EJ, Kirkwood CD, Wang D, Holmes EC (2008) Metagenomic analysis of human diarrhea: viral detection and discovery. PLoS Pathog 4(2):e1000011
Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G (2012) The shared antibiotic resistome of soil bacteria and human pathogens. Science 337(6098):1107–1111. https://doi.org/10.1126/science.1220761
Gerlach W, Stoye J (2011) Taxonomic classification of metagenomic shotgun sequences with CARMA3. Nucleic Acids Res 39(14):e91. https://doi.org/10.1093/nar/gkr225
Gillespie DE, Brady SF, Bettermann AD, Cianciotto NP, Liles MR, Rondon MR, Clardy J, Goodman RM, Handelsman J (2002) Isolation of antibiotics turbomycin A and B from a metagenomic library of soil microbial DNA. Appl Environ Microbiol 68(9):4301–4306. https://doi.org/10.1128/AEM.68.9.4301-4306.2002
Glass EM, Wilkening J, Wilke A, Antonopoulos D, Meyer F (2010) Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb Protoc 2010(1):pdb.prot5368. https://doi.org/10.1101/pdb.prot5368
Goll J, Rusch DB, Tanenbaumdm TM, Li K, Methé B, Yooseph S (2010) METAREP: JCVI metagenomics reports–an open source tool for high-performance comparative metagenomics. Bioinformatics 26(20):2631–2632. https://doi.org/10.1093/bioinformatics/btq455
Gottfried A, Singhal N, Elliot R, Swift S (2010) The role of salicylate and biosurfactant in inducing phenanthrene degradation in batch soil slurries. Appl Microbiol Biotechnol 86(5):1563–1571. https://doi.org/10.1007/s00253-010-2453-2
Greninger AL, Chen EC, Sittler T, Scheinerman A, Roubinian N, Guixia Y, Kim E, Pillai DR, Guyard C, Mazzulli T, Isa P, Arias CF, Hackett J, Schochetman G, Miller S, Tang P, Chiu CY, Tripp R (2010) A metagenomic analysis of pandemic influenza a (2009 H1N1) infection in patients from North America. PLoS One 5(10):e13381
Gutierrez T, Singleton DR, Berry D, Yang T, Aitken MD, Teske A (2013) Hydrocarbon-degrading bacteria enriched by the Deepwater horizon oil spill identified by cultivation and DNA-SIP. ISME J 7(11):2091–2104
Haddad NI (2008) Isolation and characterization of a biosurfactant producing strain, Brevibacillus brevis HOB1. J Ind Microbiol Biotechnol 35(12):1597–1604. https://doi.org/10.1007/s10295-008-0403-0
Hajer R, Nuno S, Patrícia P, Carmen T, Susana C, Gilberto I (2014) Potential impact of antimicrobial resistance in wild life, environment and human health. Front Microbiol 5(23). https://doi.org/10.3389/fmicb.2014.00023
Handelsman J (2004) Metagenomics: application of genomics to uncultured microorganisms. Microbiol Mol Biol Rev 68:669–685. https://doi.org/10.1128/MMBR.68.4.669-685.2004
Hassan M, Kjos M, Nes IF, Diep DB, Lotfipour F (2012) Natural antimicrobial peptides from bacteria: characteristics and potential applications to fight against antibiotic resistance. J Appl Microbiol 113(4):723–736. https://doi.org/10.1111/j.1365-2672.2012.05338.x
Hazen TC, Prince RC, Mahmoudi N (2016) Marine oil biodegradation. Environ Sci Technol 50(5):2121–2129
Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, Jansson JK, Probst A, Borglin SE, Fortney JL, Stringfellow WT, Bill M, Conrad ME, Tom LM, Chavarria KL, Alusi TR, Lamendella R, Joyner DC, Spier C, Baelum J, Auer M, Zemla ML, Chakraborty R, Sonnenthal EL, D’Haeseleer P, Holman H-YN, Osman S, Lu Z, Van Nostrand JD, Deng Y, Zhou J, Mason OU (2010) Deep-Sea oil plume enriches indigenous oil-degrading Bacteria. Science 330(6001):204–208
Hettich RL, Pan C, Chourey K, Giannone RJ (2013) Metaproteomics: harnessing the power of high performance mass spectrometry to identify the suite of proteins that control metabolic activities in microbial communities. Anal Chem 85(9):4203–4214
Hoff KJ, Lingner T, Meinicke P, Tech M (2009) Orphelia: predicting genes in metagenomic sequencing reads. Nucleic Acids Res 37(Web Server issue):W101–W105. https://doi.org/10.1093/nar/gkp327
Hosokawa T, Kaiwa N, Matsuura Y, Kikuchi Y, Fukatsu T (2015) Infection prevalence of Sodalis symbionts among stinkbugs. Zool Lett 1(1)
Howe AC, Jansson JK, Malfatti SA, Tringe SG, Tiedje JM, Brown CT (2014) Tackling soil diversity with the assembly of large, complex metagenomes. Proc Natl Acad Sci U S A 111(13):4904–4909. https://doi.org/10.1073/pnas.1402564111
Hugenholtz P, Pace NR (1996) Identifying microbial diversity in the natural environment: a molecular phylogenetic approach. Trends Biotechnol 14(190–197):10025–10021. https://doi.org/10.1016/0167-7799(96)
Hultberg M, Bergstrand KJ, Khalil S, Alsanius B (2008) Characterization of biosurfactant-producing strains of fluorescent pseudomonads in a soilless cultivation system. Antonie Van Leeuwenhoek 94(2):329–334. https://doi.org/10.1007/s10482-008-9250-2
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21(9):1552–1560. https://doi.org/10.1101/gr.120618.111
Iliev ID, Funari VA, Taylor KD, Nguyen Q, Reyes CN, Strom SP, Brown J, Becker CA, Fleshner PR, Dubinsky M, Rotter JI, Wang HL, McGovern DPB, Brown GD, Underhill DM (2012) Interactions between commensal fungi and the C-type Lectin receptor dectin-1 influence colitis. Science 336(6086):1314–1317
Jacobsen CS, Rasmussen OF (1992) Development and application of a new method to extract bacterial DNA from soil based on separation of bacteria from soil with cation–exchange resine. Appl Environ Microbiol 58:2458–2462
Jacques RJS, Bento FM, de Oliveira CFA (2007) Biodegradação de hidrocarbonetos aromáticos policíclicos. Cienc Nat 29(1):7–24
Jean C, Peter C, Don G, Herman G, Gyssens Inge C, Stephan H, Vincent J, Levy Stuart B, Doye Babacar N, Didier P, Rosana R, Seto Wing H, van der Meer Jos WM, Andreas V (2011) Society’s failure to protect a precious resource: antibiotics. Lancet 378:369–371. https://doi.org/10.1016/S0140-6736(11)60401-7
Jeong YS, Na HB, Kim SK, Kim YH, Kwon EJ, Kim J, Yun HD, Lee JK, Kim H (2012) Characterization of xyn10J, a novel family 10 xylanase from a compost metagenomic library. Appl Biochem Biotechnol 166(5):1328–1339. https://doi.org/10.1007/s12010-011-9520-8
Kang S-W, Kim Y-B, Shin J-D, Kim E-K (2010) Enhanced biodegradation of hydrocarbons in soil by microbial biosurfactant, sophorolipid. Appl Biochem Biotechnol 160(3):780–790
Kamal Kumar B, Balakrishnan H, Rele MV (2004) Compatibility of alkaline xylanases from an alkaliphilic Bacillus NCL (87-6-10) with commercial detergents and proteases. J Ind Microbiol Biotechnol 31(2):83–87. https://doi.org/10.1007/s10295-004-0119-8
Kashyap R, Monika, Subudhi E (2014) A novel thermoalkaliphilic xylanase from Gordonia sp. is salt, solvent and surfactant tolerant. J Basic Microbiol 54:1342–1349. https://doi.org/10.1002/jobm.201400097
Katapodis P, Vardakas M, Kalogeris E, Kekos D, Bj M, Christakopoulos P (2003) Enzymic production of a feruloylated oligosaccharide with antioxidant activity from wheat flour arabinoxylan. Eur J Nutr 42(1):55–60. https://doi.org/10.1007/s00394-003-0400-z
Kelley DR, Liu B, Delcher AL, Pop M, Salzberg SL (2012) Gene prediction with Glimmer for metagenomic sequences augmented by classification and clustering. Nucleic Acids Res 40(1):e9. https://doi.org/10.1093/nar/gkr1067
Kerepesi C, Bánky D, Grolmusz V (2014) Amphora net: the web server implementation of the AMPHORA2metagenomicworkflowsuite. Gene 533:538–540. https://doi.org/10.1016/j.gene.2013.10.015
Kim PI, Bai H, Bai D, Chae H, Chung S, Kim Y, Park R, Chi YT (2004) Purification and characterization of a lipopeptide produced by Bacillus thuringiensis CMB26. J Appl Microbiol 97:942–949. https://doi.org/10.1111/j.1365-2672.2004.02356.x
Kim PI, Ryu J, Kim YH, Chi YT (2010) Production of biosurfactant lipopeptides Iturin A, fengycin and surfactin A from Bacillus subtilis CMB32 for control of Colletotrichum gloeosporioides. J Microbiol Biotechnol 20(1):138–145. https://doi.org/10.4014/jmb.0905.05007
Kim SK, Kim YC, Lee S, Kim JC, Yun MY, Kim IS (2011) Insecticidal activity of rhamnolipid isolated from Pseudomonas sp. EP-3 against green peach aphid (Myzus persicae). J Agric Food Chem 59(3):934–938. https://doi.org/10.1021/jf104027x
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H et al (2012) Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol 62(Pt 3):716–721. https://doi.org/10.1093/nar/gkr1067
King GM, Kostka JE, Hazen TC, Sobecky PA (2015) Microbial responses to the oil spill: from coastal wetlands to the deep sea. Annu Rev Mar Sci 7(1):377–401
Kirk O, Borchert TV, Fuglsang CC (2002) Industrial enzyme applications. Curr Opin Biotechnol 13(4):345–351. https://doi.org/10.1016/S0958-1669(02)00328-2
Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, Glöckner FO (2013) Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res 41(1):e1–e1
Krzyzanowska DM, Potrykus M, Golanowska M, Polonis K, Gwizdek-Wisniewska A, Lojkowska E, Jafra S (2012) Rhizosphere bacteria as potential biocontrol agents against soft rot caused by various Pectobacterium and Dickeya spp. strains. J Plant Pathol 94(2):367–378. https://doi.org/10.4454/JPP.FA.2012.042
LaMontagne MG, Michel FC, Holden PA, Reddy CA (2002) Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J Microbiol Methods 49(3):255–264
Lee CC, Kibblewhite-Accinelli RE, Wagschal K, Robertson GH, Wong DW (2006) Cloning and characterization of a cold-active xylanase enzyme from an environmental DNA library. Extremophiles 10(4):295–300. https://doi.org/10.1007/s00792-005-0499-3
Leng J, Zhong X, Zhu RJ, Yang SL, Gou X, Mao HM (2011) Assessment of protozoa in Yunnan yellow cattle rumen based on the 18S rRNA sequences. Mol Biol Rep 38(1):577–585
Li W (2009) Comparison of very large metagenomes with fast clustering and functional annotation. BMC Bioinf 10(1):359. https://doi.org/10.1186/1471-2105-10-359
Lim HK, Chung EJ, Kim JC, Choi GJ, Jang KS, Chung YR, Cho KY, Lee SW (2005) Characterization of a forest soil metagenome clone that confers indirubin and indigo production on Escherichia coli. Appl Environ Microbiol 71(12):7768–7777. https://doi.org/10.1128/AEM.71.12.7768-7777.2005
Links MG, Dumonceaux TJ, Hemmingsen SM, Hill JE, Neufeld J (2012) The chaperonin-60 universal target is a barcode for bacteria that enables de novo assembly of metagenomic sequence data. PLoS One 7(11):e49755
Lindgreen S, Adair KL, Gardner PP (2016) An evaluation of the accuracy and speed of metagenome analysis tools. Sci Rep 6:19233. https://doi.org/10.1038/srep19233
Lingner T, Asshauer KP, Schreiber F, Meinicke P (2011) CoMet–a web server for comparative functional profiling of metagenomes. Nucleic Acids Res 39(Web Server issue):W518–W523. https://doi.org/10.1093/nar/gkr388
Liu B, Gibbons T, Ghodsi M, Pop M (2010a) MetaPhyler: taxonomic profiling for metagenomic sequences, Bioinformatics and Biomedicine (BIBM). In 2010 IEEE International Conference; IEEE: pp 95100. https://doi.org/10.1109/BIBM.2010.5706544
Liu WW, Yin R, Lin XG, Zhang J, Chen XM, Li XZ, Yang T (2010b) Interaction of biosurfactant-microorganism to enhance phytoremediation of aged polycyclic aromatic hydrocarbons (PAHS) contaminated soils with alfalfa (Medicago sativa L.). Huan Jing Ke Xue 31(4):1079–1084. https://www.ncbi.nlm.nih.gov/pubmed/20527195
Loman NJ, Constantinidou C, Christner M, Rohde H, Chan JZ-M, Quick J, Weir JC, Quince C, Smith GP, Betley JR, Aepfelbacher M, Pallen MJ (2013) A culture-independent sequence-based Metagenomics approach to the investigation of an outbreak of Shiga-toxigenic Escherichia coli O104:H4. JAMA 309(14):1502
Lorenz P, Liebeton K, Niehaus F, Eck J (2002) Screening for novel enzymes for biocatalytic processes: accessing the metagenome as a resource of novel functional sequence space. Curr Opin Biotechnol 13(6):572–577. https://doi.org/10.1016/S0958-1669(02)00345-2
Lovley DR (2003) Cleaning up with genomics: applying molecular biology to bioremediation. Nat Rev Microbiol 1(1):35–44
Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S et al (2012) Defining the core Arabidopsis thaliana root microbiome. Nature 488:86–90. https://doi.org/10.1038/nature11237
MacNeil IA, Tiong CL, Minor C, August PR, Grossman TH, Loiacono KA, . Lynch BA, Phillips T, Narula S, Sundaramoorthi R, . Tyler A, Aldredge T, Long H., Gilman M, Holt D, and Osburne MS (2001) Expression and isolation of antimicrobial small molecules from soil DNA libraries. J Mol Microbiol Biotechnol. Vol. 3(2): 301–308. https://www.ncbi.nlm.nih.gov/pubmed/11321587
Marcos MS, Lozada M, Dionisi HM (2006) Aromatic hydrocarbon degradation genes from chronically polluted Subantarctic marine sediments. Lett Appl Microbiol 49(5):602–608. https://doi.org/10.1111/j.1472-765X.2009.02711.x
Mason OU, Hazen TC, Borglin S, Chain PSG, Dubinsky EA, Fortney JL, Han J, Holman H-YN, Hultman J, Lamendella R, Mackelprang R, Malfatti S, Tom LM, Tringe SG, Woyke T, Zhou J, Rubin EM, Jansson JK (2012) Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J 6(9):1715–1727
Markowitz VM, Chen IA, Chu K, Szeto K, Palaniappan K, Grechkin Y, Ratner A, Jacob B, Pati A et al (2012) IMG/M: the integrated metagenome data management and comparative analysis system. Nucleic Acids Res 40(Database issue):D123–D129. https://doi.org/10.1093/nar/gkr975
McCliment EA, Voglesonger KM, O’Day PA, Dunn EE, Holloway JR, Cary SC (2006) Colonization of nascent, deep-sea hydrothermal vents by a novel archaeal and Nanoarchaeal assemblage. Environ Microbiol 8:114–125. https://doi.org/10.1111/j.1462-2920.2005.00874.x
McGarvey KM, Konstantin Q, Stanley F (2012) Wide variation in antibiotic resistance protein identified by functional metagenomic screening of a soil DNA library. Appl Environ Microbiol 78(6):1708–1714. 10.1128/AEM.067 59–6711
McLean SJ et al (2013) Genome of the pathogen Porphyromonas gingivalis recovered from a biofilm in a hospital sink using a high-throughput single-cell genomics platform. Genome Res. https://doi.org/10.1101/gr.150433.112
Medvedev P, Georgiou K, Myers G et al (2007) Computability of models for sequence assembly. Gene 4645:289–301. https://springerlink.bibliotecabuap.elogim.com/content/pdf/10.1007/978-3-540-74126-8.pdf#page=300
Mitchell A, Bucchini F, Cochrane G et al (2015) EBI metagenomics in 2016 – an expanding and evolving resource for the analysis and archiving of metagenomic data. In: Nucleic acids research. https://doi.org/10.1093/nar/gkv1195
Moldes AB, Paradelo R, Rubinos D, Devesa-Rey R, Cruz JM, Barral MT (2011) Ex situ treatment of hydrocarbon-contaminated soil using biosurfactants from Lactobacillus pentosus. J Agric Food Chem 59:9443–9447. https://doi.org/10.1021/jf201807r
Mokili JL, Dutilh BE, Lim YW, Schneider BS, Taylor T, Haynes MR, Metzgar D, Myers CA, Blair PJ, Nosrat B, Wolfe ND, Rohwer F, Burk RD (2013) Identification of a novel human papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness. PLoS One 8(3):e58404
Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV et al (2012) Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13:R79. https://doi.org/10.1186/gb-2012-13-9-r79
Myers EW (1995) Toward simplifying and accurately formulating fragment assembly. J Comput Biol 2:275–290. https://doi.org/10.1089/cmb.1995.2.275
National Academy of Sciences (2005) Mineral tolerance of animals. 2nd Revised Edition
Nielsen TH, Sorensen D, Tobiasen C, Andersen JB, Christophersen C, Givskov M, Sorensen J (2002) Antibiotic and biosurfactant properties of cyclic Lipopeptides produced by fluorescent pseudomonas spp. from the sugar beet Rhizosphere. Appl Environ Microbiol 68(7):3416–3423
Nihorimbere V, Marc Ongena M, Smargiassi M, Thonart P (2011) Beneficial effect of the rhizosphere microbial community for plant growth and health. Biotechnol Agron Soc Environ 15:327–337. http://orbi.ulg.ac.be/bitstream/2268/113786/1/2011%20Nihorimbere%20Base.pdf
Noguchi H, Taniguchi T, Itoh T (2008) MetaGeneAnnotator: detecting species specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA research : an international journal for rapid publication of reports on genes and genomes 15(6):387–396. https://doi.org/10.1093/dnares/dsn027
Ogram A, Sayler GS, Barbay T (1987) The extraction and purification of microbial DNA from sediments. J Microbiol Methods 7:57–66. https://doi.org/10.1016/0167-7012(87)90025-X
Pace NR, Stahl DA, Lane DJ, Olsen GJ (1986) The analysis of natural microbial populations by ribosomal RNA sequences. Adv Microb Ecol 9:1–55. https://doi.org/10.1007/978-1-4757-0611-6
Pacwa-Płociniczak M, Płaza GA, Piotrowska-Seget Z, Cameotra SS (2010) Environmental applications of biosurfactants: recent advances. Int J Mol Sci 12(1):633–654. https://doi.org/10.3390/ijms12010633
Parada AE, Needham DM, Fuhrman JA (2016) Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol 18(5):1403–1414
Partovinia A, Naeimpoor F, Hejazi P (2010) Carbon content reduction in a model reluctant clayey soil: slurry phase n-hexadecane bioremediation. J Hazard Mater 181(1–3):133–139. https://doi.org/10.1016/j.jhazmat.2010.04.106
Patel RK, Jain M (2012) NGS QC Toolkit: a Toolkit for quality control of next generation sequencing data. PLoS One 7:e30619. https://doi.org/10.1371/journal.pone.0030619
Patel RN, Banerjee A, Ko RY, Howell JM, Li WS, Comezoglu FT, Partyka RA, Szarka FT (1994) Enzymic preparation of (3R-cis)-3-(acetyloxy)-4-phenyl-2-azetidinone: a taxol side-chain synthon. Biotechnol Appl Biochem 20(1):23–33. https://doi.org/10.1111/j.1470-8744.1994.tb00304.x
Payne RB, May HD, Sowers KR (2011) Enhanced reductive Dechlorination of polychlorinated biphenyl impacted sediment by bioaugmentation with a Dehalorespiring bacterium. Environ Sci Technol 45(20):8772–8779
Pevzner PA, Tang H, Tesler G (2004) De novo repeat classification and fragment assembly. Genome Res 14:1786–1796. https://doi.org/10.1101/gr.2395204
Pop M (2009) Genome assembly reborn: recent computational challenges. Brief Bioinform 10:354–366. https://doi.org/10.1093/bib/bbp026
Pop M, Salzberg SL (2008) Bioinformatics challenges of new sequencing technology. Trends Genet 24:142–149. https://doi.org/10.1016/j.tig.2007.12.006
Prosser JI (2015) Dispersing misconceptions and identifying opportunities for the use of “omics” in soil microbial ecology. Nat Rev Microbiol 13:439–446
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. https://doi.org/10.1093/nar/gks1219
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SSK, McCulle SL, Karlebach S, Gorle R, Russell J, Tacket CO, Brotman RM, Davis CC, Ault K, Peralta L, Forney LJ (2011) Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci 108(Supplement_1):4680–4687
Redmond MC, Valentine DL (2012) Natural gas and temperature structured a microbial community response to the Deepwater horizon oil spill. Proc Natl Acad Sci 109(50):20292–20297
Reich JG, Drabsch H, Dimuler A (1984) On the statistical assessment of similarities in DNA sequences. Nucleic Acids Res 12(13):5529–5543. https://doi.org/10.1093/nar/12.13.5529
Rho M, Tang H, Ye Y (2010) FragGeneScan: predicting genes in short and error prone reads. Nucleic Acids Res 38(20):e191. https://doi.org/10.1093/nar/gkq747
Richter DC, Schuster SC, Oslay HDH (2007) Optimal syntenic layout of unfinished assemblies. Bioinformatics 23(13):1573–1579. https://doi.org/10.1093/bioinformatics/btm153
Salanitro JP, Johnson PC, Spinnler GE, Maner PM, Wisniewski HL, Bruce C (2000) Field-scale demonstration of enhanced MTBE bioremediation through aquifer bioaugmentation and oxygenation. Environ Sci Technol 34(19):4152–4162
Schloss PD, Handelsman J (2003) Biotechnological prospects from metagenomics. Curr Opin Biotechnol 14(3):303–310. https://doi.org/10.1016/S0958-1669(03)00067-3
Schloss PD (2008) Evaluating different approaches that test whether microbial communities have the same structure. ISME J 2(3):265–275
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. https://doi.org/10.1128/AEM.01541-09
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. https://doi.org/10.1093/bioinformatics/btr026
Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C (2012) Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 9(8):811–814. https://doi.org/10.1038/nmeth.2066
Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M (2007) CAMERA: a community resource for metagenomics. PLoS Biol 5(3):e75. https://doi.org/10.1371/journal.pbio.0050075
Seth-Smith HMB, Harris SR, Skilton RJ, Radebe FM, Golparian D, Shipitsyna E, Duy PT, Scott P, Cutcliffe LT, O’Neill C, Parmar S, Pitt R, Baker S, Ison CA, Marsh P, Jalal H, Lewis DA, Unemo M, Clarke IN, Parkhill J, Thomson NR (2013) Whole-genome sequences of chlamydia trachomatis directly from clinical samples without culture. Genome Res 23(5):855–866
Sha R, Jiang L, Meng Q, Zhang G, Song Z (2011) Producing cell-free culture broth of rhamnolipids as a cost-effective fungicide against plant pathogens. J Basic Microbiol 52(4):458–466. https://doi.org/10.1002/jobm.201100295
Shakya M, Gottel N, Castro H, Yang ZK, Gunter L, Labbé J, Muchero W, Bonito G, Vilgalys R, Tuskan G, Podar M, Schadt CW, Shah V (2013) A multifactor analysis of fungal and bacterial community structure in the root microbiome of mature populus deltoides trees. PLoS One 8(10):e76382
Sharma S, Khan FG, Qazi GN (2010) Molecular cloning and characterization of amylase from soil metagenomic library derived from Northwestern Himalayas. Appl Microbiol Biotechnol 86(6):1821–1828. https://doi.org/10.1007/s00253-009-2404-y
Sharma PK (2012) Optimization of process parameters for fruit juice clarification using silica immobilized xylanase from Pseudomonas sp. Pure Appl Biol 1(2):52–55
Sharpton TJ (2014) An introduction to the analysis of shotgun metagenomic data. Front Plant Sci 5:209
Sharpton TJ, Riesenfeld SJ, Kembel SW, Ladau J, O’Dwyer JP, Green JL et al (2011) PhylOTU: a high-throughput procedure quantifies microbial community diversity and resolves novel taxa from metagenomic data. PLoS Comput Biol 7:e1001061. https://doi.org/10.1371/journal.pcbi.1001061
Sierra-García IN, Alvarez JC, de Vasconcellos SP, de Souza AP, Neto EV d S, de Oliveira VM, Mormile MR (2014) New hydrocarbon degradation pathways in the microbial metagenome from Brazilian petroleum reservoirs. PLoS One 9(2):e90087
Silva CC, Hayden H, Sawbridge T, Mele P, Paula SOD, Silva LCF, Vidigal PMP, Vicentini R, Sousa MP, Torres APR, Santiago VMJ, Oliveira VM (2013) Identification of genes and pathways related to phenol degradation in metagenomic libraries from petroleum refinery wastewater. PLoS One 8(4):1–11. https://doi.org/10.1371/journal.pone.0061811
Singh A, Van Hamme JD, Ward OP (2007) Surfactants in microbiology and biotechnology: Part 2 Application aspects. Biotechnol Adv 25(2):99–121. https://doi.org/10.1016/j.biotechadv.2006.10.004
Sirohi SK, Singh N, Dagar SS, Puniya AK (2012) Molecular tools for deciphering the microbial community structure and diversity in rumen ecosystem. Appl Microbiol Biotechnol 95:1135–1154
Soni R, Nazir A, Chaddha BS, Saini HS (2008) Novel sources of fungal cellulases for efficient deinking of composite paper waste. Bio Resources 3(1):234–246. http://152.1.0.246/index.php/BioRes/article/view/BioRes_03_1_0234_Soni_CS_FungalCellulases/179
Soo RM, Wood SA, Grzymski JJ, McDonald IR, Cary SC (2009) Microbial biodiversity of thermophilic communities in hot mineral soils of Tramway Ridge, Mount Erebus. Antarctica Environ Microbiol 11:715–728. https://doi.org/10.1111/j.1462-2920.2009.01859.x
Straube WL, Nestler CC, Hansen LD, Ringleberg D, Pritchard PH, Jones-Meehan J (2003) Remediation of polyaromatic hydrocarbons (PAHs) through landfarming with biostimulation and bioaugmentation. Acta Biotechnol 23(2–3):179–196. https://doi.org/10.1002/abio.200390025
Sun X, Wu L, Luo Y (2006) Application of organic agents in remediation of heavy metals- contaminated soil. Ying Yong Sheng Tai Xue Bao 17(6):1123–1128. PMID:16964954
The NIH HMP Working Group, Peterson J, Garges S, Giovanni M, Mc Innes P, Wang L et al (2009) The NIH human microbiome project. Genome Res 19(12):2317–2323. https://doi.org/10.1101/gr.096651.109
Thomas CM, Nielsen KM (2005) Mechanisms of and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol 3(9):711–721. https://doi.org/10.1038/nrmicro1234
Thomas T, Gilbert J, Meyer F (2012) Metagenomics - a guide from sampling to data analysis. Microb Inf Exp 2(1):3
Uchiyama T, Miyazaki K (2009) Functional metagenomics for enzyme discovery: challenges to efficient screening. Curr Opin Biotechnol 20(6):616–622
Van Hijum SAFT, Zomer AL, Kuipers OP, Kok J (2005) Projector 2: contig mapping for efficient gap-closure of prokaryotic genome sequence assemblies. Nucleic Acids Res 33(SUPPL. 2):560–566. https://doi.org/10.1093/nar/gki356
Van EJD, Mantynen V, Wolters AC (1997) Soil DNA extraction and assessment of the fate of Mycobacterium cholorophenolicum strain PC-1 in different soils by 16S ribosomal gene sequence based most probable number PCR and immunofluorescence. Biol Fertil Soils 24:188–195. https://doi.org/10.1007/s003740050230
Vatsa P, Sanchez L, Clement C, Baillieul F, Dorey S (2010) Rhamnolipid biosurfactants as new players in animal and plant defense against microbes. Int J Mol Sci 11(12):5095–5108. https://doi.org/10.3390/ijms11125095
Velho RV, Medina LF, Segalin J, Brandelli A (2011) Production of lipopeptides among Bacillus strains showing growth inhibition of phytopathogenic fungi. Folia Microbiol 56(4):297–303. https://doi.org/10.1007/s12223-011-0056-7
Victor M, et al. (2005) Biological Data Management and Technology Center. Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley and Microbial Genomics and Metagenomics Program, Department of Energy Joint Genome Institute, 2800 Mitchell Drive, Walnut Creek, USA: https://doi.org/10.1093/nar/gkm869
Virgin HW, Todd JA (2011) Metagenomics and personalized medicine. Cell 147(1):44–56
Vorholt JA (2012) Microbial life in the phyllosphere. Nat Rev Microbiol 10:828–840. https://doi.org/10.1038/nrmicro2910
Wan X-F, Barnett JL, Cunningham F, Chen S, Yang G, Nash S, Long L-P, Ford L, Blackmon S, Zhang Y, Hanson L, He Q (2013) Detection of African swine fever virus-like sequences in ponds in the Mississippi Delta through metagenomic sequencing. Virus Genes 46(3):441–446
Wang L, Wang S, Li W (2012) RSeQC: quality control of RNA-seq experiments. Bioinformatics 28(16):2184–2185. https://doi.org/10.1093/bioinformatics/bts356
Wattanaphon HT, Kerdsin A, Thammacharoen C, Sangvanich P, Vangnai AS (2008) A biosurfactant from BSP3 and its enhancement of pesticide solubilization. J Appl Microbiol 105(2):416–423
Wilke A, Bischof J et al (2016) The MG-RAST metagenomics database and portal. Nucleic Acids Res 44(D1):D590–D594. https://doi.org/10.1093/nar/gkv1322
Wilson DB (2009) Cellulases and biofuels. Curr Opin Biotechnol 20(3):295–299. https://doi.org/10.1016/j.copbio.2009.05.007
Wilson MC, Piel J (2013) Metagenomic approaches for exploiting uncultivated bacteria as are source for novel biosynthetic enzymology. Chem Biol 20(5):636–647. https://doi.org/10.1016/j.chembiol.2013.04.011
Wooley JC, Godzik A, Friedberg I (2010) A primer on metagenomics. PLoS Comput Biol. https://doi.org/10.1371/journal.pcbi.1000667
Wrighton KC, Thomas BC, Sharon I, Miller CS, Castelle CJ, VerBerkmoes NC, Wilkins MJ, Hettich RL, Lipton MS, Williams KH, Long PE, Banfield JF (2012) Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337(6102):1661–1665
Wu S, Zhu W, Fu L, Niu B, Li W (2011) WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics 12(1):444. https://doi.org/10.1186/1471-2164-12-444
Xu B, Liu L, Huang X, Ma H, Zhang Y, Du Y, Wang P, Tang X, Wang H, Kang K, Zhang S, Zhao G, Wu W, Yang Y, Chen H, Mu F, Chen W, Palacios G (2011) Metagenomic analysis of fever, thrombocytopenia and leukopenia syndrome (FTLS) in Henan province, China: discovery of a new bunya virus. PLoS Pathog 7(11):e1002369
Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M et al (2012) Human gut microbiome viewed across age and geography. Nature 486:222–227. https://doi.org/10.1038/nature11053
Yozwiak NL, Skewes-Cox P, Stenglein MD, Balmaseda A, Harris E, DeRisi JL, Rico-Hesse R (2012) Virus identification in unknown tropical febrile illness cases using deep sequencing. PLoS Negl Trop Dis 6(2):e1485
Zerbino DR, Velvet BE (2008) Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829. https://doi.org/10.1101/gr.074492.107
Zhou Q, Su X, Ning K (2014) Assessment of quality control approaches for metagenomic data analysis. Sci Rep 4:6957. https://doi.org/10.1038/srep06957
Zhu W, Lomsadze A, Borodovsky M (2010) Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38(12):e132. https://doi.org/10.1093/nar/gkq275
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Yadav, B.S., Yadav, A.K., Singh, S., Singh, N.K., Mani, A. (2019). Methods in Metagenomics and Environmental Biotechnology. In: Gothandam, K., Ranjan, S., Dasgupta, N., Lichtfouse, E. (eds) Nanoscience and Biotechnology for Environmental Applications. Environmental Chemistry for a Sustainable World, vol 22. Springer, Cham. https://doi.org/10.1007/978-3-319-97922-9_4
Download citation
DOI: https://doi.org/10.1007/978-3-319-97922-9_4
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-97921-2
Online ISBN: 978-3-319-97922-9
eBook Packages: Earth and Environmental ScienceEarth and Environmental Science (R0)