
Abstract
For anti-tumor therapy different strategies have been employed, e.g., radiotherapy, chemotherapy, or immunotherapy. Notably, these approaches do not only address the tumor cells themselves, but also the tumor stroma cells, e.g., endothelial cells, fibroblasts, and macrophages. This is of advantage, since these cells actively contribute to the proliferative and invasive behavior of the tumor cells via secretion of growth factors, angiogenic factors, cytokines, and proteolytic enzymes. In addition, tumor stroma cells take part in immune evasion mechanisms of cancer. Thus, approaches targeting the tumor stroma attract increasing attention as anti-cancer therapy. Several molecules including growth factors (e.g., VEGF, CTGF), growth factor receptors (CD105, VEGFRs), adhesion molecules (αvβ3 integrin), and enzymes (CAIX, FAPα, MMPs, PSMA, uPA) are induced or upregulated in the tumor microenvironment which are otherwise characterized by a restricted expression pattern in differentiated tissues. Consequently, these molecules can be targeted by inhibitors as well as by active and passive immunotherapy to treat cancer. Here we discuss the results of these approaches tested in preclinical models and clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
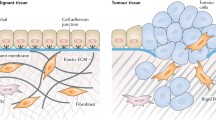
The development and progression of cancer depends on genetic and epigenetic alterations in the transformed cells. However, many steps in cancerogenesis, e.g., proliferation, invasion, angiogenesis, and metastasis are promoted by microenvironmental factors such as growth factors and proteolytic enzymes produced by stromal cells (Fig. 1). Indeed, the reciprocal interactions between tumor and tumor stroma cells, i.e., cancer-associated fibroblasts (CAFs), tumor endothelial cells (TECs), and tumor-associated macrophages (TAMs), result in tumor progression. CAFs are reactive fibroblasts with a distinctive phenotype as compared to quiescent fibroblasts in differentiated adult tissue. They are present in the close vicinity of tumor cells and enhance tumor growth by secreting growth factors such as transforming growth factor β (TGF-β), matrix degrading enzymes, e.g., matrix metalloproteinases (MMPs), and angiogenic factors, like vascular endothelial growth factor (VEGF). TECs also support the neoplastic cells by production of growth factors, but more importantly they are mandatory for tumor hem- and lymphangiogenesis. TAMs represent a major component of the leukocyte infiltrate in solid tumors. They secrete both growth factors and proteolytic enzymes, moreover, they generate an immune privileged state of the tumor microenvironment. Thus, both tumor stromal cells and their products are promising targets for cancer therapy.

Influence of the tumor microenvironment on tumor development. Tumor stromal cells, e.g., cancer associated fibroblasts (CAFs), tumor endothelial cells (TECs), and tumor-associated macrophages (TAMs) express growth factors sustaining tumor growth, angiogenic factors promoting angiogenesis, and proteolytic enzymes catalyzing the degradation of the ECM facilitating tumor cell invasion and finally metastasis. Tumor cells are depicted in brown, CAFs in orange, TECs in red, and TAMs in yellow
Tumor stroma cells differ from their normal counterparts by upregulation or induction of various molecules (Table 1). Their upregulation is selective for the tumor microenvironment and occurs in a broad spectrum of solid tumors. Corresponding to the various interactions of tumor and stroma cells anti-stromal therapies fall into different categories. To date primarily molecules contributing to angiogenesis, e.g., vascular endothelial growth factor (VEGF) and its receptors have been targeted. Moreover, proteins involved in remodeling of the extra-cellular matrix (ECM) (e.g., MMPs, uPA/PAR system) have been silenced or functionally inhibited. However, due to redundancy of signals the inhibition of one factor or even a group of molecules is often not sufficient to significantly influence tumor growth and progression. Therefore, alternative strategies aim at modulating the tumor microenvironment by eliminating the stromal cells, e.g., by antibodies or by cytotoxic effector cells. Besides targeting stroma cells immunotherapeutically [41], new strategies are currently in preclinical or clinical testing. In the following sections we describe a selection of these approaches for the treatment of cancer (Table 2).
Strategies to prevent tumor angiogenesis
Angiogenesis, the development of new blood vessels, plays a central role in the pathogenesis of cancer. It is crucial for maintaining the supply of oxygen and nutrients and the removal of waste products to support tumor growth beyond a few mm3 (Fig. 1). Consequently, TECs play a major role in tumor progression. Their growth is sustained by autocrine and paracrine secretion of growth factors by stroma and tumor cells, respectively. It should be further noted, that TECs are essential to maintain the oncogenic phenotype of tumor cells by production of several soluble factors. Consequently, growth and angiogenic factors, e.g., VEGF or TGF-β, and their receptors as well as TECs themselves have been targeted for cancer therapy (Fig. 2).

Targeting endothelial cells for anti cancer therapy. A variety of proteins are targeted to prevent angiogenesis. a The function of molecules expressed on endothelial cells, e.g., VEGFRs, PSMA, TEM8, and CD105 is inhibited either directly by inhibitors and antibodies or alternatively their expression is prevented by ribozymes. In addition, they are exploited to eliminate tumor endothelial cells, e.g., by bispecific antibodies or specific expression or accumulation of toxins at endothelial cells. b CTGF is targeted by siRNA, inhibitors or antibodies. αvβ3 integrin expression is prevented by siRNA, its function is inhibited by antibodies, and effector molecules are targeted to αvβ3 integrin positive cells using inhibitors binding to αvβ3 integrin. Expression/secretion of target molecules is indicated by green arrows, binding to receptors by brown arrows, and their effects by black arrows. Therapeutical substances are depicted in red and their effects by blue arrows
The potent pro-angiogenic growth factor VEGF and its tyrosine kinase receptors VEGFR-1 (Flt-1) and VEGFR-2 (KDR/Flk-1) play a fundamental role in tumor vessel formation. VEGF is abundantly expressed in most tumors due to hypoxic conditions, acidic pH, hypoglycemia and several inducing cytokines, as well as genetic and epigenetic changes in tumor cells. Its expression, however, is not restricted to the tumor cells. VEGF is also expressed in CAFs, TECs, TAMs, and the inflammatory infiltrate (Table 1). Its expression is associated with increased angiogenesis, tumor growth, invasion, metastasis, and a greater risk of recurrence. Hypoxia and increased VEGF levels enhance expression of VEGFR-1 and VEGFR-2 on the vascular endothelium of the tumor, but both receptors are also expressed on tumor cells such as melanoma, thyroid, ovarian, breast carcinoma or cutaneous T cell lymphoma. In consequence, several VEGF- and VEGFR-antagonists have been developed and several of these inhibitors have already entered the clinic (Table 2; Fig. 2a).
In this regard the most advanced therapy consists of the recombinant humanized anti-VEGF mab Bevacizumab. It neutralizes VEGF, thereby blocking signal transduction mediated by VEGFR-1 and -2. Indeed, more then 200 clinical studies with Bevacizumab are currently being initiated or in progress (http://www.clinicaltrials.gov). Bevacizumab significantly inhibits growth of different tumor types (e.g., colorectal, renal cell, NSCL cancer) already as single-agent therapy, which can be additively or synergistically enhanced by chemo- or radiotherapy [42, 107]. Hence, a combined regimen has been approved by the FDA [46]. Alternatively VEGF signaling may also be inhibited by receptor-specific antibodies. Anti-VEGFR-2 antibodies act synergistically with chemo- or radiation therapy [48, 55]. The growth of established human tumor xenografts of different origin was inhibited due to decreased angiogenesis, reduced tumor cell proliferation and enhanced tumor cell apoptosis by treatment with antibodies. The VEGFR-2 antibody IMC-1C11 is currently being tested in clinical studies [81]. Soluble receptors intercept the growth factor before it can bind to the cell surface receptor. To this end, an adenoviral vector encoding the extra-cellular domain of VEGFR-1 showed promising activity in different tumor models and a reduction of vascular density was also reached by soluble VEGFR-2 [53, 61]. A soluble receptor comprising portions of VEGFR-1 and -2 demonstrated an even higher efficacy [21]. Another approach to reduce VEGF level in vivo has been tested in corneal neovascularization but may be used in anti-tumor therapy as well; transfection with plasmids encoding VEGFR-1 coupled with an endoplasmatic retention signal reduced injury-induced VEGF secretion [96]. Other approaches investigated include VEGF antisense oligonucleotides or ribozymes. In this regard, VEGFR-1 ribozymes were well tolerated in patients and objective tumor responses were reached in combination with carboplatin and paclitaxel [49].
An endothelial cell surface molecule of particular interest for vascular targeting is CD105 (endoglin), a 95 kDa cell surface protein expressed as a homodimer. CD105 functions as an accessory protein for kinase receptor complexes of the TGF-β superfamily and modulates TGF-β signaling, i.e., it antagonizes the inhibitory effects of TGF-β1, e.g., the suppression of growth, migration and capillary tube formation. In addition, CD105 has anti-apoptotic effects under hypoxic conditions. CD105 expression is not restricted to TECs (Table 1). However, only on TECs CD105 is expressed in significant amounts. CD105 is upregulated by TGF-β2 and hypoxia. Its expression has been described for a multitude of solid tumors (Table 1). Expression of CD105 is correlated with vascular density and poor prognosis. These properties render CD105 an attractive target for therapeutic interventions (Table 2; Fig. 2a). In preclinical studies, CD105-specific antibodies—either alone or conjugated with different effector molecules—react only weakly or not at all with quiescent endothelium but specifically bind to proliferating endothelial cells during tumor angiogenesis [31]. Correspondingly, side effects were low [68]. To test the effect of CD105-specific antibodies on human blood vessels, chimeric vasculature of human and murine origin was generated by transplantation of human foreskin onto immunodeficient mice: The growth of tumors induced in these tissues is reduced by anti-CD105 antibodies [99]. Recently, the safety of a humanized anti-CD105-antibody has been demonstrated in a primate model [95]. Lysis of endothelial cells is also achieved by retargeting CTL to CD105-positive cells by means of CD3/CD105-bispecific single-chain diabodies in vitro [54].
Prostate-specific membrane antigen (PSMA) is a 110 kDa glycoprotein with glutamate carboxypeptidase activity. PSMA is the prototype cell surface marker of prostate cancer as it is expressed on malignant prostate cancer cells. In addition, PSMA is expressed on some normal tissues (Table 1). Nevertheless, it has been widely used as therapeutic target in preclinical prostate cancer models [29, 37, 66]. PSMA is also abundantly expressed on TECs of many solid tumors (Table 1). Thus, targeting PSMA seems to be a feasible therapeutic option for different kinds of tumors apart from prostate carcinoma (Table 2; Fig. 2a). Currently, the fully human anti-PSMA antibody MDX-070 is tested in phase II trial for prostate cancer (Medarex).Footnote 1 While PSMA-specific antibodies alone often did not inhibit tumor growth, immunotoxins significantly delayed tumor progression in preclinical models [37]. A single-chain antibody fused to Pseudomonas exotoxin A specifically binds to PSMA-positive prostate cancer cells and reduces their viability [103]. Auristatin conjugated to a human monoclonal antibody to PSMA enhanced survival in a murine xenograft prostate model [66]. Furthermore, in clinical studies PSMA-specific antibodies labeled with radioactive isotopes induced objective responses [71]. Similarly, docetaxel is targeted to PSMA expressing cells by nanoparticle-aptamer bioconjugates which exhibited anticancer efficiency in a xenograft model [29].
TEM8 (ATR = the anthrax-toxin-receptor) is a transmembrane receptor belonging to the tumor endothelial markers (TEMs). It binds to the collagen subunit α3 (VI) but its cellular function is still unknown. TEM8 is strongly expressed on vascular endothelial cells during embryo- and carcinogenesis. Its expression on endothelial cells is associated with enhanced cell–matrix-interaction and migration. TEM8 protein has been found on TECs of different cancer types but also on cancer cells themselves (Table 1). Recently, TEM8 mRNA has been detected in a cell population, coexpressing DC and endothelial markers, capable of generating functional blood vessels; thus, indicating that TEM8 has a function during vasculogenesis [25]. Since TEM8 functions as a docking protein for the Bacillus anthracis toxin, its upregulation on tumor endothelial cells has been exploited to target anthrax toxin to tumor endothelial and tumor cells [15] (Table 2). Fusion proteins that have been designed to allow the active toxin to be released by cleavage by metalloproteinases which are present in the tumor microenvironment, have demonstrated therapeutic activity, thereby illustrating the feasibility of an anti-TEM8 therapy (Fig. 2a) [62].
The connective tissue growth factor (CTGF, CCN2) is a 38 kDa extra-cellular matricellular protein that belongs to the CCN cysteine-rich family of proteins. CTGF has multiple functions. It interacts with integrin receptors, including αvβ3, and several growth factors, e.g., TGF-β. In addition, it serves as biostore for angiogenic factors such as VEGF. The VEGF signaling can be restored by cleavage of CTGF by MMP. CTGF is upregulated by estrogen, TGF-β, EGF (epidermal growth factor), PDGF (platelet-derived growth factor), VEGF and hypoxia, for example. CTGF modulates cell adhesion, migration, proliferation, chemotaxis, apoptosis, ECM deposition, and angiogenesis and is important in reproduction, embryonic development, wound repair, inflammation, fibrosis disorders, and tumorigenesis. CTGF expression levels are elevated in a multitude of cancers where it is produced by several types of stromal cells including TECs, vascular smooth muscle cells, and CAFs, and can also be expressed in cancer cells (Table 1). This over-expression correlates with tumor stage and/or prognosis [105, 106]. Moreover, addition of CTGF to the medium increases proliferation and invasiveness of pancreatic cancer cells in vitro [5]. Nevertheless in some cancers such as esophageal, NSCL, and colorectal carcinoma high CTGF expression has been associated with improved survival, reduced angiogenesis, and metastasis. To this end, transfection of lung adenocarcinoma cell lines with CTGF-over-expressing vectors reduced VEGF expression, microvessel density, tumor growth, and metastasis in xenograft models [23]. As CTGF frequently is upregulated in fibrosis, CTGF mainly has been targeted in fibrotic diseases [60]. However, these approaches may also be effective in cancer. A CTGF-specific antibody, FG-3019, is currently being tested in a preclinical pancreatic cancer model (FibroGen, Fig. 2b), where it decreases tumor growth, metastasis, as well as angiogenesis and enhances the therapeutic effect of gemcitabine [5, 27]. Alternatively, stabilization of CTGF mRNA induced by VEGF can be inhibited by the angiogenesis inhibitor DN-9693 (Table 2) [51].
On TECs several integrins, e.g., αvβ3 and α5β1, are upregulated. These cell surface adhesion molecules are involved in cell growth, migration, tumor invasion, proliferation, metastasis, angiogenesis, and survival. The integrin αvβ3, is minimally expressed in mature vasculature and found on new blood vessels as well as the surface of many solid tumors (Table 1). Its expression is associated with an aggressive tumor phenotype. In consequence, it represents a target for anti-cancer therapy (Table 2; Fig. 2b). Several antagonists, including antibodies, Arg–Gly–Asp (RGD) peptides, and recombinant proteins induce tumor regression by reduction of angiogenesis and enhanced apoptosis of TEC [6, 84]. The RGD peptide cilengitide, for example, is tested in several clinical phase II studies for the treatment of glioblastoma (NCT00112866, NCT00085254) and prostate cancer (NCT00121238, NCT00103337) alone or in combination with radiation therapy. Similarly, the RGD peptidomimetic S247 and the cyclic peptides RGD4C and RGDfK decrease angiogenesis, tumor growth, and metastasis and improve survival in mice [86, 98]. In addition, radiosensitivity of endothelial cells is synergistically enhanced [1]. Recently, efforts were made to improve the affinity and pharmacological properties of these antagonists, e.g., by binding them to HPMA [N-2-hydroxypropyl) methacrylamide] polymers thereby enhancing tumor to background ratios of the antagonist’s distribution in the body [72, 73]. The RGD4C peptide has also been chosen to target αv integrin positive cells by integration of this peptide into virus capsids or liposomes. An adeno-associated virus bacteriophage hybrid vector encoding the herpex simplex thymidine kinase that activates the prodrug ganciclovir proved effective in preclinical models [36]. Alternatively, combretastatin has been targeted to tumors by liposomes linked to RGD peptides which delayed tumor growth in a melanoma model [79]. Such liposomes have also been used to deliver siRNAs resulting in reduced prostate cancer bone metastases [10]. Fusion of truncated tissue factor to an αv-targeting peptide induced thrombosis in tumor vessels, thereby inhibiting growth or inducing even regression of tumors in adenocarcinoma, melanoma and fibrosarcoma models [47].
In murine models, antibodies to αvβ3 integrin block angiogenesis, tumor formation, and metastasis. In clinical studies the αvβ3 inegrin-specific antibody Vitaxin (MEDI-523) proved to be safe, however, only limited efficacy was observed, e.g., disease stabilization in a leiomyosarcoma patient [35, 78]. Vitaxin is now being investigated in phase II trials in melanoma and prostate cancer (MedImmune, Inc.). The antibody Abergrin (MEDI-522), that has a greater affinity to αvβ3 integrin, was similarly well tolerated in patients and is currently being tested in prostate (NCT00072930) and colorectal cancer (NCT00284817) as well as metastatic melanoma alone or in combination therapy (NCT00111696, NCT00066196, MedImmune, Inc.) [69].
Interference with the remodeling of the extra-cellular matrix
Malignant progression of tumors is a complex process in which cells need to gain the ability to invade into surrounding tissues. For this purpose, cancer cells need to attach to, degrade and invade the ECM. These events are followed by invasion of the wall of blood or lymphatic vessels; after transport within the blood or lymph circulation, extravasation of cancer cells again involves degradation of the basement membrane and the ECM. For all these steps structural changes of the ECM are essential. Several proteases are involved in these processes including the MMPs and the urokinase plasminogen activator (uPA).
MMPs are a family of membrane-anchored and secreted zinc-dependent endopeptidases. Collectively, they are capable of degrading all ECM and basement membrane components. Their activity is important for matrix remodeling in biological processes such as embryonic development, tissue regeneration, and wound healing. In the tumor micromilieu MMPs are frequently upregulated in response to growth factors, cytokines and membrane-anchored molecules. They are produced by both tumor and tumor stroma cells, predominantly at the invasive front of the tumor (Table 1). Indeed, MMP expression correlates with an invasive phenotype of tumor cells. MMPs have been implicated in tumor growth, invasion, metastasis, angiogenesis, and cancer cell survival/apoptosis as well as the clinical course in a variety of cancer types. Thus, several approaches to inhibit MMP expression and function have been tested for cancer therapy (Table 2; Fig. 3a). A wide variety of substances has been used to block MMP activity, e.g., hydroxamates, bisphosphonates, tetracyclines, and tissue inhibitors of metalloproteinases (TIMPs). The therapeutic efficacy of synthetic MMP inhibitors has been demonstrated in vitro and in preclinical studies. Moreover, several drugs have proceeded into the clinic and even advanced to phase III clinical trials [102]. Pseudopeptidic hydroxamate inhibitors, for example, bind the zinc atom in the catalytic domain of MMPs and have broad specificities. Batimastat (BB-94), iIlomastat (Galardin, GM-6001) and the orally bioavailable derivative marimastat (BB-2516) inhibit the activity of many MMPs. Marimastat clinically improved progression free and overall survival of patients with advanced gastric cancer [16]. However, neither progression free nor overall survival was prolonged in metastatic breast cancer or small cell lung (SCL) cancer patients after first line chemotherapy or in pancreatic carcinoma patients in combination with gemcitabine [17, 94, 97]. The dose-limiting side effect of marimastat treatment is musculoskeletal pain. The non-peptidic biphenyl MMP inhibitor tanomastat (BAY 12-9566) has higher specificity towards MMP-2, -3, and -9. It demonstrated anti-invasive, anti-metastatic, and anti-angiogenic activity in a variety of tumor models and has also been evaluated in clinical studies. Although clinical effects were seen in combination with chemotherapy toxicity limits its usage [74]. Moreover, phase III trials revealed that tanomastat had a negative impact on progression free and overall survival in ovarian, pancreatic, and SCL cancer [39, 75]. A small molecule, broad spectrum inhibitor that lacks musculoskeletal side effects is rebimastat (BMS-275291) [88]. However, it could not demonstrate any beneficial effect if used as monotherapy for treatment of patients with breast and colorectal cancer or in combination with paclitaxel and carboplatin in non small cell lung (NSCL) cancer [59, 70, 88]. A nonpeptidic inhibitor that has also been tested in phase III clinical studies is prinomastat (AG3340). It was examined in prostate and NSCL cancer in combination with chemotherapy, but it also lacked efficacy [11]. Another group of MMP inhibitors are chemically modified tetracyclines, like metastat (CMT-3, Col-3) which inhibits several MMPs, e.g., MMP-1, -2, -8, -9, and -13. In rodent models, it inhibits malignant cell invason and angiogenesis [65]. Metastat has been examined in a phase II clinical trial in patients with Kaposi sarcoma where it reduced MMP-2 and MMP-9 plasma levels [26]. Apart from these synthetic inhibitors endogenous MMP inhibitors have also been evaluated. Under physiological conditions these TIMPs are essential for the regulation of MMP activity. TIMPs have antitumoral function since they can inhibit cell invasion, tumor growth, metastasis and angiogenesis. This notion has been confirmed by adenoviral expression of TIMPs in tumor tissues [4, 18]. However, TIMPs do not act selectively on the MMPs promoting tumor growth. Furthermore, TIMPs are associated with the upregulation of the antiapoptotic protein Bcl-XL [43].

Therapy approaches influencing the ECM degradation and targeting CAFs. a MMPs and the uPA/uPAR system influence the ECM degradation in the tumor microenvironment and promote invasion and metastasis of tumors. They are targeted by inhibiting their expression, e.g., by siRNA, antisense constructs, ribozymes or DNAzymes or their function by inhibitors, antibodies or soluble receptors, for example. In addition the uPA/uPAR system is used to induce apoptosis by recruitment or activation of toxins. b Tenascin expression is downregulated by siRNA approaches or targeted by antibodies or aptamers labeled with radioisotopes. Molecules expressed on the cell surface of CAFs, e.g., c FAPα and d CAIX, are also used to directly destroy these stroma cells mediated by antibodies (labeled with radioactive isotopes or different effector molecules) or cellular immune responses. Expression/secretion of target molecules is indicated by green arrows, binding to receptors by brown arrows, and their effects by black arrows. Therapeutical substances are depicted in red and their effects by blue arrows
As described above, despite some clinical effects of MMP inhibitors, the results of clinical trials have been rather disappointing. In most studies the MMP inhibitors had no beneficial and sometimes even adverse effects [11, 17, 94, 97]. This may partly be explained by the usage of broad spectrum inhibitors and the treatment of patients without knowledge about the expression of the respective MMPs in the individual tumors. MMPs are over-expressed in most tumors, however, their expression pattern varies in each tumor type and even in metastases of the same tumor to different organs. As broad spectrum inhibitors may have both inhibitory and promoting effects on tumor development and progression, more selective inhibitors for tumor-progression-associated MMPs have been developed. Ro-28-2653 inhibitis MMP-2, -9 and -14 and shows high anti-invasive activity in vitro. In preclinical models, it inhibits growth of MMP producing tumor cells as well as the tumor growth promoting effect of stromal cells [67]. The anti tumor effect is enhanced by combination with chemotherapy [2]. SB-3CT, a mechanism-based inhibitor that is formed only within the active site of the targeted enzymes, i.e., MMP-2 and MMP-9, inhibits liver metastases and increases survival in an aggressive murine model of T-cell lymphoma [56].
Several approaches to selectively inhibit the expression of a specific MMP have been examined, e.g., antisense constructs, RNA interference, and antisense ribozymes. However, even if single MMPs are targeted, the results are not always predictable as some MMPs may have a dual effect on tumor progression. Notably, MMPs cleave a diverse variety of substrates, resulting in opposing effects on tumor growth [30]. Indeed, activation of growth and angiogenic factors, and cleavage of FasL promote tumor development whereas the generation of angiogenesis inhibitors, e.g., angiostatin, arrestin, canstatin, endostatin, tumstatin, and ADAMTSs may inhibit tumor progression.
The serine protease urokinase plasminogen activator (uPA) converts plasminogen to plasmin which is able to degrade many ECM proteins such as collagen IV, laminin, and fibronectin either directly or through activation of other proteases. uPA, its inhibitor plasminogen activator inhibitor-1 (PAI-1), and its receptor uPAR (CD87) are involved in cell migration, tissue degradation, and angiogenesis under normal and pathological conditions. They do not only regulate ECM degradation but also cell adhesion and migration mediated by the interaction between uPAR and integrins, as well as ECM components, such as vitronectin. Over-expression of uPA and uPAR is a characteristic of various human cancers (Table 1). Whereas uPAR is particularly detected on tumor cells, uPA is mainly expressed by TAMs and CAFs, and to lesser extent on TECs. Studies in uPA knock out mice have confirmed the role of stromal uPA expression for tumor progression. Consequently, the uPA system has been addressed for anti-cancer therapy. Several approaches to interfere with the uPA system, ranging from neutralizing antibodies, soluble receptors, catalytically inactive uPA fragments, and synthetic peptides/peptidomimetics to antisense approaches, RNAi vectors, and DNAzymes have been tested to date (Table 2; Fig. 3a). The inhibitory effects of these substances have been conclusively demonstrated in preclinical models. Treatment with 213Bi labeled PAI-2 inhibits tumor growth in a pancreatic cancer xenograft model [83]. In murine tumor models intratumoral injection of small interfering RNA constructs for uPA and uPAR abrogated growth of established tumors [52, 82]. Adenoviral delivery of a chimeric protein composed of the receptor binding part of uPA linked to the plasmin inhibitor BPTI (aprotinin) or the treatment with hybrid proteins consisting of the uPAR binding part and urinary trypsin inhibitor (UT1) also reduced tumor growth and metastases in tumor models [50, 58]. In addition, toxins or bioactive molecules binding to components of the uPA system or being activated after cleavage by uPA in the tumor microenvironment have been developed [32, 63, 90, 101]. In all these systems the selective targeting of cells expressing uPA/uPAR, was not associated with major side effects. Recently, an uPA-derived peptide, Å6, which in animal models reduced tumor growth, metastasis, and angiogenesis alone or in combination with other therapies was evaluated in a phase I clinical study in patients with gynaecologic, especially ovarian, cancer. This study demonstrated the safety of Å6. Moreover, this trial suggested some clinical potential [9]. Thus, Å6 is currently being tested in a phase II study (NCT00083928, Ångstrom Pharmaceuticals).
Tenascin-C is an extra-cellular hexameric glycoprotein expressed during embryonic development and adult tissue remodeling. Alternative splicing results in monomers of different sizes and the large isoform is virtually undetectable in differentiated tissues but is abundantly expressed in the stroma of most solid tumors (Table 1). Tenascin-C is expressed by both cancer cells and CAFs, especially at the invasive front. Tenascin-C is upregulated by hypoxia, mechanical stress, and various cytokines including TGF-β and CTGF. Tenascin-C promotes tumor growth by several mechanisms including enhanced proliferation, invasion, and migration, as well as escape from immune surveillance, and it is associated with a poor prognosis. Antibodies to tenascin-C delay tumor growth and induce apparent cures in xenograft models (Table 2; Fig. 3b). Treatment of patients with recurrent brain tumors, i.e., glioma and astrocytoma, with the I131 labeled antibody 81C6 demonstrated limited toxicity and induced a prolonged survival [85]. This antibody has also induced clinical responses in patients with non-Hodgkin lymphoma [89]. Additive tumor targeting was obtained by combining two different antitenascin antibodies [80]. In another approach, fluorescence and radiolabeled aptamers to tenascin-C were used in xenograft models for tumor imaging, suggesting that labeling of aptamer conjugates may be used to deliver radioisotopes and chemotherapeutics [38]. Affinity matured human antibodies to tenascin-C in a small immunoprotein format (scFv disulfide linked homodimer) have been generated and their specific accumulation in glioblastoma of a murine model has been demonstrated [14]. In glioblastoma multiforme and astrocytoma patients following brain resection tenascin-C specific RNA interference was applied to suppress tumor growth [108].
Fibroblast activation protein α (FAPα, seprase) is another enzyme participating in ECM degradation. FAPα is a serine protease with dual function, i.e., gelatinase/collagenase and dipeptidyl peptidase (N-terminal, post-prolyl amino peptidase) activity. Its natural substrate has not yet been identified. FAPα is expressed during embryonic development and wound healing. While it is hardly present in differentiated adult tissues, it is selectively induced on reactive stromal fibroblasts of more than 90% of common solid tumors. Besides its primary localization in fibroblasts, FAPα mRNA has also been detected in endothelial cells undergoing reorganization and capillary morphogenesis. Endothelial expression of FAPα protein in complexes with dipeptidyl peptidase IV (DPPIV) has been described in capillary-like blood vessels in breast ductal carcinoma. The biological function of FAPα in the tumor microenvironment still remains elusive. It has been suggested that FAPα functions via degradation of the ECM and/or processing of soluble factors (such as chemokines, hormones or bioactive peptides). FAPα over-expression is associated with reduced dependency on exogenous growth factors, enhanced tumor growth, invasion, angiogenesis, and metastasis. However, the role of FAPα in cancerogenesis is still controversial as in some studies a beneficial effect of FAPα expression has been demonstrated: (i) expression of FAPα in a mouse melanoma model decreased tumorigenicity, restored contact inhibition, induced cell cycle arrest and growth factor dependence; (ii) in human breast carcinoma expression of FAPα in fibroblasts is associated with longer overall and disease-free survival. Nevertheless, the differential expression of FAPα in cancer versus normal tissues makes it a promising therapeutic target (Fig. 3c). PT-100, an inhibitor of FAPα and DPPIV dipeptidyl peptidase activity, upregulates cytokine and chemokine production by stroma cells, and thereby augments the anti-tumor immune response, and it was well tolerated in a phase I clinical study [3, 76]. Other peptide inhibitors are also in preclinical development. Dipeptide proline diphenyl phosphonates exert an anti-invasive effect on the FAPα positive melanoma cell line LOX [33]. Recently, an FAPα-specific small molecule inhibitor has been developed which specifically targets FAPα but not DPPIV [28]. FAPα-directed anti-catalytic antibodies have been demonstrated to reduce the growth of FAP+ tumor cells [24]. However, some of the biological effects of FAPα are mediated independently from its catalytic function as catalytic mutants still influence tumor growth. In consequence, several antibody constructs have been tested not only to inhibit the enzymatic activity of FAPα but also to mediate the destruction of FAPα+ cells. The humanized version of the mab F19 to FAPα, Sibrotuzumab (BIBH 1) has been demonstrated to be safe and well tolerated in clinical studies [40, 93]. The 131I-labeled BIBH 1 was used to characterize its biodistribution in humans, demonstrating that it selectively accumulated in primary and metastatic colorectal as well as NSCL cancer [93]. However, the FAPα-specific antibody alone lacked therapeutic efficacy. Thus, FAPα-specific antibodies have been used to target bioactive molecules to the tumor stroma (reviewed in [92]): (i) An FAPα-CD3-bispecific single chain antibody was used to recruit CTLs to the tumor stroma [104]; (ii) TNFα was directed to FAPα expressing cells by an anti-FAPα-TNFα fusion protein; binding of the construct mimicks membrane-integrated TNFα signaling and leads to apoptosis and tissue factor production in vitro and reduced tumor growth in vivo [8]; (iii) the function of tissue factor in the coagulation cascade was also used in another approach where the human tissue factor was fused with a single chain anti-FAPα antibody; after binding to the cell surface coagulation was triggered in vitro [87]; finally (iv) a FasL fusion protein regained full activity upon cell-surface binding and induced apoptosis of FAPα positive cells in vitro and prevented growth of FAPα-positive tumor cells without systemic toxicity in vivo [91].
For the selective destruction of CAFs antigens not directly implicated in the ECM degradation may also be considered (Fig. 3d). The carbonic anhydrase IX (CAIX, MN, G250) is a marker for tumor hypoxia and is important for pH regulation. CAIX is over-expressed in CAFs of renal cell, colorectal, cervix, NSCL, bladder, and kidney cancer as well as on some malignant cells themselves (Table 1). High CAIX expression in tumors is associated with an unfavorable prognosis. It should be noted, however, that CAIX is also expressed in normal gastric epithelium. Efforts are in progress to design carbonic anhydrase inhibitors specifically binding to CAIX [77, 100]. Sulfonamide derivatives, for example show efficacy at inhibiting hypoxia-induced acidosis in vitro [7, 22]. With respect to passive immune therapy, the CAIX-specific antibody WX-G250 (Rencarex) efficiently mediated ADCC in vitro [64]. Moreover, treatment of renal cell carcinoma patients with this antibody resulted in clinical responses [12, 13]. Conjugates of this antibody with radioactive isotopes improved survival in preclinical studies, and currently are being tested in the clinic [19, 20]. Spontaneous T cell responses against CAIX are only rarely detected in cancer patients even if TIL are used for analysis [34]. Thus, in a recently initiated clinical trial the adoptive transfer of autologous T lymphocytes transduced to express a single-chain antibody to CAIX is being investigated [57].
Conclusion
Stroma cells influence tumor initiation and progression. They allow vasculo- and angiogenesis, as well as recruitment of additional stromal cells; secrete growth factors and proteolytic enzymes, and modify the ECM to make it more suitable for the tumor cells. A variety of methods to inhibit these tumor-promoting interactions of tumor and tumor stroma cells are currently being tested in preclinical and clinical studies. These approaches target one or multiple molecules to inhibit their expression, to interfere with their function or to destroy the tumor stroma cells. Several of these strategies achieved promising results in early clinical trials demonstrating that they may become an effective tool to treat cancer. In consequence, efforts are in progress to advance tumor stroma-directed therapies both to be applied by themselves or to complement conventional treatments.
Abbreviations
- CAIX:
-
Carbonic anhydrase IX
- CAF:
-
Cancer-associated fibroblast
- CTGF:
-
Connective tissue growth factor
- DPPIV:
-
Dipeptidyl peptidase IV
- ECM:
-
Extra-cellular matrix
- FAPα:
-
Fibroblast activation protein α
- MHC:
-
Major histocompatibility complex
- MMP:
-
Matrix metalloproteinase
- (N)SCL(C):
-
(Non) small cell lung (cancer)
- PAGRIT:
-
Pretargeted antibody guided radioimmunotherapy
- PSMA:
-
Prostate-specific membrane antigen
- SIP:
-
Small immunoprotein format
- TAM:
-
Tumor-associated macrophage
- TEC:
-
Tumor endothelial cell
- TEM:
-
Tumor endothelial marker
- TIMP:
-
Tissue inhibitor of metalloproteinases
- uPA(R):
-
Urokinase plasminogen activator (receptor)
- VEGF(R):
-
Vascular endohelial growth factor (receptor)
References
Abdollahi A, Griggs DW, Zieher H, Roth A, Lipson KE, Saffrich R, Grone HJ, Hallahan DE, Reisfeld RA, Debus J, Niethammer AG, Huber PE (2005) Inhibition of alpha(v)beta3 integrin survival signaling enhances antiangiogenic and antitumor effects of radiotherapy. Clin Cancer Res 11:6270–6279
Abramjuk C, Lein M, Rothaug W, Krell HW, Loening SA, Jung K (2006) Enhanced inhibitory effect of the matrix metalloproteinase inhibitor Ro 28–2653 in combination with estramustine and etoposide on the prostate carcinoma in the rat Dunning orthotopic tumor model. Cancer Chemother Pharmacol 59:275–282
Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP (2004) PT-100, a small molecule dipeptidyl peptidase inhibitor, has potent antitumor effects and augments antibody-mediated cytotoxicity via a novel immune mechanism. Cancer Res 64:5471–5480
Ahonen M, Ala-aho R, Baker AH, George SJ, Grenman R, Saarialho-Kere U, Kahari VM (2002) Antitumor activity and bystander effect of adenovirally delivered tissue inhibitor of metalloproteinases-3. Mol Ther 5:705–715
Aikawa T, Gunn J, Spong SM, Klaus SJ, Korc M (2006) Connective tissue growth factor-specific antibody attenuates tumor growth, metastasis, and angiogenesis in an orthotopic mouse model of pancreatic cancer. Mol Cancer Ther 5:1108–1116
Albert JM, Cao C, Geng L, Leavitt L, Hallahan DE, Lu B (2006) Integrin alpha(v)beta(3) antagonist Cilengitide enhances efficacy of radiotherapy in endothelial cell and non-small-cell lung cancer models. Int J Radiat Oncol Biol Phys 65:1536–1543
Alterio V, Vitale RM, Monti SM, Pedone C, Scozzafava A, Cecchi A, De Simone G, Supuran CT (2006) Carbonic anhydrase inhibitors: X-ray and molecular modeling study for the interaction of a fluorescent antitumor sulfonamide with isozyme II and IX. J Am Chem Soc 128:8329–8335
Bauer S, Adrian N, Williamson B, Panousis C, Fadle N, Smerd J, Fettah I, Scott AM, Pfreundschuh M, Renner C (2004) Targeted bioactivity of membrane-anchored TNF by an antibody-derived TNF fusion protein. J Immunol 172:3930–3939
Berkenblit A, Matulonis UA, Kroener JF, Dezube BJ, Lam GN, Cuasay LC, Brunner N, Jones TR, Silverman MH, Gold MA (2005) A6, a urokinase plasminogen activator (uPA)-derived peptide in patients with advanced gynecologic cancer: a phase I trial. Gynecol Oncol 99:50–57
Bisanz K, Yu J, Edlund M, Spohn B, Hung MC, Chung LW, Hsieh CL (2005) Targeting ECM-integrin interaction with liposome-encapsulated small interfering RNAs inhibits the growth of human prostate cancer in a bone xenograft imaging model. Mol Ther 12:634–643
Bissett D, O’Byrne KJ, von Pawel J, Gatzemeier U, Price A, Nicolson M, Mercier R, Mazabel E, Penning C, Zhang MH, Collier MA, Shepherd FA (2005) Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J Clin Oncol 23:842–849
Bleumer I, Knuth A, Oosterwijk E, Hofmann R, Varga Z, Lamers C, Kruit W, Melchior S, Mala C, Ullrich S, De Mulder P, Mulders PF, Beck J (2004) A phase II trial of chimeric monoclonal antibody G250 for advanced renal cell carcinoma patients. Br J Cancer 90:985–990
Bleumer I, Oosterwijk E, Oosterwijk-Wakka JC, Voller MC, Melchior S, Warnaar SO, Mala C, Beck J, Mulders PF (2006) A clinical trial with chimeric monoclonal antibody WX-G250 and low dose interleukin-2 pulsing scheme for advanced renal cell carcinoma. J Urol 175:57–62
Brack SS, Silacci M, Birchler M, Neri D (2006) Tumor-targeting properties of novel antibodies specific to the large isoform of tenascin-C. Clin Cancer Res 12:3200–3208
Bradley KA, Young JA (2003) Anthrax toxin receptor proteins. Biochem Pharmacol 65:309–314
Bramhall SR, Hallissey MT, Whiting J, Scholefield J, Tierney G, Stuart RC, Hawkins RE, McCulloch P, Maughan T, Brown PD, Baillet M, Fielding JW (2002) Marimastat as maintenance therapy for patients with advanced gastric cancer: a randomised trial. Br J Cancer 86:1864–1870
Bramhall SR, Schulz J, Nemunaitis J, Brown PD, Baillet M, Buckels JA (2002) A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br J Cancer 87:161–167
Brand K, Baker AH, Perez-Canto A, Possling A, Sacharjat M, Geheeb M, Arnold W (2000) Treatment of colorectal liver metastases by adenoviral transfer of tissue inhibitor of metalloproteinases-2 into the liver tissue. Cancer Res 60:5723–5730
Brouwers AH, Mulders PF, de Mulder PH, van den Broek WJ, Buijs WC, Mala C, Joosten FB, Oosterwijk E, Boerman OC, Corstens FH, Oyen WJ (2005) Lack of efficacy of two consecutive treatments of radioimmunotherapy with 131I-cG250 in patients with metastasized clear cell renal cell carcinoma. J Clin Oncol 23:6540–6548
Brouwers AH, van Eerd JE, Frielink C, Oosterwijk E, Oyen WJ, Corstens FH, Boerman OC (2004) Optimization of radioimmunotherapy of renal cell carcinoma: labeling of monoclonal antibody cG250 with 131I, 90Y, 177Lu, or 186Re. J Nucl Med 45:327–337
Byrne AT, Ross L, Holash J, Nakanishi M, Hu L, Hofmann JI, Yancopoulos GD, Jaffe RB (2003) Vascular endothelial growth factor-trap decreases tumor burden, inhibits ascites, and causes dramatic vascular remodeling in an ovarian cancer model. Clin Cancer Res 9:5721–5728
Cecchi A, Hulikova A, Pastorek J, Pastorekova S, Scozzafava A, Winum JY, Montero JL, Supuran CT (2005) Carbonic anhydrase inhibitors. Design of fluorescent sulfonamides as probes of tumor-associated carbonic anhydrase IX that inhibit isozyme IX-mediated acidification of hypoxic tumors. J Med Chem 48:4834–4841
Chang CC, Lin MT, Lin BR, Jeng YM, Chen ST, Chu CY, Chen RJ, Chang KJ, Yang PC, Kuo ML (2006) Effect of connective tissue growth factor on hypoxia-inducible factor 1alpha degradation and tumor angiogenesis. J Natl Cancer Inst 98:984–995
Cheng JD, Dunbrack RL Jr, Valianou M, Rogatko A, Alpaugh RK, Weiner LM (2002) Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res 62:4767–4772
Conejo-Garcia JR, Buckanovich RJ, Benencia F, Courreges MC, Rubin SC, Carroll RG, Coukos G (2005) Vascular leukocytes contribute to tumor vascularization. Blood 105:679–681
Dezube BJ, Krown SE, Lee JY, Bauer KS, Aboulafia DM (2006) Randomized phase II trial of matrix metalloproteinase inhibitor COL-3 in AIDS-related Kaposi’s sarcoma: an AIDS Malignancy Consortium Study. J Clin Oncol 24:1389–1394
Dornhofer N, Spong S, Bennewith K, Salim A, Klaus S, Kambham N, Wong C, Kaper F, Sutphin P, Nacalumi R, Hockel M, Le Q, Longaker M, Yang G, Koong A, Giaccia A (2006) Connective tissue growth factor-specific monoclonal antibody therapy inhibits pancreatic tumor growth and metastasis. Cancer Res 66:5816–5827
Edosada CY, Quan C, Wiesmann C, Tran T, Sutherlin D, Reynolds M, Elliott JM, Raab H, Fairbrother W, Wolf BB (2006) Selective inhibition of fibroblast activation protein protease based on dipeptide substrate specificity. J Biol Chem 281:7437–7444
Farokhzad OC, Cheng J, Teply BA, Sherifi I, Jon S, Kantoff PW, Richie JP, Langer R (2006) Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy in vivo. Proc Natl Acad Sci USA 103:6315–6320
Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C (2004) Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol 48:411–424
Fonsatti E, Jekunen AP, Kairemo KJ, Coral S, Snellman M, Nicotra MR, Natali PG, Altomonte M, Maio M (2000) Endoglin is a suitable target for efficient imaging of solid tumors: in vivo evidence in a canine mammary carcinoma model. Clin Cancer Res 6:2037–2043
Gerspach J, Nemeth J, Munkel S, Wajant H, Pfizenmaier K (2006) Target-selective activation of a TNF prodrug by urokinase-type plasminogen activator (uPA) mediated proteolytic processing at the cell surface. Cancer Immunol Immunother 55:1590–1600
Glimore BF, Lynas JF, Scott CJ, McGoohan C, Martin L, Walker B (2006) Dipeptide proline diphenyl phosphonates are potent, irreversible inhibitors of seprase (FAPalpha). Biochem Biophys Res Commun 346:436–446
Grabmaier K, Vissers JL, De Weijert MC, Oosterwijk-Wakka JC, Van Bokhoven A, Brakenhoff RH, Noessner E, Mulders PA, Merkx G, Figdor CG, Adema GJ, Oosterwijk E (2000) Molecular cloning and immunogenicity of renal cell carcinoma-associated antigen G250. Int J Cancer 85:865–870
Gutheil JC, Campbell TN, Pierce PR, Watkins JD, Huse WD, Bodkin DJ, Cheresh DA (2000) Targeted antiangiogenic therapy for cancer using Vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res 6:3056–3061
Hajitou A, Trepel M, Lilley CE, Soghomonyan S, Alauddin MM, Marini FC III, Restel BH, Ozawa MG, Moya CA, Rangel R, Sun Y, Zaoui K, Schmidt M, von Kalle C, Weitzman MD, Gelovani JG, Pasqualini R, Arap W (2006) A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 125:385–398
Henry MD, Wen S, Silva MD, Chandra S, Milton M, Worland PJ (2004) A prostate-specific membrane antigen-targeted monoclonal antibody-chemotherapeutic conjugate designed for the treatment of prostate cancer. Cancer Res 64:7995–8001
Hicke BJ, Stephens AW, Gould T, Chang YF, Lynott CK, Heil J, Borkowski S, Hilger CS, Cook G, Warren S, Schmidt PG (2006) Tumor targeting by an aptamer. J Nucl Med 47:668–678
Hirte H, Vergote IB, Jeffrey JR, Grimshaw RN, Coppieters S, Schwartz B, Tu D, Sadura A, Brundage M, Seymour L (2006) A phase III randomized trial of BAY 12–9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: a National Cancer Institute of Canada Clinical Trials Group Study. Gynecol Oncol 102:300–308
Hofheinz RD, al Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, Stehle G (2003) Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie 26:44–48
Hofmeister V, Vetter C, Schrama D, Brocker EB, Becker JC (2006) Tumor stroma-associated antigens for anti-cancer immunotherapy. Cancer Immunol Immunother 55:481–494
Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F (2004) Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 350:2335–2342
Jiang Y, Wang M, Celiker MY, Liu YE, Sang QX, Goldberg ID, Shi YE (2001) Stimulation of mammary tumorigenesis by systemic tissue inhibitor of matrix metalloproteinase 4 gene delivery. Cancer Res 61:2365–2370
Johnson DH, Fehrenbacher L, Novotny WF, Herbst RS, Nemunaitis JJ, Jablons DM, Langer CJ, DeVore RF III, Gaudreault J, Damico LA, Holmgren E, Kabbinavar F (2004) Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 22:2184–2191
Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, Griffing S, Bergsland E (2003) Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol 21:60–65
Kabbinavar FF, Schulz J, McCleod M, Patel T, Hamm JT, Hecht JR, Mass R, Perrou B, Nelson B, Novotny WF (2005) Addition of bevacizumab to bolus fluorouracil and leucovorin in first-line metastatic colorectal cancer: results of a randomized phase II trial. J Clin Oncol 23:3697–3705
Kessler T, Bieker R, Padro T, Schwoppe C, Persigehl T, Bremer C, Kreuter M, Berdel WE, Mesters RM (2005) Inhibition of tumor growth by RGD peptide-directed delivery of truncated tissue factor to the tumor vasculature. Clin Cancer Res 11:6317–6324
Klement G, Baruchel S, Rak J, Man S, Clark K, Hicklin DJ, Bohlen P, Kerbel RS (2000) Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 105:R15–R24
Kobayashi H, Eckhardt SG, Lockridge JA, Rothenberg ML, Sandler AB, O’Bryant CL, Cooper W, Holden SN, Aitchison RD, Usman N, Wolin M, Basche ML (2005) Safety and pharmacokinetic study of RPI.4610 (ANGIOZYME), an anti-VEGFR-1 ribozyme, in combination with carboplatin and paclitaxel in patients with advanced solid tumors. Cancer Chemother Pharmacol 56:329–336
Kobayashi H, Sugino D, She MY, Ohi H, Hirashima Y, Shinohara H, Fujie M, Shibata K, Terao T (1998) A bifunctional hybrid molecule of the amino-terminal fragment of urokinase and domain II of bikunin efficiently inhibits tumor cell invasion and metastasis. Eur J Biochem 253:817–826
Kondo S, Tanaka N, Kubota S, Mukudai Y, Yosimichi G, Sugahara T, Takigawa M (2006) Novel angiogenic inhibitor DN-9693 that inhibits post-transcriptional induction of connective tissue growth factor (CTGF/CCN2) by vascular endothelial growth factor in human endothelial cells. Mol Cancer Ther 5:129–137
Kondraganti S, Gondi CS, McCutcheon I, Dinh DH, Gujrati M, Rao JS, Olivero WC (2006) RNAi-mediated downregulation of urokinase plasminogen activator and its receptor in human meningioma cells inhibits tumor invasion and growth. Int J Oncol 28:1353–1360
Kong HL, Hecht D, Song W, Kovesdi I, Hackett NR, Yayon A, Crystal RG (1998) Regional suppression of tumor growth by in vivo transfer of a cDNA encoding a secreted form of the extracellular domain of the flt-1 vascular endothelial growth factor receptor. Hum Gene Ther 9:823–833
Korn T, Muller R, Kontermann RE (2004) Bispecific single-chain diabody-mediated killing of endoglin-positive endothelial cells by cytotoxic T lymphocytes. J Immunother 27:99–106
Kozin SV, Boucher Y, Hicklin DJ, Bohlen P, Jain RK, Suit HD (2001) Vascular endothelial growth factor receptor-2-blocking antibody potentiates radiation-induced long-term control of human tumor xenografts. Cancer Res 61:39–44
Kruger A, Arlt MJE, Gerg M, Kopitz C, Bernardo MM, Chang M, Mobashery S, Fridman R (2005) Antimetastatic activity of a novel mechanism-based gelatinase inhibitor. Cancer Res 65:3523–3526
Lamers CHJ, Sleijfer S, Vulto AG, Kruit WHJ, Kliffen M, Debets R, Gratama JW, Stoter G, Oosterwijk E (2006) Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol 24:e20–e22
Lefesvre P, Attema J, van Bekkum D (2002) Adenoviral gene transfer of angiostatic ATF-BPTI inhibits tumour growth. BMC Cancer 2:17
Leighl NB, Paz-Ares L, Douillard JY, Peschel C, Arnold A, Depierre A, Santoro A, Betticher DC, Gatzemeier U, Jassem J, Crawford J, Tu D, Bezjak A, Humphrey JS, Voi M, Galbraith S, Hann K, Seymour L, Shepherd FA (2005) Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR. 18. J Clin Oncol 23:2831–2839
Li G, Xie Q, Shi Y, Li D, Zhang M, Jiang S, Zhou H, Lu H, Jin Y (2006) Inhibition of connective tissue growth factor by siRNA prevents liver fibrosis in rats. J Gene Med 8:889–900
Lin P, Sankar S, Shan S, Dewhirst MW, Polverini PJ, Quinn TQ, Peters KG (1998) Inhibition of tumor growth by targeting tumor endothelium using a soluble vascular endothelial growth factor receptor. Cell Growth Differ 9:49–58
Liu S, Netzel-Arnett S, Birkedal-Hansen H, Leppla SH (2000) Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res 60:6061–6067
Liu S, Redeye V, Kuremsky JG, Kuhnen M, Molinolo A, Bugge TH, Leppla SH (2005) Intermolecular complementation achieves high-specificity tumor targeting by anthrax toxin. Nat Biotechnol 23:725–730
Liu Z, Smyth FE, Renner C, Lee FT, Oosterwijk E, Scott AM (2002) Anti-renal cell carcinoma chimeric antibody G250: cytokine enhancement of in vitro antibody-dependent cellular cytotoxicity. Cancer Immunol Immunother 51:171–177
Lokeshwar BL, Selzer MG, Zhu BQ, Block NL, Golub LM (2002) Inhibition of cell proliferation, invasion, tumor growth and metastasis by an oral non-antimicrobial tetracycline analog (COL-3) in a metastatic prostate cancer model. Int J Cancer 98:297–309
Ma D, Hopf CE, Malewicz AD, Donovan GP, Senter PD, Goeckeler WF, Maddon PJ, Olson WC (2006) Potent antitumor activity of an auristatin-conjugated, fully human monoclonal antibody to prostate-specific membrane antigen. Clin Cancer Res 12:2591–2596
Maquoi E, Sounni NE, Devy L, Olivier F, Frankenne F, Krell HW, Grams F, Foidart JM, Noel A (2004) Anti-invasive, antitumoral, and antiangiogenic efficacy of a pyrimidine-2,4,6-trione derivative, an orally active and selective matrix metalloproteinases inhibitor. Clin Cancer Res 10:4038–4047
Matsuno F, Haruta Y, Kondo M, Tsai H, Barcos M, Seon BK (1999) Induction of lasting complete regression of preformed distinct solid tumors by targeting the tumor vasculature using two new anti-endoglin monoclonal antibodies. Clin Cancer Res 5:371–382
McNeel DG, Eickhoff J, Lee FT, King DM, Alberti D, Thomas JP, Friedl A, Kolesar J, Marnocha R, Volkman J, Zhang J, Hammershaimb L, Zwiebel JA, Wilding G (2005) Phase I trial of a monoclonal antibody specific for alphavbeta3 integrin (MEDI-522) in patients with advanced malignancies, including an assessment of effect on tumor perfusion. Clin Cancer Res 11:7851–7860
Miller KD, Saphner TJ, Waterhouse DM, Chen TT, Rush-Taylor A, Sparano JA, Wolff AC, Cobleigh MA, Galbraith S, Sledge GW (2004) A randomized phase II feasibility trial of BMS-275291 in patients with early stage breast cancer. Clin Cancer Res 10:1971–1975
Milowsky MI, Nanus DM, Kostakoglu L, Vallabhajosula S, Goldsmith SJ, Bander NH (2004) Phase I trial of yttrium-90-labeled anti-prostate-specific membrane antigen monoclonal antibody J591 for androgen-independent prostate cancer. J Clin Oncol 22:2522–2531
Mitra A, Coleman T, Borgman M, Nan A, Ghandehari H, Line BR (2006) Polymeric conjugates of mono- and bi-cyclic alpha(V)beta(3) binding peptides for tumor targeting. J Control Release 114:175–183
Mitra A, Nan A, Papadimitriou JC, Ghandehari H, Line BR (2006) Polymer-peptide conjugates for angiogenesis targeted tumor radiotherapy. Nucl Med Biol 33:43–52
Molina JR, Reid JM, Erlichman C, Sloan JA, Furth A, Safgren SL, Lathia CD, Alberts SR (2005) A phase I and pharmacokinetic study of the selective, non-peptidic inhibitor of matrix metalloproteinase BAY 12–9566 in combination with etoposide and carboplatin. Anticancer Drugs 16:997–1002
Moore MJ, Hamm J, Dancey J, Eisenberg PD, Dagenais M, Fields A, Hagan K, Greenberg B, Colwell B, Zee B, Tu D, Ottaway J, Humphrey R, Seymour L (2003) Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12–9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol 21:3296–3302
Nemunaitis J, Vukelja SJ, Richards D, Cunningham C, Senzer N, Nugent J, Duncan H, Jones B, Haltom E, Uprichard MJ (2006) Phase I Trial of PT-100 (PT-100), a cytokine-inducing small molecule, following chemotherapy for solid tumor malignancy. Cancer Invest 24:553–561
Pastorekova S, Casini A, Scozzafava A, Vullo D, Pastorek J, Supuran CT (2004) Carbonic anhydrase inhibitors: the first selective, membrane-impermeant inhibitors targeting the tumor-associated isozyme IX. Bioorg Med Chem Lett 14:869–873
Patel SR, Jenkins J, Papadopolous N, Burgess MA, Plager C, Gutterman J, Benjamin RS (2001) Pilot study of vitaxin-an angiogenesis inhibitor-in patients with advanced leiomyosarcomas. Cancer 92:1347–1348
Pattillo CB, Sari-Sarraf F, Nallamothu R, Moore BM, Wood GC, Kiani MF (2005) Targeting of the antivascular drug combretastatin to irradiated tumors results in tumor growth delay. Pharm Res 22:1117–1120
Petronzelli F, Pelliccia A, Anastasi AM, D’Alessio V, Albertoni C, Rosi A, Leoni B, De Angelis C, Paganelli G, Palombo G, Dani M, Carminati P, De Santis R (2005) Improved tumor targeting by combined use of two antitenascin antibodies. Clin Cancer Res 11:7137s–7145s
Posey JA, Ng TC, Yang B, Khazaeli MB, Carpenter MD, Fox F, Needle M, Waksal H, LoBuglio AF (2003) A phase I study of anti-kinase insert domain-containing receptor antibody, IMC-1C11, in patients with liver metastases from colorectal carcinoma. Clin Cancer Res 9:1323–1332
Pulukuri SM, Gondi CS, Lakka SS, Jutla A, Estes N, Gujrati M, Rao JS (2005) RNA interference-directed knockdown of urokinase plasminogen activator and urokinase plasminogen activator receptor inhibits prostate cancer cell invasion, survival, and tumorigenicity in vivo. J Biol Chem 280:36529–36540
Qu C, Song E, Li Y, Rizvi S, Raja C, Smith R, Morgenstern A, Apostolidis C, Allen B (2005) Pre-clinical study of 213Bi labeled PAI2 for the control of micrometastatic pancreatic cancer. Clin Exp Metastasis 22:575–586
Raguse JD, Gath HJ, Bier J, Riess H, Oettle H (2004) Cilengitide (EMD 121974) arrests the growth of a heavily pretreated highly vascularised head and neck tumour. Oral Oncol 40:228–230
Reardon DA, Akabani G, Coleman RE, Friedman AH, Friedman HS, Herndon JE, McLendon RE, Pegram CN, Provenzale JM, Quinn JA, Rich JN, Vredenburgh JJ, Desjardins A, Gururangan S, Badruddoja M, Dowell JM, Wong TZ, Zhao XG, Zalutsky MR, Bigner DD (2006) Salvage radioimmunotherapy with murine iodine-131-labeled antitenascin monoclonal antibody 81C6 for patients with recurrent primary and metastatic malignant brain tumors: phase II study results. J Clin Oncol 24:115–122
Reinmuth N, Liu W, Ahmad SA, Fan F, Stoeltzing O, Parikh AA, Bucana CD, Gallick GE, Nickols MA, Westlin WF, Ellis LM (2003) Alphavbeta3 integrin antagonist S247 decreases colon cancer metastasis and angiogenesis and improves survival in mice. Cancer Res 63:2079–2087
Rippmann JF, Pfizenmaier K, Mattes R, Rettig WJ, Moosmayer D (2000) Fusion of the tissue factor extracellular domain to a tumour stroma specific single-chain fragment variable antibody results in an antigen-specific coagulation-promoting molecule. Biochem J 349(Pt 3):805–812
Rizvi NA, Humphrey JS, Ness EA, Johnson MD, Gupta E, Williams K, Daly DJ, Sonnichsen D, Conway D, Marshall J, Hurwitz H (2004) A phase I study of oral BMS-275291, a novel nonhydroxamate sheddase-sparing matrix metalloproteinase inhibitor, in patients with advanced or metastatic cancer. Clin Cancer Res 10:1963–1970
Rizzieri DA, Akabani G, Zalutsky MR, Coleman RE, Metzler SD, Bowsher JE, Toaso B, Anderson E, Lagoo A, Clayton S, Pegram CN, Moore JO, Gockerman JP, DeCastro C, Gasparetto C, Chao NJ, Bigner DD (2004) Phase 1 trial study of 131I-labeled chimeric 81C6 monoclonal antibody for the treatment of patients with non-Hodgkin lymphoma. Blood 104:642–648
Rono B, Romer J, Liu S, Bugge TH, Leppla SH, Kristjansen PEG (2006) Antitumor efficacy of a urokinase activation-dependent anthrax toxin. Mol Cancer Ther 5:89–96
Samel D, Muller D, Gerspach J, Assohou-Luty C, Sass G, Tiegs G, Pfizenmaier K, Wajant H (2003) Generation of a FasL-based proapoptotic fusion protein devoid of systemic toxicity due to cell-surface antigen-restricted activation. J Biol Chem 278:32077–32082
Schrama D, Reisfeld RA, Becker JC (2006) Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov 5:147–159
Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, Divgi CR, Hanson LH, Mitchell P, Gansen DN, Larson SM, Ingle JN, Hoffman EW, Tanswell P, Ritter G, Cohen LS, Bette P, Arvay L, Amelsberg A, Vlock D, Rettig WJ, Old LJ (2003) A phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res 9:1639–1647
Shepherd FA, Giaccone G, Seymour L, Debruyne C, Bezjak A, Hirsh V, Smylie M, Rubin S, Martins H, Lamont A, Krzakowski M, Sadura A, Zee B (2002) Prospective, randomized, double-blind, placebo-controlled trial of marimastat after response to first-line chemotherapy in patients with small-cell lung cancer: a trial of the National Cancer Institute of Canada-Clinical Trials Group and the European Organization for Research and Treatment of Cancer. J Clin Oncol 20:4434–4439
Shiozaki K, Harada N, Greco W, Haba A, Uneda S, Tsai H, Seon B (2006) Antiangiogenic chimeric anti-endoglin (CD105) antibody: pharmacokinetics and immunogenicity in nonhuman primates and effects of doxorubicin. Cancer Immunol Immunother 55:140–150
Singh N, Amin S, Richter E, Rashid S, Scoglietti V, Jani PD, Wang J, Kaur R, Ambati J, Dong Z, Ambati BK (2005) Flt-1 intraceptors inhibit hypoxia-induced VEGF expression in vitro and corneal neovascularization in vivo. Invest Ophthalmol Vis Sci 46:1647–1652
Sparano JA, Bernardo P, Stephenson P, Gradishar WJ, Ingle JN, Zucker S, Davidson NE (2004) Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group Trial E2196. J Clin Oncol 22:4683–4690
Strieth S, Eichhorn ME, Sutter A, Jonczyk A, Berghaus A, Dellian M (2006) Antiangiogenic combination tumor therapy blocking alpha(v)-integrins and VEGF-receptor-2 increases therapeutic effects in vivo. Int J Cancer 119:423–431
Takahashi N, Haba A, Matsuno F, Seon BK (2001) Antiangiogenic therapy of established tumors in human skin/severe combined immunodeficiency mouse chimeras by anti-endoglin (CD105) monoclonal antibodies, and synergy between anti-endoglin antibody and cyclophosphamide. Cancer Res 61:7846–7854
Thiry A, Ledecq M, Cecchi A, Dogne JM, Wouters J, Supuran CT, Masereel B (2006) Indanesulfonamides as carbonic anhydrase inhibitors. Toward structure-based design of selective inhibitors of the tumor-associated isozyme CA IX. J Med Chem 49:2743–2749
Vallera DA, Li C, Jin N, Panoskaltsis-Mortari A, Hall WA (2002) Targeting urokinase-type plasminogen activator receptor on human glioblastoma tumors with diphtheria toxin fusion protein DTAT. J Natl Cancer Inst 94:597–606
Vihinen P, Ala-aho R, Kahari VM (2005) Matrix metalloproteinases as therapeutic targets in cancer. Curr Cancer Drug Targets 5:203–220
Wolf P, Gierschner D, Buhler P, Wetterauer U, Elsasser-Beile U (2006) A recombinant PSMA-specific single-chain immunotoxin has potent and selective toxicity against prostate cancer cells. Cancer Immunol Immunother 55:1367–1373
Wuest T, Moosmayer D, Pfizenmaier K (2001) Construction of a bispecific single chain antibody for recruitment of cytotoxic T cells to the tumour stroma associated antigen fibroblast activation protein. J Biotechnol 92:159–168
Xie D, Yin D, Wang HJ, Liu GT, Elashoff R, Black K, Koeffler HP (2004) Levels of expression of CYR61 and CTGF are prognostic for tumor progression and survival of individuals with gliomas. Clin Cancer Res 10:2072–2081
Zeng ZJ, Yang LY, Ding X, Wang W (2004) Expressions of cysteine-rich61, connective tissue growth factor and Nov genes in hepatocellular carcinoma and their clinical significance. World J Gastroenterol 10:3414–3418
Zhu AX, Blaszkowsky LS, Ryan DP, Clark JW, Muzikansky A, Horgan K, Sheehan S, Hale KE, Enzinger PC, Bhargava P, Stuart K (2006) Phase II study of gemcitabine and oxaliplatin in combination with bevacizumab in patients with advanced hepatocellular carcinoma. J Clin Oncol 24:1898–1903
Zukiel R, Nowak S, Wyszko E, Rolle K, Gawronska I, Barciszewska MZ, Barciszewski J (2006) Suppression of human brain tumor with interference RNA specific for tenascin-C. Cancer Biol Ther 5:1002–1007
Acknowledgments
This work was supported by the DFG (Klinische Forschergruppe 124).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hofmeister, V., Schrama, D. & Becker, J.C. Anti-cancer therapies targeting the tumor stroma. Cancer Immunol Immunother 57, 1–17 (2008). https://doi.org/10.1007/s00262-007-0365-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-007-0365-5