
Abstract
Pathological angiogenesis occurs during tumor progression and leads in the formation of an abnormal vasculature in the tumor microenvironment (TME). The tumor vasculature is disorganized, tortuous and leaky, resulting in high interstitial pressure and hypoxia in the TME, all of which are events that support tumor growth and survival. Given the sustaining role of the tumor vasculature, it has become an increasingly attractive target for the development of anti-cancer therapies. Antibodies, tyrosine kinase inhibitors and cancer vaccines that target pro-angiogenic factors, angiogenesis-associated receptors or tumor blood vessel-associated antigens continue to be developed and tested for therapeutic efficacy. Preferred anti-angiogenic protocols include those that “normalize” the tumor-associated vasculature which reduce hypoxia and improve tumor blood perfusion, resulting in tumor cell apoptosis, decreased immunosuppression, and enhanced effector immune cell infiltration/tumoricidal action within the TME.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
13.1 The Abnormal Tumor Vasculature
Pathological angiogenesis is a hallmark of cancer [1]. Solid tumors, like normal tissues, require nutrients and oxygen as well as a mechanism to expel waste and carbon dioxide. These requirements necessitate the formation of neovascular networks for tumor growth and maintenance. In adult normal tissues, angiogenesis is quiescent, occurring only in specific physiological events such as wound healing. Notably, in such physiological events, new vessels mature and stabilize rapidly because of the tightly regulated balance of pro- and anti-angiogenic factors. On the other hand, in tumors, the production of pro-angiogenic factors is favored over anti-angiogenic factors, turning on the “angiogenic switch.” Tumorigenic conditions such as hypoxia [2], oncogene activation and tumor-suppressor mutation [3] contribute to the skewing of the balance towards the expression of pro-angiogenic factors. This imbalance leads to the sustained growth of tumor blood vessels that are very distinct from their normal counterparts.
Tumors employ several methods to grow blood vessels, the most common and most studied of which is sprouting angiogenesis, in which new capillaries grow from pre-existing ones. During sprouting angiogenesis, vascular endothelial growth factor (VEGF) induces pre-existing capillaries or post-capillary venules to dilate and become leaky [4], allowing plasma proteins to extravasate and form a provisional matrix for activated vascular endothelial cells (VECs). Angiotensin-2 (Ang-2) loosens the association between VECs and abluminal pericytes and, along with proteinases, dissolves the extracellular matrix [5]. Various factors, including VEGF and fibroblast growth factor (FGF), promote the proliferation, migration, and assembly of VECs [6, 7] into tubular structures, followed by the recruitment of perivascular cells and then the production of basement membrane around the new blood vessel [8, 9]. During tumor angiogenesis, pericytes atypically remain only loosely-associated with the blood vessels [10] and the basement membrane is abnormally thick or thin [11]. Aside from sprouting angiogenesis, other mechanisms of tumor vessel growth include: [12] vasculogenesis, in which endothelial progenitor cells are recruited from bone marrow or peripheral blood into the TME to form blood vessels, [13] intussusception, in which VECs re-organize causing blood vessels to split and give rise to daughter vessels, [14] vessel co-option, in which tumor cells grow along existing blood vessels and [15] vasculogenic mimicry, in which tumor cells may de-differentiate into endothelial-like cells and form tubes [16].
When compared to blood vessels in normal somatic tissues, the tumor vasculature is architecturally and functionally abnormal. The tumor vascular network is highly tortuous, disorganized and lacks the normal hierarchical arrangement of arterioles, capillaries and venules [17, 18]. The tumor vessels are also morphologically dilated and leaky with chaotic and variable blood flow, resulting in regions of tumor that are hypoxic and acidic [17]. In conjunction, the cellular components of the tumor blood vessels are also abnormal in their phenotype/function. VECs and pericytes exhibit altered gene expression profiles and elicit a defective basement membrane [11, 13, 19]. These alterations in angiogenesis and vascular components in progressor tumors may be targeted therapeutically in order to treat patients with cancer.
13.2 Vascular Endothelial Growth Factor (VEGF)
The VEGF gene family is comprised of VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor (PIGF). VEGF-A, usually referred to as VEGF, is the main member of the VEGF family and is a major driver of angiogenesis. The importance of VEGF in the development and differentiation of the vascular system has been demonstrated in a study wherein the inactivation of a single VEGF allele led to embryonic lethality [20]. Alternative splicing of VEGF mRNA leads to the generation of soluble isoforms and ECM- or cell membrane-bound isoforms of the protein [21]. Various metalloproteinases (MMPs) cleave bound VEGF to its diffusible form [22]. The angiogenic functions of VEGF are primarily mediated via VEGF receptor 2 (VEGFR2)-dependent signaling in endothelial cells. In this regard, mice deficient in VEGFR2 fail to develop blood islands and organized blood vessels [23].
VEGF is intrinsically overexpressed in a number of tumors [24]. Transcription of VEGF mRNA is driven by hypoxia but may also be induced by transforming events [21]. Mutations in K-ras are associated with the upregulation of VEGF, with disruption of the mutant K-ras allele shown to decrease VEGF activity [25]. Mutations in the Wnt signaling pathway in colonic adenomas also lead to an increase in VEGF expression [26]. In tumors, VEGF induces neoangiogenesis, increases vascular permeability, induces VEC migration and division, maintains the endothelium and reprograms VEC gene expression profiles [24].
13.3 Components of the Tumor Vasculature
13.3.1 Vascular Endothelial Cells (VECs)
VECs arise from hemangioblast precursors that are derived from the ventral floor of the dorsal aorta in the aorta-gonad-mesonephros region [27, 28]. During de novo organization of VECs into vessels (also known as vasculogenesis or angiogenesis), newly formed VECs express growth factor receptors like VEGFR1 and VEGFR2 that allow VECs to proliferate, migrate and form tubal structures in response VEGF and FGF. Once the formed vessels mature, VECs down-regulate their expression of these growth factor receptors. In developing tumor blood vessels, however, VEGFR-1, VEGFR-2 and even VEGFR-3 (which is usually restricted to lymphatic vessels in adults) are re-activated and elevated in the VECs, allowing for the unrestricted proliferation and formation of blood vessels in the VEGF-rich TME [29,30,31]. Aside from growth factor receptors, tumor VECs also upregulate other genes that are typically not expressed on normal VECs [32, 33]. Most of these genes play a role in the angiogenic process but some are induced by hypoxia or under conditions of abnormal blood flow [32, 34].
VECs are held together by cell-to-cell junctions to form a continuous monolayer of cells that lines the blood vessel. The normal endothelium acts as a selective barrier that tightly controls the exchange of substances between the blood and the surrounding tissue [28, 35]. Different membrane bound receptors present in the endothelium allow the active transport of large molecules such as proteins, metabolites and hormones from blood to tissue [36]. The endothelial cells have junctional sections that are very similar to adherens and tight junctions in epithelial cells. The tight junctions are localized in the apical regions to seal the clefts between cells in the luminal surface, therefore, functionally restricting paracellular permeability. On the other hand, adherens junctions occupy the basal position and function to limit paracellular permeability, as well as, to control vessel morphogenesis and stability [37,38,39]. The adherens and tight junctions also transfer intracellular signals that mediate contact inhibition of cell growth, cell polarity and VEC-pericyte interaction and, hence, allow VECs to adapt quickly to evolving changes in their microenvironment, such as release of growth factors, angiogenic cues and inflammatory conditions [39, 40]. Gap junctions are also present in the endothelial cells [38].
The endothelium in tumors is structurally defective, composed of irregularly-shaped endothelial cells that have long, fragile cytoplasmic projections that sometimes extend across the vessel lumen [41] and is characterized by intercellular gaps between endothelial cells, transendothelial holes and endothelial fenestrae (pores) [42,43,44]. Increased production of VEGF in the TME dissolves the VE-cadherins and other adherens junction complexes, disrupting VEC cell-to-cell interactions [45]. These abnormalities make the tumor vessels unusually leaky and highly-permeable to plasma proteins, and even erythrocytes. The leakiness and uncontrolled extravasation of blood and plasma results in increased interstitial (fluid) pressure within the tumor lesion [46]. Structural abnormalities and high interstitial pressure contribute to irregular blood flow resulting in the uneven and impeded distribution of nutrients, oxygen, and (systemically administered) chemotherapeutic drugs within tumors. This also leads to areas in the tumor that are highly-acidic and hypoxic. The leaky tumor vessel can also promote the traffic of tumor cells into the blood stream and the eventual formation of distal metastases [43].
13.3.2 Pericytes
Pericytes and vascular smooth muscle cells (vSMCs), collectively called the mural cells, cover and support the endothelial tubules to maintain vascular integrity. Pericytes are usually found as single cells or a discontinuous single-cell layer around arterioles, capillaries, and post-capillary venules [47]. On the other hand, vSMCs are generally found around arteries and veins as multiple concentric layers of cells to mediate vascular tone and contraction [14, 47]. However, classification between pericytes and vSMCs are not always clear-cut and it has been suggested that there is a continuum of phenotypes ranging from the canonical vSMCs and classical pericytes instead of just two distinct populations of mural cells [14].
Pericytes have a number of different developmental origins such that with a single region of a given blood vessel may identify pericytes that have arisen from a range of precursors [48]. Furthermore, no single universal pericyte marker has been identified to date. As such, pericytes are typically identified using a combination of different characteristics including location (i.e. cells embedded in a basement membrane that is shared with the endothelium [49]), in addition to morphology and gene expression profile.
During physiological angiogenesis, pericytes are recruited to the developing vessel via signals such as platelet-derived growth factor β (PDGFβ), sphingosine 1-phospate (S1P), Ang-1 and transforming growth factor-β1 (TGFβ1). The recruited pericytes form tight connections with the underlying endothelial cells with one pericyte usually in contact with multiple endothelial cells. Such pericyte interaction is very important in maintaining the integrity of the endothelium, playing a central role in regulating endothelial proliferation, differentiation, contractility, tone, stability, and permeability [14, 15, 50,51,52,53]. However, the pericyte-endothelial cell interaction is perturbed in the TME, with poor pericyte investment usually observed in a large proportion of tumor-associated blood vessels. Tumor pericytes have cytoplasmic processes that extend into the tumor tissue [10] and are loosely associated with the endothelium [8, 10]. This abnormal pericyte-endothelial cell association contributes to increased vascular permeability [43], which results in poor perfusion and hypoxia, and modified basement membrane [54]. Vessels are prone to be more hemorrhagic, with a disrupted basement membrane [54]. The pericyte coverage of the endothelium is also very variable, depending on the type of tumor. For example, islet carcinomas have dense pericyte coverage while glioblastomas have reduced numbers of pericytes per unit venule area [8]. Variable coverage can also occur within the same tumor [54]. The pericyte cover is thought to help maintain and stabilize the tumor vessels [55], with “naked” endothelium seemingly more dependent on VEGF for VEC survival [56]. Moreover, the tumor-associated pericytes exhibit an altered marker and protein expression profile, making them attractive targets for therapeutic intervention. Indeed, in tumors that overexpress PDGFRβ and thus have high pericyte density, blocking the PDGFRβ signaling results in detachment of pericytes from the endothelium and results in restricted tumor growth [57,58,59].
13.3.3 Fibroblasts
Cancer-associated fibroblasts (CAFs) are spindle-shaped cells embedded within the extracellular matrix (ECM) that originate from resident fibroblasts and bone marrow-derived mesenchymal precursors [60, 61]. They are phenotypically and functionally distinct from normal fibroblasts and comprise a significant component of the tumor stroma [62].
Although technically not part of the blood vessels, cancer-associated fibroblasts still play a major role in angiogenesis and in the promotion of the tumor blood vessel formation, mostly by producing pro-angiogenic factors. Although cancer cells themselves can release VEGF, the principal source of VEGF in the tumor microenvironment is the CAFs [63]. In cancers such as pancreatic cancer and breast cancer, CAFs produce stromal-derived factor 1 (SDF-1) or CXCL12, which contribute to tumor vascularization by recruiting endothelial precursor cells from the bone marrow [64,65,66]. CAFs could also indirectly promote tumor angiogenesis by secreting chemokines like CXCL1 and CXCL2 to recruit pro-angiogenic macrophages and neutrophils into the tumor microenvironment [67]. Furthermore, CAFs release matrix metalloproteinases that degrade the ECM, thus spatially accommodating the growing blood vessel and, at the same time, releasing VEGF previously sequestered in the ECM [68].
13.3.4 Basement Membrane
The basement membrane (BM) is a complex of proteins, glycoproteins and proteoglycans. The BM is similar to the ECM but has a different density and is always in contact with cells [11, 69]. The general role of the BM is to serve as boundary between tissues compartments, provide structural support, and regulate cell behavior [70].
The vascular basement membrane envelops the VECs and pericytes and is primarily composed of Type IV collagen, laminin, fibronectin and heparin sulfate proteoglycan [69]. The BM is integral in the initiation and resolution of angiogenesis, as it possesses both pro- and anti-angiogenic activities. Quiescent endothelial cells are bound to BM, indicating that the primary signals from BM inhibit VEC proliferation [71]. During active angiogenesis, the BM is degraded leading to the detachment of VECs and pericytes and the release of sequestered growth factors. Pro-angiogenic factors induce VEC to produce and embed in a provisional matrix composed of vitronectin, fibronectin, type I collagen and thrombin. Cryptic domains of Type IV collagen in partially degraded BM and the provisional matrix provides proliferative cues to the VECs [69, 72].
Deposition of basement membrane is dependent on VEC-pericyte interaction and disrupting this interaction by blocking pericyte recruitment, for example, results in perturbed or reduced basement membrane deposition [73,74,75]. Therefore, the abnormalities in tumor basement membrane can be partially explained by the disrupted VEC-pericyte interaction in the tumor. It was previously believed that BM was absent in tumor vasculature. However, more recent studies show that BM is present and can even fully cover tumor vessels albeit in an abnormal manner. Tumor vascular BM has variable thickness and multiple layers, suggesting that the BM has undergone several rounds of remodeling. In addition, it has loose association with VECs and pericytes, which is characteristic of degenerating or forming blood vessels [11].
13.4 Tumor Vasculature in Cancer Progression
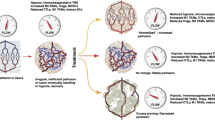
Several classical studies have shown that tumor blood vessels and the process of angiogenesis are necessary for tumor progression. When tumor cells were implanted in avascular tissues, tumor growth was inhibited and limited until sprouting vessels were able to access an existing blood supply [76, 77]. Since the importance of the tumor blood vasculature was demonstrated, numerous studies have shown that it not only supports the tumor by delivering nutrients, it also promotes tumor cell survival, confers drug resistance, helps to limit immune system surveillance, aids in metastasis and promotes stemness (Fig. 13.1).

The aberrant tumor vascular system promotes cancer progression. Tumor vasculogenesis is driven by the release of pro-angiogenic factors within the tumor microenvironment (TME) that fosters a state of locoregional hypoxia, acidosis and high interstitial pressure that is contraindicated for the delivery of protective T cells, but which fosters recruitment and immunosuppression mediated by regulatory cell populations including Treg and MDSCs. Cancer stem cells may also be stabilized in the perivascular niche, with metastatic tumor seeding into the blood as a consequence of “leaky” tumor blood vessels
13.4.1 Tumor Vasculature Supports Tumor Growth and Survival
The tumor vasculature actively supports tumor growth by secreting factors that promote tumor survival. For example, the CAFs in the tumor microenvironment produce epidermal growth factor (EGF), hepatocyte growth factor (HGF) and heregulin that promote tumor proliferation and vitality [78]. VECs in head and neck squamous cell carcinomas (HNSCC) overexpress the anti-apoptotic molecule Bcl-2, which induces the production of VEGF, IL-6, CXCL8/IL-8, and EGF [79, 80]. VEC-derived VEGF binds the VEGFR1 expressed by tumor cells, triggering the production of Bcl-2 in the HNSCC cells and enhancing their survival. IL-6, CXCL8, and epidermal growth factor (EGF) derived from VECs also activate STAT3, Akt, and extracellular signal-regulated kinase (ERK) in the tumor cells, enhancing migration and preventing apoptosis. Interruption of these signaling cascades by silencing Bcl-2 or the factors secreted by the VECs inhibits tumor growth [80].
13.4.2 Aberrant Tumor-Associated Vasculature Confers Drug Resistance
The tumor vasculature intrinsically confers therapy resistance by impeding drug delivery into the tumor lesion. The structural aberrations of the vessels cause excessive leakiness that leads to abnormal plasma accumulation and retention. This results in heterogeneous blood flow and high-interstitial fluid pressure in tumor tissue, preventing uniform distribution of systemically-administered anti-cancer drugs. Indeed, the erratic blood flow in the tumor promotes the concentrated delivery of drugs to the perfused outer (cortical) regions of the tumor while delivery to the poorly-perfused medullary tumor regions requires efficient perfusion/diffusion [81]. Furthermore, extravasation of therapeutic drugs is dependent on fluid movement across the vessel wall, which is influenced by proper pressure gradient between the vessels and the interstitial space. The high interstitial fluid pressure in the tumor disrupts the development of an optimal gradient, thus hindering drug delivery to the tumor core [82]. The tumor-associated vasculature may also actively prevent drug delivery by overexpressing efflux pumps that promote drug removal/detoxification [83, 84].
13.4.3 Tumor-Associated Vasculature Contributes to Immune Escape
The abnormal tumor vasculature not only impedes drug delivery, it also obstructs anti-cancer immune response by limiting entry of effector cells while simultaneously contributing to an immunosuppressive TME. The adaptive T cell response is critical to protective anti-tumor immunity and the endothelium plays an important role in regulating the trafficking of T cells and other leukocytes into peripheral (inflamed) tissues. In normal tissues, resting VECs express little to no adhesion molecules on their luminal surface interfacing with the blood supply. However, during infection or under conditions of tissue injury, pro-inflammatory signals such as lipopolysaccharide (LPS), tumor necrosis factor (TNF)-α and interleukin (IL)-1β induce the expression of adhesion molecules to allow the extravasation of leukocytes in the blood stream [85, 86]. E-selectin and P-selectin on the endothelial cells bind with their ligands Sialyl LewisX and P-selectin glycoprotein ligand 1 on the activated T cells and other leukocytes [87, 88]. This binding event is a weak affinity interaction that induces the tethering and rolling of the T cells on the inflamed endothelium. The inflamed endothelium also expresses chemokines that induce surface integrins, such as lymphocyte function-associated antigen-1 (LFA-1) and very late antigen-4 (VLA-4), on rolling T cells to spread and cluster. The LFA-1 and VLA-4 bind strongly to intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1 (VCAM-1) on activated VECs and, hence, mediate the arrest of interactive T cells on the inflamed endothelium, thus facilitating the extravasation of T cells into tissue. Expression of ICAM-1 and VCAM-1 are down-regulated by an overabundance of angiogenic factors and other VEC-produced proteins (i.e. Egfl7) in the TME [89], resulting in the reduced flux of rolling leukocytes and density of adhering leukocytes in tumor-associated blood vessels [90].
Most studies on leukocyte trafficking into tissues focus on VECs, however, a growing literature suggests that pericytes also play significant roles in the extravasation and activation of effector immune cells. During inflammation, pericytes upregulate adhesion molecules, chemokines and cytokines [91,92,93,94] and may also display characteristics that are usually associated with antigen-presenting cells [95, 96]. Innate immune cells, specifically neutrophils, have been observed to crawl on pericytes [97]. It was later demonstrated that the pericyte-expressed molecule NG2 (aka chondroitin-sulfate glycoprotein-4; CSGP4) interacts with neutrophils and macrophages, instructing them to migrate into inflammatory tissue sites [93]. It is currently unknown whether pericytes provide the same homing signals to transmigrating adaptive immune cells in the TME. Moreover, a recent study indicates that pericytes have a direct role in tumor immune escape by inhibiting CD4 T cell activation and by promoting T cell anergy through Regulator of G-protein Signaling-5 (RGS-5)- and IL-6-dependent pathways [98].
13.4.4 Tumor-Associated Vasculature Promotes the evelopment of Immunosuppressive TMEs
Poor blood perfusion in the tumor leads to hypoxia, which has been shown to promote an immunosuppressive tumor microenvironment. Hypoxia promotes the differentiation of tumor-infiltrating myeloid cells to M2-like tumor-associated macrophages (TAMs). These M2-like TAMs adapt to the hypoxic environment by expressing hypoxia inducible factor (HIF)-1. HIFs are transcription factors that are stabilized under hypoxic conditions and are responsible for the expression of factors that allow cells to survive in a low oxygen environment. M2-like TAMs express high levels of IL-10, arginase I (Arg1) and TGFβ, which are factors that suppress protective type-1 T cell-mediated immunity, and VEGF, which exerts pro-angiogenic effects [99, 100]. Furthermore, M2-like TAMs preferentially localize within hypoxic regions and TAMs that are situated near functional blood vessels express lower levels of the aforementioned M2-associated immunosuppressive gene products [100].
Hypoxic conditions also promote tumor cells to produce immunosuppressive molecules that inhibit effector T cell and NK cell survival/function, while coordinately supporting the T regulatory cells (Tregs) and myeloid-derived suppressor cells (MDSCs) [101, 102]. Hypoxia decreases the survival and activity of T cells in the TME, partly through inhibition of IL-2 in a HIF-1-dependent manner [103, 104]. HIF-1 also drives expression of FoxP3 associated with Treg differentiation [105].
13.4.5 Tumor-Associated Vasculature Contributes to Metastasis
Metastasis is a multistep process that involves dissociation of tumor cells from the primary tumor mass, invasion of surrounding stroma, intravasation to the vasculature, survival in circulation, extravasation to a distal site and subsequent colonization of new organs [106]. The tumor vasculature is primarily involved in intravasation of tumor cells but can also contribute to other metastatic steps [107, 108].
Hypoxia, due to poor blood perfusion brought about by the structural abnormalities in the tumor-associated vasculature, is a contributing factor to the acquisition of malignant phenotype [109]. During hypoxic stress, HIF-1a together with TGFβ triggers epithelial-to-mesenchymal transition (EMT) in tumor cells. EMT is the process wherein epithelial markers are down-regulated and mesenchymal markers are up-regulated, leading to the dissociation of epithelial cell-to-cell interactions and the facilitation of cell motility and invasiveness [110].
Invasion of surrounding stroma by tumor cells requires a remodeling of the ECM and the migration of cancer cells through it. CAFs “lead” the metastatic tumor cells and utilize protease- and force-mediated activities to remodel the ECM [111]. Specifically, fibroblasts implement RhoA and Rho-associated protein kinase 1 to remodel the ECM in an MMP14-dependent manner [112, 113]. Cancer cells then follow these CAF-instructed microtracks in a CDC42-dependent manner [112].
The lack of an intact basement membrane and the disorganized VEC and pericyte interactions make the tumor blood vessels more accessible to motile tumor cells, easing the intravasation process [114]. Studies have shown that a high prevalence of abluminal pericytes limits tumor metastasis [115, 116]. In some tumor models, deficient pericyte coverage of blood vessels, due to a deficiency in pericyte recruitment, can be associated with increased metastatic potential [117]. Conversely, tumor cell intravasation is decreased when pericyte coverage of blood vessels is high within the TME [118].
Cancer cells actively disrupt tumor blood vessel cell interactions in order to facilitate the intravasation process. For example, breast cancer cells express MMP17 that disturbs blood vessel integrity, leading to enhanced tumor cell migration and intravasation [119, 120]. Malignant cells also secrete TGFβ1, which causes endothelial junction retraction [121]. Some cancer cells also express Notch receptors that bind to Notch ligands displayed on VECs, assisting their transmigration through the endothelial junctions [122].
In a complementary manner VECs in the TME also express factors that assist cancer cell intravasation. Breast cancer-associated VECs express the enzyme A Disintegrin And Metalloproteinase 12 (ADAM12) that cleaves vascular endothelial cadherin (VE-cadherin) and TIE2, both of which are also expressed on VECs [123, 124]. VE-cadherin is a component of cell-to-cell adherens junctions that maintains the integrity of the endothelium, hence the ADAM12-induced shedding of this molecule promotes enhanced vascular instability/permeability [124]. TIE2 is the receptor for Ang1 and Ang1/TIE2 ligation is important for pericyte recruitment to nascent blood vessels. ADAM12-mediated disruption of this interaction contributes to vessel destabilization, which is a prerequisite for intravasation [123].
13.4.6 Tumor-Associated Vasculature Promotes Cancer Stemness
Although the existence of cancer stem cells (CSC) remains a subject of active debate, some research suggests that within a tumor mass, a minority of cells retain the ability to give rise to all cell types found in the heterogeneous tumor lesion, much like a normal stem cell has the potential to differentiate into many different types of cells. CSCs usually localize in certain specialized areas in the TME. These niches, composed of cells and matrix components, promote the maintenance of CSCs via direct cell contacts and secreted factors [125]. In HNSCC and glioblastoma, such CSC niches are commonly located in close proximity to tumor blood vessels, defining a perivascular niche [126, 127]. In these cancers, tumor-associated VECs appear directly involved in the maintenance of the CSC population through the release of soluble factors that support CSC self-renewal [128,129,130].
13.5 Therapies Targeting the Tumor Vasculature
Targeting the tumor vasculature rather than tumor cells themselves has several conceptual advantages. First, vascular cells are more readily accessible to circulating effector cells, antibodies and other therapeutic molecules than tumor cells. Also, each vessel supplies hundreds of tumor cells, thus amplifying the tumoricidal efficiency of vascular targeting strategies. When compared to tumor cells, tumor-associated vascular cells are genetically (antigenically) more stable and they exhibit more consistent expression of major histocompatibility complex (MHC) class I (and class II) molecules required for specific T cell recognition. Lastly, anti-angiogenic treatment is not restricted to a specific tumor type, making the general approach applicable to all solid (vascularized) forms for cancer. Since Folkman first proposed anti-angiogenesis as a potential therapeutic strategy in a landmark paper in 1971, numerous antiangiogenic drugs have since been developed and applied clinically (Fig. 13.2; [131, 132]).

Therapeutic intervention strategies to target tumor-associated blood vessels. The development of tumor-associated blood vessels is heavily VEGF-dependent, supporting the evolution and then clinical application of VEGFR-signaling antagonists, including (a) neutralizing anti-VEGF antibodies (such as bevacizumab) and (b) small molecule tyrosine kinase inhibitors (TKI) that block VEGFR-mediated signals (such as sunitinib and pazopanib, among others). TKI have also been developed that block signaling pathways important for pericyte survival/function, such as PDGFR-signaling antagonists (such as dasatinib, among others). It has also recently been appreciated that the TME alters the epigenetic programming of host stromal cell populations, including tumor-associated vascular endothelial cells and pericytes, leading to changes in protein expression that allow for specific T cell targeting (vs. blood vessel cells found in normal tissues). Such anti-tumor blood vessel T cells can be elicited by active vaccination or be adoptively transferred as a therapeutic modality after ex vivo expansion (i.e. as an adoptive cellular therapy; ACT)
13.5.1 Anti-VEGF
Given the major role of VEGF in angiogenesis, an array of anti-angiogenic therapies have been developed to antagonize VEGF/VEGFR-mediated signaling. Bevacizumab (Avastin, Genetech), a humanized anti-VEGF-A monoclonal antibody [133], was the first anti-angiogenic drug approved by the US Food and Drug Administration (FDA). It binds all VEGF isoforms within the TME, therefore blocking VEGF-VEGFR interaction [134,135,136]. In pre-clinical studies of bevacizumab, it was shown that tumor blood vessels that survived this anti-angiogenic treatment exhibited a more mature phenotype, resulting in a more functional vasculature that improved blood perfusion and reduced hypoxia in the TME. However, bevacizumab-induced vascular “normalization” was determined to be transient in nature, hence, necessitating the co-administration of conventional chemotherapy agents to provide enhanced therapeutic efficacy [137, 138]. Bevacizumab has been approved as first- or second-line therapy in the setting of metastatic colorectal cancer, non-small cell lung cancer (NSCLC), recurrent glioblastoma and metastatic renal cell carcinoma, usually in combination with chemotherapeutic drugs [139].
Aflibercept (Zaltrap, Regeneron and Sanofi Aventis) is a recombinant fusion protein consisting of the extracellular domains of VEGFR1 and VEGFR2 and the Fc portion of human IgG. Aflibercept, also known also VEGF-trap, is a decoy receptor that binds VEGF-A, VEGF-B and PIGF. It has been approved for second-line treatment for metastatic colorectal carcinoma in combination with chemotherapy [140]. Both bevacizumab and aflibercept inhibit the pro-angiogenic effects of VEGF, resulting in reduced VEC proliferation, vessel permeability, and blood vessel density, and inhibited tumor growth [135, 141,142,143].
13.5.2 Tyrosine Kinase Inhibitors
Most angiogenic factors (VEGF, PDGF and FGF) contribute pro-tumor signals through specific receptor-type tyrosine kinase (RTKs). In mammals, there are three VEGFR genes VEGFR1, VEGFR2, and VEGFR3 that produce four proteins VEGFR1, VEGFR2, VEGFR3 and sFlt-1 (sVEGFR) [144]. PDGFR has two subtypes (α and β) that bind the four PDGF types (A, B, C, and D) [145]. The FGFR family consists of FGFR1b, FGFR1c, FGFR2b, FGFR2c, FGFR3c, and FGFR4 [146]. RTKs generally consist of extracellular ligand-binding region, a transmembrane helix, and a cytoplasmic tyrosine kinase (TK) domain. Ligand binding activates RTK by inducing receptor dimerization/oligomerization. One receptor in the complex then phosphorylates one or more tyrosines in adjacent RTK then the phosphorylated receptor serves as a site for assembly and activation of intracellular signaling proteins [147]. Tyrosine kinase inhibitors (TKI) are small molecules that interrupt ATP binding to the tyrosine kinase catalytic domain through competition, thereby, inhibiting the downstream receptor signaling [148].
Sunitinib (SU11248, Sutent, Pfizer) targets VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-α, PDGFR-β, FLT3, stem cell factor receptor (c-Kit), RET and CSF-1R [149, 150]. In vitro, sunitinib inhibits VEGF-dependent migration and capillary-tube formation by cultured human umbilical vein endothelial cells (HUVECs), while simultaneously promoting VEC apoptosis [151, 152]. In vivo efficacy studies performed in mouse xenograft models demonstrated that administration of sunitinib inhibits tumor growth, suppresses angiogenesis and decreases microvessel density [153,154,155]. Sunitinib has been approved by the FDA as a first-line therapy in the setting of renal cell carcinoma (RCC) and unresectable pancreatic neuroendocrine tumors (PNET), and as a second-line therapy for patients with gastrointestinal stromal tumors (GIST) as a single agent or in combination with chemotherapy or radiation [149, 156]. Although RCC patient outcome has been reported to be improved after treatment with sunitinib, the best observed clinical responses have tended to be temporary disease stabilization. Furthermore, a treatment refractory RCC usually develops, that may be overcome in some instances by increasing the dosage of sunitinib being administered to patients [12, 157]. Objective clinical response to sunitinib has also been reported for other types of cancer, such as small cell lung carcinoma (SCLC), breast cancer, thyroid cancer, and chondrosarcoma. However, sunitinib has reportedly failed as a therapy in the setting of NSCLC, metastatic breast cancer, and advanced-stage esophageal cancer [149].
Sorafenib (BAY439006, Nexavar, Bayer, Inc.) inhibits Raf-1, wild type BRAF, V599E mutant BRAF, VEGFR-2, VEGFR-3, PDGFR-β, FLT3, and c-Kit [150, 158]. In several murine tumor models, sorafenib treatment resulted in decreased microvessel density, decreased vessel arborization and increased pericyte coverage of blood vessels in the TME. Treatment also blocked neovascularization and ERK activation in VECs [159]. Sorafenib may also target tumor cells directly via disruption of the ERK pathway, since ERK phosphorylation and the proliferation of some tumor cell lines could be effectively inhibited by this TKI. Tumor growth inhibition was observed in some but not all colon, breast, ovarian, thyroid, melanoma, RCC, hepatocellular carcinoma (HCC) and NSCLC xenograft models. These results suggest that the anti-proliferative action of sorafenib is dependent on blocking oncogenic signaling pathways driving tumor proliferation [158, 160, 161]. Sorafenib has been approved for the treatment of patients with advanced RCC, unresectable HCC, and recurrent or metastatic differentiated thyroid carcinoma [162, 163].
Pazopanib (GW786034, Votrient, GlaxoSmithKline) is a second-generation TKI that inhibits VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-α, PDGFR-β and c-Kit. Pazopanib also moderately binds FGFR1, FGFR3 and c-fms receptor tyrosine kinases [164]. In vitro, pazopanib blocked the VEGF-induced phosphorylation of VEGFR2, and thus prevented the proliferation of HUVECs [165]. FGF-induced proliferation of HUVECs was also blocked by pazopanib [164]. Pazopanib was shown to have anti-tumor efficacy in a range of Hu-SCID tumor xenograft models including those using RCC, NSCLC, colorectal cancer, and multiple myeloma cell lines. However, it is reportedly less effective in inhibiting the progression of melanoma, breast cancer, and prostate cancer growth in xenograft models [166]. Pazopanib has been approved by the FDA as a therapeutic agent for patients with RCC or soft tissue sarcoma after standard-of-care chemotherapy [167, 168].
Axitinib (Inlyta, AG-013736, Pfizer) is a second generation TKI that targets VEGFR1, VEGFR2 and VEGFR3 [169]. Cell assays showed that axitinib inhibited VEGF-mediated survival, tube formation and ERK, NOS and Akt signaling in HUVECs. Axitinib also inhibited tumor growth in orthotopic and xenograft tumor models when applied as a single agent or in combination with chemotherapy [170]. Preclinical data in RCC models strongly support the anti-angiogenic action of axitinib, which however, has proven to be reversible upon withdrawal of this drug [171]. Axitinib has been FDA-approved as a second-line treatment option for patients with RCC [169].
Drug dosage and schedule are two critical variables when administering anti-angiogenic small molecule inhibitors and antibodies as therapeutic agents. High doses of such agents may promote excessive pruning of the blood vessels in the TME, leading to tumor necrosis and delayed tumor growth, however, they may also promote a local state of hypoxia, that could in turn lead to a compensatory activation of alternative pro-angiogenic signaling pathways that promote tumorigenicity and metastasis. Conversely, lower doses of anti-angiogenic agents could prune some abnormal vessels while remodeling others, leading to a more normalized vasculature that reduces hypoxia and IFP within the TME, resulting in an improvement in the delivery and efficacy of co-therapies [100, 172, 173].
13.5.3 Immunotherapeutic Targeting of Tumor-Associated Vascular Cells
As previously mentioned, tumor vascular cells are considered genetically stable and highly-accessible to circulating immune cell populations, making them highly-visible for immune surveillance. Based on altered epigenetic programming instigated by cellular stressors (i.e. hypoxia, acidosis, high-IFP), tumor-associated vascular cells exhibit molecular phenotypes that distinguish them from their counterparts in normal tissues [174, 175]. Such molecular differences may be targeted immunologically via active vaccination to develop specific T cells capable of selectively reacting against and normalizing the vasculature in the TME.
Peptide-based and recombinant vaccines based on the VEGFR1 and VEGFR2 gene products have been shown to limit tumor-associated angiogenesis and disease progression, via elicitation of antigen-specific CD8+ T cell responses in pre-clinical animal models [176,177,178,179]. Phase I clinical trials of vaccines that included peptides derived from the VEGFR1 and VEGFR2 proteins demonstrated safety and some degree of efficacy (i.e. they were capable of inducing immune responses that restrict tumor angiogenesis [180,181,182,183]. However, VEGFR2 peptide-based vaccines, when applied in combination with the chemotherapeutic drug gemcitabine, did not improve overall patient survival when compared to a placebo control in a phase II/III trial in the setting of advanced-stage pancreatic cancer [184].
Although early tumor vascular vaccines targeted VEGFRs, a range of additional tumor blood vessel-associated antigens (TBVA) are important to vascular formation and maintenance in the TME, and hence, represent cogent vaccine targets. In this regard, Regulator of G-protein signaling-5 (RGS-5) is overexpressed by tumor-associated (immature) pericytes in the RIP1-Tag5 model of pancreatic islet cell cancer ([185, 186]. Genetic deletion of RGS-5 resulted in pericyte maturation, vasculature normalization and enhancement of adoptive T cell therapy benefits [186]. Expression of the Notch antagonist, Delta-like homolog-1 (DLK-1), is upregulated in pericytes isolated from murine melanoma, colorectal carcinoma and renal cell carcinomas (unpublished). Notably, peptide- and gene-based vaccines targeting DLK1 resulted in delayed tumor progression and the enhancement of CD8+ T cell infiltration in the TME of murine renal carcinomas. Tumors in DLK1-vaccinated mice displayed decreased vascular permeability and hypoxia, reduced expression of hypoxia-inducible proteins in the stroma, and enhanced levels of tumor cell apoptosis in vivo [187]. Tumor endothelial marker 1 (TEM-1; aka CD248) is a TBVA upregulated in VECs, pericytes and other stromal cell populations isolated from a range of tumor histotypes. A DNA vaccine composed of the TEM1 gene fused with the C fragment of tetanus toxoid was shown to increase CD3+ T cell infiltrates in the tumors of treated animals in association with slowed tumor growth [188]. Furthermore, dendritic cell (DC)-based vaccines integrating HLA-A2 class I-presented peptides derived from TBVA including DLK1, EphA2, hemoglobin-β (HBB), NG2, neuropilin (NRP)-1, PDGFRβ, RGS5 and TEM1 were shown to be effective in providing protective or therapeutic immunity against HLA-A2neg melanoma and colon carcinomas in HLA-A2 transgenic mice. Specific vaccination induced the generation of HLA-A2-restricted CD8+ T effector cells that recognized relevant antigen-expressing pericytes and/or VECs isolated from tumors, but not normal tissue [189].
Vaccination against TBVAs may also potentially promote the broadening of a therapeutic immune response via a progressive process termed “epitope spreading”. In such a paradigm, anti-TBVA T cells would promote vascular normalization and tumor apoptosis, leading to tumor antigen uptake by recruited dendritic cells that could subsequently activate (i.e. crossprime) secondary waves of tumor-reactive T cells yielding added therapeutic benefit based on combined immune targeting of both TBVA+ vascular cells and tumor cells [188].
13.5.4 Combination (Immune)therapies Targeting Tumor-Associated Blood Vessels
Anti-angiogenic antibodies and small molecules have been shown to be efficacious in pre-clinical tumor models and a growing number of human cancers. However, the therapeutic benefits of these agents are commonly transient in nature, with the subsequent progression of (same) treatment-refractory disease. Developed drug-resistance may be attributed to tumor adaptation events and the adoption of alternative pro-angiogenic/pro-survival pathways [Reviewed in [190]]. As a consequence, extensive research is being directed at developing novel second-line or co-first-line (combination) therapeutic regimens that will complement angiogenesis inhibition, leading to improved treatment outcomes.
Notably, many anti-angiogenic agents/treatments create a transient state of vascular normalization in the TME, during which tumor are more receptive to delivery of chemotherapeutic drugs and immune cell populations [191]. This has prompted logical second-set clinical protocols integrating anti-angiogenic drugs such as bevacizumab, along with chemotherapeutic drugs, such as paclitaxel, cisplatin, and carboplatin, amongst others [139]. In an ovarian carcinoma xenograft model, combined treatment with pazopanib and low doses of topotecan exhibited superior anti-tumor efficacy when compared with either single modality [192]. In another example, the combination of sunitinib and gemcitabine inhibited the growth and metastasis of murine pancreatic carcinoma in an orthotopic model to a greater degree than individual treatment with either sunitinib or gemcitabine [193].
However, there are some reports that under some conditions, anti-angiogenic drugs might actually impede the delivery of chemotherapeutic drugs into the TME. Drug uptake and retention of docetaxel was evaluated in patients with advanced NSCLC treated with bevacizumab and it was shown that VEGF inhibition decreased the penetration of water and the chemotherapeutic agent into tumor sites [194]. This strongly suggests that temporal interval wherein a given angiogenesis inhibitor normalizes the tumor vasculature must first be determined in order to optimize the delivery benefit to the co-therapeutic agent. Unfortunately, there appears to be a high-degree of variation in the temporal window of vascular normalization for various anti-angiogenic modalities likely necessitating the need to develop non-invasive imaging techniques to monitor tumor blood perfusion in patients to define an optimal combination treatment schedule [195, 196].
Tumor hypoxia is known to limit the efficacy of radiation therapy in the cancer setting, hence, the application of angiogenesis inhibitors with radiation has the potential to improve treatment outcomes. Indeed, the combination of VEGF inhibitors with irradiation has consistently demonstrated superior therapeutic benefits (i.e. prolonged delay in tumor growth) despite differences in the combinations of radiation schedules and drugs and doses used [197].
Anti-angiogenic agents may also improve the efficacy of cancer vaccines, that have typically proven to be immunogenic but of limited clinical benefit in the vast majority of trials thus far evaluated [198, 199]. A major limitation in vaccine efficacy likely involves the inefficient delivery of vaccine-induced T cells into the TME [200]. Since angiogenesis inhibitors can normalize blood vessels in the TME and promote preferred changes in T cell recruiting chemokine gradients and vascular adhesion molecules, in addition to reducing hypoxia and immunosuppressive MDSC and Treg content populations [200, 201], anti-angiogenic agents should serve as an effective adjuvant for cancer vaccines. Indeed, DC-based vaccines against the OVA257–264 peptide epitope in combination with the TKIs axitinib, sunitinib or dasatinib (a BCR-ABL, SRC, c-KIT, PDGFR, and ephrin tyrosine kinases inhibitor), each demonstrated superior therapeutic benefit in the murine M05 (B16.OVA) melanoma model when compared to treatment with vaccine alone or TKI alone. In vaccine + TKI treated mice, the expression of VCAM-1 and CXCR3 ligand chemokines in the tumor endothelium were augmented in association with robust type-1 T cell infiltration and extended overall survival [201,202,203].
13.6 Conclusions and Future Directions
The tumor vasculature is an excellent target for developing improved cancer therapeutics. The cells of the tumor vasculature are more genetically stable than tumor cells, express antigens that are not found in their normal cell counterparts, and are more accessible to therapeutic molecules and immune cells. Indeed, anti-angiogenic agents that target tumor blood vessels have proven capable of pruning immature vessels and normalizing the vasculature, resulting in reduced hypoxia and IFP, and increased tumor cell apoptosis. Furthermore, these approaches facilitate the delivery of drugs and immune effector cells into the therapeutic TME. However, since anti-angiogenic agent-induced vascular remodeling is only transient and subject to the development of drug-refractory progressive disease, it fully expected that these modalities will prove most clinically effective only when provided in combination (chemo-, radio-, immuno-therapy) approaches that will require careful optimization of each modality’s dose and schedule. In this regard, it will also likely be very important to identify biomarkers that could be used to predict clinical outcome of patients treated with anti-angiogenic drugs as monotherapies and in combination protocols [173].
Therapeutic vaccines promoting immune targeting of tumor vascular cells are an emerging anti-cancer strategy. Ideally, these vaccines should target antigens that are exclusively expressed (or grossly overexpressed) by cells in tumor blood vessels to minimize concerns for negative consequences in normal tissues (including the eye, blood-brain barrier) or upon normal angiogenic processes (i.e. wound-healing, pregnancy). A number of studies have shown that vaccination based on TBVAs is effective in normalizing the vasculature, inducing a T-cell effector response, and inhibiting tumor growth in murine models, without affecting the wound healing process or aspects of animal husbandry [177]. The continued search for novel TBVAs that are broadly shared across solid cancers is clearly of great interest for prospective clinical targeting. As with any vaccine involving the targeting of non-mutated “self” antigens, adjuvants might have to be administered with TBVA-based vaccines in order to effectively break operational immune tolerance in patients. Furthermore, the ability of vaccine-induced anti-TBVA T cells to be recruited into the TME will require implementation of conditioning regimens that may be accommodated via the use of TKIs that both improve T effector cell delivery into tumor sites and coordinately mitigate the immunosuppressive influence of MDSC and Treg regulatory cell populations. Such potentiation of anti-TBVA T cell vasculocidal/tumoricidal action might also be evidenced in combination regimens integrating immune checkpoint inhibitors, such as anti-CTLA-4 and/or anti-PD-1/PD-L1, which mediate profound anti-tumor clinical activity in the setting of multiple forms of human cancer [204].
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol Cell Physiol. 2001;280:C1367–74.
Bottos A, Bardelli A. Oncogenes and angiogenesis: a way to personalize anti-angiogenic therapy? Cell Mol Life Sci. 2013;70:4131–40.
Bates D, Hillman N, Williams B, Neal C, Pocock T. Regulation of microvascular permeability by vascular endothelial growth factors. J Anat. 2002;200:581–97.
Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, Yancopoulos GD, Wiegand SJ. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science. 1999;284:1994–8.
Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201–7.
Gavalas NG, Liontos M, Trachana SP, Bagratuni T, Arapinis C, Liacos C, Dimopoulos MA, Bamias A. Angiogenesis-related pathways in the pathogenesis of ovarian cancer. Int J Mol Sci. 2013;14:15885–909.
Bergers G, Song S. The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol. 2005;7:452–64.
Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–93.
Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM. Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2002;160:985–1000.
Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–15.
Adelaiye R, Ciamporcero E, Miles KM, Sotomayor P, Bard J, Tsompana M, Conroy D, Shen L, Ramakrishnan S, Ku SY, et al. Sunitinib dose escalation overcomes transient resistance in clear cell renal cell carcinoma and is associated with epigenetic modifications. Mol Cancer Ther. 2015;14:513–22.
Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. 2012;74:13–40.
Armulik A, Abramsson A, Betsholtz C. Endothelial/pericyte interactions. Circ Res. 2005;97:512–23.
Armulik A, Genove G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215.
Plate KH, Scholz A, Dumont DJ. Tumor angiogenesis and anti-angiogenic therapy in malignant gliomas revisited. Acta Neuropathol. 2012;124:763–75.
Baluk P, Hashizume H, McDonald DM. Cellular abnormalities of blood vessels as targets in cancer. Curr Opin Genet Dev. 2005;15:102–11.
McDonald DM, Choyke PL. Imaging of angiogenesis: from microscope to clinic. Nat Med. 2003;9:713–25.
Wragg JW, Durant S, McGettrick HM, Sample KM, Egginton S, Bicknell R. Shear stress regulated gene expression and angiogenesis in vascular endothelium. Microcirculation. 2014;21:290–300.
Ferrara N, Davis-Smyth T. The biology of vascular endothelial growth factor. Endocr Rev. 1997;18:4–25.
Ferrara N, Mass RD, Campa C, Kim R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu Rev Med. 2007;58:491–504.
Lee S, Jilani SM, Nikolova GV, Carpizo D, Iruela-Arispe ML. Processing of VEGF-A by matrix metalloproteinases regulates bioavailability and vascular patterning in tumors. J Cell Biol. 2005;169:681–91.
Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6.
Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol Off J Am Soc Clin Oncol. 2002;20:4368–80.
Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–80.
Zhang X, Gaspard JP, Chung DC. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 2001;61:6050–4.
Marshall CJ, Moore RL, Thorogood P, Brickell PM, Kinnon C, Thrasher AJ. Detailed characterization of the human aorta-gonad-mesonephros region reveals morphological polarity resembling a hematopoietic stromal layer. Dev Dyn. 1999;215:139–47.
Sumpio BE, Riley JT, Dardik A. Cells in focus: endothelial cell. Int J Biochem Cell Biol. 2002;34:1508–12.
Ruoslahti E. Specialization of tumour vasculature. Nat Rev Cancer. 2002;2:83–90.
Schwartz JD, Rowinsky EK, Youssoufian H, Pytowski B, Wu Y. Vascular endothelial growth factor receptor-1 in human cancer: concise review and rationale for development of IMC-18F1 (human antibody targeting vascular endothelial growth factor receptor-1). Cancer. 2010;116:1027–32.
Smith NR, Baker D, James NH, Ratcliffe K, Jenkins M, Ashton SE, Sproat G, Swann R, Gray N, Ryan A, et al. Vascular endothelial growth factor receptors VEGFR-2 and VEGFR-3 are localized primarily to the vasculature in human primary solid cancers. Clin Cancer Res. 2010;16:3548–61.
Otsubo T, Hida Y, Ohga N, Sato H, Kai T, Matsuki Y, Takasu H, Akiyama K, Maishi N, Kawamoto T, et al. Identification of novel targets for antiangiogenic therapy by comparing the gene expressions of tumor and normal endothelial cells. Cancer Sci. 2014;105:560–7.
St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B, et al. Genes expressed in human tumor endothelium. Science. 2000;289:1197–202.
Nanda A, St Croix B. Tumor endothelial markers: new targets for cancer therapy. Curr Opin Oncol. 2004;16:44–9.
Michiels C. Endothelial cell functions. J Cell Physiol. 2003;196:430–43.
Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, Pober JS, Wick TM, Konkle BA, Schwartz BS, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood. 1998;91:3527–61.
Dejana E, Orsenigo F, Molendini C, Baluk P, McDonald DM. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009a;335:17–25.
Dejana E, Tournier-Lasserve E, Weinstein BM. The control of vascular integrity by endothelial cell junctions: molecular basis and pathological implications. Dev Cell. 2009b;16:209–21.
Lampugnani MG. Endothelial cell-to-cell junctions: adhesion and signaling in physiology and pathology. Cold Spring Harb Perspect Med. 2012;2(10):a006528.
Wallez Y, Huber P. Endothelial adherens and tight junctions in vascular homeostasis, inflammation and angiogenesis. Biochim Biophys Acta. 2008;1778:794–809.
Dudley AC. Tumor endothelial cells. Cold Spring Harb Perspect Med. 2012;2(10):a006536.
Dvorak HF, Nagy JA, Dvorak JT, Dvorak AM. Identification and characterization of the blood vessels of solid tumors that are leaky to circulating macromolecules. Am J Pathol. 1988;133:95–109.
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM. Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol. 2000;156:1363–80.
Roberts WG, Delaat J, Nagane M, Huang S, Cavenee WK, Palade GE. Host microvasculature influence on tumor vascular morphology and endothelial gene expression. Am J Pathol. 1998;153:1239–48.
Claesson-Welsh L. Blood vessels as targets in tumor therapy. Ups J Med Sci. 2012;117:178–86.
Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. 2002;2:266–76.
Barlow KD, Sanders AM, Soker S, Ergun S, Metheny-Barlow LJ. Pericytes on the tumor vasculature: jekyll or hyde? Cancer microenvironment: official journal of the international cancer microenvironment. Society. 2013;6:1–17.
Birbrair A, Zhang T, Wang ZM, Messi ML, Mintz A, Delbono O. Pericytes at the intersection between tissue regeneration and pathology. Clin Sci (Lond). 2015;128:81–93.
Sims DE. The pericyte—a review. Tissue Cell. 1986;18(2):153–74. PMID 3085281
Gerhardt H, Betsholtz C. Endothelial-pericyte interactions in angiogenesis. Cell Tissue Res. 2003;314:15–23.
Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–53.
Lindahl P, Hellstrom M, Kalen M, Betsholtz C. Endothelial-perivascular cell signaling in vascular development: lessons from knockout mice. Curr Opin Lipidol. 1998;9:407–11.
von Tell D, Armulik A, Betsholtz C. Pericytes and vascular stability. Exp Cell Res. 2006;312:623–9.
Diaz-Flores L, Gutierrez R, Madrid JF, Varela H, Valladares F, Acosta E, Martin-Vasallo P, Diaz-Flores L Jr. Pericytes. Morphofunction, interactions and pathology in a quiescent and activated mesenchymal cell niche. Histol Histopathol. 2009;24:909–69.
Hall AP. Review of the pericyte during angiogenesis and its role in cancer and diabetic retinopathy. Toxicol Pathol. 2006;34:763–75.
Benjamin LE, Golijanin D, Itin A, Pode D, Keshet E. Selective ablation of immature blood vessels in established human tumors follows vascular endothelial growth factor withdrawal. J Clin Invest. 1999;103:159–65.
Bergers G, Song S, Meyer-Morse N, Bergsland E, Hanahan D. Benefits of targeting both pericytes and endothelial cells in the tumor vasculature with kinase inhibitors. J Clin Invest. 2003;111:1287–95.
Reinmuth N, Liu W, Jung YD, Ahmad SA, Shaheen RM, Fan F, Bucana CD, McMahon G, Gallick GE, Ellis LM. Induction of VEGF in perivascular cells defines a potential paracrine mechanism for endothelial cell survival. FASEB J. 2001;15:1239–41.
Shaheen RM, Tseng WW, Davis DW, Liu W, Reinmuth N, Vellagas R, Wieczorek AA, Ogura Y, McConkey DJ, Drazan KE, et al. Tyrosine kinase inhibition of multiple angiogenic growth factor receptors improves survival in mice bearing colon cancer liver metastases by inhibition of endothelial cell survival mechanisms. Cancer Res. 2001;61:1464–8.
Kharaishvili G, Simkova D, Bouchalova K, Gachechiladze M, Narsia N, Bouchal J. The role of cancer-associated fibroblasts, solid stress and other microenvironmental factors in tumor progression and therapy resistance. Cancer Cell Int. 2014;14:41.
Ohlund D, Elyada E, Tuveson D. Fibroblast heterogeneity in the cancer wound. J Exp Med. 2014;211:1503–23.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6:392–401.
Fukumura D, Xavier R, Sugiura T, Chen Y, Park EC, Lu N, Selig M, Nielsen G, Taksir T, Jain RK, et al. Tumor induction of VEGF promoter activity in stromal cells. Cell. 1998;94:715–25.
Cirri P, Chiarugi P. Cancer associated fibroblasts: the dark side of the coin. Am J Cancer Res. 2011;1:482–97.
Kryczek I, Wei S, Keller E, Liu R, Zou W. Stroma-derived factor (SDF-1/CXCL12) and human tumor pathogenesis. Am J Physiol Cell Physiol. 2007;292:C987–95.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121:335–48.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient Neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–47.
Lederle W, Hartenstein B, Meides A, Kunzelmann H, Werb Z, Angel P, Mueller MM. MMP13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis. 2010;31:1175–84.
Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–33.
Paulsson M. Basement membrane proteins: structure, assembly, and cellular interactions. Crit Rev Biochem Mol Biol. 1992;27:93–127.
Form DM, Pratt BM, Madri JA. Endothelial cell proliferation during angiogenesis. In vitro modulation by basement membrane components. Lab Invest. 1986;55:521–30.
Xu J, Rodriguez D, Petitclerc E, Kim JJ, Hangai M, Moon YS, Davis GE, Brooks PC. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J Cell Biol. 2001;154:1069–79.
Geevarghese A, Herman IM. Pericyte-endothelial crosstalk: implications and opportunities for advanced cellular therapies. Translational research. J Lab Clin Med. 2014;163:296–306.
Stratman AN, Davis GE. Endothelial cell-pericyte interactions stimulate basement membrane matrix assembly: influence on vascular tube remodeling, maturation, and stabilization. Microsc Microanal. 2012;18:68–80.
Stratman AN, Malotte KM, Mahan RD, Davis MJ, Davis GE. Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood. 2009;114:5091–101.
Ausprunk DH, Knighton DR, Folkman J. Vascularization of normal and neoplastic tissues grafted to the chick chorioallantois. Role of host and preexisting graft blood vessels. Am J Pathol. 1975;79:597–618.
Gimbrone MA Jr, Cotran RS, Leapman SB, Folkman J. Tumor growth and neovascularization: an experimental model using the rabbit cornea. J Natl Cancer Inst. 1974;52:413–27.
Micke P, Ostman A. Exploring the tumour environment: cancer-associated fibroblasts as targets in cancer therapy. Expert Opin Ther Targets. 2005;9:1217–33.
Kaneko T, Zhang Z, Mantellini MG, Karl E, Zeitlin B, Verhaegen M, Soengas MS, Lingen M, Strieter RM, Nunez G, et al. Bcl-2 orchestrates a cross-talk between endothelial and tumor cells that promotes tumor growth. Cancer Res. 2007;67:9685–93.
Neiva KG, Zhang Z, Miyazawa M, Warner KA, Karl E, Nor JE. Cross talk initiated by endothelial cells enhances migration and inhibits anoikis of squamous cell carcinoma cells through STAT3/Akt/ERK signaling. Neoplasia. 2009;11:583–93.
Narang AS, Varia S. Role of tumor vascular architecture in drug delivery. Adv Drug Deliv Rev. 2011;63:640–58.
Boucher Y, Baxter LT, Jain RK. Interstitial pressure gradients in tissue-isolated and subcutaneous tumors: implications for therapy. Cancer Res. 1990;50:4478–84.
Azzi S, Hebda JK, Gavard J. Vascular permeability and drug delivery in cancers. Front Oncol. 2013;3:211.
Cordon-Cardo C, O'Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. The journal of histochemistry and cytochemistry: official journal of the Histochemistry. Society. 1990;38:1277–87.
Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10:826–37.
Scott DW, Patel RP. Endothelial heterogeneity and adhesion molecules N-glycosylation: implications in leukocyte trafficking in inflammation. Glycobiology. 2013;23:622–33.
Bevilacqua MP. Endothelial-leukocyte adhesion molecules. Annu Rev Immunol. 1993;11:767–804.
von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med. 2000;343:1020–34.
Delfortrie S, Pinte S, Mattot V, Samson C, Villain G, Caetano B, Lauridant-Philippin G, Baranzelli MC, Bonneterre J, Trottein F, et al. Egfl7 promotes tumor escape from immunity by repressing endothelial cell activation. Cancer Res. 2011;71:7176–86.
Wu NZ, Klitzman B, Dodge R, Dewhirst MW. Diminished leukocyte-endothelium interaction in tumor microvessels. Cancer Res. 1992;52:4265–8.
Balabanov R, Washington R, Wagnerova J, Dore-Duffy P. CNS microvascular pericytes express macrophage-like function, cell surface integrin alpha M, and macrophage marker ED-2. Microvasc Res. 1996;52:127–42.
Edelman DA, Jiang Y, Tyburski JG, Wilson RF, Steffes CP. Cytokine production in lipopolysaccharide-exposed rat lung pericytes. J Trauma. 2007;62:89–93.
Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML, Gartner F, Khandoga AG, Legate KR, Pless R, et al. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and 'instruct' them with pattern-recognition and motility programs. Nat Immunol. 2013;14:41–51.
Verbeek MM, Westphal JR, Ruiter DJ, de Waal RM. T lymphocyte adhesion to human brain pericytes is mediated via very late antigen-4/vascular cell adhesion molecule-1 interactions. J Immunol. 1995;154:5876–84.
Balabanov R, Beaumont T, Dore-Duffy P. Role of central nervous system microvascular pericytes in activation of antigen-primed splenic T-lymphocytes. J Neurosci Res. 1999;55:578–87.
Maier CL, Pober JS. Human placental pericytes poorly stimulate and actively regulate allogeneic CD4 T cell responses. Arterioscler Thromb Vasc Biol. 2011;31:183–9.
Proebstl D, Voisin MB, Woodfin A, Whiteford J, D'Acquisto F, Jones GE, Rowe D, Nourshargh S. Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med. 2012;209:1219–34.
Bose A, Barik S, Banerjee S, Ghosh T, Mallick A, Bhattacharyya Majumdar S, Goswami KK, Bhuniya A, Banerjee S, Baral R, et al. Tumor-derived vascular pericytes anergize Th cells. J Immunol. 2013;191:971–81.
Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel). 2014;6:1670–90.
Huang Y, Goel S, Duda DG, Fukumura D, Jain RK. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013;73:2943–8.
Barsoum IB, Koti M, Siemens DR, Graham CH. Mechanisms of hypoxia-mediated immune escape in cancer. Cancer Res. 2014;74:7185–90.
Chouaib S, Messai Y, Couve S, Escudier B, Hasmim M, Noman MZ. Hypoxia promotes tumor growth in linking angiogenesis to immune escape. Front Immunol. 2012;3:21.
Lukashev D, Klebanov B, Kojima H, Grinberg A, Ohta A, Berenfeld L, Wenger RH, Ohta A, Sitkovsky M. Cutting edge: hypoxia-inducible factor 1alpha and its activation-inducible short isoform I.1 negatively regulate functions of CD4+ and CD8+ T lymphocytes. J Immunol. 2006;177:4962–5.
Zuckerberg AL, Goldberg LI, Lederman HM. Effects of hypoxia on interleukin-2 mRNA expression by T lymphocytes. Crit Care Med. 1994;22:197–203.
Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, de Zoeten EF, Cambier JC, Stenmark KR, Colgan SP, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci U S A. 2012;109:E2784–93.
Jin X, Zhu Z, Shi Y. Metastasis mechanism and gene/protein expression in gastric cancer with distant organs metastasis. Bull Cancer. 2014;101:E1–12.
Cao Z, Shang B, Zhang G, Miele L, Sarkar FH, Wang Z, Zhou Q. Tumor cell-mediated neovascularization and lymphangiogenesis contrive tumor progression and cancer metastasis. Biochim Biophys Acta. 2013;1836:273–86.
Moserle L, Casanovas O. Anti-angiogenesis and metastasis: a tumour and stromal cell alliance. J Intern Med. 2013;273:128–37.
Tsai YP, Wu KJ. Hypoxia-regulated target genes implicated in tumor metastasis. J Biomed Sci. 2012;19:102.
Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54.
Friedl P, Gilmour D. Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol. 2009;10:445–57.
Gaggioli C, Hooper S, Hidalgo-Carcedo C, Grosse R, Marshall JF, Harrington K, Sahai E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol. 2007;9:1392–400.
Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS, Friedl P. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9:893–904.
Beerling E, Ritsma L, Vrisekoop N, Derksen PW, van Rheenen J. Intravital microscopy: new insights into metastasis of tumors. J Cell Sci. 2011;124:299–310.
Gerhardt H, Semb H. Pericytes: gatekeepers in tumour cell metastasis? J Mol Med (Berl). 2008;86:135–44.
Xian X, Hakansson J, Stahlberg A, Lindblom P, Betsholtz C, Gerhardt H, Semb H. Pericytes limit tumor cell metastasis. J Clin Invest. 2006;116:642–51.
Yonenaga Y, Mori A, Onodera H, Yasuda S, Oe H, Fujimoto A, Tachibana T, Imamura M. Absence of smooth muscle actin-positive pericyte coverage of tumor vessels correlates with hematogenous metastasis and prognosis of colorectal cancer patients. Oncology. 2005;69:159–66.
Welen K, Jennbacken K, Tesan T, Damber JE. Pericyte coverage decreases invasion of tumour cells into blood vessels in prostate cancer xenografts. Prostate Cancer Prostatic Dis. 2009;12:41–6.
Chabottaux V, Ricaud S, Host L, Blacher S, Paye A, Thiry M, Garofalakis A, Pestourie C, Gombert K, Bruyere F, et al. Membrane-type 4 matrix metalloproteinase (MT4-MMP) induces lung metastasis by alteration of primary breast tumour vascular architecture. J Cell Mol Med. 2009;13:4002–13.
Sohail A, Marco M, Zhao H, Shi Q, Merriman S, Mobashery S, Fridman R. Characterization of the dimerization interface of membrane type 4 (MT4)-matrix metalloproteinase. J Biol Chem. 2011;286:33178–89.
Reymond N, d'Agua BB, Ridley AJ. Crossing the endothelial barrier during metastasis. Nat Rev Cancer. 2013;13:858–70.
Garcia A, Kandel JJ. Notch: a key regulator of tumor angiogenesis and metastasis. Histol Histopathol. 2012;27:151–6.
Frohlich C, Klitgaard M, Noer JB, Kotzsch A, Nehammer C, Kronqvist P, Berthelsen J, Blobel C, Kveiborg M, Albrechtsen R, et al. ADAM12 is expressed in the tumour vasculature and mediates ectodomain shedding of several membrane-anchored endothelial proteins. Biochem J. 2013;452:97–109.
Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell. 2013;26:441–54.
Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008;132:598–611.
Charles N, Holland EC. The perivascular niche microenvironment in brain tumor progression. Cell Cycle. 2010;9:3012–21.
Ritchie KE, Nor JE. Perivascular stem cell niche in head and neck cancer. Cancer Lett. 2013;338:41–6.
Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, Oh EY, Gaber MW, Finklestein D, Allen M, et al. A perivascular niche for brain tumor stem cells. Cancer Cell. 2007;11:69–82.
Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman JI, Prince ME, Wicha MS, Nor JE. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. 2010;70:9969–78.
Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, Temple S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–40.
Fens MH, Storm G, Schiffelers RM. Tumor vasculature as target for therapeutic intervention. Expert Opin Investig Drugs. 2010;19:1321–38.
Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6.
Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, Winkler M, Ferrara N. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57:4593–9.
Rosen LS. Angiogenesis inhibition in solid tumors. Cancer J. 2001;7(suppl 3):S120–8.
Shih T, Lindley C. Bevacizumab: an angiogenesis inhibitor for the treatment of solid malignancies. Clin Ther. 2006;28:1779–802.
Wang Y, Fei D, Vanderlaan M, Song A. Biological activity of bevacizumab, a humanized anti-VEGF antibody in vitro. Angiogenesis. 2004;7:335–45.
Ma J, Waxman DJ. Combination of antiangiogenesis with chemotherapy for more effective cancer treatment. Mol Cancer Ther. 2008;7:3670–84.
Zhou Q, Guo P, Gallo JM. Impact of angiogenesis inhibition by sunitinib on tumor distribution of temozolomide. Clin Cancer Res. 2008;14:1540–9.
De Falco S. Antiangiogenesis therapy: an update after the first decade. Korean J Intern Med. 2014;29:1–11.
Ciombor KK, Berlin J, Chan E. Aflibercept. Clin Cancer Res. 2013;19:1920–5.
Byrne AT, Ross L, Holash J, Nakanishi M, Hu L, Hofmann JI, Yancopoulos GD, Jaffe RB. Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin Cancer Res. 2003;9:5721–8.
Rosen LS. VEGF-targeted therapy: therapeutic potential and recent advances. Oncologist. 2005;10:382–91.
Verheul HM, Hammers H, van Erp K, Wei Y, Sanni T, Salumbides B, Qian DZ, Yancopoulos GD, Pili R. Vascular endothelial growth factor trap blocks tumor growth, metastasis formation, and vascular leakage in an orthotopic murine renal cell cancer model. Clin Cancer Res. 2007;13:4201–8.
Shibuya M. Vascular endothelial growth factor and its receptor system: physiological functions in angiogenesis and pathological roles in various diseases. J Biochem. 2013;153:13–9.
Chen PH, Chen X, He X. Platelet-derived growth factors and their receptors: structural and functional perspectives. Biochim Biophys Acta. 2013;1834:2176–86.
Katoh M, Nakagama H. FGF receptors: cancer biology and therapeutics. Med Res Rev. 2014;34:280–300.
Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–34.
Hojjat-Farsangi M. Small-molecule inhibitors of the receptor tyrosine kinases: promising tools for targeted cancer therapies. Int J Mol Sci. 2014;15:13768–801.
Kim S, Ding W, Zhang L, Tian W, Chen S. Clinical response to sunitinib as a multitargeted tyrosine-kinase inhibitor (TKI) in solid cancers: a review of clinical trials. Onco Targets Ther. 2014;7:719–28.
Nguewa PA, Calvo A, Pullamsetti SS, Banat GA, Grimminger F, Savai R. Tyrosine kinase inhibitors with antiangiogenic properties for the treatment of non-small cell lung cancer. Expert Opin Investig Drugs. 2011;20:61–74.
Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, Vandenbeldt K, Qian CN, Teh BT. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res. 2010;70:1053–62.
Osusky KL, Hallahan DE, Fu A, Ye F, Shyr Y, Geng L. The receptor tyrosine kinase inhibitor SU11248 impedes endothelial cell migration, tubule formation, and blood vessel formation in vivo, but has little effect on existing tumor vessels. Angiogenesis. 2004;7:225–33.
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–37.
O'Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood. 2003;101:3597–605.
Young E, Miele L, Tucker KB, Huang M, Wells J, Gu JW. SU11248, a selective tyrosine kinases inhibitor suppresses breast tumor angiogenesis and growth via targeting both tumor vasculature and breast cancer cells. Cancer Biol Ther. 2010;10:703–11.
Vinik AI, Raymond E. Pancreatic neuroendocrine tumors: approach to treatment with focus on sunitinib. Ther Adv Gastroenterol. 2013;6:396–411.
Joosten SC, Hamming L, Soetekouw PM, Aarts MJ, Veeck J, van Engeland M, Tjan-Heijnen VC. Resistance to sunitinib in renal cell carcinoma: from molecular mechanisms to predictive markers and future perspectives. Biochim Biophys Acta. 2015;1855:1–16.
Iyer R, Fetterly G, Lugade A, Thanavala Y. Sorafenib: a clinical and pharmacologic review. Expert Opin Pharmacother. 2010;11:1943–55.
Murphy DA, Makonnen S, Lassoued W, Feldman MD, Carter C, Lee WM. Inhibition of tumor endothelial ERK activation, angiogenesis, and tumor growth by sorafenib (BAY43-9006). Am J Pathol. 2006;169:1875–85.
Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Sorafenib blocks the RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer Res. 2006;66:11851–8.
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099–109.
Ibrahim N, Yu Y, Walsh WR, Yang JL. Molecular targeted therapies for cancer: sorafenib mono-therapy and its combination with other therapies (review). Oncol Rep. 2012;27:1303–11.
Thomas L, Lai SY, Dong W, Feng L, Dadu R, Regone RM, Cabanillas ME. Sorafenib in metastatic thyroid cancer: a systematic review. Oncologist. 2014;19:251–8.
Sonpavde G, Hutson TE, Sternberg CN. Pazopanib, a potent orally administered small-molecule multitargeted tyrosine kinase inhibitor for renal cell carcinoma. Expert Opin Investig Drugs. 2008;17:253–61.
Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, Davis-Ward RG, Epperly AH, Hinkle KW, Hunter RN 3rd, Johnson JH, et al. Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-b enzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51:4632–40.
Hamberg P, Verweij J, Sleijfer S. (Pre-)clinical pharmacology and activity of pazopanib, a novel multikinase angiogenesis inhibitor. Oncologist. 2010;15:539–47.
Deeks ED. Pazopanib: in advanced soft tissue sarcoma. Drugs. 2012;72:2129–40.
Drabkin HA. Pazopanib and anti-VEGF therapy. Open Access J Urol. 2010;2:35–40.
Dabney R, Devine R, Sein N, George B. New agents in renal cell carcinoma. Target Oncol. 2014;9:183–93.
Hu-Lowe DD, Zou HY, Grazzini ML, Hallin ME, Wickman GR, Amundson K, Chen JH, Rewolinski DA, Yamazaki S, Wu EY, et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin Cancer Res. 2008;14:7272–83.
Rixe O, Bukowski RM, Michaelson MD, Wilding G, Hudes GR, Bolte O, Motzer RJ, Bycott P, Liau KF, Freddo J, et al. Axitinib treatment in patients with cytokine-refractory metastatic renal-cell cancer: a phase II study. Lancet Oncol. 2007;8:975–84.
Jain RK. Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science. 2005;307:58–62.
Jain RK. Normalizing tumor microenvironment to treat cancer: bench to bedside to biomarkers. J Clin Oncol Off J Am Soc Clin Oncol. 2013;31:2205–18.
Ghilardi C, Chiorino G, Dossi R, Nagy Z, Giavazzi R, Bani M. Identification of novel vascular markers through gene expression profiling of tumor-derived endothelium. BMC Genomics. 2008;9:201.
Hofmeister V, Schrama D, Becker JC. Anti-cancer therapies targeting the tumor stroma. Cancer Immunol Immunother. 2008;57:1–17.
Dong Y, Qian J, Ibrahim R, Berzofsky JA, Khleif SN. Identification of H-2Db-specific CD8+ T-cell epitopes from mouse VEGFR2 that can inhibit angiogenesis and tumor growth. J Immunother. 2006;29:32–40.
Ishizaki H, Tsunoda T, Wada S, Yamauchi M, Shibuya M, Tahara H. Inhibition of tumor growth with antiangiogenic cancer vaccine using epitope peptides derived from human vascular endothelial growth factor receptor 1. Clin Cancer Res. 2006;12:5841–9.
Nair S, Boczkowski D, Moeller B, Dewhirst M, Vieweg J, Gilboa E. Synergy between tumor immunotherapy and antiangiogenic therapy. Blood. 2003;102:964–71.
Wada S, Tsunoda T, Baba T, Primus FJ, Kuwano H, Shibuya M, Tahara H. Rationale for antiangiogenic cancer therapy with vaccination using epitope peptides derived from human vascular endothelial growth factor receptor 2. Cancer Res. 2005;65:4939–46.
Hazama S, Nakamura Y, Takenouchi H, Suzuki N, Tsunedomi R, Inoue Y, Tokuhisa Y, Iizuka N, Yoshino S, Takeda K, et al. A phase I study of combination vaccine treatment of five therapeutic epitope-peptides for metastatic colorectal cancer; safety, immunological response, and clinical outcome. J Transl Med. 2014;12:63.
Okamoto I, Arao T, Miyazaki M, Satoh T, Okamoto K, Tsunoda T, Nishio K, Nakagawa K. Clinical phase I study of elpamotide, a peptide vaccine for vascular endothelial growth factor receptor 2, in patients with advanced solid tumors. Cancer Sci. 2012;103:2135–8.
Okuyama R, Aruga A, Hatori T, Takeda K, Yamamoto M. Immunological responses to a multi-peptide vaccine targeting cancer-testis antigens and VEGFRs in advanced pancreatic cancer patients. Oncoimmunology. 2013;2:e27010.
Suzuki H, Fukuhara M, Yamaura T, Mutoh S, Okabe N, Yaginuma H, Hasegawa T, Yonechi A, Osugi J, Hoshino M, et al. Multiple therapeutic peptide vaccines consisting of combined novel cancer testis antigens and anti-angiogenic peptides for patients with non-small cell lung cancer. J Transl Med. 2013;11:97.
Yamaue H, Tsunoda T, Tani M, Miyazawa M, Yamao K, Mizuno N, Okusaka T, Ueno H, Boku N, Fukutomi A, et al. Randomized phase II/III clinical trial of elpamotide for patients with advanced pancreatic cancer; PEGASUS-PC study. In:Cancer Sci; 2015.
Berger M, Bergers G, Arnold B, Hammerling GJ, Ganss R. Regulator of G-protein signaling-5 induction in pericytes coincides with active vessel remodeling during neovascularization. Blood. 2005;105:1094–101.
Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, Rabie T, Kaden S, Grone HJ, Hammerling GJ, et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature. 2008;453:410–4.
Chi Sabins N, Taylor JL, Fabian KP, Appleman LJ, Maranchie JK, Stolz DB, Storkus WJ. DLK1: a novel target for immunotherapeutic remodeling of the tumor blood vasculature. Mol Ther. 2013;21:1958–68.
Facciponte JG, Ugel S, De Sanctis F, Li C, Wang L, Nair G, Sehgal S, Raj A, Matthaiou E, Coukos G, et al. Tumor endothelial marker 1-specific DNA vaccination targets tumor vasculature. J Clin Invest. 2014;124:1497–511.
Zhao X, Bose A, Komita H, Taylor JL, Chi N, Lowe DB, Okada H, Cao Y, Mukhopadhyay D, Cohen PA, et al. Vaccines targeting tumor blood vessel antigens promote CD8(+) T cell-dependent tumor eradication or dormancy in HLA-A2 transgenic mice. J Immunol. 2012;188:1782–8.
Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603.
Griffioen AW, Weiss A, Berndsen RH, Abdul UK, te Winkel MT, Nowak-Sliwinska P. The emerging quest for the optimal angiostatic combination therapy. Biochem Soc Trans. 2014;42:1608–15.
Hashimoto K, Man S, Xu P, Cruz-Munoz W, Tang T, Kumar R, Kerbel RS. Potent preclinical impact of metronomic low-dose oral topotecan combined with the antiangiogenic drug pazopanib for the treatment of ovarian cancer. Mol Cancer Ther. 2010;9:996–1006.
Tran Cao HS, Bouvet M, Kaushal S, Keleman A, Romney E, Kim G, Fruehauf J, Imagawa DK, Hoffman RM, Katz MH. Metronomic gemcitabine in combination with sunitinib inhibits multisite metastasis and increases survival in an orthotopic model of pancreatic cancer. Mol Cancer Ther. 2010;9:2068–78.
Van der Veldt AA, Lubberink M, Bahce I, Walraven M, de Boer MP, Greuter HN, Hendrikse NH, Eriksson J, Windhorst AD, Postmus PE, et al. Rapid decrease in delivery of chemotherapy to tumors after anti-VEGF therapy: implications for scheduling of anti-angiogenic drugs. Cancer Cell. 2012;21:82–91.
Franco M, Man S, Chen L, Emmenegger U, Shaked Y, Cheung AM, Brown AS, Hicklin DJ, Foster FS, Kerbel RS. Targeted anti-vascular endothelial growth factor receptor-2 therapy leads to short-term and long-term impairment of vascular function and increase in tumor hypoxia. Cancer Res. 2006;66:3639–48.
Moserle L, Jimenez-Valerio G, Casanovas O. Antiangiogenic therapies: going beyond their limits. Cancer Discov. 2014;4:31–41.
Nieder C, Wiedenmann N, Andratschke N, Molls M. Current status of angiogenesis inhibitors combined with radiation therapy. Cancer Treat Rev. 2006;32:348–64.
Vujanovic L, Butterfield LH. Melanoma cancer vaccines and anti-tumor T cell responses. J Cell Biochem. 2007;102:301–10.
Yu Z, Restifo NP. Cancer vaccines: progress reveals new complexities. J Clin Invest. 2002;110:289–94.
Shi S, Chen L, Huang G. Antiangiogenic therapy improves the antitumor effect of adoptive cell immunotherapy by normalizing tumor vasculature. Med Oncol. 2013;30:698.
Bose A, Lowe DB, Rao A, Storkus WJ. Combined vaccine+axitinib therapy yields superior antitumor efficacy in a murine melanoma model. Melanoma Res. 2012;22:236–43.
Bose A, Taylor JL, Alber S, Watkins SC, Garcia JA, Rini BI, Ko JS, Cohen PA, Finke JH, Storkus WJ. Sunitinib facilitates the activation and recruitment of therapeutic anti-tumor immunity in concert with specific vaccination. Int J Cancer. 2011;129:2158–70.
Lowe DB, Bose A, Taylor JL, Tawbi H, Lin Y, Kirkwood JM, Storkus WJ. Dasatinib promotes the expansion of a therapeutically superior T-cell repertoire in response to dendritic cell vaccination against melanoma. Oncoimmunology. 2014;3:e27589.
Baksh K, Weber J. Immune checkpoint protein inhibition for cancer: preclinical justification for CTLA-4 and PD-1 blockade and new combinations. Semin Oncol. 2015;42:363–77.
Acknowledgements
The authors wish to thank Drs. Ronald Fecek and Jennifer Taylor for their helpful comments provided during review of this document. This work was supported by NIH R01 grant CA169118 (to W.J.S.).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter

Cite this chapter
Fabian, K.L., Storkus, W.J. (2017). Immunotherapeutic Targeting of Tumor-Associated Blood Vessels. In: Kalinski, P. (eds) Tumor Immune Microenvironment in Cancer Progression and Cancer Therapy. Advances in Experimental Medicine and Biology, vol 1036. Springer, Cham. https://doi.org/10.1007/978-3-319-67577-0_13
Download citation
DOI: https://doi.org/10.1007/978-3-319-67577-0_13
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-67575-6
Online ISBN: 978-3-319-67577-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)