
Abstract
The innate immune response comprises the initial events that occur during tissue insult, causing cellular activation and triggering inflammation. Innate immune cells, including resident and early migrated cells from the bloodstream, sense a plethora of molecules called molecular patterns, that are derived from microorganisms or host cells. Once activated, pattern recognition receptor (PRR) signalling is triggered intracellularly and promotes the synthesis and release of vasoactive molecules, which target endothelial cells and cause inflammation. In addition, circulating molecules and pathogens also activate PRRs that are expressed on endothelial cells. These events modify endothelial cell metabolism, changing their conformational state and promoting the expression of pro-inflammatory molecules. Importantly, gain-of-function mutations in PRRs are associated with continuous cellular activation, leading to the development of autoinflammatory diseases. Here, we discuss the relationship among the cellular and humoral arms of the innate immune system in inflammatory processes, with special attention given to endothelial cell activation.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Innate immunity
- Inflammation
- Pattern recognition receptors
- Cellular metabolism
- Autoinflammatory diseases
1 Introduction
Inflammation is a protective response of the body to ensure removal of harmful stimuli and to stimulate a healing process for tissue repair [1]. Inflammation is closely associated with the innate immune response to microbial infection, tissue injury and other sterile stimuli [2]. Resident cells are key elements that orchestrate the release of potent pro-inflammatory mediators by sensing a plethora of stimuli, ranging from pattern molecules to cellular stresses. The produced cytokines and other classical vasoactive molecules in the tissue bind to and activate the nearby blood vessels, causing profound changes in endothelial cell (EC) metabolism , conformational structures, and the synthesis of molecules that create a local environment that facilitates the translocation of blood molecules and cells to the affected tissue. These early events are the basis of the clinical signs of acute inflammation, characterized by redness, swelling, heat, pain and loss of tissue function [3].
EC activation is the basis of the inflammatory response. ECs are monolayers of cells that form the inner lining of blood and lymphatic vessels. These cells form the endothelium, a barrier between the vascular space and the interstitium [4]. In adults, the endothelium weighs approximately 1 kg, comprising 1.6 × 1013 cells, and has a surface area of 1–7 m2 [5]. During homeostasis, ECs control blood fluidity in different ways, and they inhibit coagulation and platelet adhesion throughout the vascular system [6, 7]. ECs also regulate the muscular tonus by releasing vasodilators, such as nitric oxide (NO) and prostacyclins, or vasoconstrictors, such as endothelin [8, 9]. Protein transport and endothelium permeability occur mainly through interendothelial junctions that connect the ECs into a continuous monolayer. In that way, plasma proteins are prevented from moving from the blood to tissues through endothelium in non-inflamed tissues [10]. In addition, the interactions among ECs and leukocytes are minimal during the EC resting phase since they sequester the proteins necessary for these interactions, such as selectins and chemokines, in specialized secretory vesicles [11].
ECs participate and regulate different steps of inflammation during the innate immune response [12, 13]. ECs express pattern recognition receptors (PRRs) and complement protein receptors. Once activated, ECs increase the permeability of the endothelium, facilitating the leakage of serum components and extravasation of leukocytes, and they are also a source of cytokines, chemokines, acute phase proteins, and reactive oxygen species [14,15,16,17]. Here we discuss the contributions of key innate immune elements during inflammatory responses, focusing on how ECs sense a plethora of stimuli by innate receptors, change their metabolism, and drive inflammation. In addition, we discuss the involvement of innate receptors in the development of autoinflammatory diseases.
2 Endothelial Cell Heterogeneity and Tissue Specialization During Inflammation
The circulatory system comprises the blood and lymphatic vasculature and plays an essential role in physiology, interconnecting and transporting gases, nutrients, metabolites and cells. The blood vasculature, consisting of arteries, veins and capillaries that exhibit distinct architectures, molecular and functional properties, is essential for normal organ function and disease [18]. The endothelium of blood vessels forms a continuous monolayer, whereas capillary endothelial cells can be classified as continuous, fenestrated or discontinuous, depending on the tissue-specific type in which they reside [18]. Leukocyte migration from the blood flow of post-capillary venules and influx into tissue is a coordinated process involving multistep signals in endothelial and leukocytes via a hierarchy of adhesion molecule activity that mediates the steps of leukocyte tethering, rolling, arrest, activation, firm adhesion and transmigration [19, 20]. Leukocyte tethering and rolling on the endothelial surface is mediated by selectins [19]. Inflammation causes endothelial cell expression of P-selectin and E-selectin, whereas L-selectin is expressed by leukocytes [19,20,21,22]. Leukocytes express selectin ligands, including P-selectin glycoprotein ligand-1 (PSGL-1), E-selectin ligand-1 and CD44, which interact with the endothelial selectins [20,21,22]. L-selectin mediates leukocyte rolling interactions by binding to glycosylated proteins on activated ECs, such as GlyCAM-1 and CD34 [20]. Interestingly, Pentraxin 3 (PTX3) , a protein with well-known functions in innate immunity, reduces neutrophil migration and inflammation in vivo by binding to P-selectin, impairing the rolling of neutrophils on vessels [23]. Rolling brings the leukocyte into close proximity with the endothelium, allowing them to respond to chemotactic arrest signals from chemokines present at the endothelial surface [20, 22]. A wide variety of molecules possess this chemoattractant function , including proteins such as chemokines. Leukocytes respond to chemoattractants by rapidly upregulating the affinity of their β2 and α4 integrins [19, 20, 22]. This enables these adhesion molecules to bind to their endothelial-expressed ligands, with β2 integrins binding to molecules, such as ICAM-1 and fibrinogen, and α4 integrins interacting with VCAM-1 and MAdCAM-1 [19, 20, 22]. This interaction causes arrest of the rolling leukocytes, which then firmly attach to the endothelial surface. After arrest, leukocytes migrate across the endothelial surface [19, 20, 22] to identify an optimal location.
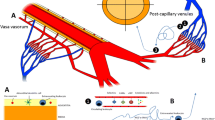
The leukocyte recruitment established in post-capillary venules is not exactly the same in all organs. The microvasculature of the lung, liver and kidney are characterized by structural specializations that are required for their functions. Recruitment of leukocytes into the diverse tissues through the specialized capillary network contrasts with the recruitment of leukocytes through post-capillary venules at sites of inflammation (Fig. 1).

The multistep cascade of leukocyte recruitment in post-capillary venules (a) and lungs (b): (a) Leukocyte extravasation from the blood into inflamed tissues follows a multistep cascade that involves the sequential action of molecular signals and adhesion molecules. Selectins (such as P-selectin and E-selectin) initiate leukocyte tethering and rolling along the inflamed endothelium. Rolling slows down circulating leukocytes, bringing them into close proximity with endothelial cells and allowing the binding of chemokines that are displayed on the inflamed endothelium to specific G-protein-coupled chemokine receptors on leukocytes. The activation of chemokine receptors triggers intracellular signalling pathways that activate leukocyte integrins. Interactions between integrins expressed on the endothelium mediate the firm adhesion of leukocytes. The interactions between β2-integrins and their ligand intercellular adhesion molecule 1 (ICAM1) and between integrin very late antigen 4 (VLA4) and its ligand vascular cell-adhesion molecule 1 (VCAM1) are of crucial importance for leukocyte adhesion. Leukocytes are directed by Fig. 1 (continued) immobilized chemokines under flow (chemotaxis) or by chemokine gradients (haptotaxis) to migrate across the endothelium and into the inflamed tissue. (b) The initial steps of leukocyte migration, such as tethering and rolling, are absent in tissues during inflammation. Neutrophil migration across the alveolar capillary wall can occur through endothelial-independent selectin activity; however, the pulmonary capillary architecture sequesters leukocytes in small capillaries, and leukocytes marginate and deform to pass through this very small vasculature. Chemokines secreted by alveolar macrophages or endothelial cells activate more endothelial cells and induce leukocyte arrest. Leukocytes that have crossed the endothelium traverse the basement membrane and, through interactions with fibroblasts and neutrophils in the interstitium, adhere to the fibroblast surface. Adhesion is known to involve leukocyte β2-integrin (CD18) and fibroblast intercellular adhesion molecule 1 (ICAM-1). Motility is regulated by CD18 and leukocyte β1-integrins. The unique positioning of the fibroblasts within the interstitium provides directional information, guiding neutrophils toward type II pneumocytes and, thus, leading them to emergence between the margins of two type I pneumocytes and one type II pneumocyte in airways
2.1 Leukocyte Recruitment in Alveolar Capillaries
The dense capillary network of the lung is a major site of physiological sequestration of leukocytes from the systemic circulation [18, 24, 25]. Compared with blood in the large vessels of most vascular beds, the blood from alveolar capillaries contains circa 50-fold more neutrophils, lymphocytes and monocytes [24], which contribute to the innate immune response and haematopoiesis in the lungs [24, 26, 27]. The alveolar capillaries provide a vascular defensive niche whereby the endothelium and neutrophils cooperate for immediate detection and capture of disseminating pathogens during host defence, mediating vascular protection [24, 27], and crawl throughout the endothelium via a TLR4/CD11b-dependent process [27]. Pulmonary ECs also constitutively express ICAM-1 at much higher levels than in other organs [25]. This process of margination is induced in part by the delay of neutrophils as they undergo the deformation required to pass through the narrow lung microvasculature [24]. Marginated leukocytes are in a dynamic equilibrium with those in the circulation, a situation that is maintained by the ongoing entry and exit of leukocytes from the marginated pool. During inflammation, much of the sequestration and infiltration occurs through vessels so narrow that physical trapping is sufficient to stop the flowing neutrophils [24, 25].
In comparison to post-capillary venules, the tethering mechanisms required to capture neutrophils from flowing blood in larger vessels is not necessary in the alveolar capillary bed. The diameters of neutrophils are larger than the diameters of many capillary segments, and 50% of the capillary segments therefore require changes in the shape of neutrophils for transmigration [24]. The events following the initial sequestration of neutrophils within alveolar capillary beds are apparently influenced by adhesion molecules. Systemic activation of neutrophils by intravenous injection of the chemokine CXCL8 results in rapid neutropenia with massive sequestration of neutrophils within alveolar capillaries. This event is not dependent on L-selectin or β2-integrins, but the retention times within this capillary bed are influenced by these adhesion molecules [24, 28], and this type of adhesion is likely orchestrated by the interaction of leukocyte adhesion molecules and endothelial adhesion molecules, such as VAP-1 [29]. These studies demonstrate that leukocyte recruitment to the pulmonary microvasculature does not necessarily follow the conventional paradigm in that the requirement for archetypal adhesion molecules is variable and recruitment to the lung can occur in the absence of both β2 integrins and the selectin family of adhesion molecules.
2.2 Leukocyte Recruitment in Sinusoid Capillaries
ECs lining hepatic sinusoids are also unique in that they are highly fenestrated and lack a basal lamina [18, 25, 30]. These openings in the endothelial layer allow plasma to flow freely into the sub-endothelium, where it comes into contact with hepatocytes. Sinusoidal endothelial cells also have a limited capacity to express P-selectin, E-selectin and VCAM-1, while they constitutively express high levels of ICAM-1, as well as less commonly expressed adhesion molecules, such as vascular adhesion protein-1 (VAP-1) [25, 30]. During the mechanisms regulating leukocyte adhesion in the sinusoids in the model of focal necrosis, Mac-1 and ICAM-1 are central to neutrophil recruitment [31]. Moreover, neutrophil recruitment occurs via a sequential process initiated by arrest induced by ATP released from damaged cells. Subsequently, intravascular migration is induced by the CXC family of chemokines, finally resulting in migration into necrotic tissue occurred via formylated peptides [31,32,33]. In addition, the steps of tethering and rolling of leukocytes in liver sinusoids also occur independently of selectins, but integrins mediate leukocyte arrest and transmigration, as observed in lung tissue.
2.3 Leukocyte Recruitment in Glomerular Capillaries
Glomerular capillaries are lined by specialized ECs that are highly fenestrated [18]. The luminal surface of glomerular ECs is covered by an endothelial surface layer consisting of negatively charged glycoproteins, glycosaminoglycans and proteoglycans, a structure that contributes to the barrier function of the glomerulus [18, 25, 34]. Regarding adhesion molecule expression, evidence suggests that glomerular endothelial cells do not express pre-formed P-selectin but use platelets as a mechanism for expression of P-selectin under inflammatory conditions [35]. In contrast, these cells constitutively express ICAM-1 and can, under inflammatory conditions, express VCAM-1 and E-selectin de novo, as well as increase ICAM-1 expression [36, 37]. In experiments, leukocytes could be observed undergoing adhesion in glomerular capillaries, but no rolling interactions were observed [25, 38]. In accordance with this finding, a selectin inhibitor failed to reduce glomerular leukocyte recruitment [38]. In contrast, inhibition of the β2-integrin Mac-1 prevented inflammation-associated adhesion of leukocytes in glomeruli [25, 36]. Taken together, these findings indicate that while leukocyte adhesion in glomerular capillaries requires an adhesion molecule-mediated interaction , selectins are not essential for this process.
3 Cellular Innate Immune Response: Role of Pattern Recognition Receptors on Endothelial Cells
In 1989, Charles Janeway Jr. proposed that innate immune cells should present germ line-encoded pattern recognition receptors (PRRs) to recognize conserved microbial components named pathogen associated molecular patterns (PAMPs) [39, 40]. It was later demonstrated that PRRs also recognize endogenous molecules released from damaged cells, called damage-associated molecular patterns (DAMPs ) , or alarmins [16, 41]. There are different classes of PRRs that can be subdivided according to their structure, localization, and binding and signalling properties. The transmembrane PRRs include Toll-like receptors (TLR) and C-type lectin receptors (CLR), while the cytoplasmic PRRs are represented by retinoic acid-inducible gene (RIG)-I-like receptors, nucleotide-binding oligomerization domain-like NOD-like receptors (NLR) [42], AIM2-like receptors (ALR) [43], and cyclic GMP-AMP synthase (cGAS) [44].
PRRs are present in virtually all cell types. However, most knowledge about the association of PRRs with tissue inflammation has been obtained from activated innate immune cell recognition of exogenous and endogenous patterns . Once activated, resident and early migrated cells release a plethora of vasoactive molecules, including prostanoids, histamine, cytokines, and chemokines, which cause profound changes in ECs, contributing to inflammation [45]. Here we explore the mechanisms used by ECs to sense pathogens and endogenous molecules and how these interactions result in tissue inflammation and disease (Fig. 2).

Endothelial cells sense endogenous and microbial molecules that contribute to tissue inflammation. Different molecules derived from tissue resident cells, microorganisms, and plasma components interact with innate immune-associated receptors expressed on endothelial cells. The intracellular signalling cascades result in endothelial cell activation, promoting adhesion molecule expression and morphologic changes in the endothelium, which facilitate interactions with circulating leukocytes. Furthermore, activated endothelial cells produce and release biologically active mediators that contribute to tissue inflammation and are important for pathogen control and for the development of inflammatory diseases
3.1 Toll-Like Receptors (TLR)
The TLR family is the major and most extensively studied class of PRRs [46]. TLRs were first described in 1997 and were originally discovered based on homology to the Drosophila melanogaster Toll protein, which plays a role in dorso-ventral patterning during embryogenesis as well as in the antifungal response [47]. TLRs are glycoproteins characterized by an extracellular or luminal ligand-binding domain containing leucine-rich repeat (LRR) motifs and a cytoplasmic signalling Toll/interleukin-1 (IL-1) receptor homology (TIR) domain [48, 49]. TLRs detect distinct patterns derived from bacteria, viruses, parasites and self-components. In humans, 11 TLRs have been identified, while 13 are described in mice [50]. TLRs are expressed in different cell compartments, favouring the recognition of extra and intracellular patterns. While TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10 are expressed on the cell surface and are important for interactions with extracellular patterns, the intracellular compartments contain TLR3, TLR7, TLR8, and TLR9 and are important for binding to nucleic acids [51]. TLR11 is non-functional in humans but present in murine endosomal compartments together with TLR12 and 13 [52], and it plays a role in the recognition of T. gondii profilin [53, 54]. Almost all TLRs signal through the adaptor molecule myeloid differentiating factor 88 (MyD88), mainly by activating the nuclear factor kappa-B (NFκB) and mitogen-activated protein kinase (MAPK) transcription factors [55]. In addition, signalling by TIR domain-containing adapter-inducing interferon-β (TRIF) activates interferon regulatory factors (IRFs) responsible for type I interferon production [56]. Of note, only TLR3 and TLR4 signal via TRIF, although the latter also signals via MyD88 [57, 58]. This ligand-receptor interaction induces receptor oligomerization and triggers a signalling cascade into the cytosol that culminates in the transcription of several genes involved in immune and inflammatory responses.
All TLRs have been detected in ECs [59]. ECs present in the aorta, subclavian, carotid, mesenteric, iliac, and temporal arteries express basal levels of TLR1, TLR2, TLR4, and TLR6. TLR3 is predominantly expressed in human aorta and carotid macrovessels [60] and human brain ECs [61]. However, TLR7 and TLR9 have only been detected in iliac ECs, although at low levels [62, 63]. It has been shown that in humans, ECs from lymph nodes express TLR 1–6 and TLR9 [64, 65]. In addition, human endothelial colony forming cells (ECFCs), comprising a subpopulation of endothelial progenitor cells characterized by their ability to differentiate into mature ECs, express detectable mRNA of all TLRs, albeit higher levels of TLR4 [66, 67].
In general, TLR ligands profoundly alter EC homeostasis, interfering in the coagulation cascade, vascular permeability, and synthesis of pro-inflammatory molecules [68, 69]. TLR activation upregulates the expression of adhesion molecules, such as E- and P-selectins, favouring platelet and leukocyte attachment on the EC surface [70, 71]. Additionally, TLR2 and TLR4 agonists lead to cytokine and chemokine expression by cultured ECs [72, 73]. Intravenous injection of TLR3 agonist long double-stranded RNA in mice impairs EC function, causing vasodilation, increased vascular permeability, and the production of reactive oxygen species [74]. During sepsis, TLR2 and TLR4 activation on ECs increases endothelial permeability and modulates the expression of coagulation factor molecules [66, 75]. In addition, TLR9 activation in human coronary artery endothelial cells by bacterial DNA shifts the balance of tissue factor and tissue factor pathway inhibitor toward a procoagulant phenotype and activates blood coagulation in mice, representing an important mechanism during the coagulation cascade in various pathologies [76].
Although much attention has been given to the participation of TLRs in the context of infectious diseases, non-infectious stimuli also activate TLRs on ECs, contributing to sterile inflammatory diseases. In vitro, shear stress modulates TLR expression on human coronary artery ECs [77]. During hypertension, elevated blood pressure can cause tissue damage resulting in DAMP and EC activation [78]. Importantly, angiotensin II infusion in mice cause an upregulation of TLR4 in the aorta, which is associated with EC dysfunction and activation, releasing pro-inflammatory molecules [79]. Oxidized low-density lipoprotein (oxLDL) activates TLR2 and TLR4 in human coronary artery ECs and stimulates the synthesis of bone morphogenetic protein-2 (BMP-2), which plays an important role in atherosclerotic vascular calcification [80]. Vascular dysfunction is an important event in diabetic complications. Hyperglycaemia upregulates TLR2 and TLR4 in human macrovascular aortic ECs and blocks TLR signalling, attenuating hyperglycaemia-induced inflammation, leukocyte adhesion and glycocalyx dysfunction in these cells [81]. Closely related, hyperglycaemia also induces TLR2 and TLR4 expression in human retinal ECs, contributing to the pathogenesis of diabetic retinopathy [82]. Taken together, the control of TLR signalling in ECs must still be considered as a potential strategy to regulate EC activation, facilitating the reduction of infectious and non-infectious inflammatory diseases.
3.2 NOD-Like Receptors (NLR)
The NLR belongs to a class of intracellular receptors/sensors consisting of 23 representatives in humans and 34 in mice [83]. Sequence homology revealed NOD1 as the first NLR member [84, 85]. NOD1 encodes an intracellular multi-domain scaffolding protein consisting of a caspase activation and recruitment domain (CARD), a nucleotide-binding oligomerization domain (NOD), and multiple leucine rich repeats (LRRs). NOD2 is a closely related protein with an additional CARD domain [86]. Both NOD1 and NOD2 recognize different pathogen and endogenous patterns, triggering activation of the NF-κB family of transcriptional regulators [87, 88]. Different ECs express NOD1, including HUVECs, HAECs and microvascular ECs. Incubation of ECs with the Gram-negative bacterium Chlamydophila pneumoniae contributes to the inflammatory response by increasing the synthesis of CXCL8 via a mechanism that is dependent on NOD1/NF-κB activation [89]. In a similar way, HUVECs incubated with Gram-positive bacteria Listeria monocytogenes also produce NOD1-dependent CXCL8. In addition, inhibition of p38 MAPK blocks NOD1-induced CXCL8 production [90].
Although NOD2 is only marginally expressed in ECs, it is quickly upregulated in the presence of microorganisms and pro-inflammatory cytokines, facilitating or potentiating the immune response and tissue inflammation. Under LPS, IL-1β, or TNF-α stimulation, HUVECs show increased NOD2 expression and become responsive to its agonist muramyl dipeptide (MDP), resulting in NF-κB activation [91]. In response to different stimulation, human aortic ECs incubated with Gram-positive bacteria Streptococcus mutans promotes the expression of IL-6 and the chemokines CXCL8 and CCL2 via a mechanism that is dependent on NOD2 and TLR2 [92]. Furthermore, IL-6 secretion by ECs under NOD2 activation is associated with CD4+ T helper cell-17 (Th17) polarization while inhibiting CD4+ Th1 and Th2 responses [13, 93].
In an infectious context, Porphyromonas gingivalis also stimulates NOD2, NOD1, and TLR2 expression in HUVEC cells. P. gengivalis are Gram-negative bacteria that are associated with periodontal disease and atherosclerosis. P. gengivalis stimulates NF-κB activation and E-selectin synthesis in HUVECs via a mechanism that is dependent on the three receptors [94]. On the other hand, NOD2 can also contribute to non-infectious inflammatory diseases, such as diabetes . High concentrations of glucose induce NOD2 expression in glomerular endothelial cells (GEnCs). In addition, overexpression of NOD2 is positively associated with the severity of diabetic nephropathy since it is associated with the loss of EC and gain of mesenchymal characteristics, a phenomenon called endothelial-to-mesenchymal transition, resulting in albuminuria and subsequent renal disorder [95].
The other branch of NLRs is represented by the inflammasomes, a group of intracellular multimeric protein complexes that activate pro-inflammatory caspases, leading to pro-IL-1β and pro-IL-18 cleavage and inducing, in particular, a type of cell death called pyroptosis [96]. The best-known inflammasome prototypes comprise NLRP1, NLRP3, NLRP6, NLRC4, and Pyrin. In several cases, the activated inflammasome engages the adaptor molecule ASC, which, in turn, activates caspase-1 [97]. NLRP1, NLRP3, ASC and caspase-1 are expressed in ECs [98]. Lipopolysaccharide (LPS) and ATP induce activation of the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs) [99]. In a mouse model of Kawasaki disease, Lactobacillus casei cell wall fragments (LCWE) can activate the NLRP3 inflammasome in coronary arteries, resulting in EC dysfunction [100]. Likewise, DAMPs can also activate the NLRP3 inflammasome by lysosomal destabilization in HUVECs, leading to the production of IL-1β, that, in turn, induces IL-6 and CXCL8 in an autocrine manner in HUVECs [101]. High levels of glucose can also activate NLRP3 in ECs, and NLRP3 ablation prevents inflammasome activation and tight junction disassembly in the coronary arterial endothelium of diabetic mice [100]. Similarly, NLRP3 gene silencing prevents high glucose-induced down-regulation of tight junction proteins in cultured mouse vascular endothelial cells (MVECs) [102]. In atherosclerosis, plasma triglycerides and VLDL cholesterol obtained from patients promote in vitro NLRP1 inflammasome expression in HAECs [103].
Inflammasome activation also induces cell death via pyroptosis. It has been demonstrated that cadmium, an important and common environmental pollutant that has been linked to cardiovascular diseases, induces pyroptosis in HUVECs through NLRP3 activation in a mechanism that is dependent on mitochondrial ROS generation [104]. Additionally, systemic exposure to LPS causes severe human lung microvascular EC (hMVECs) pyroptosis via a mechanism that is mediated by the activation of human caspase 4/5 or its homolog caspase-11 in mice in vivo [105]. Furthermore, hyperhomocysteinaemia (HHcy) is an independent risk factor for cardiovascular disease (CVD). HHcy preferentially induces EC pyroptosis via caspase-1-dependent inflammasome activation, leading to EC dysfunction [106]. Thus, EC dysfunction can occur in response to different types of inflammasome stimulation, revealing inflammatory disease mechanisms and identifying new opportunities for therapies.
3.3 Absent in Melanoma 2-Like Receptors (AIM2)
Absent in melanoma 2 (AIM2) is cytoplasmic dsDNA sensor belonging to PYHIN (IFI20X/IFI16 protein) family. Once binding microbial and host dsDNA, it complexes with ASC for caspase-1 activation, leading IL-1β maturation and release [107]. As part of the innate immune response, AIM2 activation helps host protection against different pathogens [108]. Few studies had investigated the role of AIM2 in EC activation. HUVEC cells stimulated with cell-free DNA increase the expression of AIM2 and also activate NOX4 in an AIM2-dependent manner [109]. dsDNA, IFN-γ, and TNF-α also induce AIM2 synthesis in human aortic ECs, smooth muscle cells, and T/G-human aortic vascular smooth muscle cells. Interestingly, AIM2 is overexpressed in lesions derived from abdominal aortic aneurism and atherosclerotic carotid artery when compared to intact aortic wall, suggesting a possible role of AIM2 in ECs activation and vascular inflammation in these diseases [110]. Another representative of PYHIN group, IFI16, also causes EC activation. Human dermal microvascular endothelial cells infected with Kaposi’s sarcoma-associated herpesvirus promote oligimerization of a complex formed by IFI16, ASC, and caspase -1, leading to IL-1β maturation [111].
3.4 RIG-I-Like Receptors (RLR)
The RNA helicases retinoic acid inducible gene-I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) constitute a further PRR family of receptors called RIG-I-like receptors (RLR) [93]. Both proteins are localized in the cell cytosol and consist of a DexD/H box RNA helicase domain as well as two CARDs motifs [112]. These receptors have major function during viral infections, detecting replicating viruses in cytoplasm, particularly at early phases of viral infection [113]. Activation of this innate immune response lead to the induction of type I and III interferons (IFN) and inflammatory cytokines, whose antiviral activity blocks viral replication and facilitate the activation of antigen-presenting cells to activate antigen-specific immunity against viral pathogens [114]. RIG-I and MDA5 are constitutively expressed in ECs and they are upregulated during viral infection and other pro-inflammatory stimuli [112, 115]. However, RIG-I activation cause EC dysfunction. Intravenous injection of a RIG-I agonist in mice impairs EC vasodilatation and increases aortic oxidative stress. Similar events occurred using different EC lineages [116].
Measles virus-infected HUVEC cells synthesize chemokines and IFN-β, which, in turn, upregulates RLR synthesis in these cells [117]. Using glomerular ECs (GEnC), the RIG-I agonist poly I:C RNA stimulated the synthesis of several pro-inflammatory molecules, including IL-6, CCL2, CCL5, and CXCL10, demonstrating a potential involvement of GEnC for the pathogenesis of glomerulonephritis caused by viral infections [118]. Dengue virus also stimulates ECs via RIG-I. It is well known that Dengue virus infection leads to structural and functional alterations in the vascular endothelium [119]. In addition, Dengue virus serotype 2 replicated in human brain microvascular ECs. In addition to increasing the synthesis of RIG-I in these cells, Dengue virus 2 induced the production of IFN-β, IL-6, different chemokines, and the adhesion molecule ICAM-1 in a RIG-I-dependent manner [120]. Furthermore, activated ECs during viral infections not only amplify tissue inflammation but also could be associated with sickness behaviour. Type I IFN production from brain endothelial cells during RIG-I stimulation was associated to mouse cognitive impairment and depression-like behaviour [121]. Thus, activated RLR in ECs have crucial contribution to tissue inflammation during viral infections.
Activation of RLR in ECs seems to contribute to inflammation also in bacterial infections and non-infectious diseases [122] suggesting an important function for RIG-I during sepsis, where there is EC dysfunction. LPS and TNF-α induced the synthesis of pro-inflammatory and adhesion molecules by HUVEC cells and promoted leukocyte adhesion in a mechanism controlled by RIG-I and mitochondrial antiviral signalling (MAVS), the RIG-I downstream molecule [122]. 25-hydroxycholesterol increased RIG-I levels in HUVEC cytosol and stimulated CXCL8 expression through RIG-I-dependent mechanisms, which could favour neutrophil and T cell recruitment into the subendothelial space during atherogenesis [116].
3.5 Cyclic GMP-AMP Synthase-Stimulator of Interferon Genes (cGAS- STING)
Cyclic GMP-AMP synthase (cGAS ) is a cytosolic protein that senses dsDNA. The downstream signalling in response to this interaction leads to the synthesis of the second messenger cyclic GMP-AMP, which binds to the protein stimulator of IFN genes (STING) present on the endoplasmic reticulum membrane. This cascade culminates in the recruitment of the transcription factor IRF3, which mediates the transcription of IFN-β [123]. STING is expressed in ECs [124]. During viral infections, ECs actively participate in the innate immune response, creating local conditions for tissue inflammation and viral removal. In vitro studies have demonstrated that HUVECs infected with the dsDNA of virus Kaposi’s sarcoma-associated herpesvirus or cytomegalovirus produce high levels of IFN-β in a mechanism that is dependent on cGAS-STING activation [125, 126].
cGAS-STING has been demonstrated to play different roles in other innate immune response to infectious diseases, regulating EC function and activation. ECs in biopsy samples from patients who present a gain-of-function mutation of the TMEM173 gene (which encodes STING) express inflammatory EC markers, such as inducible nitric oxide synthase, tissue factor, E selectin, and intercellular adhesion molecule 1. Similarly, HUVECs incubated with the STING-binding molecule cGAMP produce the same inflammatory markers but also engage apoptosis [124]. In human aortic ECs, palmitic acid (PA) leads to cellular stress, releasing mitochondrial DNA (mtDNA) into the cytosol, which activates cGAS-STING, resulting in IRF3 phosphorylation and nuclear translocation. PA is used in studies investigating diabetes since it negatively regulates insulin activity. cGAS-STING has been found to be important for ICAM-1 synthesis and monocyte–endothelial cell adhesion [127]. Using STING-deficient mice, researchers have demonstrated a dependence of the STING pathway on diet-induced obesity, adipose tissue inflammation and insulin resistance [127]. In another study, human aortic ECs incubated with a high concentration of PA showed impaired proliferation, migration, and angiogenesis capacities via a mechanism that was dependent on cGAS-STING activation by mtDNA. These events culminated in the synthesis of MST1 (mammalian Ste20-like kinases 1), a pro-apoptotic protein kinase [128]. Thus, cGAS-STING plays a very important role during tissue inflammation by controlling EC activation during infection and sterile disorders.
4 Role of the Complement System in Endothelial Cell Activation and Inflammation
Pre-formed and newly synthesized soluble molecules during the innate immune response are fundamental to initiate the control of pathogen invaders by activating host cells and creating an inflammatory environment or via the direct lysis of pathogens. However, these molecules also trigger or amplify tissue inflammation in non-infectious disorders , such as autoimmune, autoinflammatory, and metabolic diseases [129]. Complement system and acute phase proteins are examples of the humoral arm of the innate immune response and strongly influence EC activation. The complement system is an integral part of the innate immune response and acts as a bridge between innate and acquired immunity. It consists of a series of proteins that are mostly synthesized in the liver and exists in plasma and on cell surfaces as inactive precursors (zymogens) [130]. Complement mediates responses to inflammatory triggers through a coordinated sequential enzyme cascade leading to the clearance of foreign cells through pathogen recognition, opsonisation and lysis. There are three known pathways leading to complement activation: classical, alternative and lectin , which vary according to the initial cascade and its components [131].
ECs express complement factors, regulators, and receptors. Complement deposition in ECs leads to cell activation, the expression of adhesion molecules, release of pro-inflammatory cytokines and chemokines, and promotion of membrane attack complex (MAC) formation and cytolysis [132,133,134]. C3a and C5a are well-known molecules that cause profound inflammatory changes in endothelium and also present chemotactic properties. Several studies have reported that targeting C3a and C5a receptors results in a reduction of tissue inflammation, which leads the development of several compounds for clinical use [129, 135]. Human microvascular and umbilical ECs (hMUECs) have been demonstrated to activate the classical complement pathway when exposed to shear stress by the continuous flow loop [136]. In a complementary manner, activation of this pathway in ECs promotes neutrophil adhesion to the endothelium [137]. ECs can also activate the lectin pathway: MASP-1, the key protease in this pathway, induces IL-6 and CXCL8 production by ECs, which leads to neutrophil chemotaxis [138]. In addition, HUVECs that are activated by MASP-1 can decrease ICAM-2 and increase E-selectin expression, leading to adherence between neutrophils and endothelial cells [139]. Thus, both the cellular and humoral arms of the innate immune system have potential effects on diverse ECs, leading to EC modification and activation and actively contributing to the inflammatory process.
5 Cellular Metabolism Reprogramming and Its Contribution to Inflammation
Traditionally, cell metabolism has been considered as a series of pathways that are responsible for extracting energy from fuel sources , such as glucose, fatty acids, ketones and amino acids. However, in the past few years, cellular metabolism and its by-products have demonstrated much wider implications, including the outcomes of inflammatory responses and of several pathologic processes [140,141,142,143]. Hence, metabolic processes , such as glycolysis, the tricarboxylic acid (TCA) cycle, and fatty acid metabolism, have highly specific effects on the function of macrophages and ECs. The manipulation of these pathways can interfere dramatically in the function of these cells in very specific manners, impacting their ability to produce inflammatory mediators and to exert their effector functions rather than simply being involved in energy generation or general biosynthesis [140,141,142,143]. In the next section, we will address how shifts in the cellular metabolism of macrophages and ECs, termed metabolism reprogramming, govern the outcome of inflammatory processes by determining cellular activation upon inflammatory stimulation.
5.1 Metabolism in Resting Macrophages
All cell types derive energy from the catabolism of three major biomass sources, glucose, fatty acids, and amino acids, using mainly two processes, glycolysis and oxidative phosphorylation (OXPHOX). In brief, one molecule of glucose can yield up to 38 molecules of ATP, of which 2 are derived from glycolysis, 2 from the TCA cycle, and 34 from OXPHOX. Based on this amount of ATP produced per glucose molecule, OXPHOX would be the most efficient bioenergetic pathway and should be preferred. This is the case for several cell types, including resting macrophages, which, under normoxic conditions, use OXPHOX to generate ATP. The major components of the electron transport chain (ETC) utilize the NADH and FADH generated during reactions in the TCA cycle, which is fuelled by the above-described biomass sources. Glycolysis is low due to expression of the PFKFB1 gene, resulting in higher levels of the liver isoform of the phosphofructokinase (PFK) 2 enzyme, which yields low levels of the glycolysis activator fructose-2,6-bisphosphate (F-2,6-BP) and slows down the rate of the reactions involved in pyruvate generation from glucose [144]. In fact, rather than glucose, macrophages obtain much of their energy in the resting state from fatty acid oxidation (FAO) and OXPHOX because they express high levels of fatty acid transporters and catabolic enzymes, as well as proteins involved in ETC and those that drive Acetyl-CoA into the TCA cycle [145]. This oxidative metabolism is usually controlled by specific transcription factors, such as PGC-1β, which are present at higher levels in quiescent macrophages [141, 145]. Therefore, macrophages in the resting state obtain much of their energy from FAO and oxidative metabolism, which can efficiently sustain their basal activities for long periods of time.
The metabolic profiles of resting macrophages are usually shifted during cellular activation, in an event called metabolic reprogramming [142, 146]. Metabolic reprogramming can be simply considered as a response of cells to critical changes in the environment. For example, when oxygen tension is low, cells can switch their metabolic profile to enable the proper generation of energy, even in this altered environment. Hence, under hypoxic conditions, cells usually generate ATP through glycolysis independently from OXPHOX, but this pathway is highly dependent on glucose as the sole fuel source [142, 143]. The core metabolic pathways are integrated to interchange carbons between sugars, fatty acids, nucleic acids and proteins, and therefore metabolic flexibility can play an important role due to the changes in prevailing nutrient and oxygen conditions. This metabolic flexibility also seems to be important when cells are faced with distinct functional demands. Hence, recent work has emphasized that changes in key metabolic regulatory events in macrophages (and to a lesser extent in ECs) are initiated not only by shifts in nutrient or oxygen availability but also by downstream activation of PRRs and cytokine receptors [142, 143]. Thus, these cells present the potential to switch their metabolic activities in response to signals from other cells or from changes in the environment, such as those present in the inflammatory milieu. Importantly, metabolic reprogramming has been shown to govern the phenotype of macrophages by controlling transcriptional and post-transcriptional events that are central to their activation status [142]. The mechanisms involved in metabolic reprogramming in these cell types are outlined in the following section.
5.2 Metabolic Reprogramming in Activated Macrophages
Approximately 50 years ago, early work on leukocyte metabolism indicated an increase in oxygen consumption during phagocytosis [147]. In addition, it was shown around this time that monocytes engage glycolytic metabolism during phagocytosis [148]. Thirty years later, Fukuzumi and co-workers showed that LPS-activated macrophages present increased glucose uptake via a mechanism involving the upregulation of the GLUT1 glucose transporter [149]. More recent work has established that, upon activation by TLR ligands, such as LPS, a shift towards glycolysis and fatty acid synthesis and down-modulation of OXPHOX characterizes proinflammatory macrophages [150, 151]. Inflammatory macrophages also upregulate the pentose phosphate pathway (PPP), which branches from glycolysis and generates NADPH for redox balance [150, 152]. Importantly, this metabolic reprogramming is essential for some effector functions of activated macrophages, such as the production of IL-1β, lipid mediators and reactive oxygen and nitrogen species [142, 146]. Glycolysis also seems to be pivotal for macrophage migration to inflammatory sites [153].
This metabolic shift towards glycolysis in activated macrophages is termed aerobic glycolysis because it occurs even under normoxic conditions. The shift towards aerobic glycolysis seems to be optimally suited to the fast, short-term burst of activation that is required at infectious or sterile inflammatory sites [142, 146]. Recent work has provided extensive evidence that changes in the metabolites associated with macrophage metabolic reprogramming are able to facilitate or promote the specialized activities of these cells. One of the changes in gene expression that increases glycolytic capacity in activated macrophages is the expression of the ubiquitous u-PFK2, the highly active isoform of phosphofructokinase 2, which generates higher quantities of the glycolysis activator F-2,6-BP [144]. In addition, increased expression of the PKM2 isoform of the pyruvate kinase enzyme favours the generation of lactate from pyruvate and diverts it from entry in the TCA cycle [154]. In fact, the TCA cycle is broken at two points in activated macrophages, after citrate and after succinate, leading to an accumulation of these metabolites [150, 151, 155]. Citrate accumulates in activated macrophages as a result of two major causes: increased citrate synthase expression under these conditions [151] and reduced citrate catabolism by isocitrate dehydrogenase due to expression of the Idh1 gene encoding this enzyme, which is downregulated after inflammatory activation [150]. The concentration of succinate accumulates as a result of other three phenomena. First, glutamine-dependent anaplerosis generates succinate, whereby glutamine is used to generate glutamate and subsequently α-KG in the TCA cycle [156]. Second, succinate is generated through the γ-aminobutyric acid (GABA) shunt, which involves transamination of α-KG by the enzyme GABA α-oxoglutarate transaminase, generating L-glutamic acid (L-GA). Glutamic acid-decarboxylase catalyses the conversion of L-GA to GABA, which is then converted to succinic semialdehyde, a source of succinate [155]. Third, expression of the immunoresponsive gene 1 (Irg1) gene during inflammatory activation of macrophages leads to conversion of cis-aconitate (generated from citrate) to itaconate, a metabolite that inhibits succinate dehydrogenase (SDH), diminishing succinate catabolism and thereby linking citrate and succinate accumulation [157]. The accumulation of these two metabolites is essential for several effector functions of activated macrophages.
Citrate accumulation seems to be a key event for the production of three important classes of inflammatory mediators: prostaglandins, nitric oxide (NO) and reactive oxygen species (ROS). To achieve this goal, citrate must be transported from mitochondria to the cytoplasm, and interestingly, LPS induces expression of the mitochondrial citrate carrier (CIC) [158]. Citrate is used for synthesis of phospholipids, which are a source of the arachidonic acid precursor of prostaglandins and other lipid mediators. Citrate can also lead to NADPH generation via malic enzyme and pyruvate. The generated NADPH is used by the inducible nitric oxide synthase enzyme to catalyse NO generation from arginine. Finally, NADPH is also used by NADPH oxidase to produce ROS. Is important to note that NADPH can also be generated by PPP , which is strongly upregulated in activated macrophages [150]. Nevertheless, inhibition of CIC expression by gene silencing decreases the production of NO, ROS and prostaglandins [158], emphasizing how a single TCA intermediate is involved in the production of key inflammatory mediators. Additionally, the generation of itaconate from citrate improves the antibacterial activities of macrophages because itaconate inhibits microbial metabolism, affecting their viability [159].
Succinate accumulation, on the other hand, is a key event leading to enhanced IL-1β production [155] and greater mitochondrial ROS production by activated macrophages. Succinate controls IL-1β production by enhancing the activity of the transcription factor HIF1α. HIF1α then directly induces expression of the IL-1β gene because its promoter region contains HIF1α-binding sites [155]. Succinate oxidation by SDH and enhanced mitochondrial membrane potential favour ROS production [160]. Enhanced mitochondrial ROS production by SDH amplifies IL-1β production [161]. Finally, succinate may also act as an alarmin because it can be released from inflammatory macrophages and lead to autocrine and paracrine activation of the GPR91 receptor, further increasing HIF1α-induced IL-1β production [162]. GPR91 has also been shown to drive leukocyte migration [163]. In conclusion, metabolic reprogramming of activated macrophages towards glycolysis and consequent accumulation of the two cited TCA cycle intermediates is a key event for the proper effector function of macrophages during inflammation.
5.3 Metabolic Reprogramming in ECs
Much less is known about metabolic reprogramming in ECs, especially in inflammatory contexts. However, there is now sufficient evidence showing that ECs in hypoxic or pro-angiogenic environments also adapt their metabolism to sustain their effector functions in these conditions. This phenomenon has been extensively revised elsewhere [140, 143, 164]. Here we will summarize the metabolic alterations documented in activated ECs that are potentially involved in their responses to mediators or cells present in inflammatory environments.
In contrast to resting macrophages, quiescent ECs (independently of the subtype) are highly glycolytic [165]. Even in healthy quiescent vasculature with plenty of oxygen availability, ECs show high rates of glycolysis, although the rates of other metabolic pathways remain to be well characterized [143]. ECs present a relatively low mitochondrial content [166], and their glycolytic rates are approximately 200-fold higher than their OXPHOX activity [165, 167, 168]. Although the lower ATP yield per molecule, glycolysis might provide more ATP in a shorter period of time than OXPHOX when available glucose is unlimited and has the advantage of shunting to glycolysis side branches (such as the PPP) for macromolecule synthesis [152]. Other advantages of glycolytic metabolism in ECs are thought to be the reduction in OXPHOX-generated ROS, diminishing oxidative stress, the preservation of maximal amounts of oxygen for use by perivascular cells, the adequate adaptation of ECs to hypoxic environments during angiogenesis, and the observed ability of the glycolytic by-product lactate to exert pro-angiogenic activity [140].
Interestingly, EC activation by hypoxic or angiogenic factors (such as VEGF) further enhances glycolytic flux by upregulating PFKFB3. Indeed, when PFKFB3 is silenced, the levels of its product F-2,6-BP, are decreased, leading to a reduction of the glycolytic flux by approximately 35% [165]. Glycolytic intermediates are also shunted to PPP, which provide the ribose units necessary for nucleotide synthesis and generates NADPH, which is used for the redox balance [152]. Decreasing the activity of PPP by inhibiting the expression of its rate-limiting enzyme glucose-6-phosphate dehydrogenase (G6PD) reduces effector responses in ECs activated by VEGF [169]. In addition to ROS scavenging, NADPH generated by PPP is involved in endothelial NOS (eNOS)-mediated NO production, which is involved in vasodilation [169] .
Another metabolic driver of NO production in endothelial cells is the glycolytic transcription factor HIF1α. HIF1α directly drives eNOS transcription through binding to hypoxia-responsive elements present in the promoter region of the eNOS gene [170] and through tax-responsive elements [171]. Hypoxia or VEGF-activated ECs upregulate HIF1α, further increasing glycolytic metabolism [172, 173] and enhancing NO production, which leads to vasodilation [174], an important event in the inflammatory milieu. Reduced shear stress present in the vasodilated vasculature also seems to regulate glycolytic metabolism in ECs. Laminar shear stress elevates the expression of Kruppel-like factor 2 (KLF2) in ECs, downregulating glycolytic enzymes, including PFKFB3 [175]. As the vasculature in inflammatory sites is often subjected to a reduction in blood flow, it is conceivable that these changes lead to enhanced PFKFB3 expression and glycolytic flux in ECs. Finally, pro-inflammatory mediators also upregulate glycolysis in ECs [176] and glycolysis further promotes EC pro-inflammatory activity [177]. Therefore, glycolytic metabolism is further induced in activated ECs and seems to be involved in their inflammatory functions in a manner similar to that observed in inflammation-activated macrophages (Fig. 3).

The metabolism of activated macrophages and endothelial cells control their effector functions: Upon activation by TLR ligands, such as LPS, macrophages reprogramme their metabolism by enhancing glycolytic activity and reducing OXPHOX. The series of glycolytic reactions depends on glucose availability, which is increased by GLUT1-mediated glucose uptake, and by increased transcription of the glycolytic enzymes HK, PFKFB3 and PKM2. Glycolysis also provides energy and substrates for the PPP flux, which is important for the redox balance. This metabolic shift leads to the accumulation of TCA intermediates. Citrate accumulation depends on reduced citrate catabolism in the TCA cycle and increased citrate synthase activity. The citrate carrier transports citrate to the cytoplasm where it is used for lipid synthesis and prostaglandin production. NADPH generation is important for NOS2-mediated NO production and for NOX-mediated ROS generation. Succinate accumulation is induced by reduced SDH activity due to conversion mediated by the IRG1 enzyme of citrate to itaconate, which inhibits SDH activity. Additionally, via a series of reactions, glutamine is converted to succinate. Succinate accumulation leads to mitochondrial ROS production, HIF1α activation and consequent IL-1β production, in addition to chemotaxis mediated by the GPR91 receptor. Endothelial cells are usually glycolytic, but upon activation, this metabolism is further enhanced. HIF1α activation by hypoxia and angiogenic factors, such as VEGF, fosters this glycolytic programme, and the PFKFB3 enzyme is key for glycolysis-mediated endothelial cell effector function. In addition to energy, glycolytic metabolism favours NO production and vasodilation due to PPP-generated NADPH. Inhibition of PPP by targeting the enzyme G6PD inhibits NO production. Pro-inflammatory mediators, such as IL-1β, also foster glycolytic metabolism in endothelial cells, and a reduced blood flow in inflammatory environments might promote glycolytic metabolism by inhibiting KLF2-mediated suppression of PFKFB3 transcription
6 Autoinflammatory Diseases
Dysregulation of the innate immune system is directly associated with the development of autoinflammatory diseases, such as disorders characterized by recurrent episodes of fever, rash, and swelling that affects different tissues, and are dissociated with infectious and autoimmune components (revised by [178]). The causative agents for autoinflammatory diseases are diverse, including those that present mutations of PRR platforms, impaired cytokine signalling, and altered cell metabolism. However, the development of several of these diseases occurs due to gain-of-function mutations in the inflammasome, causing increased production and maturation of IL-1β with consequent neutrophilia, which are potential targets for effective therapies. Nonetheless, other diseases share common features of autoinflammatory diseases despite the absence of point mutations in the inflammasome/IL-1β axis, including atherosclerosis, diabetes, gout, and osteoporosis [178,179,180]. Here we list examples of autoinflammatory disorders caused by mutated inflammasome platforms.
One of the most common and well-known autoinflammatory diseases is Familial Mediterranean Fever , which is an autosomal recessive syndrome caused by mutations in alleles of the MEFV (Mediterranean fever) gene encoding the protein Pyrin [181]. Currently, more than 300 sequence variants have been described in the MEFV gene [182]. Pyrin is a crucial molecule that controls caspase-1 activation via the interaction between molecules carrying a Pyrin domain (PYD), such as ASC and some inflammasome prototypes. Nonetheless, Pyrin can also be interpreted as a PRR since it can sense pathogens during the innate immune response [183]. In FMF, mutated Pyrin binds to ASC and causes caspase-1 activation with the consequent release of mature IL-1β [184]. The inflammatory attacks in FMF are self-limited, but recurrent attacks could lead to chronic inflammation. There are different recommendations for FMF management , but colchicine and IL-1β blockers are the most effective options. By disrupting microtubule dynamics, colchicine inhibits Pyrin-ASC aggregation and, consequently, decreases IL-1β maturation and release [185].
Some autoinflammatory diseases are associated with mutations in genes responsible for the synthesis of inflammasome regulatory molecules . For instance, a mutation in the actin regulatory gene WDR1 is characterized by periodic fever, neutrophilia, and thrombocytopenia, associated with high serum levels of IL-18 but not IL-1β [186]. In this study performed in two girls born to consanguineous parents, their monocyte-derived dendritic cells and neutrophils produced high levels of IL-18 under LPS stimulation, with no alterations in IL-1β. Mechanistically, the authors demonstrated a co-localization of the pyrin inflammasome with mutant WDR1 aggregates, culminating in caspase-1 activation [186]. Previous studies have shown that mice carrying a hypomorphic allele of Wdr1 present spontaneous autoinflammatory syndrome and thrombocytopenia [187, 188]. The mechanisms responsible for this murine disorder are similar to those identified in humans, although monocytes seemed to be the most active cells in IL-18 release in humans. In the murine model, the depletion of monocytes in vivo or the inhibition of actin polymerization prevented the development of autoinflammatory-related symptoms [188].
Distinct autoinflammatory disorders resulting from a gain-of-function mutation in the NLRP3 gene occur in cryopyrin-associated periodic syndromes (CAPS ) , comprising familial cold autoinflammatory syndrome (FCAS), Muckle–Wells syndrome (MWS) and neonatal-onset multisystem inflammatory disease (NOMID) [189]. The increased levels of tissue and serum IL-1β are associated with cutaneous, neurological, ophthalmologic, and rheumatologic manifestations of inflammation. However, the continuous activation of mutated NLPR3 in CAPS could result from poor control of NLRP3 inhibition [190], as demonstrated by the blockade of human NLRP3 activation due to phosphorylation of ser295 in NLRP3 by prostaglandin E2-induced cAMP-PKA. Interestingly, HEK293A cells transfected with plasmids encoding NLRP3 mutations previously identified in CAPS patients adjacent to ser295 NLRP3 lost the negative regulatory effect of PKA, keeping the cells in an activated state [190]
Recently, a study demonstrated that adult-onset Still disease (AOSD ) patients had high levels of the mRNA for NLRP3 in peripheral blood mononuclear cells [191]. AOSD is a rare systemic inflammatory disease that shares common autoinflammatory disease symptoms, such as fever, arthralgia (with or without synovitis), skin rash, and striking leucocytosis with neutrophilia [192]. Although that study did not investigate any mutations in the NLRP3 gene in AOSD patients, the authors also detected increased levels of caspase-1, IL-1β, and IL-18 in these patients compared with healthy volunteers [191].
Mutations in the NLRP1 inflammasome are associated with autoimmune diseases, such as rheumatoid arthritis, type 1 diabetes, and systemic lupus erythematosus [193,194,195]. However, a recent study identified a new autoinflammatory disease in patients exhibiting a systemic juvenile idiopathic arthritis phenotype. The authors identified a homozygous mutation in the NLRP1 gene in three patients from two unrelated families that positively correlated with high levels of caspase-1 and IL-18. Clinically, these patients had recurrent fever, arthritis and dyskeratosis. The authors named this disease NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis) [196]. Interestingly, the mechanisms underlying this disease had been identified previously. A gain-of-function mutation in the N-terminal PYD of NLRP1 is responsible for self-oligomerization and activation of NLRP1, which is highly expressed in skin, promoting an increase in the release of IL-1β by keratinocytes that leads to skin inflammation and epidermal hyperplasia [197].
More recently, gain-of-function mutations in the NLRC4 gene have also been associated with autoinflammatory disorders. A mutation in the nucleotide-binding domain of the NLRC4 inflammasome is associated to early-onset recurrent fever flares and macrophage activation syndrome (MAS). This phenotype was observed in a single patient who presented spontaneous inflammasome assembly with IL-1β and IL-18 overproduction and increased macrophage pyroptosis [198]. Similarly, a distinct NLRC4 point mutation was detected in a family with neonatal-onset enterocolitis, periodic fever, and fatal or near-fatal autoinflammatory attack. In this second report, overstimulation of macrophages was also observed, releasing excessive amounts of IL-1β and IL-18 associated with pyroptosis [199]. In addition [200], identified a heterozygous NLRC4 mutation in a patient diagnosed with NOMID. As mentioned previously, NOMID is an autoinflammatory disorder associated with NLPR3 mutations. However, not all NOMID patients present the NLPR3 mutation [201]. In that study, exome sequencing of the patient revealed somatic mosaicism (the occurrence of two genetically distinct populations of cells within an individual, derived from a postzygotic mutation [202]) of a novel NLRC4 mutation. Interestingly, knockout of the NLRC4 locus in a mutant-induced pluripotent stem cell clone using CRISPR/Cas9 technology abrogated the excessive IL-1β and IL-18 secretion by these cells [200].
The use of next-generation sequencing (NGS) technology has been fundamental for the discovery of different mutations in genes related to autoinflammatory disorders, improving diagnosis and directing the best options for disease management. The most frequently inflammasomopathies can be effectively treated using anti-IL-1 therapies [203,204,205]. However, in autoinflammatory diseases characterized by increased levels of IL-18, including those with gain-of-function mutations in the NLRC4 gene, therapy based on recombinant IL-18 binding protein could be the best option [206].
7 Concluding Remarks
Recent knowledge regarding the function of cells and molecules associated with the innate immune response have substantially contributed to unravelling important mechanisms in inflammatory diseases. ECs are very active during the initial events of inflammation by expressing receptors that are classically associated with the innate immune response, enabling them to recognize different exogenous and endogenous molecules. This recognition modifies their metabolism, promoting the synthesis of pro-inflammatory molecules, and changes their conformational state, establishing tissue inflammation. Nonetheless, altered cellular metabolism and the function of innate immune receptors, mainly on leukocytes, can trigger the development of innate immune-associated autoinflammatory diseases. Thus, attention should be focused on the function of innate immune elements in regard to endothelial cells and leukocytes during the course of inflammatory diseases, which will facilitate the development of novel and proper anti-inflammatory therapies.
References
Medzhitov R (2008) Origin and physiological roles of inflammation. Nature 454(7203):428–435
Newton K, Dixit VM (2012) Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol 4(3):pii: a006049
Basil MC, Levy BD (2015) Specialized pro-resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol 16(1):51–67. Nature Publishing Group
Bazzoni G, Dejana E (2004) Endothelial cell-to-cell junctions: molecular organization and role in vascular homeostasis. Physiol Rev 84(0031–9333 (Print)):869–901
Cines BDB et al (1998) Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 91(10):3527–3561
Michiels C (2003) Endothelial cell functions. J Cell Physiol 196(3):430–443
Yau JW, Teoh H, Verma S (2015) Endothelial cell control of thrombosis. BMC Cardiovasc Disord 15:130
Egan K, FitzGerald GA (2006) Eicosanoids and the vascular endothelium, the vascular endothelium I. Handb Exp Pharmacol 176(Pt 1):189–211
Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7(10):803–815
Komarova YA et al (2017) Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res 120(1):179–206
Ferraro F et al (2016) Weibel-Palade body size modulates the adhesive activity of its von Willebrand Factor cargo in cultured endothelial cells. Sci Rep 6(August):32473. Nature Publishing Group
Cook-mills JM, Deem TL (2009) Active participation of endothelial cells in inflammation. Pathology 77(4):487–495
Mai J et al (2013) An evolving new paradigm: endothelial cells--conditional innate immune cells. J Hematol Oncol 6(1):61
Fan LM et al (2017) Endothelial cell – specific reactive oxygen species production increases susceptibility to aortic dissection. Circulation 129(25):2661–2672
Muller WA (2014) How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol 184(4):886–896. American Society for Investigative Pathology
Takeuchi O, Akira S (2010) Pattern recognition receptors and inflammation. Cell 140(6):805–820. Elsevier Inc
Vestweber D (2015) How leukocytes cross the vascular endothelium. Nat Rev Immunol 15(11):692–704. Nature Publishing Group
Potente M, Makinen T (2017) Vascular heterogeneity and specialization in development and disease. Nat Rev Mol Cell Biol 18(8):477–494. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved
Ley K, Zarbock A (2017) Hold on to your endothelium: postarrest steps of the leukocyte adhesion cascade. Immunity 25(2):185–187. Elsevier
Zarbock A et al (2011) Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood 118(26):6743–6751. Washington, DC: American Society of Hematology
Zarbock A et al (2008) PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcRγ to induce slow leukocyte rolling. J Exp Med 205(10):2339–2347. The Rockefeller University Press
Zarbock A, Ley K (2008) Mechanisms and consequences of neutrophil interaction with the endothelium. Am J Pathol 172(1):1–7. American Society for Investigative Pathology
Deban L et al (2010) Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol 11(4):328–334. Nature Publishing Group
Burns AR, Smith CW, Walker DC (2003) Unique structural features that influence neutrophil emigration into the lung. Physiol Rev 83(2):309–336
Hickey MJ, Westhorpe CLV (2013) Imaging inflammatory leukocyte recruitment in kidney, lung and liver – challenges to the multi-step paradigm. Immunol Cell Biol 91(4):281–289
Lefrançais E et al (2017) The lung is a site of platelet biogenesis and a reservoir for haematopoietic progenitors. Nature 544(7648):105–109. Macmillan Publishers Limited, part of Springer Nature. All Rights Reserved
Yipp BG et al (2017) The lung is a host defense niche for immediate neutrophil-mediated vascular protection. Sci Immunol 2(10):eaam8929
Kubo H et al (1999) L- and P-selectin and CD11/CD18 in intracapillary neutrophil sequestration in rabbit lungs. Am J Respir Crit Care Med 159(1):267–274. American Thoracic Society – AJRCCM
Schilter HC et al (2015) Effects of an anti-inflammatory VAP-1/SSAO inhibitor, PXS-4728A, on pulmonary neutrophil migration. Respir Res 16(1):42. London: BioMed Central
Liu L, Kubes P (2003) Molecular mechanisms of leukocyte recruitment: organ-specific mechanisms of action. Thromb Haemostasis 89(2):213–220. Schattauer Publishers
McDonald B et al (2010) Intravascular danger signals guide neutrophils to sites of sterile inflammation. Science 330(6002):362–366
Amaral SS et al (2013) Altered responsiveness to extracellular ATP enhances acetaminophen hepatotoxicity. Cell Commun Signal 11:10. BioMed Central
Marques PE et al (2012) Chemokines and mitochondrial products activate neutrophils to amplify organ injury during mouse acute liver failure. Hepatology 56(5):1971–1982
Haraldsson B, Nyström J, Deen WM (2008) Properties of the glomerular barrier and mechanisms of proteinuria. Physiol Rev 88(2):451–487
Kuligowski MP, Kitching AR, Hickey MJ (2006) Leukocyte recruitment to the inflamed glomerulus: a critical role for platelet-derived P-selectin in the absence of rolling. J Immunol 176(11):6991–6999
Janssen ULF, Assmann JM (1998) Improved survival and amelioration in intercellular adhesion molecule- of nephrotoxic 1 knockout mice nephritis. J Am Soc Nephrol 9:1805–1814
Nagao T et al (2007) Up-regulation of adhesion molecule expression in glomerular endothelial cells by anti-myeloperoxidase antibody. Nephrol Dial Transplant 22(1):77–87
De Vriese AS et al (1999) The role of selectins in glomerular leukocyte recruitment in rat anti-glomerular basement membrane glomerulonephritis. J Am Soc Nephrol 10(12):2510–2517
Jain A, Pasare C (2017) Innate control of adaptive immunity: beyond the three-signal paradigm. J Immunol 198(10):3791–3800
Janeway CA (2013) Pillars article: approaching the asymptote? Evolution and revolution in immunology. J Immunol 191(9):4475–4487. Cold Spring Harb Symp Quant Biol 54:1–13, 1989
Grote K, Schütt H, Schieffer B (2011) Toll-like receptors in angiogenesis. Sci World J 11:981–991
Liu J, Cao X (2016) Cellular and molecular regulation of innate inflammatory responses. Cell Mol Immunol 1358(10):711–721. Nature Publishing Group
Nakaya Y et al (2017) AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. MBio 8(4):1–17
Sun L et al (2013) Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type-I interferon pathway. Science 339(6121):786–791
Kotsovolis G, Kallaras K (2010) The role of endothelium and endogenous vasoactive substances in sepsis. Hippokratia 14(2):88–93
Janeway CA, Medzhitov R (2002) Innate immune recognition. Annu Rev Immunol 20(1):197–216
Medzhitov R, Preston-Hurlburt P, Janeway CA (1997) A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature 388(6640):394–397
Achek A, Yesudhas D, Choi S (2016) Toll-like receptors: promising therapeutic targets for inflammatory diseases. Arch Pharm Res 39(8):1032–1049. Pharmaceutical Society of Korea
Gao W et al (2017) Inhibition of toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol 8(July):508
Imhof BA, Jemelin S, Emre Y (2017) Toll-like receptors elicit different recruitment kinetics of monocytes and neutrophils in mouse acute inflammation. Eur J Immunol 47(6):1002–1008
Mogensen TH (2009) Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22(2):240–273
Blasius AL, Beutler B (2010) Intracellular toll-like receptors. Immunity 32(3):305–315. Elsevier Inc
Pifer R et al (2011) UNC93B1 is essential for TLR11 activation and IL-12-dependent host resistance to Toxoplasma gondii. J Biol Chem 286(5):3307–3314
Yarovinsky F (2014) Innate immunity to Toxoplasma gondii infection. Nat Rev Immunol 14(2):109–121. Nature Publishing Group
Horng T, Medzhitov R (2001) Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc Natl Acad Sci U S A 98(22):12654–12658
Salvador B et al (2016) Modulation of endothelial function by Toll like receptors. Pharmacol Res 108:46–56
Ahmed S et al (2013) TRIF-mediated TLR3 and TLR4 signaling is negatively regulated by ADAM15. J Immunol 190(5):2217–2228
Lin X et al (2015) Effect of TLR4/MyD88 signaling pathway on expression of IL-1B and TNF- a in synovial fibroblasts from temporomandibular joint exposed to lipopolysaccharide. Mediat Inflamm 2015:329405
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11(5):373–384. Nature Publishing Group
Pryshchep O et al (2008) Vessel-specific toll-like receptor profiles in human medium and large arteries. Circulation 118(12):1276–1284
Nagyoszi P et al (2010) Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochem Int 57(5):556–564
Sturtzel C (2017) Endothelial cells. Adv Exp Med Biol 1003:71–91
Xiao L, Liu Y, Wang N (2014) New paradigms in inflammatory signaling in vascular endothelial cells. Am J Physiol Heart Circ Physiol 306(3):H317–H325
Card CM, Yu SS, Swartz MA (2014) Emerging roles of lymphatic endothelium in regulating adaptive immunity. J Clin Investig 124(3):943–952
Pegu A et al (2008) Human lymphatic endothelial cells express multiple functional TLRs. J Immunol 180(5):3399–3405
Khakpour S, Wilhelmsen K, Hellman J (2015) Vascular endothelial cell Toll-like receptor pathways in sepsis. Innate Immun 21(8):827–846
Mazzucchelli I et al (2015) Expression and function of toll-like receptors in human circulating endothelial colony forming cells. Immunol Lett 168(1):98–104. Elsevier B.V.
Opitz B et al (2007) Extra- and intracellular innate immune recognition in endothelial cells. Thromb Haemost 96(6):756–766
Zheng GJ, Sun Q, Li YP (2009) Inflammation, endothelium, coagulation in sepsis. Chin Crit Care Med 21(9):573–576
Dauphinee SM, Karsan A (2006) Lipopolysaccharide signaling in endothelial cells. Lab Investig 86(1):9–22
Gotsch U et al (1994) Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-a in vivo. Cell Adhes Commun 2:7
Gatheral T et al (2012) A key role for the endothelium in NOD1 mediated vascular inflammation: comparison to TLR4 responses. PLoS One 7(8):1–13
Wilhelmsen K et al (2012) Activation of endothelial TLR2 by bacterial lipoprotein upregulates proteins specific for the neutrophil response. Innate Immun 18(4):602–616
Zimmer S et al (2011) Activation of endothelial toll-like receptor 3 impairs endothelial function. Circ Res 108(11):1358–1366
Faure E et al (2001) Bacterial lipopolysaccharide and IFN-γ induce Toll-like receptor 2 and Toll-like receptor 4 expression in human endothelial cells: role of NF-κB activation. J Immunol 166(3):2018–2024
El Kebir D et al (2015) Toll-like receptor 9 signaling regulates tissue factor and tissue factor pathway inhibitor expression in human endothelial cells and coagulation in mice. Crit Care Med 43(6):e179–e189
Dunzendorfer S, Lee HK, Tobias PS (2004) Flow-dependent regulation of endothelial toll-like receptor 2 expression through inhibition of SP1 activity. Circ Res 95(7):684–691
Bomfim GF et al (2014) Toll like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rat. Clin Sci 122(11):535–543
Hernanz R et al (2015) Toll-like receptor 4 contributes to vascular remodelling and endothelial dysfunction in angiotensin II-induced hypertension. Br J Pharmacol 172(12):3159–3176
Su X et al (2011) Oxidized low density lipoprotein induces bone morphogenetic protein-2 in coronary artery endothelial cells via toll-like receptors 2 and 4. J Biol Chem 286(14):12213–12220
Pahwa R, Nallasamy P, Jialal I (2016) Toll-like receptors 2 and 4 mediate hyperglycemia induced macrovascular aortic endothelial cell inflammation and perturbation of the endothelial glycocalyx. J Diabetes Complications 30(4):563–572. Elsevier Inc
Rajamani U, Jialal I (2014) Hyperglycemia induces toll-like receptor-2 and -4 expression and activity in human microvascular retinal endothelial cells: implications for diabetic retinopathy. J Diabetes Res 2014:7–10. Hindawi Publishing Corporation
Franchi L et al (2010) Function of NOD-like receptors in microbial recognition and host defense. Cancer 227(1):106–128
Bertin J et al (1999) Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-ƙB*. Biochemistry 274(19):12955–12958
Inohara N et al (1999) Nod1, and Apap-1-like activator of caspase-9 and nuclear factor-kB. J Biol Chem 274(21):14560–14567
Ogura Y et al (2001) Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-??B. J Biol Chem 276(7):4812–4818
Claes A et al (2015) NOD-like receptors: guardians of intestinal mucosal barriers. Physiology (Bethesda) 30:241–250
Motta V et al (2015) NOD-like receptors: versatile cytosolic sentinels. Physiol Rev 95(1):149–178
Opitz B et al (2005) Nod1-mediated endothelial cell activation by Chlamydophila pneumoniae. Circ Res 96(3):319–326
Opitz B et al (2006) Listeria monocytogenes activated p38 MAPK and induced IL-8 secretion in a nucleotide-binding oligomerization domain 1-dependent manner in endothelial cells. J Immunol 176(1):484–490
Oh HM et al (2005) Induction and localization of NOD2 protein in human endothelial cells. Cell Immunol 237(1):37–44
Nagata E, Oho T (2016) Invasive Streptococcus mutans induces inflammatory cytokine production in human aortic endothelial cells via regulation of intracellular TLR2 and NOD2. Mol Oral Microbiol 32:131–141
Manni M et al (2011) Muramyl dipeptide induces Th17 polarization through activation of endothelial cells. J Immunol 186(6):3356–3363
Wan M et al (2015) E-selectin expression induced by Porphyromonas gingivalis in human endothelial cells via nucleotide-binding oligomerization domain-like receptors and Toll-like receptors. Mol Oral Microbiol 30(5):399–410
Lin C et al (2017) Helix B surface peptide attenuates diabetic cardiomyopathy via AMPK-dependent autophagy. Biochem Biophys Res Commun 482(4):665–671. Elsevier Ltd
Bergsbaken T, Fink SL, Cookson BT (2010) Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7(2):99–109
Ghosh S et al (2017) The PYHIN protein p205 regulates the inflammasome by controlling Asc expression. J Immunol 199:ji1700823
Schumann RR et al (1998) Lipopolysaccharide activates caspase-1 (interleukin-1-converting enzyme) in cultured monocytic and endothelial cells. Blood 91(2):577–584
Li Y et al (2017) Negative regulation of NLRP3 inflammasome by SIRT1 in vascular endothelial cells. Immunobiology 222(3):552–561. Elsevier GmbH
Chen Y et al (2015) Endothelial Nlrp3 inflammasome activation associated with lysosomal destabilization during coronary arteritis. Biochim Biophys Acta 853(2):396–408.
Kinnunen K (2017) Lysosomal destabilization activates the NLRP3 inflammasome in human umbilical vein endothelial cells (HUVECs). J Cell Commun Signal 11(3):275–279
Chen Y et al (2016b) Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J Mol Med (Berlin) 94(12):1335–1347
Bleda S et al (2016) Elevated levels of triglycerides and vldl-cholesterol provoke activation of nlrp1 inflammasome in endothelial cells. Int J Cardiol 220:52–55. Elsevier Ireland Ltd
Chen H et al (2016a) Cadmium induces NLRP3 inflammasome-dependent pyroptosis in vascular endothelial cells. Toxicol Lett 246:7–16. Elsevier Ireland Ltd
Cheng KT et al (2017) Caspase-11 – mediated endothelial pyroptosis underlies endotoxemia-induced lung injury. J Clin Invest 7(26):1–12
XI H et al (2016) Caspase-1 inflammasome activation mediates homocysteine- induced pyrop-apoptosis in endothelial cells. Circ Res 165(7):1789–1802
Franchi L et al (2009) The inflammasome: a caspase-1 activation platform regulating immune responses and disease pathogenesis. Nat Immun 10(3):241
Rathinam VAK et al (2010) The AIM2 inflammasome is essential for host-defense against cytosolic bacteria and DNA viruses. Nat Immunol 11(5):395–402
Alekseeva AY et al (2016) Multiple ways of cfDNA reception and following ROS production in endothelial cells. Adv Exp Med Biol 924:127–131
Hakimi M et al (2014) Inflammation-related induction of absent in melanoma 2 (AIM2) in vascular cells and atherosclerotic lesions suggests a role in vascular pathogenesis. J Vasc Surg 59(3):794–803.e2. Society for Vascular Surgery
Singh VV et al (2013) Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates gamma interferon-inducible protein 16-mediated inflammasomes. J Virol 87(8):4417–4431
Opitz B et al (2009) Role of Toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections. Thromb Haemost 102(6):1103–1109
Matsumiya T, Stafforini DM (2010) Function and regulation of retinoic acid-inducible gene-I. Crit Rev Immunol 30(6):489–513
Kato H, Takahasi K, Fujita T (2011) RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev 243:91–98
Imaizumi T, Yoshida H, Satoh K (2004) Regulation of CX3CL1/fractalkine expression in endothelial cells. J Atheroscler Thromb 11(1):15–21
Asdonk T et al (2012) Endothelial RIG-I activation impairs endothelial function. Biochem Biophys Res Commun 420(1):66–71. Elsevier Inc
Berghäll H et al (2006) The interferon-inducible RNA helicase, mda-5, is involved in measles virus-induced expression of antiviral cytokines. Microbes Infect 8(8):2138–2144
Hägele H et al (2009) Double-stranded RNA activates type i interferon secretion in glomerular endothelial cells via retinoic acid-inducible gene (RIG)-1. Nephrol Dial Transplant 24(11):3312–3318
Peyrefitte C et al (2006) Dengue virus infection of human microvascular endothelial cells from different vascular beds promotes both common and specific functional changes. J Med Virol 78(12):229–242
da Conceição TM et al (2013) Essential role of RIG-I in the activation of endothelial cells by dengue virus. Virology 435(2):281–292
Blank T et al (2016) Brain endothelial- and epithelial-specific interferon receptor chain 1 drives virus-induced sickness behavior and cognitive impairment. Immunity 44(4):901–912
Moser J et al (2016) Intracellular RIG-I signaling regulates TLR4-independent endothelial inflammatory responses to endotoxin. J Immunol 196(11):4681–4691
Margolis SR, Wilson SC, Vance RE (2017) Evolutionary origins of cGAS-STING signaling. Trends Immunol 38(10):733–743. Elsevier Ltd
Liu Y et al (2014) Activated STING in a vascular and pulmonary syndrome. N Engl J Med 371(6):507–518
Lio C-WJ et al (2016) cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J Virol. https://doi.org/10.1128/JVI.01040-16
Ma Z et al (2015) Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc Natl Acad Sci 112(31):E4306–E4315
Mao Y et al (2017) STING–IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler Thromb Vasc Biol 37(5):920–929
Yuan L et al (2017) Palmitic acid dysregulates the Hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling. J Biol Chem. https://doi.org/10.1074/jbc.M117.804005
Fullerton JN, Gilroy DW (2016) Resolution of inflammation: a new therapeutic frontier. Nat Rev Drug Discov 15(8):551–567. Nature Publishing Group
Fischetti F, Tedesco F (2017) Cross-talk between the complement system and endothelial cells in physiologic conditions and in vascular diseases. Autoimmunity 39(5):417–428
Carter AM (2012) Complement activation: an emerging player in the pathogenesis of cardiovascular disease. Scientifica 2012:402783
Brunn GJ, Saadi S, Platt JL (2006) Differential regulation of endothelial cell activation by complement and interleukin 1{alpha}. Circ Res 96:793–800
Karpman D et al (2015) Complement interactions with blood cells, endothelial cells and microvesicles in thrombotic and inflammatory conditions. Adv Exp Med Biol 865:19
Kilgore KS et al (1995) Enhancement by the complement membrane attack complex of tumor necrosis factor-alpha-induced endothelial cell expression of E-selectin and ICAM-1. J Immunol 155(3):1434–1441
Proctor LM et al (2004) Comparative anti-inflammatory activities of antagonists to C3a and C5a receptors in a rat model of intestinal ischaemia/reperfusion injury. Br J Pharmacol 142(4):756–764
Yin W et al (2007) Classical pathway complement activation on human endothelial cells. Mol Immunol 44(9):2228–2234
Riedl M et al (2017) Complement activation induces neutrophil adhesion and neutrophil-platelet aggregate formation on vascular endothelial cells. Kidney Int Rep 2(1):66–75. Elsevier Inc
Dobó J et al (2014) Multiple roles of complement MASP-1 at the interface of innate immune response and coagulation. Mol Immunol 61(2):69–78. Elsevier Ltd
Jani PK et al (2016) Complement MASP-1 enhances adhesion between endothelial cells and neutrophils by up-regulating E-selectin expression. Mol Immunol 75:38–47. Elsevier Ltd
Ghesquiere B et al (2014) Metabolism of stromal and immune cells in health and disease. Nature 511(7508):167–176. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved
O’Neill LAJ, Hardie DG (2013) Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493(7432):346–355. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved
O’Neill LAJ, Pearce EJ (2016) Immunometabolism governs dendritic cell and macrophage function. J Exp Med 213(1):15–23. The Rockefeller University Press
Wong BW et al (2017) Endothelial cell metabolism in health and disease: impact of hypoxia. EMBO J 36(15):2187–2203
Rodríguez-Prados J-C et al (2010) Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J Immunol 185(1):605 LP–605614
Vats D et al (2006) Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab 4(1):13–24
Galván-Peña S, O’Neill LAJ (2014) Metabolic reprograming in macrophage polarization. Front Immunol 5:420
Rossi F, Zatti M (1964) Changes in the metabolic pattern of polymorphonuclear leucocytes during phagocytosis. Br J Exp Pathol 45(5):548–559
Oren R et al (1963) Metabolic patterns in three types of phagocytizing cells. J Cell Biol 17(3):487–501. The Rockefeller University Press
Fukuzumi M et al (1996) Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT 1. Infect Immun 64(1):108–112
Jha AK et al (2017) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42(3):419–430. Elsevier
Newsholme P et al (1986) Metabolism of glucose, glutamine, long-chain fatty acids and ketone bodies by murine macrophages. Biochem J 239(1):121–125
Riganti C et al (2012) The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate. Free Radic Biol Med 53(3):421–436
Semba H et al (2016) HIF-1α-PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat Commun 7(May):11635. Nature Publishing Group
Palsson-McDermott EM et al (2015) Pyruvate Kinase M2 regulates Hif-1α activity and IL-1β induction, and is a critical determinant of the Warburg Effect in LPS-activated macrophages. Cell Metab 21(1):65–80
Tannahill GM et al (2013) Succinate is a danger signal that induces IL-1β via HIF-1α. Nature 496(7444):238–242
Palsson-Mcdermott EM, O’Neill LAJ (2013) The Warburg effect then and now: from cancer to inflammatory diseases. BioEssays 35(11):965–973
Németh B et al (2015) Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J 30:286–300
Infantino V et al (2011) The mitochondrial citrate carrier: a new player in inflammation. Biochem J 438(3):433–436
Michelucci A et al (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc Natl Acad Sci 110(19):7820–7825
Mills EL et al (2017) Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167(2):457–470.e13. Elsevier
Garaude J et al (2016) Mitochondrial respiratory chain adaptations in macrophages contribute to antibacterial host defence. Nat Immunol 17(9):1037–1045
Littlewood-Evans A et al (2016) GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med 213:1655
Rubic T et al (2008) Triggering the succinate receptor GPR91 on dendritic cells enhances immunity. Nat Immunol 9(11):1261–1269. Nature Publishing Group
Eelen G et al (2015) Endothelial cell metabolism in normal and diseased vasculature. Circ Res 116(7):1231–1244
De Bock K et al (2017) Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 154(3):651–663. Elsevier
Groschner LN et al (2012) Endothelial mitochondria – less respiration, more integration. Pflugers Arch – Eur J Physiol 464(1):63–76
Dagher Z et al (2001) Acute regulation of fatty acid oxidation and AMP-activated protein kinase in human umbilical vein endothelial cells. Circ Res 88(12):1276–1282
Mertens S et al (1990) Energetic response of coronary endothelial cells to hypoxia. Am J Physiol Heart Circ Physiol 258(3):H689 LP–H68H694
Leopold JA et al (2003) Glucose-6-phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bioavailable nitric oxide. Arterioscler Thromb Vasc Biol 23(3):411–417
Coulet F et al (2003) Identification of hypoxia-response element in the human endothelial nitric-oxide synthase gene promoter. J Biol Chem 278(47):46230–46240
Min J et al (2006) Hypoxia-induced endothelial NO synthase gene transcriptional activation is mediated through the tax-responsive element in endothelial cells. Hypertension 47(6):1189–1196
Weigand JE et al (2012) Hypoxia-induced alternative splicing in endothelial cells. PLoS One 7(8):e42697. Public Library of Science
Xu Y et al (2014) Endothelial 6-phosphofructo-2-kinase (PFKFB3) plays a critical role in angiogenesis. Arterioscler Thromb Vasc Biol 34(6):1231–1239
Chan CK et al (2013) A-FABP and oxidative stress underlie the impairment of endothelium-dependent relaxations to serotonin and the intima-medial thickening in the porcine coronary artery with regenerated endothelium. ACS Chem Neurosci 4(1):122–129. American Chemical Society
Doddaballapur A et al (2014) Laminar shear stress inhibits endothelial cell metabolism via KLF2-mediated repression of PFKFB3. Arterioscler Thromb Vasc Biol 35(1):137–145
Folco EJ et al (2011) Hypoxia but not inflammation augments glucose uptake in human macrophages: implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. J Am Coll Cardiol 58(6):603–614
Cantelmo AR et al (2017) Inhibition of the glycolytic activator PFKFB3 in endothelium induces tumor vessel normalization, impairs metastasis, and improves chemotherapy. Cancer Cell 30(6):968–985. Elsevier
Manthiram K et al (2017) The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 18(8):832–842. Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved
Klen J et al (2015) NLRP3 inflammasome polymorphism and macrovascular complications in type 2 diabetes patients. J Diabetes Res 2015:616747
Miao ZM et al (2009) NALP3 inflammasome functional polymorphisms and gout susceptibility. Cell Cycle 8(1):27–30
French FMF Consortium (1997) A candidate gene for familial Mediterranean fever. Nat Genet 17(1):25–31
Giancane G et al (2015) Evidence based recommendations for genetic diagnosis of familial Mediterranean fever. Ann Rheum Dis 74(4):635–641
Chu LH et al (2016) An updated view on the structure and function of PYRIN domains. Apoptosis 20(2):157–173
Manukyan G, Aminov R (2016) Update on pyrin functions and mechanisms of familial Mediterranean fever. Front Microbiol 7(March):1–8
Gao W et al (2016) Site-specific phosphorylation and microtubule dynamics control Pyrin inflammasome activation. Proc Natl Acad Sci 113(33):E4857–E4866
Standing ASI et al (2017) Autoinflammatory periodic fever, immunodeficiency, and thrombocytopenia (PFIT) caused by mutation in actin-regulatory gene WDR1. J Exp Med 214(1):59–71
Kile BT et al (2007) Mutations in the cofilin partner Aip1/Wdr1 cause autoinflammatory disease and macrothrombocytopenia. Blood 110(7):2371–2380
Kim ML et al (2015) Aberrant actin depolymerization triggers the pyrin inflammasome and autoinflammatory disease that is dependent on IL-18, not IL-1β. J Exp Med 212(6):927–938
Agostini L et al (2004) NALP3 forms an IL-1β-processing inflammasome with increased activity in Muckle-Wells autoinflammatory disorder. Immunity 20(3):319–325
Mortimer L et al (2016) NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol 17(10):1176–1186. Nature Publishing Group
Hsieh CW et al (2017) Elevated expression of the NLRP3 inflammasome and its correlation with disease activity in adult-onset still disease. J Rheumatol 44(8):1142–1150
Gopalarathinam R et al (2016) Adult onset Still’s disease: a review on diagnostic workup and treatment options. Case Rep Rheumatol 2016:1–6. Hindawi Publishing Corporation
Magitta NF et al (2009) A coding polymorphism in NALP1 confers risk for autoimmune Addison’s disease and type 1 diabetes. Genes Immun 10(2):120–124
Pontillo A et al (2012) Polimorphisms in inflammasome genes are involved in the predisposition to systemic lupus erythematosus. Autoimmunity 45(4):271–278. Taylor & Francis
Sui J et al (2012) NLRP1 gene polymorphism influences gene transcription and is a risk factor for rheumatoid arthritis in han chinese. Arthritis Rheum 64(3):647–654
Grandemange S et al (2017) A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1- associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis 76(7):1191–1198
Zhong FL et al (2016) Germline NLRP1 mutations cause skin inflammatory and cancer susceptibility syndromes via inflammasome activation. Cell 167(1):187–202.e17
Canna SW et al (2015) An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46(10):1140–1146
Romberg N et al (2014) Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 165(7):1789–1802
Kawasaki Y et al (2016) Pluripotent cell-based phenotypic dissection identifies a high-frequency somatic NLRC4 mutation as a cause of autoinflammation. Arthritis Rheumatol 11(10):300–308
Goldbach-Mansky R et al (2006) Neonatal-onset multisystem inflammatory disease responsive to interleukin-1β inhibition. N Engl J Med 355(6):581–592
Freed D, Stevens EL, Pevsner J (2014) Somatic mosaicism in the human genome. Genes 5(4):1064–1094
Hoffman HM et al (2008) Efficacy and safety of rilonacept (Interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 58(8):2443–2452
Lachmann HJ et al (2009) Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 360(23):2416–2425
Meinzer U et al (2017) Interleukin-1 targeting drugs in familial Mediterranean fever: a case series and a review of the literature. Semin Arthritis Rheum 41(2):265–271. Elsevier
Canna SW et al (2017) Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol 139(5):1698–1701
Conflicts of Interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Boff, D., Fagundes, C.T., Russo, R.C., Amaral, F.A. (2018). Innate Immunity and Inflammation: The Molecular Mechanisms Governing the Cross-Talk Between Innate Immune and Endothelial Cells. In: Riccardi, C., Levi-Schaffer, F., Tiligada, E. (eds) Immunopharmacology and Inflammation. Springer, Cham. https://doi.org/10.1007/978-3-319-77658-3_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-77658-3_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-77657-6
Online ISBN: 978-3-319-77658-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)