
Abstract
The knowledge of the underlying aetiology of neonatal idiopathic hepatitis and the so-called “intrahepatic” cholestasis has been rapidly expanding in the last decade, and great advances in genetic testing have clarified that the vast majority of these conditions are monogenic liver disorders. Among those referred to as progressive familial intrahepatic cholestases (PFICs), the level of serum GGT is a good discriminant to guide the initial evaluation, being low/normal in Byler disease, BSEP deficiency, TJP2 deficiency, FXR deficiency and MYO5B deficiency, and increased only in MDR3 deficiency; however genetic testing is needed to reach a definite categorisation. In bile acid synthesis defects, normal serum bile acid is a clue to the diagnosis, although mass spectrometry is required to characterise the type of defect. Other well-known conditions such as Alagille syndrome and alpha-1 antitrypsin deficiency are more common and less challenging to recognise. In this chapter we discuss the clinical features of canalicular transport and tight junction defects, bile acid synthesis defects, biliary developmental defects and metabolic disorders that can present with neonatal/infantile cholestasis, providing also a rational approach to the diagnosis of the rarest forms as well as information on their current standard of care.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Familial cholestasis
- Alagille syndrome
- Progressive familial intrahepatic cholestasis (PFIC)
- Inborn errors of bile acid synthesis
- Alpha-1 antitrypsin deficiency
- Galactosemia
- Tyrosinemia
- Congenital defects of glycosylation
- Mitochondrial cytopathies
- Liver transplantation
-
A large proportion of cholestatic disorders are caused by monogenic defects affecting the hepatocellular or biliary functions responsible for bile formation.
-
Several genetic diseases can cause transient or permanent cholestasis, including canalicular transport and tight junction defects, bile acid synthesis defects, biliary developmental defects and metabolic disorders.
-
In an infant with conjugated jaundice, once biliary atresia is ruled out, the most likely aetiology is a monogenic defect, nowadays recognisable by next-generation sequencing.
-
In the last decade, several new defects causing intrahepatic cholestasis have been discovered, such as TJP2 deficiency, FXR deficiency and MYO5B deficiency.
-
Although the diagnostic yield in this scenario is remarkably improved, no effective treatment is available for most genetic cholestatic disorders, making them candidates to some 20% of all liver transplants performed in children.
-
To improve the diagnostic yield of genetic testing for cholestatic diseases through widely adopted next-generation sequencing panels and protocols
-
To develop strategies able to modify the phenotype of these disorders, by gene therapy/editing, cell therapy, RNA interference, chaperones, etc., to avoid the progression to end-stage liver disease
-
To test different forms of medical and surgical biliary diversion, especially in conditions that are only partially corrected by liver transplantation, such as PFIC1
1 Introduction
Neonatal and infantile cholestatic liver diseases are a group of diverse hepatobiliary disorders characterised by the retention of bile components into hepatocytes and/or bile ducts, ultimately flowing back to the bloodstream. Excluding the extrahepatic causes, represented by biliary atresia or other biliary obstructive diseases, the knowledge about the underlying aetiology of the so-called “intrahepatic” cholestasis has been rapidly expanding in the last four decades. The great advances in molecular genetics have clarified that the vast majority of these conditions are monogenic liver disorders. Here the different genetic causes of cholestasis will be described and classified on the basis of the pathogenesis and of the clinical context in which they occur.
2 Genetic Cholestatic Disorders
2.1 Defects of the Biliary Canalicular Transport
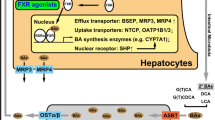
Progressive familial intrahepatic cholestasis (PFIC) is a group of heterogeneous autosomal recessive disorders causing hepatocellular cholestasis often presenting in the neonatal age or in the first year of life [1, 2]. With an incidence roughly comprised between 1:50,000 and 1:100,000, they represent about 9–12% of the causes of infantile cholestasis and a major indication for LT in children [3, 4]. Beyond the classically recognised PFIC1 (Byler disease), PFIC2 [Bile Salt Export Pump (BSEP) deficiency] and PFIC3 (MDR3 deficiency), high-throughput sequencing studies have recently identified new gene mutations associated with previously unexplained cases, thus expanding the disease spectrum to the so-called PFIC4 (TJP2 deficiency), PFIC5 (related to NR1H4) and MYO5B-associated cholestasis [5,6,7]. The different proteins and their role in canalicular transport are illustrated in Fig. 13.1.

Simplified view of the most important transporters of the biliary canaliculus. BSEP (encoded by ABCB11) provides high concentration of bile acids against gradient across the biliary pole plasma membrane; FIC1 (encoded by ATP8B1) is a flippase responsible for the aminophospholipid enrichment of the outer leaflet, protecting hepatocyte from the detergent action of bile acids; MDR3 (encoded by ABCB4) is the major phospholipid transporter, essential for bile micellar balance; TJP2 represent a connection between transmembrane claudins and actin cytoskeleton; MRP2 is a transporter for the organic anions, including conjugated bilirubin; FXR is a bile acid-sensitive nuclear receptor that enhances the expression of multiple genes including ABCB11; Myosin 5b is a cytoskeletal component directly involved in recycling BSEP-containing endosomes to the membrane maintaining polarity. Myo 5b myosin 5b, AL aminophospholipids, PC phosphatidylcholine, OA organic anions, BA bile acids. Courtesy of Mattia D’Antiga
2.1.1 Clinical Presentation
All these disorders usually present in the first months of life, with recurrent episodes of cholestatic jaundice that eventually become permanent [8,9,10,11]. Pruritus is common, often intractable, representing one of the indications to LT in later childhood. It has been reported that 15% of patients with PFIC1 and 44% of those affected by PFIC2 present by the first month of age [8]. All these entities invariably show progression to fibrosis and cirrhosis, but in the case of PFIC2, cholestasis is more severe, and the course of the disease appears to be more aggressive. Another feature of PFIC2 is the possible occurrence of early (in the first year of age) hepatocellular carcinoma (HCC) and, less commonly, cholangiocarcinoma.
Unlike the other forms, PFIC3 is usually diagnosed later in infancy and can also be identified during childhood or even in young adult age. In PFIC3 the main clinical feature is portal hypertension. Remarkably, since commonly these patients are not jaundiced and have milder pruritus, cirrhosis and its complications may not be overlooked until the age of 3 [9, 12].
In addition to cholestasis, PFIC1 is associated with other organ involvement, such as enteropathy, pancreatitis, elevated sweat electrolyte concentration, short stature and sensorineural deafness, underscoring the importance of evaluating the expression of FIC1 protein outside the liver, especially when it comes to listing to LT [13, 14].
Interestingly, milder mutations in PFIC1-PFIC2 genes (ATP8B1 and ABCB11) have been also associated with relapsing episodes of jaundice or pruritus called benign recurrent intrahepatic cholestasis (BRIC1-BRIC2) or with cholestasis of pregnancy [15].
2.1.2 Laboratory and Histological Findings
Typically, many PFICs show cholestasis with very high serum bile acids but normal GGT activity, while serum cholesterol is usually normal. PFIC2 is characterised by higher serum transaminases and alpha-fetoprotein. On the other hand, the biochemical features of the patients with PFIC3 are quite different: GGT is invariably elevated, and serum bile acids are moderately increased [16].
Liver biopsies from PFIC patients show almost invariably canalicular cholestasis and giant cell transformation, while ductular proliferation is generally absent or mild. Focal biliary metaplasia of the hepatocytes can be seen in periportal areas (Fig. 13.2a–c). Steatosis is occasionally noted in PFIC1 patients. Noteworthy, in PFIC2, the pronounced abnormalities of liver function tests are reflected by a more disrupted parenchymal structure, with lobular inflammation and early portal fibrosis. Also from the histologic point of view, PFIC3 behaves quite differently from the other familial cholestasis: ductular proliferation is seen at onset, and portal fibrosis is a feature, while giant cell transformation is mild. Other specific features of the different aetiologies are shown in Table 13.1.

Histological features of PFIC2. (a) Cirrhotic architecture with canalicular cholestasis (c) and focal giant cell transformation (arrows); (b) higher magnification showing pseudo-glandular arrangement or rosetting (*); (c) immunohistochemistry for cytokeratin 7 identifying biliary metaplasia of the hepatocytes; (d) nodule of hepatocellular carcinoma on cirrhosis in a PFIC2 patient at hepatectomy invading a blood vessel (arrowheads). Courtesy of Aurelio Sonzogni
2.2 Aetiology and Specific Features
2.2.1 PFIC1
PFIC1, also referred to as “Byler disease”, is caused by mutations in ATP8B1, encoding FIC1, a P-type ATPase acting as a “flippase”, responsible for the maintenance of the phosphatidylserine and phosphatidylethanolamine abundance of the inner leaflet of the plasma membrane. Such composition is crucial for the integrity of the canalicular membranes in presence of concentrated bile acids [17]. However, the definite pathogenesis of cholestasis in PFIC1 is unclear, and several mechanisms have been advocated. The abnormal plasma membrane structure is thought to indirectly determine an impairment in bile acids excretion, as witnessed by the very low primary bile acids that can be measured in the duodenal fluid after duodenal aspiration. This can be due to a different degree of downregulation of the farnesoid X receptor (FXR), which in turn is responsible for a reduced expression of BSEP and for a steady upregulation of the intestinal apical sodium bile salt transporter (ASBT) [18, 19]. Intriguingly, ATB8B1-deficient cells have an impaired apical localisation of the cystic fibrosis transmembrane conductance regulator (CFTR) that could contribute to the bile components retention and also account for some cystic fibrosis-like features of PFIC1. In fact, some extrahepatic manifestations include pancreatitis, abnormal chloride sweat content and even lung infections [1, 20]. ATP8B1 is widely expressed in the liver, pancreas, kidney and small intestine, where it can cause chronic diarrhoea through the alteration of the enterohepatic cycling of bile acids and where it may play a central role in the post-transplant PFIC1 allograft steatohepatitis and related metabolic complications [13]. Other extrahepatic features of the disease are short stature and sensorineural deafness [1, 21].
2.2.2 PFIC2
BSEP, encoded by the gene ABCB11, is the most important canalicular transporter, able to concentrate bile acids against extreme gradients. Its absence or reduced function can lead to bile acids retention and hepatocellular cholestasis [22]. BSEP disease spectrum is variable from BRIC to severe cholestasis with rapidly progressing chronic liver disease and possible risk of HCC (Fig. 13.2d). A genotype-phenotype correlation exists and explains part of the disease variability [23]. For instance, the two most common PFIC2-causing missense mutations of BSEP (D482G and E297G) exhibit a higher degree of impairment of the enzymatic activity and of the protein trafficking compared to those associated with a BRIC phenotype (such as A570T and R1050C). Alternatively, PFIC2-causing mutations may lead to milder, late-onset diseases if carried in compound heterozygosity with polymorphisms such as V444A. On the other hand, frameshift or protein-truncating mutations severely disrupt BSEP transport function and lead to the poorest membrane expression, and, consistently, they are associated with a poorer outcome and the highest risk of HCC [24, 25]. However, if protein-truncating mutations are likely to cause negative BSEP staining at liver biopsy, detectable BSEP expression does not preclude functional BSEP deficiency, whereas undetectable BSEP is seen also in other non-BSEP familial cholestasis, so that genetics has largely replaced immunohistochemistry for both diagnostic and prognostic purposes.
2.2.3 PFIC3
At the concentrations occurring in intrahepatic bile, bile acids could theoretically damage the apical membrane of the hepatocytes, but this is effectively counteracted by an enhanced cholesterol and phospholipid transport in the canalicular lumen [26]. The class III multidrug resistance of P-glycoprotein (MDR3, encoded by ABCB4) is the major phospholipid translocator enriching bile in phosphatidylcholine at the canalicular membrane. Loss of function of MDR3 results in inadequate phospholipid content with subsequent extracellular (canalicular) bile acid toxic effect. In addition, the imbalanced phosphatidylcholine content leads to unstable mixed micelles, favouring the cholesterol nucleation and the formation of gallstones. The result of these two phenomena is a cholangitis that represents the mechanism of liver damage in the PFIC3 [2]. PFIC3 could present with very different pictures, from neonatal cholestasis to cirrhosis in young adults. Histologically, it can mimic sclerosing cholangitis, since signs of biliary obstruction can be seen at liver biopsy and cholangiography may show abnormalities of the biliary tree [16].
2.2.4 FXR Deficiency (NR1H4-Associated Cholestasis, PFIC5)
The farnesoid X receptor (FXR, encoded by NR1H4) is a hepatocyte bile acid-sensitive nuclear receptor, involved in several hepatocyte metabolic pathways, especially biliary homeostasis. In response to high bile acids concentration, FXR suppresses bile acids biosynthesis and uptake and increases their export [27, 28]. Homozygous mutations in NR1H4 causing loss of functions of FXR have been described in a few patients presenting with low-GGT cholestasis very early in life, usually in the first month after birth or at birth with ascites, pleural effusion and coagulopathy, very high alpha-fetoprotein (trending down with time) and rapid progression to liver failure [6]. The hallmark of FXR deficiency is coagulopathy that is invariably present and is disproportionate to the liver injury. This can be explained by the fact that FXR directly regulates coagulation and complement factors. Since also BSEP expression is regulated by FXR, it is not surprising that BSEP staining was negative in all the described patients. Interestingly, some patients displayed post-transplant graft dysfunction and steatosis that could be related to the decreased production of FGF19, an intestinal growth factor that in normal conditions sends a negative feedback to the bile acid synthesis in the liver.
2.2.5 MYO5B-Associated Cholestasis
Myosin 5b (encoded by MYO5B) is a protein involved in plasma membrane recycling and transcytosis through its protein-to-protein interaction with Rab8a and Rab11a, essential for the polarisation of different epithelial cells and for the targeting of some transmembrane transporters, including CFTR and BSEP [29,30,31,32]. Until recently, the genetic defect in human pathology was known for its role as the cause of an enterocyte structural defect causing microvillous inclusion disease, an autosomal recessive disorder characterised by congenital diarrhoea often leading to intestinal failure and need for long-term parenteral nutrition. These children tend to develop cholestasis with unexpectedly low GGT and intractable pruritus, occurring before or even after intestinal transplantation, with negative BSEP staining at histology [33]. Recently, it has been clarified that mutations in MYO5B can cause isolated cholestasis, accounting for up to 20% of the neonatal cholestasis of previously indeterminate aetiology. Although the disease seems to invariably present in the first year of age, cholestasis can be transient, recurrent or persistent and progressive. Clinical features are similar to other low-GGT PFICs, with elevated serum bile acids, mild to moderate transaminases elevation and very low primary bile acids in duodenal aspiration, while BSEP and MDR staining can be absent or displaced to cytoplasm [7, 34]. These features overlap with those of PFIC2, but alpha-fetoprotein has been described as normal in these patients. MYO5B mutations causing isolated cholestasis are usually single nucleotide variants causing a single amino acid change, whereas those causing microvillous inclusion disease are commonly protein-truncating or disrupting the binding with Rab11a [7].
2.3 Tight Junction Defects
Tight junctions are the most apical cell junction complexes that determine cell polarity and create a barrier preventing and regulating the paracellular diffusion of water or small proteins. These complexes are made of transmembrane components (claudins, occludins and junction adhesion molecules) and cytosolic components such as tight junction proteins that act binding transmembrane proteins to the actin cytoskeleton [35] (Fig. 13.1). Since the tight junction system is redundant in humans, the loss of function of one of its components is critical only in unusually hostile environment, such as the canalicular membranes, which face high concentrations of detergent bile acids [36].
2.3.1 TJP2 Deficiency (PFIC4)
Homozygous mutations in TJP2 abolishing protein translation cause isolated, low-GGT, intrahepatic cholestasis that is indistinguishable from the classical PFICs on clinical ground [5]. The patients affected by this new entity present with cholestatic jaundice by the third month of life and almost invariably progress to cirrhosis needing LT; only a few of those described so far remained with stable cholestatic liver disease. Histology is similar to other forms of intrahepatic cholestasis, BSEP staining is maintained, while Claudin-1 (the major transmembrane protein binding TJP2) fails to localise at the canalicular membrane, although its expression is normal. Abnormal tight junctions can be observed at electron microscopy. Interestingly, missense TJP2 mutations were previously known to be associated with familial Amish hypercholanemia (a disorder characterised by pruritus and fat-soluble vitamins malabsorption) in a supposed oligogenic inheritance with coexistent homozygous BAAT missense mutations [37] and with autosomal dominant non-syndromic hearing loss in Korean families [38].
2.3.2 NISCH Syndrome
Neonatal sclerosing cholangitis is a severe cholangiopathy that presents early in life with a picture mimicking biliary atresia but with patent, although abnormal, bile ducts. In the very few cases described, this picture results from a rare autosomal recessive condition associated with neonatal ichthyosis (neonatal ichthyosis and sclerosing cholangitis, (NISCH)), caused by mutations in CLDN1 encoding Claudin-1. The majority of the families described are of Moroccan origin. Cholestasis usually presents in the first months of life with increased GGT, and dermatologic features are better appreciated later in life (Fig. 13.3a, b). Disproportionate pruritus, alopecia/hypotrichosis and ichthyosis are almost invariably present, and enamel abnormalities and hypodontia can be adjunctive features. Hepatocellular and canalicular cholestasis can be accompanied by a variable degree of portal fibrosis and ductular proliferation at liver biopsy (Fig. 13.3c). Cholangiography shows patent but abnormal biliary tree [39].

(a) Alopecia and (b) ichthyosis in a 6-month-old NISCH patient; (c) liver histology with interlobular septa, moderate inflammatory infiltrate and absence of interlobular bile ducts. NISCH neonatal ichthyosis sclerosing cholangitis. Courtesy of Massimiliano Paganelli
2.4 Bile Acid Synthesis Defects (BASD)
Synthesis of bile acids from cholesterol involves at least 14 enzymatic reactions. The mechanism of disease consists not only in a reduced bile flow but also in the lack of negative feedback operated by cholic and chenodeoxycholic acid on the FXR, so that hepatocytes continue to metabolise cholesterol leading to the accumulation of abnormal and toxic intermediates, ultimately determining cholestasis, fat-soluble vitamin malabsorption and, in some subtypes, neurological impairment. BASDs account for only 1–2% of all paediatric liver diseases, with a prevalence in Europe of 1–9/1,000,000, but their identification is compelling due to the availability of medical treatment [40, 41].
Of note, when BASDs present in infancy, generally with low-GGT cholestasis, pruritus is nearly absent, since there are no circulating primary bile acids. However these disorders can be diagnosed in older children or young adults presenting with neurological symptoms due to fat-soluble vitamin malabsorption. Their biochemical hallmark is the cholestasis with low serum bile acids, since standard assays detect primary bile acids (cholic and chenodeoxycholic acid) but not bile acids precursors and sterols. The detection of metabolites characterising BASDs relies on mass spectrometry. The metabolic pathway leading to endogenous primary bile acid synthesis is illustrated in Fig. 13.4.

Schematic representation of the endogenous primary bile acid synthesis. The classical pathway is represented in green, while the alternative acidic and 25-hydroxylase pathways are shown in blue and grey, respectively. In the grey boxes, the enzymes whose loss of function is associated with bile acid synthesis defects (BASDs) are indicated. The hatched red area indicates enzymatic activities requiring peroxisomal integrity. CA cholic acid, CDCA chenodeoxycholic acid, THCA 3α,7α,12α-trihydroxy-5β-cholestanoic acid
2.5 Aetiology and Specific Features
2.5.1 3β-Hydroxysteroid-C27-Steroid Oxidoreductase Deficiency (3β-HSD Deficiency)
This is the most frequent BASD, caused by the impairment of the conversion of 7α-hydroxycholesterol to 7α-hydroxy-4-cholesten-3-one. The onset is in neonatal age with low-GGT cholestasis, hypertransaminasemia, hepatomegaly with or without splenomegaly and fat-soluble vitamin malabsorption [42]. Liver biopsy is characterised by hepatocellular cholestasis and giant cell transformation. The phenotype is very heterogeneous: while the majority of the affected infants show progression to cirrhosis in absence of treatment, some of the patients transiently resolve their jaundice to present later on with a more severe derangement. Nowadays, some cases are identified investigating chronic cholestasis or cirrhosis of indeterminate aetiology.
The signature of the defect is the presence of sulphate and glycosulfate conjugates of the 3β-hydroxy-Δ5 bile acids at urine or blood mass spectrometry, while genetic testing can find mutations in the HSD3B7 gene (Fig. 13.5).

Liquid chromatography-tandem mass spectrometry spectra of full scan obtained from a patient with bile acid synthesis defect at the time of diagnosis, compared with spectra obtained from the urine of a normal control. Peaks A–D correspond to metabolites diagnostic for 3β-HSD deficiency. Adapted from [91]
2.5.2 Δ4-3-Oxosteroid 5β-Reductase Deficiency
This enzymatic defect causes the loss of conversion of the intermediates 7α-hydroxy-4-cholesten-3-one and 7α,12α dihydroxy-4-cholesten-3-one to the corresponding 3-oxo-5β(H) intermediates. Clinically it is very similar to 3β-HSD deficiency, but it presents at a younger age and is characterised by a more severe course [Clayton 1988]. Of note, GGT is elevated, while cytolysis and jaundice are remarkable. Coagulopathy is often a feature, in some cases resembling neonatal hemochromatosis [43]. Histology reveals a substantial parenchymal disturbance, with giant cell transformation, hepatocellular cholestasis and, uniquely, absent or slit-like biliary canaliculi [44]. The latter feature is thought to be a consequence of the toxicity of the relatively insoluble Δ4-3-oxo bile acids that represent the main pointer to the diagnosis when detected by urine mass spectrometry. The diagnosis can be genetically confirmed by sequencing of AKR1D1.
2.5.3 Cerebrotendinous Xanthomatosis (27-Hydroxylase Deficiency)
The name of this condition immediately evokes the presentation in adulthood, characterised by xanthomas of the brain and the tendons and by cognitive deterioration, with ataxia and, sometimes, cataracts. With the refinement of the diagnostic techniques, it has been clarified that this condition, caused by the loss of 27-sterol-hydroxylase activity (i.e. the entry step of the classical bile acid synthetic pathway), can present with a picture of neonatal cholestasis that can progress to liver failure or alternatively resolve, even spontaneously [45,46,47,48]. Since primary bile acids, especially cholic acid, can be synthesised via the alternative 25-hydroxylase pathway, total serum bile acids can be quite elevated and GGT not as low as in other BASDs. As a result of the enzymatic defect and of the increased cholesterol biosynthesis in presence of an impairment of the side chain oxidation, several bile acid glucuronides and 5α-cholestan-3β-ol (cholestanol) accumulate and can be detected by mass spectrometry.
2.5.4 Bile Acid Amidation Defects (BACL and BAAT Deficiency)
Two defects are responsible for the lack of bile acid conjugation with the amino acids glycine and taurine: bile acid-CoA ligase (BACL) deficiency and bile acid-CoA:amino acid N-acyltransferase (BAAT) deficiency. Both defects can present with low-GGT and mild cholestasis of neonatal onset [49] and almost invariably show marked fat-soluble vitamin malabsorption, but patients with BAAT deficiency have been also described to present with marked coagulopathy and no jaundice. The detected spectrum on urine mass spectrometry invariably shows the lack of conjugated primary bile acids. Recently, the treatment with glycocholic acid has been reported as safe and effective to control cholestasis and improve malabsorption of amidation defects [50].
2.5.5 2-Methylacil-CoA Racemase Deficiency
Only few patients have been described with this defect that allows the critical step of formation of the 25S-enantiomer of the 13α,7α,12α-trihydroxy-5β-cholestanoic acid (THCA). Such patients have been identified in adult age because of fat-soluble vitamin malabsorption or in infancy for cholestatic liver disease. The hallmark metabolites are the cholestanoic acids that cannot enter the peroxisomal oxidation.
2.5.6 Oxisterol-7α-Hydroxylase Deficiency
This defect has been described in only two children [51, 52]. The enzyme belongs to the alternative acidic bile acid synthesis pathway that seems to be important early in life. Of note, liver biopsy, in addition to the features commonly observed in BASDs, shows portal fibrosis and ductular proliferation. Typically, 3β-monohydroxy-Δ5 bile acids are observed in urine and blood by mass spectrometry.
2.5.7 Secondary BASDs
In peroxisomal disorders—classically presenting at different ages with dysmorphic feature, neurological abnormalities and other organ involvement—the phenotype includes also a variable degree of liver disease due to the disruption of the organelle environment in which the transformation of the THCA into cholic acid takes place. Zellweger syndrome, infantile Refsum disease and neonatal adrenoleukodystrophy show substantial trihydroxycoprostanoic and dihydroxycoprostanoic acid amounts in urine [53,54,55].
Another disorder which typically presents in neonatal age with cholestasis due to impaired bile acid biosynthesis is Smith-Lemli-Opitz syndrome (SLOS) [56]. Due to the loss of function of 7-dehydrocholesterol Δ7-reductase catalysing the last step of cholesterol synthesis, SLOS patients lack the substrate for bile acid formation, since Δ7-sterols are poorly 7α-hydroxylated. The consequent cholestasis complicates a multi-organ picture with a poor prognosis characterised by dysmorphisms; microcephaly; failure to thrive; limb, cardiac, renal and endocrine abnormalities; cataracts; and mental retardation.
2.6 Trafficking and Canalicular Targeting Defects
2.6.1 Arthrogryposis-Renal Dysfunction-Cholestasis (ARC) Syndrome
ARC syndrome is a rare autosomal recessive multisystem disorder, characterised by the presence of arthrogryposis, renal tubular acidosis and neonatal cholestatic jaundice [1]. Other possible accompanying features can be ichthyosis (~50%), platelet anomalies (~25%), agenesis of the corpus callosum (>20%), congenital cardiovascular anomalies (~10%), deafness and recurrent infections [57]. Two genes are involved in its pathogenesis, VPS33B and the one encoding its interacting protein VIPAR [58]. VPS33B protein is involved in vacuolar sorting and in the intracellular vesicular trafficking pathways, including vesicular exocytosis, synaptic transmission and general secretion. Its loss of function causes abnormal localisation or accumulation of plasma proteins in polarised cells [59]. VIPAR interacts with VPS33B and is essential to maintain polarity and apical membrane protein restriction [60]. In these newborns, cholestasis is characterised by low GGT and slightly increased transaminases, but jaundice and pruritus are marked. Treatment is supportive and the prognosis is generally poor.
2.7 Biliary Developmental Defects
2.7.1 Alagille Syndrome
Alagille syndrome (AS) is the most common monogenic cause of neonatal cholestasis, with an estimated incidence of 1:30,000. AS is an autosomal dominant multisystem disorder caused by the loss of Notch2 signalling that is crucial for developmental processes of many organs and tissues, specifically caused by mutations in JAG1 or NOTCH2 (~94% and ~2% of the cases, respectively) [61]. The syndrome is defined by a paucity of intrahepatic bile ducts associated with at least three of the following: cholestasis, cardiac disease (most commonly peripheral pulmonary artery stenosis), skeletal abnormalities (“butterfly vertebras”), ocular abnormalities (embriotoxon) and peculiar facial features (triangular face, pointed chin, prominent forehead, hypertelorism) [62]. Beyond these diagnostic criteria, affected children usually have short stature [63] and may present brain vascular anomalies at risk of bleeding or stroke (including “moyamoya” disease) [64], as well as renal dysfunction in up to 40% (Fig. 13.6) [65]. Cholestasis arises from bile duct paucity. Whatever the gene involved, the pathogenesis of ductopenia has been eminently reviewed in a recent article [66]: Jag1 protein expression in the embryo portal vein mesenchyme initiates the bile duct development, interacting with Notch2 to induce the expression of Hes1, HNF1β and Sox9, which in turn promote and regulate ductal plate and intrahepatic bile duct morphogenesis [67,68,69]. Interestingly, Notch2 is essential for prenatal but not for secondary bile duct formation, which could explain the partial recovery of cholestasis during infancy of some patients [70, 71]. Hepatic manifestations are conjugated hyperbilirubinemia with high GGT, increased serum bile acids and striking elevation in serum cholesterol and triglycerides, with early development of xanthomas. Additional features are failure to thrive and malabsorption. Cholestatic pruritus is often the main problem in children with AS, and, if intractable, it may represent an indication for LT. Histology classically shows intrahepatic bile duct paucity, but in biopsies taken in the first few months of life, ductal proliferation may be the dominant feature, leading to a possible misdiagnosis of biliary atresia. The diagnosis of AS is clinical and histological and nowadays requires genetic confirmation. Genetic testing has an important role in incomplete phenotypes that are very common in this condition and easily overlooked. Cholestasis is unremitting and progresses to cirrhosis and liver insufficiency in about 15–20% of infants, who then require LT early in life. A multi-organ evaluation is needed before listing these patients for LT, in order to assess the risk related to cardiac, renal and vascular abnormalities [72].

(a) Facial and (b) body features of Alagille syndrome. PS pulmonary stenosis, CoAo aortic coarctation
2.7.2 Neonatal Sclerosing Cholangitis Related to DCDC2
In 2016 DCDC2 (encoding the Doublecortin domain-containing protein 2) was found to cause a proportion of cases of neonatal sclerosing cholangitis that had no mutations in CLDN1 [73, 74]. The encoded protein is highly expressed as a component of cholangiocyte primary cilia, which, in affected children, fails to localise in this structure but is found in the cytoplasm. This change is supposed to cause liver disease by modifying the bile composition and/or reducing the protection of the epithelium against the bile and by disrupting the microtubular structure of the biliocyte. From the point of view of the pathogenesis, this entity should be classified as a ciliopathy, but, due to the peculiar presentation, DCDC2 disease should be considered a monogenic cholestatic disorder rather than a type of fibrocystic disease of the liver. Affected children present with cholestatic jaundice and pale stools, hepatomegaly and high GGT. The liver biopsy shows features consistent with biliary atresia such as ductular reaction and portal fibrosis, but cholangiography reveals an irregular but patent biliary tract. No specific treatment exists for these children, and LT is the only option.
2.8 Metabolic Diseases Secondarily Causing Cholestasis
2.8.1 Carbohydrate Metabolism Defects
Some defects of the carbohydrate metabolism can affect the liver early in life and are briefly treated in this chapter because, even if they do not directly impair bile components production and transport, they can present in neonatal or infantile period with cholestasis. Most of them are addressed in the chapter addressing the “practical approach to the jaundiced infant”.
Galactosemia nowadays is often diagnosed in asymptomatic children, in countries that implemented the universal newborn metabolic screening. The undiagnosed patient with a classical phenotype (related to GALT mutations that almost completely abolish the GALT enzyme activity) presents after the introduction of galactose in the diet with signs of hepatocellular damage, raised transaminases, jaundice (70%), hepatomegaly (40%) and coagulopathy, accompanied by vomiting, failure to thrive and frequent bacterial infections, typically Escherichia coli sepsis. If a galactose-free diet is not rapidly established, liver failure rapidly ensues.
Transaldolase deficiency (TALDO) is a rare disorder of the pentose phosphate pathway identified in 2001, caused by mutations in TALDO1 [75]. The lack of enzymatic activity leads to the accumulation of sugar phosphates and abnormal polyols (erythritol, d-arabitol and ribitol) in several tissues. The sugar phosphates are thought to be responsible for liver injury that can present in newborns but also with unexplained cirrhosis by 3 years of age. Hepatomegaly is almost invariably present, sometimes accompanied by splenomegaly, while liver function tests show elevated transaminases and hyperbilirubinemia in about 60% and 36% of the patients, respectively. At diagnosis, a synthetic derangement can be present in about a half of the children, and progression to cirrhosis and HCC or liver failure has been described. The systemic nature of the disease is revealed by the presence of other symptoms: dysmorphism (cutis laxa, triangular face, low-set ears, thin lips), congenital cardiac defects (aortic coarctation, bicuspid aortic valve, ventricular or atrial septum defects), renal tubular dysfunction and hematologic abnormalities such as thrombocytopenia, developmental delay and gonadal dysfunction. Treatment is supportive, while N-acetylcysteine has been used to counteract the depletion of NADP. LT is an option in advanced disease or HCC.
Congenital disorders of glycosylation (CDGs) are a group of metabolic diseases caused by abnormal protein or lipid glycosylation. Over the last 10 years, the number of CGDs has increased from about 40 to over 125 different entities, following the advent of next-generation DNA sequencing and especially with the use of whole exome sequencing [76]. CDGs are of difficult identification, since they have varied multisystem involvement. Liver involvement in CDGs occurs in approximately 20% of the known defects, and its severity is very variable. For the purposes of this chapter, only the forms with isolated or predominant liver involvement will be mentioned. MPI-CDG is caused by the lack of function of mannose phosphate isomerase, a cytosolic enzyme that catalyses the isomerisation of fructose-6-phosphate to mannose-6-phosphate as first step of the N-glycosylation [77]. Affected children present often in the postneonatal age with signs of liver dysfunction commonly associated with enteropathy, vomiting and failure to thrive. However, the picture is dominated by hepatomegaly and elevated transaminases rather than cholestasis, and liver fibrosis develops early and progressively unless mannose treatment is started promptly. Some cases present developmental abnormalities of the ductal plate, with a picture overlapping that of congenital hepatic fibrosis. CCDC115-CDG can present in the first months of life with cholestatic jaundice and hepatomegaly, failure to thrive, redundant skin, developmental delay and epilepsy. Transaminases are increased, GGT is not as elevated as alkaline phosphatase, ceruloplasmin is low when tested, and patients exhibit a type 2 CDG transferrin isoelectrofocusing profile [78]. The X-linked ATP6AP1 deficiency can present with neonatal jaundice, hepatomegaly and immunodeficiency with hypogammaglobulinemia, epilepsy, mild developmental delay and sensorineural deafness. In PMM2-CDG several dysmorphisms can be observed, including the hint of inverted nipples, and liver involvement is variable, ranging from mild hepatomegaly and hypertransaminasemia to severe disease leading to cirrhosis by the fourth month of age. COG-CDGs also can present with unremitting cholestasis and diverse degree of liver disease, with variably severe neurological involvement, as well as other organ involvement, and growth retardation of intrauterine onset.
2.8.2 Amino Acid Metabolism Defects
These diseases are more extensively treated in the Chap. 16. Here the conditions presenting predominantly with cholestasis will be briefly addressed.
Tyrosinemia type 1 (incidence 1:100,000) is caused by an impairment of the aromatic amino acid tyrosine (that is metabolised only in hepatocytes and proximal renal tubular cells) due to loss of the fumarylacetoacetate hydrolase activity (encoded by the FAH gene) [79]. The tissue damage is caused by the accumulation of the toxic intermediates fumarylacetoacetate (that is highly mutagenic) and succinylacetone (mitochondrial toxicity, hepatotoxicity), the latter being the major toxic and also a biochemical hallmark of the disease, easily found in the urine of the affected patients [80]. The disease course varies from acute to subacute and chronic forms, with jaundice, cytolysis, hepatomegaly and synthetic derangement, features predominantly characterising the neonatal presentation. Renal tubular dysfunction can be present. In the absence of treatment, the disease leads to cirrhosis and HCC. The diagnosis is easily made, if suspected, by the serum amino acid profile showing elevated tyrosine and methionine and by the urinary organic acid profile revealing high succinylacetone excretion. Low phenylalanine end tyrosine restriction diet is insufficient to prevent liver disease progression, while nitisinone, a compound inhibiting the upward step by the hydroxyphenylpyruvate dioxygenase, dramatically reduces, even though not completely, the metabolite toxicity and the oncogenic risk, especially if introduced before the sixth month of life. LT, required if HCC arises, is curative.
Neonatal intrahepatic cholestasis caused by citrin deficiency (NICCD) is the neonatal phenotype caused by the loss of function of the gene SLC25A13 encoding citrin, a mitochondrial aspartate glutamate carrier important for aerobic glycolysis and gluconeogenesis. The disease is more common in Eastern Asia. The picture is typically characterised by cholestasis (generally transient), aminoacidaemia (notably increased citrulline but also tyrosine, threonine, arginine and methionine) and galactosuria [81]. Hypoglycemia and failure to thrive coexist in most of the cases described, and evidence of clotting derangement can be present. This rare entity generally improves and completely resolves even spontaneously, but these patients may present later in young adulthood with neurological deterioration due to the secondary urea cycle defect or suddenly with hyperammonemic coma or hypoglycaemia. In these cases chronic liver dysfunction with steatosis may become evident [82]. A prompt diagnosis allows the initiation of a low-carbohydrate diet and helps in preventing the delayed complications.
2.9 Defects of the Cellular Energy Production
Mitochondrial disorders (MDSs) are mainly related to mutations in genes involved in the respiratory chain and have heterogeneous phenotypes, most commonly characterised by a defect of cellular energy production. Those presenting with predominant liver involvement are related to nuclear gene products (inherited with autosomal recessive pattern in 90% and autosomal dominant and X-linked pattern in 10%), responsible for mitochondrial DNA replication (POLG1), maintenance of deoxyribonucleoside triphosphate (dNTP) pools (DGUOK) and membrane mitochondrial integrity (MPV17) [83, 84]. Mutations in these genes cause mitochondrial DNA depletion syndromes, characterised by hepato-cerebral phenotypes, unlike those of maternal inheritance characterised by predominant neurological involvement. In addition to liver dysfunction, these children present variable degrees of hypotonia, severe developmental delay and failure to thrive; myoclonus and nystagmus are often present. Hypoglycemia, lactic acidosis and plasma amino acid profile consistent with a blocked energy production metabolism are clues to the diagnosis. In Alpers syndrome (due to mutations in POLG1) sodium valproate administered because of intractable seizures often triggers acute liver decompensation and death. The histological changes on liver biopsy include fatty degeneration, bile duct proliferation, fibrosis and lobular collapse. LT is contraindicated because of the irreversible neurological involvement [85].
2.10 Storage Diseases
The hepatic phenotype of alpha-1 antitrypsin (A1AT) deficiency is caused by homozygous mutations in the SERPINA1 gene, modifying the aggregation properties of the protein, so that loop-sheet polymers accumulate in the hepatocytes causing fibrosis and cirrhosis. Some affected children can present early in life with completely acholic stools and histological features mimicking biliary atresia. In most patients, a borderline or frankly low serum A1AT level is a good pointer to the diagnosis, although, since A1AT is an acute phase reactant, during inflammation its serum level can be found in the normal range, and the condition may be overlooked. At liver biopsy, characteristic PAS-positive, diastase-resistant hepatocyte inclusions suggest the aetiology, although these vacuoles tend to develop beyond the first few months of life, resulting an unreliable marker for an early diagnosis.
Lysosomal storage diseases (such as Niemann-Pick type C, Gaucher and Wolman disease) can present with neonatal cholestasis diseases, but hepatosplenomegaly is predominant. In particular, in neonatal/infantile cholestasis, the very early development of splenomegaly (when portal hypertension is very unlikely to be already established) should raise the suspicion of a lysosomal storage disease [86].
3 Diagnosis
In a child with neonatal/infantile cholestasis, the surgical causes (biliary atresia, choledochal cyst) should be sought first. If biliary atresia is ruled out, the picture is most likely related to a monogenic liver disease.
Careful physical examination may point out valuable features and symptoms, which might not be obvious in very young patients. Dysmorphisms are quite evident in ARC syndrome and in Zellweger disease spectrum. Triangular-shaped face, prominent forehead, pointed chin and thin hair suggest Alagille syndrome. Transaldolase deficiency, although rare, could be easily recognised by low hair implantation, hirsutism, cutis laxa, low-set ears and clitoridomegaly.
The serum GGT activity is a good discriminant to guide initial evaluation, unravelling at least part of the diagnostic conundrum. In a child with low or normal GGT for age, serum bile acids can point to a diagnosis of PFIC (when elevated) or to a much rarer BASD (if low). In case of a low-GGT PFICs (PFIC1, PFIC2, PFIC4 or TJP2 deficiency, PFIC5 or FXR deficiency, MYO5B deficiency) genetic testing is needed to reach a definite diagnosis. If a BASD is suspected, a screening procedure through urine tandem gas chromatography-mass spectrometry is preferred to characterise the type of defect, and a trio genetic testing can be used for confirmation and for counselling purposes.
In all the other cases, in which GGT is high, the differential diagnosis is rather wide. Serum A1AT is a useful screening for A1AT deficiency, and the diagnosis is confirmed looking for the SERPINA1 Z-allele mutations. A metabolic screening with urinary galactose, plasma amino acid profile, urinary succinylacetone and organic acids, lactic acid and blood gas analysis can rule out the most common metabolic causes.
When there are no clear pointers to a metabolic and genetic defect, or to confirm a suspected diagnosis, physical or virtual gene panels by next-generation sequencing are the best method to achieve a definite diagnosis in this setting [87].
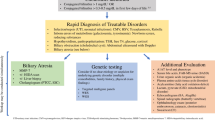
In Fig. 13.7 we report a diagnostic algorithm that implements the use of genetic testing in such scenario.

Proposed algorithm for the diagnostic workup of cholestatic infants. A1ATD alpha-1 antitrypsin deficiency, CF cystic fibrosis, GGT gamma-glutamyl transpeptidase, NICCD neonatal idiopathic cholestasis due to citrin deficiency, PFIC progressive familial intrahepatic cholestasis, NP-C Niemann-Pick type C disease, ARC arthrogryposis renal dysfunction and cholestasis syndrome, TALDO transaldolase deficiency, *In infants with acholic stools, A1AT should be tested in serum before Kasai portoenterostomy, whereas its genetic testing is included in the NGS panel we use in children with cholic stools
4 Treatment
The primary goal for the treatment of familial/genetic cholestatic diseases is the control of pruritus and possibly of the cholestasis itself. The currently available standard medical treatment is based on UDCA, which replaces the more toxic endogenous bile acids and has choleretic properties increasing the bile flow. Treatment of pruritus is based on symptomatic drugs, such as rifampicin, naloxone/naltrexone and cholestyramine, and antihistaminic drugs. Nutritional support includes fat-soluble vitamin and MCT supplementation.
In case of medical treatment failure, in patients with preserved liver synthetic function and without substantial fibrosis, surgical biliary diversion becomes the first choice. Biliary diversion can be total or partial, external (cholecysto-jejunostomy, button cholecystostomy, less frequently cholecysto-appendicostomy) and internal (generally isoperistaltic jejunal loop connecting the gallbladder and colon; ileal bypass). Whatever the type of surgery, the mechanism of action is based on the reduction of circulating bile acid pool. The overall success rate of these interventions mainly depends on the patients’ selection. Patients with PFIC1 have the greatest benefit, while those with PFIC2 should be stratified according to the genotype: patients with at least one mild mutation (p.D482G and p.E297G) are most likely to respond and bear less risk to develop hepatocellular carcinoma. With these cautions, the event-free survival of PFIC patients under biliary diversion increases from 40% to more than 80% [10, 88, 89].
Patients with Alagille syndrome have also been treated with biliary diversion, but the benefit has been much lower than in PFICs, since the progression of fibrosis in presence of uncontrolled cholestasis is the rule [90].
Coming to the new drugs, two bile acid reuptake inhibitors are in the pipeline. Both products (inhibiting the ileal bile acid transporter and the apical sodium-dependent bile acid transporter, respectively) are being tested in phase 3 studies in PFIC1-PFIC2 patients, with the rationale of realising a “medical biliary diversion”, depleting the bile acid circulating pool.
Patients with BASDs usually benefit from the administration of primary bile acids, and the specific treatment is described in Table 13.1. Of note, administered primary bile acids bear potential toxic effects; therefore they should be given at the minimum dose sufficient to produce a negative feedback to bile acid synthesis. The required dose of primary bile acid (usually much lower than that used in other cholestatic disorders) can be titrated monitoring the abnormal intermediates in the urine by mass spectrometry [91] (Fig. 13.8).

Titration of bile acid supplements based on the level of the toxic metabolite 3β,7α-dihydroxy-5-cholenoic acid (3β-D-OH-5C) in the urine of two patients affected by 3βHSD. UDCA reduces but does not normalise the level of toxic metabolites, whereas CDCA (and cholic acid) does. CDCA chenodeoxycholic acid, UDCA ursodeoxycholic acid. Adapted from [91]
5 Implications for Liver Transplantation
The following situations represent an indication to list the patients affected by genetic cholestatic disorders to LT: the uncontrolled cholestasis in presence of severe fibrosis and cirrhosis, the suspected or confirmed hepatocellular carcinoma and the failure of previous surgical biliary diversion. Overall, PFICs and Alagille syndrome account for some 20% of paediatric LT (http://www.eltr.org/Pediatric-transplantation.html), with overall excellent outcomes. However, specific transplant-related issues must be addressed.
In patients transplanted for PFIC1, the liver allograft can become affected by severe steatosis possibly progressing to steatohepatitis and cirrhosis with graft failure [92, 93]. Since this happens almost invariably in correspondence of exacerbation of diarrhoea, it is very likely that the allograft disease be the result of the interaction between the native bowel and the liver graft by diverse mechanisms. In fact, biliary diversion has been successfully used to treat diarrhoea in these patients, permanently improving graft steatofibrosis and function [94] (Fig. 13.9). The severity of post-LT steatohepatitis is a further reason to consider these patients for a primary biliary diversion before listing them for LT or at the time of LT.

Liver biopsies of a PFIC1 patient developing graft steatohepatitis 6 (a) and 24 months (b) after LT and 6 months after EBD (30 months after LT; c). Both upper and lower pictures show the diffuse macrovacuolar steatosis and the worsening fibrosis beginning from the portal tracts (*) (a, b). Six months after EBD, a dramatic improvement of the steatosis can be observed (c). LT liver transplantation, EBD external biliary diversion. Adapted from [94]
A recurrent post-LT BSEP deficiency, due to the development of anti-BSEP antibodies, has been well described in PFIC2 patients [95]. Such condition has been proved to be complement-mediated, associated with C4d deposition detectable at liver histology, and to improve under rituximab treatment [96].
In Alagille syndrome patients, the pre-LT checklist should routinely encompass a careful cardiological evaluation and a brain angio-MR. In fact, pulmonary obstruction due to stenosis and possible right ventricular dysfunction, as well as the described brain vascular abnormalities, accounts for not negligible transplant morbidity and mortality.
References
Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. Progressive familial intrahepatic cholestasis. Orphanet J Rare Dis. 2009;4(1):1. BioMed Central.
Jacquemin E. Progressive familial intrahepatic cholestasis. Clin Res Hepatol Gastroenterol. 2012;36(Suppl 1):S26–35.
Fischler B, Papadogiannakis N, Nemeth A. Aetiological factors in neonatal cholestasis. Acta Paediatr. 2007;90(1):88–92. Wiley/Blackwell (10.1111).
Kamath BM, Chen Z, Romero R, Fredericks EM, Alonso EM, Arnon R, et al. Quality of life and its determinants in a multicenter cohort of children with Alagille syndrome. J Pediatr. 2015;167(2):390–3.
Sambrotta M, Strautnieks S, Papouli E, Rushton P, Clark BE, Parry DA, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46(4):326–8.
Gomez-Ospina N, Potter CJ, Xiao R, Manickam K, Kim M-S, Kim KH, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:10713. Nature Publishing Group.
Qiu Y-L, Gong J-Y, Feng J-Y, Wang R-X, Han J, Liu T, et al. Defects in myosin VB are associated with a spectrum of previously undiagnosed low γ-glutamyltransferase cholestasis. Hepatology. 2017;65(5):1655–69.
Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51(5):1645–55.
Mehaidib Al A, Shahrani Al A. 1381 progressive familial intrahepatic cholestasis in ARABS. J Hepatol. 2013;58:S555–6.
Englert C, Grabhorn E, Richter A, Rogiers X, Burdelski M, Ganschow R. Liver transplantation in children with progressive familial intrahepatic cholestasis. Transplantation. 2007;84(10):1361–3.
Wanty C, Joomye R, Van Hoorebeek N, Paul K, Otte JB, Reding R, et al. Fifteen years single center experience in the management of progressive familial intrahepatic cholestasis of infancy. Acta Gastroenterol Belg. 2004;67(4):313–9.
Jacquemin E. Role of multidrug resistance 3 deficiency in pediatric and adult liver disease: one gene for three diseases. Semin Liver Dis. 2001;21(4):551–62. Copyright © 2001 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA. Tel.: +1(212) 584-4662.
Lykavieris P, van Mil S, Cresteil D, Fabre M, Hadchouel M, Klomp L, et al. Progressive familial intrahepatic cholestasis type 1 and extrahepatic features: no catch-up of stature growth, exacerbation of diarrhea, and appearance of liver steatosis after liver transplantation. J Hepatol. 2003;39(3):447–52.
Demeilliers C, Jacquemin E, Barbu V, Mergey M, Paye F, Fouassier L, et al. Altered hepatobiliary gene expressions in PFIC1: ATP8B1 gene defect is associated with CFTR downregulation. Hepatology. 2006;43(5):1125–34. Wiley-Blackwell.
Pauli-Magnus C, Meier PJ. Hepatobiliary transporters and drug-induced cholestasis. Hepatology. 2006;44(4):778–87. Wiley-Blackwell.
Davit-Spraul A, Gonzales E, Baussan C, Jacquemin E. The spectrum of liver diseases related to ABCB4Gene mutations: pathophysiology and clinical aspects. Semin Liver Dis. 2010;30(2):134–46. © Thieme Medical Publishers.
Paulusma CC, Groen A, Kunne C, Ho-Mok KS, Spijkerboer AL, Rudi de Waart D, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44(1):195–204. Wiley-Blackwell.
Chen F, Ananthanarayanan M, Emre S, Neimark E, Bull LN, Knisely AS, et al. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology. 2004;126(3):756–64.
Alvarez L, Jara P, Sánchez-Sabaté E, Hierro L, Larrauri J, Diaz MC, et al. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum Mol Genet. 2004;13(20):2451–60.
van der Mark VA, de Jonge HR, Chang J-C, Ho-Mok KS, Duijst S, Vidović D, et al. The phospholipid flippase ATP8B1 mediates apical localization of the cystic fibrosis transmembrane regulator. Biochim Biophys Acta. 2016;1863(9):2280–8.
Egawa H, Yorifuji T, Sumazaki R, Kimura A, Hasegawa M, Tanaka K. Intractable diarrhea after liver transplantation for Byler’s disease: successful treatment with bile adsorptive resin. Liver Transpl. 2002;8(8):714–6.
Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203–14.
Thompson R, Strautnieks S. BSEP: function and role in progressive familial intrahepatic cholestasis. Semin Liver Dis. 2001;21(4):545–50. Copyright © 2001 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA. Tel.: +1(212) 584-4662.
Kagawa T, Watanabe N, Mochizuki K, Numari A, Ikeno Y, Itoh J, et al. Phenotypic differences in PFIC2 and BRIC2 correlate with protein stability of mutant Bsep and impaired taurocholate secretion in MDCK II cells. Am J Physiol Gastrointest Liver Physiol. 2008;294(1):G58–67. American Physiological Society.
Lam P, Pearson CL, Soroka CJ, Xu S, Mennone A, Boyer JL. Levels of plasma membrane expression in progressive and benign mutations of the bile salt export pump (Bsep/Abcb11) correlate with severity of cholestatic diseases. Am J Physiol Cell Physiol. 2007;293(5):C1709–16. American Physiological Society.
Amigo L, Mendoza H, Zanlungo S, Miquel JF, Rigotti A, González S, et al. Enrichment of canalicular membrane with cholesterol and sphingomyelin prevents bile salt-induced hepatic damage. J Lipid Res. 1999;40(3):533–42.
Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013;368(1–2):17–29.
Kuipers F, Bloks VW, Groen AK. Beyond intestinal soap—bile acids in metabolic control. Nat Rev Endocrinol. 2014;10(8):488–98. Nature Publishing Group.
Lapierre LA, Kumar R, Hales CM, Navarre J, Bhartur SG, Burnette JO, et al. Myosin Vb is associated with plasma membrane recycling systems. Guidotti G, editor. Mol Biol Cell. 2001;12(6):1843–57.
Roland JT, Kenworthy AK, Peranen J, Caplan S, Goldenring JR. Myosin Vb interacts with Rab8a on a tubular network containing EHD1 and EHD3. Brennwald P, editor. Molecular Biology of the Cell. 2007;18(8):2828–37.
Swiatecka-Urban A, Talebian L, Kanno E, Moreau-Marquis S, Coutermarsh B, Hansen K, et al. Myosin Vb is required for trafficking of the cystic fibrosis transmembrane conductance regulator in Rab11a-specific apical recycling endosomes in polarized human airway epithelial cells. J Biol Chem. 2007;282(32):23725–36. American Society for Biochemistry and Molecular Biology.
Wakabayashi Y, Dutt P, Lippincott-Schwartz J, Arias IM. Rab11a and myosin Vb are required for bile canalicular formation in WIF-B9 cells. Proc Natl Acad Sci U S A. 2005;102(42):15087–92.
Girard M, Lacaille F, Verkarre V, Mategot R, Feldmann G, Grodet A, et al. MYO5B and bile salt export pump contribute to cholestatic liver disorder in microvillous inclusion disease. Hepatology. 2014;60(1):301–10. Wiley-Blackwell.
Gonzales E, Taylor SA, Davit-Spraul A, Thébaut A, Thomassin N, Guettier C, et al. MYO5B mutations cause cholestasis with normal serum gamma-glutamyl transferase activity in children without microvillous inclusion disease. Hepatology. 2017;65(1):164–73.
Sawada N. Tight junction-related human diseases. Pathol Int. 2013;63(1):1–12. Wiley/Blackwell (10.1111).
Grosse B, Cassio D, Yousef N, Bernardo C, Jacquemin E, Gonzales E. Claudin-1 involved in neonatal ichthyosis sclerosing cholangitis syndrome regulates hepatic paracellular permeability. Hepatology. 2012;55(4):1249–59. Wiley-Blackwell.
Carlton VEH, Harris BZ, Puffenberger EG, Batta AK, Knisely AS, Robinson DL, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34(1):91–6. Nature Publishing Group.
Kim M-A, Kim Y-R, Sagong B, Cho H-J, Bae JW, Kim J, et al. Genetic analysis of genes related to tight junction function in the Korean population with non-syndromic hearing loss. Weber CR, editor. PLoS One. 2014;9(4):e95646. Public Library of Science.
Paganelli M, Stéphenne X, Gilis A, Jacquemin E, Henrion-Caude A, Girard M, et al. Neonatal ichthyosis and sclerosing cholangitis syndrome: extremely variable liver disease severity from claudin-1 deficiency. J Pediatr Gastroenterol Nutr. 2011;53(3):350–4.
Setchell KDR, Heubi JE. Defects in bile acid biosynthesis-diagnosis and treatment. J Pediatr Gastroenterol Nutr. 2006;43(Suppl 1):S17–22.
Heubi J, Setchell K, Bove K. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27(3):282–94.
Jacquemin E, Setchell KDR, O’Connell NC, Bernard O. A new cause of progressive intrahepatic cholestasis: 3β-Hydroxy-C27-steroid dehydrogenase/isomerase deficiency. J Pediatr. 1994;125(3):379–84.
Shneider BL, Setchell KDR, Whitington PF, Neilson KA, Suchy FJ. Δ4-3-Oxosteroid 5β-reductase deficiency causing neonatal liver failure and hemochromatosis. J Pediatr. 1994;124(2):234–8.
Daugherty CC, Setchell KD, Heubi JE, Balistreri WF. Resolution of liver biopsy alterations in three siblings with bile acid treatment of an inborn error of bile acid metabolism (delta 4-3-oxosteroid 5 beta-reductase deficiency). Hepatology. 1993;18(5):1096–101.
Pierre G, Setchell K, Blyth J, Preece MA, Chakrapani A, McKiernan P. Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis. 2008;31(S2):241–5.
Vaz FM, Bootsma AH, Kulik W, Verrips A, Wevers RA, Schielen PC, et al. A newborn screening method for cerebrotendinous xanthomatosis using bile alcohol glucuronides and metabolite ratios. J Lipid Res. 2017;58(5):1002–7. American Society for Biochemistry and Molecular Biology.
Clayton PT, Verrips A, Sistermans E, Mann A, Mieli-Vergani G, Wevers R. Mutations in the sterol 27-hydroxylase gene (CYP27A) cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. J Inherit Metab Dis. 2002;25(6):501–13.
Gong J-Y, Setchell KDR, Zhao J, Zhang W, Wolfe B, Lu Y, et al. Severe neonatal cholestasis in cerebrotendinous xanthomatosis: genetics, immunostaining, mass spectrometry. J Pediatr Gastroenterol Nutr. 2017;65(5):561–8.
Heubi J, Setchell K, Bove K. Inborn errors of bile acid metabolism. Semin Liver Dis. 2007;27(3):282–94. Copyright © 2007 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA.
Heubi JE, Setchell KDR, Jha P, Buckley D, Zhang W, Rosenthal P, et al. Treatment of bile acid amidation defects with glycocholic acid. Hepatology. 2015;61(1):268–74. 1st ed. Wiley-Blackwell.
Setchell KD, Schwarz M, O’Connell NC, Lund EG, Davis DL, Lathe R, et al. Identification of a new inborn error in bile acid synthesis: mutation of the oxysterol 7alpha-hydroxylase gene causes severe neonatal liver disease. J Clin Invest. 1998;102(9):1690–703. American Society for Clinical Investigation.
Ueki I, Kimura A, Nishiyori A, Chen H-L, Takei H, Nittono H, et al. Neonatal cholestatic liver disease in an Asian patient with a homozygous mutation in the oxysterol 7α-hydroxylase gene. J Pediatr Gastroenterol Nutr. 2008;46(4):465–9.
Goldfischer S, Moore CL, Johnson AB, Spiro AJ, Valsamis MP, Wisniewski HK, et al. Peroxisomal and mitochondrial defects in the cerebro-hepato-renal syndrome. Science. 1973;182(4107):62–4.
Poll-The BT, Saudubray JM, Ogier H, Schutgens RB, Wanders RJ, Schrakamp G, et al. Infantile Refsum’s disease: biochemical findings suggesting multiple peroxisomal dysfunction. J Inherit Metab Dis. 1986;9(2):169–74.
Goldfischer S, Collins J, Rapin I, Coltoff-Schiller B, Chang CH, Nigro M, et al. Peroxisomal defects in neonatal-onset and X-linked adrenoleukodystrophies. Science. 1985;227(4682):67–70.
Smith DW, Lemli L, Opitz JM. A newly recognized syndrome of multiple congenital anomalies. J Pediatr. 1964;64(2):210–7.
Zhou Y, Zhang J. Arthrogryposis–renal dysfunction–cholestasis (ARC) syndrome: from molecular genetics to clinical features. Ital J Pediatr. 2014;40(1):1. BioMed Central.
Gissen P, Tee L, Johnson CA, Genin E, Caliebe A, Chitayat D, et al. Clinical and molecular genetic features of ARC syndrome. Hum Genet. 2006;120(3):396–409.
Peterson MR, Emr SD. The class C Vps complex functions at multiple stages of the vacuolar transport pathway. Traffic. 2001;2(7):476–86. Wiley/Blackwell (10.1111).
Cullinane AR, Straatman-Iwanowska A, Zaucker A, Wakabayashi Y, Bruce CK, Luo G, et al. Mutations in VIPAR cause an arthrogryposis, renal dysfunction and cholestasis syndrome phenotype with defects in epithelial polarization. Nat Genet. 2010;42(4):303–12. Nature Publishing Group.
Saleh M, Kamath BM, Chitayat D. Alagille syndrome: clinical perspectives. Appl Clin Genet. 2016;9:75–82. Dove Press.
Danks DM, Campbell PE, Jack I, Rogers J, Smith AL. Studies of the aetiology of neonatal hepatitis and biliary atresia. Arch Dis Child. 1977;52(5):360–7.
Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29(3):822–9. Wiley-Blackwell.
Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, et al. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109(11):1354–8. American Heart Association, Inc.
Kamath BM, Spinner NB, Rosenblum ND. Renal involvement and the role of Notch signalling in Alagille syndrome. Nat Rev Nephrol. 2013;9(7):409–18.
Mašek J, Andersson ER. The developmental biology of genetic Notch disorders. Development. 2017;144(10):1743–63. Oxford University Press for the Company of Biologists Limited.
Antoniou A, Raynaud P, Cordi S, Zong Y, Tronche F, Stanger BZ, et al. Intrahepatic bile ducts develop according to a new mode of tubulogenesis regulated by the transcription factor SOX9. Gastroenterology. 2009;136(7):2325–33.
Geisler F, Nagl F, Mazur PK, Lee M, Zimber-Strobl U, Strobl LJ, et al. Liver-specific inactivation of Notch2, but not Notch1, compromises intrahepatic bile duct development in mice. Hepatology. 2008;48(2):607–16. Wiley-Blackwell.
Kodama Y, Hijikata M, Kageyama R, Shimotohno K, Chiba T. The role of notch signaling in the development of intrahepatic bile ducts. Gastroenterology. 2004;127(6):1775–86.
Walter TJ, Vanderpool C, Cast AE, Huppert SS. Intrahepatic bile duct regeneration in mice does not require Hnf6 or notch signaling through Rbpj. Am J Pathol. 2014;184(5):1479–88.
Riely CA. Arteriohepatic dysplasia: a benign syndrome of intrahepatic cholestasis with multiple organ involvement. Ann Intern Med. 1979;91(4):520–7. American College of Physicians.
Pavanello M, Severino M, D’Antiga L, Castellan L, Calvi A, Colledan M, et al. Pretransplant management of basilar artery aneurysm and moyamoya disease in a child with Alagille syndrome. Liver Transpl. 2015;21(9):1227–30. Wiley-Blackwell.
Grammatikopoulos T, Sambrotta M, Strautnieks S, Foskett P, Knisely AS, Wagner B, et al. Mutations in DCDC2 (doublecortin domain containing protein 2) in neonatal sclerosing cholangitis. J Hepatol. 2016;65(6):1179–87.
Girard M, Bizet AA, Lachaux A, Gonzales E, Filhol E, Collardeau-Frachon S, et al. DCDC2 mutations cause neonatal sclerosing cholangitis. Hum Mutat. 2016;37(10):1025–9. Wiley-Blackwell.
Verhoeven NM, Huck JHJ, Roos B, Struys EA, Salomons GS, Douwes AC, et al. Transaldolase deficiency: liver cirrhosis associated with a new inborn error in the pentose phosphate pathway. Am J Hum Genet. 2001;68(5):1086–92.
Ng BG, Freeze HH. Perspectives on glycosylation and its congenital disorders. Trends Genet. 2018;34(6):466–76.
Marques-da-Silva D, Reis Ferreira dos V, Monticelli M, Janeiro P, Videira PA, Witters P, et al. Liver involvement in congenital disorders of glycosylation (CDG). A systematic review of the literature. J Inherit Metab Dis. 2017;40(2):195–207. 6th ed. Springer Netherlands.
Jansen JC, Cirak S, van Scherpenzeel M, Timal S, Reunert J, Rust S, et al. CCDC115 deficiency causes a disorder of Golgi homeostasis with abnormal protein glycosylation. Am J Hum Genet. 2016;98(2):310–21.
Morrow G, Tanguay RM. Biochemical and clinical aspects of hereditary tyrosinemia type 1. Adv Exp Med Biol. 2017;959(11):9–21. 8th ed. Cham: Springer International Publishing.
Grompe M. The pathophysiology and treatment of hereditary tyrosinemia type 1. Semin Liver Dis. 2001;21(4):563–71. Copyright © 2001 by Thieme Medical Publishers, Inc., 333 Seventh Avenue, New York, NY 10001, USA. Tel.: +1(212) 584-4662.
Dimmock D, Kobayashi K, Iijima M, Tabata A, Wong LJ, Saheki T, et al. Citrin deficiency: a novel cause of failure to thrive that responds to a high-protein, low-carbohydrate diet. Pediatrics. 2007;119(3):e773–7.
Fiermonte G, Soon D, Chaudhuri A, Paradies E, Lee PJ, Krywawych S, et al. An adult with type 2 citrullinemia presenting in Europe. N Engl J Med. 2008;358(13):1408–9.
Cui H, Li F, Chen D, Wang G, Truong CK, Enns GM, et al. Comprehensive next-generation sequence analyses of the entire mitochondrial genome reveal new insights into the molecular diagnosis of mitochondrial DNA disorders. Genet Med. 2013;15(5):388–94. Springer Nature.
Dames S, Chou L-S, Xiao Y, Wayman T, Stocks J, Singleton M, et al. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J Mol Diagn. 2013;15(4):526–34. Elsevier.
Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, Dirocco M, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. 2008;32(2):143–58. Springer Netherlands.
Gotti G, Marseglia A, De Giacomo C, Iascone M, Sonzogni A, D’Antiga L. Neonatal Jaundice with splenomegaly: not a common pick. Fetal Pediatr Pathol. 2016;35(2):108–11.
Nicastro E, D’Antiga L. Next generation sequencing in pediatric hepatology and liver transplantation. Liver Transpl. 2018;24(2):282–93. Wiley-Blackwell.
Arnell H, Papadogiannakis N, Zemack H, Knisely AS, Nemeth A, Fischler B. Follow-up in children with progressive familial intrahepatic cholestasis after partial external biliary diversion. J Pediatr Gastroenterol Nutr. 2010;51(4):494–9.
Davit-Spraul A, Fabre M, Branchereau S, Baussan C, Gonzales E, Stieger B, et al. ATP8B1 and ABCB11 analysis in 62 children with normal gamma-glutamyl transferase progressive familial intrahepatic cholestasis (PFIC): phenotypic differences between PFIC1 and PFIC2 and natural history. Hepatology. 2010;51(5):1645–55. Wiley-Blackwell.
Emerick KM, Elias MS, Melin-Aldana H, Strautnieks S, Thompson RJ, Bull LN, et al. Bile composition in Alagille syndrome and PFIC patients having partial external biliary diversion. BMC Gastroenterol. 2008;8:47.
Riello L, D’Antiga L, Guido M, Alaggio R, Giordano G, Zancan L. Titration of bile acid supplements in 3beta-hydroxy-Delta 5-C27-steroid dehydrogenase/isomerase deficiency. J Pediatr Gastroenterol Nutr. 2010;50(6):655–60.
Miyagawa-Hayashino A, Egawa H, Yorifuji T, Hasegawa M, Haga H, Tsuruyama T, et al. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transpl. 2009;15(6):610–8. Wiley-Blackwell.
Usui M, Isaji S, Das BC, Kobayashi M, Osawa I, Iida T, et al. Liver retransplantation with external biliary diversion for progressive familial intrahepatic cholestasis type 1: a case report. Pediatr Transplant. 2009;13(5):611–4. Wiley/Blackwell (10.1111).
Nicastro E, Stéphenne X, Smets F, Fusaro F, de Magnée C, Reding R, et al. Recovery of graft steatosis and protein-losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child. Pediatr Transplant. 2012;16(5):E177–82.
Jara P, Hierro L, Martínez-Fernández P, Alvarez-Doforno R, Yánez F, Diaz MC, et al. Recurrence of bile salt export pump deficiency after liver transplantation. N Engl J Med. 2009;361(14):1359–67. Massachusetts Medical Society.
Patel KR, Harpavat S, Finegold M, Eldin K, Hicks J, Firan M, et al. Post-transplant recurrent bile salt export pump disease: a form of antibody-mediated graft dysfunction and utilization of C4d. J Pediatr Gastroenterol Nutr. 2017;65(4):364–9.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Nicastro, E., D’Antiga, L. (2019). Genetic Cholestatic Disorders. In: D'Antiga, L. (eds) Pediatric Hepatology and Liver Transplantation. Springer, Cham. https://doi.org/10.1007/978-3-319-96400-3_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-96400-3_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-96399-0
Online ISBN: 978-3-319-96400-3
eBook Packages: MedicineMedicine (R0)