
Abstract
Purpose of Review
Cholestasis is characterized by a conjugated hyperbilirubinemia secondary to impaired bile synthesis, transport, or excretion from the liver. It is always pathologic and can be indicative of an underlying hepatobiliary, genetic, or metabolic disorder, several of which require timely diagnosis to ensure proper management and optimal outcomes. This review provides an overview of the evaluation of cholestasis with a focus on current and emerging treatment strategies.
Recent Findings
Increased accessibility of next generation sequencing (NGS) allows for utilization of genetic testing early in the diagnostic process. This may alter the clinical algorithm for diagnosis of cholestatic disorders. An enhanced understanding of the underlying pathophysiology may help guide future development of targeted therapies, such as ileal bile acid transporter (IBAT) inhibitors. These were recently approved for treatment of cholestatic pruritus in patients with Alagille syndrome and Progressive Familial Intrahepatic Cholestasis.
Summary
Current management of cholestasis is aimed at the biochemical consequences of impaired bile flow, including malnutrition, pruritus, and progressive fibrosis. NGS has led to an enhanced understanding of biliary pathology and may guide development of future treatment modalities based on specific gene mutations. Rapid discernment of the underlying etiology is essential as new treatment modalities emerge.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cholestasis is defined as a decrease or absence of bile flow from the liver to the small intestine. This can be caused by defects in bile production, transmembrane transport, or a mechanical obstruction to bile flow. Neonatal cholestasis affects roughly 1 in 2500 term births and early identification is critical as this may impact treatment outcomes [1, 2]. Practice guidelines endorsed by the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition (NASPGHAN) in 2017 can help guide evaluation of the pediatric patient with cholestasis. Any infant presenting with jaundice beyond two to three weeks of life should undergo evaluation. An elevated serum conjugated or direct bilirubin level > 1.0 mg/dL warrants further diagnostic evaluation. A threshold of 2.0 may be appropriate in infants with intestinal failure-associated liver disease [1]. Recent reports have also suggested that a lower conjugated/direct bilirubin level of > 0.3–0.5 mg/dL and > 10% of the total bilirubin in the first few days of life should also trigger suspicion for cholestasis and consideration for further investigation [3,4,5].
The diagnostic algorithm for neonatal cholestasis has evolved in recent years with the advent of next generation sequencing (NGS) enabling rapid identification of disorders that cannot be directly diagnosed from routine serologic tests or liver biopsy. Genetic sequencing is being utilized earlier in the diagnostic pathway in concert with evaluation for biliary atresia and other treatable disorders.
The biochemical features of cholestasis reflect the retention of bile components in the serum, namely bilirubin, bile acids and cholesterol. These may lead to clinical manifestations of jaundice, pruritus, xanthomas, and malnutrition. Damage to the liver parenchyma can also lead to progressive fibrosis and cirrhosis with resultant portal hypertension and loss of hepatic synthetic function over time. The pattern and severity of each of these abnormalities varies with the underlying disorder. This review presents an overview of cholestasis in infants and children with a focus on current and emerging treatment strategies.
Clinical Presentation
Infants with cholestatic jaundice often present with scleral icterus and may also have symptoms of acholic stool, dark urine, and increased irritability secondary to pruritus, which may be helpful in distinguishing cholestasis from physiologic jaundice. Family history of liver disease, especially in siblings, suggests an underlying genetic abnormality, early emphysema in family members raises suspicion for alpha-1 antitrypsin (A1AT) deficiency, and history of consanguinity increases the likelihood of an autosomal recessive pathology [6].
If there is progression of the underlying liver disease, physical exam may also reveal hepatosplenomegaly, ascites, and signs of malnutrition and frailty with low muscle mass, temporal wasting, and delayed growth. Physical exam findings that may provide additional diagnostic clues include hypotonia (mitochondrial or peroxisomal disorder), heart murmur (Alagille syndrome or biliary atresia), dysmorphic facies (Alagille syndrome or underlying chromosomal abnormality), cataracts, chorioretinitis, and microcephaly (congenital infection).
Diagnostic Evaluation
The first step in evaluating for cholestasis is to fractionate the total bilirubin level. Cholestatic jaundice is defined as a serum conjugated bilirubin concentration greater than 1.0 mg/dL. Once established, further evaluation should be performed in a stepwise approach with a focus on rapid identification of treatable disorders (Fig. 1). Conditions such as sepsis/infection, hypothyroidism, inborn errors of metabolism (i.e., galactosemia), and panhypopituitarism must be diagnosed and treated promptly to avoid progression of systemic illness. Biliary obstruction, as seen with choledochal cysts, may be identified with ultrasound [7].

Diagnostic Approach to Evaluation of Cholestasis in Neonates and Young Infants
Biliary atresia (BA) must also be identified promptly as early surgical intervention has been associated with better long-term outcomes. The work-up often includes a series of laboratory tests, imaging, and liver histology to exclude other causes of cholestasis. Definitive diagnosis is made with cholangiogram. Hepatoportoenterostomy (HPE) or Kasai procedure, is a surgical attempt to restore bile flow from the liver to the intestine. During this procedure, a Roux-en-Y loop of bowel is created and directly anastomosed to the hilum of the liver, following excision of the biliary remnant and portal fibrous plate. Younger age at the time of HPE, specifically when performed before 45 days of age, is associated with improved outcomes. Native liver survival declines when HPE is performed beyond 60 days of life thereby emphasizing the importance of timely diagnosis [8,9,10].
Serum measurement of matrix metalloproteinase-7 (MMP-7) has recently been explored as a potential biomarker of BA that may help to guide decisions regarding the pursuit of more invasive studies. MMP-7 is highly expressed in the intrahepatic bile ducts and an elevated serum MMP-7 has been noted around 1–2 months of age in patients with BA, with positive and negative predictive values exceeding 90 and 95% respectively [11, 12]. Further validation is needed to clarify the role of MMP-7 in the diagnostic algorithm of neonatal cholestasis.
The remainder of the workup for cholestasis is guided by the overall clinical presentation of the patient. Hospitalized infants with complex comorbidities may also have multifactorial cholestasis in the setting of infection, cardiac dysfunction, delays in enteral stimulation, and use of parenteral nutrition. Family and maternal history along with physical exam findings can help guide next steps in patients without a clear etiology to cholestasis (Table 1) [13,14,15,16,17,18,19,20,21].
Role of Next-Generation Sequencing
Genetic disorders account for more than 25–50% of all cases of neonatal cholestasis [22••, 23]. Identification of the precise etiology can be challenging due to the extensive overlap of clinical presentations. Routine serologic and other diagnostic assessments are often insufficient to distinguish between these disorders. Historically, genetic testing was limited to analysis of a single gene based on clinical observation and initial evaluation; however, given the large number of potential genetic causes of cholestasis, single-gene testing has become impractical, and moreover, may result in delays in timely diagnosis. Next-generation sequencing (NGS), including multigene panels, whole exome sequencing (WES), and whole genome sequencing (WGS), now enables rapid testing of multiple genes simultaneously, with available results within days to weeks. This allows for the utilization of genetic testing early in the diagnostic process. A recent study using a 66 gene panel in 716 infants and older children with cholestasis or liver disease of unknown etiology reported a positive or likely positive molecular diagnosis in 11.7% and a single pathogenic or likely pathogenic variant in another 12.7% of patients [22••]. The analysis of multiple genes in a single panel reduces the time to identify, or perhaps exclude, specific genes as a cause of cholestasis. While the increased accessibility and affordability of NGS has allowed for rapid evaluation of these patients, wide-spread testing also presents the need for additional considerations. The ongoing discovery of new gene variants, single mutations in known pathogenic genes, unclear pathogenicity and variable phenotypes have made interpretation of these results challenging at times. The use of NGS without selection may give rise to unnecessary or uninterpretable information which may complicate rather than clarify the diagnosis [23] With continued implementation, multigene panels may alter the clinical algorithm for diagnosis of cholestatic disorders, including the necessity of liver biopsy.
Common Causes of Cholestasis (Table 1)
A brief overview of common causes of neonatal cholestasis is presented here. A more detailed review and approach to evaluation are presented in Table 1.
Biliary Atresia
Biliary atresia (BA) is a progressive, idiopathic, fibro-obliteration of the bile ducts which can lead to progressive fibrosis, cirrhosis, and eventually end-stage liver disease. It is universally fatal if not treated and is the most common identifiable cause of neonatal cholestasis and the leading indication for pediatric liver transplant. The etiology of BA remains elusive and is likely multifactorial in origin with contributing factors including exposure to viral or toxic pathogens in utero, immunologic mechanisms and possible predisposing genetic factors [24]. Biliary atresia may occur in isolation (70%), with laterality malformations (10% to 15%) also known as biliary atresia splenic malformation syndrome (BASM), or with other congenital anomalies (10% to 15%). Those with laterality malformations often have a worse prognosis. HPE outcomes vary widely between institutions and geographically, and many patients will ultimately require liver transplantation. The most important prognostic factors impacting long-term outcomes include younger age at the time of HPE (< 60 days), the experience of the surgeon and health-care center at which the procedure is performed, and the decrease in total bilirubin level in the months following HPE. Patients with a total bilirubin > 2 mg/dL at 3 months following HPE should be consider for liver transplant evaluation [25].
Medical care following HPE consists of choleretic medications, antibiotics for cholangitis prophylaxis, fat-soluble vitamin (FSV) and nutritional supplementation. Those with BA require life-long care for the evaluation and management of complications including progressive liver disease with portal hypertension, recurrent cholangitis, and hepatocellular carcinoma (HCC).
Alagille Syndrome
Alagille syndrome is an autosomal dominant disorder characterized by a paucity of intrahepatic bile ducts. Mutations in the JAG1 gene account for approximately 90% of patients; a smaller percentage of patients have mutations in NOTCH2 (2.5%). The incidence of disease is likely underestimated given the incomplete penetrance and variable phenotypic presentation.
This is a multisystemic disorder which may also involve cardiac, skeletal, renal, (neuro)vascular, and ocular abnormalities as well as characteristic dysmorphic facies (Table 1). Bile duct paucity is seen on liver histology which can result in chronic cholestasis, the accumulation of bile acids and subsequent liver injury. Treatment is generally aimed at the consequences of cholestasis, namely pruritus, xanthomas, and nutritional deficiencies. Liver transplantation is reserved for those with end stage liver disease or debilitating pruritus significantly impacting quality of life [26].
A recent large, international cohort study of Alagille patients revealed that only 40.3% of patients survived to adulthood with their native liver. A total bilirubin level < 5.0 mg/dL between 6 and 12 months of age appears to be associated with improved hepatic outcomes. [27••]
Progressive Familial Intrahepatic Cholestasis
Progressive familial intrahepatic cholestasis (PFIC) disorders consist of a growing selection of autosomal recessive conditions characterized by bile acid transport defects with varying underlying genetic mutations (see Table 1). These disorders typically present during infancy and childhood. Intractable pruritus is a dominant feature, especially in PFIC 1 and 2.
Patients with PFIC 1 and 2 present during infancy with low-gamma-glutamyl transpeptidase (ggt) cholestasis, whereas those with PFIC 3 often present later in childhood and have an elevation in ggt. Individuals with PFIC 1 secondary to FIC1 deficiency and mutations in ATP8B1 often have extrahepatic manifestations such as diarrhea, hearing loss, pancreatic insufficiency, and ocular abnormalities. Liver transplantation does not address these additional manifestations and patients with PFIC 1 may develop graft steatosis and worsening diarrhea following transplant. Patients with PFIC 2 or severe BSEP deficiency secondary to mutations in ABCB11, have an increased risk for the development of hepatocellular carcinoma (HCC).
PFIC 3 involves mutations in the ABCB4 gene also known as multidrug resistance protein-3 P-glycoprotein (MDR3). As previously stated, these patients tend to present later in childhood or even adulthood and have an elevation in ggt. Many patients are predisposed to cholesterol precipitation and gallstone formation. PFICs 4–6 are more recently discovered, rare cholestatic disorders with broad extrahepatic systemic manifestations likely related to complexity in the expression of their underlying genetic mutations [28, 29].
Complications of Cholestasis: Monitoring and Treatment Strategies
The management of cholestasis is often aimed at the biochemical consequences of impaired bile flow. These include malnutrition, pruritus, xanthoma formation, and progressive fibrosis. Together, these complications can significantly impact the quality of life of both patients and caretakers. Monitoring recommendations and disease specific therapies are outlined in Table 2.
Malnutrition and Fat-soluble Vitamin (FSV) Deficiencies
The nutritional evaluation should include a detailed investigation of dietary intake, nutritional supplementation, and ongoing assessments to help guide long-term management. In children with chronic cholestatic and/or end-stage liver disease, nutritional deficiencies should be identified without delay to permit early interventions aimed at the optimization of infant development and the prevention of further complications.
Routine assessment of anthropometrics is a critical component of the nutritional assessment. An adequately measured length (< 2 years) or height (≥ 2 years) may be a more accurate reflection of nutritional status than weight, especially in the context of ascites and/or organomegaly. Mid upper arm circumference (MUAC) and triceps skin fold (TSF) are also useful in the assessment of malnutrition as they are less likely to be influenced by fluid overload. They have also been shown to be a more accurate reflection of short-term changes in nutritional status and can provide important information regarding body composition. MUAC measures both muscle mass and adipose tissue, whereas TSF reflects adiposity. These anthropometric measures are helpful predictors of growth in infants and children with chronic liver diseases [30, 31]. The frequency at which these measurements should be performed is variable and depends largely on the degree of malnutrition, ranging from every 2 weeks to 3 months [31].
Impaired bile salt excretion can adversely affect nutrition, resulting in fat malabsorption and FSV deficiencies. These include vitamins A, D, E and K. For lipids and FSV to be absorbed they must be emulsified and incorporated into micelles which are directly absorbed across enterocytes through passive diffusion or specific transporters. They are then reorganized into chylomicrons and released into the lymphatic system for later use or storage. Cholestasis leads to an insufficient concentration of intraluminal bile acids being excreted from the liver which are necessary for micelle formation. Additionally, enteropathy secondary to underlying liver disease with portal hypertension, decreased intake, endocrine dysfunction, and increased energy needs in these infants further complicate nutritional needs [31]. Malnutrition and vitamin deficiencies can impact long-term outcomes. The goals of treatment are to correct the underlying deficiencies thereby reducing morbidity and preventing further complications. This is particularly important in patients with end-stage liver disease necessitating transplantation. Optimization of nutritional status prior to surgery may help to hasten post-transplant recovery and decrease the rate of complications.
Infants with chronic cholestasis often require 130–150% of the daily caloric requirements due to increased metabolic demands compounding concerns for fat malabsorption. Formulas enriched in medium-chain triglycerides (MCTs) are often utilized as a lipid supplement in cholestatic infants and young children. Although MCTs have a lower energy content than long-chain triglycerides (LCTs) their shorter chains allow for passive diffusion through the gastrointestinal tract and thereby directly into the portal circulation. Unlike LCTs, MCTs do not require micellar solubilization because they bypass the lymphatic system, with approximately 95% bioavailability, even in the setting of severe cholestasis [31]. The optimal proportion of total lipids as MCTs for nutritional management is between 30 and 50%. MCT content in the diet in excess of 80% may result in essential fatty acid deficiency and should be avoided. If unable to achieve adequate weight gain and growth despite caloric fortification with MCT-enriched formulas, supplemental nasogastric tube feeds or parental nutrition should be considered [31].
FSV deficiencies can lead to complications such as rickets and bone fractures, coagulopathy with hemorrhage, cerebellar ataxia, and impaired vision [32]. The prevalence of these deficiencies increases with the severity of cholestasis. Vitamin levels are inversely correlated with total bilirubin [32]. Initial supplementation is provided through water-soluble ADEK multivitamin preparations. These aqueous formulations, such as DEKAs Essential, are specifically designed for cholestasis and can be transported directly into the portal circulation independent of bile salts. They contain amounts of Vitamins A, D, E and K in significant excess of typical recommended daily values. Depending on the severity of cholestasis, additional supplementation of individual FSVs may also be necessary [33].
The chemical modification of a vitamin also helps to promote greater water solubility. The conjugation of α tocopheryl succinate with polyethylene glycol-1000 creates a water-soluble molecule (TPGS). This formulation of tocopherol (or Vitamin E) has excellent intestinal absorption even in the setting of severe cholestasis [32]. When TPGS is co-administered with other FSVs, it helps to enhance the absorption of the unmodified vitamins. Alternatively, periodic intramuscular administration of vitamins can help circumvent poor intestinal absorption. This approach is generally not favored by patients or their families. In the United States, preparations are available for parenteral administration of vitamins A, D, and K, but not for vitamin E. Vitamin levels should be monitored closely until stable levels are achieved, or cholestasis has improved (Table 2).
Pruritus
The impairment of bile flow leads to a buildup of bile acids in the liver which eventually spill over into the bloodstream. Increased serum bile acids are associated with significant pruritus. Cholestatic pruritus often presents as an intense, unrelenting itch and has frequently been described in patients with Alagille syndrome and PFIC. Clinical manifestations may include visible scratch marks, skin excoriations and scaring, as well as poor weight gain, impaired sleep, and growth. Pruritus may have a profound effect on quality life with interruptions in sleep, and irritability leading to difficulties concentrating thereby impacting school performance [34].
The treatment of choice for pruritus associated with cholestasis is correction of the underlying hepatobiliary disease, when possible. If the underlying etiology cannot be corrected, treatment is aimed at the pruritus itself. Pharmacologic agents used in the treatment of cholestatic pruritus include choleretics, bile acid sequestrants, pregnane X receptor (PXR) agonists, opioid antagonists, and selective serotonin reuptake inhibitors (SSRIs) [35, 36]. These are summarized in Table 2.
When pruritus is refractory to medical therapies surgical treatments should be explored. For cholestatic infants without evidence of advanced fibrosis or portal hypertension, a partial external biliary diversion (PEBD), internal diversion or ileal exclusion procedure aimed at interrupting enterohepatic circulation may be considered to help ameliorate symptoms of pruritus and cholestasis [37, 38]. PEBD disrupts the enterohepatic circulation of bile salts by partially diverting bile from the gallbladder through a loop of bowel which connects the gallbladder with the abdominal skin through a stoma. The influx of bile salts into the intestine and their subsequent reuptake are consequently diminished. Long-term efficacy is uncertain, and the burden of a lifelong stoma is also significant [34, 39]. When surgical interruption is ineffective or with advanced liver disease, liver transplantation may be considered to address ongoing consequences of cholestasis including pruritus, xanthomas, growth failure and poor quality of life.
Progressive Liver Disease and Hepatocellular Carcinoma
The accumulation of bile acids in the liver may contribute to ongoing inflammation and liver injury. Damage to the liver parenchyma can also lead to progressive fibrosis and cirrhosis, portal hypertension and loss of hepatic synthetic function over time. Progressive fibrosis and early signs of portal hypertension can be detected by the presence of splenomegaly on physical examination. Thrombocytopenia and confirmation of splenomegaly on abdominal imaging such as ultrasound also help to corroborate physical exam findings. The presence of abdominal ascites and bleeding secondary to esophageal varices signify hepatic decompensation and may be indications for liver transplantation. Noninvasive markers of liver fibrosis have largely replaced the need for invasive procedures such as liver biopsy. Liver stiffness may be evaluated over time by transient elastography (TE), shear wave elastography (SWE), and magnetic resonance elastography (MRE) [40,41,42].
Hepatocellular carcinoma (HCC) can occur in anyone in whom cirrhosis develops, but persons with BSEP deficiency, tyrosinemia and A1AT deficiency also carry an increased risk. Individuals with advanced fibrosis and cirrhosis require lifelong screening with serum alpha fetal protein (AFP) and abdominal ultrasound [43, 44].
Emerging Treatments
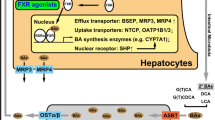
Bile acids are reabsorbed from the ileum via specific transporters. Inhibitors of the apical sodium-dependent bile acid transporter (ASBT), also known as ileal bile acid transporter (IBAT) inhibitors, are novel therapies aimed at blocking entry of bile acids into enterocytes through selective binding of these transporters to ameliorate symptoms of pruritus. These minimally absorbed oral therapies interrupt the enterohepatic circulation of bile acids by increasing fecal excretion and reducing the hepatic uptake of bile acids. This also reduces activation of both the hepatic and ileal farnesoid X receptor (FXR) thereby increasing bile acid synthesis from cholesterol in the liver. Thus, IBAT inhibitors both reduce bile acid reabsorption while increasing bile acid synthesis and secretion in the hepatocytes. In pediatric trials of patients with Alagille syndrome and PFIC, this resulted in the reduction of pruritus, serum bile acids and xanthomas. Clinically meaningful improvements in growth and quality of life were also observed. [45••, 46, 47••]
These therapies have a favorable safety profile and are generally well-tolerated. The most common adverse reactions reported in clinical trials included diarrhea, abdominal pain, vomiting, and liver test abnormalities. Serum bilirubin, alanine and aspartate transaminases, alkaline phosphatase, ggt, and prothrombin time (INR) should be determined before initiation of treatment and intermittently during the course of therapy. Additionally, bile salt depletion can cause malabsorption of fat and fat-soluble vitamins, which may require supplementation.
There are currently two IBAT inhibitors approved for use in children. Odevixibat has been approved for the treatment of cholestatic pruritus in patients with PFIC ≥ 3 months of age and Alagille syndrome ≥ 12 months, while maralixibat has been approved for patients with Alagille syndrome and age ≥ 3 months. Though there is emerging data to suggest that these therapies may delay the progression of liver disease and prolong transplant-free survival, long-term data and further exploration of these findings are necessary [48••]. At present, investigation of the use of IBAT inhibitors for treatment of other cholestatic liver diseases such as biliary atresia and primary sclerosing cholangitis are ongoing.
Conclusions
Cholestasis in infants and young children requires early recognition and a thoughtful approach to timely diagnosis. Though BA accounts for a majority of these cases, an increasing number of genetic etiologies are being recognized. Current treatment strategies are aimed at addressing the biochemical consequences of cholestasis including pruritus, xanthomas, nutritional deficiencies, and improved quality of life. The increased availability of NGS may help reduce the time to identify and eliminate specific genes thereby reshaping the diagnostic algorithm of cholestasis. The widespread use of rapid multigene testing has also led to newly identified genetic variants and an improved understanding of biliary physiology. This enhanced understanding may help to guide the future development of targeted therapies and new treatment modalities based on specific gene mutations.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Fawaz R, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017;64(1):154–68.
Dick MC, Mowat AP. Hepatitis syndrome in infancy–an epidemiological survey with 10 year follow up. Arch Dis Child. 1985;60(6):512–6.
Harpavat S, Finegold MJ, Karpen SJ. Patients with biliary atresia have elevated direct/conjugated bilirubin levels shortly after birth. Pediatrics. 2011;128(6):e1428–33.
Harpavat S, Garcia-Prats JA, Shneider BL. Newborn Bilirubin Screening for Biliary Atresia. N Engl J Med. 2016;375(6):605–6.
Harpavat S, et al. Newborn Direct or Conjugated Bilirubin Measurements As a Potential Screen for Biliary Atresia. J Pediatr Gastroenterol Nutr. 2016;62(6):799–803.
Feldman AG, Sokol RJ. Recent developments in diagnostics and treatment of neonatal cholestasis. Semin Pediatr Surg. 2020;29(4): 150945.
Feldman AG, Sokol RJ. Neonatal Cholestasis: Updates on Diagnostics, Therapeutics, and Prevention. NeoReviews. 2021;22(12):e819–36.
Schreiber RA, et al. Biliary atresia: the Canadian experience. J Pediatr. 2007;151(6):659-665.e1.
Serinet MO, et al. Impact of age at Kasai operation on its results in late childhood and adolescence: a rational basis for biliary atresia screening. Pediatrics. 2009;123(5):1280–6.
Chardot C, et al. Is the Kasai operation still indicated in children older than 3 months diagnosed with biliary atresia? J Pediatr. 2001;138(2):224–8.
Yang L, et al. Diagnostic Accuracy of Serum Matrix Metalloproteinase-7 for Biliary Atresia. Hepatology. 2018;68(6):2069–77.
Wu JF, et al. Quantification of Serum Matrix Metallopeptide 7 Levels May Assist in the Diagnosis and Predict the Outcome for Patients with Biliary Atresia. J Pediatr. 2019;208:30-37.e1.
Amendola M, Squires JE. Pediatric genetic cholestatic liver disease overview. 2022 Sep 15 [updated 2023 May 25]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2023.
Townsend SA, et al. Systematic review: the natural history of alpha-1 antitrypsin deficiency, and associated liver disease. Aliment Pharmacol Ther. 2018;47(7):877–85.
Patterson M. Niemann-pick disease type C. 2000 Jan 26 [updated 2020 Dec 10]. In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington, Seattle; 1993–2023.
Gonzales E, et al. Cholic acid for primary bile acid synthesis defects: a life-saving therapy allowing a favorable outcome in adulthood. Orphanet J Rare Dis. 2018;13(1):190.
Pfister ED, et al. Extrahepatic manifestations of progressive familial intrahepatic cholestasis syndromes: Presentation of a case series and literature review. Liver Int. 2022;42(5):1084–96.
Bolia R, et al. Biliary diversion in progressive familial intrahepatic cholestasis: a systematic review and meta-analysis. Expert Rev Gastroenterol Hepatol. 2022;16(2):163–72.
Vinayagamoorthy V, Srivastava A, Sarma MS. Newer variants of progressive familial intrahepatic cholestasis. World J Hepatol. 2021;13(12):2024–38.
Gomez-Ospina N, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7:10713.
Chinsky JM, et al. Diagnosis and treatment of tyrosinemia type I: a US and Canadian consensus group review and recommendations. Genet Med. 2017;19(12):1380–95.
Karpen SJ, et al. Use of a Comprehensive 66-Gene Cholestasis Sequencing Panel in 2171 Cholestatic Infants, Children, and Young Adults. J Pediatr Gastroenterol Nutr. 2021;72(5):654–60. Examinated the use of targed multigene panels in the diagnosis of cholestasis in neonates and young infants.
Jeyaraj R, et al. The Genetics of Inherited Cholestatic Disorders in Neonates and Infants: Evolving Challenges. Genes (Basel). 2021;12(11):1837.
Davenport M, Kronfli R, Makin E. Advances in understanding of biliary atresia pathogenesis and progression - a riddle wrapped in a mystery inside an enigma. Expert Rev Gastroenterol Hepatol. 2023;17(4):343–52.
Shneider BL, et al. Total Serum Bilirubin within 3 Months of Hepatoportoenterostomy Predicts Short-Term Outcomes in Biliary Atresia. J Pediatr. 2016;170:211–7.
Kohut TJ, Gilbert MA, Loomes KM. Alagille Syndrome: A Focused Review on Clinical Features, Genetics, and Treatment. Semin Liver Dis. 2021;41(4):525–37.
Vandriel SM, et al. Natural history of liver disease in a large international cohort of children with Alagille syndrome: Results from the GALA study. Hepatology. 2023;77(2):512–29. Large, international retrospective review of the natural history of patients with Alagille syndrome.
Bull LN, Thompson RJ. Progressive Familial Intrahepatic Cholestasis. Clin Liver Dis. 2018;22(4):657–69.
Gomez-Ospina N, et al. Mutations in the nuclear bile acid receptor FXR cause progressive familial intrahepatic cholestasis. Nat Commun. 2016;7(1):10713.
Young S, et al. Nutrition assessment and support in children with end-stage liver disease. Nutr Clin Pract. 2013;28(3):317–29.
Tessitore M, et al. Malnutrition in Pediatric Chronic Cholestatic Disease: An Up-to-Date Overview. Nutrients. 2021;13(8):2785.
Shneider BL, et al. Efficacy of fat-soluble vitamin supplementation in infants with biliary atresia. Pediatrics. 2012;130(3):e607–14.
Kamath BM, et al. Fat Soluble Vitamin Assessment and Supplementation in Cholestasis. Clin Liver Dis. 2022;26(3):537–53.
Kamath BM, et al. Systematic Review: The Epidemiology, Natural History, and Burden of Alagille Syndrome. J Pediatr Gastroenterol Nutr. 2018;67(2):148–56.
Zellos A, Roy A, Schwarz KB. Use of oral naltrexone for severe pruritus due to cholestatic liver disease in children. J Pediatr Gastroenterol Nutr. 2010;51(6):787–9.
Cies JJ, Giamalis JN. Treatment of cholestatic pruritus in children. Am J Health Syst Pharm. 2007;64(11):1157–62.
Yang H, et al. Partial external biliary diversion in children with progressive familial intrahepatic cholestasis and Alagille disease. J Pediatr Gastroenterol Nutr. 2009;49(2):216–21.
Van Vaisberg V, et al. Ileal exclusion for pruritus treatment in children with progressive familial intrahepatic cholestasis and other cholestatic diseases. J Pediatr Surg. 2020;55(7):1385–91.
Emerick KM, Whitington PF. Partial external biliary diversion for intractable pruritus and xanthomas in Alagille syndrome. Hepatology. 2002;35(6):1501–6.
Banc-Husu AM, Bass LM. Transient Elastography in Pediatric Liver Disease. J Pediatr Gastroenterol Nutr. 2021;73(2):141–4.
Shneider BL, et al. Nonfasted Liver Stiffness Correlates with Liver Disease Parameters and Portal Hypertension in Pediatric Cholestatic Liver Disease. Hepatol Commun. 2020;4(11):1694–707.
Chongsrisawat V, et al. Transient elastography for predicting esophageal/gastric varices in children with biliary atresia. BMC Gastroenterol. 2011;11:41.
Knisely AS, et al. Hepatocellular carcinoma in ten children under five years of age with bile salt export pump deficiency. Hepatology. 2006;44(2):478–86.
Hadžić N, et al. Hepatocellular carcinoma in biliary atresia: King’s College Hospital experience. J Pediatr. 2011;159(4):617-22.e1.
Gonzales E, et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): a randomised phase 2 study. Lancet. 2021;398(10311):1581–92. Demonstrated safety and efficacy of maraliximab, an IBAT inhibitor, for the treatment of cholestatic pruritus in patients with Alagille syndrome.
Shneider BL, et al. Placebo-Controlled Randomized Trial of an Intestinal Bile Salt Transport Inhibitor for Pruritus in Alagille Syndrome. Hepatol Commun. 2018;2(10):1184–98.
Thompson RJ, et al. Odevixibat treatment in progressive familial intrahepatic cholestasis: a randomised, placebo-controlled, phase 3 trial. Lancet Gastroenterol Hepatol. 2022;7(9):830–42. Demonstrated safety and efficacy of odevixibat for the treatment of cholestatic pruritus in patients with PFIC.
Sokol RJ, Gonzales EM, Kamath BM, Baker A, Vig P, Mogul DB, Garner W, Hansen BE, Jacquemin E, Thompson RJ. Predictors of 6-year event-free survival in alagille syndrome patients treated with maralixibat, an ileal bile acid transporter inhibitor. Hepatology. https://doi.org/10.1097/HEP.0000000000000502. Six year study providing early evidence that use of maralixibat, an IBAT inhibitor, may prolong transplant-free survival in patients with Alagille syndrome.
Author information
Authors and Affiliations
Contributions
J.V. was responsible for the design of the manuscript. Both J.V. and N.H participated in the writing and review of the main manuscript as well as the prepared tables and figures.
Corresponding author
Ethics declarations
Conflicts of Interest
Nicole Heinz has no conflicts of interest to disclosure.
Jennifer Vittorio is a consultant for Mirum Pharma and Albireo Pharma.
Human and Animal Rights
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Heinz, N., Vittorio, J. Treatment of Cholestasis in Infants and Young Children. Curr Gastroenterol Rep 25, 344–354 (2023). https://doi.org/10.1007/s11894-023-00891-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11894-023-00891-8