
Abstract
The glutamine–glutamate/GABA cycle is an astrocytic-neuronal pathway transferring precursors for transmitter glutamate and GABA from astrocytes to neurons. In addition, the cycle carries released transmitter back to astrocytes, where a minor fraction (~25 %) is degraded (requiring a similar amount of resynthesis) and the remainder returned to the neurons for reuse. The flux in the cycle is intense, amounting to the same value as neuronal glucose utilization rate or 75–80 % of total cortical glucose consumption. This glucose:glutamate ratio is reduced when high amounts of β-hydroxybutyrate are present, but β-hydroxybutyrate can at most replace 60 % of glucose during awake brain function. The cycle is initiated by α-ketoglutarate production in astrocytes and its conversion via glutamate to glutamine which is released. A crucial reaction in the cycle is metabolism of glutamine after its accumulation in neurons. In glutamatergic neurons all generated glutamate enters the mitochondria and its exit to the cytosol occurs in a process resembling the malate-aspartate shuttle and therefore requiring concomitant pyruvate metabolism. In GABAergic neurons one half enters the mitochondria, whereas the other one half is released directly from the cytosol. A revised concept is proposed for the synthesis and metabolism of vesicular and nonvesicular GABA. It includes the well-established neuronal GABA reuptake, its metabolism, and use for resynthesis of vesicular GABA. In contrast, mitochondrial glutamate is by transamination to α-ketoglutarate and subsequent retransamination to releasable glutamate essential for the transaminations occurring during metabolism of accumulated GABA and subsequent resynthesis of vesicular GABA.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- Astrocyte
- β-Hydroxybutyrate
- Branched chain amino acids
- GABAergic neuron
- Glucose
- Glutamatergic neuron
- Glutamate–glutamine cycle
- Neuronal GABA uptake
- Succinate
2.1 Introduction
The brain is almost unique among organs in having an obligate requirement of glucose for function. However, the brain can use a variety of other substrates including lactate, β-hydroxybutyrate , acetate, and medium and long chain fatty acids. Although the brain’s relative preference for substrates was well known from the pioneering AV difference studies of Kety and colleagues (Siesjö 1978), the reason for requirement for glucose has remained unexplained. In this paper, we examine evidence ranging from isolated enzymes, to cell culture, to in vivo studies to address this question. Our overall hypothesis is that the obligate requirement for glucose is due to the special needs for supporting the large glutamate and γ-aminobutyric acid (GABA) neurotransmitter cycling fluxes in the awake brain. We describe a model for glutamate–glutamine cycling that can explain the observed glucose dependence, as well as a revised model that explains the reduced dependence on glucose in GABAergic neurons . These models are consistent with the present results over a wide range of systems. Although the definitive in vivo studies remain to be done to test these models they will hopefully be useful in guiding experimentation.
2.2 Brain Metabolism of Glucose, Lactate, and β-Hydroxybutyrate
2.2.1 Glucose
The adult mammalian brain consumes about 20 % of total bodily oxygen and thus calorie utilization (Raichle and Gusnard 2002), and it normally uses glucose as its only or main substrate (Sokoloff et al. 1977). Studies in the rodent and human brain in vivo using 13C-based magnetic resonance spectroscopy (MRS) and labeled glucose or acetate, which is oxidized in astrocytes but not in neurons (Muir et al. 1986; Waniewski and Martin 1998) showed that in the awake human brain astrocytes contribute ~25 % of the oxidative metabolism in brain gray matter (Gruetter et al. 2001; Lebon et al. 2002; Mason et al. 2007). Most of this glucose is metabolized via formation of two molecules of pyruvate in the cytosol from one molecule of glucose (glycolysis ) followed by mitochondrial formation of acetyl coenzyme A (ac.CoA) from pyruvate (Fig. 2.1). The latter reaction is catalyzed by the pyruvate dehydrogenase complex (PDH) , which is present in all brain cells. In the tricarboxylic acid (TCA) cycle the acetyl part of acetyl CoA (containing two carbon atoms) condenses with a four-carbon TCA intermediate, oxaloacetate (OAA), to form a six-carbon TCA cycle constituent (citrate), which via two decarboxylations regenerates OAA (4 carbon atoms), ready to condense with another molecule of acetyl CoA.

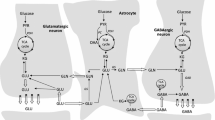
Cartoon of glucose metabolism via pyruvate in neurons (left—N) and astrocytes (right—A) and of glutamine–glutamate (GABA) cycling. One molecule glucose is metabolized by glycolysis in the cytosol to two molecules of pyruvate in a complex and strictly regulated pathway, where one oxidative process requires transfer of reducing equivalents to the mitochondria (see, e.g., Hertz and Dienel 2002). In both neurons and astrocytes pyruvate metabolism via acetyl Coenzyme A (ac.CoA) leads to formation of citrate by condensation with preexisting oxaloacetate (OAA) in the tricarboxylic acid (TCA) , an end result of the previous turn of the cycle. Citrate oxidation in the TCA cycle includes two decarboxylations, leading to reformation of oxaloacetate , ready for another turn of the cycle, and to reduction of NADH, creating large amounts of energy (ATP) via reoxidation in the electron transport chain. Pyruvate carboxylation, which is active in astrocytes , but absent in neurons (reviewed in Hertz 2013), creates a new molecule of oxaloacetate , which after condensation with acetyl Coenzyme A , derived from a second molecule of pyruvate, forms a new molecule of citrate. α-Ketoglutarate (α-KG), one of the intermediates of the TCA cycle can leave the cycle to form glutamate (glu), catalyzed by either aspartate aminotransferase or glutamate dehydrogenase . Further metabolism by the cytosolic and astrocyte-specific enzyme glutamine synthetase leads to the formation of glutamine (gln). In glutamatergic neurons all glutamate formed by deamidation of glutamine enters the mitochondria (mit) and is returned to the cytosol in a complex process, which requires simultaneous glucose metabolism. In GABAergic neurons this is only the case for some of the glutamate, whereas the remainder enters the cytosol directly. This mechanism, which is further described in Fig. 2.6, its legend, and associated text may make GABA production less sensitive to replacement of glucose as the substrate with β-hydroxybutyrate in patients receiving very high amounts of ketone bodies to prevent seizures. Released glutamate is almost quantitatively reaccumulated in astrocytes , together with at least part of the released GABA [upper line of the glutamine–glutamate/GABA cycle (glu–gln cycle)] and reaccumulated in the astrocytic cytosol. Here, about 75 % is converted to glutamine and reenters the glutamine–glutamate/GABA cycle. The remaining ~25 % is oxidatively degraded, via one of two partly different pathways. In both α-ketoglutarate is reconverted to malate. In one malate exits to the cytosol, is decarboxylated by cytosolic malic enzyme to pyruvate, which is oxidized in the TCA cycle via acetyl Coenzyme A . In the other malate does not exit the TCA cycle but may be further metabolized to α-ketoglutarate after condensation with acetyl Coenzyme A, allowing resynthesis of another molecule of glutamate from only one molecule pyruvate. In either case the degraded glutamate must in the long term be replaced by a quantitatively similar production of glutamate from glucose , in the first case by complete de novo synthesis from one molecule glucose, in the second from one half of a glucose molecule. However, temporary fluctuations in the content of glutamate occur. The initial part of GABA metabolism is different, as all GABA is metabolized via succinic semialdehyde , succinate , and α-ketoglutarate to glutamate. Modified from Hertz (2013)
TCA cycle activity followed by oxidative phosphorylation in the respiratory chain creates a large amount of energy in the form of adenosine triphosphate (ATP) in both astrocytes and neurons. Since OAA is consumed at the beginning of the TCA cycle and regenerated at the end of the cycle, this process continues as long as ac.CoA is available and energy is consumed, but it cannot give rise to a new molecule of any TCA cycle constituent. Nevertheless, it does lead to labeling of glutamate and its derivatives, a labeling that forms the basis of 13C-NMR spectroscopy. However, this labeling is due to a transamination-mediated bidirectional exchange between α-KG and glutamate (Fitzpatrick et al. 1990).
Recently, experiments by Patel et al. (2005, 2015) and Duarte and Gruetter (2013) have shown a considerable flow of neurotransmitter GABA to astrocytes via the GABA/glutamine cycle. GABA after its release from inhibitory neurons is metabolized in astrocytes via succinic semialdehyde and succinate . It will later be discussed that a similar reaction may occur in GABAergic neurons . Succinate is a TCA cycle intermediate between α-ketoglutarate (α-KG) and malate (Fig. 2.1) and in the TCA cycle it is metabolized via OAA to citrate, requiring uptake of one molecule ac.CoA. Citrate is further metabolized to α-KG and transaminated to glutamate. Subsequently, the generated glutamate can be returned to neurons via astrocytes in the conventional glutamine–glutamate/GABA cycle via glutamine (Fig. 2.1). Duarte and Gruetter (2013) showed using metabolic modeling that the ac.CoA flow needed in the astrocyte to accommodate GABA metabolism constitutes a considerable fraction of total astrocyte ac.CoA production. The formation of ac.CoA needed for metabolism to citrate of OAA derived from GABA is included in the 25 % of total brain glucose consumption assigned to astrocytes; however, from the standpoint of ATP production the flow from succinate also needs to be considered.
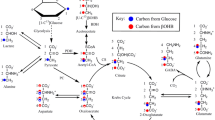
Although pyruvate formation from glucose takes place in the cytosol one oxidative process, formation of diphosphoglycerate from glyceraldehyde 3-phosphate is involved. As an oxidation this process is coupled to formation of nicotinamide dinucleotide NADH from NAD+. Since NADH is unable to cross the mitochondrial membrane for reoxidation in the mitochondria a reducing equivalent must be carried across the mitochondrial membrane for oxidation with return of the corresponding oxidized equivalent. In the brain this transport is mediated by the malate-aspartate shuttle (MAS) . In the MAS (Fig. 2.2), cytosolic NADH is reoxidized to NAD+ by reduction of cytosolic OAA to malate. Malate traverses the mitochondrial membrane but not in direct exchange for OAA (which cannot leave the mitochondria), but in exchange for α-KG, using the malate/α-ketoglutarate carrier (OGC-Slc25a11), and it is reoxidized to OAA in the TCA cycle. In the mitochondria OAA is transaminated by aspartate transaminase (AAT) to the corresponding amino acid, aspartate, which then exits the mitochondrial membrane in exchange for glutamate, using the glutamate/aspartate exchanger (AGC) , in brain AGC1 or aralar (Slc25a12). This exchanger is abundantly expressed not only in neurons but also in astrocytes (Lovatt et al. 2007; Li et al. 2012). The glutamate that enters the mitochondria is simultaneously transaminated to α-KG, its corresponding keto acid. α-KG exits the mitochondria in exchange with incoming malate in the process catalyzed by malate/α-ketoglutarate carrier. This carrier is also operating in brain (Passarella et al. 1987), including astrocytes (Pardo et al. 2011). In the cytosol α-KG is transaminated to glutamate with simultaneous conversion of aspartate to OAA, closing the cycle (Fig. 2.2). AAT activity is high in most cells and in retina, a cerebral tissue, it is expressed both in the cytosol and in mitochondria (Ross and Godfrey 1987). Due to the dependence of oxidative glycolysis on MAS activity, complete and direct oxidation of glucose requires not only that pyruvate enters the mitochondria as ac.CoA, but also that not only malate and aspartate, but also glutamate and α-KG are transported across the mitochondrial membrane. Note also that the dependency between these processes is mutual: the MAS would not be able to operate without NAD+ reduction.

The malate-aspartate shuttle (MAS) . In the malate-aspartate shuttle (MAS), which serves to transfer reducing equivalents across the mitochondrial membrane, cytosolic malate dehydrogenase (MDHc) oxidizes NADH and converts oxaloacetate (OAA) to malate (top right of figure), which enters the mitochondria in exchange with α-ketoglutarate (α-KG). The mitochondrial malate dehydrogenase (MDHm) reoxidizes malate to OAA, which is transaminated to aspartate by the mitochondrial aspartate aminotransferase (AATm) . Aspartate leaves the mitochondria in exchange with glutamate, requiring aspartate-glutamate exchanger (aralar in brain). In the mitochondria glutamate conversion to α-KG is essential for AATm activity forming aspartate from OAA and delivering α-KG for mitochondrial export. The glutamate imported into the mitochondria had been formed by cytosolic aspartate aminotransferase (AATc) from α-KG after its entry into the cytosol. From Hertz and Dienel (2002)
In addition to entering the TCA cycle via ac.CoA, pyruvate can also be carboxylated to OAA, an anaplerotic process , which expands the amount of TCA cycle intermediates and is catalyzed by the astrocyte-specific pyruvate carboxylase (PC) . This enzyme is virtually absent in neurons both in the brain in vivo (Shank et al. 1984; Hutson et al. 2008) and in cultured cells (Yu et al. 1983). Condensation of one molecule OAA with one molecule of acetyl-CoA forms a new molecule of citrate. This molecule as well as all molecules derived from it, including α-KG and glutamate will be labeled from [13C]glucose in different positions than corresponding molecules generated by PDH activity alone, which is the key principle used for determination of rate of pyruvate carboxylation in the brain by 13C-MRS (Gruetter et al. 2001; Sibson et al. 2001; Mason et al. 2007). Although pyruvate carboxylation is necessary to replace lost TCA intermediates, by far its main role in the brain is to produce glutamate which is then transferred to neurons for use as transmitter glutamate and, after decarboxylation, transmitter GABA. A minor part of synthesized glutamate is also used by the astrocytes themselves as gliotransmitter, for synthesis of glutathione or for detoxification of ammonia to glutamine . Since carboxylation represents ~10 % of total pyruvate metabolism and it must be combined with a similar PDH-mediated metabolism, a large fraction of the 25 % of total pyruvate metabolism occurring in astrocytes must serve to produce glutamate. This does not mean that no energy is produced since combined production and oxidative degradation of glutamate, which also mainly occurs in astrocytes, produces almost as much ATP as direct oxidation of glucose (Gruetter et al. 2001; Hertz et al. 2007).
Besides the pathway shown in Fig. 2.1 a small amount of glucose (2–5 % of total glucose utilization in brain) is metabolized via the pentose phosphate pathway (Gaitonde et al. 1987; Hostetler and Landau 1967; Ben-Yoseph et al. 1995). This pathway generates pentoses, necessary for synthesis of DNA and RNA. It also produces NADPH , a reducing equivalent not replaceable by NADH, used for synthesis of lipids and in a few metabolic pathways, including reduction of oxidized glutathione (Vogel et al. 1999). Although this pathway is relatively small it has been recently suggested as responsible for part of the 5–10 % mismatchFootnote 1 between total brain glucose uptake and oxidation (Raichle 2015).
2.2.2 Lactate
Pyruvate is rapidly reduced to lactate and vice versa and most normal cells contain considerably higher concentrations of lactate than of pyruvate. Both can also cross the cell membrane in either direction catalyzed by equilibrating monocarboxylate transporters (MCTs) . Lactate uptake is followed by its oxidation to pyruvate with concomitant reduction of NAD+ to NADH. This has the consequences that lactate oxidation competes with glucose oxidation and that metabolism of lactate, like that of glucose activates MAS. MCT transporters have different kinetics in astrocytes (displaying MCT1 and 3) and neurons (displaying MCT2). It is important that these transporters, like the glucose transporters, are equilibrating, not concentrative, because that means that they are unable to accumulate (or release) the unaltered compound to a concentration beyond that from which it is accumulated or released (reviewed by Hertz and Dienel 2005). However, once the native compound is removed by metabolism, uptake or release can continue. It has been suggested that astrocytically released lactate is taken up and metabolized largely by neurons after transfer by the astrocyte neuron lactate shuttle ANLS (Pellerin and Magistretti 2012) and this may be critical for complex functions such as memory formation (Steinman et al. 2016), but considerable evidence is available against this hypothesis (e.g., Dienel 2012; Patel et al. 2014). Furthermore, astrocytes have a high ability to transport lactate between themselves through gap junctions which may substantially reduce transport to neurons (Gandhi et al. 2009). Nevertheless a small amount of glycogenderived lactate seems to be transferred from astrocytes to neurons during learning (Steinman et al. 2016). If this lactate is accumulated into the minute dendritic spines which have no mitochondria it may create ATP by oxidation to pyruvate without interfering with glucose metabolism (Hertz and Chen 2016). The established importance of glial cells for memory formation depends upon glycogen but is most likely independent of a major lactate transfer between cell types (Hertz and Chen 2016).
The unlikelihood of astrocytes supplying neurons with glucose-derived lactate does not mean that lactate metabolism is unable to cover a substantial fraction of the brain’s energy demand. Thus, Van Hall et al. (2009) showed that lactate at a plasma level of 6.9 mM contributed 27 % of energy demand in human brain; high exercise intensity can decrease brain glucose uptake by almost 40 % (Kemppainen et al. 2005), and a similar decrease occurs in the anesthetized rat when plasma lactate concentration is increased to 5.8 % (Wyss et al. 2011). Boumezbeur et al. (2010) used 13C magnetic resonance spectroscopy (13C MRS) to estimate the relationship between plasma lactate level and cortical lactate metabolism. They found under resting awake conditions that plasma lactate at physiological levels (low mM range) accounted for ~10 % of consumption by the TCA cycle. However, based on the transport kinetics determined it was concluded that it could account for up to 60 % under maximally elevated conditions, a value comparable with those cited earlier. The labeling patterns observed were the same as with glucose, suggesting a similar percentage contribution to neuronal and glial metabolism. In mature cultured cortical astrocytes , glucose utilization can similarly be reduced by 60–70 % in the presence of lactate (Swanson and Benington 1996; Rodrigues et al. 2009), but in very young cultures of cortical neurons lactate can cover even more of total energy consumption (Bouzier-Sore et al. 2003, 2006). In this context it should be remembered that lactate oxidation to pyruvate competes with the oxidation of glyceraldehyde 3-phosphate for cytosolic NADH and thereby may inhibit glucose utilization. Nevertheless, a small increase in lactate is under certain conditions capable of increasing glucose uptake into brain at low glucose concentrations without major effect on lactate contribution to metabolism (De Feyter et al. 2013; Herzog et al. 2013). This effect appears to be more marked in neurons than in astrocytes . It is unknown whether it might be related to the signaling effect of physiological concentrations of lactate on noradrenergic neurons (Tang et al. 2014).
As a substrate lactate is able to sustain all the functions of glucose shown in Fig. 2.1 except glycolysis. It can support both PDH and PC activity and lactate and pyruvate can even give rise to glycogen synthesis (Dringen et al. 1993; Huang et al. 1994; Dringen et al. 2005). Lactate interacts with MAS. It cannot be used as a substrate for the pentose phosphate shunt; however, cytosolic NADPH can be produced from lactate conversion by pyruvate carboxylase into OAA and by malic enzyme into pyruvate with the conversion of an NADP+ to an NADPH .
2.2.3 Acetate, Fatty Acids, and Ketone Bodies
Most dietary lipids are triglycerides . After lipolysis glycerol is converted via glyceraldehyde 3-phosphate to pyruvate (Zabłocki and Bryła 1988), and fatty acids are β-oxidized leading to the formation of ac.CoA (Lynen 1953). The formed ac.CoA can either be metabolized as such by condensation with OAA (e.g., in the heart), or two molecules of ac.CoA can be converted to the ketone bodies acetoacetate and its reduced metabolite β-hydroxybutyrate, a reaction occurring in the liver (McPherson and McEneny 2012). After release from liver cells circulating ketone bodies are transported by MCTs (Vannucci and Simpson 2003) and utilized as metabolic substrates in several organs, but normally not to an major degree in adult brain. This is in contrast to brain energy metabolism in young animals where MCT activity in the blood–brain barrier is high (Cremer et al. 1976) and ketone bodies plentiful in blood. However, ketone bodies, mainly in the reduced form of β-hydroxybutyrate, accumulate when glucose metabolism is failing, e.g., in diabetes. Ketone bodies also accumulate during fasting (due to metabolism of fatty acids), and Owen et al. (1967) showed (1) a considerable arteriovenous difference for ketone bodies across the brain of obese patients treated with starvation and (2) that ketone bodies accounted for ~60 % of brain oxygen consumption in these patients. An increased ketone body concentration in the systemic circulation and upregulation of blood–brain barrier permeability may have contributed to the increase (Robinson and Williamson 1980), but an elevation in d-β-hydroxybutyrate dehydrogenase activity has also been described in the brains of fasting rats (Smith et al. 1969).
That ketone body oxidation to approximately 50–60 % can replace glucose oxidation in intact brain has repeatedly been confirmed (Zhang et al. 2013). Chowdhury et al. (2014) compared rate of β-hydroxybutyrate metabolism in awake, mildly anaesthetized (halothane), and in deeply anaesthetized (pentobarbital) rats with isoelectric brain activity. In the awake animals ketone body oxidation increased with its plasma concentration and at a saturating plasma β-hydroxybutyrate concentration of 17 mM reached a plateau of ~60 % of total substrate oxidation in brain cortex, a similar value as that observed by Owen et al. (1967). However, in the deeply anaesthetized rats the much less active neuronal TCA cycle was fully supported by ketone bodies at a β-hydroxybutyrate plasma level of ~12 mM, in spite of the fact that the animals were euglycemic. Accordingly, ketone bodies can provide complete energetic support of basal (nonsignaling) processes in brain when available in sufficient concentration, but they are unable to fully support signaling processes even at the highest concentrations. By subtracting the nonsignaling dependent component Chowdhury et al. (2014) concluded that in the awake rat cerebral cortex approximately 50 % of neuronal energetics required for signaling cannot be replaced by β-hydroxybutyrate and has to derive from glucose oxidation (Fig. 2.4).
The functional capabilities of ketone bodies compared to glucose are likely to be related to the different metabolic pathways followed in their oxidative degradation. β-Hydroxybutyrate is oxidized to acetoacetate intramitochondrially, and acetoacetate is converted to two molecules of ac.CoA which then condense with OAA to form citrate. Since the oxidation of β-hydroxybutyrate is intramitochondrial it does not compete with oxidation of glyceraldehyde 3-phosphate . This is in contrast to lactate, and metabolism of β-hydroxybutyrate does also not share the ability of glucose and lactate to interact with MAS. In contrast to lactate, it can only support PDH -mediated activity, not pyruvate carboxylation. It supports not only neuronal (Amaral 2013; Chowdhury et al. 2014) but also astrocytic PDH activity (Melø et al. 2006; Gibbs et al. 2009; Jiang et al. 2011; McKenna 2012). Similarly, Pan et al. (2002) showed in awake human subjects that the labeling pattern from β-hydroxybutyrate was similar to that of glucose and calculated a similar relative percentage contribution to neuronal and glial oxidative metabolism as has been found with glucose . This is in contrast to acetate, which in cell culture is almost exclusively metabolized (although only via ac.CoA) by astrocytes (Waniewski and Martin 1998), confirming previous findings in brain and retina (Muir et al. 1986). In 13C MRS studies on humans and rodents the labeling kinetics have also shown glial localization of acetate metabolism (Lebon et al. 2002; Jiang et al. 2013). Although acetate is normally found in low levels in the blood stream it can be substantially elevated by alcohol consumption to levels over 1 mM. At these levels it has been measured using 13C MRS to account for as much as 50 % of glial oxidative ATP needs, particularly in subject groups exposed to hypoglycemia such as intensively insulin-treated type 1 diabetes patients (Gulanski et al. 2013). Acetate has even been reported to have protective effect against hypoglycemic seizures (Urion et al. 1979), which would suggest a major contribution by astrocytes to the seizures. Consumers of large amounts of alcohol also show increased acetate metabolism, reflecting that alcohol is metabolized to acetate (Jiang et al. 2013; Volkow et al. 2013, 2015).
2.3 The Glutamine–Glutamate/GABA Cycle
2.3.1 Synthesis and Degradation of Glutamate and GABA
In the adult brain perfusion techniques have shown very limited transport of glutamate and its precursor glutamine from blood to the brain, and changes in plasma concentration of glutamine have only little influence on brain glutamate content (Smith 2000; Patel et al. 2015). Accordingly, neurotransmitter glutamate and its decarboxylation product GABA must be synthesized within the brain. As discussed in Sect. 2.2.2 formation of glutamate requires synthesis of a new molecule of a TCA cycle intermediate and both PDH and PC activities. It therefore occurs in astrocytes but not in neurons. Transport to neurons is mediated by the glutamine–glutamate/GABA cycle where formation of neuronal glutamate accounts for about 75–80 % of the flux (measured from neurons toward astrocytes) and the remaining 20–25 % is made up by GABA (Patel et al. 2005, 2015; Duarte and Gruetter 2013). A recent study of glutamate amino decarboxylase 67 KD isoform GAD67 knock out mice suggested lower contributions by GABA, but the possibility of an underestimate was mentioned (Walls et al. 2015).
The cycle matures slowly, i.e. after 1 month in rats (reviewed by Chowdhury et al. 2007; Hertz 2013; Brekke and Morken 2015) and in the adult brain it has a flux equal to rate of glucose utilization in neurons (Sibson et al. 1998; Chowdhury et al. 2007; Hyder et al. 2013a, b) or ~75 % of the total rate of glucose utilization in brain. However, only a minor part (25 % or less) of the glutamate flux from astrocytes to neurons represents newly synthesized glutamate, whereas the remainder is made up of glutamate that has already been released as transmitter but subsequently is accumulated by astrocytes and returned to neurons in the cycle (Rothman et al. 2011). Almost all released transmitter glutamate is treated in this manner, whereas only little is reaccumulated directly into glutamatergic neurons (Danbolt 2001; Zhou and Danbolt 2013). Some neuronal reuptake has been described in cultured hippocampal neurons (Coco et al. 1997) but might be due to the young age of the cultures (see Hertz 2013).
In the long term the glutamate concentration in adult healthy brain must stay unaltered (Lebon et al. 2002; Sonnewald 2014), meaning that a fraction similar to that synthesized de novo (~25 %) must be oxidatively degraded in astrocytes , or to a much lesser extent lost from the brain as glutamine, a process mainly occurring during ammonia detoxification (Zielińska et al. 2014). For oxidative degradation glutamate is converted to α-KG, which after circling in the TCA cycle to malate can exit into the cytosol and be converted by the astrocyte-enriched (Kurz et al. 1993) cytosolic malic enzyme to pyruvate. Pyruvate can then enter the TCA cycle and be oxidatively degraded (Fig. 2.1). However, Sonnewald (2014) hypothesized that a relatively low degree of pyruvate recycling (turnover of pyruvate or one of its metabolites in the TCA cycle to regenerate pyruvate) suggested that instead pyruvate was released from the cells as lactate, which would constitute a similarly effective cataplerosis . This hypothesis is supported by her previous observation that lactate generated from TCA cycle intermediates is found in the medium (Sonnewald et al. 1993), but these experiments were performed in cultured cells known to release very large amounts of lactate. On the other hand, labeled ac.CoA was derived from TCA cycle intermediates in brain mitochondria, demonstrating malic enzyme activity and pyruvate recycling by Bakken et al. (1997) from the same group. Also, release from brain of specifically glutamate-derived lactate would cause a very considerable decrease in the amount of ATP produced by astrocytes . A complication in determining pyruvate recycling using labeling methods is also that the astrocyte-derived pattern of labeling it produces in lactate will be substantially diluted by the large amount of lactate from neuronal metabolism. Furthermore, similar scrambling occurs in glucose released by the liver which can make determination ambiguous.
A third possibility is that some of the ~25 % of returning glutamate which is converted to α-KG is only partly degraded. This would happen if malate did not leave the astrocytic TCA cycle but was converted to OAA, condensed with a new molecule of ac.CoA and further metabolized via α-KG to glutamate. Such a process would allow formation of another molecule of glutamate and glutamine using only one pyruvate molecule and requiring no pyruvate carboxylation . Accordingly it would not require glucose or lactate utilization but could also use β-hydroxybutyrate as the source. Maciejewski and Rothman (2008) described several alternate pathways of glutamate neurotransmitter cycling that involved glutamate oxidation but did not require pyruvate carboxylation, but at present there is no evidence of their importance for the glutamine–glutamate/GABA cycle in the brain in vivo, at least under normal conditions (Rothman et al. 2012).
In the short term glutamate content in brain can transiently increase, e.g., during learning (Gibbs et al. 2007), in some cases of epileptic seizures and during visual stimulation (Bjørnsen et al. 2007; Peca et al. 2010; Mangia et al. 2010, 2012; Perez et al. 2012). The involvement of astrocytic metabolism during anaplerosis/cataplerosis provides huge possibilities for astrocytic regulation. However, even astrocytic involvement only during return of previously released transmitter without conversion of glutamate to α-KG enables some astrocytic regulation, because glutamine can move within the astrocytic syncytium (Cruz et al. 2007). Perhaps glutamate released from neurons might even be redirected to GABA-ergic neurons and vice versa. Formation of a new molecule of glutamate by conversion of glutamate-derived malate to α-KG and glutamate in the TCA cycle and incorporation of ac.CoA during this process would increase the astrocytic influence .
2.3.2 Individual Steps of Glutamate Formation and Degradation
An important, debated question is whether the initial conversion of α-KG to glutamate in astrocytes is catalyzed by glutamate dehydrogenase (GDH) or by aspartate aminotransferase (AAT) . Westergaard et al. (1996) showed that in cultured astrocytes conversion of a-KG (Greek alpha) to glutamate is catalyzed by AAT wheres glutamate oxidation is mediated by GDH. The equilibrium constant for AAT is close to unity (Krebs 1953), making the reaction easily reversible. In contrast, in the GDH reaction the reductive amination to glutamate is thermodynamically favored (Engel and Dalziel 1967). However, a high K m for ammonia together with a high NAD+:NADH ratio in brain may enable oxidative deamination of glutamate (Zaganas et al. 2001, 2009). GDH is highly regulated by allosteric activators and inhibitors (Schousboe et al. 2013), whereas AAT is dependent upon the presence of suitable transactivation partners, for example, conversion of aspartate to OAA concomitant with glutamate formation from α-KG.
One of us (L.H) bears some responsibility for the concept of a large involvement of GDH in glutamate oxidation, since this process was first suggested to depend on GDH in his laboratory, based on findings in cultured astrocytes (Yu et al. 1982). This finding has repeatedly been confirmed (e.g., McKenna et al. 1996), and GDH is also the enzyme involved in glutamate degradation in astrocytes obtained from brain tissue (Whitelaw and Robinson 2013). However, in both of these situations the astrocytes are isolated without the possibility of functional interactions with neurons and a complete glutamine–glutamate/GABA cycle. A suggested, but not proven, functional interaction between glutamate anaplerosis and cataplerosis (Hertz 2011a, 2013) is therefore not able to operate. The suggestion of such a correlation between glutamate formation and degradation (Fig. 2.3) was triggered by a large stimulation of glutamate/glutamine formation in astrocytes in intact brain in the presence of aspartate found by Pardo et al. (2011). As previously mentioned, a high concentration of aspartate facilitates glutamate formation from α-KG by AAT but it will not affect the corresponding GDH-mediated reaction. The model proposed in Fig. 2.3 represents an attempt to find an endogenous source of aspartate capable of exerting this important stimulation. As seen in this figure synthesis and degradation along the suggested pathways, which are described in detail in the legend of this Fig and in Hertz (2011a, b, 2013), would be able to supply aspartate for glutamate synthesis. Moreover, both cytosolic and mitochondrial aspartate aminotransferase activity are very high in brain (Cooper 2013), allowing rapid nitrogen exchange between glutamate and aspartate. Furthermore, in brain mitochondria glutamate oxidation depends mainly on AAT (Balazs 1965; Dennis et al. 1977), and in GDH knockout mice, most functions remain unchanged except for a reduced glutamate oxidation in cultured, and thus isolated astrocytes (Frigerio et al. 2012).

Proposed pathway for coupled production and metabolism of transmitter glutamate using aspartate transamination for exchange between α-ketoglutarate and glutamate. Joint pyruvate carboxylase and pyruvate dehydrogenase activation generates a “new” molecule of citrate (lower left corner) as detailed in Fig. 2.1. Citrate-derived α-ketoglutarate exiting the mitochondrial membrane leaves the astrocytic TCA cycle and is transaminated with aspartate to form glutamate, with concomitant oxaloacetate (OAA) formation from aspartate. The mitochondrial exit of α-ketoglutarate occurs via the ketoglutarate/malate exchanger, generally acknowledged to be expressed in astrocytes , and the cytosolic malate with which is exchanged, is generated via NADH-supported reduction of oxaloacetate formed from aspartate (Pardo et al. 2011). Glutamate is amidated to glutamine (pathway 1), which is transferred to glutamatergic neurons (without indication of any extracellular space in the figure) and extracellular release as transmitter glutamate (pathway 2), and subsequent reuptake of glutamate and oxidative metabolism in astrocytes (pathway 3). During oxidative metabolism transamination of glutamate to α-ketoglutarate generates aspartate that can be used in the transamination of α-ketoglutarate to glutamate in pathway 1 after transfer via pathway 4. Metabolism of α-ketoglutarate via malate is only shown to pyruvate . Biosynthesis of glutamine is shown in brown and metabolic degradation of glutamate in blue. Redox shuttling and astrocytic release of glutamine and uptake of glutamate are shown in black, and neuronal uptake of glutamine, hydrolysis to glutamate, and its release is shown in red. Reactions involving or resulting from transamination between aspartate and oxaloacetate (OAA) are shown in green. Small blue circle shows pyruvate carrier into mitochondria and small purple circle malate carrier out from mitochondria. AGC1 aspartate/glutamate exchanger, aralar; α-KG α-ketoglutarate ; Glc glucose ; Pyr pyruvate; OGC malate/α-ketoglutarate exchanger (From Hertz 2013)
Nevertheless, a recent paper (Karaca et al. 2015) shows abnormalities in glutamate oxidation in the knockout mice. Those found in astrocytes and mitochondria may be related to the fact that no concomitant synthesis and degradation of glutamate can occur in these isolated preparations, but an increase in ADP/ATP ratio in brain cannot. However, GDH1, the enzyme studied in the mice, shows a reduced K0.5 for ammonia when ADP is lowered from 1 to 0.1 mM (Zaganas et al. 2013), which may enhance glutamate oxidation when GDH is operating. Elimination of this mechanism in the knockout animals might possibly explain the increased ADP/ATP ratio. However, the findings do suggest that also in the brain in vivo GDH does contribute to glutamate formation/oxidation although the contribution might be minor. This would be consistent with a ten times higher activity of AAT than of GDH in mitochondria (compare Zaganas et al. 2001 with McKenna et al. 2006), and it would not disagree with the observations by Balazs (1965) and Dennis et al. (1977). They also confirm that glutamate is an important metabolic substrate (McKenna 2012, 2013) perhaps more because of its direct access to the TCA cycle than of the amount oxidized which must be much smaller than that of glucose , since brain glutamate exclusively originates from its astrocytic production from glucose and astrocytes only account for about one quarter of brain volume and metabolism (reviewed by Hertz 2011b). Finally, although the model suggested in Fig. 2.3 requires that glutamate formation from α-KG and glutamate degradation to α-KG are catalyzed by the same enzyme that enzyme may not catalyze 100 % of the interconversion.
An alternative mechanism for anaplerotic resynthesis of glutamate from α-KG was originally proposed by Yudkoff et al. (1996), who found a rapid uptake of the branched chain amino acid (BCAA) leucine into cultured astrocytes (V max 54 nmol/mg of protein/min and K m 450 μM). Astrocytic accumulation of leucine was three times greater in the presence of alpha-aminooxyacetic acid (AOAA) , a transamination inhibitor, suggesting that these isolated astrocytes rapidly transaminate leucine to alpha-ketoisocaproic acid (KIC) , which they then release into the extracellular fluid. This finding was linked to glutamate oxidation by Hutson and coworkers (Hutson et al. 1998, 2001), who suggested that mitochondrial branched chain amino acid transferase (BCATm) donates an amino nitrogen group to α-KG, forming glutamate which is then converted by glutamine synthase to glutamine. In their original model the amino nitrogen was fixed in the neuron by GDH and transferred by cytosolic neuronal BCAT (BCATn) to a branched chain keto acid, which then shuttled from the neuron to the astrocyte as a BCAA . However, it is now believed that this is more likely an internal glial reaction, based on lack of evidence that neuronal GDH can mediate glutamate synthesis. Rothman et al. (2012) determined the flux via BCAT in brain tissue vivo to ~0.1 μmol/nmol/mg, which would just be enough to catalyze transamination of the 20 % of glutamate synthesis that represents anaplerosis. Although it is unusual that a transaminase activity is similar to the net rate of the actual process (usually being several to many times greater) BCAT activity might therefore catalyze anaplerotic synthesis of glutamate. An advantage of using BCAT is that BCAAs are continuously supplied to the brain from the blood, and the nitrogen all four BCAAs together bring is almost stoichiometrically equivalent to the amount that leaves the brain as glutamine (Rothman et al. 2012 and see later). This means that the reaction described by Yudkoff et al. (1996) would suffice to generate glutamate from α-KG. The rate of α-KG synthesis (20–25 % of ~0.6 nmol/mg of protein/min) is at least as high, and in the pathways suggested in Fig. 2.3 the amount of aspartate supplied equals the glutamate uptake and metabolism in astrocytes (Hertz et al. 1978; McKenna 2012), which is much more intense than those of the BCAAs . Important evidence against participation of BCAT in glutamine–glutamate/GABA cycle glutamate synthesis is that the 50 % stimulation of glutamate synthesis caused by administration of aspartate could not be replicated by administration of the branched chain amino acid leucine (Pardo et al. 2011). Thus, use of BCAT for glutamate synthesis related to neurotransmitter cycling , as opposed to nitrogen balance, in intact tissue under normal conditions is not likely. However, in the situations where synthesis of glutamate exceeds its metabolic degradation (and thus rate of aspartate formation) BCATs could play a role in glutamate synthesis by converting BCAAs to branched chain keto acids. The latter could be reconverted to BCAAs when cataplerosis catches up with the increased anaplerosis. GDH maight also play a role. Glutamate is converted to glutamine by the astrocyte-specific (Martinez-Hernandez et al. 1977; Norenberg and Martinez-Hernandez 1979; Hutson et al. 2008; Anlauf and Derouiche 2013) enzyme glutamine synthetase (GS) , which is localized in the cytosol. Inhibition of this enzyme in retinal Müller cells (an astrocyte-like cell) almost immediately interrupts glutamatergic activity in the retina (Barnett et al. 2000). The inhibition also increases retinal cell death (Gorovits et al. 1997; Bringmann et al. 2013), whereas a hormonally induced increase in Müller cell glutamine synthase GS protects against neuronal injury (Ola et al. 2011). Similarly acute administration of MSO to inhibit GS in vivo leads to rapid elevation of extracellular glutamate and can result in seizure and chronic administration will lead to the development of epilepsy (Eid et al. 2008).
Enhanced metabolism of BCAAs also counteract ammonia toxicity (Watanabe et al. 1986; Muto et al. 2005), and a nonnegligible oxidative metabolism (following transamination) and incorporation of BCAAs into protein in cultured astrocytes is strongly inhibited by a toxic concentration of ammonia (Murthy and Hertz 1987). At one time ammonia detoxification was believed to be the major pathway of glutamine synthesis but it was shown to be extremely slow compared to that in the glutamate–glutamine cycle , even during hepatic encephalopathy (Shen et al. 1998; Sibson et al. 2001; Keiding et al. 2006; Cudalbu et al. 2012). Under hyperammonemic conditions the glutamate formed for detoxification is probably formed by glutamate dehydrogenase GDH working in the glutamate synthesis direction due to the elevated NH4 + concentration (Shen et al. 1998; Cooper 2012; Cudalbu et al. 2012; Kanamori and Ross 1995), but the ammonia interactions with BCAAs may suggest that they are also involved.
Glutamine exit from astrocytes and entry intro neurons are equally important as glutamine synthesis for regulation of de novo synthesis of glutamate and GABA and it is essential even during the return of released transmitter glutamate to neurons after its initial uptake in astrocytes. The amino acid transporter SN1 mediates electroneutral and bidirectional glutamine transport in astrocytes (Nissen-Meyer and Chaudhry 2013). Its activity is regulated by many factors, e.g., by extracellular pH, because protons compete with Na+, which is required for its transport activity. There are also consistent observations that SN1 is down-regulated by protein kinase C phosphorylation , probably by internalization (Nissen-Meyer and Chaudhry 2013). After its release to the extracellular space glutamine is accumulated into glutamatergic neurons by a different transporter which is coupled to Na+, SAT1, or at some places SAT2. It is subsequently converted to glutamate by phosphate-activated glutaminase (PAG) .
As discussed in more detail by Hertz (2013) and Schousboe et al. (2013), the conversion of glutamine to glutamate in glutamatergic neurons is remarkably complex. After PAG-mediated deamidation within the mitochondrial membrane glutamate enters the mitochondrial matrix and is returned from here to the cytoplasm in a “pseudomalate-aspartate shuttle .” This has been demonstrated both in cultured astrocytes (Palaiologos et al. 1988, 1989) and in mitochondria (Ziemińska et al. 2004; Bak et al. 2008). The “pseudomalate-aspartate shuttle” is identical to the ‘real’ MAS (Fig. 2.2) with the exception that glutamate on the cytosolic side is generated by deamidation of glutamine by PAG, abolishing the need for reaccumulation into mitochondria of cytosolic glutamate formed from α-KG by transamination. This glutamate thus becomes freely available in the cytosol for use as transmitter glutamate. Since cytosolic malate formation is an indispensable part of MAS (Fig. 2.2) and thus also of pseudo-MAS, formation of transmitter glutamate must also coincide with cytosolic reduction of NAD+ to NADH. This will in general mean formation of one molecule of pyruvate from glucose , i.e., utilization of half a molecule of glucose. The energy obtained from this process and subsequent oxidation of glucose does not need to be used in the glutamine–glutamate cycle, which only requires little ATP to function (for glutamate and glutamine uptake and glutamine synthesis). However, it contributes substantially (1/2 molecule glucose per molecule glutamate formed) to the ratio between glucose (i.e., 2 pyruvate) utilization and glutamate flux , which as shown in Fig. 2.4 equals approximately 85 % of cerebralcortical neuronal glucose oxidation in the resting awake state and 60–70 % of total cerebralcortical glucose oxidation (with the remainder due to glia and GABAergic neurons primarily) (Sibson et al. 1998; Rothman et al. 2011). Since the “pseudo-MAS ” only operates in glutamate formation which accounts for 80 % of the total flux the contribution must be about 40 %. Anaplerotic astrocytic glutamate formation from glucose which accounts for about one quarter of total flux must account for another 20 %.

Rate of the glutamate/glutamine cycle versus glucose oxidation and ability of ketone oxidation to displace glucose. Left: Measured relation between the rate of the glutamate/glutamine cycle in rat cerebral cortex versus neuronal glucose oxidation based on 12 published studies in rodents and 9 in humans (adapted from Hyder et al. 2013b). The relation is close to linear throughout the range with a slope of close to 1:1. Right: Saturating ketones dark part of columns can displace all of the glucose oxidation under isoelectric conditions but only approximately 50 % of the glucose oxidation needed to support neuronal signaling in glutamatergic neurons (adapted from Chowdhury et al. 2014). This percentage is in agreement with the prediction of the pseudo malate asparate shuttle model which is discussed in the text (and see Figs. 2.1, 2.2, and 2.3) VAcCoA-kbN, rate of neuronal acetyl-CoA utilization from ketone bodies; VpdhN, neuronal pyruvate dehydrogenase flux (i.e., rate of acetyl-CoA utilization from glucose)
After glutamate has been released from neurons as a transmitter most glutamate is accumulated in astrocytes by the powerful glutamate transporters Glut-1 and GLAST (Danbolt 2001; Zhou and Danbolt 2013). Although some glutamate may also be taken up by axonal terminals in glutamatergic neurons this is not sufficient to maintain glutamate homeostasis (Bjørnsen et al. 2014). In astrocytes a part (20–25 %) is oxidized as already discussed, whereas the remaining 75–80 % is again converted to glutamine and returned to neurons for reuse as transmitter (Rothman et al. 2011). However, total flux in the glutamine–glutamate/GABA cycle is so large and the association between glutamate uptake and metabolism so close that the fraction of glutamate which is oxidized in astrocytes is more than high enough to support its own energy-consuming uptake (McKenna 2012). Metabolic utilization of glutamate is strongly supported by a close association between astrocytic glutamate transporters and metabolic enzymes demonstrated by the Robinson group (Genda et al. 2011; Bauer et al. 2012; Whitelaw and Robinson 2013; Jackson et al. 2014). A close functional interaction between formation and degradation of glutamate (Fig. 2.3) is also consistent with the quantitative similarity between anaplerosis and cataplerosis and the evidence that the latter also mainly occurs in astrocytes . However, the conclusion that glutamate is a metabolizable substrate is in distinct contrast to a view expressed in a recent “Neuron ” review by Magistretti and Allaman (2015), despite the studies discussed earlier and findings that human, cat, and guinea pig brain slices show at least as high rates of oxygen uptake with glutamate alone as the substrate as with glucose alone (McIlwain 1953; Kratzing 1953; Takagaki and Tsukada 1957).
The presumed pathway for oxidative metabolism in intact tissue (Fig. 2.3) is relatively simple: Glutamate enters the mitochondria in exchange with aspartate , formed from OAA generated during glutamate synthesis. It is subsequently transaminated to α-KG in the same transamination process that formed aspartate from OAA, and α-KG enters the TCA cycle. One problem with this hypothesis is that glutamate may not necessarily be synthesized and degraded in the same astrocyte . One potential solution to this problem may be that lactate, which is rapidly transported between astrocytes (Gandhi et al. 2009), may be metabolized and facilitate glutamate synthesis when return of aspartate is deficient as a result of reduced glutamate oxidation (Hertz et al. 2014). For example, lactate might support production of OAA and thus of aspartate in pathway 4 in Fig. 2.3 if insufficient glutamate is being oxidized in a specific astrocyte . As already mentioned BCAA transamination or GDH activity may also alleviate this problem .
2.3.3 Individual Steps of GABA Formation and Degradation
It has been mentioned that GABA-derived glutamate accounts for ~20 % of the total glutamine–glutamate/GABA cycle flux from neurons toward astrocytes in the brain in vivo (Patel et al. 2005), and an approximately similar magnitude was found by Duarte and Gruetter (2013) and Patel et al. (2015). This means that in spite of a considerable rate of GABA reuptake into GABA-ergic neurons (the functional importance of which will be discussed later) GABA synthesis ultimately requires operation of the glutamine–glutamate/GABA cycle. It is likely that glutamine production and release in astrocytes for GABA production proceeds along similar pathways as those for glutamate production. It is therefore of interest to establish whether astrocytic formation and degradation of GABA might be interconnected in a similar manner as suggested for glutamate. Figure 2.5 shows that this might well be the case, although the needed conversion of GABA to glutamate before it can be returned to astrocytes in the glutamine–glutamate/GABA cycle makes the pathway for astrocytic return of GABA as glutamate more complex than that of transmitter glutamate.

Proposed pathway for coupled production and metabolism of transmitter GABA using aspartate transamination for exchange between α-ketoglutarate and glutamate. Pathways, color coding, and most abbreviations are as in Fig. 2.3. Most differences are in the lower right corner of the figure. The suggested glutamate-GABA exchange at the mitochondrial membrane has not been described in mammalian brain, but metabolism of GABA via succinic semialdehyde (SSA) to succinate (Succ.), a TCA cycle constituent is a well-established pathway. Metabolism of succinate via malate is only shown to pyruvate. As in Fig. 2.3 glutamate metabolism provides aspartate that can be used in the transamination of α-ketoglutarate to glutamate in pathway 1. It also generates α-ketoglutarate needed for the transamination of GABA to SSA
GABA uptake has repeatedly been demonstrated in cultured astrocytes (Schousboe et al. 1977b; Hertz et al. 1978), and high GABA transporter currents have been measured in striatal astrocytes by patch-clamp intracellular recording (Goubard et al. 2011). Initially released GABA must after its cytosolic uptake be transferred to the mitochondria, since the conversion of GABA to succinic semialdehyde (SSA) is initiated by GABA transaminase (GABA-T) (Wong et al. 1974), which is a mitochondrial enzyme (Schousboe et al. 1977a). Figure 2.5 suggests a glutamate/GABA antiporter at the mitochondrial membrane, although such a transporter has presently not been demonstrated in mitochondria. It has, however, been shown in the bacterial membrane (Sa et al. 2015).
SSA is further oxidized to succinate , which enters the mitochondria. Figure 2.5 also suggests that glutamate reenters the mitochondria through the glutamate-aspartate exchanger (requiring aralar) and that the simultaneous exit of aspartate from mitochondria to cytosol as in Fig. 2.3 may secure the AAT substrate needed for transamination-mediated glutamate production at the initial step of GABA production. As in the metabolism of accumulated glutamate this aspartate is formed from OAA generated during the initial formation of precursor glutamate (Pathway 4) in the transamination process converting glutamate to α-KG. Instead of being oxidized α-KG retransamination to glutamate enables concomitant GABA transamination to SSA and at the same time creates the glutamate suggested to exit the mitochondria in exchange with GABA. McKenna and Sonnewald (2005) have provided some experimental evidence for these suggestions . In a study of metabolism in cultured astrocytes they found increased aspartate content and larger formation of [U-13C]aspartate from added [U-13C]glutamate when the cells were incubated with nonlabeled GABA. This was tentatively interpreted as caused by increased entry of [U-13C] glutamate into the TCA cycle to allow for the transamination of GABA. This explanation is strikingly similar to what is shown in Fig. 2.5 for GABA degradation, but in intact tissue, where GABA production and degradation may be linked, extracellular glutamate is not needed for this purpose, since cytosolic glutamate can be derived from mitochondrial glutamate formed as described earlier.
Duarte and Gruetter (2013) described a complete metabolic scheme for the return of GABA taken up by the astrocyte in the glutamine–glutamate/GABA cycle , where malate stayed in the TCA cycle to ultimately give rise to α-KG and glutamate. The GABA-derived carbon could also undergo complete oxidation by conversion to pyruvate by malic enzyme, which may be reintroduced and completely metabolized in the TCA cycle (Fig. 2.5). The possibility of GABA oxidation, which would then require anaplerosis for replacement is supported by the observation by Zhang et al. (1995) that small amounts of CO2 are formed from methyl esters of succinate in rat brain cells. It is possible that glutamate and succinate are accumulated in different types of mitochondria , since astrocytes show mitochondrial heterogeneity (Sonnewald et al. 1993; Collins et al. 2002; Waagepetersen et al. 2006).
In spite of the assumed similarities between the GABA/glutamine and glutamate/glutamine cycles in astrocytic formation of glutamate and glutamine and the transfer of the latter to neurons, deamidation of glutamine to glutamate in neurons may occur in a very different manner. Part of the glutamate produced by PAG in the intermembrane space of the mitochondria is released directly to the cytosol (the direct pathway) whereas another fraction is transaminated to α-KG in the mitochondria (the indirect pathway) (Waagepetersen et al. 2001; Leke et al. 2011). The contribution of each pathway to GABA synthesis is about the same. Both pathways were concluded to contribute to vesicular GABA release and to be involved in determining GABAergic tone resulting from exit of cytosolic GABA via reversal of uptake (Waagepetersen et al. 2001; Walls et al. 2011; Leke et al. 2011), for example, by exposure to elevated extracellular K+ (Romei et al. 2014). The glutamate decarboxylase GAD 67 is functioning in both pools, whereas GAD 65 operates only in the vesicular pool (Waagepetersen et al. 2001) and is crucial for biosynthesis of synaptic GABA (Kaufman et al. 1991; Tian et al. 1999; Walls et al. 2011) and is the primary isoform in vivo responsible for activity-dependent GABA metabolism (Patel et al. 2006).
Waagepetersen et al. (1999, 2001) and Schousboe et al. (2013) have described that the α-KG entering the mitochondria undergoes a full turn of TCA cycle flux, regenerating α-KG. This conclusion was based on the finding of incorporation of isotope from 0.5 mM [U-13C]glucose into GABA during a 4 h incubation of GABAergic cerebrocortical neurons . An important consequence of this complete turn in the TCA cycle is introduction of ac.CoA between OAA and citrate. If this ac.CoA is derived from glucose or lactate it can enable exit of α-KG from the mitochondria to the cytosol and its conversion to glutamate similar to what was described for astrocytes (pathway 1 in Figs. 2.3 and 2.5). This will, however, not be the case if the ac.CoA is derived from β-hydroxybutyrate , which enters the mitochondria directly. Cytosolic glutamate can then be decarboxylated by glutamate decarboxylase (GAD) , a cytosolic enzyme (Balazs et al. 1966) to GABA. Thus, cycling of α-KG within the mitochondria during GABA synthesis via the indirect pathway may enable exit of glutamate from mitochondria, but it requires that the ac.CoA required to metabolize OAA to citrate is formed from glucose or lactate.
In contrast to the low rate of reaccumulation of glutamate in glutamatergic neurons , GABA is reaccumulated into GABA-ergic neurons in brain slices (Iversen and Neal 1968). Cell culture experiments have indicated that neuronal uptake exceeds that into astrocytes (Larsson et al. 1983; Yu et al. 1984; Schousboe et al. 2013). Müller cells in intact retina also show a high rate of GABA uptake, and in addition GABA-T is activated and NAD(P)H fluorescence increased at the mitochondrial location after GABA uptake (Biedermann et al. 2002). By aid of immunocytochemistry it has been shown that the transporter 3 (GAT 3) is expressed in astrocytes and in some animals also oligodendrocytes , whereas GAT 1 is expressed in neurons with very little astrocytic expression (Scimemi 2014a). In the mouse GABA from vesicular sources is accumulated by GAT1, while that from nonvesicular sources is taken up by the astrocytic GAT3 and 4 (Song et al. 2013). Blockade of uptake by the GAT1-specific inhibitor tiagabine during continued release of GABA should therefore be expected to reduce the neuronal content of GABA. Nevertheless, tiagabine has no effect on tissue amino acid levels and on 13C enrichments from [2-13C]acetate in lightly anesthetized rats (Patel et al. 2015). This is the case although similar concentrations of tiagabine cause a large increase in extracellular GABA (Fink-Jensen et al. 1992; Ipponi et al. 1999). Rather than enhancing neurotransmitter cycling tiagabine tended to reduce rates of neurotransmitter cycling in both glutamatergic and GABAergic neurons slightly without any change in the ratio between the two (Patel et al. 2015). These observations suggest that GABA taken up into GABAergic neurons from the extracellular space is either metabolized or released. In this context it is relevant that Romei et al. (2015) concluded that high-affinity uptake of GABA into nerve terminals seems to have functions other than recapture of the transmitter. The small impact of tiagabine on the astrocytic GABA/glutamine cycle may also reflect different K m’s of the transporters with much higher transient levels of GABA anticipated from vesicular release than back transport of GABA during depolarization. The high affinity of the neuronal GABA transporter is consistent with this possibility.
Romei et al. (2015) used synaptosomes from mouse cerebellum prelabeled with [3H]GABA and superfused with GABA. Influx of GABA through GAT1 transporters stimulated efflux of [3H]GABA. The authors suggested that this was partly by homoexchange, although such a process had not been found for GABA in cultured neurons by Hertz et al. (1978). Similarly Scimemi (2014a, b) presented evidence that not only GABA biosynthesis but also GABA uptake is essential to sustain GABAergic synaptic transmission. Based on these results, those by Duarte and Gruetter (2013) and Chowdhury et al. (2014) and the observation by Biedermann et al. (2002) that Müller cells show enhanced GABA-T expression and stimulation of mitochondrial function after GABA uptake we hypothesize that metabolism of accumulated GABA in neurons may facilitate the transport between mitochondrial and cytosolic compartments needed for production of GABA via the indirect pathway. This suggestion is not in disagreement with the experimental findings by Waagepetersen et al. (1999, 2001), since the 4-h incubation period used by these authors provides ample time for GABA release, reuptake, and metabolism.
In accordance with the observations by Waagepetersen et al. (2001) the suggested pathway assumes that for every two glutamine molecule one is converted in the cytosol to glutamate (light blue pathway in Fig. 2.6) from which GABA is formed by decarboxylation and released by arrow 1. The other glutamine molecule enters the mitochondria where it is deamidated to glutamate and converted by transamination to α-KG, contingent upon the presence of OAA for simultaneous conversion to aspartate (sea-blue pathway). This OAA becomes available when released GABA, which at the beginning originates exclusively from the cytosol, is reaccumulated (gray arrow and arrow 2) and metabolized. As in astrocytes , GABA is assumed to enter the mitochondria in exchange with glutamate. Here it is transaminated to SSA (in contrast to the pathways shown in Figs. 2.3 and 2.5 this process has to be a transamination ), with concomitant transamination of the α-KG, formed from the glutamine that initially entered the mitochondria, to glutamate. This glutamate exits to the cytosol via a glutamate carrier (sea-blue pathway) and from there the extracellular space, from where it is accumulated by astrocytes for return in the glutamine–glutamate/GABA cycle or for metabolic degradation. Accordingly, the glutamine accumulated into the mitochondria does not give rise to any formation of GABA, but it is essential for the transamination of incoming GABA to SSA, and it also delivers aspartate for the subsequent conversion of succinate -derived alpha-KG to glutamate and glutamate for return to astrocytes . SSA is oxidized to succinate which is metabolized via OAA, condensing with ac.CoA to alpha-KG which is transaminated to glutamate, and conversion of aspartate to OAA. This glutamate molecule exits to the cytosol (in exchange with GABA) and is again decarboxylated to a molecule of GABA which is released as synaptic GABA (arrow 3). Once the vesicular release is established, it is accordingly this GABA which is reaccumulated (arrow 4 and the pathway shown in red in Fig. 2.6), since Song et al. (2013) have shown that it is vesicularly released GABA which is accumulated by the neuronal GAT1. Thus, although the released and reaccumulated GABA does not give rise to any increase in GABA concentration it is used as the source for production of another glutamate molecule which can enter the cytosol and form releasable GABA. The whole process (direct and indirect pathways together) produces two molecules of GABA from two molecules of glutamine, but they are both formed from the glutamine molecule that entered the cytosol by the direct pathway. The indirect pathway is not used for direct production of GABA, but it facilitates resynthesis of GABA from accumulated and metabolized GABA. According to the proposed mechanism it is thus not the mitochondrial α-KG formed from astrocytically delivered glutamine which is circled a full turn in the TCA cycle but succinate (which is formed in the TCA cycle by decarboxylation of α-KG) which is converted via almost a complete turn of the cycle to α-KG. An important difference from the original concept by Waagepetersen et al. (1999, 2001) is that the exogenous ac.CoA which is needed to convert mitochondrial OAA to citrate can equally well be supplied by glucose and β-hydroxybutyrate . This would make GABA synthesis much less sensitive to replacement of most glucose with a ketogenic diet than glutamate synthesis. This conclusion is in agreement with a better metabolic efficiency of β-hydroxybutyrate of rats on ketogenic diet found by Roy et al. (2015). The same authors found a 44 % increase in brain content of the BCAAs leucine and isoleucine associated with the presence of free leucine and isoleucine in the liver that was never seen in control rats. Along similar lines Chowdhury et al. (2014, see description earlier and Fig. 2.4) had found a very drastic reduction of glucose :glutamate ratio in the animals treated with high doses of β-hydroxybutyrate (from 1:1 in controls to 0.4:1). Together, the findings by these two groups suggest more drastic changes in glutamine–glutamate/GABA cycling than only replacement of glucose with β-hydroxybutyrate in the processes indicated in this review.

Proposed pathways for production of cytosolically and vesicularly released GABA in GABAergic neurons from glutamine (Gln) released from astrocytes and accumulated into the astrocytic cytosol. Since initially one half of accumulated Gln deamidated to glutamate by glutaminase (PAG) located in the intramembranaceous space enters directly in the cytosol (light blue), whereas another one half enters the mitochondria in a similar manner as in glutamatergic neurons (Waagepetersen et al. 1999, 2001) the figure depicts the metabolic fate of two molecules of glutamine (Italics), whereas each of the two pathways follows the metabolism of one molecule Glu. Glu released directly to the cytosol is decarboxylated to GABA and released to the extracellular space (light blue and arrow 1). When GABAergic signaling is initiated it is reaccumulated into the cytosol of the GABAergic neuron (red and arrow 2) and from there into the mitochondria, probably in exchange with glutamate. Here it is metabolized via succinic semialdehyde (SSA) to succinate which is accumulated into the TCA cycle, where it is metabolized to α-ketoglutarate (α-KG), probably explaining the incorporation of radioactivity from the TCA cycle in released GABA shown by Waagepetersen et al. (1999, 2001). This cycling requires condensation of oxaloacetate (OAA) with acetyl coenzyme A (co.A), which may be derived from either glucose , lactate, or β-hydroxybutyrate (green arrow). α-KG is transaminated to Glu, which exits from the mitochondria (probably in exchange with incoming GABA), is decarboxylated to GABA, which is released to the extracellular space (red and arrow 3). Results by Song et al. (2013) show that during established GABAergic signaling in vivo it is probably mainly or exclusively the GABA formed by this mechanism (released via arrow 3) that becomes reaccumulated into the GABAergic neuron (arrow 4). Thus during established GABAergic signaling formation of vesicularly released GABA sustains itself by the neuronal uptake of GABA and its metabolism to “new” releasable GABA, whereas neuronal reuptake of GABA formed from glutamate released directly to the cytosol (unlabelled gray arrow) ceases. This allows uptake of this GABA (Italics) into astrocytes in the glutamine–glutamate/GABA cycle . The Gln initially released into the mitochondria is converted to α-KG (as also concluded by Waagepetersen et al. 1999, 2001), and this α-KG is reconverted to Glu in the transamination of GABA to SSA. This has the consequence that the cycle of vesicular release and neuronal reuptake and production of “new” GABA only is possible when another molecule of Gln simultaneously is converted to mitochondrial Glu and α-KG. Glu is released to the cytosol andFig. 2.6 (continued) from there to the extracellular fluid and probably accumulated into astrocytes (Italics) in the glutamine–glutamate/GABA cycle. Exchange of OAA, α-KG, and Asp between the two pathways is indicated in the figure, and there is stoichiometric balance between synthesis and utilization. Thus for each 2 Gln molecules the neurons receive from the astrocytes they return 1 Glu and 1 GABA molecule in the glutamine/glutamate/GABA cycle. These pathways are more complex than that originally suggested by Waagepetersen et al. (1999, 2001) but fully compatible with the experimental data obtained by these authors since the 4-h. incubation period used allows time for GABA uptake and metabolism and release via arrow 3. In vivo data by other authors supporting the proposed pathways are cited in the text. To prove or disprove this pathway it will be necessary to study the effect of extracellular GABA on GABA content in GABAergic neurons in the presence of extracellular glutamine
The proposed pathway might explain how the neuron disposes of excess TCA cycle constituents although it shows no pyruvate recycling, indicating lack of formation of pyruvate from TCA cycle constituents (Waagepetersen et al. 2002) and express little if any malic enzyme (Kurz et al. 1993). Of the two glutamine molecules followed in Fig. 2.6 one is returned to astrocytes as released glutamate, the other by the GABA released by arrow 1 after it is no longer reaccumulated by the neurons (cessation of the process shown by the gray arrow) after the reaccumulation of synaptically released GABA has been established. This GABA is converted to glutamate as described by Duarte and Gruetter (2013), but if the pathway scheduled in Fig. 2.6 is followed only one half of this conversion (one GABA molecule) occurs in astrocytes, the other (the GABA molecule metabolized in the pathway shown in red) in the GABAergic neurons . The mechanism seems to be consistent with the conclusion by Van den Berg et al. (1974) ‘that there are two metabolic spaces involved in the degradation of GABA…. One of the GABA degradation metabolic spaces leads to a high labeling of glutamine, the other not.’ The space leading to the high labeling of glutamine is astrocytic. The other neuronal, where it can be seen that no glutamine is formed in the pathway shown in red. It can probably also be concluded that GAD 67 , which participates in the formation of both cytosolic and vesicular GABA catalyzes the formation of the GABA released by nonvesicular means (arrow 1). GAD 65 , which selectively leads to formation of vesicular GABA the release of which is shown in arrow 3.
GABA production and degradation also contribute to the 1:1 ratio between glutamate flux and neuronal glucose utilization in individuals on a normal diet. It was concluded earlier that the requirement for pyruvate formation during conversion of glutamine to glutamate in glutamatergic neurons and pyruvate utilization for anaplerosis together could account for 60 % of the glucose consumption. De novo GABA production depends also on glucose metabolism because newly synthesized astrocytic glutamate normally will be derived from glucose, but this is already included in the 60 %. However, an additional 10–12 % (one half of the 20–25 % of the glutamine–glutamate/GABA cycle used for GABA synthesis) can be explained by the complex processes occurring both in astrocytes (Duarte and Gruetter 2013) and according to Fig. 2.6 also in GABAergic neurons , when GABA is converted to glutamate for recycling (see green arrow in Fig. 2.6). In addition, the rates of GABAergic neuronal glucose oxidation and glutamate/glutamine cycling increase proportionate to glutamatergic neuron glucose oxidation and glutamate/glutamine cycling indicating that the proportionality will be maintained across changes in activity. Thus, under normal conditions with glucose as the only important substrate the combined turnover of glutamate and GABA can account for 70 % of the 1:1 ratio between glutamate cycling and neuronal glucose metabolism .
2.4 Concluding Remarks
The glutamine–glutamate/GABA cycle is an astrocytic-neuronal pathway which is essential for production of transmitter glutamate and GABA and thus for normal brain function. Glucose or lactate is essential precursor for some processes involved in the cycle, whereas they can be replaced by ketone bodies, a BCAA , or acetate at other steps. Formation of glutamate from glutamine is complex in both glutamatergic and GABAergic neurons . It is completely dependent upon concomitant glucose metabolism in glutamatergic neurons , whereas β-hydroxybutyrate can replace glucose in GABAergic neurons . Accordingly, formation of GABA may be less sensitive than that of glutamate to inhibition by diets highly enriched in fats. Energetically glutamate and GABA production via the glutamine–glutamate/GABA cycle is much more expensive than neuronal production, reuptake, and oxidation of glutamate (and GABA) would have been. However, a higher neuronal reuptake capacity would mean that the levels of glutamate and GABA in the extracellular space would be influenced to a greater extent by membrane depolarization as opposed to vesicular release, which could interfere with neuronal communication especially in the case of glutamate. Such a process seems to occur in the very immature brain and the development of the glutamine–glutamate/GABA cycle may be a key factor explaining the difference between the functional capability of the immature and mature brain. It also provides astrocytes with the possibility to coregulate glutamatergic and GABAergic transmission. The modified pathway for synthesis of GABA shown in Fig. 2.6 seems to be able to explain several previous findings of association of specific GAD isozymes and transporters with cytosolic and vesicular released GABA and it may also be relevant for tonic and phasic GABA signaling.
Notes
- 1.
Considerable work has been done studying this mismatch and the larger mismatches that occur during functional activation. There are several theories to explain apparent uncoupling of glucose uptake and oxidation including the astrocyte neuron lactate shuttle (ANLS) , discussed again later, which has received a great deal of attention. As this chapter focuses on oxidative metabolism we refer the reader to two recent reviews on opposite sides of the ANLS controversy (Pellerin and Magistretti 2012; Dienel 2012).
Abbreviations
- 13C MRS:
-
13C magnetic resonance spectroscopy
- α-KG:
-
α-ketoglutarate
- AAT:
-
Aspartate aminotransferase
- Ac.CoA:
-
Acetyl coenzyme A
- ADP:
-
Adenosine diphosphate
- AGC:
-
Glutamate/aspartate exchanger (in brain AGC1 or aralar)
- ANLS:
-
Astrocyte neuron lactate shuttle
- AOAA:
-
Alpha-aminooxyacetic acid
- ATP:
-
Adenosine triphosphate
- AV:
-
Arteriovenous
- BCAA:
-
Branched chain amino acid
- BCAT:
-
Branched chain amino acid transaminase
- BCATc:
-
Cytosolic branched chain amino acid transferase
- BCATm:
-
Mitochondrial branched chain amino acid transferase
- DNA, RNA:
-
Deoxyribonucleic acid, ribonucleic acid
- GABA:
-
γ-aminobutyric acid
- GABA-T:
-
GABA transaminase
- GAD65, GAD67:
-
Glutamate amino decarboxylase 65 and 67 KD isoforms
- GAT1, GAT2, GAT3:
-
GABA transporters 1, 2, 3
- GDH:
-
Glutamate dehydrogenase
- GLAST:
-
Glutamate aspartate transporter
- GLT-1:
-
Glutamate type 1 transporter
- GS:
-
Glutamine synthase
- KIC:
-
Alpha-ketoisocaproic acid
- MAS:
-
Malate-aspartate shuttle
- MCT1:
-
Monocarboxylic acid transporter 1
- MCT2, MCT3:
-
Monocarboxylic acid transporters 2, 3
- MCTs:
-
Monocarboxylic acid transporters
- NADH/NAD+ :
-
Nicotinamide adenine dinucleotide
- NADPH:
-
Nicotinamide adenine dinucleotide phosphate
- OAA:
-
Oxaloacetate
- PAG:
-
Phosphate activated glutaminase
- PC:
-
Pyruvate carboxylase
- PDH:
-
Pyruvate dehydrogenase complex
- SAT1, SAT2:
-
System A glutamine transporters 1 and 2
- SN1:
-
System N glutamine transporter 1
- SSA:
-
Succinic semialdehyde
- TCA cycle:
-
Tricarboxylic acid cycle
References
Amaral AI (2013) Effects of hypoglycaemia on neuronal metabolism in the adult brain: role of alternative substrates to glucose. J Inherit Metab Dis 36(4):621–634
Anlauf E, Derouiche A (2013) Glutamine synthetase as an astrocytic marker: its cell type and vesicle localization. Front Endocrinol (Lausanne) 4:144 (Hertz L, Rodrigues TB (Eds). eCollection)
Bak LK, Ziemńska E, Waagepetersen HS, Schousboe A, Albrecht J (2008) Metabolism of [U-13C]glutamine and [U-13C]glutamate in isolated rat brain mitochondria suggests functional phosphate-activated glutaminase activity in matrix. Neurochem Res 233(2):273–278
Bakken IJ, Sonnewald U, Clark JB, Bates TE (1997) [U-13C]glutamate metabolism in rat brain mitochondria reveals malic enzyme activity. Neuroreport 8(7):1567–1570
Balazs R (1965) Control of glutamate oxidation in brain and liver mitochondrial systems. Biochem J 95:497–508
Balazs R, Dahl D, Harwood JR (1966) Subcellular distribution of enzymes of glutamate metabolism in rat brain. J Neurochem 13:897–905
Barnett NL, Pow DV, Robinson SR (2000) Inhibition of Müller cell glutamine synthetase rapidly impairs the retinal response to light. Glia 30(1):64–73
Bauer DE, Jackson JG, Genda EN, Montoya MM, Yudkoff M, Robinson MB (2012) The glutamate transporter, GLAST, participates in a macromolecular complex that supports glutamate metabolism. Neurochem Int 61(4):566–574
Ben-Yoseph O, Camp DM, Robinson TE, Ross BD (1995) Dynamic measurements of cerebral pentose phosphate pathway activity in vivo using [1,6-13C2,6,6-2H2]glucose and microdialysis. J Neurochem 64(3):1336–1342
Biedermann B, Bringmann A, Reichenbach A (2002) High-affinity GABA uptake in retinal glial (Müller) cells of the guinea pig: electrophysiological characterization, immunohistochemical localization, and modeling of efficiency. Glia 39(3):217–228
Bjørnsen LP, Eid T, Holmseth S, Danbolt NC, Spencer DD, de Lanerolle NC (2007) Changes in glial glutamate transporters in human epileptogenic hippocampus: inadequate explanation for high extracellular glutamate during seizures. Neurobiol Dis 25(2):319–330
Bjørnsen LP, Hadera MG, Zhou Y, Danbolt NC, Sonnewald U (2014) The GLT-1 (EAAT2; slc1a2) glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse. J Neurochem 128(5):641–649
Boumezbeur F, Petersen KF, Cline GW, Mason GF, Behar KL, Shulman GI, Rothman DL (2010) The contribution of blood lactate to brain energy metabolism in humans measured by dynamic 13C nuclear magnetic resonance spectroscopy. J Neurosci 30(42):13983–13991
Bouzier-Sore AK, Voisin P, Canioni P, Magistretti PJ, Pellerin L (2003) Lactate is a preferential oxidative energy substrate over glucose for neurons in culture. J Cereb Blood Flow Metab 23(11):1298–12306
Bouzier-Sore AK, Voisin P, Bouchaud V, Bezancon E, Franconi JM, Pellerin L (2006) Competition between glucose and lactate as oxidative energy substrates in both neurons and astrocytes: a comparative NMR study. Eur J Neurosci 24(6):1687–1694
Brekke E, Morken TS (2015) Sonnewald U (2015) Glucose metabolism and astrocyte-neuron interactions in the neonatal brain. Neurochem Int 82:33–41
Bringmann A, Grosche A, Pannicke T, Reichenbach A (2013) GABA and glutamate uptake and metabolism in retinal glial (Müller) cells. Front Endocrinol (Lausanne) 4:48 (Hertz L, Rodrigues TB (Eds). eCollection)
Chowdhury GM, Patel AB, Mason GF, Rothman DL, Behar KL (2007) Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J Cereb Blood Flow Metab 27(12):1895–1907
Chowdhury GM, Jiang L, Rothman DL, Behar KL (2014) The contribution of ketone bodies to basal and activity-dependent neuronal oxidation in vivo. J Cereb Blood Flow Metab 34(7):1233–1242
Coco S, Verderio C, Trotti D, Rothstein JD, Volterra A, Matteoli M (1997) Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur J Neurosci 9:1902–1910
Collins TJ, Berridge MJ, Lipp P, Bootman MD (2002) Mitochondria are morphologically and functionally heterogeneous within cells. EMBO J 21(7):1616–1627
Cooper AJL (2012) The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem Res 37(11):2439–2455
Cooper AJ (2013) Quantitative analysis of neurotransmitter pathways under steady state conditions—a perspective. Front Endocrinol (Lausanne) 4:179 (Hertz L, Rodrigues TB (Eds). eCollection)
Cremer JE, Braun LD, Oldendorf WH (1976) Changes during development in transport processes of the blood-brain barrier. Biochim Biophys Acta 448(4):633–637
Cruz NF, Ball KK, Dienel GA (2007) Functional imaging of focal brain activation in conscious rats: impact of [(14)C]glucose metabolite spreading and release. J Neurosci Res 85(15):3254–3266
Cudalbu C, Lanz B, Duarte JM, Morgenthaler FD, Pilloud Y, Mlynárik V, Gruetter R (2012) Cerebral glutamine metabolism under hyperammonemia determined in vivo by localized (1)H and (15)N NMR spectroscopy. J Cereb Blood Flow Metab 32(4):696–708
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65(1):1–105
De Feyter HM, Mason GF, Shulman GI, Rothman DL, Petersen KF (2013) Increased brain lactate concentrations without increased lactate oxidation during hypoglycemia in type 1 diabetic individuals. Diabetes 62(9):3075–3080
Dennis SC, Lai JC, Clark JB (1977) Comparative studies on glutamate metabolism in synaptic and non-synaptic rat brain mitochondria. Biochem J 164(3):727–736
Dienel GA (2012) Brain lactate metabolism: the discoveries and the controversies. J Cereb Blood Flow Metab 32(7):1107–1138
Dringen R, Schmoll D, Cesar M, Hamprecht B (1993) Incorporation of radioactivity for gluconeogenesis in brain cells. Biol Chem Hoppe Seyler
Dringen R, Schmoll D, Cesar M, Hamprecht B (2005) Incorporation of radioactivity from [14C]lactate into the glycogen of cultured mouse astroglial cells. Evidence for functional roles in brain cells. J Neurosci Res 79(1–2):11–18
Duarte JM, Gruetter R (2013) Glutamatergic and GABAergic energy metabolism measured in the rat brain by (13) C NMR spectroscopy at 14.1 T. J Neurochem 126(5):579–590
Eid T, Ghosh A, Wang Y, Beckström H, Zaveri HP, Lee TS, Lai JC, Malthankar-Phatak GH, de Lanerolle NC (2008) Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 131(Pt 8):2061–2070
Engel PC, Dalziel K (1967) The equilibrium constants of the glutamate dehydrogenase systems. Biochem J 105(2):691–695
Fink-Jensen A, Suzdak PD, Swedberg MD, Judge ME, Hansen L, Nielsen PG (1992) The gamma-aminobutyric acid (GABA) uptake inhibitor, tiagabine, increases extracellular brain levels of GABA in awake rats. Eur J Pharmacol 220(2–3):197–201
Fitzpatrick SM, Hetherington HP, Behar KL, Shulman RG (1990) The flux from glucose to glutamate in the rat brain in vivo as determined by 1H-observed, 13C-edited NMR spectroscopy. J Cereb Blood Flow Metab 10(2):170–179
Frigerio F, Karaca M, De Roo M, Mlynárik V, Skytt DM, Carobbio S, Pajęcka K, Waagepetersen HS, Gruetter R, Muller D, Maechler P (2012) Deletion of glutamate dehydrogenase 1 (Glud1) in the central nervous system affects glutamate handling without altering synaptic transmission. J Neurochem 123(3):342–348
Gaitonde MK, Jones J, Evans G (1987) Metabolism of glucose into glutamate via the hexose monophosphate shunt and its inhibition by 6-aminonicotinamide in rat brain in vivo. Proc R Soc Lond B Biol Sci 231(1262):71–90
Gandhi GK, Cruz NF, Ball KK, Dienel GA (2009) Astrocytes are poised for lactate trafficking and release from activated brain and for supply of glucose to neurons. J Neurochem 111(2):522–536
Genda EN, Jackson JG, Sheldon AL, Locke SF, Greco TM, O’Donnell JC, Spruce LA, Xiao R, Guo W, Putt M, Seeholzer S, Ischiropoulos H, Robinson MB (2011) Co-compartmentalization of the astroglial glutamate transporter, GLT-1, with glycolytic enzymes and mitochondria. J Neurosci 31(50):18275–18288
Gibbs ME, Lloyd HG, Santa T, Hertz L (2007) Glycogen is a preferred glutamate precursor during learning in 1-day-old chick: biochemical and behavioral evidence. J Neurosci Res 85(15):3326–3333
Gibbs ME, Gibbs Z, Hertz L (2009) Rescue of Abeta(1-42)-induced memory impairment in day-old chick by facilitation of astrocytic oxidative metabolism: implications for Alzheimer’s disease. J Neurochem 109(Suppl 1):230–236
Gorovits R, Avidan N, Avisar N, Shaked I, Vardimon L (1997) Glutamine synthetase protects against neuronal degeneration in injured retinal tissue. Proc Natl Acad Sci U S A 94(13):7024–7029
Goubard V, Fino E, Venance L (2011) Contribution of astrocytic glutamate and GABA uptake to corticostriatal information processing. J Physiol 589(Pt9):2301–2319
Gruetter R, Seaquist ER, Ugurbil K (2001) A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab 281(1):E100–E112
Gulanski BI, De Feyter HM, Page KA, Belfort-DeAguiar R, Mason GF, Rothman DL, Sherwin RS (2013) Increased brain transport and metabolism of acetate in hypoglycemia. J Clin Endocrinol Metab 98(9):3811–3820
Hertz L (2011a) Brain glutamine synthesis requires neuronal aspartate: a commentary. J Cereb Blood Flow Metab 31(1):384–387
Hertz L (2011b) Astrocytic energy metabolism and glutamate formation—relevance for 13C-NMR spectroscopy and importance of cytosolic/mitochondrial trafficking. Magn Reson Imaging 29(10):1319–1329
Hertz L (2013) The glutamate-glutamine (GABA) cycle: importance of late postnatal development and potential reciprocal interactions between biosynthesis and degradation. Front Endocrinol (Lausanne) 4:59, Hertz L, Rodrigues TB (Eds). eCollection
Hertz L, Dienel GA (2002) Energy metabolism in the brain. Int Rev Neurobiol 51:1–102
Hertz L, Dienel GA (2005) Lactate transport and transporters: general principles and functional roles in brain cells. J Neurosci Res 79(1–2):11–18
Hertz L, Wu PH, Schousboe A (1978) Evidence for net uptake of GABA into mouse astrocytes in primary cultures—its sodium dependence and potassium independence. Neurochem Res 3(3):313–323
Hertz L, Peng L, Dienel GA (2007) Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J Cereb Blood Flow Metab 27(2):219–249
Hertz L, Gibbs ME, Dienel GA (2014) Fluxes of lactate into, from, and among gap junction-coupled astrocytes and their interaction with noradrenaline. Front Neurosci 8:261
Hertz L, Chen Y. (2016) Editorial: all 3 types of glial cells are important for memory formation. Front Integr Neurosci (in press)
Herzog RI, Jiang L, Herman P, Zhao C, Sanganahalli BG, Mason GF, Hyder F, Rothman DL, Sherwin RS, Behar KL (2013) Lactate preserves neuronal metabolism and function following antecedent recurrent hypoglycemia. J Clin Invest 123(5):1988–1998
Hostetler KY, Landau BR (1967) Estimation of the pentose cycle contribution to glucose metabolism in tissue in vivo. Biochemistry 6(10):2961–2964
Huang R, Peng L, Chen Y, Hajek I, Zhao Z, Hertz L (1994) Signaling effect of elevated potassium concentrations and monoamines on brain energy metabolism at the cellular level. Dev Neurosci 16(5–6):337–351
Hutson SM, Berkich D, Drown P, Xu B, Aschner M, LaNoue KF (1998) Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J Neurochem 71(2):863–874
Hutson SM, Lieth E, LaNoue KF (2001) Function of leucine in excitatory neurotransmitter metabolism in the central nervous system. J Nutr 131(3):846S–850S
Hutson SM, Cole JT, Sweatt AJ, LaNoue KF (2008) Is the anaplerotic enzyme pyruvate carboxylase (PC) only expressed in astrocytes? J Neurochem 104(Suppl 1):58–59
Hyder F, Fulbright RK, Shulman RG, Rothman DL (2013a) Glutamatergic function in the resting awake human brain is supported by uniformly high oxidative energy. J Cereb Blood Flow Metab 33(3):339–347
Hyder F, Rothman DL, Bennett MR (2013b) Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci U S A 110(9):3549–3554
Ipponi A, Lamberti C, Medica A, Bartolini A, Malmberg-Aiello P (1999) Tiagabine antinociception in rodents depends on GABA(B) receptor activation: parallel antinociception testing and medial thalamus GABA microdialysis. Eur J Pharmacol 368(2–3):205–211
Iversen LL, Neal MJ (1968) The uptake of [3H]GABA by slices of rat cerebral cortex. J Neurochem 15:1141–1910
Jackson JG, O’Donnell JC, Takano H, Coulter DA, Robinson MB (2014) Neuronal activity and glutamate uptake decrease mitochondrial mobility in astrocytes and position mitochondria near glutamate transporters. J Neurosci 34(5):1613–1624
Jiang L, Mason GF, Rothman DL, de Graaf RA, Behar KL (2011) Cortical substrate oxidation during hyperketonemia in the fasted anesthetized rat in vivo. J Cereb Blood Flow Metab 31(12):2313–2323
Jiang L, Gulanski BI, De Feyter HM, Weinzimer SA, Pittman B, Guidone E, Koretski J, Harman S, Petrakis IL, Krystal JH, Mason GF (2013) Increased brain uptake and oxidation of acetate in heavy drinkers. J Clin Invest 123(4):1605–1614
Kanamori K, Ross BD (1995) Steady-state in vivo glutamate dehydrogenase activity in rat brain measured by 15N NMR. J Biol Chem 270(42):24805–24809
Karaca M, Frigerio F, Migrenne S, Martin-Levilain J, Skytt DM, Pajecka K, Martin-del-Rio R, Gruetter R, Tamrit-Rodriguez J, Waagepetersen HS, Magnan C, Maechler P (2015) GDH-dependent glutamate oxidation in the brain dictates peripheral energy substrate distribution. Cell Rep 13:365–375
Kaufman DL, Houser CR, Tobin AJ (1991) Two forms of the gamma-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem 56(2):720–723
Keiding S, Sørensen M, Bender D, Munk OL, Ott P, Vilstrup H (2006) Brain metabolism of 13N-ammonia during acute hepatic encephalopathy in cirrhosis measured by positron emission tomography. Hepatology 43(1):42–50
Kemppainen J, Aalto S, Fujimoto T, Kalliokoski KK, Långsjö J, Oikonen V, Rinne J, Nuutila P, Knuuti J (2005) High intensity exercise decreases global brain glucose uptake in humans. J Physiol 568(Pt 1):323–332
Kratzing CC (1953) The ability of some carboxylic acids to maintain phosphate level and support electrical stimulation in cerebral tissues. Biochem J 54(2):312–317
Krebs HA (1953) Equilibria in transamination systems. Biochem J 54(1):82–86
Kurz GM, Wiesinger H, Hamprecht B (1993) Purification of cytosolic malic enzyme from bovine brain, generation of monoclonal antibodies, and immunocytochemical localization of the enzyme in glial cells of neural primary cultures. J Neurochem 60(4):1467–1474
Larsson OM, Johnston GA, Shousboe A (1983) Differences in uptake kinetics of cis-3-aminocyclohexane carboxylic acid into neurons and astrocytes in primary cultures. Brain Res 260(2):279–285
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22(5):1523–1531
Leke R, Bak LK, Iversen P, Sørensen M, Keiding S, Vilstrup H, Ott P, Portela LV, Schousboe A, Waagepetersen HS (2011) Synthesis of neurotransmitter GABA via the neuronal tricarboxylic acid cycle is elevated in rats with liver cirrhosis consistent with a high GABAergic tone in chronic hepatic encephalopathy. J Neurochem 117(5):824–832
Li B, Hertz L, Peng L (2012) Aralar mRNA and protein levels in neurons and astrocytes freshly isolated from young and adult mouse brain and in maturing cultured astrocytes. Neurochem Int 61(8):1325–1332
Lovatt D, Sonnewald U, Waagepetersen HS, Schousboe A, He W, Lin JH, Han X, Takano T, Wang S, Sim FJ, Goldman SA, Nedergaard M (2007) The transcriptome and metabolic gene signature of protoplasmic astrocytes in the adult murine cortex. J Neurosci 27(45):12255–12266
Lynen F (1953) Mechanism of beta oxidation of fatty acids. Bull Soc Chim Biol (Paris) 35(10):1061–1083
Maciejewski PK, Rothman DL (2008) Proposed cycles for functional glutamate trafficking in synaptic neurotransmission. Neurochem Int 52(4–5):809–825
Magistretti PJ, Allaman I (2015) A cellular perspective on brain energy metabolism and functional imaging. Neuron 86(4):883–901
Mangia S, Garreffa G, Maraviglia B, Giove F (2010) Metabolic correlatives of brain activity in a FOS epilepsy patient. NMR Biomed 23(2):170–178
Mangia S, Giove F, Dinuzzo M (2012) Metabolic pathways and activity-dependent modulation of glutamate concentration in the human brain. Neurochem Res 37(11):2554–2561
Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195:1356–1358
Mason GF, Petersen KF, de Graaf RA, Shulman GI, Rothman D (2007) Measurements of the anaplerotic rate in the human cerebral cortex using 13C magnetic resonance spectroscopy and [1-13C] and [2-13C] glucose. J Neurochem 100:73–86
McIlwain H (1953) Substances which support respiration and metabolic response to electrical impulses in human cerebral tissues. J Neurol Neurosurg Psychiatry 16(4):257–266
McKenna MC (2012) Substrate competition studies demonstrate oxidative metabolism of glucose, glutamate, glutamine, lactate and 3-hydroxybutyrate in cortical astrocytes from rat brain. Neurochem Res 37(11):2613–2626
McKenna MC (2013) Glutamate pays its own way in astrocytes. Front Endocrinol (Lausanne) 16(4):191
McKenna MC, Sonnewald U (2005) GABA alters the metabolic fate of [U-13C]glutamate in cultured cortical astrocytes. J Neurosci Res 79(1–2):81–87
McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66(1):386–393
McKenna MC, Hopkins IB, Lindauer SL, Bamford P (2006) Aspartate aminotransferase in synaptic and nonsynaptic mitochondria: differential effect of compounds that influence transient hetero-enzyme complex (metabolon) formation. Neurochem Int 48(6–7):629–636
McPherson PA, McEneny J (2012) The biochemistry of ketogenesis and its role in weight management, neurological disease and oxidative stress. J Physiol Biochem 68(1):141–151
Melø TM, Nehlig A, Sonnewald U (2006) Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int 48(6–7):498–507
Muir D, Berl S, Clarke DD (1986) Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res 380:336–340
Murthy CR, Hertz L (1987) Comparison between acute and chronic effects of ammonia on branched-chain amino acid oxidation and incorporation into protein in primary cultures of astrocytes and of neurons. J Neurosci Res 17(3):271–276
Muto Y, Sato S, Watanabe A, Moriwaki H, Suzuki K, Kato A, Kato M, Nakamura T, Higuchi K, Nishiguchi S, Kumada H (2005) Long-Term Survival Study Group. Effects of oral branched-chain amino acid granules on event-free survival in patients with liver cirrhosis. Clin Gastroenterol Hepatol 3(7):705–713
Nissen-Meyer LS, Chaudhry FA (2013) Protein kinase C phosphorylates the system N glutamine transporter SN1 (Slc38a3) and regulates its membrane trafficking and degradation. Front Endocrinol (Lausanne) 4:138
Norenberg MD, Martinez-Hernandez A (1979) Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res 161(2):303–310
Ola MS, Hosoya K, LaNoue KF (2011) Regulation of glutamate metabolism by hydrocortisone and branched chain keto acids in cultured rat retinal Müller cells (TR-MUL). Neurochem Int 59(5):656–663
Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF Jr (1967) Brain metabolism during fasting. J Clin Invest 46(10):1589–1595
Palaiologos G, Hertz L, Schousboe A (1988) Evidence that aspartate aminotransferase activity and ketodicarboxylate carrier function are essential for biosynthesis of transmitter glutamate. J Neurochem 51(1):317–320
Palaiologos G, Hertz L, Schousboe A (1989) Role of aspartate aminotransferase and mitochondrial dicarboxylate transport for release of endogenously and exogenously supplied neurotransmitter in glutamatergic neurons. Neurochem Res 14(4):359–366
Pan JW, de Graaf RA, Petersen KF, Shulman GI, Hetherington HP, Rothman DL (2002) [2,4-13 C2]-beta-Hydroxybutyrate metabolism in human brain. J Cereb Blood Flow Metab 22(7):890–898
Pardo B, Rodrigues TB, Contreras L, Garzón M, Llorente-Folch I, Kobayashi K, Saheki T, Cerdan S, Satrústegui J (2011) Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J Cereb Blood Flow Metab 31(1):90–101
Passarella S, Atlante A, Barile M, Quagliariello E (1987) Anion transport in rat brain mitochondria: fumarate uptake via the dicarboxylate carrier. Neurochem Res 12(3):255–264
Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL (2005) The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Natl Acad Sci U S A 102(15):5588–5593
Patel AB, de Graaf RA, Martin DL, Battaglioli G, Behar K (2006) Evidence that GAD65 mediates increased GABA synthesis during intense neuronal activity in vivo. J Neurochem 97(2):385–396
Patel AB, Lai JC, Chowdhury GM, Hyder F, Rothman DL, Shulman RG, Behar KL (2014) Direct evidence for activity-dependent glucose phosphorylation in neurons with implications for the astrocyte-to-neuron lactate shuttle. Proc Natl Acad Sci U S A 111(14):5385–5390
Patel AB, de Graaf RA, Rothman DL, Behar KL (2015) Effects of GAT1 inhibition by tiagabine on brain glutamate and GABA metabolism in the anesthetized rat in vivo. J Neurosci Res 93(7):1101–1108
Peca S, Carnì M, Di Bonaventura C, Aprile T, Hagberg GE, Giallonardo AT, Manfredi M, Mangia S, Garreffa G, Maraviglia B, Giove F (2010) Metabolic correlatives of brain activity in a FOS epilepsy patient. NMR Biomed 23(2):170–178
Pellerin L, Magistretti PJ (2012) Sweet sixteen for ANLS. J Cereb Blood Flow Metab 32(7):1152–1166
Perez EL, Lauritzen F, Wang Y, Lee TS, Kang D, Zaveri HP, Chaudhry FA, Ottersen OP, Bergersen LH, Eid T (2012) Evidence for astrocytes as a potential source of the glutamate excess in temporal lobe epilepsy. Neurobiol Dis 47(3):331–337
Raichle ME (2015) The restless brain: how intrinsic activity organizes brain function. Philos Trans R Soc Lond B Biol Sci 19(370):20140172
Raichle ME, Gusnard DA (2002) Appraising the brain’s energy budget. Proc Natl Acad Sci U S A 99(16):10237–10239
Robinson AM, Williamson DH (1980) Physiological role of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev 60(1):143–187
Rodrigues TB, López-Larrubia P, Cerdán S (2009) Redox dependence and compartmentation of [13C]pyruvate in the brain of deuterated rats bearing implanted C6 gliomas. J Neurochem 109(Suppl 1):237–245
Romei C, Sabolla C, Raiteri L (2014) GABA release provoked by disturbed Na(+), K(+) and Ca(2+) homeostasis in cerebellar nerve endings: roles of Ca(2+) channels, Na(+)/Ca(2+) exchangers and GAT1 transporter reversal. Neurochem Int 72:1–9
Romei C, Sabolla C, Raiteri L (2015) High-affinity GABA uptake by neuronal GAT1 transporters provokes release of [(3)H]GABA by homoexchange and through GAT1-independent Ca(2+)-mediated mechanisms. Neuropharmacology 88:164–170
Ross CD, Godfrey DA (1987) Distribution of activities of aspartate aminotransferase isoenzymes and malate dehydrogenase in guinea pig retinal layers. J Histochem Cytochem 35(6):669–674
Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL (2011) 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed 24(8):943–957
Rothman DL, De Feyter HM, Maciejewski PK, Behar KL (2012) Is there in vivo evidence for amino acid shuttles carrying ammonia from neurons to astrocytes? Neurochem Res 37(11):2597–25612
Roy M, Beauvieux MC, Naulin J, El Hamrani D, Gallis JL, Cunnane SC, Bouzier-Sore AK (2015) Rapid adaptation of rat brain and liver metabolism to a ketogenic diet: an integrated study using (1)H- and (13)C-NMR spectroscopy. J Cereb Blood Flow Metab 25(7):1153–1162
Sa HD, Park JY, Jeong SJ, Lee KW, Kim JH (2015) Characterization of glutamate decarboxylase (GAD) from Lactobacillus sakei A156 isolated from Jeot-gal. J Microbiol Biotechnol 25(5):696–703
Schousboe I, Bro B, Schousboe A (1977a) Intramitochondrial localization of the 4-aminobutyrate-2-oxoglutarate transaminase from ox brain. Biochem J 162(2):303–307
Schousboe A, Hertz L, Svenneby G (1977b) Uptake and metabolism of GABA in astrocytes cultured from dissociated mouse brain hemispheres. Neurochem Res 2(2):217–229
Schousboe A, Bak LK, Waagepetersen HS (2013) Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol (Lausanne) 4:102
Scimemi A (2014a) Plasticity of GABA transporters: an unconventional route to shape inhibitory synaptic transmission. Front Cell Neurosci 8:128, eCollection
Scimemi A (2014b) Structure, function, and plasticity of GABA transporters. Front Cell Neurosci 8:161. doi:10.3389/fncel.2014.00161, eCollection
Shank RP, Bennett GS, Freytag SO, Campbell GL (1984) Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res 29(1–2):364–367
Shen J, Sibson NR, Cline G, Behar KL, Rothman DL, Shulman RG (1998) 15N-NMR spectroscopy studies of ammonia transport and glutamine synthesis in the hyperammonemic rat brain. Dev Neurosci 20(4–5):434–443
Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG (1998) Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Natl Acad Sci U S A 95(1):316–321
Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG (2001) In vivo 13C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during [2-13C]glucose infusion. J Neurochem 76(4):975–989
Siesjö BK (1978) Brain energy metabolism. Wiley, New York
Smith QR (2000) Transport of glutamate and other amino acids at the blood-brain barrier. J Nutr 130(4S Suppl):1016S–1022S
Smith AL, Satterthwaite HS, Sokoloff L (1969) Induction of brain D(--)-beta-hydroxybytrate dehydrogenase activity by fasting. Science 163(3862):79–81
Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M (1977) The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem 28(5):897–916
Song I, Volynski K, Brenner T, Ushkaryov Y, Walker M, Semyanov A (2013) Different transporter systems regulate extracellular GABA from vesicular and non-vesicular sources. Front Cell Neurosci 7:23
Sonnewald U (2014) Glutamate synthesis has to be matched by its degradation—where do all the carbons go? J Neurochem 131(4):399–406
Sonnewald U, Westergaard N, Hassel B, Müller TB, Unsgård G, Fonnum F, Hertz L, Schousboe A, Petersen SB (1993) NMR spectroscopic studies of 13C acetate and 13C glucose metabolism in neocortical astrocytes: evidence for mitochondrial heterogeneity. Dev Neurosci 15(3–5):351–358
Steinman MQ, Gao V, Alberini CM (2016) The role of lactate-mediated metabolic coupling between astrocytes and neurons in long term memory formation. Front Intergr Neurosci 3(10):10
Swanson RA, Benington JH (1996) Astrocyte glucose metabolism under normal and pathological conditions in vitro. Dev Neurosci 18(5–6):515–521
Takagaki G, Tsukada Y (1957) The effect of some inorganic ions on brain slices metabolizing glucose or pyruvate. J Neurochem 2(1):21–29
Tang F, Lane S, Korsak A, Paton JF, Gourine AV, Kasparov S, Teschemacher AG (2014) Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat Commun 5:3284
Tian N, Petersen C, Kash S, Baekkeskov S, Copenhagen D, Nicoll R (1999) The role of the synthetic enzyme GAD65 in the control of neuronal gamma-aminobutyric acid release. Proc Natl Acad Sci U S A 96(22):12911–12916
Urion D, Vreman HJ, Weiner MW (1979) Effect of acetate on hypoglycemic seizures in mice. Diabetes 28(11):1022–1026
Van den Berg CJ, Matheson DF, Ronda G (1974) A model of glutamate metabolism in brain. A biochemical analysis of a heterogeneous structure. In: Berl S, Clarke DD, Schneider D (eds) Metabolic compartmentation and neurotransmission. Plenum Press, New York, pp 515–540
Van Hall G, Strømstad M, Rasmussen P, Jans O, Zaar M, Gam C, Quistorff B, Secher NH, Nielsen HB (2009) Blood lactate is an important energy source for the human brain. J Cereb Blood Flow Metab 29(6):1121–1129
Vannucci SJ, Simpson IA (2003) Developmental switch in brain nutrient transporter expression in the rat. Am J Physiol Endocrinol Metab 285(5):E1127–E1134
Vining EP, Freeman JM, Ballaban-Gil K, Camfield CS, Camfield PR, Holmes GL, Shinnar S, Shuman R, Trevathan E, Wheless JW (1998) A multicenter study of the efficacy of the ketogenic diet. Arch Neurol 55(11):1433–1437
Vogel R, Wiesinger H, Hamprecht B, Dringen R (1999) The regeneration of reduced glutathione in rat forebrain mitochondria identifies metabolic pathways providing the NADPH required. Neurosci Lett 275(2):97–100
Volkow ND, Kim SW, Wang GJ, Alexoff D, Logan J, Muench L, Shea C, Telang F, Fowler JS, Wong C, Benveniste H, Tomasi D (2013) Acute alcohol intoxication decreases glucose metabolism but increases acetate uptake in the human brain. Neuroimage 64:277–283
Volkow ND, Wang GJ, Shokri Kojori E, Fowler JS, Benveniste H, Tomasi D (2015) Alcohol decreases baseline brain glucose metabolism more in heavy drinkers than controls but has no effect on stimulation-induced metabolic increases. J Neurosci 35(7):3248–3255
Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A (1999) Synthesis of vesicular GABA from glutamine involves TCA cycle metabolism in neocortical neurons. J Neurosci Res 57(3):342–349
Waagepetersen HS, Sonnewald U, Gegelashvili G, Larsson OM, Schousboe A (2001) Metabolic distinction between vesicular and cytosolic GABA in cultured GABAergic neurons using 13C magnetic resonance spectroscopy. J Neurosci Res 63(4):347–355
Waagepetersen HS, Qu H, Hertz L, Sonnewald U, Schousboe A (2002) Demonstration of pyruvate recycling in primary cultures of neocortical astrocytes but not in neurons. Neurochem Res 27(11):1431–1437
Waagepetersen HS, Hansen GH, Fenger K, Lindsay JG, Gibson G, Schousboe A (2006) Cellular mitochondrial heterogeneity in cultured astrocytes as demonstrated by immunogold labeling of alpha-ketoglutarate dehydrogenase. Glia 53(2):225–231
Walls AB, Nilsen LH, Eyjolfsson EM, Vestergaard HT, Hansen SL, Schousboe A, Sonnewald U, Waagepetersen H (2011) Knockout of GAD65 has major impact on synaptic GABA synthesized from astrocyte-derived glutamine. J Cereb Blood Flow Metab 31(2):494–503
Walls AB, Waagepetersen HS, Bak LK, Schousboe A, Sonnewald U (2015) The glutamine/glutamate/GABA cycle: function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem Res 40(2):402–409
Waniewski RA, Martin DL (1998) Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci 18(14):5225–5233
Watanabe A, Shiota T, Takei N, Fujiwara M, Nagashima H (1986) Ammonia detoxification by accelerated oxidation of branched chain amino acids in brains of acute hepatic failure rats. Biochem Med Metab Biol 3:367–375
Westergaard N, Drejer J, Scjhouseboe A, Sonnewald U (1996) Evaluation of the importance of transamination versus deamination in astrocytic metabolism of [U-13C]glutamate. Glia 17(2):160–168
Whitelaw BS, Robinson MB (2013) Inhibitors of glutamate dehydrogenase block sodium-dependent glutamate uptake in rat brain membranes. Front Endocrinol (Lausanne) 4:123 (Hertz L, Rodrigues TB (Eds). eCollection)
Wong E, Schousboe A, Saito K, Wu JY, Roberts E (1974) Immunochemical studies of brain glutamate decarboxylase and GABA-transaminase of six inbred strains of mice. Brain Res 68(1):133–142
Wyss MT, Jolivet R, Buck A, Magistretti PJ, Weber B (2011) In vivo evidence for lactate as a neuronal energy source. J Neurosci 31(20):7477–7485
Yu AC, Schousboe A, Hertz L (1982) Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem 4:954–960
Yu AC, Drejer J, Hertz L, Schousboe A (1983) Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem 41(5):1484–1487
Yu AC, Hertz E, Hertz L (1984) Alterations in uptake and release rates for GABA, glutamate, and glutamine during biochemical maturation of highly purified cultures of cerebral cortical neurons, a GABAergic preparation. J Neurochem 42(4):951–960
Yudkoff M, Daikhin Y, Grunstein L, Nissim I, Stern J, Pleasure D, Nissim I (1996) Astrocyte leucine metabolism: significance of branched-chain amino acid transamination. J Neurochem 66(1):378–385
Zabłocki K, Bryła J (1988) Effect of glycerol on gluconeogenesis in isolated rabbit kidney cortex tubules. Biochim Biophys Acta 970(3):231–240
Zaganas I, Waagepetersen HS, Georgopoulos P, Sonnewald U, Plaitakis A, Schousboe A (2001) Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J Neurosci Res 66(5):909–913
Zaganas I, Kanavouras K, Mastorodemos V, Latsoudis H, Spanaki C, Plaitakis A (2009) The human GLUD2 glutamate dehydrogenase: localization and functional aspects. Neurochem Int 55(1–3):52–63
Zaganas I, Pajęcka K, Wendel Nielsen C, Schousboe A, Waagepetersen HS, Plaitakis A (2013) The effect of pH and ADP on ammonia affinity for human glutamate dehydrogenases. Metab Brain Dis 28(2):127–131
Zhang TM, Rasschaert J, Malaisse WJ (1995) Metabolism of succinic acid methyl esters in neural cells. Biochem Mol Med 54(2):112–116
Zhang Y, Kuang Y, Xu K, Harris D, Lee Z, LaManna J, Puchowicz MA (2013) Ketosis proportionately spares glucose utilization in brain. J Cereb Blood Flow Metab 33(8):1307–1311
Zhou Y, Danbolt NC (2013) GABA and glutamate transporters in brain. Front Endocrinol (Lausanne) 4:165 (Hertz L, Rodrigues TB (Eds). eCollection)
Zielińska M, Popek M, Albrecht J (2014) Roles of changes in active glutamine transport in brain edema development during hepatic encephalopathy: an emerging concept. Neurochem Res 39(3):599–604
Ziemińska E, Hilgier W, Waagepetersen HS, Hertz L, Sonnewald U, Schousboe A, Albrecht J (2004) Analysis of glutamine accumulation in rat brain mitochondria in the presence of a glutamine uptake inhibitor, histidine, reveals glutamine pools with a distinct access to deamidation. Neurochem Res 29(11):2121–2123
Acknowledgements
D.L.R. acknowledges support from the National Institute of Health grant R01NS087568A.
Conflicts of interest
The authors declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Hertz, L., Rothman, D.L. (2016). Glucose, Lactate, β-Hydroxybutyrate, Acetate, GABA, and Succinate as Substrates for Synthesis of Glutamate and GABA in the Glutamine–Glutamate/GABA Cycle. In: Schousboe, A., Sonnewald, U. (eds) The Glutamate/GABA-Glutamine Cycle. Advances in Neurobiology, vol 13. Springer, Cham. https://doi.org/10.1007/978-3-319-45096-4_2
Download citation
DOI: https://doi.org/10.1007/978-3-319-45096-4_2
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-45094-0
Online ISBN: 978-3-319-45096-4
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)