
Abstract
The health of living organisms is constantly challenged by bacterial and viral threats. The recognition of pathogenic microorganisms by diverse receptors triggers a variety of host defense mechanisms, leading to their eradication. Toll-like receptors (TLRs), which are type I transmembrane proteins, recognize specific signatures of the invading microbes and activate a cascade of downstream signals inducing the secretion of inflammatory cytokines, chemokines, and type I interferons. The TLR response not only counteracts the pathogens but also initiates and shapes the adaptive immune response. Under normal conditions, inflammation is downregulated after the removal of the pathogen and cellular debris. However, a dysfunctional TLR-mediated response maintains a chronic inflammatory state and leads to local and systemic deleterious effects in host cells and tissues. Such inappropriate TLR response has been attributed to the development and progression of multiple diseases such as cancer, autoimmune, and inflammatory diseases. In this review, we discuss the emerging role of TLRs in the pathogenesis of inflammatory diseases and how targeting of TLRs offers a promising therapeutic strategy for the prevention and treatment of various inflammatory diseases. Additionally, we highlight a number of TLR-targeting agents that are in the developmental stage or in clinical trials.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
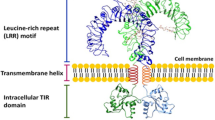
The innate immune response provides the first line of host defense against invading pathogens. This response is triggered by the activation of pattern recognition receptors (PRRs), which represent a crucial link between pathogen detection and the induction of a pro-inflammatory cascade aimed at suppressing the infectious agent (Ospelt and Gay 2010). Among the growing family of PRRs, toll-like receptors (TLRs) were the first to be identified and are the most widely studied. Various immune and non-immune cells express these evolutionarily conserved receptors (De Nardo 2015). TLRs play a fundamental role in the primary response against invaders connecting both innate and adaptive immune responses. To date, 10 and 12 TLRs have been discovered in human and mice, respectively, and their specific ligands have been largely characterized. Human cells ubiquitously express ten functional TLRs (TLR1–10), whereas twelve TLRs (TLR1–9 and TLR11–13) are expressed in mice (De Nardo 2015; Ospelt and Gay 2010). TLRs are classified as members of the toll/interleukin (IL)-1 receptor (TIR) superfamily based on the similarities between their structural features (O’neill et al. 2009). They are composed of extracellular leucine-rich repeat motifs, which facilitate pathogen identification and ligand binding, a single transmembrane helix, and a highly conserved cytoplasmic TIR domain. The TIR domain is involved in the recruitment of adaptor molecules such as myeloid differentiation 88 (MyD88), MyD88-adaptor-like (MAL), TIR-domain-containing adaptor-inducing interferon-β (TRIF), and TRIF-related adaptor molecule (TRAM), and thus in the activation of the downstream signaling cascade (Jin and Lee 2008; Kawai and Akira 2010).
Despite their structural and functional similarities, TLRs mainly differ in their ligand specificity, usage of adaptor proteins, and cellular localization. The arrangement of TLRs differs according to the ligand recognized. Therefore, TLRs involved in the recognition of cell-surface molecules, such as TLRs 1, 2, 4, 5, 6, and 10, are expressed on the cell surface, whereas TLRs 3, 7, 8, and 9, involved in nucleic acid recognition, are located intracellularly, anchored to the endosome (Jin and Lee 2008; Kawai and Akira 2010; Blasius and Beutler 2010). TLRs enable the host to identify not only a diverse repertoire of conserved pathogen-derived fragments known as pathogen-associated molecular patterns (PAMPs) (Janeway and Medzhitov 1998) such as bacterial lipopolysaccharides (LPS), viral RNA, CpG-containing DNA, and flagellin, but also various molecules released from damaged cells known as danger-associated molecular patterns (DAMPs) (Bianchi 2007) (Table 1). The specific detection of PAMPs and DAMPs by host receptors drives a cascade of signaling that converges at nuclear factor-κB (NF-κB) and interferon regulatory factors (IRFs) and induces the secretion of pro-inflammatory cytokines, type I interferon (IFN), and chemokines, which promote direct killing of the pathogen (Ospelt and Gay 2010; Anwar et al. 2013). The wide variety of ligands sensed by these receptors and the complexity of the immune responses triggered by their activation justify in part their connection with the onset of several infectious, autoimmune, and inflammatory diseases. The binding of TLRs to their ligands results in the activation and maturation of antigen-presenting cells (APCs) such as macrophages or dendritic cells, which are responsible for the initiation of the adaptive immune responses through the stimulation of T- and B cell-mediated immune signals (Schnare et al. 2001).
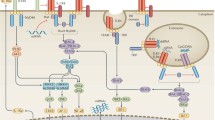
TLRs can trigger roughly two different series of signaling events (Fig. 1). In one pathway, MyD88 is the main adaptor protein, triggering of which leads to early-phase activation of NF-κB and mitogen-activated protein kinases (MAPKs) (Lee et al. 2012). In the MyD88-independent pathway, the recruitment of TRIF conveys signals to downstream adaptor molecules and leads to an ensuing late-phase activation of IRFs and NF-κB (Akira and Hoshino 2003; Kawai and Akira 2005). The inflammatory response induced by TLR activation is a protective response that ensures not only the removal of harmful stimuli but also the repair of damaged tissues (Mudaliar et al. 2013). This response is usually rapidly terminated once the tissues are repaired and the pathogens are eradicated. However, inappropriate activation of TLR signaling due to failure of their regulatory mechanisms might disrupt the homeostasis by creating a feedback loop of inflammatory cytokine secretion leading to the development of autoimmune and inflammatory diseases. Therefore, TLR-mediated responses are to be tightly regulated for optimal and balanced performance of the immune system (Piccinini and Midwood 2010; Anwar et al. 2013). In this review, we will cover the roles played by TLRs during inflammation. Additionally, we aimed at providing new insights into the functions that TLRs might have in the development and the progression of inflammatory diseases, and more importantly, into the potential of TLRs as therapeutic targets.

Overview of the TLR signaling pathway. Cell-surface and endosomal TLRs are triggered by their representative ligands originating from invading pathogens to start downstream signaling. With the exception of TLR3, all TLRs activate the MyD88-dependent pathway. Following the activation of TLRs, MyD88 recruits the IRAK family of proteins along with the adapter protein TRAF6. The phosphorylation of IRAK proteins passes the signal to the TAK1 complex, subsequently activating the IKK complex. The activated IKK complex phosphorylates IκB and marks it for degradation. The phosphorylated IκB induces the release of NF-κB and its translocation into the nucleus, resulting in the production and release of pro-inflammatory cytokines. TLR3 and TLR4 use a TRIF-dependent pathway, with the recruitment of TRAM adaptor protein for TLR4 activation. The TRIF pathway needs TRAF3 to pass the signals to TBK1 and IKKε complexes, which activate IRF3 and IRF7, respectively. The IRF proteins enter the nucleus and start the transcription of type I IFN
Toll-like receptor signaling
Given the essential role of TLRs in the initiation of an immune response, their involvement in the development and/or maintenance of various diseases is not surprising. In their inactive forms, TLRs are present in a monomeric state, whereas upon exposure to their specific ligand, most of them form active homodimers, while a few are likely to form heterodimers, depending on the ligand specificity (Medzhitov 2007; Brown et al. 2011). For instance, TLR2 forms heterodimers with TLR1 or TLR6 upon binding of lipoprotein or lipopeptides, respectively, whereas the binding of bacterial flagellin to TLR5 is known to be responsible for its homodimerization (Yoon et al. 2012). The cell-surface TLR4 forms a complex with its co-receptor cluster of differentiation 14 (CD14) and its accessory molecule myeloid differentiation factor 2 (MD2) that provides a hydrophobic core where the acyl chains of lipid A (the biologically active constituent of LPS) can be accommodated (Piazza et al. 2012; Cighetti et al. 2014). Endosomal TLRs recognize both self and foreign nucleic acid structures. Thus, while TLR3 recognizes dsRNA, TLR7 and TLR8 respond to ssRNA (Cook et al. 2004), and TLR9 responds to CpG-rich DNA in a conformation-specific mechanism (Blasius and Beutler 2010). Pathogen recognition activates TLR dimers, which in return stimulate the recruitment of TIR-domain containing adaptor proteins. The TIR domains act as a scaffold for downstream signaling molecules. The engagement of adaptor protein promotes the formation of higher-order complexes functioning in MyD88-dependent NF-κB activation and TRIF-dependent IFN regulatory factor (IRF) activation (Gay et al. 2014).
The MyD88-dependent pathway is utilized by all TLRs except TLR3, which uses a TRIF-dependent pathway. Exceptionally, TLR4 can activate both pathways. TLRs 1, 2, 4, 6, and 10 recruit, in addition to MyD88, the TIR domain-containing adapter protein (TIRAP), which serves as a link between the TIR domain of the TLR and MyD88. Later, MyD88 recruits interleukin-1 receptor-associated kinase 4 (IRAK4), which phosphorylates IRAK1 and IRAK2 that are responsible for early and late phase TLR responses, respectively (Meylan and Tschopp 2008). IRAK4 is the master regulator of the IRAK family proteins (Qian and Cao 2013). The phosphorylated IRAKs dissociate from MyD88 and bind to the tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6), which in turn activate transforming-growth-factor-β-activated kinase 1 (TAK1) complex and TGF-β-activated kinase (TAB) 2, and 3 through polyubiquitination and trigger early-phase activation of NF-κB and MAPKs (Kumar et al. 2011). The activation of NF-κB typically involves phosphorylation of nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor (IκB) by the inhibitor of nuclear factor-κB kinase (IKK) complex, which consists of IKKα, β, and γ. The phosphorylation of IκB leads to its ubiquitylation and subsequent degradation, which allows the release of NF-κB and its translocation to the nucleus. Furthermore, MAPKs pass the signals to p38 and c-Jun N-terminal kinases (JNKs) to activate cAMP-responsive element (CREB) and activator protein-1 (AP-1) transcription factors inducing the transcription of inflammatory cytokines and chemokines (Kawai and Akira 2007, 2010; Fig. 1).
TRIF has been identified as a protein fundamental to the MyD88-independent pathway. Once it recognizes its specific ligand, TLR3 recruits TRIF adapter protein, whereas the binding of TLR4 to its specific ligand recruits TRIF through TRAM adaptor proteins (Kawasaki and Kawai 2014). The interaction of TRIF with TRAF3 activates non-canonical IKKs, such as serine/threonine-protein kinase (TBK1) and IKKε, to phosphorylate IRF3 and IRF7 (Kumar et al. 2011). The phosphorylated IRFs translocate to the nucleus where they initiate the transcription of type I IFN. Of note, the C-terminal region of TRIF contains a receptor-interacting protein homotypic interaction motif (RHIM), which interacts with receptor-interacting serine/threonine-protein kinase 1 (RIP1) to activate TRAF6 and results in the late phase activation of NF-κB and MAPKs (Kumar et al. 2011; Kawasaki and Kawai 2014).
Negative regulators in TLR signaling
Following TLR activation and the elimination of danger signals, signaling is considered to be terminated at a checkpoint, and the system returns to its homeostatic state to avoid host damage. To this end, a number of molecules act in synergy and play vital roles in regulating the TLR-induced inflammation. Some of these negative regulators downregulate TLR expression, whereas others may restrict the signaling by blocking TLR activation (Fig. 2). Radioproductive 105 (RP105), a TLR homolog lacking a signaling domain that is present in B cells and dendritic cells specifically inhibits TLR4 signaling by preventing the binding of LPS (Liew et al. 2005; Kawai and Akira 2007). The transmembrane protein ST2L blocks the recruitment of MyD88 and TIRAP required for TLR4 activation (Brint et al. 2004; Liu et al. 2010). Single immunoglobulin and toll–interleukin 1 receptor (SIGIRR) fits in the IL-1 receptor family and acts together with IRAKs and TRAF6 to block TLR signaling. However, the mechanism by which SIGIRR suppresses TLR function is not fully understood (Wald et al. 2003; Sham et al. 2013). Suppressor of cytokine signaling 1 (SOCS1), an E3 ligase initially identified as an inhibitor of the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathways, also suppresses signaling downstream of TLR2 and TLR4 (Brown et al. 2011). Furthermore, SOCS1 is capable of directly inhibiting LPS-induced (but not TNF-induced) NF-κB activation in a TLR4- and MD2-dependent manner. TRAIL receptor (TRAILR) inhibits TLR signaling by stabilizing IκBα, thereby restricting the nuclear translocation of NF-κB (Liew et al. 2005; Mansell et al. 2006).

Negative regulation of TLR signaling. Binding of TLRs with their cognate ligands induces the inflammatory response through the activation of NF-κB. Sustained activation of TLRs can cause the development of inflammatory diseases. Some of the negative regulators for TLR signaling are shown in the figure. The transmembrane proteins ST2L and SIGIRR act as negative regulators of TIRAP/MyD88 and IRAK proteins, respectively. IRAK-M prevents the dissociation of IRAKs from MyD88, restricting further signaling. A20, β-arrestins, and SOCS3 regulators block the downstream signaling of TRAF6 and TRAF3 by inhibiting their ubiquitination. TRAILR blocks NF-κB activation by stabilizing IκB and preventing its degradation. TAG protein displaces TRIF from the TRAM adaptor protein, while SARM interacts directly with TRIF and blocks subsequent signaling. The regulator proteins SOCS3 and DUBA limit TRAF3 ubiquitination, whereas Pin1 and RAUL speed up the ubiquitination and degradation of IRF3/7
Additionally, several intracellular proteins have been identified as negative regulators of TLR signaling. For instance, the overexpression of IRAK-M, a member of the IRAK family, prevents the dissociation of IRAK4 and IRAK1 from MyD88, thereby blocking the MyD88-dependent pathway (Kobayashi et al. 2002). Following the binding of LPS to TLR4, β-arrestins interact with TRAF6 to hinder its oligomerization, resulting in the inhibition of TRAF6 polyubiquitination and subsequent activation of NF-κB and MAPKs (Wang et al. 2006; Kawai and Akira 2007). Furthermore, the inducible deubiquitination enzyme A20 is capable of removing ubiquitin moieties from TRAF6 in order to end TLR signaling (Boone et al. 2004). Meanwhile, TRAM adaptor with GOLD domain (TAG) removes the adaptor protein TRIF from TRAM and prevents the TRIF-dependent pathway (Palsson-Mcdermott et al. 2009; Han et al. 2010). Sterile α- and armadillo-motif-containing protein (SARM) is a TLR adaptor protein that can interfere with TRIF-dependent gene expression by directly interacting with TRIF (Baral and Utaisincharoen 2013). SOCS3 (Rothlin et al. 2007) and deubiquitinating enzyme A (DUBA) act as negative regulator by controlling the ubiquitination process of TRAF3 (Kayagaki et al. 2007). Phosphorylated IRF3 will bind with peptidyl-prolyl cis–trans isomerase NIMA-interacting 1 (Pin1) for polyubiquitination resulting in its degradation. Although the mechanism underlying this reaction is unclear, experiments have proved that the reduced expression of Pin1 enhanced IRF-3-dependent production of INF-β (Saitoh et al. 2006). Replication and transcription activator-associated ubiquitin ligase (RAUL), a ubiquitin E3 ligase, catalyzes lysine 48-linked polyubiquitination of IRF7 and IRF3 followed by proteasome-dependent degradation (Yu and Hayward 2010).
TLRs in inflammatory diseases
TLRs act as a double-edged sword: deficient TLR signaling might render the organism vulnerable to exposure to pathogenic attack, while an excessive TLR response results in uncontrolled release of a range of pro-inflammatory cytokines and chemokines, which might result in the emergence of inflammatory disease. Available evidence for the involvement of TLRs in the pathogenesis of a certain set of diseases will be discussed in the following.
TLRs in rheumatoid arthritis (RA)
Rheumatoid arthritis (RA) is an autoimmune, chronic inflammatory joint disease characterized by hyperactivation of synovial fibroblasts, which leads to synovitis. The pathogenesis starts with angiogenesis during which lymphocytes and macrophages find their way to the joint cavity causing the expansion of the synovial tissue into a pannus (Davis et al. 2015). The hyperplastic synovium invades and destroys both cartilage and bone via proteolytic cleavage of aggrecan and collagen. At this level, synovial fibroblasts secrete pro-inflammatory cytokines and matrix-degrading effectors providing a perfect storm for chronic inflammation (Mcinnes and O’dell 2010; Shotorbani et al. 2011). Although the etiology of this abnormal activation of synovial cells remains unidentified, multiple studies have pointed out a key role for TLRs in the development of this disease.
A study analyzing blood cells obtained from RA patients reported that the peripheral blood monocytes of RA patients present increased surface expression of TLR2 and TLR4 (Iwahashi et al. 2004; Sorensen et al. 2008). Multiple studies reported an abnormal increase in the expression of TLRs 3, 7, and 9, together with TLR2 and TLR4 in RA synovial tissues (Goh and Midwood 2012; Bhinder et al. 2014). Particularly, TLR3 and 4 were shown to be highly expressed in both early and long-standing RA, emphasizing their potential involvement in the pathogenesis and persistence of the disease (Radstake et al. 2004; Roelofs et al. 2005; Ospelt et al. 2008; Goh and Midwood 2012). Furthermore, an increasing body of data indicates the presence of various endogenous TLR ligands in inflammatory synovial fluid obtained from patients with RA. Among those ligands, fibrinogen, hyaluronan, heat shock protein B8 (HSP22), and high-mobility group protein B1 (HMGB1) were the most abundant (Taniguchi et al. 2003; Roelofs et al. 2006; Huang et al. 2009; Goh and Midwood 2012). HSP22, along with a fragment of hyaluronic acid, was recognized as a potential endogenous TLR4 ligand responsible for the activation of the myeloid dendritic cells (Termeer et al. 2002; Roelofs et al. 2006). Further, HMGB1, a nuclear protein that stabilizes nucleosome formation, is released after cell damage and induces NF-κB activation following its binding to TLR2 and 4 (Park et al. 2004; He et al. 2012).
To gain an in-depth understanding of the role played by TLRs during RA, various animal models have been established. In a streptococcal cell wall (SCW)-induced arthritis model, a single injection of SCW into murine joints induced joint inflammation mediated by TLR2 and MyD88 (Joosten et al. 2003; Abdollahi-Roodsaz et al. 2008). Repeated injections of SCW led to chronic, destructive arthritis, which was reported to be mediated by TLR4 (Abdollahi-Roodsaz et al. 2008). These results justify in part the involvement of endogenous activation of TLR4 in the emergence of cartilage erosion during the late phases of the disease. Supporting the same findings, inhibition of TLR4 in a collagen-induced arthritis (CIA) model suppressed arthritis clinically and histologically, while the addition of LPS enhanced its severity (Divanovic et al. 2005; Pierer et al. 2011). While most studies examined the implication of cell-surface TLRs in the pathogenesis of RA, only few have focused on endosomal TLRs and their role in RA. It is worth mentioning that the findings reported are seemingly contradictory. In a CIA mouse model, the activation of TLR3 suppressed arthritis and reduced the inflammation (Yarilina et al. 2007). However, stimulation of TLR3 in vitro induced angiogenic and osteoclastogenic factors in human RA synovial fibroblasts (Moon et al. 2010). Accordingly, knockdown of TLR3 in a rat model with pristine-induced arthritis (PIA) resulted in disease improvement (Meng et al. 2010). Similarly, data on the role of TLR9 in RA are contradictory. In some studies, direct injection of CpG DNA motifs into the joints of healthy mice caused the development of mild arthritis (Batsford et al. 2011). In agreement with these findings, the inhibition of TLR9 in PIA rat alleviated the disease (Herman et al. 2011). However, systemic administration of CpG into mice induced an anti-inflammatory response and reined the arthritis (Wu et al. 2007; Thwaites et al. 2014). On the other hand, TLR7 activation has been suggested to contribute to CIA pathology and the worsening of the inflammatory state during the disease (Alzabin et al. 2012).
Overall, the data available for TLR4 and 2 strongly support their role in the pathogenesis of RA. Yet, more straightforward data concerning the role of endogenous TLRs in experimental models of RA are needed.
TLRs in inflammatory bowel disease (IBD)
Crohn’s disease (CD) and ulcerative colitis (UC) are chronic inflammatory disorders of the gastrointestinal tract often referred to as IBD. Ample evidence supports the role of innate and adaptive immune responses in the onset of IBD (Geremia et al. 2014). The disease is characterized by an imbalance between pro-inflammatory and regulatory-T-cell (Treg) responses (Frosali et al. 2015). Although the precise etiology of IBD remains unknown, the inflammation observed during its pathogenesis is assumed to result from inappropriate activation of mucosal immunity by environmental factors in genetically susceptible individuals (Xavier and Podolsky 2007). TLRs, together with other PRRs, are expressed by intestinal epithelial cells. They play a crucial role in driving a basal immune response indispensable for the protection of host-barrier integrity (Yesudhas et al. 2014; Frosali et al. 2015).
In healthy conditions, TLRs uphold a perpetual intestinal homeostasis between the tolerance of commensal microflora and the recognition of pathogens. Normally, the TLR response to commensal bacteria triggers the secretion of protective factors such as transforming growth factor β (TGFβ), defensin, keratinocyte growth factor, and cyclooxygenase-2, which help to maintain the proliferation as well as differentiation of intestinal epithelial cells (Uehara et al. 2007). However, in susceptible individuals, alteration of TLR signaling may lead to failure of commensal-mucosal homoeostasis maintenance, facilitating tissue injury and resulting in the development of gut inflammation (Franchimont et al. 2004; Frosali et al. 2015). In the gut, the type of response induced by TLR activation and the subsequent secretion of pro/anti-inflammatory response are mainly influenced by the intestinal immunological niche and local homeostasis. The expression of TLRs is regulated depending on their location and the milieu they are found in. For instance, upon heterodimerization with TLR6, TLR2 induces the secretion of IL-10 by dendritic cells and promotes the activation of Tregs (Depaolo et al. 2008). On the other hand, TLR2 associated to TLR1 induces a shift toward IL-12 or IL-17 promoting the differentiation of T helper 1 (TH1) and T helper 17 (TH17) cells, respectively (Watanabe et al. 2004; Depaolo et al. 2012).
An in vitro study using intestinal epithelial cells from patients with Crohn’s disease revealed that stimulation of TLR2 led to excessive secretion of inflammatory and TH1-related cytokines (Watanabe et al. 2004). Similar findings were reported in a mouse model of colitis, in which the expression levels of both TLR2 and TLR4 were considerably increased, and the activation of both receptors caused aggravation of the disease (Heimesaat et al. 2007). Furthermore, data on the role of TLR4 in IBD are controversial: while some studies pled for a protective role of TLR4 activation in IBD, others found its activation to worsen pathogenesis. In this regard, TLR4 expression was elevated in patients suffering from IBD and the polymorphisms Asp299Gly and Thr399Ile were highly related to the onset of disease (Franchimont et al. 2004; Senhaji et al. 2014). Furthermore, different studies have reported that in the presence of inflammatory cytokines such as IFN-γ and TNF-α, TLR4 expression in intestinal epithelial cells was upregulated, thereby worsening the inflammation and tissue damage (Suzuki et al. 2003; Frolova et al. 2008; Tan et al. 2014). In a chimeric mouse model, it has been reported that TLR4 signaling in colonic epithelial cells worsened intestinal inflammation (Ungaro et al. 2009). However, TLR4 signaling was shown to protect against epithelial disruption as it promotes the induction of chemokines and cytokines as well as the recruitment of adaptive immune cells needed to limit pathogen invasion (Furuta et al. 2006). In a murine model of acute colitis, the absence of TLR4 during injury caused severe mucosal damage along with impaired epithelial proliferation and a reduced inflammatory response (Fukata et al. 2005). A study in a MyD88−/− mouse model of Typhimurium-induced gastroenteritis colitis reported similar findings. The MyD88−/− mice presented accelerated tissue damage with decreased induction of inflammatory cytokine TNF-α transcription (Bhinder et al. 2014). Furthermore, the protective roles of TLR9 and TLR3 signaling in experimental colitis have been reported (Vijay-Kumar et al. 2007; Rose et al. 2012). The administration of CpG in a murine model of colitis reduced the secretion of pro-inflammatory cytokines and apoptosis, which reduced disease severity (Rose et al. 2012).
Overall, the data available show that the role of TLR activation during IBD differs depending on a variety of factors such as intestinal microenvironment, the localization of TLRs in the gastrointestinal tract, and genetic predisposition for bowel diseases.
TLRs in psoriasis
The skin, like the gastrointestinal tract, is constantly challenged by a massive amount of bacteria. Psoriasis is a long-lasting, relapsing inflammatory disease of the skin identified by epidermal hyperproliferation and infiltration of inflammatory cells throughout the dermis and epidermis causing severe skin lesions (Flutter and Nestle 2013). Physiologically, TLRs are expressed by three epidermal cells: keratinocytes, melanocytes, and Langerhans cells. Keratinocytes are reported to express all TLRs except TLR7 and TLR8, and their activation upon exposure to TLR ligands results mainly in the production of IL-8, nitric oxide synthase, chemokines and matrix metalloproteases (MMPs) 2 and 9, all needed for the inflammatory response and remodeling of the damaged tissue (Mcinturff et al. 2005; Lee et al. 2010). Human melanocytes express TLRs 2, 3, 4, 7, and 9. Upon stimulation by ligands, melanocytes secrete IL-6 and IL-8, increase chemokine mRNA production, and upregulate the phosphorylation of IκBα, promoting the activation of NF-κB (Yu et al. 2009). Langerhans cells play the role of cutaneous APCs and have been reported to express high levels of TLRs 2, 3, 4, 8, and 10 and low levels of TLRs 1, 5, 6, 7, and 9 (Renn et al. 2006; Hari et al. 2010). Additionally, Langerhans cells play an important role in the anti-viral immune response owing to their ability to induce the secretion of IFN-α and IL-18 and subsequent initiation of a TH1 response (Lebre et al. 2003; Renn et al. 2006).
In the skin of patients presenting psoriasis, increased T cell proliferation, TH1 and TH17 cytokine secretion, antigen presentation, and high production of type I IFNs have been reported (Gilliet et al. 2004). Skin deletion observed during psoriasis stimulates keratinocytes to release antimicrobial peptide, which forms aggregates with extracellular self-DNA, enters plasmacytoid dendritic cells, and activates TLR9 and therefore, sustains Type I IFN secretion, myeloid dendritic cell maturation, and subsequent activation of autoreactive T cells (Farkas et al. 2008). Moreover, several studies have reported that in pathological conditions, keratinocytes heavily express TGF-α and heat shock proteins, which upregulate the expression of TLRs 2, 4, 5, and 9, resulting in excessive activation of NF-κB, stimulation of inflammatory cytokines, antigen presentation to autoreactive T cells, and therefore, disease aggravation (Miller et al. 2005; Seung et al. 2007). Similar findings were reported in an immunohistochemistry-based study of psoriatic skin, where the expression levels of both TLR2 and TLR4 were elevated in dermal and epidermal dendritic cells and in keratinocytes when compared to normal skin (Curry et al. 2003).
Interestingly, the study of psoriasis has been limited by the lack of informative yet easy to utilize animal models. Despite the use of transgenic models in numerous studies, these models failed to mimic the cellular mechanism of the disease, and in some cases were unable to present drug-specific responses that are known to occur during psoriasis, thus limiting their usability (Nestle et al. 2009). In the past few years, an imiquimod-induced model of psoriasis has been widely used as an alternative. This model provided a better understanding of the development of skin inflammatory responses (Van Der Fits et al. 2009; Walter et al. 2013). Using this model, the activation of TLR7 and TLR8 signaling was shown to be involved in the exacerbation of plaque psoriasis as imiquimod (a TLR7/8-specific agonist) was responsible for the aggravation of the pathological symptoms (Gaspari 2006; Walter et al. 2013).
TLRs in asthma
One of the most common diseases linked with TLR-induced cytokine is asthma. This disease is identified by the dominant presence of mast cells, eosinophils, and CD4+ T cells in the airways (Zuo et al. 2015). A study aimed at identifying the sites of action of TLRs reported that all tissues expressed at least one TLR, confirming their expression in the lungs (Zarember and Godowski 2002; Tirumurugaan et al. 2010). As the respiratory tract is continuously exposed to environmental elements, a defect in the function of airway dendritic cells and airway epithelial cells (AECs) in response to allergens or viral infection might lead to chronic respiratory disease (Bezemer et al. 2012). House dust mite (HDM) antigen from Dermatophagoides farinae and fungal protease from Aspergillus oryzae were reported to activate TLR4 signaling in AECs, thereby causing allergic responses (Trompette et al. 2009; Millien et al. 2013; Holtzman et al. 2014). In accordance with these findings, suppression of TLR4 activation was shown to be involved in the immunological mechanism of asthma by controlling the anti-inflammatory cytokine IL-10 and restricting the release of TH1-related cytokine IL-1 (Lun et al. 2009; Bezemer et al. 2012).
TLR3 activation by double-stranded RNA from the respiratory syncytial virus (RSV), human rhinovirus (HRV), and influenza A viruses was reported to play a major role in the pathogenesis of severe asthma (Holtzman et al. 2014). Thymic stromal lymphopoietin proteins along with TLR3 ligands boost the differentiation of TH17 by activating human dendritic cells (Shannon et al. 2008; Tanaka et al. 2009). TH17 cells belong to the group of CD4 T cells, which take part in the production of inflammatory cytokines IL-5, 17, and 25. These cytokines are involved in pathogenic process of asthma; especially, IL-5, which is the main cytokine involved in the differentiation and survival of eosinophils (Tesmer et al. 2008; Wakashin et al. 2008; Cosmi et al. 2011; Bezemer et al. 2012). Other TLRs such as TLR7, TLR8, and TLR9 are induced by virus-associated nucleic acids, and they play a role in disease pathogenesis (Zarember and Godowski 2002; Shannon et al. 2008). The effects of single nucleotide polymorphisms (SNPs) have been mainly studied for TLR1-10 and the evaluation of SNPs in the genes of TLRs 1, 6, and 10 emphasized the protective effect of these receptors in childhood asthma patients; they lowered the risk for atopic asthma by almost half (Kormann et al. 2008). Ser249Pro in the extracellular domain of TLR6 showed significant positive results in a case–control study. In contrast, other work revealed that Ser249Pro is associated with childhood asthma (Tantisira et al. 2004; Hoffjan et al. 2005).
In a recent study, curcumin was identified as an effective drug for ovalbumin (OVA)-induced allergic inflammation in a mouse model of allergic asthma. Curcumin efficiently suppressed IL-17A and improved IL-10, thereby inhibiting eosinophil recruitment and mucus overproduction (Ma et al. 2013). Another study revealed that an underacylated form of Rhodobacter sphaeroides LPS (a TLR4 antagonist) acts as an inhibitor of HDM and strongly reduces airway lymphocytosis and eosinophilia in mice (Hammad et al. 2009).
TLRs in Alzheimer’s disease (AD)
TLR-induced inflammation is associated with various neurodegenerative diseases, including AD. AD is characterized by the accumulation of amyloid fibrous protein in the cerebrum (plaques) and the formation of tangles of Tau protein in the neurons (Heneka et al. 2015). Neuro-inflammation may not be the starting point of AD; nonetheless, disease progression always depends on it. Sustained inflammatory responses involving both microglia and astrocytes contribute to disease progression (Glass et al. 2010). Microglia are macrophages that act as “warriors” of the immune system and are located in both the human brain and spinal cord (Streit 2002). The major functions of microglia are surveying of the microenvironment and maintaining homeostasis. Activated microglia interact with astrocytes (glial cells) and neurons to fight pathogen invasion and promote the inflammatory response (Landreth and Reed-Geaghan 2009). Nonetheless, a sustained inflammatory response might alarm cell surface receptors, triggering a continuous activation of TLRs.
TLR4 present on the surface of microglia is recognized as the major sensor of danger signals and its activation results in the secretion of TNF-α and IL-1β. Both cytokines stimulate astrocyte activation triggering secondary inflammatory responses (Saijo et al. 2009). The inflammatory reaction in AD is governed mainly by the interaction between microglia and cell surface receptors (SCARA1, CD36, CD14, α6β1 integrin, and CD47), including TLRs (2, 4, 6, and 9) (Landreth and Reed-Geaghan 2009). The binding of microglia soluble Aβ fibrils through CD36, TLR4, or TLR6, causes the secretion of a range of immunostimulatory cytokines and chemokines. In a mouse model of AD, disruption of the complex CD36-TLR4 and TLR6 through the targeting of CD36 resulted in low production of nitric oxide, reactive oxygen species, and IL-1β by microglia, thereby protecting the neurons from amyloid beta (Aβ)-induced cell death (Walter et al. 2007; Jin et al. 2008; Stewart et al. 2010). TLR2 and TLR4 mediate cell death not only in immune cells (Lehnardt et al. 2007) but also in Aβ-affected neurons, suggesting that TLRs act as death receptors during AD (Tang et al. 2008).
TLRs have been detected in neurons as well as in the central nervous system (Prehaud et al. 2005). The microRNA let-7 activates TLR7 and induces neurodegeneration. Cerebrospinal fluid from AD patients presented increased amounts of let-7b (Diebold et al. 2004; Lehmann et al. 2012). Extracellular induction of let-7b into cerebrospinal fluid showed that wild-type mice lacking TLR7 receptors were resistant to neurodegeneration (Lehmann et al. 2012). However, the exact mechanism of TLRs in neurodegenerative disease remains partially unclear.
TLRs in systemic lupus erythematosus (SLE)
SLE is a complex auto-inflammatory disease characterized by the loss of tolerance to self-nuclear antigens along with continuous and repetitive stimulation of innate and adaptive immune responses by endogenous nucleic acids released upon cell death by apoptosis or necrosis (Clancy et al. 2016).
Recently, numerous studies have focused on the involvement of activation of endosomal TLRs in the onset of SLE (Clancy et al. 2016; Celhar and Fairhurst 2014). Endosomal TLRs expressed in B cells and plasmacytoid dendritic cells, are suggested to be responsible for the generation of anti-nuclear antibodies with the production of type I IFNs (Clancy et al. 2016). The roles of both TLR7 and TLR9 in SLE were studied in a lupus-prone mouse model wherein the overexpression of TLR7 is suggested to be linked to the disease onset (Guiducci et al. 2010). Additionally, overexpression of TLR7 in mouse models was reported to be correlated with the production of RNA-associated antibodies, an increase in autoreactive B cell response, and the development of auto-inflammatory phenotypes by mice (Nundel et al. 2015). Furthermore, mouse models lacking TLR7 displayed amelioration of disease symptoms with a significant reduction in levels of circulating autoantibodies and a decrease in IL-6 and INF-α secretion (Lee et al. 2008).
While the pathological role of TLR7 in SLE is relatively known, the role of TLR9 remains controversial. In vivo studies on mice have reported the important role of TLR9 in the production of autoantibodies by B cells (Nundel et al. 2015). Moreover, B cells and monocytes from patients with active disease showed higher TLR9 expression compared to those from patients with inactive disease (Lamphier et al. 2014). However, deletion of TLR9 in lupus-prone mouse models did not ameliorate disease symptoms but contrastingly exacerbated the disease, suggesting a protective role for TLR9 in SLE (Nickerson et al. 2010).
In addition, TLR2 and TLR4 were shown to be related to SLE pathogenesis. Recently, it has been reported that the expression levels of TLR2 and TLR4 mRNA in peripheral blood mononuclear cells (PBMCs) of patients with SLE were much higher than those in PBMCs of healthy subjects (Lee et al. 2016). In a lupus mouse model, the deficiency of TLR4 and/or TLR2 limited the production of autoantibodies and therefore attenuated the development of SLE symptoms (Chen et al. 2016). Similarly, the TLR4−/− mouse model showed a decrease in the secretion of inflammatory cytokines and anti-dsDNA and anti-RNP antibodies, resulting in the amelioration of lupus symptoms in a pristane-induced experimental lupus model (Fagone et al. 2014).
TLRs in other inflammatory diseases
Several studies have focused on the role of TLRs in atherosclerosis, suggesting the association of the D299G polymorphism in human TLR4 with a reduced risk for carotid artery atherosclerosis (Cooke et al. 2002; Lu et al. 2015). These studies showed that patients with the D299G polymorphism presented lower concentrations of circulating proinflammatory cytokines such as IL-6, soluble vascular cell adhesion molecule 1, and fibrinogen. Other genotypic studies have reported that the Agr753Gln single-nucleotide polymorphism in human TLR2 was also a risk factor for SLE (Balistreri et al. 2008; Bielinski et al. 2011). In atherosclerosis mouse models, knockout of MyD88 resulted in significantly decreased plaque size and a lower expression of proinflammatory cytokines and chemokines, thus ameliorating disease symptoms in mice (Subramanian et al. 2013).
Another disease wherein TLRs seem to be involved at the onset is multiple sclerosis (MS). MS is a chronic debilitating disease of the central nervous system (CNS), characterized by inflammation and subsequent immune responses, which result in progressive demyelination and axonal/neuronal injury (Derkow et al. 2013). The expression of TLRs was reported to be increased in active MS lesions. In order to study the disease in depth, murine experimental autoimmune encephalitis (EAE) was used as an experimental model for human MS in several studies. An in vivo study reported that MyD88−/− mice were completely EAE resistant, whereas TLR9−/− mice developed the disease with significantly reduced severity. Therefore, both TLR9 and MyD88 are known to be required for EAE induction and progression (Miranda-Hernandez et al. 2011).
TLRs have also been associated with the pathogenesis of sepsis. The D299G polymorphism in human TLR4 has been linked with increased risk of Gram-negative infections and systemic inflammatory syndrome. In addition, the R753Q polymorphism in human TLR2 has been associated with a decreased response to bacterial peptides, resulting in increased susceptibility to staphylococcal infections (Montes et al. 2006; Xiong et al. 2012).
Therapeutic targeting of TLRs during inflammatory disease
TLR signaling seems to be involved in the development of numerous human diseases. This perspective has been strengthened by a wealth of data obtained from in vitro and in vivo studies which support the roles of particular TLRs in disease initiation and/or progression (Perkins and Vogel 2016). Activation of TLRs in inflammatory diseases underpins their pathophysiology via aberrant secretion of pro-inflammatory cytokines and chemokines, which in turn creates an inflammatory feedback loop. Breakage of this feedback loop should dampen the inflammation and re-establish an appropriate immune response to pathogens. Thus, the development of TLR inhibitors might hold promise for a powerful therapeutic strategy (Basith et al. 2011). In general, strategies utilized to downmodulate the overactivation of TLRs involve the use of antagonists that block the binding of ligands or protein–ligand complexes to the receptors, or antagonists that interfere with the intracellular adaptor molecules of the common signaling pathways, neutralizing their effects and blocking the activation of TLR signaling cascade. Given their pivotal role in orchestrating the immune response, the main challenge for the development of TLR-blocking agents is to reduce the excessive inflammation without affecting innate immunity. The antagonists developed to date comprise small molecules, oligonucleotides, peptides, proteins, and antibodies. Some of the TLRs inhibitors tested for their efficacies in some inflammatory diseases are summarized in Table 2 (Li et al. 2011; Kandimalla et al. 2013; Ramani et al. 2013).
Antagonizing TLR2
Hyperactivation of TLR2 is involved in various inflammatory diseases including chronic inflammatory joint disease and Gram-positive sepsis (Keogh and Parker 2011), rendering it a favorable target for the control of inflammation. In a recent study, Mistry et al. identified a small molecule C16H15NO4, designated “C29”, as a potential TLR2-specific inhibitor. The molecule acts specifically by blocking the dimerization of the TLR and TIR domains, disrupting TLR2 activation, thus repressing TLR2-mediated inflammation. Human embryonic kidney 293-TLR2 (HEK293-TLR2) and human acute monocytic leukemia (THP-1) cells showed decreased production of IL-8 mRNA upon treatment with C29. These findings were confirmed in vivo, as the treatment of mice with C29 blocked the TLR2-induced IL-12 p40 and TNF-α liver cytokine mRNA and serum protein (Mistry et al. 2015). Another study showed that the inhibition of TLR2 dimerization by using a TLR2 transmembrane peptide (TLR2-p) decreased the levels of both extracellular signal-regulated kinases (ERK) and pro-inflammatory cytokines. Moreover, it drastically ameliorated dextran sulfate sodium-induced colitis by interfering specifically with the inflammatory cytokine Ly6C+, blocking its aggregation without affecting recruitment to the colon. This study suggested that TLR2-p might have therapeutic potential for acute gut inflammation (Shmuel-Galia et al. 2016).
Furthermore, blocking of TLRs with neutralizing antibodies seems to be a promising therapeutic route. In this regard, OPN-305 (Arslan et al. 2008), a humanized TLR2-specific antibody that blocks the production of inflammatory cytokines mediated by TLR2 (Arslan et al. 2012). OPN-305 is now undergoing phase-II of clinical trial (NCT01794663) to evaluate its effect and safety in delayed graft function.
Antagonizing TLR4
TLR4 overactivation has been reported to play a potent role in sepsis, RA, psoriasis, asthma, etc. (Murad 2014). Hence, inhibiting TLR4 signaling may prevent the onset of these diseases. The most well-known antagonist is Eisai’s Eritoran (E5564) derived from R. sphaeroides. It is a synthetic lipid A analog that blocks the biding of LPS to the MD2 pocket, thus preventing the activation of TLR4 (Mullarkey et al. 2003; Gearing 2007; Shirey et al. 2013). E5564 reached phase-III clinical trials (NCT00334828); however, the study was suspended as this molecule failed to protect against severe sepsis and to improve septic conditions in patients (Opal et al. 2013).
Furthermore, Ramani et al. (2013) identified a peptide referred to as SPA4, which interacts with the interface between surfactant protein-A and TLR4 and thus blocks the subsequent activation of TLR4 signaling. Cell-based assays showed reduced NF-κB activity and suppressed TNF-α secretion after treatment with SPA4. Moreover, SPA4 inhibited LPS-induced inflammation and alleviated the endotoxic shock-like symptoms in a mouse model, suggesting that the anti-inflammatory activity of this peptide might have a therapeutic benefit on TLR4-mediated inflammatory diseases (Ramani et al. 2013).
TAK-242 (Resatorvid) is a small-molecule-specific inhibitor of TLR4 signaling. It inhibits the production of LPS-induced inflammatory mediators by binding to Cys747 in the intracellular TIR domain of TLR4 and by blocking the protein–protein interactions between TLR4 and its adaptor molecules (Matsunaga et al. 2011). TAK-242 showed promising results in vivo, such as reducing inflammation and relieving the endotoxic shock-like symptoms in mice, thus protecting them from endotoxic death (Yamada et al. 2005). However, despite its exceptional inhibitory properties in vitro and in vivo, TAK-242 failed to improve symptoms of sepsis in phase III clinical trials (NCT00633477).
In addition, NI-0101 (NCT01808469), a humanized monoclonal anti-TLR4 antibody developed by Novimmune, showed therapeutic potential for RA. In a synovial explant culture model, NI-0101 inhibited TNF-α and IL-6 secretion and it halted disease progression in a murine model of inflammatory arthritis (Elson et al. 2011). NI-0101 showed effective inhibition of LPS-induced cytokine release without raising safety or tolerability concerns in a phase-I clinical trial (Elson et al. 2011). In a mouse model of sepsis, the use of the monoclonal antibody 1A6 to target the complex TLR4–MD2 protected against disease (Spiller et al. 2008). The antibody hindered the progression of dextran sulfate sodium-induced colitis and reduced the inflammatory response. Nonetheless, 1A6 caused an impairment of the mucosal healing process (Ungaro et al. 2009).
Antagonizing TLRs 7, 8, and 9
Multiple studies have emphasized the important role of endosomal TLRs in the anti-viral response. However, prolonged activation of these TLRs may render the system prone to the development of detrimental disorder. In this regard, blockade of endosomal TLRs can present a promising strategy for the treatment of several autoimmune and inflammatory diseases (Anwar et al. 2013; Thwaites et al. 2014).
IMO-3100 (NCT01622348) antagonizes TLR7 and TLR9 and is a lead drug candidate in development by Idera for the treatment of autoimmune and inflammatory diseases. The preclinical studies suggested that IMO-3100 inhibits the secretion of TLR7/9-induced pro-inflammatory cytokines (Suarez-Farinas et al. 2013). The results of phase-II clinical trials showed that treatment with IMO-3100 for 4 weeks, improved the clinical symptoms of psoriasis. Furthermore, Sioud et al. (2010) reported that 2′-methyl-modified RNA significantly reduced TLR7-mediated secretion of IFN-α and IL-6 in murine dendritic cells and human peripheral blood mononuclear cells, as well as in animal models, indicating it potential utility as an inhibitor of pathogenic inflammatory reactions mediated by TLR7 activation.
CpG-ODN c41 from the Pseudomonas aeruginosa genome was able to inhibit the immunostimulatory activity induced by TLR9 in murine 264.7 cells and in human monocytes, thereby preventing the TLR9-mediated inflammatory response. Additionally, CpG-ODN c41-mediated protection was able to reduce the lethal response in vivo. These findings suggest that this compound could be used as a novel therapeutic agent for CpG-ODN-mediated inflammatory diseases (Li et al. 2011).
IMO-8400 is a synthetic DNA compound that inhibits TLR7-, TLR8-, and TLR9-mediated immune responses. When tested in cell-based assays and in vivo, this compound showed noticeable inhibitory properties. IMO-8400 was evaluated in preclinical mouse models of psoriasis. Mice treated with this molecule presented a reduction in epidermal hyperplasia and leukocyte infiltration along with reduced inflammatory cytokine secretion, resulting in suppression of lesion development in psoriasis mouse models (Jiang et al. 2012). IMO-8400 is currently undergoing phase II clinical trials to determine its safety and efficacy in patients with plaque psoriasis (NCT01899729) and dermatomyositis (NCT02612857).
The synthetic oligoribonucleotide (ODN) antagonists of TLRs 7, 8, and 9 were tested in murine and human cell-based assays, demonstrating inhibitory properties with reduced secretion of a broad range of inflammatory cytokines and chemokines. The same results were also confirmed in vivo in mice and monkeys (Kandimalla et al. 2013).
Conclusion
Because of their ability to recognize both microbial products and endogenous molecules released from injured tissues, TLRs play a critical role linking both innate and adaptive immune responses and protecting the host against invading pathogens and endogenous danger signals. With the compendium of information present to date, the involvement of TLRs in the development of inflammatory diseases continues to be unveiled, which highlights that overactivation of these receptors causes disruption of homeostasis, increasing the risk for several diseases. Thus, TLR-driven responses need to be tightly regulated to prevent any detrimental effect from their aberrant activation. Although preclinical studies reported promising inhibitory effects of some antagonistic ligands on TLRs signaling, the clinical success is still limited. The direct targeting of TLRs can be avoided by targeting a combination of downstream signaling adaptors utilized by these receptors to reach the anticipated therapeutic goals. Of note, the numerous signaling pathways involved in innate immunity as well as the complexity of their interactions pose a great challenge to the development of negative or positive regulators. Thus, additional studies are required to provide new insights into therapeutic targeting of TLRs not only in inflammatory diseases but also in diseases related to immune disorders.
References
Abdollahi-Roodsaz S, Joosten LA, Helsen MM, Walgreen B, Van Lent PL, Van Den Bersselaar LA, Koenders MI, Van Den Berg WB (2008) Shift from toll-like receptor 2 (TLR-2) toward TLR-4 dependency in the erosive stage of chronic streptococcal cell wall arthritis coincident with TLR-4-mediated interleukin-17 production. Arthritis Rheum 58:3753–3764
Akira S, Hoshino K (2003) Myeloid differentiation factor 88-dependent and -independent pathways in toll-like receptor signaling. J Infect Dis 187(Suppl 2):S356–S363
Alzabin S, Kong P, Medghalchi M, Palfreeman A, Williams R, Sacre S (2012) Investigation of the role of endosomal toll-like receptors in murine collagen-induced arthritis reveals a potential role for TLR7 in disease maintenance. Arthritis Res Ther 14:R142
Anwar MA, Basith S, Choi S (2013) Negative regulatory approaches to the attenuation of toll-like receptor signaling. Exp Mol Med 45:e11
Arslan F, De Kleijn DP, Timmers L, Doevendans PA, Pasterkamp G (2008) Bridging innate immunity and myocardial ischemia/reperfusion injury: the search for therapeutic targets. Curr Pharm Des 14:1205–1216
Arslan F, Houtgraaf JH, Keogh B, Kazemi K, De Jong R, Mccormack WJ, O’neill LA, Mcguirk P, Timmers L, Smeets MB, Akeroyd L, Reilly M, Pasterkamp G, De Kleijn DP (2012) Treatment with OPN-305, a humanized anti-toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv 5:279–287
Balistreri CR, Candore G, Mirabile M, Lio D, Caimi G, Incalcaterra E, Caruso M, Hoffmann E, Caruso C (2008) TLR2 and age-related diseases: potential effects of Arg753Gln and Arg677Trp polymorphisms in acute myocardial infarction. Rejuvenation Res 11:293–296
Baral P, Utaisincharoen P (2013) Sterile-α- and armadillo motif-containing protein inhibits the TRIF-dependent downregulation of signal regulatory protein α to interfere with intracellular bacterial elimination in Burkholderia pseudomallei-infected mouse macrophages. Infect Immun 81:3463–3471
Basith S, Manavalan B, Lee G, Kim SG, Choi S (2011) Toll-like receptor modulators: a patent review (2006–2010). Expert Opin Ther Pat 21:927–944
Batsford S, Dunn J, Mihatsch M (2011) Induction of experimental arthritis by borrelial lipoprotein and CpG motifs: are toll-like receptors 2, 4, 9 or CD-14 involved? Open Rheumatol J 5:18–23
Bezemer GF, Sagar S, Van Bergenhenegouwen J, Georgiou NA, Garssen J, Kraneveld AD, Folkerts G (2012) Dual role of toll-like receptors in asthma and chronic obstructive pulmonary disease. Pharmacol Rev 64:337–358
Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, Ma C, Jacobson K, Vallance BA (2014) Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun 82:3753–3763
Bianchi ME (2007) DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol 81:1–5
Bielinski SJ, Hall JL, Pankow JS, Boerwinkle E, Matijevic-Aleksic N, He M, Chambless L, Folsom AR (2011) Genetic variants in TLR2 and TLR4 are associated with markers of monocyte activation: the atherosclerosis risk in communities MRI study. Hum Genet 129:655–662
Blasius AL, Beutler B (2010) Intracellular toll-like receptors. Immunity 32:305–315
Boone DL, Turer EE, Lee EG, Ahmad RC, Wheeler MT, Tsui C, Hurley P, Chien M, Chai S, Hitotsumatsu O, Mcnally E, Pickart C, Ma A (2004) The ubiquitin-modifying enzyme A20 is required for termination of toll-like receptor responses. Nat Immunol 5:1052–1060
Brint EK, Xu D, Liu H, Dunne A, Mckenzie AN, O’neill LA, Liew FY (2004) ST2 is an inhibitor of interleukin 1 receptor and toll-like receptor 4 signaling and maintains endotoxin tolerance. Nat Immunol 5:373–379
Brown J, Wang H, Hajishengallis GN, Martin M (2011) TLR-signaling networks: an integration of adaptor molecules, kinases, and cross-talk. J Dent Res 90:417–427
Celhar T, Fairhurst AM (2014) Toll-like receptors in systemic lupus erythematosus: potential for personalized treatment. Front Pharmacol 5:265
Chen JQ, Szodoray P, Zeher M (2016) Toll-like receptor pathways in autoimmune diseases. Clin Rev Allergy Immunol 50:1–17
Cighetti R, Ciaramelli C, Sestito SE, Zanoni I, Kubik L, Arda-Freire A, Calabrese V, Granucci F, Jerala R, Martin-Santamaria S, Jimenez-Barbero J, Peri F (2014) Modulation of CD14 and TLR4.MD-2 activities by a synthetic lipid A mimetic. ChemBioChem 15:250–258
Clancy RM, Markham AJ, Buyon JP (2016) Endosomal toll-like receptors in clinically overt and silent autoimmunity. Immunol Rev 269:76–84
Cook DN, Pisetsky DS, Schwartz DA (2004) Toll-like receptors in the pathogenesis of human disease. Nat Immunol 5:975–979
Cooke GS, Segal S, Hill AV, Tuberculosis G, Environment Study G, Oxford Pneumococcal Surveillance Study G (2002) Toll-like receptor 4 polymorphisms and atherogenesis. N Engl J Med 347:1978–1980
Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F (2011) Th17 cells: new players in asthma pathogenesis. Allergy 66:989–998
Curry JL, Qin JZ, Bonish B, Carrick R, Bacon P, Panella J, Robinson J, Nickoloff BJ (2003) Innate immune-related receptors in normal and psoriatic skin. Arch Pathol Lab Med 127:178–186
Davis ML, Levan TD, Yu F, Sayles H, Sokolove J, Robinson W, Michaud K, Thiele GM, Mikuls TR (2015) Associations of toll-like receptor (TLR)-4 single nucleotide polymorphisms and rheumatoid arthritis disease progression: an observational cohort study. Int Immunopharmacol 24:346–352
De Nardo D (2015) Toll-like receptors: activation, signalling and transcriptional modulation. Cytokine 74:181–189
Depaolo RW, Tang F, Kim I, Han M, Levin N, Ciletti N, Lin A, Anderson D, Schneewind O, Jabri B (2008) Toll-like receptor 6 drives differentiation of tolerogenic dendritic cells and contributes to LcrV-mediated plague pathogenesis. Cell Host Microbe 4:350–361
Depaolo RW, Kamdar K, Khakpour S, Sugiura Y, Wang W, Jabri B (2012) A specific role for TLR1 in protective T(H)17 immunity during mucosal infection. J Exp Med 209:1437–1444
Derkow K, Bauer JM, Hecker M, Paap BK, Thamilarasan M, Koczan D, Schott E, Deuschle K, Bellmann-Strobl J, Paul F, Zettl UK, Ruprecht K, Lehnardt S (2013) Multiple sclerosis: modulation of toll-like receptor (TLR) expression by interferon-beta includes upregulation of TLR7 in plasmacytoid dendritic cells. PLoS One 8:e70626
Diebold SS, Kaisho T, Hemmi H, Akira S, E Sousa CR (2004) Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303:1529–1531
Divanovic S, Trompette A, Atabani SF, Madan R, Golenbock DT, Visintin A, Finberg RW, Tarakhovsky A, Vogel SN, Belkaid Y, Kurt-Jones EA, Karp CL (2005) Negative regulation of toll-like receptor 4 signaling by the toll-like receptor homolog RP105. Nat Immunol 6:571–578
Elson G, Page T, Buatois V, Daubeuf B, Chatel L, Cons L, Lippens C, Salgado-Pires S, Ferlin W, Kosco-Vilbois M, Midwood K, Shang L (2011) NI-0101, a therapeutic TLR 4 monoclonal antibody for rheumatoid arthritis. Arthritis Rheum 63:977
Fagone P, Muthumani K, Mangano K, Magro G, Meroni PL, Kim JJ, Sardesai NY, Weiner DB, Nicoletti F (2014) VGX-1027 modulates genes involved in lipopolysaccharide-induced toll-like receptor 4 activation and in a murine model of systemic lupus erythematosus. Immunology 142:594–602
Farkas A, Tonel G, Nestle FO (2008) Interferon-alpha and viral triggers promote functional maturation of human monocyte-derived dendritic cells. Br J Dermatol 158:921–929
Flutter B, Nestle FO (2013) TLRs to cytokines: mechanistic insights from the imiquimod mouse model of psoriasis. Eur J Immunol 43:3138–3146
Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Deviere J, Rutgeerts P (2004) Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut 53:987–992
Frolova L, Drastich P, Rossmann P, Klimesova K, Tlaskalova-Hogenova H (2008) Expression of toll-like receptor 2 (TLR2), TLR4, and CD14 in biopsy samples of patients with inflammatory bowel diseases: upregulated expression of TLR2 in terminal ileum of patients with ulcerative colitis. J Histochem Cytochem 56:267–274
Frosali S, Pagliari D, Gambassi G, Landolfi R, Pandolfi F, Cianci R (2015) How the intricate interaction among toll-like receptors, microbiota, and intestinal immunity can influence gastrointestinal pathology. J Immunol Res 2015:489821
Fukata M, Michelsen KS, Eri R, Thomas LS, Hu B, Lukasek K, Nast CC, Lechago J, Xu R, Naiki Y, Soliman A, Arditi M, Abreu MT (2005) Toll-like receptor-4 is required for intestinal response to epithelial injury and limiting bacterial translocation in a murine model of acute colitis. Am J Physiol Gastrointest Liver Physiol 288:G1055–G1065
Furuta T, Kikuchi T, Akira S, Watanabe N, Yoshikawa Y (2006) Roles of the small intestine for induction of toll-like receptor 4-mediated innate resistance in naturally acquired murine toxoplasmosis. Int Immunol 18:1655–1662
Gaspari AA (2006) Innate and adaptive immunity and the pathophysiology of psoriasis. J Am Acad Dermatol 54:S67–S80
Gay NJ, Symmons MF, Gangloff M, Bryant CE (2014) Assembly and localization of toll-like receptor signalling complexes. Nat Rev Immunol 14:546–558
Gearing AJ (2007) Targeting toll-like receptors for drug development: a summary of commercial approaches. Immunol Cell Biol 85:490–494
Geremia A, Biancheri P, Allan P, Corazza GR, Di Sabatino A (2014) Innate and adaptive immunity in inflammatory bowel disease. Autoimmun Rev 13:3–10
Gilliet M, Conrad C, Geiges M, Cozzio A, Thurlimann W, Burg G, Nestle FO, Dummer R (2004) Psoriasis triggered by toll-like receptor 7 agonist imiquimod in the presence of dermal plasmacytoid dendritic cell precursors. Arch Dermatol 140:1490–1495
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934
Goh FG, Midwood KS (2012) Intrinsic danger: activation of toll-like receptors in rheumatoid arthritis. Rheumatology 51:7–23
Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, Chan JH, Wright T, Punaro M, Bolland S, Soumelis V, Banchereau J, Coffman RL, Pascual V, Barrat FJ (2010) TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature 465:937–941
Hammad H, Chieppa M, Perros F, Willart MA, Germain RN, Lambrecht BN (2009) House dust mite allergen induces asthma via toll-like receptor 4 triggering of airway structural cells. Nat Med 15:410–416
Han C, Jin J, Xu S, Liu H, Li N, Cao X (2010) Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol 11:734–742
Hari A, Flach TL, Shi Y, Mydlarski PR (2010) Toll-like receptors: role in dermatological disease. Mediat Inflamm 2010:437246
He Z, Shotorbani SS, Jiao Z, Su Z, Tong J, Liu Y, Shen P, Ma J, Gao J, Wang T, Xia S, Shao Q, Wang S, Xu H (2012) HMGB1 promotes the differentiation of Th17 via up-regulating TLR2 and IL-23 of CD14+ monocytes from patients with rheumatoid arthritis. Scand J Immunol 76:483–490
Heimesaat MM, Fischer A, Siegmund B, Kupz A, Niebergall J, Fuchs D, Jahn HK, Freudenberg M, Loddenkemper C, Batra A, Lehr HA, Liesenfeld O, Blaut M, Gobel UB, Schumann RR, Bereswill S (2007) Shift towards pro-inflammatory intestinal bacteria aggravates acute murine colitis via toll-like receptors 2 and 4. PLoS One 2:e662
Heneka MT, Golenbock DT, Latz E (2015) Innate immunity in Alzheimer’s disease. Nat Immunol 16:229–236
Herman S, Fischer A, Pfatschbacher J, Hoffmann M, Steiner G (2011) A TLR 9 antagonist diminishes arthritis severity in a rat model of rheumatoid arthritis. Ann Rheum Dis 70:A39
Hoffjan S, Stemmler S, Parwez Q, Petrasch-Parwez E, Arinir U, Rohde G, Reinitz-Rademacher K, Schultze-Werninghaus G, Bufe A, Epplen JT (2005) Evaluation of the toll-like receptor 6 Ser249Pro polymorphism in patients with asthma, atopic dermatitis and chronic obstructive pulmonary disease. BMC Med Genet 6:34
Holtzman MJ, Byers DE, Alexander-Brett J, Wang X (2014) The role of airway epithelial cells and innate immune cells in chronic respiratory disease. Nat Rev Immunol 14:686–698
Huang QQ, Sobkoviak R, Jockheck-Clark AR, Shi B, Mandelin AM 2nd, Tak PP, Haines GK 3rd, Nicchitta CV, Pope RM (2009) Heat shock protein 96 is elevated in rheumatoid arthritis and activates macrophages primarily via TLR2 signaling. J Immunol 182:4965–4973
Iwahashi M, Yamamura M, Aita T, Okamoto A, Ueno A, Ogawa N, Akashi S, Miyake K, Godowski PJ, Makino H (2004) Expression of toll-like receptor 2 on CD16+ blood monocytes and synovial tissue macrophages in rheumatoid arthritis. Arthritis Rheum 50:1457–1467
Janeway CA Jr, Medzhitov R (1998) Introduction: the role of innate immunity in the adaptive immune response. Semin Immunol 10:349–350
Jiang A, Zhu F, Tang J, Kandimalla ER, La Monica N, Agrawal S (2012) IMO-8400, a novel TLR7, TLR8 and TLR9 antagonist, inhibits disease development in mouse models of psoriasis (119.8). J Immunol Res 188(119):8
Jin MS, Lee JO (2008) Structures of the toll-like receptor family and its ligand complexes. Immunity 29:182–191
Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K (2008) Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer’s disease. J Neuroinflamm 5:23
Joosten LA, Koenders MI, Smeets RL, Heuvelmans-Jacobs M, Helsen MM, Takeda K, Akira S, Lubberts E, Van De Loo FA, Van Den Berg WB (2003) Toll-like receptor 2 pathway drives streptococcal cell wall-induced joint inflammation: critical role of myeloid differentiation factor 88. J Immunol 171:6145–6153
Kandimalla ER, Bhagat L, Wang D, Yu D, Sullivan T, La Monica N, Agrawal S (2013) Design, synthesis and biological evaluation of novel antagonist compounds of toll-like receptors 7, 8 and 9. Nucleic Acids Res 41:3947–3961
Kawai T, Akira S (2005) Toll-like receptor downstream signaling. Arthritis Res Ther 7:12–19
Kawai T, Akira S (2007) TLR signaling. Semin Immunol 19:24–32
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol 11:373–384
Kawasaki T, Kawai T (2014) Toll-like receptor signaling pathways. Front Immunol 5:461
Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O’rourke KM, Eby M, Pietras E, Cheng G, Bazan JF, Zhang Z, Arnott D, Dixit VM (2007) DUBA: a deubiquitinase that regulates type I interferon production. Science 318:1628–1632
Keogh B, Parker AE (2011) Toll-like receptors as targets for immune disorders. Trends Pharmacol Sci 32:435–442
Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, Flavell RA (2002) IRAK-M is a negative regulator of toll-like receptor signaling. Cell 110:191–202
Kormann MS, Depner M, Hartl D, Klopp N, Illig T, Adamski J, Vogelberg C, Weiland SK, Von Mutius E, Kabesch M (2008) Toll-like receptor heterodimer variants protect from childhood asthma. J Allergy Clin Immunol 122:86–92
Kumar H, Kawai T, Akira S (2011) Pathogen recognition by the innate immune system. Int Rev Immunol 30:16–34
Lamphier M, Zheng W, Latz E, Spyvee M, Hansen H, Rose J, Genest M, Yang H, Shaffer C, Zhao Y, Shen Y, Liu C, Liu D, Mempel TR, Rowbottom C, Chow J, Twine NC, Yu M, Gusovsky F, Ishizaka ST (2014) Novel small molecule inhibitors of TLR7 and TLR9: mechanism of action and efficacy in vivo. Mol Pharmacol 85:429–440
Landreth GE, Reed-Geaghan EG (2009) Toll-like receptors in Alzheimer’s disease. Curr Top Microbiol Immunol 336:137–153
Lebre MC, Antons JC, Kalinski P, Schuitemaker JH, Van Capel TM, Kapsenberg ML, De Jong EC (2003) Double-stranded RNA-exposed human keratinocytes promote Th1 responses by inducing a Type-1 polarized phenotype in dendritic cells: role of keratinocyte-derived tumor necrosis factor alpha, type I interferons, and interleukin-18. J Invest Dermatol 120:990–997
Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, Kellner ES, Nacionales D, Barker T, Kelly-Scumpia K, Van Rooijen N, Kumar H, Kawai T, Satoh M, Akira S, Reeves WH (2008) TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med 205:2995–3006
Lee Y, Kim H, Kim S, Kim KH, Chung JH (2010) Activation of toll-like receptors 2, 3 or 5 induces matrix metalloproteinase-1 and -9 expression with the involvement of MAPKs and NF-kappaB in human epidermal keratinocytes. Exp Dermatol 19:e44–e49
Lee CC, Avalos AM, Ploegh HL (2012) Accessory molecules for toll-like receptors and their function. Nat Rev Immunol 12:168–179
Lee YH, Choi SJ, Ji JD, Song GG (2016) Association between toll-like receptor polymorphisms and systemic lupus erythematosus: a meta-analysis update. Lupus 25:593–601
Lehmann SM, Kruger C, Park B, Derkow K, Rosenberger K, Baumgart J, Trimbuch T, Eom G, Hinz M, Kaul D, Habbel P, Kalin R, Franzoni E, Rybak A, Nguyen D, Veh R, Ninnemann O, Peters O, Nitsch R, Heppner FL, Golenbock D, Schott E, Ploegh HL, Wulczyn FG, Lehnardt S (2012) An unconventional role for miRNA: let-7 activates toll-like receptor 7 and causes neurodegeneration. Nat Neurosci 15:827–835
Lehnardt S, Wennekamp J, Freyer D, Liedtke C, Krueger C, Nitsch R, Bechmann I, Weber JR, Henneke P (2007) TLR2 and caspase-8 are essential for group B Streptococcus-induced apoptosis in microglia. J Immunol 179:6134–6143
Li Y, Cao H, Wang N, Xiang Y, Lu Y, Zhao K, Zheng J, Zhou H (2011) A novel antagonist of TLR9 blocking all classes of immunostimulatory CpG-ODNs. Vaccine 29:2193–2198
Liew FY, Xu D, Brint EK, O’neill LA (2005) Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol 5:446–458
Liu J, Buckley JM, Redmond HP, Wang JH (2010) ST2 negatively regulates TLR2 signaling, but is not required for bacterial lipoprotein-induced tolerance. J Immunol 184:5802–5808
Lu Z, Zhang X, Li Y, Lopes-Virella MF, Huang Y (2015) TLR4 antagonist attenuates atherogenesis in LDL receptor-deficient mice with diet-induced type 2 diabetes. Immunobiology 220:1246–1254
Lun SW, Wong CK, Ko FW, Hui DS, Lam CW (2009) Expression and functional analysis of toll-like receptors of peripheral blood cells in asthmatic patients: implication for immunopathological mechanism in asthma. J Clin Immunol 29:330–342
Ma C, Ma Z, Fu Q, Ma S (2013) Curcumin attenuates allergic airway inflammation by regulation of CD4+ CD25+ regulatory T cells (Tregs)/Th17 balance in ovalbumin-sensitized mice. Fitoterapia 87:57–64
Mansell A, Smith R, Doyle SL, Gray P, Fenner JE, Crack PJ, Nicholson SE, Hilton DJ, O’neill LA, Hertzog PJ (2006) Suppressor of cytokine signaling 1 negatively regulates toll-like receptor signaling by mediating Mal degradation. Nat Immunol 7:148–155
Matsunaga N, Tsuchimori N, Matsumoto T, Ii M (2011) TAK-242 (resatorvid), a small-molecule inhibitor of toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol Pharmacol 79:34–41
Mcinnes IB, O’dell JR (2010) State-of-the-art: rheumatoid arthritis. Ann Rheum Dis 69:1898–1906
Mcinturff JE, Modlin RL, Kim J (2005) The role of toll-like receptors in the pathogenesis and treatment of dermatological disease. J Investig Dermatol 125:1–8
Medzhitov R (2007) Recognition of microorganisms and activation of the immune response. Nature 449:819–826
Meng L, Zhu W, Jiang C, He X, Hou W, Zheng F, Holmdahl R, Lu S (2010) Toll-like receptor 3 upregulation in macrophages participates in the initiation and maintenance of pristane-induced arthritis in rats. Arthritis Res Ther 12:R103
Meylan E, Tschopp J (2008) IRAK2 takes its place in TLR signaling. Nat Immunol 9:581–582
Miller LS, Sorensen OE, Liu PT, Jalian HR, Eshtiaghpour D, Behmanesh BE, Chung W, Starner TD, Kim J, Sieling PA, Ganz T, Modlin RL (2005) TGF-alpha regulates TLR expression and function on epidermal keratinocytes. J Immunol 174:6137–6143
Millien VO, Lu W, Shaw J, Yuan X, Mak G, Roberts L, Song LZ, Knight JM, Creighton CJ, Luong A, Kheradmand F, Corry DB (2013) Cleavage of fibrinogen by proteinases elicits allergic responses through toll-like receptor 4. Science 341:792–796
Miranda-Hernandez S, Gerlach N, Fletcher JM, Biros E, Mack M, Korner H, Baxter AG (2011) Role for MyD88, TLR2 and TLR9 but not TLR1, TLR4 or TLR6 in experimental autoimmune encephalomyelitis. J Immunol 187:791–804
Mistry P, Laird MH, Schwarz RS, Greene S, Dyson T, Snyder GA, Xiao TS, Chauhan J, Fletcher S, Toshchakov VY, Mackerell AD Jr, Vogel SN (2015) Inhibition of TLR2 signaling by small molecule inhibitors targeting a pocket within the TLR2 TIR domain. Proc Natl Acad Sci USA 112:5455–5460
Montes AH, Asensi V, Alvarez V, Valle E, Ocana MG, Meana A, Carton JA, Paz J, Fierer J, Celada A (2006) The toll-like receptor 4 (Asp299Gly) polymorphism is a risk factor for Gram-negative and haematogenous osteomyelitis. Clin Exp Immunol 143:404–413
Moon SJ, Park MK, Oh HJ, Lee SY, Kwok SK, Cho ML, Ju JH, Park KS, Kim HY, Park SH (2010) Engagement of toll-like receptor 3 induces vascular endothelial growth factor and interleukin-8 in human rheumatoid synovial fibroblasts. Korean J Intern Med 25:429–435
Mudaliar H, Pollock C, Komala MG, Chadban S, Wu H, Panchapakesan U (2013) The role of toll-like receptor proteins (TLR) 2 and 4 in mediating inflammation in proximal tubules. Am J Physiol Renal Physiol 305:F143–F154
Mullarkey M, Rose JR, Bristol J, Kawata T, Kimura A, Kobayashi S, Przetak M, Chow J, Gusovsky F, Christ WJ, Rossignol DP (2003) Inhibition of endotoxin response by e5564, a novel toll-like receptor 4-directed endotoxin antagonist. J Pharmacol Exp Ther 304:1093–1102
Murad S (2014) Toll-like receptor 4 in inflammation and angiogenesis: a double-edged sword. Front Immunol 5:313
Nestle FO, Di Meglio P, Qin JZ, Nickoloff BJ (2009) Skin immune sentinels in health and disease. Nat Rev Immunol 9:679–691
Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, Shlomchik MJ (2010) TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol 184:1840–1848
Nundel K, Green NM, Shaffer AL, Moody KL, Busto P, Eilat D, Miyake K, Oropallo MA, Cancro MP, Marshak-Rothstein A (2015) Cell-intrinsic expression of TLR9 in autoreactive B cells constrains BCR/TLR7-dependent responses. J Immunol 194:2504–2512
O’neill LA, Bryant CE, Doyle SL (2009) Therapeutic targeting of toll-like receptors for infectious and inflammatory diseases and cancer. Pharmacol Rev 61:177–197
Opal SM, Laterre PF, Francois B, Larosa SP, Angus DC, Mira JP, Wittebole X, Dugernier T, Dugernier D, Tidswell M, Jauregui L, Krell K, Pachl J, Takahashi T, Peckelsen C, Cordasco E, Chang CS, Oeyen S, Aikawa N, Maruyama T, Schein R, Kalil AC, Van Nuffelen M, Lynn M, Rossignol DP, Gogate J, Roberts MB, Wheeler JL, Vincent JL, Vincent AS (2013) Effect of eritoran, an antagonist of MD2-TLR4, on mortality in patients with severe sepsis: the ACCESS randomized trial. JAMA 309:1154–1162
Ospelt C, Gay S (2010) TLRs and chronic inflammation. Int J Biochem Cell Biol 42:495–505
Ospelt C, Brentano F, Rengel Y, Stanczyk J, Kolling C, Tak PP, Gay RE, Gay S, Kyburz D (2008) Overexpression of toll-like receptors 3 and 4 in synovial tissue from patients with early rheumatoid arthritis: toll-like receptor expression in early and longstanding arthritis. Arthritis Rheum 58:3684–3692
Palsson-Mcdermott EM, Doyle SL, Mcgettrick AF, Hardy M, Husebye H, Banahan K, Gong M, Golenbock D, Espevik T, O’neill LA (2009) TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat Immunol 10:579–586
Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279:7370–7377
Perkins DJ, Vogel SN (2016) Inflammation: species-specific TLR signalling—insight into human disease. Nat Rev Rheumatol 12:198–200
Piazza M, Calabrese V, Damore G, Cighetti R, Gioannini T, Weiss J, Peri F (2012) A synthetic lipid A mimetic modulates human TLR4 activity. ChemMedChem 7:213–217
Piccinini AM, Midwood KS (2010) DAMPening inflammation by modulating TLR signalling. Mediators Inflamm 2010:672395
Pierer M, Wagner U, Rossol M, Ibrahim S (2011) Toll-like receptor 4 is involved in inflammatory and joint destructive pathways in collagen-induced arthritis in DBA1 J mice. PLoS One 6:e23539
Prehaud C, Megret F, Lafage M, Lafon M (2005) Virus infection switches TLR-3-positive human neurons to become strong producers of beta interferon. J Virol 79:12893–12904
Qian C, Cao X (2013) Regulation of toll-like receptor signaling pathways in innate immune responses. Ann N Y Acad Sci 1283:67–74
Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, Van Riel PL, Barrera P, Joosten LA, Van Den Berg WB (2004) Expression of toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-gamma. Arthritis Rheum 50:3856–3865
Ramani V, Madhusoodhanan R, Kosanke S, Awasthi S (2013) A TLR4-interacting SPA4 peptide inhibits LPS-induced lung inflammation. Innate Immun 19:596–610
Renn CN, Sanchez DJ, Ochoa MT, Legaspi AJ, Oh CK, Liu PT, Krutzik SR, Sieling PA, Cheng G, Modlin RL (2006) TLR activation of Langerhans cell-like dendritic cells triggers an antiviral immune response. J Immunol 177:298–305
Roelofs MF, Joosten LA, Abdollahi-Roodsaz S, Van Lieshout AW, Sprong T, Van Den Hoogen FH, Van Den Berg WB, Radstake TR (2005) The expression of toll-like receptors 3 and 7 in rheumatoid arthritis synovium is increased and costimulation of toll-like receptors 3, 4, and 7/8 results in synergistic cytokine production by dendritic cells. Arthritis Rheum 52:2313–2322
Roelofs MF, Boelens WC, Joosten LA, Abdollahi-Roodsaz S, Geurts J, Wunderink LU, Schreurs BW, Van Den Berg WB, Radstake TR (2006) Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J Immunol 176:7021–7027
Rose WA 2nd, Sakamoto K, Leifer CA (2012) TLR9 is important for protection against intestinal damage and for intestinal repair. Sci Rep 2:574
Rothlin CV, Ghosh S, Zuniga EI, Oldstone MBA, Lemke G (2007) TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131:1124–1136
Saijo K, Winner B, Carson CT, Collier JG, Boyer L, Rosenfeld MG, Gage FH, Glass CK (2009) A Nurr1/CoREST pathway in microglia and astrocytes protects dopaminergic neurons from inflammation-induced death. Cell 137:47–59
Saitoh T, Tun-Kyi A, Ryo A, Yamamoto M, Finn G, Fujita T, Akira S, Yamamoto N, Lu KP, Yamaoka S (2006) Negative regulation of interferon-regulatory factor 3-dependent innate antiviral response by the prolyl isomerase Pin1. Nat Immunol 7:598–605
Schnare M, Barton GM, Holt AC, Takeda K, Akira S, Medzhitov R (2001) Toll-like receptors control activation of adaptive immune responses. Nat Immunol 2:947–950
Senhaji N, Diakite B, Serbati N, Zaid Y, Badre W, Nadifi S (2014) Toll-like receptor 4 Asp299Gly and Thr399Ile polymorphisms: new data and a meta-analysis. BMC Gastroenterol 14:206
Seung NR, Park EJ, Kim CW, Kim KH, Kim KJ, Cho HJ, Park HR (2007) Comparison of expression of heat-shock protein 60, toll-like receptors 2 and 4, and T-cell receptor gammadelta in plaque and guttate psoriasis. J Cutan Pathol 34:903–911
Sham HP, Yu EYS, Gulen MF, Bhinder G, Stahl M, Chan JM, Brewster L, Morampudi V, Gibson DL, Hughes MR, Mcnagny KM, Li X, Vallance BA (2013) SIGIRR, a negative regulator of TLR/IL-1R signalling promotes microbiota dependent resistance to colonization by enteric bacterial pathogens. PLoS Pathog 9:e1003539
Shannon J, Ernst P, Yamauchi Y, Olivenstein R, Lemiere C, Foley S, Cicora L, Ludwig M, Hamid Q, Martin JG (2008) Differences in airway cytokine profile in severe asthma compared to moderate asthma. Chest 133:420–426
Shirey KA, Lai W, Scott AJ, Lipsky M, Mistry P, Pletneva LM, Karp CL, Mcalees J, Gioannini TL, Weiss J, Chen WH, Ernst RK, Rossignol DP, Gusovsky F, Blanco JC, Vogel SN (2013) The TLR4 antagonist Eritoran protects mice from lethal influenza infection. Nature 497:498–502
Shmuel-Galia L, Aychek T, Fink A, Porat Z, Zarmi B, Bernshtein B, Brenner O, Jung S, Shai Y (2016) Neutralization of pro-inflammatory monocytes by targeting TLR2 dimerization ameliorates colitis. EMBO J 35:685–698
Shotorbani SS, Su ZL, Xu HX (2011) Toll-like receptors are potential therapeutic targets in rheumatoid arthritis. World J Biol Chem 2:167–172
Sioud M (2010) Development of TLR7/8 small RNA antagonists. Methods Mol Biol 629:387–394
Sorensen LK, Havemose-Poulsen A, Sonder SU, Bendtzen K, Holmstrup P (2008) Blood cell gene expression profiling in subjects with aggressive periodontitis and chronic arthritis. J Periodontol 79:477–485
Spiller S, Elson G, Ferstl R, Dreher S, Mueller T, Freudenberg M, Daubeuf B, Wagner H, Kirschning CJ (2008) TLR4-induced IFN-gamma production increases TLR2 sensitivity and drives Gram-negative sepsis in mice. J Exp Med 205:1747–1754
Stewart CR, Stuart LM, Wilkinson K, Van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, Lacy-Hulbert A, El Khoury J, Golenbock DT, Moore KJ (2010) CD36 ligands promote sterile inflammation through assembly of a toll-like receptor 4 and 6 heterodimer. Nat Immunol 11:155–161
Streit WJ (2002) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40:133–139
Suarez-Farinas M, Arbeit R, Jiang W, Ortenzio FS, Sullivan T, Krueger JG (2013) Suppression of molecular inflammatory pathways by toll-like receptor 7, 8, and 9 antagonists in a model of IL-23-induced skin inflammation. PLoS One 8:e84634
Subramanian M, Thorp E, Hansson GK, Tabas I (2013) Treg-mediated suppression of atherosclerosis requires MYD88 signaling in DCs. J Clin Investig 123:179–188
Suzuki M, Hisamatsu T, Podolsky DK (2003) Gamma interferon augments the intracellular pathway for lipopolysaccharide (LPS) recognition in human intestinal epithelial cells through coordinated up-regulation of LPS uptake and expression of the intracellular toll-like receptor 4-MD-2 complex. Infect Immun 71:3503–3511
Tan Y, Zou KF, Qian W, Chen S, Hou XH (2014) Expression and implication of toll-like receptors TLR2, TLR4 and TLR9 in colonic mucosa of patients with ulcerative colitis. J Huazhong Univ Sci Technol Med Sci 34:785–790
Tanaka J, Watanabe N, Kido M, Saga K, Akamatsu T, Nishio A, Chiba T (2009) Human TSLP and TLR3 ligands promote differentiation of Th17 cells with a central memory phenotype under Th2-polarizing conditions. Clin Exp Allergy 39:89–100
Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A, Siler DA, Markesbery WR, Arumugam TV, Mattson MP (2008) Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol 213:114–121
Taniguchi N, Kawahara K, Yone K, Hashiguchi T, Yamakuchi M, Goto M, Inoue K, Yamada S, Ijiri K, Matsunaga S, Nakajima T, Komiya S, Maruyama I (2003) High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum 48:971–981
Tantisira K, Klimecki WT, Lazarus R, Palmer LJ, Raby BA, Kwiatkowski DJ, Silverman E, Vercelli D, Martinez FD, Weiss ST (2004) Toll-like receptor 6 gene (TLR6): single-nucleotide polymorphism frequencies and preliminary association with the diagnosis of asthma. Genes Immun 5:343–346
Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC (2002) Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med 195:99–111
Tesmer LA, Lundy SK, Sarkar S, Fox DA (2008) Th17 cells in human disease. Immunol Rev 223:87–113
Thwaites R, Chamberlain G, Sacre S (2014) Emerging role of endosomal toll-like receptors in rheumatoid arthritis. Front Immunol 5:1
Tirumurugaan KG, Dhanasekaran S, Raj GD, Raja A, Kumanan K, Ramaswamy V (2010) Differential expression of toll-like receptor mRNA in selected tissues of goat (Capra hircus). Vet Immunol Immunopathol 133:296–301
Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL (2009) Allergenicity resulting from functional mimicry of a toll-like receptor complex protein. Nature 457:585–588
Uehara A, Fujimoto Y, Fukase K, Takada H (2007) Various human epithelial cells express functional toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol 44:3100–3111
Ungaro R, Fukata M, Hsu D, Hernandez Y, Breglio K, Chen A, Xu R, Sotolongo J, Espana C, Zaias J, Elson G, Mayer L, Kosco-Vilbois M, Abreu MT (2009) A novel toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am J Physiol Gastrointest Liver Physiol 296:G1167–G1179
Van Der Fits L, Mourits S, Voerman JS, Kant M, Boon L, Laman JD, Cornelissen F, Mus AM, Florencia E, Prens EP, Lubberts E (2009) Imiquimod-induced psoriasis-like skin inflammation in mice is mediated via the IL-23/IL-17 axis. J Immunol 182:5836–5845
Vijay-Kumar M, Wu H, Aitken J, Kolachala VL, Neish AS, Sitaraman SV, Gewirtz AT (2007) Activation of toll-like receptor 3 protects against DSS-induced acute colitis. Inflamm Bowel Dis 13:856–864
Wakashin H, Hirose K, Maezawa Y, Kagami S, Suto A, Watanabe N, Saito Y, Hatano M, Tokuhisa T, Iwakura Y, Puccetti P, Iwamoto I, Nakajima H (2008) IL-23 and Th17 cells enhance Th2-cell-mediated eosinophilic airway inflammation in mice. Am J Respir Crit Care Med 178:1023–1032
Wald D, Qin J, Zhao Z, Qian Y, Naramura M, Tian L, Towne J, Sims JE, Stark GR, Li X (2003) SIGIRR, a negative regulator of toll-like receptor-interleukin 1 receptor signaling. Nat Immunol 4:920–927
Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, Bode B, Manietta N, Walter J, Schulz-Schuffer W, Fassbender K (2007) Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell Physiol Biochem 20:947–956
Walter A, Schafer M, Cecconi V, Matter C, Urosevic-Maiwald M, Belloni B, Schonewolf N, Dummer R, Bloch W, Werner S, Beer HD, Knuth A, Van Den Broek M (2013) Aldara activates TLR7-independent immune defence. Nat Commun 4:1560
Wang Y, Tang Y, Teng L, Wu Y, Zhao X, Pei G (2006) Association of beta-arrestin and TRAF6 negatively regulates toll-like receptor-interleukin 1 receptor signaling. Nat Immunol 7:139–147
Watanabe T, Kitani A, Murray PJ, Strober W (2004) NOD2 is a negative regulator of toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol 5:800–808
Wu HJ, Sawaya H, Binstadt B, Brickelmaier M, Blasius A, Gorelik L, Mahmood U, Weissleder R, Carulli J, Benoist C, Mathis D (2007) Inflammatory arthritis can be reined in by CpG-induced DC-NK cell cross talk. J Exp Med 204:1911–1922
Xavier RJ, Podolsky DK (2007) Unravelling the pathogenesis of inflammatory bowel disease. Nature 448:427–434
Xiong Y, Song C, Snyder GA, Sundberg EJ, Medvedev AE (2012) R753Q polymorphism inhibits toll-like receptor (TLR) 2 tyrosine phosphorylation, dimerization with TLR6, and recruitment of myeloid differentiation primary response protein 88. J Biol Chem 287:38327–38337
Yamada M, Ichikawa T, Ii M, Sunamoto M, Itoh K, Tamura N, Kitazaki T (2005) Discovery of novel and potent small-molecule inhibitors of NO and cytokine production as antisepsis agents: synthesis and biological activity of alkyl 6-(N-substituted sulfamoyl)cyclohex-1-ene-1-carboxylate. J Med Chem 48:7457–7467
Yarilina A, Dicarlo E, Ivashkiv LB (2007) Suppression of the effector phase of inflammatory arthritis by double-stranded RNA is mediated by type I IFNs. J Immunol 178:2204–2211
Yesudhas D, Gosu V, Anwar MA, Choi S (2014) Multiple roles of toll-like receptor 4 in colorectal cancer. Front Immunol 5:334
Yoon SI, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, Wilson IA (2012) Structural basis of TLR5-flagellin recognition and signaling. Science 335:859–864
Yu Y, Hayward GS (2010) The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33:863–877
Yu N, Zhang S, Zuo F, Kang K, Guan M, Xiang L (2009) Cultured human melanocytes express functional toll-like receptors 2–4, 7 and 9. J Dermatol Sci 56:113–120
Zarember KA, Godowski PJ (2002) Tissue expression of human toll-like receptors and differential regulation of toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol 168:554–561
Zuo L, Lucas K, Fortuna CA, Chuang CC, Best TM (2015) Molecular regulation of toll-like receptors in asthma and COPD. Front Physiol 6:312
Acknowledgments
This work was supported by the Mid-Career Researcher Program through the National Research Foundation of Korea, funded by the Ministry of Education, Science, and Technology (NRF-2015R1A2A2A09001059), and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (HI14C1992). This work was also partially supported by a grant from the Priority Research Centers Program (NRF 2012-0006687).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Rights and permissions
About this article

Cite this article
Achek, A., Yesudhas, D. & Choi, S. Toll-like receptors: promising therapeutic targets for inflammatory diseases. Arch. Pharm. Res. 39, 1032–1049 (2016). https://doi.org/10.1007/s12272-016-0806-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0806-9