
Abstract
Mammalian Toll-like receptors (TLRs) represent pattern recognition receptors of the immune system and are related to the Toll protein of Drosophila. Pathogen-associated molecular patterns (PAMPs) of microbial and viral origin bind to TLRs and initiate the innate and adaptive immune response. However, TLRs are not solely found on cells of the immune system but also on non-myeloid cells in various tissues, e.g., on vascular cells. In addition to PAMPs, there is increasing evidence that TLRs also recognize endogenous ligands. Recent studies demonstrate the contribution of distinct TLRs in different inflammatory disorders such as cardiovascular diseases, rheumatoid arthritis, systemic lupus erythematosus, and cancer. Many of these disorders are characterized by enhanced angiogenesis which is mainly trigged by inflammation. However, this inflammation-induced angiogenesis is not only important for pathogen defense during acute infection or chronic inflammatory disorders but as well involved in regenerative processes during wound healing and tissue repair. There is cumulative evidence that TLR activation by exogenous as well as endogenous ligands especially contributes to the angiogenic process in this scenario. The present chapter will summarize the current understanding of TLR-linked signal transduction in angiogenesis during inflammatory processes with future prospects for pro- or antiangiogenic therapy.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Angiogenesis
- Toll-like receptors
- Pathogen-associated pattern
- Damage-associated molecular patterns
- Inflammation
1 Introduction: Toll! Everything Started in Drosophila
A group of maternal effect genes are necessary for the embryo patterning of the fruit fly Drosophila melanogaster including the Toll gene. Lack of function experiments revealed that the Toll gene product provides the source for a morphogen gradient in the dorsal-ventral axis of the Drosophila embryo [1, 2]. Mutants in this gene were originally identified in 1985 by the group of Christiane Nüsslein-Volhard at the Max-Planck-Institute in Tübingen/Germany. The name of the gene derives from her exclamation “Das ist ja toll!” which translates as “That’s amazing!” during microscopic observation of the drosophila mutants. Three years later, the Toll gene of Drosophila was cloned in the lab of Kathryn Anderson, the first author of the initial studies [3]. In 1992, Christiane Nüsslein-Volhard was awarded with the Nobel Prize for her groundbreaking research. Later on, Toll was found to play an important role in the fly’s immune response by the group of Jules Hoffmann [4, 5]. In total, nine Toll receptors are encoded in the Drosophila genome, including the Toll pathway receptor Toll. The induction of the Toll pathway by fungi or by gram-positive bacteria leads to the activation of antimicrobial peptides. After proteolytical cleavage, binding of the extracellular ligand Spaetzle to the Toll receptor controls the expression of the antifungal peptide gene drosomycin. Mutations in the Toll signaling pathway dramatically reduce survival after fungal infection demonstrating the importance of this pathway for immune response.
2 Toll-Like Receptors in Mammalians
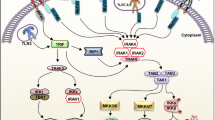
The identification of the Drosophila Toll pathway and the subsequent characterization of Toll-like receptor (TLR) function have reshaped the current understanding of the immune system. Mammalian homologues of the Drosophila Toll protein have been discovered 10 years later in the mid-1990s of last century which were consequently named TLRs [6, 7]. A scientific highlight in mammalian TLR discovery was the identification of TLR4 as the functional receptor for bacterial lipopolysaccharide (LPS) in mice carrying a mutation in the TLR4 gene by Bruce Beutler [8]. Based on this important discovery, Bruce Beutler was awarded with the Nobel Prize in 2011 which he shared with Jules Hoffmann for his findings in Drosophila and Ralph Steinmann who discovered dendritic cells. The discoveries of Hoffmann and Beutler triggered an explosion of research in innate immunity. Around a dozen different TLRs have now been identified in humans and mice comprising an entire receptor protein family [9]. All of them have initially been described as guardians of the innate immunity recognizing invading pathogens in the front line on the plasma membrane or after phagocytosis and processing on endosomal membranes, respectively. TLRs represent cognate pattern recognition receptors (PRRs) of the innate immunity and recognizing a high diversity of molecules common in pathogens of bacterial and viral origin referred to as pathogen-associated molecular patterns (PAMPs) . The specificity of TLRs for their ligands was mainly investigated in mice with functional mutations carrying an increased risk of infection. TLR ligation induces the activation of inflammatory pathways such as the mitogen-activated protein kinase (MAPK) cascade or nuclear factor κB (NFκB) and finally leads to the expression of cytokines and co-stimulatory molecules [10]. Thus, TLRs activate a potent immunostimulatory response and the signal that is transmitted from TLRs must therefore be tightly controlled. Structurally, all TLRs are type I integral membrane proteins consisting of an ectodomain comprised of leucine-rich repeats (LRRs) and a cytoplasmic domain containing a Toll/interleukin-1 receptor homology domain (TIR) , which is required for signaling. TLRs occur as dimers; different receptor assemblies as mono- or heterodimers are known [11,12,13]. TLR2 builds heterodimers; in this regard TLR2/1 dimers sense bacterial triacylated lipopeptides were as TLR2/6 dimers sense bacterial diacylated lipopeptides. The LPS receptor TLR4 and TLR9, the receptor for unmethylated CpG-motifs in bacterial and viral DNA homodimerize and TLR4 may additional forms heterodimers with TLR2 in microglial cells in response to ethanol [14]. Homodimerization is presumed to be the case for TLR3 which senses synthetic and double-stranded RNA of viral origin (dsRNA) as well as for TLR5 which detects flagellin from bacteria. TLR7 and TLR8 recognize synthetic imidazoquinolines components and single-stranded RNA (ssRNA) and TLR8 has been shown to dimerize with TLR7 and TLR9. TLR10 is the only pattern-recognition receptor without known ligand specificity and biological function and maybe is a modulatory receptor with mainly inhibitory effects [15]. It has been demonstrated that TLR10 can heterodimerize with TLR1 or TLR2 [16]. More recently, TLR11, 12 and 13 have been identified in mouse. Interestingly, an eukaryotic ligand have been described for TLR11, namely a profilin-like molecule from the obligate intracellular protozoan parasite Toxoplasma gondii [17], which is recognized in cooperation with TLR12 [18]. Finally, it has been shown that TLR13 is the functional receptor for a conserved sequence in the 23S ribosomal RNA (rRNA) from bacteria [19]. The number of putative TLR interaction partners and identified PAMPs that bind to TLRs is already large and diverse and is still growing (Fig. 3.1, Table 3.1) [20].

TLRs form homo- and heterodimers
A fundamental basis of TLR signaling is dependent upon the recruitment and association of adaptor molecules that contain the structurally conserved TIR domain. Signaling by TLRs involves five so far identified adaptor proteins known as myeloid differentiation primary response gene 88 (MyD88) , MyD88-adaptor-like (MAL, also known as TIRAP), TIR-domain-containing adaptor protein inducing interferon-β (TRIF; also known as TICAM1), TRIF-related adaptor molecule (TRAM; also known as TICAM2), and sterile α- and armadillo-motif-containing protein (SARM) . These adaptor molecules provide the necessary framework to recruit and activate downstream kinases and transcription factors that regulate the host inflammatory response. The canonical TIR pathway is dependent on MyD88 , the immediate adapter molecule that is common to all TLRs, except TLR3. An alternative MyD88-independent pathway is controlled by TRIF, the only TLR3 adaptor, whereas TLR4 binds both MyD88 and TRIF. The remaining three adaptor proteins serve as co-adaptors (MAL, TRAM) or even as a negative regulator (SARM) . MAL and TRAM are just used by few TLRs. MAL recruits MyD88 to TLR2 and TLR4, whereas TRAM recruits TRIF to TLR4 [13]. After ligand binding to the specific TLR and assembly of the adaptor proteins, the activated membrane receptor complex induces the interleukin-1 receptor-associated kinase (IRAK) and tumor necrosis factor receptor-associated factor (TRAF) family members. The IRAK family – with their four members: IRAK1, IRAK2, IRAK4, and IRAKM – plays a pivotal role in mediating almost all TLR-mediated functions. All IRAK family members contain an amino-terminal death domain and a serine/threonine kinase domain. IRAK4 is known to be essential for TLR-mediated cellular responses. After TLR ligation, IRAK4 phosphorylates IRAK1 [21]. IRAK activation results in the recruitment/activation of TRAF family members such as TRAF3 and TRAF6, along with other E2 ubiquitin protein ligases which activate a complex containing transforming growth factor-β-activated kinase 1 (TAK1), TAK1-binding protein 1 (TAB1), TAB2, and TAB3 [22]. MyD88dependent TAK1 activation induces the NFκB pathway and MAPK members such as the extracellular signal-regulated kinase (ERK)1/2, p38, and stress-activated protein kinase/c-Jun N-terminal kinase (SAPK/JNK) to initiate the expression of inflammatory cytokines [9,10,11]. The TLR3 pathway is MyD88-indendent but TRIF-dependent that activates TRAF6 and NFκB, resulting in the expression of inflammatory cytokines [23]. But TLR3 engagement also induces the expression of type I interferons (IFNs) via interferon regulatory transcription factor (IRF) 3 [24]. TLR7 and TLR9 engagement induces the secretion of inflammatory cytokines through the activation of NFκB via MyD88. However, TLR7 and TLR9 can also induce the expression of type I IFNs through the activation of IRF7 [25]. Taken together, PAMP ligation to TLRs leads to the expression of effector molecules which finally organize the body’s immune responds to pathogens (Fig. 3.2).

TLR adaptor molecules and signaling pathways
There is accumulating evidence from recent research that TLRs have distinct different functions beyond simple pathogen recognition. In a more complex immunologic view, an important role in dendritic cell maturation and T cell activation established TLRs as a link between innate and adaptive immunity [26]. Furthermore, the detection of several TLR members in multiple tissues and cell types – besides cells of the immune system – led to a more wide-ranging view on TLRs. Especially inflammatory disorders such as ischemic coronary artery disease [27] and liver disease [28] but also autoimmune diseases [29] are critically influenced by TLRs. Moreover, an involvement of TLRs in allograft acceptance/rejection during transplantation [30] or contact allergy to nickel [31] has been shown. Of interest, an interaction of TLRs with endogenous ligands released during tissue damage and fibrosis or from apoptotic cells has recently been discovered and seems to regulate many sterile inflammatory processes [32]. In this regard the term danger or damage-associated molecular patterns (DAMPs) has been introduced. These ligands include proteins and peptides, polysaccharides and proteoglycans, nucleic acids, and phospholipids, which are cellular components or extracellular matrix (ECM) degradation products (Table 3.2). Recent studies provided clear evidence that endogenous ligand-mediated TLR signaling is involved in pathological conditions such as tissue injury, autoimmune diseases, and tumorigenesis. The ability of TLRs to recognize endogenous ligands appears to be essential for their function in regulating noninfectious inflammation. Furthermore, a novel role for TLRs in wound healing [33, 34] and liver regeneration [35] also in response to endogenous ligands [32] has been reported, suggesting even a regenerative aspect in TLR biology.
3 Angiogenesis: General Remarks
Physiological tissue function depends on adequate supply of nutrients and oxygen through blood vessels. Consequently, the cardiovascular system is the first organ system that develops during embryogenesis. Blood vessels in the embryo form the hemangioblast by differentiation of common mesodermal progenitor cells . The hemangioblast forms aggregates that evolve into hematopoietic precursor cells and angioblasts which further assemble the primary capillary plexus as differentiated endothelial cells. The formation of this primitive network on the basis of progenitor cells is called vasculogenesis . On the contrary, angiogenesis describes the enlargement of capillaries which sprout or become divided by pillars of periendothelial cells (intussusception) or by transendothelial cell bridges followed by remodeling and maturation processes that transform the primary capillary plexus into a complex network of functional vessels [36]. Further covering and stabilization of vessels by smooth muscle cells as well as the enlargement of preexisting collateral arterioles is summarized as arteriogenesis. Although in the adult most vessels arise through angiogenesis, vasculogenesis may also be involved to some extent. Therefore, both processes are summarized in the hypernym neovascularization , which is involved in organ growth and wound healing but also contributes to pathological processes in malignant and inflammatory disorders [37,38,39].
Angiogenesis through sprouting and subsequent remodeling of capillaries into larger vessels has been extensively studied, and several essential steps have been described. Vascular endothelial growth factor (VEGF) and its receptors (VEGFR) have been identified as central regulators of both vasculogenesis and angiogenesis [40]. Until now, five VEGF ligands have been identified which occur in different spliced and processed variants and all of them represent secreted dimeric glycoproteins of ~40 kDa. In addition to VEGF A–D, also placenta growth factor (PLGF) belongs to the VEGF family as well. These ligands bind to the three receptor tyrosine kinases VEGFR1–3 with an overlapping pattern and co-receptors such as heparan sulfate, proteoglycans, and neuropilins. Different VEGFRs have distinct different functions; VEGFR1 is involved in the recruitment of hematopoietic progenitor cells and migration of monocytic cells whereas VEGFR2 and 3 are essential for the function of endothelial cells, especially during angiogenesis. Initially, VEGF was described to increases vascular permeability [41], thereby permitting extravasation of plasma proteins that establish a preliminary scaffold for migrating endothelial cells. For the emigration of endothelial cells from their resident site, interendothelial cell contacts and periendothelial cell support have to be dissolved, leading to destabilization of the mature vessel. Angiopoietin (Ang) 2, an inhibitor for tyrosine kinase with Ig and epidermal growth factor (EGF) homology domains (Tie) 2 signaling are involved in detaching smooth muscle cells and breaking up the ECM [42, 43]. Especially during angiogenesis, the interaction of the Ang-Tie system with the VEGF system becomes apparent. Capillaries sprout and subsequently grow alongside a VEGF gradient. Endothelial cells at the leading edge of the migration front, so-called tip cells, exhibit numerous filopodia and express members of the VEGFR family. Subjacent endothelial cells could be subdivided in highly proliferative and differentiating stalk cells and resting phalanx cells which both express components of the Ang/Tie system [43]. Furthermore, proteinases of the plasminogen activator, matrix metalloproteinases (MMPs), and chymase families influence angiogenesis by degrading ECM and by liberating growth factors, e.g., VEGF, basic fibroblast growth factor (bFGF), and insulin-like growth factor (IGF)-1, sequestered within the ECM. When the path has been cleared, endothelial cells can proliferate and migrate to remote sites [39].
Angiogenic sprouting is controlled by a tightly regulated balance of activators and inhibitors. In addition to VEGF, Tie2 phosphorylation by Ang1 is chemotactic for endothelial cells and stabilizes VEGF-initiated endothelial networks by stimulating the interaction between endothelial cells and periendothelial cells [43]. Members of the FGF and platelet-derived growth factor (PDGF) family support angiogenesis presumably by recruitment of mesenchymal or inflammatory cells. Another key component of sprouting angiogenesis by regulating tip cell vs. stalk cell communication is the highly conserved Delta/Notch signaling pathway . Mammalians possess four different notch receptors, referred to as Notch1–4. Notch receptors are single-pass transmembrane receptors and capable of binding the membrane-bound ligands Delta-like (Dll) 1–4 and Jagged. Notch signaling in the stalk cells induces a quiescent and non-sprouting phenotype in endothelial cells whereas adjacent tip cells express Dll4, therefore promoting sprouting activity [44]. In addition, molecules are involved which mediate cell-cell or cell-matrix interactions, e.g., αvβ3 which localizes MMP-2 at the endothelial cell surface and promotes endothelial cell spreading. Moreover, a continuously number of molecules are discovered which are proangiogenic upon exogenous administration, including erythropoietin, leptin, hepatocyte growth factor (HGF), EGF, IGF-1, tissue factor (TF), and several other cytokines, chemokines, and growth factors [39, 45]. Even hematopoietic growth factors such as granulocyte colony-stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF) have been shown to exhibit proangiogenic potential [46]. On the contrary, angiogenesis inhibitors suppress endothelial cell proliferation and migration, e.g., angiostatin, endostatin, antithrombin III, IFN-β, leukemia inhibitory factor (LIF), and platelet factor 4 [39, 47]. Thus, various pro- and antiangiogenic factors cooperate to regulate the angiogenic process.
Finally, vessel maturation finalizes the angiogenic process. Proliferating endothelial cells initially assemble as solid cords which acquire additional lumen formation. Lumen formation is accomplished by thinning of endothelial cells or fusion of preexisting vessels, mediated by VEGF, Ang1, and integrins such as αvβ3 or α5 and controlled by the inhibitory effects of thrombospondin-1. Additional important steps after lumen establishment involve the differentiation of endothelial cells according to the environmental demands, maturation into a functional three-dimensional endothelial network, and the protection of quiescent endothelial cells against apoptosis [39, 45]. Periendothelial cells are essential for vascular maturation and completion of angiogenesis. Nascent vessels are stabilized by pericytes in case of capillaries. In case of arteries, arterioles, veins, and venules, smooth muscle cell recruitment and growth mediated by VEGF or PDGF are crucial for vessel stabilization. They thereby provide hemostatic control and protect the new endothelium-lined vessel against rupture and regression [39, 47].
Blood vessel formation in the adult includes vasculogenesis, angiogenesis, and arteriogenesis. Impaired neovascularization represents a therapeutic target in several pathologies associated with insufficient blood supply, e.g., acute myocardial infarction or chronic peripheral artery disease. Formation of new vessels and remodeling of the preexisting vasculature are essential for a successful therapy. Therefore, different treatment strategies involving administration of growth factors, cytokines, or progenitor cells are considered [48]. Important for the understanding of angiogenic mechanisms in these pathologies is the knowledge of variations from physiologic angiogenesis. In contrast to the physiological processes, pathologic angiogenesis is often promoted by inflammation. Monocytes, platelets, mast cells, and other leukocytes are recruited to sites of inflammation or wound healing, partly by proangiogenic factors such as VEGF [37, 39]. Moreover, development of solid tumors strictly depends on a growing capillary network – termed as tumor angiogenesis – ensuring sufficient supply with oxygen and nutrients. Accordingly, antiangiogenic concepts aim at the inhibition of tumor angiogenesis and thereby tumor nutrient supply [49]. In this regard, the first antitumor therapy with a VEGF-neutralizing monoclonal antibody for the treatment of metastasizing bowel cancer was approved by the US Food and Drug Administration in 2004.
4 Inflammation-Induced Angiogenesis
Disorders associated with perpetuated angiogenesis are considered to be angiogenic inflammatory diseases. Inflammation plays not only a key role in pathogen defense during infection; it also plays a key role in repair mechanisms, e.g., wound healing and subsequent tissue regeneration. Physiological wound healing requires the integration of complex cellular and molecular events. The repair process is tightly controlled involving different cell types during the phases of initial inflammation as well as the successive cell migration, cell proliferation, and angiogenesis. Several angiogenic mediators, including growth factors, cytokines, MMPs, matrix macromolecules, cell adhesion receptors, chemokines, and chemokine receptors, have been implicated in the process of capillary formation [50]. Of note, cytokines and growth factors released at the site of injury are essential for the repair process [51]. In this regard, angiogenesis, the reestablishment of a capillary network by endothelial cells, is mainly initiated and maintained by the major proangiogenic factor VEGF. Besides endothelial cells, the angiogenic process involves also other cell types including inflammatory cells which represent a major source of growth factors and critically contribute to angiogenesis [52]. Platelets, mast cells, primarily monocytes/macrophages, neutrophils, and other leukocytes are recruited to sites of wound healing, partly by the action of the proangiogenic factors such as VEGF. All these cells in turn release proangiogenic factors such as VEGF, bFGF, TGF-β, PDGF, tumor necrosis factor (TNF)-α, insulin-like growth factor (IGF)-1, monocyte chemotactic protein (MCP)-1, interleukin (IL)-6, IL-8, and many more. All these factors finally attract endothelial cells, smooth muscle cells, pericytes, and fibroblasts to accomplish vessel growth in order to restore sufficient blood supply [39]. Newly formed blood vessels again enhance inflammatory cell recruitment setting up a stimulating forward loop. In this regard, inflammation often promotes angiogenesis establishing the term inflammation-induced angiogenesis.
In inflamed tissues a regulatory network is involved in the control of angiogenesis. Accumulating evidence suggests an association between angiogenesis and inflammation in pathological situations. Therefore, angiogenesis and inflammation seem to be intimately involved in many chronic inflammatory disorders with distinct etiopathogenic origin, including rheumatoid arthritis, diabetes, cancer, and many more. For example, there is considerable evidence of an interrelationship between the mechanisms of angiogenesis and chronic inflammation in inflammatory bowel disease (IBD) . The increased expression of endothelial junction adhesion molecules found in IBD patients indicates the presence of active angiogenesis. Evidence that angiogenesis is involved in IBD was also obtained from animal models of colitis, most notably from studies of angiogenesis inhibition. Moreover, serum levels of VEGF correlate with disease activity in human IBD [53]. This concept has been further supported by the finding that several previously established non inflammatory disorders, such as obesity, display both inflammation and angiogenesis in an exacerbated manner [54]. In addition, the interplay between recruited inflammatory cells and local endothelial cells and fibroblasts at sites of chronic inflammation, together with the fact that inflammation and angiogenesis can actually be triggered by the same molecular events, further strengthen this association. Angiogenesis might be targeted by several specific approaches that could be therapeutically used to control inflammatory diseases.
5 Toll-Like Receptors in Inflammation-Induced Angiogenesis
It is experimentally well established that angiogenesis and inflammation represent two prominent processes involved in normal physiologic responses and pathological states. Emerging evidence also suggests that TLRs have an important role in maintaining tissue homeostasis by regulating the inflammatory and tissue repair responses to injury. Infectious disorders result in inflammation which in turn promotes angiogenesis mainly by the action of growth factors released by different leucocytes. Even though the association of inflammation and angiogenesis has been established for a while the knowledge about the role of TLRs in this context is still limited [55]. However, a significant number of publications demonstrate that several TLR agonists are able to induce the expression and secretion of angiogenic factors from different cell types in vitro. The majority of these studies remain rather descriptive in this context and are very much focused on LPS and VEGF. Up to know, only few data document a direct involvement of TLRs in angiogenesis, both in physiological and in pathophysiological settings .
6 Toll-Like Receptors in Infection-Induced Angiogenesis
Accumulating evidence points to a direct contribution of TLRs to the angiogenic process following bacterial infections, also referred to as infection-induced angiogenesis. In this regard, PAMPs from various bacterial species – super abound in an infection setting – are known to act via different TLRs. A possible influence of TLRs on angiogenic processes was first discovered in the context of adenosine and its A2A receptor (A2AR). The nucleoside adenosine was found to stimulate angiogenesis through upregulation of VEGF, thereby participating in tissue protection following ischemic events. In 2002 Leibovich et al. described a synergistic interaction of A2AR agonists with LPS through the TLR4 pathway. This interaction resulted in a strong upregulation of VEGF and downregulation of TNF-α in macrophages [56] and could also be demonstrated for TLR2, 7, and 9 [57], representing an angiogenic switch. This synergy observed in vitro seems to play an important role in vivo, too. Given the fact that MyD88-deficient mice showed markedly slower wound healing and reduced generation of new capillaries in response to an A2AR agonist [34]. In terms of TLR4, it is very likely that LPS induces adenosine which in turn promotes angiogenesis through A2AR by the upregulation of VEGF expression in macrophages [58].
Independent of the A2AR system, Pollet et al. showed that the TLR4 ligand LPS directly stimulates endothelial sprouting in vitro via a TRAF6-, NFκB-, and JNK-dependent mechanism. However, the responsible angiogenic growth factors remained elusive in this context [59]. Furthermore, a so far unidentified TLR ligand seems to be involved in the formation of angiogenic lesions resulting from infection with the facultative intracellular bacterium Bartonella henselae. This bacterial infection leads to the activation of hypoxia-inducible factor-1 (HIF-1) and thus to an enhanced MCP-1 production in endothelial cells which in turn induces chemotaxis of monocytes in order to initiate angiogenesis by VEGF production. Interestingly, MCP-1 production was independent of LPS/TLR4 but dependent on NFκB [60, 61]. A serious problem of severe ocular infection is pathological corneal neovascularization which could finally lead to visual disorders. In this regard, it has been shown that VEGF and TLR4 expression are upregulated in response to LPS and that VEGF expression is TLR4-dependent [62].
But angiogenesis also contributes to the regeneration process during liver fibrosis which is associated with increased endotoxin levels in the gut and portal circulation. Jagavelu et al. recently demonstrated a key role for the TLR4/MyD88 axis during VEGF production and the subsequent angiogenic process in liver endothelial cells following LPS stimulation [63]. Likewise, mycoplasma infections could be accompanied by enhanced angiogenesis and microvascular remodeling which are features of the chronic inflammation as elicited by Mycoplasma pulmonis infections of the respiratory tract [64]. In this regard, we recently investigated the highly angiogenic properties of the specific TLR2/6 agonist macrophage-activating lipopeptide of 2 kDa (MALP-2), a diacylated lipopeptide which occurs in Mycoplasma species and gram-positive bacteria. Interestingly, this process seems to be independent of VEGF. We discovered a TLR2/6-dependent induction of the MAPK cascade and NFκB and a strong secretion of GM-CSF in particular from endothelial cells and to a lesser degree from monocytes. Accordingly, MALP-2-induced angiogenesis in vitro and in vivo could be suppressed by inhibition of GM-CSF [65]. Similarly, human bone marrow mesenchymal stem cells (MSCs) secreted growth factors in response to a TLR2/6-dependent stimulation by MALP-2 . This process in turn promoted proangiogenic properties of endothelial cells such as migration, proliferation, and tube formation in vitro in a paracrine manner. MSCs isolated from the bone marrow of sheep and co-cultivated with MALP-2 ex vivo significantly enhanced capillary density of skeletal muscle after autogenic implantation of these MSCs [66]. This renders MALP-2 potentially eligible for therapeutic angiogenesis or cell therapy.
In addition to an acute infection upon injury, there are different acute or chronic inflammatory disorders which are also associated with bacterial infection independent of an initial injury. Arthritis is characterized by inflammatory cell infiltration into the concerned joint. Progression of the disease includes self-perpetuating destruction of articular cartilage and extensive angiogenesis in the synovial membrane. Especially TLR2 ligands of gram-positive bacteria such as peptidoglycan (PGN) seem to be responsible for this angiogenic phenotype characterized by the induction of VEGF in chondrocytes [67] and accordingly VEGF and IL-8 in fibroblasts [68]. In light of immune defense, infection-induced angiogenesis might represent a general mechanism to restore blood flow in order to recruit immune cells for pathogen clearance and tissue regeneration with implication for future angiogenic therapy.
7 Toll-Like Receptors in Tumor Angiogenesis
The development of cancer has been associated with microbial infection, injury, inflammation, angiogenesis, and tissue repair. The role of TLRs in tumor angiogenesis is quite diverse just as cancer itself. Tumor inflammation could promote tumor angiogenesis, immunosuppression, and finally tumor growth. However, the mechanism controlling inflammatory cell recruitment to the tumor is not well understood. Cyclooxygenase (COX)-2 is known to play a crucial role in Helicobacter pylori-associated gastric cancer. In this regard, Chang et al. demonstrated that H. pylori acts through TLR2 and TLR9 to activate the MAPK cascade leading to COX-2-dependent prostaglandin E2 (PGE2) release and thereby contributing to cancer cell invasion and angiogenesis [69]. Furthermore, extracellular HSP70 peptide complexes are able to promote the proliferation of hepatocellular carcinoma cells in a TLR2/4 dependent manner [70]. Besides exogenous ligands that contribute to TLR-mediated tumor angiogenesis, also parts of the extracellular matrix (ECM) which are implicated in a variety of human cancers can induce VEGF expression in endothelial cells. Biglycan as one component of the ECM increases the interaction of NFκB and the HIF-1α promotor in a TLR2- and TLR4-dependent manner resulting in VEGF secretion, enhanced cell proliferation and tube formation. VEGF released by endothelial cells in turn promotes cancer cell migration and metastasis [71]. On the other hand, stimulation of TLRs with particular agonists can also cause antitumor activity, interfering with cancer proliferation and angiogenesis by mechanism still incompletely understood. For instance, the immunomodulatory TLR9 agonist IMO inhibited microvessel formation and tumor growth [72]. Likewise, TLR3 agonists not only affect tumor microenvironment by suppressing angiogenesis but also directly induce tumor cell apoptosis and inhibit tumor cell migration [73]. Interestingly, siRNAs may produce therapeutic effects in a target-independent manner through the stimulation of the TLR3/interferon pathway and suppression of angiogenesis. Injection of siRNAs against different targets led to a comparable reduction in liver tumors and to an inhibition of tumor vasculature remodeling. In addition, polyI:C treatment reduced liver tumors and decreased hepatic arterial blood flow, indicating that TLR3 may mediate antiangiogenic and antitumor properties [74].
In all likelihood, there are two different possibilities for TLR agonists to limit tumor growth. First, by altering the tumor microenvironment and inhibiting angiogenesis and second, by clearing tumor cells due to enhanced activity of natural killer and tumor-reactive T cells. In this regard, the TLR7 agonist imidazoquinoline and the TLR9 agonist unmethylated CpG oligonucleotides were shown to exhibit strong local activity against leukemia, and respective phase I trials are currently in progress at different centers [75]. We recently identified proangiogenic properties for the TLR2/6 ligand MALP-2 [65]. Interestingly, there are also antitumor activities reported for MALP-2 [76,77,78]. However, whether MALP-2 affects tumor angiogenesis is currently unknown. Concanavalin-A (ConA) is another TLR2/6 agonist that promotes endothelial cell proliferation trough a JAK/STAT3-mediated increase in the expression of colony-stimulating factor-(CSF) 2 and -3 in human mesenchymal stromal cells [79]. TLR4 expression in the tumor microenvironment was found to be associated with adenocarcinoma in human samples and in the murine model. Adenocarcinoma patients with higher TLR4 expression in stromal compartment had a significantly increased risk in disease progression. These data suggest that high TLR4 expression in the tumor microenvironment represents a possible marker for disease progression in colon cancer [80]. So far, there are different polymorphisms in several TLR gene clusters known which may shift balance between pro- and anti-inflammatory cytokines, modulating the risk of infection, chronic inflammation, and cancer. This may offer the possibility for improved diagnostics in patients. Future studies in large populations should shed light on the significance of TLR polymorphisms for cancer prevention [81].
8 Endogenous Toll-Like Receptor Ligands in Angiogenesis
Sustained pro-inflammatory responses in diseases such as rheumatoid arthritis, atherosclerosis, diabetic retinopathy, and cancer are often associated with increased angiogenesis that contributes to tissue disruption and disease progression. In recent years, there was accumulating evidence that also endogenous ligands which are released during ECM breakdown or by apoptotic cells could bind to different TLRs (Table 3.2). In this context, the high-mobility group B1 (HMGB1) which is released by necrotic cells has been recognized to signal through the receptor for advanced glycation end products (RAGE) and via TLR2 and TRL4. Activation of these receptors resulted in the activation of NFκB and the upregulation of angiogenic factors like VEGF in both hematopoietic and endothelial cells [82]. HMGB1 released at wound sites initiates TLR4-dependent responses that contribute to angiogenesis by regulating endothelial permeability and vascular growth [83, 84]. The interaction of HMGB1 and TLR4 also mediates the recruitment of endothelial progenitor cells to the sites of neovascularization by upregulation of stromal cell-derived factor-1 (SDF-1) [85]. Recent data by van Beijnum et al. identified HMGB1 even as an important modulator of tumor angiogenesis [86]. Thus, targeting the HMGB1 signaling cascade may constitute a novel therapeutic approach to angiogenesis-related diseases. Following this line, inflammation-induced oxidative stress and angiogenesis is emerging as an important mechanism underlying numerous processes from tissue regeneration and remodeling to cancer progression. Interestingly, West et al. recently reported that end products of lipid oxidation such as ω-(2-carboxyethyl)pyrrole (CEP) are generated and accumulate during inflammation, wound healing, and in tumors. CEP is specifically recognized by TLR2 but not TLR4 or scavenger receptors in endothelial cells, leading to a MyD88-dependent angiogenic response that is independent of VEGF [87]. In this regard, stress-sensing by TLR2 seems to be a major driver of angiogenesis [88, 89]. Apparently, also endogenous ligands , which accumulate during inflammatory tissue disruption and enhanced oxidative stress conditions, are capable of promoting angiogenesis via a TLR-dependent pathway. Thus, TLRs are activated not only in response to tissue-invading pathogens but also pathogen-independent. In both cases TLRs have important functions in the recruitment of immune cells in order to initiate a regenerative program: in the first case mainly to eliminate invading pathogens and in the second case to clear the affected tissue from apoptotic cells and cellular debris. Obviously, angiogenic processes are involved in both scenarios (Fig. 3.3).

Tissue regeneration through TLRs
9 Oxidative Stress and Toll-Like Receptor-Dependent Angiogenesis
Increased oxidative stress is closely related to many disease pattern, e.g. to the pathology of cardiovascular diseases like atherosclerosis, myocardial infarction and stroke. If the well-balanced homeostasis of oxidative and anti-oxidative processes is shifted towards increased formation of reactive oxygen species (ROS) , the resulting oxidative stress leads to the onset of various inflammatory processes. However, there is growing evidence of a potential regenerative crosstalk between oxidative stress and TLRs in angiogenesis in recent years. In this regard, Chen et al. reported that decreased NADPH oxidase (NOX)1 and 4 expression, ROS formation as well as increased vascularization in fat grafts after enrichment with adipose-derived stem cells is TLR4-dependent [90]. In addition, Menden and colleagues described a new mechanism which could be involved in microvascular remodeling after sepsis in the lung. They reported that NOX2 inhibition attenuated LPS-mediated Ang2 signaling and capillary network formation in human pulmonary endothelial cells in vitro [91]. This signaling axis involves the NFκB and MAPK pathways and suggests a tied cooperation of TLR- and NADPH oxidase-dependent signaling to coordinate endothelial regeneration after infection. Interestingly, we observed a related cooperation of TLRs and the NADPH oxidase . In an ongoing project, we identified NOX2-derived superoxide anions as important regulators for GM-CSF release from endothelial cells in response to TLR2/6 stimulation (unpublished data). In this regard, we have already shown that TLR2/6-induced GM-CSF release mediates endothelial proliferation, migration and angiogenesis [65]. The already in the last section mentioned study by West et al. additionally suggested that endogenous end products of augmented oxidative stress could represent a new class of TLR ligands [87]. Since ROS do not only accelerate pathological processes, but are also important for many signaling transduction pathways, an interaction with TLRs in a regenerative aspect seems logical and opens up new possibilities for future research. Particularly because the number of studies in the field is still small .
10 A Side Glance on NOD Receptors and Angiogenesis
Although this chapter is focused on the role of TLRs in angiogenesis we want to take a brief look at a subfamiliy of the nucleotide-binding oligomerization domain (NOD)-like receptors at this point. The NOD receptors NOD1 and NOD2 are intracellular receptors, which sense conserved motifs of bacterial peptidoglycan and are the founders of the entire NOD-like receptor family. Just as TLRs, they belong to the class of PRRs with important functions in immune defense [92]. They were identified at the turn of the millennium by sequence homolog searches. Receptor ligation leads to recruitment of the receptor-interacting protein kinase 2 (RIPK2) followed by the activation of the MAPK and NFκB pathway and subsequently to the induction of many well-known inflammatory genes such as IL-6 and TNF-α. Besides immune defense, recent work showed the involvement of NOD1 and NOD2 in many inflammatory diseases, e.g. IBD, cardiovascular disease and metabolic disease [93]. Campbell and colleagues reported, that NOD2-deficient mice displayed a substantial delay in acute wound repair [94] pointing to an even regenerative role of NOD receptors. However, reports on angiogenesis are very limited so far and to our knowledge there is up to now only one single study existing. Interestingly, Schirbel et al. demonstrated that NOD1 and NOD2 agonist – just like TLR agonists – are capable of inducing proliferation, migration, transmigration and tube formation of human intestinal microvascular endothelial cells in vitro as well as angiogenesis in a mouse model in vivo [95]. These processes were consequently mediated via RIPK2, MAPK and NFκB signaling. Different to TLRs, no endogenous NOD ligands have been identified so far which might play a role in angiogenesis. This new research field as well offers many opportunities to shed more light on the overall picture of PRRs in angiogenic processes .
11 Summary and Therapeutic Perspectives
Accumulating evidence points to a crucial role of TLRs in angiogenesis. However, the mode of action of TLRs in this context is quite diverse. TLR activation consistently promotes angiogenesis in various inflammatory settings in response to both exogenous and endogenous ligands . In regard to an acute local infectious scenario, the angiogenic process seems to be important for sufficient blood supply and the recruitment of immune competent cells for pathogen clearance and subsequent tissue regeneration. In contrast, chronic local infection or prolonged pathogen-independent inflammation leads to excessive angiogenesis with eventually pathological consequences. It should not be left unmentioned here that TLRs may also prevent angiogenesis. In endothelial progenitor cells isolated from umbilical cord blood, TLR3 activation specifically inhibits their proangiogenic properties [96]. Similar, blocking TLR2 in endothelial cells has been shown to promote angiogenesis by a crosstalk between TLR2 and CXCR4 and the activation of proangiogenic kinases downstream of CXCR4 [97]. Moreover, TLR2-deficient mice undergoing hindlimb-ischemia exhibit an augmented capacity to stimulate angiogenesis. A process, that seems to be mediated by immune cells rather than endothelial cells [98]. However, specific ligands for these antiangiogenic effects have not been described yet. Pro- and antiangiogenic properties of TLRs are likewise reported in tumor angiogenesis.
In the future, modulation of TLR signaling could provide the basis for the development of novel therapeutic approaches in diverse settings. Stimulation of TLRs with specific ligands could be used for future therapeutic angiogenesis. However, beneficial effects of therapeutic angiogenesis may be negatively impacted by side effects of pharmacological substances such as statins or non-pharmacologic hormones such as erythropoietin. Moreover, certain requirements for this therapeutic process are warranted. First, as simple as it may sound but no harm should be induced especially tumor induction or tumor growth should be avoided. Second, in order to promote a sustained recovery, endogenous mechanisms of angiogenesis should be induced rather using an excessive administration of exogenous factors which may also act as antigens or inducing tolerance when applied over a long period of time. Finally, organ-specific requirements for recovery should be considered, e.g., for cerebral reconstitution angiogenesis, neurogenesis, synaptogenesis, and neuronal and synaptic plasticity should be induced in parallel [99].
Thus, therapeutic modulation of TLR signaling is a very attractive and novel but also sophisticated therapeutic approach to promote angiogenesis. In order to induce long-term organ repair and restoration after ischemic events, for example, detrimental TLR signaling should be inhibited and in parallel beneficial TLR signaling should be induced. From this point of view, inhibitory strategies targeting TLR signaling seem to be plausible in chronic and persistent infectious situations such as rheumatoid arthritis. Small molecules or siRNA against specific TLRs or their downstream targets may provide novel tools to combat local inflammation via inhibition of angiogenesis. Especially advanced tissue penetration properties of those engineered molecules render them applicable and superior for the use in tissues which are inaccessible for antibiotics. Likewise, inhibitory strategies targeting TLRs could be used to inhibit pathological tumor angiogenesis in order to limit tumor growth. In particular, modulation of TLR3, TLR7, and TLR9 activity seems to be a potential future therapeutic target [72,73,74]. However, great caution is required since pro- and antiangiogenic properties with subsequent pro- or antitumorigenic properties of different agonists recognized by the same TLR are reported.
The potentially most promising future therapeutic approach is the application of specific TLR agonists in damaged ischemic or hypoxic tissues in order to promote angiogenesis and subsequent tissue regeneration, especially when the tissue damage is not initiated or accompanied by severe infection, e.g., in peripheral arterial occlusive disease. In such settings, a single application of TLR agonists mimics an infectious scenario without prolonged local pathogen presence. Such an initial therapeutic boost of the immune system with specific TLR agonist aims to launch a defined regenerative program including enhanced angiogenesis. Of note, the application of single proangiogenic growth factors has already been tested in clinical trials. However, in the case of VEGF monotherapy, large-scale trials have not yet yielded consistent beneficial results [100, 101]. This may be related to recent observations that several other potent proangiogenic factors act in concert with VEGF for proper vessel formation and maturation [43, 44, 47]. In this regard, stimulation of specific TLRs (e.g., TLR2/6) may provide an opportunity to induce a specific pattern of proangiogenic growth factors for sufficient vessel growth and tissue regeneration. Thus, we raised our hope on biologicals such as the lipopeptide and TLR2/6 agonist MALP-2. Recent results from our group indicated that the proangiogenic properties of MALP-2 critically depended on the induction of the growth factor GM-CSF in endothelial cells and monocytes [65]. Additional experiments in a vascular endothelial denudation model in mice revealed promising effects of MALP-2 on endothelial regeneration after vascular injury [102]. Those experimental data are the basis for studies in larger experimental animals and future applications using MALP-2 or related agonists in patients, who suffer from peripheral vascular damage or occlusion in diabetes or post percutaneous vascular interventions or even following stroke. Nevertheless, the question remains how to apply such substances since local delivery is preferred in order to avoid side effects and promote endogenous proangiogenic restoration effects downstream of the site of application. Therefore, we aim to test coating procedures on traditional devices such as drug-eluting stents or coated balloons widely used in interventional cardiovascular medicine. However more innovative devices/treatment approaches such as nanofibers, polymer biodegradable soaked stents with TLR ligands, or endovascular patches placed in the occluded vessel or as seal on the balloon-disrupted vascular segment are in the focus of our interest.
In summary, modulation of TLR activity may offer the possibility for different future therapeutic concepts. Inhibition of TLRs is maybe favorable in settings of prolonged infection/inflammation to rescue the inflamed tissue or to inhibit pathological tumor angiogenesis to limit tumor growth. The contrary concept, TLR stimulation, offers a promising option to promote therapeutic angiogenesis for tissue regeneration.
References
Anderson KV, Jürgens G, Nüsslein-Volhard C (1985) Establishment of dorsal-ventral polarity in the Drosophila embryo: genetic studies on the role of the Toll gene product. Cell 42:779–789
Anderson KV, Bokla L, Nüsslein-Volhard C (1985) Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell 42:791–798
Hashimoto C, Hudson KL, Anderson KV (1988) The Toll gene of Drosophila, required for dorsal-ventral embryonic polarity, appears to encode a transmembrane protein. Cell 52:269–279
Lemaitre B, Nicolas E, Michaut L et al (1996) The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell 86:973–983
Valanne S, Wang JH, Rämet M (2011) The Drosophila Toll signaling pathway. J Immunol 186:649–656
Taguchi T, Mitcham JL, Dower SK et al (1996) Chromosomal localization of TIL, a gene encoding a protein related to the Drosophila transmembrane receptor Toll, to human chromosome 4p14. Genomics 32:486–488
Medzhitov R, Preston-Hurlburt P, Janeway CA Jr (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388:394–397
Poltorak A, He X, Smirnova I et al (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 282:2085–2088
Takeda K, Kaisho T, Akira S (2003) Toll-like receptors. Annu Rev Immunol 21:335–376
Oda K, Kitano H (2006) A comprehensive map of the toll-like receptor signaling network. Mol Syst Biol 2:2006.0015
Brikos C, O’Neill LA (2008) Signaling of toll-like receptors. In: Bauer S, Hartmann G (eds) Toll- like receptors (TLRs) and innate immunity, Handbook of experimental pharmacology, vol 183. Springer, Heidelberg, pp 21–50
Brown J, Wang H, Hajishengallis GN, Martin M (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 90:417–427
O’Neill LA, Bowie AG (2007) The family of five: TIR-domain-containing adaptors in Tolllike receptor signalling. Nat Rev Immunol 7:353–364
Fernandez-Lizarbe S, Montesinos J, Guerri C (2013) Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J Neurochem 126:261–273
Oosting M, Cheng SC, Bolscher JM et al (2014) Human TLR10 is an anti-inflammatory pattern-recognition receptor. Proc Natl Acad Sci U S A 111:E4478–E4484
Hasan U, Chaffois C, Gaillard C, Saulnier V, Merck E, Tancredi S, Guiet C, Brière F, Vlach J, Lebecque S, Trinchieri G, Bates EE (2005) Human TLR10 is a functional receptor, expressed by B cells and plasmacytoid dendritic cells, which activates gene transcription through MyD88. J Immunol 174:2942–2950
Yarovinsky F, Zhang D, Andersen JF et al (2005) TLR11 activation of dendritic cells by a protozoan profilin-like protein. Science 308:1626–1629
Neal LM, Knoll LJ (2014) Toxoplasma gondii profilin promotes recruitment of Ly6Chi CCR2+ inflammatory monocytes that can confer resistance to bacterial infection. PLoS Pathog 10:e1004203
Oldenburg M, Krüger A, Ferstl R et al (2012) TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 2012(337):1111–1115
Lee CC, Avalos AM, Ploegh HL (2012) Accessory molecules for Toll-like receptors and their function. Nat Rev Immunol 12:168–179
Li S, Strelow A, Fontana EJ et al (2002) IRAK-4: a novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci U S A 99:5567–5572
Chen ZJ (2005) Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol 7:758–765
Sato S, Sugiyama M, Yamamoto M et al (2003) Toll/IL-1 receptor domain-containing adaptor inducing IFN-beta (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-kappa B and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol 171:4304–4310
Oshiumi H, Matsumoto M, Funami K et al (2003) TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat Immunol 4:161–167
Honda K, Yanai H, Mizutani T et al (2004) Role of a transductional-transcriptional processor complex involving MyD88 and IRF-7 in toll-like receptor signaling. Proc Natl Acad Sci U S A A101:15416–15421
Kaisho T, Akira S (2003) Regulation of dendritic cell function through toll-like receptors. Curr Mol Med 3:759–771
Satoh M, Ishikawa Y, Minami Y et al (2008) Role of toll like receptor signaling pathway in ischemic coronary artery disease. Front Biosci 13:6708–6715
Seki E, Brenner DA (2008) Toll-like receptors and adaptor molecules in liver disease: update. Hepatology 48:322–335
Marshak-Rothstein A (2006) Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol 6:823–835
Obhrai J, Goldstein DR (2006) The role of toll-like receptors in solid organ transplantation. Transplantation 81:497–502
Schmidt M, Raghavan B, Muller V et al (2010) Crucial role for human Toll-like receptor 4 in the development of contact allergy to nickel. Nat Immunol 11:814–819
Yu L, Wang L, Chen S (2010) Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med 14:2592–2603
Deiters U, Barsig J, Tawil B et al (2004) The macrophage-activating lipopeptide-2 accelerates wound healing in diabetic mice. Exp Dermatol 13:731–739
Macedo L, Pinhal-Enfield G, Alshits V et al (2007) Wound healing is impaired in MyD88- deficient mice: a role for MyD88 in the regulation of wound healing by adenosine A2A receptors. Am J Pathol 171:1774–1788
Seki E, Tsutsui H, Iimuro Y et al (2005) Contribution of Toll-like receptor/myeloid differentiation factor 88 signaling to murine liver regeneration. Hepatology 41:443–450
Ribatti D (2010) The seminal work of Werner Risau in the study of the development of the vascular system. Int J Dev Biol 54:567–572
Carmeliet P (2000) Mechanisms of angiogenesis and arteriogenesis. Nat Med 6:389–395
Risau W (1997) Mechanisms of angiogenesis. Nature 386:671–674
Carmeliet P (2005) Angiogenesis in life, disease and medicine. Nature 438:932–936
Olsson AK, Dimberg A, Kreuger J et al (2006) VEGF receptor signalling—in control of vascular function. Nat Rev Mol Cell Biol 7:359–371
Senger DR, Galli SJ, Dvorak AM et al (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219:983–985
Partanen J, Armstrong E, Makela TP et al (1992) A novel endothelial cell surface receptor tyrosine kinase with extracellular epidermal growth factor homology domains. Mol Cell Biol 12:1698–1707
Augustin HG, Koh GY, Thurston G et al (2009) Control of vascular morphogenesis and homeostasis through the angiopoietin-tie system. Nat Rev Mol Cell Biol 10:165–177
Gridley T (2010) Notch signaling in the vasculature. Curr Top Dev Biol 92:277–309
Karamysheva AF (2008) Mechanisms of angiogenesis. Biochemistry (Mosc) 73:751–762
Bussolino F, Ziche M, Wang JM et al (1991) In vitro and in vivo activation of endothelial cells by colony-stimulating factors. J Clin Invest 87:986–995
Distler JH, Hirth A, Kurowska-Stolarska M et al (2003) Angiogenic and angiostatic factors in the molecular control of angiogenesis. Q J Nucl Med 47:149–161
Vandervelde S, van Luyn MJ, Tio RA et al (2005) Signaling factors in stem cell- mediated repair of infarcted myocardium. J Mol Cell Cardiol 39:363–376
Cao Y (2009) Tumor angiogenesis and molecular targets for therapy. Front Biosci 14:3962–3973
Yamaguchi Y, Yoshikawa K (2001) Cutaneous wound healing: an update. J Dermatol 28:521–534
Gharaee-Kermani M, Phan SH (2001) Role of cytokines and cytokine therapy in wound healing and fi brotic diseases. Curr Pharm Des 7:1083–1103
Frantz S, Vincent KA, Feron O et al (2005) Innate immunity and angiogenesis. Circ Res 96:15–26
Koutroubakis IE, Tsiolakidou G, Karmiris K et al (2006) Role of angiogenesis in inflammatory bowel disease. Inflamm Bowel Dis 12:515–523
Costa C, Incio J, Soares R (2007) Angiogenesis and chronic inflammation: cause or consequence? Angiogenesis 10:149–166
Grote K, Schuett H, Schieffer B (2011) Toll-like receptors in angiogenesis. ScientificWorldJournal 11:981–991
Leibovich SJ, Chen JF, Pinhal-Enfield G et al (2002) Synergistic up-regulation of vascular endothelial growth factor expression in murine macrophages by adenosine A(2A) receptor agonists and endotoxin. Am J Pathol 160:2231–2244
Pinhal-Enfield G, Ramanathan M, Hasko G et al (2003) An angiogenic switch in macrophages involving synergy between Toll-like receptors 2, 4, 7, and 9 and adenosine A(2A) receptors. Am J Pathol 163:711–721
Hara Y, Kuroda N, Inoue K et al (2009) Up-regulation of vascular endothelial growth factor expression by adenosine through adenosine A2 receptors in the rat tongue treated with endotoxin. Arch Oral Biol 54:932–942
Pollet I, Opina CJ, Zimmerman C et al (2003) Bacterial lipopolysaccharide directly induces angiogenesis through TRAF6-mediated activation of NF-kappaB and c-Jun N-terminal kinase. Blood 102:1740–1742
Riess T, Andersson SG, Lupas A et al (2004) Bartonella adhesin a mediates a proangiogenic host cell response. J Exp Med 200:1267–1278
McCord AM, Burgess AW, Whaley MJ et al (2005) Interaction of Bartonella henselae with endothelial cells promotes monocyte/macrophage chemoattractant protein 1 gene expression and protein production and triggers monocyte migration. Infect Immun 73:5735–5742
Rodriguez-Martinez S, Cancino-Diaz ME, Miguel PS et al (2006) Lipopolysaccharide from Escherichia coli induces the expression of vascular endothelial growth factor via toll-like receptor 4 in human limbal fibroblasts. Exp Eye Res 83:1373–1377
Jagavelu K, Routray C, Shergill U et al (2010) Endothelial cell toll-like receptor 4 regulates fibrosis-associated angiogenesis in the liver. Hepatology 52:590–601
McDonald DM (2001) Angiogenesis and remodeling of airway vasculature in chronic inflammation. Am J Respir Crit Care Med 164:S39–S45
Grote K, Schuett H, Salguero G et al (2010) Toll-like receptor 2/6 stimulation promotes angiogenesis via GM-CSF as a potential strategy for immune defense and tissue regeneration. Blood 115:2543–2552
Grote K, Sonnenschein K, Kapopara PR et al (2013) Toll-like receptor 2/6 agonist macrophage-activating lipopeptide-2 promotes reendothelialization and inhibits neointima formation after vascular injury. Arterioscler Thromb Vasc Biol 33:2097–2104
Varoga D, Paulsen F, Mentlein R et al (2006) TLR-2-mediated induction of vascular endothelial growth factor (VEGF) in cartilage in septic joint disease. J Pathol 210:315–324
Cho ML, Ju JH, Kim HR et al (2007) Toll-like receptor 2 ligand mediates the upregulation of angiogenic factor, vascular endothelial growth factor and interleukin-8/CXCL8 in human rheumatoid synovial fibroblasts. Immunol Lett 108:121–128
Chang YJ, Wu MS, Lin JT, Chen CC (2005) Helicobacter pylori-induced invasion and angiogenesis of gastric cells is mediated by cyclooxygenase-2 induction through TLR2/TLR9 and promoter regulation. J Immunol 175:8242–8252
Zhe Y, Li Y, Liu D et al (2016) Extracellular HSP70-peptide complexes promote the proliferation of hepatocellular carcinoma cells via TLR2/4/JNK1/2MAPK pathway. Tumour Biol 37:13951–13959
Hu L, Zang MD, Wang HX et al (2016) Biglycan stimulates VEGF expression in endothelial cells by activating the TLR signaling pathway. Mol Oncol 10:1473–1484
Damiano V, Caputo R, Bianco R et al (2006) Novel toll-like receptor 9 agonist induces epidermal growth factor receptor (EGFR) inhibition and synergistic antitumor activity with EGFR inhibitors. Clin Cancer Res 12:577–583
Guo Z, Chen L, Zhu Y et al (2012) Double-stranded RNA-induced TLR3 activation inhibits angiogenesis and triggers apoptosis of human hepatocellular carcinoma cells. Oncol Rep 27:396–402
Bergé M, Bonnin P, Sulpice E et al (2010) Small interfering RNAs induce target-independent inhibition of tumor growth and vasculature remodeling in a mouse model of hepatocellular carcinoma. Am J Pathol 177:3192–3201
Spaner DE, Masellis A (2007) Toll-like receptor agonists in the treatment of chronic lymphocytic leukemia. Leukemia 21:53–60
Shingu K, Kruschinski C, Lührmann A et al (2003) Intratracheal macrophage-activating lipopeptide-2 reduces metastasis in the rat lung. Am J Respir Cell Mol Biol 28:316–321
Schneider C, Schmidt T, Ziske C et al (2004) Tumour suppression induced by the macrophage activating lipopeptide MALP-2 in an ultrasound guided pancreatic carcinoma mouse model. Gut 53:355–361
Schmidt J, Welsch T, Jäger D et al (2007) Intratumoural injection of the toll-like receptor-2/6 agonist ‘macrophage-activating lipopeptide-2’ in patients with pancreatic carcinoma: a phase I/II trial. Br J Cancer 97:598–604
Zgheib A, Pelletier-Bonnier É, Levros LC Jr et al (2013) Selective JAK/STAT3 signalling regulates transcription of colony stimulating factor-2 and -3 in Concanavalin-A-activated mesenchymal stromal cells. Cytokine 63:187–193
Cammarota R, Bertolini V, Pennesi G et al (2010) The tumor microenvironment of colorectal cancer: stromal TLR4 expression as a potential prognostic marker. J Transl Med 8:112
Kutikhin AG (2011) Association of polymorphisms in TLR genes and in genes of the Toll-like receptor signaling pathway with cancer risk. Hum Immunol 72:1095–1116
van Beijnum JR, Buurman WA, Griffioen AW (2008) Convergence and amplification of toll- like receptor (TLR) and receptor for advanced glycation end products (RAGE) signaling pathways via high mobility group B1 (HMGB1). Angiogenesis 11:91–99
Huang W, Liu Y, Li L et al (2012) HMGB1 increases permeability of the endothelial cell monolayer via RAGE and Src family tyrosine kinase pathways. Inflammation 35:350–362
Lin Q, Yang XP, Fang D et al (2011) High-mobility group box-1 mediates toll-like receptor 4-dependent angiogenesis. Arterioscler Thromb Vasc Biol 31:1024–1032
Yang S, Yang TS, Wang F et al (2015) High-mobility group box-1-Toll-Like receptor 4 axis mediates the recruitment of endothelial progenitor cells in alkali-induced corneal neovascularization. Int Immunopharmacol 28:450–458
van Beijnum JR, Nowak-Sliwinska P, van den Boezem E et al (2012) Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene 32:363–374
West XZ, Malinin NL, Merkulova AA et al (2010) Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature 467:972–976
Wang XY, Sarkar D, Fisher PB (2011) Stress-sensing toll-like receptor as a driver of angiogenesis. Pigment Cell Melanoma Res 24:7–9
Xu Y, Zhou Y, Lin H et al (2013) Toll-like receptor 2 in promoting angiogenesis after acute ischemic injury. Int J Mol Med 31:555–560
Chen X, Yan L, Guo Z et al (2016) Adipose-derived mesenchymal stem cells promote the survival of fat grafts via crosstalk between the Nrf2 and TLR4 pathways. Cell Death Dis 7(9):e2369
Menden H, Welak S, Cossette S et al (2015) Lipopolysaccharide (LPS)-mediated angiopoietin-2-dependent autocrine angiogenesis is regulated by NADPH oxidase 2 (Nox2) in human pulmonary microvascular endothelial cells. J Biol Chem 290:5449–5461
Caruso R, Warner N, Inohara N et al (2014) NOD1 and NOD2: signaling, host defense, and inflammatory disease. Immunity 41:898–908
Feerick CL, McKernan DP (2016) Understanding the regulation of pattern recognition receptors in inflammatory diseases - a ‘Nod’ in the right direction. Immunology 150:237. [Epub ahead of print]
Campbell L, Williams H, Crompton RA et al (2013) Nod2 deficiency impairs inflammatory and epithelial aspects of the cutaneous wound-healing response. J Pathol 229:121–131
Schirbel A, Kessler S, Rieder F et al (2013) Pro-angiogenic activity of TLRs and NLRs: a novel link between gut microbiota and intestinal angiogenesis. Gastroenterology 144:613–623
Grelier A, Cras A, Balitrand N et al (2013) Toll-like receptor 3 regulates cord blood-derived endothelial cell function in vitro and in vivo. Angiogenesis 16:821–836
Wagner NM, Bierhansl L, Nöldge-Schomburg G et al (2013) Toll-like receptor 2-blocking antibodies promote angiogenesis and induce ERK1/2 and AKT signaling via CXCR4 in endothelial cells. Arterioscler Thromb Vasc Biol 33:1943–1951
Wagner NM, Bierhansl L, Butschkau A et al (2013) TLR2-deficiency of cKit+ bone marrow cells is associated with augmented potency to stimulate angiogenic processes. Int J Clin Exp Pathol 6:2813–2823
Ergul A, Alhusban A, Fagan SC (2012) Angiogenesis: a harmonized target for recovery after stroke. Stroke 43:2270–2274
Freedman SB, Vale P, Kalka C et al (2002) Plasma vascular endothelial growth factor (VEGF) levels after intramuscular and intramyocardial gene transfer of VEGF-1 plasmid DNA. Hum Gene Ther 13:1595–1603
Henry TD, Annex BH, McKendall GR et al (2003) The VIVA trial: vascular endothelial growth factor in ischemia for vascular angiogenesis. Circulation 107:1359–1365
Grote K, Petri M, Liu C et al (2013) Toll-like receptor 2/6-dependent stimulation of mesenchymal stem cells promotes angiogenesis by paracrine factors. Eur Cell Mater 26:66–79
Acknowledgments
The work of KG and BS is supported by grants from the German Research Foundation (DFG) KFO 136 and SFB 566/b9 and from the Federal Ministry of Education and Research (BMBF) 01GU0711.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Grote, K., Schuett, J., Schuett, H., Schieffer, B. (2017). Toll-Like Receptors in Angiogenesis. In: Mehta, J., Mathur, P., Dhalla, N. (eds) Biochemical Basis and Therapeutic Implications of Angiogenesis. Advances in Biochemistry in Health and Disease, vol 6. Springer, Cham. https://doi.org/10.1007/978-3-319-61115-0_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-61115-0_3
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-61114-3
Online ISBN: 978-3-319-61115-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)