
Abstract
Angiogenic dormancy can be defined as the condition in which cancer cell proliferation is counterbalanced by apoptosis owing to poor vascularization. Indeed, the lack of tumor angiogenesis impedes tumor mass expansion beyond a microscopic size, resulting in an asymptomatic and non-metastatic state. Thus, the tumor angiogenic switch is essential to promote fast-growing and expansion of tumor masses and to develop the metastatic process. In the avascular tumor lesion, angiogenesis process results blocked from the equilibrium between pro- and anti-angiogenic factors, such as vascular endothelial growth factor (VEGF) and thrombospondin-1 (TSP-1), respectively. The angiogenic switch of non-dormant tumors mainly depends on the disruption of the balance in the tumor microenvironment between anti-angiogenic and pro-angiogenic factors, in favor of the latter. Moreover, this tumors activate and recruit the circulating endothelial progenitors (CEPs) that facilitate the shift toward the generation of new blood vessels. Metronomic chemotherapy—a regular administration of drug doses able to maintain low but active concentrations of chemotherapeutic drugs during prolonged periods of time—is a promising therapeutic approach that can induce or re-induce the angiogenic tumor dormancy. Metronomic chemotherapy upregulates TSP-1 and decreases pro-angiogenic factors such as VEGF, and suppresses the proangiogenic cells such as CEPs both in adjuvant setting or in the treatment of metastatic disease. In this perspective, metronomic chemotherapy may be able to play a main role in the modulation of the angiogenic tumor dormancy, but further preclinical and clinical studies are needed to better investigate this particular aspect of this interesting therapeutic tool.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
- Tumor dormancy
- Angiogenesis
- Metronomic chemotherapy
- Thrombospondin-1
- Vascular endothelial growth factor
- Circulating endothelial progenitor
Introduction
The concept of tumor dormancy refers to the presence of asymptomatic and temporarily non-invasive cancer cells which are diagnostically undetected for months or decades. Indeed, autopsies of several people who did not die for tumors revealed the accidental presence of non-expanding microscopic primary cancer and the occurrence of these non-invasive dormant cells can be considered “normal” in healthy subjects [1]. Nevertheless, this condition of occult cancer can represent the early stage of tumor development, but it can also account for tumor recurrence after a successful treatment, as well as micro-metastases. Accordingly, cancer progression can be regarded as a multistep process. Both experimental and clinical studies suggest that cancer cell dissemination occur at very early stages of tumor growth and that these disseminated cells can remain dormant for a long time. The phenomenon of metastatic tumor dormancy is on the basis of tumor metastasis which can occur also several years after an apparently effective therapy. Then, tumor dormancy is clinically relevant in both primary and secondary tumors, which arise from residual disseminated cancer cells. However, the peculiar features of tumor dormancy impede to have appropriate experimental models and clinical accessibility and the problem remains poorly investigated [2,3,4].
It is important to distinguish between quiescent solitary cells (tumor cell dormancy) and small-sized asymptomatic cancerous lesions (tumor dormancy). Indeed, these two types of dormancy represent completely distinct conditions that significantly differ in their characteristics and underlying regulatory mechanisms [3].
It is particularly noteworthy to identify those factors that are able either to maintain cancer cells in an occult state or to promote the escape from dormancy. Taking into account that preventing screening tests are unable to reveal such undetectable abnormal solitary cells, the knowledge of tumor dormancy pathophysiology is essential to understand cancer development and to design therapeutic strategies. Tumor dormancy would involve quiescence, consisting of reversible cell cycle arrest. However, some tumor cells seem to be also able to reverse senescence, consisting of permanent cell cycle arrest, and a combination of both mechanisms might lead to tumor dormancy [4,5,6].
The complex interactions occurring in the tumor microenvironment, in particular the relationship of the cancer cell with the extracellular matrix and other normal cell types (e.g., endothelial cells, fibroblast), appear determinant in contrasting tumor growth. The expression of some receptors, including urokinase and epidermal growth factor (EGF) receptors, has been associated to the regulation of the quiescence-based dormancy of tumors [7, 8]. Considering the ability of different organs to support disseminated tumor cells growth, microenvironments have been classified as dormancy-permissive or dormancy-restrictive and this distinction might account for the different incidence of metastases and disseminated tumor cells in the same organ (“seed and soil” theory) [9, 10].
The immune surveillance plays a pivotal role in suppressing cancer growth and both cellular and humoral responses are needed to maintain the occult state of cancer mass [11]. However, the inflammatory state that associates to the release of cytokines during some immune reactions can trigger angiogenesis-mediated escape from dormancy [12].
Specific changes in cytoskeleton architecture, involving fibronectin production and activation of integrin β-1 signalling pathway, may be associated to the pattern of the dormant cell [13].
Other factors that facilitate the permanence of the tumor in a dormant state include the hormonal withdrawal and the inhibition of angiogenesis [3, 14, 15]. Apart from adjuvant chemotherapy strategies, various dietary components, in terms of food intake, energy balance and physical activity, might also influence cancer cells and their microenvironment. Indeed, several dietary phytochemicals can affect the behavior and gene expression patterns of both tumor cells and host tissues [16].
The Role of Angiogenesis in Tumor Dormancy
Angiogenesis is a biologic process consisting in the formation of new capillaries from pre-existing blood vessels [17]. This process occurs in several physiological conditions, including embryogenesis, ovulation, wound healing and repair. However, it can be also observed in pathological conditions, such as arthritis, diabetic retinopathy, endometriosis and tumors. The growth of solid tumors includes an avascular and a subsequent vascular phase. Most tumors seem to begin as small sized and non-angiogenic cellular aggregates which cannot grow until vascular network is established. Indeed, an important mechanism behind tumor dormancy is the ability of cancer cells to induce angiogenesis. In solid tumors the transition from the avascular to the vascular phase is critical for the proliferation of cancer cells in situ and at distant sites, as metastases. In this respect, tumor growth is thought to be angiogenesis-dependent and the inhibitors of angiogenesis have been then proposed as anticancer therapy [18, 19].
Not surprisingly, a pivotal mechanism behind tumor dormancy is represented by the ability of tumor cells to induce angiogenesis and, more importantly, to realize successfully and correctly the complete process of new blood vessel formation. Indeed, failure in one or more of the angiogenic steps leads to dormancy [3].
Tumor dormancy may be referred to a single cancer cell (tumor cell dormancy) which lies in cell cycle arrest (G0-G1 arrest), or to active proliferating tumor cells (tumor mass dormancy) whose growth is significantly limited by efficient immune surveillance, or insufficient blood supply (angiogenic dormancy), leading to dynamic equilibrium between cell proliferation and apoptotic death. In fact, dormant tumor cells are generally considered in an arrested state, but a debate exists whether micrometastatic disease consists in a balance of cell proliferation and death that only appears as an arrested state [6, 10].
Aguirre-Ghiso [6] just defined angiogenic dormancy the condition in which cancer cell proliferation is counterbalanced by apoptosis owing to poor vascularization. As a consequence, the cancer cells are unable to grow. Then, malignant properties of cancer cells are not enough to develop a tumor that becomes lethal: a cancer without disease! Tumor angiogenesis is strictly necessary to promote fast-growing and expansion. The lack of tumor angiogenesis impedes tumor mass expansion beyond a microscopic size, resulting in an asymptomatic and non-metastatic state [20].
The normal angiogenesis results from the equilibrium between pro- and anti-angiogenic factors. The first pro-angiogenic factor, named tumor angiogenic factor (TAF), was hypothesized 45 years ago by Judah Folkman [21], who also suggested that tumor growth strongly depends on angiogenesis and proposed anti-angiogenic therapy as a new approach to treat cancer disease [22]. In subsequent years, other receptor-mediated agents activating and regulating angiogenesis were identified, among which fibroblast growth factors 1 and 2 (FGF-1 and 2) [23], the most important vascular endothelial growth factor (VEGF) family (VEGF A to D) [17], placenta growth factor (PLGF) [24], platelet-derived growth factors (PDGFs), insulin-like growth factors (IGFs), angiopoietin 1, EGF, hepatocyte growth factor (HGF), hypoxia-inducible factor-1 α and β (HIF-1 α and β), transforming growth factor-α and β (TGF-α and β), tumor necrosis factor-α (TNF-α), interleukins (IL)-1 β, 3, 6, 8, neuropilin 1 and 2, angiogenin, adrenomedullin, stromal cell-derived factor-1 (SDF-1) [18, 25, 26].
Apart from hypoxia, other environmental stressors are able to induce the expression of pro-angiogenic factors, including glucose deprivation, accumulation of reactive oxygen species (ROS), cellular acidosis or iron deficiency, or the activation of oncogenes, such as Ras [27] and Myc, or the loss of the function of tumor suppressor genes [3].
Dormant cancer cells appear to undergo a stable genetic reprogramming process during their escape towards the fast-growing phenotype and this would occur during the angiogenic switch: progress from non-angiogenic to angiogenic phenotype, with recruitment of new blood vessels. This condition is considered an early marker of neoplastic transformation. Although dysfunctional, with irregular shape and architecture, tumor blood vessels are essential for the growth of malignant cancer cells. An important concept is that the acquisition of angiogenic capacity is required a long time before the emergence of an invasive malignancy [3].
Human tumors contain cancer cell subpopulations with different angiogenic potential. In a human liposarcoma cell line (SW-872), three different clone patterns of growth have been isolated and observed: highly angiogenic clones with rapid tumor growth; weakly angiogenic clones with slow tumor growth; non-angiogenic clones corresponding to vital but dormant tumors and also named “non-tumorigenic” or “no-take”. This concept has been also explored in animal models of tumor dormancy, especially by inoculating human cancer cells in immunocompromised mice [20, 28].
Environmental hypoxia in cancer cell proliferation appears to be a crucial factor inducing angiogenic switch, this expression indicating the transition from the non-angiogenic to the angiogenic tumor phenotype, with subsequent disease progression. When a small-sized tumor mass attempts to grow, central cancer cells remain too distant from normal surrounding blood vessels to benefit of oxygen diffusion and tend to necrosis. This hypoxic condition might trigger compensatory mechanisms in suffering cells, with an increased expression and activation of the transcription factor HIF-1 pathway or HIF-1-independent pathways, as well. Subsequently, other pro-angiogenic factors are recruited, including VEGF, PDGF and nitric oxide (NO) synthase. The angiogenic switch would depend on the disruption of the normal equilibrium in the microenvironment between anti-angiogenic and pro-angiogenic factors, in favor of the latter. The initial step is represented by hyperemic reaction at the periphery of the tumor, due to vasodilation, followed by a process of angiogenesis. A transient angiogenic switch delivered by factors of the tumor microenvironment can also convey tumorigenic properties to cancer cells [12, 14, 29].
In particular, the switch of dormant cancer cells was associated with down-regulation of the angiogenesis inhibitor thrombospondin-1 (TSP-1) and decreased sensitivity to angiostatin. Cancer cell secretion and intracellular levels of TSP-1 in non-angiogenic and angiogenic tumor cell populations isolated from the human breast cancer cell line MDAMB-436 were compared, indicating that angiogenic cancer cells contain significantly lower levels of TSP-1 than non-angiogenic tumor cells and secretion of TSP-1 from non-angiogenic tumor cells was 20-fold higher than angiogenic cells. The decrease in TSP-1 levels seems to be mediated by phosphatidylinositol 3-kinase (PI3K) [30, 31].
It was shown that in the endothelium the expression of the angiogenesis inducers epoxyeicosatrienoic acids (EETs) stimulated escape from tumor dormancy in mice. In line with this, EETs stimulated metastasis of various xenograft tumors, including Lewis lung carcinomas (LLC) and B16-F10 melanomas [32].
The Notch signaling pathway is largely used by endothelial cells to coordinate cellular activities during the blood vessel formation that occurs in angiogenesis. Then, not surprisingly, an interactive cross-talk between cancer and endothelial cells has been shown to favor the escape of tumors from dormancy, this transition being mediated by the Notch ligand Dll4 on endothelial cells and Notch 3 signaling in tumor cells, promoting a tumorigenic phenotype. In agreement with this, Notch 3 levels are low in dormant tumors. These data provide a novel angiogenesis-driven mechanism involving the Notch pathway in controlling tumor dormancy. Metabolic features also participate in the regulation of tumor dormancy. The activity of the LKB1/AMPK system, deputed to monitor cellular ATP levels, is enhanced by anti-VEGF therapy, leading to glucose depletion and reduction of ATP levels, with tumor regression [10, 33, 34].
It was reported that local traumas, injuries, wounds, burns and surgery can cause a permissive microenvironmental niche for tumor growth. These conditions are unlike to induce the onset of malignant cells, rather they promote the escape from tumor dormancy. The occurrence of an inflammatory state and the ability to attract circulating cancer cells or to mobilize circulating endothelial progenitors (CEPs), with an increase in VEGF plasma levels, might explain such a transition to a non-dormant state [7, 14].
Apart from cancer cells themselves and local stromal microenvironment, distant bone marrow cells, once recruited into tumor masses, also participate to the induction of the angiogenic switch. The stromal cells that surround tumor masses mainly include fibroblasts, lymphocytes, neutrophils, macrophages and mast cells, which communicate through intercellular signalling pathways, mediated by surface adhesion molecules, cytokines and their receptors. Paradoxically, infiltrating cells of the immune system are important constituents of tumors and can represent a fundamental source of growth stimulatory signals. Although at a different extent and with data still debated, several bone marrow-derived cell (BMDC) types have been implicated in the escape from tumor dormancy, as well as in the metastatic dissemination. These cells include endothelial progenitor cells, Tie-2 expressing monocytes, the heterogeneous family of immature myeloid cells, hemangiocytes, M1 and M2 tumor associated macrophages, dendritic cells, and mast cells. As in other pathophysiologic conditions such as healing, infection, inflammation or ischemia, several cytokines and chemokines would be released by cancer cells to recruit a large body of BMDC types which contribute to the angiogenic switch. In this respect, inflammation is regarded as a strong promoter for angiogenic switch, and also circulating platelets have been implicated in the transport and dissemination of such pro-angiogenic factor [3, 7, 35]. These data suggest that a lot of angiogenic factors are required to trigger tumor angiogenesis. On this basis, Indraccolo et al. [36] proposed the “spike hypothesis”, according to which a transient but consistent supply of angiogenic factors is able to promote the angiogenic switch.
The exosomes released by cancer cells contain soluble cytokines, growth factors, integrins, mRNA and microRNA which are able to reprogram bone marrow progenitor cells with pro-angiogenic and pro-metastatic activity [37]. In particular, exosomes released by renal carcinoma cells were able to activate an angiogenic phenotype in normal endothelial cells in vitro and tumor cell colonization of the lung and angiogenesis in vivo [38].
Some data indicate that certain tumors are able to transform bone marrow cells into pro-tumorigenic even prior to their mobilization into the circulation. This process through which humoral signals released from certain tumors stimulate bone marrow cells, which are mobilized into the circulation and subsequently induce the growth of otherwise dormant cancer cells residing at distant anatomical sites, is defined systemic instigation. Certain breast tumors (instigators) release the cytokine osteopontin (OPN) into the circulation and tumor-derived OPN programs hematopoietic progenitor cells to adopt a pro-tumorigenic state, in part, by inducing their over-expression of the secreted glycoprotein, granulin (GRN) [39].
Autophagy is a highly conserved self-degradative mechanism that plays an important role in removing dysfunctional cellular components and takes part in several physiopathological processes, including starvation, infections, programmed cell death, repair and degenerative mechanisms. Its role in cancer is dual. From one hand, it promotes survival of cancer cells, and from the other hand, it behaves as a tumor suppressor. In this respect, autophagy seems to favour tumor dormancy by inducing growth arrest with consequent prevention of programmed cell death, according to the dormant stem-like state of cancer cells. Accordingly, stimulation of autophagy induces quiescence and growth arrest in cancer cells, whereas inhibition of autophagy causes rapid cell death. In M2 tumor associated macrophages and fibroblasts, autophagy seems to promote pro-invasive, pro-angiogenic and pro-metastatic phenotype [37].
Considering that the transition from dormant to fast-growing tumor is angiogenesis-dependent and requires a stable transcriptional reprogramming, this phenomenon has been also evaluated by genome analysis. Cancer cells expressing microRNA cluster 126 (miR-126) have been shown to reduce the recruitment of endothelial cells to the tumor site by blocking GAS6/MER signaling [40]. It was also observed that suppression of the heat shock protein (HSP) 27 associates the non-angiogenic pattern with the inhibition of endothelial cell proliferation leading to long-term dormancy in human breast cancer [41]. Almog et al., [42] evaluated 19 microRNAs dealing with the phenotypic switch to fast-growth of four human dormant tumors: breast carcinoma, glioblastoma, osteosarcoma, and liposarcoma. Loss of expression of dormancy-associated microRNAs was the prevailing regulation pattern correlating with the switch of dormant tumors to fast-growth. Reconstitution of a single dormant microRNA led to phenotypic reversal of fast-growing angiogenic tumors towards long-lasting tumor dormancy. Furthermore, transcriptional reprogramming of tumors by means of dormant microRNAs over-expression led to down-regulation of pro-angiogenic factors, such as bFGF and TGF-α. Anti-angiogenic and dormancy promoting pathways such as EphA5 and angiomotin were up-regulated in dormant microRNA over-expressing tumors [42].
Taking into account the above-mentioned features of angiogenesis in tumor dormancy, non-angiogenic/dormant tumors can be defined as reported in Table 1, whereas angiogenic/non-dormant tumors can be defined as shown in Table 2 [14, 20].
Metronomic Chemotherapy as an Inducer of Angiogenic Tumor Dormancy: A Promising Research Field
Metronomic chemotherapy could be defined as a frequent, regular administration of drug doses designed to maintain a low, but active, range of concentrations of chemotherapeutic drugs during prolonged periods of time without inducing excessive toxicities [43]. Various mechanisms of action of metronomic chemotherapy have been suggested for different chemotherapeutic drugs and for the same drugs but at different plasma concentrations (e.g. cyclophosphamide) [43]. This is consistent with the numerous evidences that this type of therapy is a complex approach involving both tumor cells and their microenvironment, including microvessels and cells of immune system [44].
Numerous findings support the hypothesis that metronomic chemotherapy caused antitumor effects by inhibiting tumor angiogenesis [45] because of a preferential antiendothelial activity [46]. But this is not the sole mechanism for it causing antitumor effects. There is a growing scientific literature that indicates low doses of certain chemotherapeutic drugs—especially cyclophosphamide—could cause stimulation of cytotoxic T cells by targeting T regulatory cells [47]. Metronomic chemotherapy may also have significant direct antitumor cell effects [48], also through an activity on the putative cancer stem cell (CSC) or tumor-initiating cell (TIC) subpopulation [49, 50].
Metronomic Chemotherapy and the Anti/pro-Angiogenic Growth Factor Equilibrium
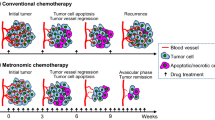
As described in the previous section of this chapter, the unbalanced expression of endogenous inhibitors (e.g., TSP-1) of angiogenesis and pro-angiogenic factors (e.g. VEGF or bFGF) toward the first ones is an important characteristic of the maintenance of a dormant tumor angiogenesis [25]. Metronomic chemotherapy may be a therapeutic approach that can induce or re-induce the tumor dormancy through a marked modulation of anti- and pro-angiogenic factors (Fig. 1) both in adjuvant setting or in the treatment of metastatic disease.

Angiogenic tumor dormancy, angiogenesis-dependant tumor growth, and metronomic chemotherapy-induced angiogenic tumor dormancy. Metronomic chemotherapy modulates the equilibrium of anti- and pro-angiogenic factors in tumor microenvironment, upregulating thrombospondin-1 (TSP-1) and decreasing the vascular endothelial growth factor (VEGF). Moreover, the low-dose chemotherapy blocks the recruitment of circulating endothelial progenitors (CEPs) in the tumor mass
Bocci and colleagues in 2003 [51] reported, for the first time, that the metronomic chemotherapy (e.g., paclitaxel, epothilones and cyclophosphamide) could induce expression of TSP-1 in vitro and in vivo. In particular, the authors demonstrated that the in vivo antiangiogenic and antitumor effects of daily oral metronomic cyclophosphamide were lost in TSP-1-null C57BL/6 mice affected by LLC, whereas, in contrast, these effects were maintained in TSP-1 wild type mice. More importantly, higher increases in circulating TSP-1 were detected in the plasma of responder human prostate (PC3) tumor xenograft-bearing mice treated with metronomic low-dose cyclophosphamide [51]. These findings were later confirmed using metronomic cyclophosphamide by Hamano et al. in in vivo models of murine cancers such as LLC and B16F10 melanoma [52] and by the group of Norrby in rats bearing a malignant prostate tumor (Dunning AT-1) [53], as well as by Vives and colleagues in mice with a human xenograft of ovarian cancer cell lines [54]. Moreover, also other metronomically-administered drugs were able to increase both the gene expression and the protein secretion of TSP-1 in preclinical in vitro and in vivo model. Metronomic gemcitabine was successfully used in human pancreatic adenocarcinomas xenografts, causing the reduction of tumor growth and the significant increase of TSP-1 [55, 54], whereas low dose capecitabine determined an antiangiogenic effect on human colorectal cancer COL-1 xenografts inducing TSP-1 expression in tumor tissues [56] and decreased microvessel density (MVD) in colon cancer elevating TSP-1 expression [57]. Metronomic S-1 (a 5-FU-based drug) and metronomic S-1 with vandetanib (a dual tyrosine kinase inhibitor of VEGFR-2 and EGFR) decreased MVDs and increased apoptosis in hepatocellular carcinoma tissues, upregulating the expression of TSP-1 [58]. Other examples of this phenomenon were the metronomic ceramide analogs (eg., C2 and AL6) that inhibited angiogenesis and tumor growth in pancreatic cancer through up-regulation of TSP-1 and caveolin-1 [59], whereas long term, low concentrations of SN-38 (the active metabolite of irinotecan) increased both TSP-1 gene expression and secretion by HT-29 colorectal cancer cells [60]. Finally, tubulin inhibitors such as paclitaxel and docetaxel have shown strong antiangiogenic characteristics at low concentrations [61, 62]. In particular, low doses of paclitaxel and of its different pharmaceutical formulations (e.g. nanoparticles) determined antiangiogenic effects through the marked increase of TSP-1 levels in tumor vascular endothelial cells [63] and in different tumor types such as ovarian carcinoma [64], colon cancer [65], breast cancer [66]. Furthermore, docetaxel increased the expression of TSP-1 in a gastric cancer model [67], blocking the angiogenic process and the tumor growth.
Interestingly, this significant upregulation of TSP-1 during metronomic chemotherapy was not only limited to preclinical findings but it was also found in patients enrolled in various phase II, metronomic chemotherapy clinical trials, involving different types of cancer. Indeed, Allegrini and colleagues described an increase of TSP-1 plasma levels in metastatic colorectal cancer patients at day 49 of treatment with a continuous low dose infusion of irinotecan (1.4 and 2.8 mg/m2/day) [68]. This finding was later confirmed in metastatic gastrointestinal cancer patients treated with a combination of metronomic cyclophosphamide (50 mg/day), UFT (100 mg/day) and celecoxib (200 mg/twice a day). Patients with a stable disease, during the metronomic schedule, had higher values of TSP-1 Area Under Curves (AUCs) if compared with patients with a progressive disease [69]. Recently, a similar result was obtained in metastatic castration-resistant prostate cancer patients treated with metronomic vinorelbine (30 mg/day p.o. thrice a week) plus 1 mg/day dexamethasone. Indeed, responder patients maintained higher plasma TSP-1 AUCs if compared to the non-responder ones [70]. Long-term oral administration of daily low-dose mercaptopurine and weekly low-dose methotrexate are used as maintenance chemotherapy in the treatment of acute lymphoblastic leukemia in children. Also this metronomic-like treatment have been described to determine a significant increase in TSP-1 plasma levels [71].
Besides the increased levels of the endogenous inhibitor of angiogenesis TSP-1, metronomic chemotherapy determine, in parallel, a well-described decrease of pro-angiogenic factor levels, such as VEGF, both in preclinical studies and in clinical trials. As such, the investigation of the antiangiogenic effects of the metronomic chemotherapy has focused on the modulation of the balance between angiogenic stimuli and natural inhibitors of angiogenesis. Indeed, another way that metronomic chemotherapy can conceivably cause an antiangiogenic effect, at least with certain drugs such as topotecan, a topoisomerase 1 inhibitor, or the anthracycline adriamycin is by suppression of the expression of HIF-1α—as originally reported by the group of Melillo and colleagues [72, 73]. HIF-1α is a known driver of VEGF-angiogenesis because it stimulates the VEGF production and secretion by hypoxic tumor cells [74]. Therefore, the pro-angiogenic VEGF levels were reduced in vitro in ovarian HeyA8 and SKOV3ip1 cancer cells by low concentrations of topotecan, independently of proteasome degradation and topoisomerase I inhibition [75]. Moreover, another camptothecin such as irinotecan have shown to reduce the expression of VEGF and HIF-1α in malignant glioma xenografts [76]. It has been also demonstrated that metronomic etoposide impaired the angiogenic equilibrium in tumors by inhibiting VEGF-A and FGF-2 secretion from tumor cells and by increasing endostatin plasma levels [77]. In another preclinical research performed in pancreatic cancer xenografts, metronomic gemcitabine decreased tumor levels of various proangiogenic molecules such as EGF, IL-1α, IL-8, ICAM-1, and VCAM-1 [78]. Moreover, Aktas and colleagues [79] tested lower doses of chemotherapeutic drugs such as 5-florouracil (5-FU), irinotecan, oxaliplatin, paclitaxel and docetaxel in different tumor cell lines, showing that these drugs decreased VEGF secretion from tumor cells without causing substantial cell killing. Both the expression and secretion of VEGF significantly decreased in BGC-823 gastric cancer cells treated with metronomic docetaxel [67], whereas the long term (144 h), continuous treatment with SN-38 of colon cancer cells (HT-29 and SW620) determined a significant decrease of secreted VEGF in cell media [60]. The 5-FU prodrug capecitabine metronomically administered decreased VEGF levels in in vivo colon cancer [57], and in gastric cancer [80] models. Furthermore, metronomic GMX1777, a chemotherapeutic drug affecting cellular energy metabolism, in a mouse model of neuroblastoma decreased stromal VEGF-A and PDGF-B mRNA in response to treatment [81]. These effects were also achieved combining different metronomic chemotherapy schedules. Mainetti and colleagues investigated the therapeutic efficacy of a combined treatment including metronomic cyclophosphamide and doxorubicin in two mouse mammary adenocarcinoma models. Interestingly, the combination was more effective than each monotherapy to decrease the VEGF serum concentration and increase tumor apoptosis [82].
Numerous phase I-II clinical studies in different types of cancer, using various chemotherapeutic drugs, have clearly suggested that plasma or serum VEGF is decreased during or after metronomic chemotherapy schedules, also combined with other drugs. In metastatic castration-resistant prostate cancer patients treated with metronomic cyclophosphamide (50 mg/day), celecoxib, and dexamethasone, the VEGF levels markedly increased in non responder subjects and remained significantly higher than in responders for more than 3 months [83]. In contrast, VEGF concentrations in responder patients constantly decreased to values corresponding to the half of the baseline [83]. Moreover, another phase II clinical trial performed in the same type of prostate cancer patients but treated with a combination of metronomic vinorelbine (30 mg/day p.o. thrice a week) plus dexamethasone, showed a plasma VEGF AUC0–24day significantly increased in non-responders if compared to the subjects with a PSA decrease [70]. Interestingly, germline VEGF-A polymorphisms predicted progression-free survival among advanced castration-resistant prostate cancer patients treated with metronomic cyclophosphamide [84]. In particular, patients harboring the VEGF-634CC genotype had a median progression-free survival (PFS) of 2.2 months whereas patients with the genotype -634CG/GG had a median PFS of 6.25 months (P = 0.0042) [84]. The decrease of plasma VEGF levels during metronomic chemotherapy have been also well described in metastatic breast cancer patients. Calleri and colleagues found out that patients affected by breast cancer with lower VEGF levels after 2 months of metronomic cyclophosphamide (50 mg/day) treatment had higher PFS, whereas at the time of progression there was a significant increase of VEGF [85]. A similar drop of serum VEGF was found in 171 metastatic breast cancer patients treated with metronomic cyclophosphamide (50 mg/day) combined with methotrexate or thalidomide after 2 months of therapy [86], whereas EL-Arab and co-workers treated the same type of patients with the combination of capecitabine (500 mg twice daily) together with oral cyclophosphamide (50 mg once daily), causing a significant decline of the median serum VEGF level after 2 and 6 months of therapy among subjects with a complete or partial response and a stable disease [87]. Interestingly, also in primary breast cancer, there was a significant suppression of VEGF-A expression in the letrozole/metronomic cyclophosphamide-treated group (50 mg/day) of patients if compared to the letrozole-treated group [88] with a lower VEGF expression at post-treatment residual histology. These data were later confirmed by Bazzola et al. who found that VEGF expression declined in tumor tissues in response to treatment with metronomic cyclophosphamide and letrozole [89]. Recently, the metronomic therapy including etoposide and cyclophosphamide determined the significant decrease of serum VEGF levels in relapsed or refractory non-Hodgkin’s lymphoma patients with overall response and disease control during different cycles of therapy [90].
Metronomic Chemotherapy and the Circulating Endothelial Progenitors
Although the role of BMDC in the early stages of tumor progression is still debated, it is well accepted that the induction of angiogenesis is a key step in the progression of microtumors. Therefore, this tumors have to activate ad recruit distant and normal cells such as CEPs that will facilitate the shift toward the generation of new blood vessels [3]. The development of therapeutic approaches that are able to block and inhibit the mobilization and viability of CEPs and other pro-angiogenic BMDCs will maintain or prolong the tumor dormancy due to the angiogenesis stoppage. In this perspective, the metronomic chemotherapy could be a perfect tool to achieve this aim (Fig. 1). Indeed, it has been described that low dose chemotherapy is able to suppress the BMDC proangiogenic cells such as CEPs [44]. In 2003, there was the first evidence of this effect in mice affected by lymphoma that underwent cycles of oral low-dose cyclophosphamide therapy [91]. The metronomic schedule markedly suppressed the number of CEPs during the therapy whereas, at the end of the drug administration, the number of endothelial progenitors increased again and tumors started to grow [91]. Furthermore, 2 years later the Kerbel’s team showed a clear correlation between the maximal suppression of CEP levels and the maximum antiangiogenic activity in mice treated with different drugs metronomically administered such as cyclophosphamide, vinblastine, cisplatin, or vinorelbine [92, 93]. For this reason, it has also been suggested that CEP suppression could be one of the main mechanisms of action of metronomic chemotherapy [94] and that this decline in blood of CEP levels could be used as pharmacodynamic biomarker of therapeutic efficacy [95]. Also oral metronomic topotecan in combination with pazopanib determined a significant reduction in viable CEPs as well as circulating endothelial cells (CECs), reducing the tumor MVD in several preclinical models of pediatric solid tumors [96]. The CEP percentage was found to be decreased in the peripheral blood of gastric tumor-bearing mice after the treatment with metronomic 5-FU or capecitabine [80]. Interestingly, Daenen et al. found that daily oral low-dose metronomic cyclophosphamide was capable of preventing the CEP spike and tumor colonization induced by a vascular disrupting agent if administered simultaneously [97].
Clinically, after the administration of trofosfamide-based conventional schedules of chemotherapy the numbers of circulating CEPs increased, whereas, in sharp contrast, under low-dose metronomic trofosfamide, the numbers of circulating CEPs declined significantly in blood of tumor patients [98]. Calleri and colleagues showed that in a group of 15 long-term responders to metronomic chemotherapy, there were significant trends toward lower levels of CEPs and CECs [85]. In a population of gastrointestinal cancer patients, the levels of progenitor or stem cell mRNA (i.e. CD133), during the metronomic combined treatment of UFT and cyclophosphamide, were consistently lower in those with stable disease whereas a substantial increase of CD133 gene expression was found in the progressive disease [69].
Conclusions
Angiogenic tumor dormancy occurs as a result of a dynamic equilibrium state in which antiangiogenic and pro-angiogenic stimuli are balanced and angiogenesis process is blocked. It can take place at the primary site of cancer, but also in metastatic lesions. Thus, a therapeutic approach that can achieve an induction or a “re-induction” of the angiogenic tumor dormancy in primary and/or metastatic tumors is highly welcomed in the clinical oncology field. In this perspective, metronomic chemotherapy, by upregulating the endogenous inhibitor TSP-1 and, parallely, decreasing pro-angiogenic factors or blocking CEPs, may be able to play a main role in the modulation of the angiogenic tumor dormancy. Further studies are needed to better investigate this particular aspect of this promising therapeutic tool.
References
Goss PE, Chambers AF (2010) Does tumour dormancy offer a therapeutic target? Nat Rev Cancer 10(12):871–877. doi:10.1038/nrc2933
Udagawa T (2008) Tumor dormancy of primary and secondary cancers. APMIS 116(7–8):615–628. doi:10.1111/j.1600-0463.2008.01077.x
Shaked Y, McAllister S, Fainaru O, Almog N (2014) Tumor dormancy and the angiogenic switch: possible implications of bone marrow-derived cells. Curr Pharm Des 20(30):4920–4933
Osisami M, Keller ET (2013) Mechanisms of metastatic tumor dormancy. J Clin Med 2(3):136–150. doi:10.3390/jcm2030136
Uhr JW, Pantel K (2011) Controversies in clinical cancer dormancy. Proc Natl Acad Sci U S A 108(30):12396–12400. doi:10.1073/pnas.1106613108
Aguirre-Ghiso JA (2007) Models, mechanisms and clinical evidence for cancer dormancy. Nat Rev Cancer 7(11):834–846. doi:10.1038/nrc2256
Favaro E, Amadori A, Indraccolo S (2008) Cellular interactions in the vascular niche: implications in the regulation of tumor dormancy. APMIS 116(7–8):648–659. doi:10.1111/j.1600-0463.2008.01025.x
Almog N (2010) Molecular mechanisms underlying tumor dormancy. Cancer Lett 294(2):139–146. doi:10.1016/j.canlet.2010.03.004
Fidler IJ (2003) The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer 3(6):453–458. doi:10.1038/nrc1098
Sosa MS, Bragado P, Aguirre-Ghiso JA (2014) Mechanisms of disseminated cancer cell dormancy: an awakening field. Nat Rev Cancer 14(9):611–622. doi:10.1038/nrc3793
Koebel CM, Vermi W, Swann JB, Zerafa N, Rodig SJ, Old LJ, Smyth MJ, Schreiber RD (2007) Adaptive immunity maintains occult cancer in an equilibrium state. Nature 450(7171):903–907. doi:10.1038/nature06309
Moserle L, Amadori A, Indraccolo S (2009) The angiogenic switch: implications in the regulation of tumor dormancy. Curr Mol Med 9(8):935–941
Barkan D, Kleinman H, Simmons JL, Asmussen H, Kamaraju AK, Hoenorhoff MJ, Liu ZY, Costes SV, Cho EH, Lockett S, Khanna C, Chambers AF, Green JE (2008) Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res 68(15):6241–6250. doi:10.1158/0008-5472.can-07-6849
Naumov GN, Folkman J, Straume O (2009) Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin Exp Metastasis 26(1):51–60. doi:10.1007/s10585-008-9176-0
Naumov GN, Folkman J, Straume O, Akslen LA (2008) Tumor-vascular interactions and tumor dormancy. APMIS 116(7–8):569–585. doi:10.1111/j.1600-0463.2008.01213.x
Chambers AF (2009) Influence of diet on metastasis and tumor dormancy. Clin Exp Metastasis 26(1):61–66. doi:10.1007/s10585-008-9164-4
Siveen KS, Prabhu K, Krishnankutty R, Kuttikrishnan S, Tsakou M, Alali FQ, Dermime S, Mohammad RM, Uddin S (2017) Vascular endothelial growth factor (VEGF) signaling in tumour vascularization: potential and challenges. Curr Vasc Pharmacol. doi:10.2174/1570161115666170105124038
Ribatti D, Vacca A, Dammacco F (1999) The role of the vascular phase in solid tumor growth: a historical review. Neoplasia 1(4):293–302
Jayson GC, Kerbel R, Ellis LM, Harris AL (2016) Antiangiogenic therapy in oncology: current status and future directions. Lancet 388(10043):518–529. doi:10.1016/s0140-6736(15)01088-0
Naumov GN, Akslen LA, Folkman J (2006) Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle 5(16):1779–1787. doi:10.4161/cc.5.16.3018
Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285(21):1182–1186. doi:10.1056/nejm197111182852108
Natale G, Bocci G, Lenzi P (2017) Looking for the word “angiogenesis” in the history of health sciences: from ancient times to the first decades of the twentieth century. World J Surg 41(6):1625–1634. doi:10.1007/s00268-016-3680-1
Katoh M (2016) Therapeutics targeting FGF signaling network in human diseases. Trends Pharmacol Sci 37(12):1081–1096. doi:10.1016/j.tips.2016.10.003
Dewerchin M, Carmeliet P (2014) Placental growth factor in cancer. Expert Opin Ther Targets 18(11):1339–1354. doi:10.1517/14728222.2014.948420
Kang SY, Watnick RS (2008) Regulation of tumor dormancy as a function of tumor-mediated paracrine regulation of stromal tsp-1 and VEGF expression. APMIS 116(7–8):638–647. doi:10.1111/j.1600-0463.2008.01138.x
Gacche RN, Meshram RJ (2013) Targeting tumor micro-environment for design and development of novel anti-angiogenic agents arresting tumor growth. Prog Biophys Mol Biol 113(2):333–354. doi:10.1016/j.pbiomolbio.2013.10.001
Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS (1995) Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res 55(20):4575–4580
Achilles EG, Fernandez A, Allred EN, Kisker O, Udagawa T, Beecken WD, Flynn E, Folkman J (2001) Heterogeneity of angiogenic activity in a human liposarcoma: a proposed mechanism for "no take" of human tumors in mice. J Natl Cancer Inst 93(14):1075–1081
Pugh CW, Ratcliffe PJ (2003) Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med 9(6):677–684. doi:10.1038/nm0603-677
Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N (2006) A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst 98(5):316–325. doi:10.1093/jnci/djj068
Almog N, Ma L, Raychowdhury R, Schwager C, Erber R, Short S, Hlatky L, Vajkoczy P, Huber PE, Folkman J, Abdollahi A (2009) Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res 69(3):836–844. doi:10.1158/0008-5472.can-08-2590
Panigrahy D, Edin ML, Lee CR, Huang S, Bielenberg DR, Butterfield CE, Barnes CM, Mammoto A, Mammoto T, Luria A, Benny O, Chaponis DM, Dudley AC, Greene ER, Vergilio JA, Pietramaggiori G, Scherer-Pietramaggiori SS, Short SM, Seth M, Lih FB, Tomer KB, Yang J, Schwendener RA, Hammock BD, Falck JR, Manthati VL, Ingber DE, Kaipainen A, D’Amore PA, Kieran MW, Zeldin DC (2012) Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J Clin Invest 122(1):178–191. doi:10.1172/jci58128
Indraccolo S (2013) Insights into the regulation of tumor dormancy by angiogenesis in experimental tumors. Adv Exp Med Biol 734:37–52. doi:10.1007/978-1-4614-1445-2
Indraccolo S, Minuzzo S, Masiero M, Pusceddu I, Persano L, Moserle L, Reboldi A, Favaro E, Mecarozzi M, Di Mario G, Screpanti I, Ponzoni M, Doglioni C, Amadori A (2009) Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res 69(4):1314–1323. doi:10.1158/0008-5472.can-08-2791
Rak J, Milsom C, Yu J (2008) Vascular determinants of cancer stem cell dormancy—do age and coagulation system play a role? APMIS 116(7–8):660–676. doi:10.1111/j.1600-0463.2008.01058.x
Indraccolo S, Favaro E, Amadori A (2006) Dormant tumors awaken by a short-term angiogenic burst: the spike hypothesis. Cell Cycle 5(16):1751–1755. doi:10.4161/cc.5.16.2985
Mowers EE, Sharifi MN, Macleod KF (2017) Autophagy in cancer metastasis. Oncogene 36(12):1619–1630. doi:10.1038/onc.2016.333
Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G (2011) Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res 71(15):5346–5356. doi:10.1158/0008-5472.can-11-0241
Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, Bray MA, Carpenter AE, Jirstrom K, Magnusson K, Ebert BL, Ponten F, Weinberg RA, McAllister SS (2011) Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest 121(2):784–799. doi:10.1172/jci43757
Png KJ, Halberg N, Yoshida M, Tavazoie SF (2011) A microRNA regulon that mediates endothelial recruitment and metastasis by cancer cells. Nature 481(7380):190–194. doi:10.1038/nature10661
Straume O, Shimamura T, Lampa MJ, Carretero J, Oyan AM, Jia D, Borgman CL, Soucheray M, Downing SR, Short SM, Kang SY, Wang S, Chen L, Collett K, Bachmann I, Wong KK, Shapiro GI, Kalland KH, Folkman J, Watnick RS, Akslen LA, Naumov GN (2012) Suppression of heat shock protein 27 induces long-term dormancy in human breast cancer. Proc Natl Acad Sci U S A 109(22):8699–8704. doi:10.1073/pnas.1017909109
Almog N, Ma L, Schwager C, Brinkmann BG, Beheshti A, Vajkoczy P, Folkman J, Hlatky L, Abdollahi A (2012) Consensus micro RNAs governing the switch of dormant tumors to the fast-growing angiogenic phenotype. PLoS One 7(8):e44001. doi:10.1371/journal.pone.0044001
Bocci G, Kerbel RS (2016) Pharmacokinetics of metronomic chemotherapy: a neglected but crucial aspect. Nat Rev Clin Oncol 13(11):659–673. doi:10.1038/nrclinonc.2016.64
Pasquier E, Kavallaris M, Andre N (2010) Metronomic chemotherapy: new rationale for new directions. Nat Rev Clin Oncol 7(8):455–465. doi:10.1038/nrclinonc.2010.82
Penel N, Adenis A, Bocci G (2012) Cyclophosphamide-based metronomic chemotherapy: after 10 years of experience, where do we stand and where are we going? Crit Rev Oncol Hematol 82(1):40–50. doi:10.1016/j.critrevonc.2011.04.009
Bocci G, Nicolaou KC, Kerbel RS (2002) Protracted low-dose effects on human endothelial cell proliferation and survival in vitro reveal a selective antiangiogenic window for various chemotherapeutic drugs. Cancer Res 62(23):6938–6943
Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B (2007) Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother 56(5):641–648. doi:10.1007/s00262-006-0225-8
Fioravanti A, Canu B, Ali G, Orlandi P, Allegrini G, Di Desidero T, Emmenegger U, Fontanini G, Danesi R, Del Tacca M, Falcone A, Bocci G (2009) Metronomic 5-fluorouracil, oxaliplatin and irinotecan in colorectal cancer. Eur J Pharmacol 619(1–3):8–14. doi:10.1016/j.ejphar.2009.08.020
Folkins C, Shaked Y, Man S, Tang T, Lee CR, Zhu Z, Hoffman RM, Kerbel RS (2009) Glioma tumor stem-like cells promote tumor angiogenesis and vasculogenesis via vascular endothelial growth factor and stromal-derived factor 1. Cancer Res 69(18):7243–7251. doi:10.1158/0008-5472.can-09-0167
Chan TS, Hsu CC, Pai VC, Liao WY, Huang SS, Tan KT, Yen CJ, Hsu SC, Chen WY, Shan YS, Li CR, Lee MT, Jiang KY, Chu JM, Lien GS, Weaver VM, Tsai KK (2016) Metronomic chemotherapy prevents therapy-induced stromal activation and induction of tumor-initiating cells. J Exp Med 213(13):2967–2988. doi:10.1084/jem.20151665
Bocci G, Francia G, Man S, Lawler J, Kerbel RS (2003) Thrombospondin 1, a mediator of the antiangiogenic effects of low-dose metronomic chemotherapy. Proc Natl Acad Sci U S A 100(22):12917–12922. doi:10.1073/pnas.2135406100
Hamano Y, Sugimoto H, Soubasakos MA, Kieran M, Olsen BR, Lawler J, Sudhakar A, Kalluri R (2004) Thrombospondin-1 associated with tumor microenvironment contributes to low-dose cyclophosphamide-mediated endothelial cell apoptosis and tumor growth suppression. Cancer Res 64(5):1570–1574
Damber JE, Vallbo C, Albertsson P, Lennernas B, Norrby K (2006) The anti-tumour effect of low-dose continuous chemotherapy may partly be mediated by thrombospondin. Cancer Chemother Pharmacol 58(3):354–360. doi:10.1007/s00280-005-0163-8
Vives M, Ginesta MM, Gracova K, Graupera M, Casanovas O, Capella G, Serrano T, Laquente B, Vinals F (2013) Metronomic chemotherapy following the maximum tolerated dose is an effective anti-tumour therapy affecting angiogenesis, tumour dissemination and cancer stem cells. Int J Cancer 133(10):2464–2472. doi:10.1002/ijc.28259
Laquente B, Lacasa C, Ginesta MM, Casanovas O, Figueras A, Galan M, Ribas IG, Germa JR, Capella G, Vinals F (2008) Antiangiogenic effect of gemcitabine following metronomic administration in a pancreas cancer model. Mol Cancer Ther 7(3):638–647. doi:10.1158/1535-7163.mct-07-2122
Ooyama A, Oka T, Zhao HY, Yamamoto M, Akiyama S, Fukushima M (2008) Anti-angiogenic effect of 5-fluorouracil-based drugs against human colon cancer xenografts. Cancer Lett 267(1):26–36. doi:10.1016/j.canlet.2008.03.008
Shi H, Jiang J, Ji J, Shi M, Cai Q, Chen X, Yu Y, Liu B, Zhu Z, Zhang J (2014) Anti-angiogenesis participates in antitumor effects of metronomic capecitabine on colon cancer. Cancer Lett 349(2):128–135. doi:10.1016/j.canlet.2014.04.002
Iwamoto H, Torimura T, Nakamura T, Hashimoto O, Inoue K, Kurogi J, Niizeki T, Kuwahara R, Abe M, Koga H, Yano H, Kerbel RS, Ueno T, Sata M (2011) Metronomic S-1 chemotherapy and vandetanib: an efficacious and nontoxic treatment for hepatocellular carcinoma. Neoplasia 13(3):187–197
Bocci G, Fioravanti A, Orlandi P, Di Desidero T, Natale G, Fanelli G, Viacava P, Naccarato AG, Francia G, Danesi R (2012) Metronomic ceramide analogs inhibit angiogenesis in pancreatic cancer through up-regulation of caveolin-1 and thrombospondin-1 and down-regulation of cyclin D1. Neoplasia 14(9):833–845
Bocci G, Falcone A, Fioravanti A, Orlandi P, Di Paolo A, Fanelli G, Viacava P, Naccarato AG, Kerbel RS, Danesi R, Del Tacca M, Allegrini G (2008) Antiangiogenic and anticolorectal cancer effects of metronomic irinotecan chemotherapy alone and in combination with semaxinib. Br J Cancer 98(10):1619–1629. doi:10.1038/sj.bjc.6604352
Bocci G, Di Paolo A, Danesi R (2013) The pharmacological bases of the antiangiogenic activity of paclitaxel. Angiogenesis 16(3):481–492. doi:10.1007/s10456-013-9334-0
Pasquier E, Andre N, Braguer D (2007) Targeting microtubules to inhibit angiogenesis and disrupt tumour vasculature: implications for cancer treatment. Curr Cancer Drug Targets 7(6):566–581
Luan X, Guan YY, Lovell JF, Zhao M, Lu Q, Liu YR, Liu HJ, Gao YG, Dong X, Yang SC, Zheng L, Sun P, Fang C, Chen HZ (2016) Tumor priming using metronomic chemotherapy with neovasculature-targeted, nanoparticulate paclitaxel. Biomaterials 95:60–73. doi:10.1016/j.biomaterials.2016.04.008
Lee SJ, Ghosh SC, Han HD, Stone RL, Bottsford-Miller J, Shen DY, Auzenne EJ, Lopez-Araujo A, Lu C, Nishimura M, Pecot CV, Zand B, Thanapprapasr D, Jennings NB, Kang Y, Huang J, Hu W, Klostergaard J, Sood AK (2012) Metronomic activity of CD44-targeted hyaluronic acid-paclitaxel in ovarian carcinoma. Clin Cancer Res 18(15):4114–4121. doi:10.1158/1078-0432.ccr-11-3250
Zhang M, Tao W, Pan S, Sun X, Jiang H (2009) Low-dose metronomic chemotherapy of paclitaxel synergizes with cetuximab to suppress human colon cancer xenografts. Anti-Cancer Drugs 20(5):355–363. doi:10.1097/CAD.0b013e3283299f36
Tao WY, Liang XS, Liu Y, Wang CY, Pang D (2015) Decrease of let-7f in low-dose metronomic paclitaxel chemotherapy contributed to upregulation of thrombospondin-1 in breast cancer. Int J Biol Sci 11(1):48–58. doi:10.7150/ijbs.9969
Wu H, Xin Y, Zhao J, Sun D, Li W, Hu Y, Wang S (2011) Metronomic docetaxel chemotherapy inhibits angiogenesis and tumor growth in a gastric cancer model. Cancer Chemother Pharmacol 68(4):879–887. doi:10.1007/s00280-011-1563-6
Allegrini G, Falcone A, Fioravanti A, Barletta MT, Orlandi P, Loupakis F, Cerri E, Masi G, Di Paolo A, Kerbel RS, Danesi R, Del Tacca M, Bocci G (2008) A pharmacokinetic and pharmacodynamic study on metronomic irinotecan in metastatic colorectal cancer patients. Br J Cancer 98(8):1312–1319. doi:10.1038/sj.bjc.6604311
Allegrini G, Di Desidero T, Barletta MT, Fioravanti A, Orlandi P, Canu B, Chericoni S, Loupakis F, Di Paolo A, Masi G, Fontana A, Lucchesi S, Arrighi G, Giusiani M, Ciarlo A, Brandi G, Danesi R, Kerbel RS, Falcone A, Bocci G (2012) Clinical, pharmacokinetic and pharmacodynamic evaluations of metronomic UFT and cyclophosphamide plus celecoxib in patients with advanced refractory gastrointestinal cancers. Angiogenesis 15(2):275–286. doi:10.1007/s10456-012-9260-6
Di Desidero T, Derosa L, Galli L, Orlandi P, Fontana A, Fioravanti A, Marconcini R, Giorgi M, Campi B, Saba A, Lucchesi S, Felipetto R, Danesi R, Francia G, Allegrini G, Falcone A, Bocci G (2016) Clinical, pharmacodynamic and pharmacokinetic results of a prospective phase II study on oral metronomic vinorelbine and dexamethasone in castration-resistant prostate cancer patients. Investig New Drugs 34(6):760–770. doi:10.1007/s10637-016-0385-0
Andre N, Cointe S, Barlogis V, Arnaud L, Lacroix R, Pasquier E, Dignat-George F, Michel G, Sabatier F (2015) Maintenance chemotherapy in children with ALL exerts metronomic-like thrombospondin-1 associated anti-endothelial effect. Oncotarget 6(26):23008–23014. doi:10.18632/oncotarget.3984
Calvani M, Rapisarda A, Uranchimeg B, Shoemaker RH, Melillo G (2006) Hypoxic induction of an HIF-1alpha-dependent bFGF autocrine loop drives angiogenesis in human endothelial cells. Blood 107(7):2705–2712. doi:10.1182/blood-2005-09-3541
Rapisarda A, Zalek J, Hollingshead M, Braunschweig T, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP, Hewitt SM, Shoemaker RH, Melillo G (2004) Schedule-dependent inhibition of hypoxia-inducible factor-1alpha protein accumulation, angiogenesis, and tumor growth by topotecan in U251-HRE glioblastoma xenografts. Cancer Res 64(19):6845–6848. doi:10.1158/0008-5472.can-04-2116
Masoud GN, Li W (2015) HIF-1alpha pathway: role, regulation and intervention for cancer therapy. Acta Pharm Sin B 5(5):378–389. doi:10.1016/j.apsb.2015.05.007
Merritt WM, Danes CG, Shahzad MM, Lin YG, Kamat AA, Han LY, Spannuth WA, Nick AM, Mangala LS, Stone RL, Kim HS, Gershenson DM, Jaffe RB, Coleman RL, Chandra J, Sood AK (2009) Anti-angiogenic properties of metronomic topotecan in ovarian carcinoma. Cancer Biol Ther 8(16):1596–1603
Takano S, Kamiyama H, Mashiko R, Osuka S, Ishikawa E, Matsumura A (2010) Metronomic treatment of malignant glioma xenografts with irinotecan (CPT-11) inhibits angiogenesis and tumor growth. J Neuro-Oncol 99(2):177–185. doi:10.1007/s11060-010-0118-8
Panigrahy D, Kaipainen A, Butterfield CE, Chaponis DM, Laforme AM, Folkman J, Kieran MW (2010) Inhibition of tumor angiogenesis by oral etoposide. Exp Ther Med 1(5):739–746. doi:10.3892/etm.2010.127
Cham KK, Baker JH, Takhar KS, Flexman JA, Wong MQ, Owen DA, Yung A, Kozlowski P, Reinsberg SA, Chu EM, Chang CW, Buczkowski AK, Chung SW, Scudamore CH, Minchinton AI, Yapp DT, Ng SS (2010) Metronomic gemcitabine suppresses tumour growth, improves perfusion, and reduces hypoxia in human pancreatic ductal adenocarcinoma. Br J Cancer 103(1):52–60. doi:10.1038/sj.bjc.6605727
Aktas SH, Akbulut H, Akgun N, Icli F (2012) Low dose chemotherapeutic drugs without overt cytotoxic effects decrease the secretion of VEGF by cultured human tumor cells: a tentative relationship between drug type and tumor cell type response. Cancer Biomark 12(3):135–140. doi:10.3233/cbm-130301
Yuan F, Shi H, Ji J, Cai Q, Chen X, Yu Y, Liu B, Zhu Z, Zhang J (2015) Capecitabine metronomic chemotherapy inhibits the proliferation of gastric cancer cells through anti-angiogenesis. Oncol Rep 33(4):1753–1762. doi:10.3892/or.2015.3765
Fuchs D, Rodriguez A, Eriksson S, Christofferson R, Sundberg C, Azarbayjani F (2010) Metronomic administration of the drug GMX1777, a cellular NAD synthesis inhibitor, results in neuroblastoma regression and vessel maturation without inducing drug resistance. Int J Cancer 126(12):2773–2789. doi:10.1002/ijc.25206
Mainetti LE, Rico MJ, Fernandez-Zenobi MV, Perroud HA, Roggero EA, Rozados VR, Scharovsky OG (2013) Therapeutic efficacy of metronomic chemotherapy with cyclophosphamide and doxorubicin on murine mammary adenocarcinomas. Ann Oncol 24(9):2310–2316. doi:10.1093/annonc/mdt164
Fontana A, Galli L, Fioravanti A, Orlandi P, Galli C, Landi L, Bursi S, Allegrini G, Fontana E, Di Marsico R, Antonuzzo A, D’Arcangelo M, Danesi R, Del Tacca M, Falcone A, Bocci G (2009) Clinical and pharmacodynamic evaluation of metronomic cyclophosphamide, celecoxib, and dexamethasone in advanced hormone-refractory prostate cancer. Clin Cancer Res 15(15):4954–4962. doi:10.1158/1078-0432.ccr-08-3317
Orlandi P, Fontana A, Fioravanti A, Di Desidero T, Galli L, Derosa L, Canu B, Marconcini R, Biasco E, Solini A, Francia G, Danesi R, Falcone A, Bocci G (2013) VEGF-A polymorphisms predict progression-free survival among advanced castration-resistant prostate cancer patients treated with metronomic cyclophosphamide. Br J Cancer 109(4):957–964. doi:10.1038/bjc.2013.398
Calleri A, Bono A, Bagnardi V, Quarna J, Mancuso P, Rabascio C, Dellapasqua S, Campagnoli E, Shaked Y, Goldhirsch A, Colleoni M, Bertolini F (2009) Predictive potential of angiogenic growth factors and circulating endothelial cells in breast cancer patients receiving metronomic chemotherapy plus bevacizumab. Clin Cancer Res 15(24):7652–7657. doi:10.1158/1078-0432.ccr-09-1493
Colleoni M, Orlando L, Sanna G, Rocca A, Maisonneuve P, Peruzzotti G, Ghisini R, Sandri MT, Zorzino L, Nole F, Viale G, Goldhirsch A (2006) Metronomic low-dose oral cyclophosphamide and methotrexate plus or minus thalidomide in metastatic breast cancer: antitumor activity and biological effects. Ann Oncol 17(2):232–238. doi:10.1093/annonc/mdj066
El-Arab LR, Swellam M, El Mahdy MM (2012) Metronomic chemotherapy in metastatic breast cancer: impact on VEGF. J Egypt Natl Canc Inst 24(1):15–22. doi:10.1016/j.jnci.2011.12.002
Bottini A, Generali D, Brizzi MP, Fox SB, Bersiga A, Bonardi S, Allevi G, Aguggini S, Bodini G, Milani M, Dionisio R, Bernardi C, Montruccoli A, Bruzzi P, Harris AL, Dogliotti L, Berruti A (2006) Randomized phase II trial of letrozole and letrozole plus low-dose metronomic oral cyclophosphamide as primary systemic treatment in elderly breast cancer patients. J Clin Oncol 24(22):3623–3628. doi:10.1200/jco.2005.04.5773
Bazzola L, Foroni C, Andreis D, Zanoni V, RC M, Allevi G, Aguggini S, Strina C, Milani M, Venturini S, Ferrozzi F, Giardini R, Bertoni R, Turley H, Gatter K, Petronini PG, Fox SB, Harris AL, Martinotti M, Berruti A, Bottini A, Reynolds AR, Generali D (2015) Combination of letrozole, metronomic cyclophosphamide and sorafenib is well-tolerated and shows activity in patients with primary breast cancer. Br J Cancer 112(1):52–60. doi:10.1038/bjc.2014.563
Zeng J, Yang L, Huang F, Hong T, He Z, Lei J, Sun H, Lu Y, Hao X (2016) The metronomic therapy with prednisone, etoposide, and cyclophosphamide reduces the serum levels of VEGF and circulating endothelial cells and improves response rates and progression-free survival in patients with relapsed or refractory non-Hodgkin’s lymphoma. Cancer Chemother Pharmacol 78(4):801–808. doi:10.1007/s00280-016-3136-1
Bertolini F, Paul S, Mancuso P, Monestiroli S, Gobbi A, Shaked Y, Kerbel RS (2003) Maximum tolerable dose and low-dose metronomic chemotherapy have opposite effects on the mobilization and viability of circulating endothelial progenitor cells. Cancer Res 63(15):4342–4346
Shaked Y, Bertolini F, Man S, Rogers MS, Cervi D, Foutz T, Rawn K, Voskas D, Dumont DJ, Ben-David Y, Lawler J, Henkin J, Huber J, Hicklin DJ, D’Amato RJ, Kerbel RS (2005) Genetic heterogeneity of the vasculogenic phenotype parallels angiogenesis: implications for cellular surrogate marker analysis of antiangiogenesis. Cancer Cell 7(1):101–111. doi:10.1016/j.ccr.2004.11.023
Shaked Y, Emmenegger U, Francia G, Chen L, Lee CR, Man S, Paraghamian A, Ben-David Y, Kerbel RS (2005) Low-dose metronomic combined with intermittent bolus-dose cyclophosphamide is an effective long-term chemotherapy treatment strategy. Cancer Res 65(16):7045–7051. doi:10.1158/0008-5472.can-05-0765
Shaked Y, Emmenegger U, Man S, Cervi D, Bertolini F, Ben-David Y, Kerbel RS (2005) Optimal biologic dose of metronomic chemotherapy regimens is associated with maximum antiangiogenic activity. Blood 106(9):3058–3061. doi:10.1182/blood-2005-04-1422
Shaked Y, Bocci G, Munoz R, Man S, Ebos JM, Hicklin DJ, Bertolini F, D’Amato R, Kerbel RS (2005) Cellular and molecular surrogate markers to monitor targeted and non-targeted antiangiogenic drug activity and determine optimal biologic dose. Curr Cancer Drug Targets 5(7):551–559
Kumar S, Mokhtari RB, Sheikh R, Wu B, Zhang L, Xu P, Man S, Oliveira ID, Yeger H, Kerbel RS, Baruchel S (2011) Metronomic oral topotecan with pazopanib is an active antiangiogenic regimen in mouse models of aggressive pediatric solid tumor. Clin Cancer Res 17(17):5656–5667. doi:10.1158/1078-0432.ccr-11-0078
Daenen LG, Shaked Y, Man S, Xu P, Voest EE, Hoffman RM, Chaplin DJ, Kerbel RS (2009) Low-dose metronomic cyclophosphamide combined with vascular disrupting therapy induces potent antitumor activity in preclinical human tumor xenograft models. Mol Cancer Ther 8(10):2872–2881. doi:10.1158/1535-7163.mct-09-0583
Stoelting S, Trefzer T, Kisro J, Steinke A, Wagner T, Peters SO (2008) Low-dose oral metronomic chemotherapy prevents mobilization of endothelial progenitor cells into the blood of cancer patients. In Vivo 22(6):831–836
Acknowledgements
G.B.’s research is currently supported by grants from the Italian Association of Cancer Research (AIRC, IG 17672) and the Istituto Toscano Tumori (ITT).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2017 Springer International Publishing AG
About this chapter
Cite this chapter
Natale, G., Bocci, G. (2017). Tumor Dormancy, Angiogenesis and Metronomic Chemotherapy. In: Wang, Y., Crea, F. (eds) Tumor Dormancy and Recurrence. Cancer Drug Discovery and Development. Humana Press, Cham. https://doi.org/10.1007/978-3-319-59242-8_3
Download citation
DOI: https://doi.org/10.1007/978-3-319-59242-8_3
Published:
Publisher Name: Humana Press, Cham
Print ISBN: 978-3-319-59240-4
Online ISBN: 978-3-319-59242-8
eBook Packages: MedicineMedicine (R0)