
Abstract
The acquisition of oncogenic mutations and promotion of angiogenesis are key hallmarks of cancer. These features are often thought of as separate events in tumor progression and the two fields of research have frequently been considered as independent. However, as we highlight in this review, activated oncogenes and deregulated angiogenesis are tightly associated, as mutations in cancer cells can lead to perturbation of the pro- and anti-angiogenic balance thereby causing aberrant angiogenesis. We propose that normalization of the vascular network by targeting oncogenes in the tumor cells might lead to more efficient and sustained therapeutic effects compared to therapies targeting tumor vessels. We discuss how pharmacological inhibition of oncogenes in tumor cells restores a functional vasculature by bystander anti-angiogenic effect. As genetic alterations are tumor-specific, targeted therapy, which potentially blocks the angiogenic program activated by individual oncogenes may lead to personalized anti-angiogenic therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Anti-angiogenic therapy: where are we?
Angiogenesis is the biological process that drives the formation of new blood vessels from a pre-existing vasculature. Throughout embryonic development, physiological angiogenesis allows for expansion of the primitive vascular network formed by vasculogenesis, thanks to branching, remodeling, and maturation of the vascular bed [1]. During adulthood, angiogenesis normally occurs in only a few processes, such as in the female reproductive apparatus, and in pathological situations including wound healing, diabetic retinopathy, rheumatoid arthritis, and cancer [2].
More than 50 years ago, angiogenesis was described as a hallmark of tumor biology, and for the first time anti-angiogenic therapy was proposed as a cancer cure. Folkman and colleagues were pioneers in demonstrating that growing tumors need neovascularization when reaching a critical volume (around 1–2 mm3) in order to continue their expansion [3, 4]. The induction of tumor vasculature, also known as “angiogenic switch”, represents a complex and time-regulated process in cancer progression during which both the cancer cells and the tumor microenvironment secrete signals that recruit and expand the vascular network [5].
Tumor angiogenesis is central for tumor progression since blood vessels provide essential nutrients as well as oxygen to the proliferating malignant cells. Beyond the importance of angiogenesis for primary tumor expansion, blood vessels are an important route for cancer cell dissemination to distant organs. Indeed, the vascular system provides the motorway through which cancer cells disseminate, a process facilitated by the fact that the integrity of the endothelial layer is significantly impaired in the tumor vasculature [6, 7]. Tumor blood vessels were initially spotted as attractive targets in cancer at two levels: to inhibit tumor growth by cutting nutrient delivery to proliferating tumors, and to prevent metastases formation by blocking the main route for cancer cell dissemination. As angiogenesis is a peculiar feature of a growing tumor mass, its blockade was considered highly tumor selective, with limited potential for side effects. Moreover, most solid cancers are dependent on angiogenesis for their expansion and for this reason anti-angiogenic therapy was envisioned as broadly applicable in clinical oncology.
Among the angiogenic factors, vascular endothelial growth factor, or vascular permeability factor (VEGF/VPF) has historically been considered the main target for the development of anti-angiogenic drugs [8–10]. Indeed, the most widely used anti-angiogenic inhibitor, bevacizumab, is a humanized monoclonal antibody that binds and selectively neutralizes the biological function of VEGF [11]. Over the years, other kinase inhibitors, such as sorafenib and sunitinib, have been developed to target the VEGFR pathway and interfere with VEGF-driven angiogenesis [12–14]. Multiple preclinical studies showed that anti-VEGF therapy that blocks tumor angiogenesis could delay tumor growth in animal models [15, 16].
In 2004, bevacizumab became the first anti-angiogenic FDA-approved drug for the treatment of metastatic colon cancer. This decision followed a phase III clinical trial where patients with metastatic colorectal cancer had an improvement in progression-free survival from 6.2 to 10.6 months when treated with bevacizumab in combination with chemotherapy, compared to chemotherapy alone [17]. Afterwards, bevacizumab therapy was extended to other malignancies such as non-small cell lung cancer, HER2-negative breast cancer, renal cell cancer, and glioblastoma [18–21]. In parallel, small-molecule tyrosine kinase inhibitors blocking the VEGFR pathway, like sunitinib, sorafenib, and pazopanib showed efficacy in the treatment of renal cell cancer and hepatocellular carcinoma [22–25].
While the initial results were regarded as highly promising, clinical evidence indicated that anti-VEGF therapy also had limitations. Bevacizumab was rarely successful as a single agent and the clinical benefits, reached only in combination with chemotherapy, were shown to be only transitory. Notably, multiple clinical studies quickly established that even within the same tumor subtype (i.e., colorectal and lung cancer) not all patients display the same rate of response to anti-angiogenic drugs and importantly, demonstrated that anti-angiogenic therapy can be overcome [17, 21, 26].
Several reasons can contribute to primary or secondary resistance to anti-VEGF clinical approaches [27]. An important consideration is that, in contrast to predictions, not all tumors are addicted to angiogenesis for their expansion and treatments that merely target blood vessels cannot provide benefit in these types of cancer [28, 29]. A relevant observation was revealed by the transcriptional analyses of angiogenic factors in primary breast tumors compared to adjacent normal tissue. In this study, the majority of pro-angiogenic molecules appeared to be down-regulated in the tumor tissue and thus leading the authors to suggest that breast cancer primary tumor is not a site of active angiogenesis [28]. We now know that tumors can use alternative ways to become vascularized and in this context a therapy directed to sprouting angiogenesis would exert a limited effect. Tumor vascularization can occur by multiple mechanisms including: co-option of pre-existing vessels, tumor cells can surround pre-existing tissues vasculature, vascular mimicry, when dedifferentiated tumor cells contribute to the formation of blood vessels, and postnatal vasculogenesis through the recruitment of bone marrow-derived endothelial precursors [30–34]. Of note, the different processes are often mixed within the tumor mass and can provide an alternative route of vascularization exploited by tumors to escape anti-angiogenic treatment [35, 36]. A second mechanism that can lead to refractoriness to anti-VEGF therapy is that tumors are biologically diverse and angiogenesis can be stimulated by alternative molecules. A clear example came from the analyses of biopsies from different grades of primary breast cancer. As previously shown, VEGF expression was correlated with poor prognosis, but its expression was higher in the early stage of the disease, while in high-grade breast cancer a wide range of other angiogenic factors, like FGF2, were more prominent [37]. This suggests not only that different tumors use diverse stimuli but also that within the same malignancy the tumor stage influences angiogenic pathway activation and thereby the response to therapy.
The most common mechanism of resistance to anti-angiogenic drugs appears to be tumor adaptation to the loss of vessels. The process involves the onset of compensatory stimuli that drive neovascularization, thereby evading anti-angiogenic approach [27]. Several angiogenic molecules secreted in the tumor microenvironment are thought to be involved in the angiogenesis rebound, such as FGF2, ephrins, angiopoietins, PDGF, SDF1, and G-CSF. These factors can directly stimulate angiogenesis or act by recruiting inflammatory cells, as it has been described for SDF1 and G-CSF, like tumor-associated macrophages (TAM) and bone marrow-derived cells (BMBC) that in turn provide angiogenic stimuli [27, 38–40]. In the RIP-Tag pancreatic mouse tumor model, treatment with a VEGFR-blocking antibody causes an initial response followed by tumor regrowth and rebound angiogenesis. A broad analysis revealed that several hypoxia-mediated genes are up-regulated in the environment and, among them, FGF2 was shown to have an essential role in driving tumor revascularization [41]. A similar phenomenon has been observed in the clinic; glioblastoma patients treated with VEGFR2 small-molecule inhibitors display elevated FGF2 and SDF1 levels at the time when tumors became refractory to treatment [42].
Several reports have pointed to the tumor stroma as an important mediator of secondary resistance to anti-angiogenic therapy. For instance, tumor-associated fibroblasts release growth factors like PDGFC, Angptl2, and SDF1 that are involved in resistance to anti-VEGF treatment [43, 44]. BMDCs are recruited to the tumor microenvironment and promote cancer growth and angiogenesis rebound by providing alternative growth factors like Bv8 (or prokineticin-2) and HGF in anti-VEGF therapy refractory tumors [45–47].
Recent observations obtained in mouse model systems indicate that anti-angiogenic drugs might have concurrent deleterious side effects due to the generation of hypoxic stress. In preclinical models, preconditioning of the so-called metastatic niche or short-term treatment with anti-VEGF targeted therapy have been shown to enhance tumor invasiveness and metastatic burden in response to hypoxic stress [48–50]. These unexpected outcomes suggest that the generation of hypoxia resulting from blood vessels pruning may increase tumor aggressiveness, raising concerns about the clinical application of classic anti-angiogenic therapy. Importantly, the clinical relevance of these findings is still under investigation as evidence that patients treated with bevacizumab have a shorter time of progression-free survival are lacking [51]. However, glioblastoma tumors that relapse after bevacizumab treatment can have a more infiltrative phenotype [52, 53]. Similar observations have been made in renal cell cancer patients, where, after interruption of VEGFR tyrosine kinase inhibitor treatment, tumor growth rebounded with a concomitant increase in metastases [54].
The negative impact of the hypoxic stress can be extended to other aspects of tumor maintenance. The generation of a hypoxic niche is pivotal to support cancer stem cell population and to increase the expression of stem cell markers, at least in glioblastoma [55, 56]. Anti-angiogenic therapy is associated with this phenomenon since, as demonstrated in breast cancer preclinical models, the onset of hypoxia as consequence of anti-VEGF therapy increases the population of cancer stem cell within the tumor [57]. This is a new challenging aspect to be considered as cancer stem cells have been proposed as key actors in resistance to therapy and tumor recurrence [58–60].
Emerging concepts of tumor angiogenesis
In recent years, the process of tumor angiogenesis has been further detailed. We have learned, for example, that tumor neovascularization does not merely reflect an increase in capillary number but also a general modification in the physiology of the vasculature [6, 61]. Secretion of pro-angiogenic signals by both tumor cells and the microenvironment causes the formation of an abnormal vasculature, with chaotic and tortuous blood vessels and aberrant function, due to vasodilatation and increased vascular permeability [6, 62]. An aberrant vasculature results in hypoxia and necrosis that negatively impacts tumor progression, and vessel leakiness causes suboptimal blood flow and tumor perfusion. Impaired tumor perfusion affects the response to standard therapies due to reduced cytotoxic drug delivery within the tumor mass and defective production of oxygen radical species required for successful radiotherapy [63].
This additional knowledge must now be incorporated in the definition of new anti-angiogenic therapies. The latter should be implemented also considering the goal of reverting abnormal vasculature and to restore normal blood flow. In principle, this strategy should improve tumor perfusion while decreasing interstitial pressure and hypoxia-driven tumor aggressiveness [63, 64].
As mentioned above, another important consideration is that the tumor mass is a complex environment composed not only of cancerous cells but also of many other cell types such as fibroblasts, endothelial, and BMDCs. Considering the continuous and dynamic cross-talk among these cell lineages, current treatments are unlikely selective for only one cell type. Indeed, targeting blood vessels affects tumor cell proliferation and migration, and similarly targeting cancer cells deeply impinges on the tumor environment. This phenomenon, which has been referred to as “accidental anti-angiogenic therapy”, can be a consequence of chemotherapy or small molecules inhibitors [65]. In the case of small-molecule inhibitors, possible off-target effects are an important consideration. For example, it has been reported that targeting oncogenic BRAF with the tyrosine kinase inhibitor sorafenib not only inhibits cell proliferation in mutated tumor cells but directly affects angiogenesis because it blocks VEGFR2 and PDGFR activity [12]. A different mechanism acts in bystander anti-angiogenic therapy where targeting oncogenic events in the epithelial cell compartment indirectly impinges on the tumor environment [66].
Oncogenes in tumor angiogenesis
Genetic modifications occurring in the genome initiate the transformation process that leads to cancer. Two types of genetic aberration can drive tumorigenesis: mutation/amplification of oncogenes, and inactivation/deletion of oncosuppressor genes. The discovery that cancer cells rely on specific genetic alterations for their survival has driven the development of targeted therapy with the aim of inhibiting the proliferation of tumor cells [67–70]. Activation of oncogenic pathways triggers profound modifications in the expression profile of tumor cells. Among others, the expression of several cytokines and growth factors is directly affected by the activity of individual oncogenes, thereby influencing the tumor environment [66, 71, 72].
Angiogenesis, just like most biological processes, relies on an appropriate balance of factors to maintain an optimal physiological condition. In cancer, the delicate equilibrium between pro- and anti-angiogenic factors is lost, with the abundance of pro-angiogenic cytokines being the main driver of tumor angiogenesis. A therapeutic approach that depletes or inactivates an angiogenic pathway, such as anti-VEGF therapy, causes vessel pruning and an inadequate vasculature with necrosis and hypoxic stress that negatively effects the tumor environment [63]. We suggest that an alternative strategy to target tumor angiogenesis could be to rescue the equilibrium of angiogenic signals by targeting the mutated oncogenes, which play a central role in this process.
Several examples of oncogene-driven angiogenesis have been described [65, 66]. Indeed, activation of MAPK and PI3K-AKT pathways, which are usually deregulated in cancer, enhances the expression of pro-angiogenic factors by acting at both the transcriptional and translational levels [73–75]. These findings may explain why targeted therapy, which usually has a cytostatic effect on tumor cells, also affects the tumor environment and normalize tumor vasculature (Table 1).
We recently described how targeting oncogenic BRAF, a serine threonine kinase, affects angiogenesis [71]. BRAF is frequently mutated in human cancer and the BRAFV600E mutation can influence the tumor environment by increasing expression of HIF1α, VEGFA, IL1β, and IL8, and by lowering levels of the angiogenic blocker thrombospondin 1 [71, 76–80]. We found that the most common BRAF variant (the BRAFV600E mutation) modulates the production of angiogenic molecules by cancer cells. Furthermore, we evaluated the effect of the specific BRAFV600E inhibitor PLX4720 on tumor angiogenesis and demonstrated that targeting BRAF stabilizes the tumor vasculature and abrogates hypoxia in tumor xenografts. Intriguingly, we found that PLX4720 acts by specifically switching-off the MAPK pathway in BRAF-mutated cells, thereby decreasing the expression of angiogenic molecules. These data led us to suggest that pharmacological inhibition of oncogenes in tumor cells can restore a functional vasculature and potentially blocks the specific angiogenic program activated by individual tumors. This mechanism of action provides a clear example of bystander anti-angiogenic therapy.
Similarly to BRAF, the RAS oncogene is a master regulator of the MAPK pathway that has been directly linked to induction of tumor angiogenesis [81]. Activated RAS increases the expression of VEGF and other angiogenic chemokines like CXCL1, CXCL5, and IL8 while suppressing expression of the angiogenesis inhibitor thrombospondin 1 [82–86]. A concomitant mechanism by which oncogenic RAS stimulates the angiogenic program is by up-regulating proteases important for matrix remodeling, such as uPA, MMP2, and MMP9 [87, 88]. The role of KRAS in driving angiogenesis is supported by clinical data in non-small cell lung cancer and in pancreatic tumor showing that KRAS activating mutations correlate with high VEGF expression [89, 90]. Inhibition of RAS activity by gene silencing suppresses VEGF expression. Moreover, decreased VEGF expression in KRAS-mutated colon cancer cells reduces the tumorigenic potential in vivo, highlighting the importance of VEGF expression in KRAS-driven tumors [91].
The PI3K-AKT-mTOR axis, which is often deregulated in human cancer, is another important pathway that controls the angiogenic program in tumor cells. The activation of this signaling pathway up-regulates the expression of HIF1α and VEGF and consequently promotes tumor angiogenesis [92–94]. Small-molecule inhibitors blocking different signaling nodes of this pathway have shown important effects on vascular normalization with consequent improvement in vascular blood flow and tumor oxygenation [95, 96].
Myc and p53 also act as master regulators of angiogenic factors. C-Myc triggers the expression of VEGF while it down-regulates thrombospondin 1 [97, 98]. p53 is an oncosuppressor gene involved in the down-regulation of pro-angiogenic factors like VEGF and FGF as well as in the up-regulation of thrombospondin 1 [99–101]. For example the expression of p53 is required to reverse the angiogenic program in hematopoietic malignancy. In this type of tumor, Myc inactivation is sufficient to induce tumor regression, but its effect is less prominent when p53 is lost. Expression of p53 reverses tumor angiogenesis by controlling the up-regulation of thrombospondin 1 and allows a sustained tumor regression after Myc inactivation [102]. A recent study shows that p53 can also repress the thrombospondin 1 promoter in prostate cancer cell lines, suggesting a context-dependent regulation of thrombospondin 1 by p53 [103].
The inhibition of EGFR or HER2, tyrosine kinase receptors that activate MAPK and PI3K, is another example of vessel normalization by bystander oncogene targeting. EGFR can be amplified and mutated in several tumors including lung, colon, and glioblastoma. Pharmacological inhibition of EGFR decreases the expression of HIF1α and VEGF by tumor cells and treatment of tumor xenografts with Erlotinib or Iressa, two different tyrosine kinase inhibitors, also lead to vessel normalization [95, 104, 105]. As mentioned above, this effect on the tumor microenvironment can improve the success of both cytotoxic chemotherapy and radiotherapy. A preclinical study has shown that pretreatment of tumor xenografts with Erlotinib increases the delivery of chemotherapeutic agents within the tumor and results in higher inhibition of tumor growth compared to the single treatment [106]. Moreover, pretreatment with Erlotinib enhances the effect of radiation therapy in vivo but not in vitro, demonstrating that EGFR targeting may positively affect the tumor microenvironment [106]. HER2 is an important oncogene overexpressed in aggressive breast cancer. It has been found that targeting HER2-positive tumors with Herceptin strongly influences vascular structure and function and cause vessel normalization. Herceptin treatment slows down the secretion of VEGF, PAI-1, TGF-α, and Angiopoietin1, all important mediators of angiogenesis and in parallel up-regulates the expression of the anti-angiogenic factor thrombospondin 1 [107].
In conclusion, multiple evidences show that oncogene-targeted drugs might also impact tumor angiogenesis, suggesting an innovative strategy to revert aberrant vasculature and positively impact tumor environment. The limitation of this approach relies in the ability of tumor cells to develop secondary resistance towards targeted therapy. It is known that a tumor can overcome the dependency on a specific oncogene through various mechanisms: by involving compensatory genes through the activation of alternative molecular pathways or by the acquisition of new genetic alterations due to the intrinsic genomic instability of cancer cells [108]. The finding that targeting oncogene addiction in tumor cells results in abrogation of pro-angiogenic signals suggests that once acquired resistance is established reactivation of oncogenic pathways may trigger an angiogenic rebound. Therefore, any anticancer therapy (be it directed to the tumor cells or to the surrounding stroma) will always be limited by secondary resistance. Overcoming the latter is key in providing long-lasting clinical benefits.
Conclusions and perspectives
Both tumor angiogenesis and oncogenic addiction are considered hallmarks of cancer [62, 109]. In this review, we highlight the connection between these two events and discuss the hypothesis that targeting oncogenes can positively affect the tumor environment. We summarized examples of therapies aimed at blocking oncogenes that concomitantly were shown to have a clear effect on vascular normalization and tumor perfusion. At the same we note that targeting oncogenes can improve blood vessel structure and tumor oxygenation without having any obvious effect on tumor cell proliferation [95]. This suggests that oncogenic pathways can be involved in the activation and maintenance of the angiogenic program even in cancer cells that are not addicted to the targeted oncogenic mutations for proliferation.
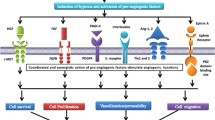
By comparing the anti-angiogenic effect of oncogene-targeted therapy with an anti-VEGF approach important considerations can be made (see Fig. 1). An anti-VEGF approach causes blood vessel pruning and hypoxic stress that can result in an adaptive response with associated rebound in neoangiogenesis and in some case enhanced tumor aggressiveness. This is a feasible explanation for the short clinical benefit observed in patients treated with anti-angiogenic drugs [110, 111]. In contrast, oncogene-targeted therapy causes blood vessel normalization by restoring the equilibrium of angiogenic molecules and it is predicted that this effect is more sustainable and should allow a prolonged response [64].

Anti-VEGF and anti-oncogene-targeted therapy in tumor angiogenesis. a Oncogenic mutations drive tumor angiogenesis by increasing the expression of pro-angiogenic factors. The unbalance between pro- and anti-angiogenic factors causes an aberrant and missfunctional vasculature. b Anti-VEGF therapy causes blood vessel pruning and hypoxia that negatively impacts tumor progression. c Oncogene-targeted therapy potentially slows down the angiogenic program in tumor cells and restores the balance between pro- and anti-angiogenic factors, thereby leading to blood vessel normalization
A second relevant consideration is that anti-VEGF therapy is directed towards a single angiogenic stimulus and acts by blocking the VEGFR signaling pathway. The first limitation of this strategy is that to obtain a functional vasculature, the maintenance of a correct amount of angiogenic cytokines and not a complete depletion is important. Moreover, tumors develop resistance to anti-VEGF treatment and drive revascularization by alternative angiogenic programs. Despite the fact that the main read-out described for oncogene-driven angiogenesis is VEGF, other angiogenic stimuli are modulated by oncogenic mutations and blocking oncogenes with targeted inhibitors has the advantage of affecting a wide range of pro- and anti-angiogenic molecules [27, 64].
All these factors suggest that standard anti-angiogenic therapy is unlikely to succeed in all tumors, but treatment strategies need to be adapted to individual cancers. Selective biomarkers are needed to predict which patients will benefit from anti-angiogenic therapy and, considering that a specific angiogenic profile can be activated in different cancers, to select the appropriate therapy. Moreover, several studies have aimed at identifying molecular changes that correlate with the response to anti-angiogenic therapy, which is an important parameter for the early identification of response or resistance to the therapy [112–114].
To date, reliable markers to predict response to anti-angiogenic treatment are not available; for example in metastatic colorectal cancer, neither VEGF nor microvessel density was predictive for response to bevacizumab [115]. Oncogenic mutations are already considered an important clinical parameter to stratify patients and identify suitable therapies. However, oncogenes have failed to be predictive for response to classical anti-angiogenic therapy [116], but it is possible that they will become useful for choosing alternative strategies. The knowledge about how the tumor microenvironment is influenced by targeted therapy will allow a better understanding of the clinical outcome and hopefully a clearer prediction of patient response.
We propose that blocking oncogenic pathways may result in inhibition of cancer cell proliferation, while concomitantly normalizing tumor vasculature. This approach opens possibilities for combinatorial treatments with chemotherapeutic agents or radiation therapy that would rely on the positive effects of vascular normalization on blood flow and tissue perfusion. Furthermore, by selectively blocking oncogenes, it should be possible to stall, at least temporarily, the angiogenic program. As oncogenes are activated in a tumor-specific fashion, we envision a personalized anti-angiogenic therapy that normalizes tumor vasculature even in cancers that are intrinsically refractory to anti-VEGF treatment thereby overcoming some of the limits of current anti-angiogenic drugs.
References
Risau W (1997) Mechanisms of angiogenesis. Nature 386(6626):671–674. doi:10.1038/386671a0
Carmeliet P, Jain RK (2000) Angiogenesis in cancer and other diseases. Nature 407(6801):249–257
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285(21):1182–1186. doi:10.1056/NEJM197111182852108
Folkman J (2006) Angiogenesis. Annu Rev Med 57:1–18. doi:10.1146/annurev.med.57.121304.131306
Bergers G, Benjamin LE (2003) Tumorigenesis and the angiogenic switch. Nat Rev Cancer 3(6):401–410
Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK, McDonald DM (2002) Abnormalities in pericytes on blood vessels and endothelial sprouts in tumors. Am J Pathol 160(3):985–1000. doi:10.1016/S0002-9440(10)64920-6
Hashizume H, Baluk P, Morikawa S, McLean JW, Thurston G, Roberge S, Jain RK, McDonald DM (2000) Openings between defective endothelial cells explain tumor vessel leakiness. Am J Pathol 156(4):1363–1380. doi:10.1016/S0002-9440(10)65006-7
Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219(4587):983–985
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935):1306–1309
Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT (1989) Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246(4935):1309–1312
Ferrara N, Hillan KJ, Gerber HP, Novotny W (2004) Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat Rev Drug Discov 3(5):391–400. doi:10.1038/nrd1381
Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, Chen C, Zhang X, Vincent P, McHugh M, Cao Y, Shujath J, Gawlak S, Eveleigh D, Rowley B, Liu L, Adnane L, Lynch M, Auclair D, Taylor I, Gedrich R, Voznesensky A, Riedl B, Post LE, Bollag G, Trail PA (2004) BAY 43–9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 64(19):7099–7109. doi:10.1158/0008-5472.CAN-04-1443
Strumberg D, Richly H, Hilger RA, Schleucher N, Korfee S, Tewes M, Faghih M, Brendel E, Voliotis D, Haase CG, Schwartz B, Awada A, Voigtmann R, Scheulen ME, Seeber S (2005) Phase I clinical and pharmacokinetic study of the Novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43–9006 in patients with advanced refractory solid tumors. J Clin Oncol 23(5):965–972. doi:10.1200/JCO.2005.06.124
Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI (2006) Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 24(1):16–24. doi:10.1200/JCO.2005.02.2574
Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N (1993) Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362(6423):841–844. doi:10.1038/362841a0
Asano M, Yukita A, Matsumoto T, Kondo S, Suzuki H (1995) Inhibition of tumor growth and metastasis by an immunoneutralizing monoclonal antibody to human vascular endothelial growth factor/vascular permeability factor121. Cancer Res 55(22):5296–5301
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350(23):2335–2342. doi:10.1056/NEJMoa032691
Rini BI, Halabi S, Rosenberg JE, Stadler WM, Vaena DA, Archer L, Atkins JN, Picus J, Czaykowski P, Dutcher J, Small EJ (2010) Phase III trial of bevacizumab plus interferon alfa versus interferon alfa monotherapy in patients with metastatic renal cell carcinoma: final results of CALGB 90206. J Clin Oncol 28(13):2137–2143. doi:10.1200/JCO.2009.26.5561
Cloughesy T (2010) FDA accelerated approval benefits glioblastoma. Lancet Oncol 11(12):1120. doi:10.1016/S1470-2045(10)70269-2
Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE (2007) Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med 357(26):2666–2676. doi:10.1056/NEJMoa072113
Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH (2006) Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med 355(24):2542–2550. doi:10.1056/NEJMoa061884
Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM (2007) Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med 356(2):125–134. doi:10.1056/NEJMoa060655
Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, Bolondi L, Greten TF, Galle PR, Seitz JF, Borbath I, Haussinger D, Giannaris T, Shan M, Moscovici M, Voliotis D, Bruix J (2008) Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 359(4):378–390. doi:10.1056/NEJMoa0708857
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, Garcia-del-Muro X, Sosman JA, Solska E, Wilding G, Thompson JA, Kim ST, Chen I, Huang X, Figlin RA (2009) Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 27(22):3584–3590. doi:10.1200/JCO.2008.20.1293
Sternberg CN, Davis ID, Mardiak J, Szczylik C, Lee E, Wagstaff J, Barrios CH, Salman P, Gladkov OA, Kavina A, Zarba JJ, Chen M, McCann L, Pandite L, Roychowdhury DF, Hawkins RE (2010) Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J Clin Oncol 28(6):1061–1068. doi:10.1200/JCO.2009.23.9764
Kerbel RS (2008) Tumor angiogenesis. N Engl J Med 358(19):2039–2049. doi:10.1056/NEJMra0706596
Bergers G, Hanahan D (2008) Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 8(8):592–603. doi:10.1038/nrc2442
Boneberg EM, Legler DF, Hoefer MM, Ohlschlegel C, Steininger H, Fuzesi L, Beer GM, Dupont-Lampert V, Otto F, Senn HJ, Furstenberger G (2009) Angiogenesis and lymphangiogenesis are downregulated in primary breast cancer. Br J Cancer 101(4):605–614. doi:10.1038/sj.bjc.6605219
Yu JL, Rak JW, Carmeliet P, Nagy A, Kerbel RS, Coomber BL (2001) Heterogeneous vascular dependence of tumor cell populations. Am J Pathol 158(4):1325–1334. doi:10.1016/S0002-9440(10)64083-7
Leenders WP, Kusters B, de Waal RM (2002) Vessel co-option: how tumors obtain blood supply in the absence of sprouting angiogenesis. Endothelium 9(2):83–87
Hendrix MJ, Seftor RE, Seftor EA, Gruman LM, Lee LM, Nickoloff BJ, Miele L, Sheriff DD, Schatteman GC (2002) Transendothelial function of human metastatic melanoma cells: role of the microenvironment in cell-fate determination. Cancer Res 62(3):665–668
Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, De Maria R (2010) Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 468(7325):824–828. doi:10.1038/nature09557
Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V (2010) Glioblastoma stem-like cells give rise to tumour endothelium. Nature 468(7325):829–833. doi:10.1038/nature09624
Peters BA, Diaz LA, Polyak K, Meszler L, Romans K, Guinan EC, Antin JH, Myerson D, Hamilton SR, Vogelstein B, Kinzler KW, Lengauer C (2005) Contribution of bone marrow-derived endothelial cells to human tumor vasculature. Nat Med 11(3):261–262. doi:10.1038/nm1200
Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, Ruiter D, Ryan A, de Waal R (2004) Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res 10(18 Pt 1):6222–6230. doi:10.1158/1078-0432.CCR-04-0823
Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, Shuman MA (2000) Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia 2(4):306–314
Relf M, LeJeune S, Scott PA, Fox S, Smith K, Leek R, Moghaddam A, Whitehouse R, Bicknell R, Harris AL (1997) Expression of the angiogenic factors vascular endothelial cell growth factor, acidic and basic fibroblast growth factor, tumor growth factor beta-1, platelet-derived endothelial cell growth factor, placenta growth factor, and pleiotrophin in human primary breast cancer and its relation to angiogenesis. Cancer Res 57(5):963–969
Pollard JW (2004) Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 4(1):71–78. doi:10.1038/nrc1256
Orimo A, Weinberg RA (2006) Stromal fibroblasts in cancer: a novel tumor-promoting cell type. Cell Cycle 5(15):1597–1601. doi:3112
Shojaei F, Ferrara N (2008) Refractoriness to antivascular endothelial growth factor treatment: role of myeloid cells. Cancer Res 68(14):5501–5504. doi:10.1158/0008-5472.CAN-08-0925
Casanovas O, Hicklin DJ, Bergers G, Hanahan D (2005) Drug resistance by evasion of antiangiogenic targeting of VEGF signaling in late-stage pancreatic islet tumors. Cancer Cell 8(4):299–309. doi:10.1016/j.ccr.2005.09.005
Batchelor TT, Sorensen AG, di Tomaso E, Zhang WT, Duda DG, Cohen KS, Kozak KR, Cahill DP, Chen PJ, Zhu M, Ancukiewicz M, Mrugala MM, Plotkin S, Drappatz J, Louis DN, Ivy P, Scadden DT, Benner T, Loeffler JS, Wen PY, Jain RK (2007) AZD2171, a pan-VEGF receptor tyrosine kinase inhibitor, normalizes tumor vasculature and alleviates edema in glioblastoma patients. Cancer Cell 11(1):83–95. doi:10.1016/j.ccr.2006.11.021
Crawford Y, Kasman I, Yu L, Zhong C, Wu X, Modrusan Z, Kaminker J, Ferrara N (2009) PDGF-C mediates the angiogenic and tumorigenic properties of fibroblasts associated with tumors refractory to anti-VEGF treatment. Cancer Cell 15(1):21–34. doi:10.1016/j.ccr.2008.12.004
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, Carey VJ, Richardson AL, Weinberg RA (2005) Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121(3):335–348. doi:10.1016/j.cell.2005.02.034
Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, Fuh G, Gerber HP, Ferrara N (2007) Tumor refractoriness to anti-VEGF treatment is mediated by CD11b + Gr1 + myeloid cells. Nat Biotechnol 25(8):911–920. doi:10.1038/nbt1323
Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, Blanchard D, Bais C, Peale FV, van Bruggen N, Ho C, Ross J, Tan M, Carano RA, Meng YG, Ferrara N (2007) Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature 450(7171):825–831. doi:10.1038/nature06348
Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, Feng J, Stewart AE, Hu-Lowe DD, Christensen JG (2010) HGF/c-Met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res 70(24):10090–10100. doi:10.1158/0008-5472.CAN-10-0489
Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS (2009) Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15(3):232–239. doi:10.1016/j.ccr.2009.01.021
Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O (2009) Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15(3):220–231. doi:10.1016/j.ccr.2009.01.027
Loges S, Mazzone M, Hohensinner P, Carmeliet P (2009) Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell 15(3):167–170. doi:10.1016/j.ccr.2009.02.007
Miles D, Harbeck N, Escudier B, Hurwitz H, Saltz L, Van Cutsem E, Cassidy J, Mueller B, Sirzen F (2011) Disease course patterns after discontinuation of bevacizumab: pooled analysis of randomized phase III trials. J Clin Oncol 29(1):83–88. doi:10.1200/JCO.2010.30.2794
Zuniga RM, Torcuator R, Jain R, Anderson J, Doyle T, Schultz L, Mikkelsen T (2010) Rebound tumour progression after the cessation of bevacizumab therapy in patients with recurrent high-grade glioma. J Neurooncol 99(2):237–242. doi:10.1007/s11060-010-0121-0
Verhoeff JJ, van Tellingen O, Claes A, Stalpers LJ, van Linde ME, Richel DJ, Leenders WP, van Furth WR (2009) Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC Cancer 9:444. doi:10.1186/1471-2407-9-444
Johannsen M, Florcken A, Bex A, Roigas J, Cosentino M, Ficarra V, Kloeters C, Rief M, Rogalla P, Miller K, Grunwald V (2009) Can tyrosine kinase inhibitors be discontinued in patients with metastatic renal cell carcinoma and a complete response to treatment? A multicentre, retrospective analysis. Eur Urol 55(6):1430–1438. doi:10.1016/j.eururo.2008.10.021
Soeda A, Park M, Lee D, Mintz A, Androutsellis-Theotokis A, McKay RD, Engh J, Iwama T, Kunisada T, Kassam AB, Pollack IF, Park DM (2009) Hypoxia promotes expansion of the CD133-positive glioma stem cells through activation of HIF-1alpha. Oncogene 28(45):3949–3959. doi:10.1038/onc.2009.252
Heddleston JM, Li Z, McLendon RE, Hjelmeland AB, Rich JN (2009) The hypoxic microenvironment maintains glioblastoma stem cells and promotes reprogramming towards a cancer stem cell phenotype. Cell Cycle 8(20):3274–3284. doi:9701
Conley SJ, Gheordunescu E, Kakarala P, Newman B, Korkaya H, Heath AN, Clouthier SG, Wicha MS (2012) Antiangiogenic agents increase breast cancer stem cells via the generation of tumor hypoxia. Proc Natl Acad Sci USA 109(8):2784–2789. doi:10.1073/pnas.1018866109
Visvader JE, Lindeman GJ (2008) Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer 8(10):755–768. doi:10.1038/nrc2499
Yu Y, Ramena G, Elble RC (2012) The role of cancer stem cells in relapse of solid tumors. Front Biosci (Elite Ed) 4:1528–1541. doi:478
Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, Van Buren G 2nd, Samuel S, Kim MP, Lim SJ, Ellis LM (2009) Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res 69(5):1951–1957. doi:10.1158/0008-5472.CAN-08-2023
Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM (2003) Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol 163(5):1801–1815. doi:10.1016/S0002-9440(10)63540-7
De Bock K, Cauwenberghs S, Carmeliet P (2011) Vessel abnormalization: another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr Opin Genet Dev 21(1):73–79. doi:10.1016/j.gde.2010.10.008
Jain RK (2005) Normalization of tumor vasculature: an emerging concept in antiangiogenic therapy. Science 307(5706):58–62
Goel S, Duda DG, Xu L, Munn LL, Boucher Y, Fukumura D, Jain RK (2011) Normalization of the vasculature for treatment of cancer and other diseases. Physiol Rev 91(3):1071–1121. doi:10.1152/physrev.00038.2010
Kerbel RS, Viloria-Petit A, Klement G, Rak J (2000) ‘Accidental’ anti-angiogenic drugs. anti-oncogene directed signal transduction inhibitors and conventional chemotherapeutic agents as examples. Eur J Cancer 36(10):1248–1257
Rak J, Yu JL (2004) Oncogenes and tumor angiogenesis: the question of vascular “supply” and vascular “demand”. Semin Cancer Biol 14(2):93–104
Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, Zimmermann J, Lydon NB (1996) Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med 2(5):561–566
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344(11):783–792. doi:10.1056/NEJM200103153441101
Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M (2004) EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 304(5676):1497–1500. doi:10.1126/science.1099314
Ciardiello F, Tortora G (2008) EGFR antagonists in cancer treatment. N Engl J Med 358(11):1160–1174. doi:10.1056/NEJMra0707704
Bottos A, Martini M, Di Nicolantonio F, Comunanza V, Maione F, Minassi A, Appendino G, Bussolino F, Bardelli A (2012) Targeting oncogenic serine/threonine-protein kinase BRAF in cancer cells inhibits angiogenesis and abrogates hypoxia. Proc Natl Acad Sci USA 109(6):E353–E359. doi:10.1073/pnas.1105026109
Ancrile BB, O’Hayer KM, Counter CM (2008) Oncogenic ras-induced expression of cytokines: a new target of anti-cancer therapeutics. Mol Interv 8(1):22–27. doi:10.1124/mi.8.1.6
Richard DE, Berra E, Gothie E, Roux D, Pouyssegur J (1999) p42/p44 mitogen-activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 274(46):32631–32637
Berra E, Pages G, Pouyssegur J (2000) MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev 19(1–2):139–145
Kevil CG, De Benedetti A, Payne DK, Coe LL, Laroux FS, Alexander JS (1996) Translational regulation of vascular permeability factor by eukaryotic initiation factor 4E: implications for tumor angiogenesis. Int J Cancer 65(6):785–790. doi:10.1002/(SICI)1097-0215(19960315)65:6<785:AID-IJC14>3.0.CO;2-3
Nucera C, Porrello A, Antonello ZA, Mekel M, Nehs MA, Giordano TJ, Gerald D, Benjamin LE, Priolo C, Puxeddu E, Finn S, Jarzab B, Hodin RA, Pontecorvi A, Nose V, Lawler J, Parangi S (2010) B-Raf(V600E) and thrombospondin-1 promote thyroid cancer progression. Proc Natl Acad Sci USA 107(23):10649–10654
Zerilli M, Zito G, Martorana A, Pitrone M, Cabibi D, Cappello F, Giordano C, Rodolico V (2010) BRAF(V600E) mutation influences hypoxia-inducible factor-1alpha expression levels in papillary thyroid cancer. Mod Pathol 23(8):1052–1060
Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP (2005) Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res 65(6):2412–2421
Kumar SM, Yu H, Edwards R, Chen L, Kazianis S, Brafford P, Acs G, Herlyn M, Xu X (2007) Mutant V600E BRAF increases hypoxia inducible factor-1alpha expression in melanoma. Cancer Res 67(7):3177–3184
Niault TS, Baccarini M (2010) Targets of Raf in tumorigenesis. Carcinogenesis 31(7):1165–1174
Kranenburg O, Gebbink MF, Voest EE (2004) Stimulation of angiogenesis by Ras proteins. Biochim Biophys Acta 1654(1):23–37. doi:10.1016/j.bbcan.2003.09.004
Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS (1995) Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res 55(20):4575–4580
Grugel S, Finkenzeller G, Weindel K, Barleon B, Marme D (1995) Both v-Ha-Ras and v-Raf stimulate expression of the vascular endothelial growth factor in NIH 3T3 cells. J Biol Chem 270(43):25915–25919
Matsuo Y, Campbell PM, Brekken RA, Sung B, Ouellette MM, Fleming JB, Aggarwal BB, Der CJ, Guha S (2009) K-Ras promotes angiogenesis mediated by immortalized human pancreatic epithelial cells through mitogen-activated protein kinase signaling pathways. Mol Cancer Res 7(6):799–808. doi:10.1158/1541-7786.MCR-08-0577
Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, Zimmer MA, Iliopoulos O, Zukerberg LR, Kohgo Y, Lynch MP, Rueda BR, Chung DC (2005) Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med 11(9):992–997. doi:10.1038/nm1294
Zabrenetzky V, Harris CC, Steeg PS, Roberts DD (1994) Expression of the extracellular matrix molecule thrombospondin inversely correlates with malignant progression in melanoma, lung and breast carcinoma cell lines. Int J Cancer 59(2):191–195
Gum R, Lengyel E, Juarez J, Chen JH, Sato H, Seiki M, Boyd D (1996) Stimulation of 92-kDa gelatinase B promoter activity by ras is mitogen-activated protein kinase kinase 1-independent and requires multiple transcription factor binding sites including closely spaced PEA3/ets and AP-1 sequences. J Biol Chem 271(18):10672–10680
Sato H, Kida Y, Mai M, Endo Y, Sasaki T, Tanaka J, Seiki M (1992) Expression of genes encoding type IV collagen-degrading metalloproteinases and tissue inhibitors of metalloproteinases in various human tumor cells. Oncogene 7(1):77–83
Konishi T, Huang CL, Adachi M, Taki T, Inufusa H, Kodama K, Kohno N, Miyake M (2000) The K-ras gene regulates vascular endothelial growth factor gene expression in non-small cell lung cancers. Int J Oncol 16(3):501–511
Ikeda N, Nakajima Y, Sho M, Adachi M, Huang CL, Iki K, Kanehiro H, Hisanaga M, Nakano H, Miyake M (2001) The association of K-ras gene mutation and vascular endothelial growth factor gene expression in pancreatic carcinoma. Cancer 92(3):488–499. doi:10.1002/1097-0142(20010801)92:3<488:AID-CNCR1347>3.0.CO;2-F
Okada F, Rak JW, Croix BS, Lieubeau B, Kaya M, Roncari L, Shirasawa S, Sasazuki T, Kerbel RS (1998) Impact of oncogenes in tumor angiogenesis: mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc Natl Acad Sci USA 95(7):3609–3614
Skinner HD, Zheng JZ, Fang J, Agani F, Jiang BH (2004) Vascular endothelial growth factor transcriptional activation is mediated by hypoxia-inducible factor 1alpha, HDM2, and p70S6K1 in response to phosphatidylinositol 3-kinase/AKT signaling. J Biol Chem 279(44):45643–45651. doi:10.1074/jbc.M404097200
Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, Giaccia AJ, Abraham RT (2002) Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 22(20):7004–7014
Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kaelin WG Jr (2003) TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4(2):147–158. doi:S1535610803001879
Qayum N, Muschel RJ, Im JH, Balathasan L, Koch CJ, Patel S, McKenna WG, Bernhard EJ (2009) Tumor vascular changes mediated by inhibition of oncogenic signaling. Cancer Res 69(15):6347–6354. doi:10.1158/0008-5472.CAN-09-0657
Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, Stumm M, Cozens R, Littlewood-Evans A, Garcia-Echeverria C, Maira SM (2008) Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res 68(16):6598–6607. doi:10.1158/0008-5472.CAN-08-1044
Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16(19):2530–2543. doi:10.1101/gad.1024602
Janz A, Sevignani C, Kenyon K, Ngo CV, Thomas-Tikhonenko A (2000) Activation of the myc oncoprotein leads to increased turnover of thrombospondin-1 mRNA. Nucleic Acids Res 28(11):2268–2275
Pal S, Datta K, Mukhopadhyay D (2001) Central role of p53 on regulation of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) expression in mammary carcinoma. Cancer Res 61(18):6952–6957
Ueba T, Nosaka T, Takahashi JA, Shibata F, Florkiewicz RZ, Vogelstein B, Oda Y, Kikuchi H, Hatanaka M (1994) Transcriptional regulation of basic fibroblast growth factor gene by p53 in human glioblastoma and hepatocellular carcinoma cells. Proc Natl Acad Sci USA 91(19):9009–9013
Dameron KM, Volpert OV, Tainsky MA, Bouck N (1994) Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 265(5178):1582–1584
Giuriato S, Ryeom S, Fan AC, Bachireddy P, Lynch RC, Rioth MJ, van Riggelen J, Kopelman AM, Passegue E, Tang F, Folkman J, Felsher DW (2006) Sustained regression of tumors upon MYC inactivation requires p53 or thrombospondin-1 to reverse the angiogenic switch. Proc Natl Acad Sci USA 103(44):16266–16271. doi:10.1073/pnas.0608017103
Su F, Pascal LE, Xiao W, Wang Z (2010) Tumor suppressor U19/EAF2 regulates thrombospondin-1 expression via p53. Oncogene 29(3):421–431. doi:10.1038/onc.2009.326
Pore N, Jiang Z, Gupta A, Cerniglia G, Kao GD, Maity A (2006) EGFR tyrosine kinase inhibitors decrease VEGF expression by both hypoxia-inducible factor (HIF)-1-independent and HIF-1-dependent mechanisms. Cancer Res 66(6):3197–3204. doi:10.1158/0008-5472.CAN-05-3090
Ciardiello F, Caputo R, Bianco R, Damiano V, Fontanini G, Cuccato S, De Placido S, Bianco AR, Tortora G (2001) Inhibition of growth factor production and angiogenesis in human cancer cells by ZD1839 (Iressa), a selective epidermal growth factor receptor tyrosine kinase inhibitor. Clin Cancer Res 7(5):1459–1465
Cerniglia GJ, Pore N, Tsai JH, Schultz S, Mick R, Choe R, Xing X, Durduran T, Yodh AG, Evans SM, Koch CJ, Hahn SM, Quon H, Sehgal CM, Lee WM, Maity A (2009) Epidermal growth factor receptor inhibition modulates the microenvironment by vascular normalization to improve chemotherapy and radiotherapy efficacy. PLoS One 4(8):e6539. doi:10.1371/journal.pone.0006539
Izumi Y, Xu L, di Tomaso E, Fukumura D, Jain RK (2002) Tumour biology: herceptin acts as an anti-angiogenic cocktail. Nature 416(6878):279–280. doi:10.1038/416279b
Ellis LM, Hicklin DJ (2009) Resistance to targeted therapies: refining anticancer Therapy in the Era of molecular oncology. Clin Cancer Res 15(24):7471–7478. doi:10.1158/1078-0432.CCR-09-1070
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi:10.1016/j.cell.2011.02.013
Allegra CJ, Yothers G, O’Connell MJ, Sharif S, Petrelli NJ, Colangelo LH, Atkins JN, Seay TE, Fehrenbacher L, Goldberg RM, O’Reilly S, Chu L, Azar CA, Lopa S, Wolmark N (2011) Phase III trial assessing bevacizumab in stages II and III carcinoma of the colon: results of NSABP protocol C-08. J Clin Oncol 29(1):11–16. doi:10.1200/JCO.2010.30.0855
Kerr DJ, Young AM (2011) Targeted therapies: bevacizumab–has it reached its final resting place? Nat Rev Clin Oncol 8(4):195–196. doi:10.1038/nrclinonc.2011.32
Murukesh N, Dive C, Jayson GC (2010) Biomarkers of angiogenesis and their role in the development of VEGF inhibitors. Br J Cancer 102(1):8–18. doi:10.1038/sj.bjc.6605483
Sessa C, Guibal A, Del Conte G, Ruegg C (2008) Biomarkers of angiogenesis for the development of antiangiogenic therapies in oncology: tools or decorations? Nat Clin Pract Oncol 5(7):378–391. doi:10.1038/ncponc1150
Shojaei F (2012) Anti-angiogenesis therapy in cancer: current challenges and future perspectives. Cancer Lett 320(2):130–137. doi:10.1016/j.canlet.2012.03.008
Jubb AM, Hurwitz HI, Bai W, Holmgren EB, Tobin P, Guerrero AS, Kabbinavar F, Holden SN, Novotny WF, Frantz GD, Hillan KJ, Koeppen H (2006) Impact of vascular endothelial growth factor-A expression, thrombospondin-2 expression, and microvessel density on the treatment effect of bevacizumab in metastatic colorectal cancer. J Clin Oncol 24(2):217–227. doi:10.1200/JCO.2005.01.5388
Ince WL, Jubb AM, Holden SN, Holmgren EB, Tobin P, Sridhar M, Hurwitz HI, Kabbinavar F, Novotny WF, Hillan KJ, Koeppen H (2005) Association of k-ras, b-raf, and p53 status with the treatment effect of bevacizumab. J Natl Cancer Inst 97(13):981–989. doi:10.1093/jnci/dji174
Acknowledgments
A warm thanks to Prof. Nancy E. Hynes, Dr. Jason W. Gill, and Dr. Alessio Noghero for critical reading and suggestions. The research leading to these results has received funding from the European Community’s Seventh Framework Programme under Grant agreement no. 259015 COLTHERES; AIRC 2010 Special Program Molecular Clinical Oncology 5xMille, Project no. 9970; Intramural Grant—5xmille 2008 Fondazione Piemontese per la Ricerca sul Cancro—ONLUS, AIRC IG Grant no. 12812. A. Bottos was supported by the Swiss Science Foundation (#310030_138417).
Conflict of interest
A. Bardelli owns stock in, and has received consultancy fees, from Horizon Discovery.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bottos, A., Bardelli, A. Oncogenes and angiogenesis: a way to personalize anti-angiogenic therapy?. Cell. Mol. Life Sci. 70, 4131–4140 (2013). https://doi.org/10.1007/s00018-013-1331-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-013-1331-3