
Summary
Cerebrotendinous xanthomatosis (CTX) OMIM#213700 is a rare disorder of bile acid synthesis caused by deficiency of the enzyme sterol 27-hydroxylase. It results in deficiency of bile acids and accumulation of abnormal bile alcohols and accelerated cholesterol synthesis. CTX usually presents in the second or third decade with slowly progressive neurological dysfunction, cerebellar ataxia and premature atherosclerosis. Treatment with bile acid supplementation improves but does not completely reverse the neurological signs and symptoms. However, CTX is now known to be associated with a period of neonatal cholestasis. If it is diagnosed at this point, treatment may prevent the onset of neurological problems. We present the case histories and developmental findings in two affected siblings treated from infancy. We plan to continue regular neurodevelopmental reviews.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
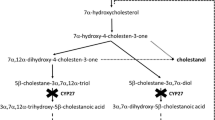
Cerebrotendinous xanthomatosis OMIM#213700 is a rare disorder of bile acid synthesis caused by mutations in the sterol 27-hydroxylase gene (CYP27A1, OMIM *606530). Sterol 27-hydroxylase (EC 1.14.13.15), a hepatic mitochondrial enzyme is important in the synthesis of bile acids from cholesterol (Andersson etal 1989). It catalyses the initial steps of side-chain oxidation in the conversion of a C27 sterol into a C24 bile acid. As a result of its deficiency, the key intermediates, 5β-cholestane-3α,7α,12α-triol and 5β-cholestane-3α,7α-diol are not hydroxylated at the C-27 position and therefore cannot undergo side-chain oxidation to the usual C24-primary bile acids. (Russell and Setchell 1992) These intermediates accumulate in the liver (Fig. 1) and become hydroxylated at multiple sites to form a series of bile alcohols. Although cholic acid is synthesized in subnormal levels by an alternative C-25 hydroxylation pathway (Shefer et al 1976), the lack of normal feedback inhibition on bile acid synthesis leads to excessive production of bile alcohol glucuronides that are diagnostic for this genetic disease. (Egestad et al 1985) Cholesterol synthesis is also upregulated and there is an increased synthesis of cholestanol (Menkes et al 1968; Salen 1971).

Block in the pathway of bile acid synthesis caused by deficiency of sterol 27-hydroxylase
Disease pathology occurs because of widespread deposition of cholestanol and cholesterol in almost all tissues, especially tendons and brain (Ko and Lee 2001; Menkes et al 1968; Salen 1971). Affected patients usually present as teenagers or adults with slowly progressive neurological dysfunction, cerebellar ataxia, premature atherosclerosis and cataracts; the last is an early feature. Spinal cord involvement and dementia can also occur, while mental retardation is seen in over 90% of patients (Ko and Lee 2001). Typical changes on brain MRI include focal or diffuse white-matter abnormalities with cerebral and cerebellar atrophy. Bilateral nonhomogeneous, hyperintense signal changes are also seen in the dentate nuclei (Muhammed et al 2006). Death usually occurs in the fifth decade due to advanced dementia or complications of atherosclerosis.
There have been a few case reports of presentation in childhood with chronic diarrhoea (Kuriyama et al 1991), but generally diagnosis of CTX before the age of 10 years is rare because clinical presentation before this age is rare. Recently, however, an association with neonatal cholestasis and CTX has been described (Setchell et al 2000; Clayton et al 2002). There is therefore a possibility that CTX could be diagnosed before the onset of irreversible disease pathology in later childhood and adulthood. The standard treatment for CTX is bile acid supplementation. It improves neurological signs and symptoms but does not completely reverse them (Berginer et al 1984). Early treatment prior to the onset of neurological symptoms may prevent the onset of irreversible pathology.
We present the case histories and developmental findings in two affected siblings treated from infancy.
Methods
The index case was diagnosed during the neonatal period when he presented with neonatal cholestasis. The other sibling was subsequently diagnosed because of family testing. She did not have neonatal cholestasis. Development assessments were carried out on both siblings. Different tools were used depending on their ages at the time of assessment: the Bayley test was used for ages 1–42 months; the Wechsler preschool and primary scale of intelligence (WPPSI) for ages 2–6 years; and the Wechsler Intelligence Scale for Children – third edition (WISC-111) for ages greater than 6 years. Development assessments were done in conjunction with an interpreter as the children were bilingual in English and Mirpuri.
Case reports
The index case, patient A, was the first child of consanguineous Asian parents. He was born after an uncomplicated pregnancy and developed unexplained neonatal jaundice shortly after birth. He was transferred to a specialist liver unit for investigation at 3 months of age. Investigation showed abnormal transaminases but relatively low gamma-glutamyltransferase (total bilirubin 83 μmol/L (<5), alkaline phosphatase 2255 IU/L (250–1000), aspartate transferase 275 IU/L (20–60), albumin 39 mmol/L (34–42), gamma-glutamyltransferase 95 IU/L (0–23)). Coagulation was normal. Liver biopsy showed chronic active hepatitis with piecemeal necrosis and moderate fibrosis. He was started on vitamins, ursodeoxycholic acid and nutritional supplements. The jaundice resolved spontaneously by 5 months of age and his liver function normalized by 8 months. As part of his ongoing investigations, urinary bile acids were analysed using negative-ion FAB-MS (Fig. 2). This showed elevated bile alcohol glucuronides and decreased primary bile acid concentrations, consistent with a diagnosis of CTX. FAB-MS is a semiquanitative technique that can give a definitive diagnosis of CTX. Heterozygotes in the neonatal period where there is physiological jaundice may show slight elevation of bile alcohol glucuronides but this is clearly differentiated from affected individuals as on repeated samples this is absent or much less evident. Mutation analysis confirmed a homozygous mutation in CYP27A1 (G changed to A in the splice donor sequence of intron 6). This mutation has been described once before in an Italian patient (Garuti etal 1997). The intron 6 mutation causes an abnormally spliced mRNA that does not encode a viable protein. At 1 year of age he had a single generalized tonic-clonic seizures but EEG, brain MRI and ophthalmological examinations were all normal. He was started on cholic acid (15 mg/kg) at age 14 months and showed good biochemical response, concomitant with a marked decrease in urinary bile alcohol excretion as monitored by follow-up FAB-MS of the urine (Fig. 2). He has had no further seizures. At age 8 years he is clinically normal without any xanthomas or cataracts. His brain MRI has been repeated and remains normal. Laboratory liver function has also remained within normal limits. His development was initially delayed, especially in his verbal skills, but repeated assessments have shown good progress and he remains low average for his age (Table 1).

Urinary negative-ion FAB-MS profiles from patient A. Pre-treatment: there is an absence of primary bile acids and predominance of bile alcohol glucuronides (ions at m/z 591, 593, 609, 611, 625, 627 and 643) and bile alcohol sulfates (m/z 513 and m/z 515), confirming cerebrotendinous xanthomatosis. Post-treatment: (4 weeks after treatment with oral cholic acid): there is a virtual disappearance of bile alcohol conjugates confirming suppression of endogenous bile acid synthesis. x = glycerol background ions used as a carrier for FAB-MS probe
The second sibling (patient B), a girl, was born one year later. Postnatal testing showed a similar profile of urinary bile acids, and DNA analysis confirmed the same mutation.
Cholic acid was started at age 5 months following the confirmatory diagnosis. However, she never developed jaundice and she maintained normal liver function throughout. She has also had repeated developmental assessments. At 3 years of age she was found to have delayed verbal skills relative to performance skills, but this has improved. At 7 years of age she has a normal brain MRI and is low average for her age (Table 2).
Discussion
As CTX usually presents in adult life, its early recognition is rare. In 2000, at the International Bile Acid Symposium, Setchell and colleagues (2000) first described neonatal cholestasis as a previously unrecognized early manifestation of CTX and was later supported by a report of a number of additional cases by Clayton and colleagues. Clayton and colleagues (2002) described a neonate who presented at the age of 7 days with hepatomegaly and cholestatic jaundice with elevated transaminases and elevated cholesterol but normal gamma glutamyltransferase. Urine analysis showed increased bile alcohol glucuronides and sulfates and mutation analysis confirmed CTX. He was started on cholic acid with improvement of his cholestasis. Clayton and colleagues (2002) then reviewed the detailed medical histories of nine adult patients with proven CTX. Six of them had a period of prolonged but self-limiting cholestatic jaundice.
CTX therefore joins a group of other inborn errors of metabolism (e.g. Niemann–Pick C, citrullinaemia type II) as a rare cause of neonatal cholestasis, and screening for these should form part of the investigation of unexplained neonatal cholestasis. This association with neonatal jaundice has now permitted the earlier diagnosis of the disorder through mass-spectrometric screening programmes for genetic defects in bile acid synthesis (Clayton 2001; Setchell and Heubi 2006; Setchell et al 2008). Our index case was diagnosed with CTX by this approach, following a period of unexplained neonatal cholestasis, but his affected sibling did not become jaundiced. Neonatal cholestasis therefore does not occur in every case of CTX even where the same mutation exists. Exactly why cholestatic jaundice may occur in some cases but not others is therefore unclear. While mutations of the CYP27A gene lead to virtually complete impairment of chenodeoxycholic acid synthesis, patients with CTX still synthesize cholic acid via an alternative C-25 hydroxylation pathway (Shefer et al 1976), albeit a low rates of synthesis. It is therefore possible that individual differences in the synthesis rates of cholic acid via this alternative pathway, and variations in the total bile acid pool size, which is normally reduced in the neonatal period, may explain why cholestasis is not observed in all CTX patients in early life. One other theory is that in patients with CTX neonatal cholestasis will only occur if some other genetic, environmental or developmental factor interacts to affect other steps in bile acid metabolism). As other pathways become established, the cholestatic jaundice then resolves (Clayton et al 2002; Russell and Setchell 1992 ). It appears, though, that in some cases the cholestasis maybe persistent and even fatal without treatment. In the case published by Clayton and colleagues (2002), resolution of the jaundice occurred only after cholic acid was started. This patient also had a previous sibling who died at the age of 13 months having been jaundiced all his life.
Standard treatment of CTX is oral bile acid supplementation with chenodeoxycholic acid (Berginer et al 1984; Koopman et al 1985). Bile acid supplementation provides a source of primary bile acid, thereby restoring the normal feedback inhibition of endogenous cholesterol and bile acid synthesis, and this leads to a reduction in the production of cholestanol (Salen et al 1975). There is therefore less deposition and accumulation of cholestanol in tissues. Adult patients treated with chenodeoxycholic acid have shown improvement in dementia, pyramidal and cerebellar signs, and peripheral neuropathy (Berginer et al 1984). More recently, cholic acid has been the choice of primary bile acid therapy for neonates as this has been effective in the treatment of other genetic defects in bile acid synthesis (Setchell and Heubi 2006). Our experience has been that chenodeoxycholic acid, because it is cathartic and intrinsically hepatotoxic (Yashuhara et al 1985), causes diarrhoea and exacerbates liver dysfunction in neonates and infants. For these reasons, cholic acid is considered the safer option. A complementary treatment option is to decrease the amount of cholesterol used in bile acid synthesis by reducing cholesterol synthesis. To this effect HMG-CoA reductase inhibitors have been tried in combination with bile acid supplementation in some cases and we plan to implement this when our cases are 10 years of age. Results have been conflicting, however, some reporting a significant lowering of cholestanol pools (Kuriyama et al 1994; Nakamura etal 1991) while other report little effect (Salen et al 1994).
Although in theory early treatment of CTX could prevent or slow the onset of neurological symptoms, there is very little experience of the early use of bile acid supplementation and there are no data on the development of those who have been treated from early childhood. The development assessments of patients A and B (Tables 1 and 2) were done in conjunction with an interpreter: Mirpuri is the primary language used at home. This may explain both children’s earlier lower scores in verbal ability compared to their non-verbal ability. So far, both children’s development is average for their age. We plan to continue regular neurodevelopment reviews and 5-yearly brain MRI scans on both siblings. Early recognition of problem areas would allow early intervention such as additional help at school and aid our knowledge of the clinical course of the illness in childhood. As more patients are identified and treated early, long-term follow-up would help answer the question of prevention versus improvement.
Abbreviations
- CTX:
-
cerebrotendinous xanthomatosis
- HMG-CoA:
-
hydroxymethylglutaryl-coenzyme A
- WISC-III:
-
Wechsler Intelligence Scale for Children – third edition
- WPPSI:
-
Wechsler preschool and primary scale of intelligence
References
Andersson S, Davis DL, Dahlback H, Jornvall H, Russell DW (1989) Cloning, structure, and expression of the mitochondrial cytochrome P- 450 sterol 26-hydroxylase, a bile acid biosynthetic enzyme. J Biol Chem 264: 8222–8229.
Berginer VM, Salen G, Shefer S (1984) Long-term treatment of cerebrotendinous xanthomatosis with chenodeoxycholic acid. N Engl J Med 311: 1649–1652.
Clayton PT (2001) Applications of mass spectrometry in the study of inborn errors of metabolism. J Inherit Metab Dis 24: 139–150.
Clayton PT, Verrips A, Sistermans E, Mann A, Mieli-Vergani G, Weverrs R (2002) Mutations in the sterol 27-hydroxylase gene cause hepatitis of infancy as well as cerebrotendinous xanthomatosis. JIMD 25: 501–513.
Egestad B, Pettersson P, Skrede S, Sjövall J (1985) Fast atom bombardment mass spectrometry in the diagnosis of cerebrotendinous xanthomatosis. Scand J Clin Lab Invest 45: 443–446.
Garuti R, Croce MA, Tiozzo R, et al (1997) Four novel mutations of sterol 27-hydroxylase gene in Italian patients with cerebrotendinous xanthomatosis. J Lipid Res 38(11): 2322–2334.
Ko KF, Lee KW (2001) Cerebrotendinous xanthomatosis in three siblings from a Chinese family. Singapore Med J 42(1): 30–32.
Koopman BJ, Wolthers BG, Van der Molen JC, Waterreus RJ (1985) Bile acid therapies applied to patients suffering from cerebrotendinous xanthomatosis. Clin Chim Acta 152: 115–122.
Kuriyama M, Fujiyama J, Yoshidome H, et al (1991) Cerebrotendinous xanthomatosis: Clinical and biochemical evaluation of eight patients and review of the literature. J Neurol Sci 102: 225–232.
Kuriyama M, Tokimaura M, Fujiyama J, Utatsu Y, Osame M (1994) Treatment of cerebrotendinous xanthomatosis: effects of chenodeoxycholic acid, pravastatin and combined use. J Neurol Sci 125(1): 22–28.
Menkes JH, Schimschock JR, Swanson PD (1968) Cerebrotendinous xanthomatosis. The storage of cholestanol within the nervous system. Arch Neurol 19: 47–53.
Muhammed K, Nandakumar G, Saritha S (2006) Cerebrotendinous xanthomatosis: Need for early diagnosis. Indian J Dermatol 72(5): 364–366.
Nakamura T, Matsuzawa Y, Takemura K, Kubo M, Miki H, Tarui S (1991) Combined treatment with chenodeoxycholic acid and pravastatin improves plasma cholestanol levels associated with marked regression of tendon xanthomas in cerebrotendinous xanthomatosis. Metabolism 40(7): 741–746.
Russell DW, Setchell KD (1992) Bile acid biosynthesis. Biochemistry 31: 4737–4749.
Salen G (1971) Cholestanol deposition in cerebrotendinous xanthomatosis. A possible mechanism. Ann Intern Med 75: 843–851.
Salen G, Meriwether TW, Nicolau G (1975) Chenodeoxycholic acid inhibits increased cholesterol and cholestanol synthesis in patients with cerebrotendinous xanthomatosis. Biochem Med 14: 57–74.
Salen G, Batta AK, Tint GS, Shefer S (1994) Comparative effects of lovastatin and chenodeoxycholic acid on plasma cholestanol levels and abnormal bile acid metabolism in cerebrotendinous xanthomatosis. Metabolism 43(8):1018–1022.
Setchell KD, Heubi JE (2006) Defects in bile acid biosynthesis–diagnosis and treatment. J Pediatr Gastroenterol Nutr 43(Supplement 1): S17–22.
Setchell KD, O’Connell N, Russell D, Kelly DA (2000) A unique case of cerebrotendinous xanthomatosis in infancy with cholestastic liver disease further highlights bile acid synthetic defects as an important category of metabolic liver disease. In: XX International Bile Acid Meeting, 13–14 June, Amsterdam. Falk Symposium 165. [Abstract].
Setchell KD, Bove K, Heubi JE (2008) Disorders of bile acid synthesis. In: Walker WA, Goulet O, Kleinman RE, Sherman PM, Shneider BL, Sanderson IR, eds. Pediatric Gastrointestinal Disease. B C Decker Inc, Hamilton, ON. [In press].
Shefer S, Cheng FW, Dayal B, et al (1976) A 25-hydroxylation pathway of cholic acid biosynthesis in man and rat. J Clin Invest 57: 897–903.
Yasuhara H, Tonooka M, Kamei K, Sakamoto K (1985) Membrane effects of various drugs on isolated rat hepatocytes and erythrocytes. Toxicol Appl Pharmacol 79: 453–460.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicating editor: Peter Clayton
Competing interests: None declared
References to electronic databases: Cerebrotendinous xanthomatosis (CTX) OMIM#213700. www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=OMIM; www.chem.qmul.ac.uk/iubmb/enzyme/; www.gene.ucl.ac.uk/nomenclature/.
Rights and permissions
About this article
Cite this article
Pierre, G., Setchell, K., Blyth, J. et al. Prospective treatment of cerebrotendinous xanthomatosis with cholic acid therapy. J Inherit Metab Dis 31 (Suppl 2), 241–245 (2008). https://doi.org/10.1007/s10545-008-0815-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10545-008-0815-z