
Abstract
The high in vivo flux of the glutamate/glutamine cycle puts a strong demand on the return of ammonia released by phosphate activated glutaminase from the neurons to the astrocytes in order to maintain nitrogen balance. In this paper we review several amino acid shuttles that have been proposed for balancing the nitrogen flows between neurons and astrocytes in the glutamate/glutamine cycle. All of these cycles depend on the directionality of glutamate dehydrogenase, catalyzing reductive glutamate synthesis (forward reaction) in the neuron in order to capture the ammonia released by phosphate activated glutaminase, while catalyzing oxidative deamination of glutamate (reverse reaction) in the astrocytes to release ammonia for glutamine synthesis. Reanalysis of results from in vivo experiments using 13N and 15N labeled ammonia and 15N leucine in rats suggests that the maximum flux of the alanine/lactate or branched chain amino acid/branched chain amino acid transaminase shuttles between neurons and astrocytes are approximately 3–5 times lower than would be required to account for the ammonia transfer from neurons to astrocytes needed for glutamine synthesis (amide nitrogen) to sustain the glutamate/glutamine cycle. However, in the rat brain both the total ammonia fixation rate by glutamate dehydrogenase and the total branched chain amino acid transaminase activity are sufficient to support a branched chain amino acid/branched chain keto acid shuttle, as proposed by Hutson and coworkers, which would support the de novo synthesis of glutamine in the astrocyte to replace the ~20 % of neurotransmitter glutamate that is oxidized. A higher fraction of the nitrogen needs of total glutamate neurotransmitter cycling could be supported by hybrid cycles in which glutamate and tricarboxylic acid cycle intermediates act as a nitrogen shuttle. A limitation of all in vivo studies in animals conducted to date is that none have shown transfer of nitrogen for glutamine amide synthesis, either as free ammonia or via an amino acid from the neurons to the astrocytes. Future work will be needed, perhaps using methods for selectively labeling nitrogen in neurons, to conclusively establish the rate of amino acid nitrogen shuttles in vivo and their coupling to the glutamate/glutamine cycle.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
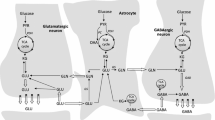
Over 90 % of neurons in the mammalian cerebral cortex are glutamatergic or GABAergic [1] and depend on neurotransmitter trafficking with astrocytes to maintain neurotransmitter homeostasis. Because neurons do not express pyruvate carboxylase [2, 3] the glutamate or GABA molecules released from neurons must be replaced by a five or four carbon molecule derived from astrocytes for neurotransmitter resynthesis. Glutamine is believed to be the main compound released by the astrocytes for neurotransmitter repletion of glutamate and GABA. The pathways in which neuronal glutamate or GABA is released in neurotransmission, taken up by astrocytes, converted to glutamine, and released for neuronal uptake are called the glutamate/glutamine and GABA/glutamine cycles [4–10]. Figure 1a shows a diagram of the glutamate/glutamine cycle. In this cycle released neurotransmitter glutamate is transported into the astrocytes using sodium coupled transporters [11], and converted to glutamine by the enzyme glutamine synthetase (GS) in a reaction requiring ATP and ammonia. The glutamine is then released by astrocytes through SN-type glutamine transporters into the extracellular fluid for uptake by neurons through SA-type glutamine transporters. In the neuron glutamine is converted into glutamate and ammonia by the action of phosphate activated glutaminase (PAG). GS and PAG catalyze the following reactions in the neurons and astrocytes respectively:
GS, which catalyzes the conversion of glutamate to glutamine, is expressed in astrocytes and not in neurons [12]. This compartmentalization of GS in astrocytes is consistent with the function of astrocytes, as consumers of glutamate, in functional glutamate trafficking [13]. PAG, which catalyzes the conversion of glutamine to glutamate and ammonia, is expressed in both neurons and astrocytes [14, 15], but is primarily active in neurons [16, 17] which is also supported by experimental data in vivo [18–21].

Proposed variants of the glutamate/glutamine cycle for shuttling ammonia between the neurons and astrocytes. a Ammonia diffusion; b BCAA/BCKA shuttle; c alanine–lactate shuttle. Reproduced with permission from Maciejewski and Rothman [36]. Descriptions of each variant are provided in the text
From the standpoint of carbon mass balance the flow of glutamate from the neuron to the astrocyte is balanced by the reverse flow of glutamine to the neuron. However due to glutamine having two nitrogens the cycle leaves the astrocyte with a shortfall of one nitrogen atom per molecule glutamate (Fig. 1a). Therefore the maintenance of nitrogen homeostasis requires a mechanism to replace the nitrogen lost by the astrocytes. Research findings over the last two decades have shown that the glutamate and GABA neurotransmitter cycles operate at high rates, approaching cellular rates of oxidative metabolism. Prior to these findings, rates of neurotransmitter cycling were considered to be relatively small compared with synthetic fluxes requiring nitrogen (e.g. the rate of ammonia entry from the blood into the brain) so that no special mechanism for transferring ammonium produced in the neuron back to the astrocyte was needed. However, studies in the 1990s of human and rodent brain by in vivo 13C MRS reported high rates of cerebral glutamine labeling from 13C glucose infused into the blood [22, 23]. Although the results were unclear at first whether the high rate of glutamine synthesis reflected neurotransmitter cycling or ammonia detoxification, subsequent experiments using a variety of approaches showed that under normal physiological conditions the large majority (~80 %) of glutamine synthesis reflected neurotransmitter cycling and the direct conversion of glutamate released by neurons into glutamine [18, 19, 21, 24–32]. As discussed in “Amino Acid Shuttles for Maintaining Nitrogen Balance” most of the remaining 20 % is likely to reflect glial glutamate oxidation and resynthesis of glutamine to maintain glutamate neurotransmitter homeostasis.
The rate of the glutamate/glutamine cycle appears tightly coupled to cerebral energy metabolism and electrical activity. In 1998 Sibson and coworkers showed that in the rodent cerebral cortex the rate of the glutamate/glutamine cycle was directly proportional to the rate of neuronal glucose oxidation above isoelectricity with a slope of ~1:1 [26]. Figure 2 shows a plot summarizing the rates of the glutamate/glutamine cycle and neuronal glucose oxidation from 11 studies where cortical activity has ranged from isoelectricity to the awake condition [33]. In the awake state the glutamate/glutamine cycle operates at close to 80 % of the rate of neuronal glucose oxidation when glucose is the fuel source for the TCA cycle. Similarly, the human cerebral cortex has been found to undergo a high rate of glutamate/glutamine cycling [19, 24, 27, 28, 34, 35]. This high rate of neurotransmitter cycling creates a significant challenge to maintain nitrogen balance between astrocytes and neurons.

Approximately 1:1 relationship between the neuronal TCA cycle (0.5 * VTCAn) and the glutamate/glutamine cycle (Vcyc) with increasing electrical activity in the rat cerebral cortex. The plot shows the mean values of 0.5 * VTCAn (equivalent to CMRglc(ox)N in Sibson et al. [26]) plotted versus Vcyc reported from 12 published studies at activity levels ranging from awake to isoelectricity [26, 31, 32, 97, 108–115]. Regression analysis yields a slope of 0.97 (R2 = 0.94) and an intercept of 0.5 * VTCAn of 0.097 at isoelectricity (Vcyc ~ 0), values similar to those found in the original 1998 study by Sibson et al. [26]. In the case of Ref. [114] for both anesthetized and awake state, values of VTCAn were calculated from the time constants reported for glutamate turnover during a glucose infusion. The ratio of glutamate to glutamine steady state fractional enrichment during [2-13C] acetate infusion was used to calculate Vcyc using the equation described in Lebon et al. [19]. Symbols: anaesthetized (filled circle), awake (open circle), seizures (filled triangle)
Several mechanisms have been proposed for the shuttling of ammonia from neurons to astrocytes from the free diffusion of ammonia as NH3 gas (Fig. 1a) to pathways where the nitrogen is carried between cell types by amino acid shuttles that work in parallel with the glutamate/glutamine neurotransmitter cycle. These shuttles include the branched chain amino acid/branched chain keto acid (BCAA/BCKA) shuttle (Fig. 1b), and the lactate/alanine shuttle (Fig. 1c). In addition hybrid cycles have been proposed in which the nitrogen shuttling function is provided by neuron-released glutamate and TCA cycle intermediates released by the astrocyte [36]. In the discussion below we describe these nitrogen transport shuttles and assess the evidence that they can account for the large nitrogen flux between neurons and astrocytes coupled to brain activity. We also evaluate whether there is in vivo evidence that these cycles can replete the portion of the glutamate/glutamine cycle coupled to glutamate oxidation as originally suggested by Hutson et al. [37].
Evidence for Ammonia Transfer Between Neurons and Astrocytes by Free Diffusion
The simplest mechanism by which ammonia can be transferred from neurons to astrocytes is by free diffusion of the gas NH3 form [38, 39]. This mechanism and its coupling to the glutamate/glutamine cycle are shown in Fig. 1a. The balance equations for the series of reaction steps depicted in Fig. 1a are shown below: A and N represent astrocytic and neuronal, respectively.
In this series of reactions ammonia released in the neuron by the action of PAG on glutamine (Eq. 4) diffuses as a gas to the astrocyte (Eq. 5) where it is used in the resynthesis of glutamine from glutamate (Eq. 7). In parallel a glutamate molecule is released by the neuron through vesicular mediated mechanisms and taken up using sodium coupled transporters which in the cerebral cortex are much higher in activity in astrocytes than neurons resulting in most of the glutamate flowing into the astrocytes (Eq. 6) [11, 40].
Under in vivo conditions (pH 6.8–7.0) and for a pKa of ~9.2 ammonia exists mainly (99 %) as the conjugate acid, NH4 + so that once in the astrocyte the NH3 will equilibrate taking up a proton (Eq. 8).
However, plasma membrane transport of NH4 + (believed to ride the K+ transporter) is commonly viewed to be much slower than diffusion of NH3, and studies suggest NH3 gas diffusion is the main transport mechanism for ammonia from blood to brain [41]. Nitrogen balance is maintained provided the rate of net ammonia diffusion (VNH3) being equal to the rate of glutamate transfer from the neuron (Vcycle), glutamine synthesis (VGS), glutamine transfer to the neuron (Vcycle) and the rate of neuronal PAG (VPAG). This equality can be written as (Eq. 9)
Ammonia diffusion from the neuron (Eq. 5), must be coupled to neuronal activity (Vcycle) (Eq. 6), in order for nitrogen mass balance to be maintained.
At present there is no direct evidence that ammonia diffusion from the neuron to the astrocyte takes place at the rate of the glutamate/glutamine cycle [42]. Indirect evidence for nitrogen transfer by diffusion of ammonia from the neurons to the astrocytes comes from studies that found an increase in astrocytic pH during stimulation [43, 44]. This increase is expected if there is an increase in ammonia gas entry because it will absorb protons in the conversion to NH4 + [45]. However intracellular pH regulation is complex and there are other potential explanations for these findings. Some evidence from cell studies under conditions of elevated (~1 mM and higher) extracellular ammonia concentrations suggests that NH4 + transport into astrocytes may occur at rates high enough to support GS for the glutamate-glutamine cycle [45]. However at present it is not known whether ammonia diffusion is sufficient at normal brain concentrations of ~0.3 mM to support the high neuron to astrocyte flux needed by neurotransmitter cycles. As described below alternate mechanisms have been proposed involving neuron-to-astrocyte shuttles using amino acids.
Amino Acid Shuttles for Maintaining Nitrogen Balance
Alternative mechanisms for transferring ammonia nitrogen released by neuronal PAG activity back to astrocytes for glutamine synthesis have been proposed which involve the incorporation of ammonia into amino acids such as alanine, branched chain amino acids, or glutamate followed by their transport to astrocytes. An advantage that amino acid shuttles have over free ammonia diffusion is that they involve specific transport of the species involved and therefore in principle can be used to more directly target the ammonia nitrogen flux from neurons to the astrocytes that require it or glutamine synthesis. All amino acid shuttles require the coordinated activity of PAG, GS, the forward activity of glutamate dehydrogenase (GDH) (reductive amination of 2-oxoglutarate (2-OG) to form glutamate) in the neuron and the reverse activity of GDH (oxidative deamination of glutamate) in the astrocyte to balance the nitrogen flows of the glutamate/glutamine cycle. In this section we describe three proposed amino acid shuttles and later review the evidence for their activity in vivo.
BCAA/BCKA Shuttle
The first amino acid shuttle proposed for transferring ammonia nitrogen released by neuronal PAG back to the astrocyte for glutamine synthesis is the BCAA/BCKA shuttle [46–48]. Figure 1b shows a schematic of this shuttle coupled to the glutamate/glutamine cycle with nitrogen and carbon flows [36]. The reaction equations for the shuttle and the glutamate/glutamine cycle are given below:
In this mechanism ammonia released by PAG is incorporated into neuronal glutamate by the forward action of GDH (Eq. 12). The ammonia is then transferred from glutamate to the amino group of a BCKA via cytosolic BCAA transaminase (BCATc) (Eq. 13) [48–50]. The resultant BCAA (isoleucine, leucine, or valine) is transported out of the neuron, using large neutral amino acid transporters [51] or specific sodium coupled transporters such as SBAT1 [52], at a rate equal to Vcycle and taken up by the astrocyte. In the astrocyte the BCAA undergoes transamination with 2-OG via the mitochondrial BCATm transferring the nitrogen to glutamate (Eq. 16) [48]. The ammonia is then released from glutamate by GDH acting in the reverse direction (Eq. 17), followed by incorporation into glutamine via GS (Eq. 18).
Examination of (Eq. 10–18) shows that the carbon and nitrogen flows between neurons and astrocytes are balanced. For every glutamine that carries two atoms of nitrogen from the astrocyte two atoms of nitrogen reenter the astrocyte in the form of glutamate and a branched-chain amino acid. Carbon balance is maintained provided that the BCKA lost by the neuron is replaced by a BCKA derived from the astrocyte (Eq. 14b). Because of the reciprocal directions of the GDH reactions in the neuron and astrocyte, for each complete cycle neurons produce and astrocytes consume one redox equivalent, respectively [36]. This process can be accommodated by the coordinated actions of the malate-aspartate shuttle and glycolysis in the neuron.
Alanine/Lactate Shuttle
An alanine-lactate shuttle [53, 54, 92] has also been proposed and a version with carbon and nitrogen flows balanced with the glutamate/glutamine cycle is illustrated in Fig. 1c [36]. The series of reactions and transfers involved in coordination with the glutamate-glutamine cycle are shown in the equations below.
The primary difference between the alanine/lactate and the BCAA/BCKA shuttles is that ammonia released by PAG is transferred to alanine by the combined action of GDH and alanine aminotransferase (AAT) (Eqs. 21 and 22). Alanine is transported out of the neurons at a rate equal to Vcycle, and taken up by the astrocytes (Eq. 23a). In the astrocytes the amino nitrogen of alanine is transferred to glutamate by the action of AAT (Eq. 25) producing pyruvate which due to the LDH equilibrium will mostly be converted to lactate. Ammonia is released from glutamate by the action of GDH working in the reverse direction (Eq. 26). Ammonia is then incorporated into glutamine through the GS reaction (Eq. 27) which will be transferred to the neurons. As with the BCAA/BCKA shuttle, nitrogen flows are intrinsically balanced by the coordinated action with the glutamate-glutamine cycle. The carbon skeleton of alanine lost by the neuron can be replaced either by transport of lactate from the astrocyte to the neuron (Eq. 23b) via monocarboxylic acid transporters [55, 56] or through glycolytic production of pyruvate and subsequent oxidation of NADH by lactate dehydrogenase in the neuron [36].
Glutamate TCA Intermediate Cycles
In addition to the glutamate-glutamine cycle other neurotransmitter cycles have been proposed in which TCA cycle intermediates, e.g., 2-OG, are transferred from the astrocyte to the neuron where glutamate is formed by GDH acting in the forward direction [9, 57–60]. Recently, several variants of these alternate cycles were proposed in which ammonia balance between neurons and astrocytes was maintained by astrocytes transferring a glutamine molecule and a TCA cycle intermediate molecule to neurons in return for two molecules of neurotransmitter glutamate [36]. A diagram of one of these cycles, the Glutamate/Glutamine/2-Oxoglutarate cycle (GGO cycle), is shown in Fig. 3 and the component reactions given below:
From the standpoint of neurotransmitter cycling the GGO cycle differs from the glutamate/glutamine cycle in that for every two molecules of glutamate released by the neuron one molecule of glutamine and one molecule of 2-OG is released by the astrocyte and taken up by the neuron (Eqs. 28 and 29) using sodium coupled transport [61]. In the neuron the ammonia released from glutamine by the action of PAG is incorporated into 2-OG via GDH working in the forward direction (Eqs. 30 and 31). For every two glutamate molecules transferred to the astrocyte (Eq. 32) one is converted to 2-OG via the reverse reaction of GDH (Eq. 33) and the released ammonia is incorporated into glutamine via GS (Eq. 34). The subsequent equimolecular release of glutamine and 2-OG completes the cycle.

Proposed alternate model for functional glutamate trafficking. The Glutamate/glutamine/oxoglutarate cycle (GGO). Reproduced with permission from Maciejewski and Rothman [36]. The cycle is described in the text
The GGO cycle can be viewed as intrinsically having an amino acid shuttle component in which glutamate and 2-OG act in analogy with alanine and lactate in the alanine/lactate shuttle and BCAAs and BCKAs in the BCAA/BCKA shuttle. As with the amino acid shuttles, the GGO cycle depends upon GDH in the neuron and astrocyte working in opposite directions at a rate that is stoichiometric with glutamate neurotransmitter release (Vcycle). Carbon balance is maintained by the transfer of one 2-OG molecule (or equivalently, another TCA intermediate such as malate) and one glutamine molecule from the astrocyte to the neuron to balance the transfer of two neurotransmitter glutamate molecules from the neuron to the astrocyte. Nitrogen balance is maintained by the transfer of two glutamate molecules from the neuron to the astrocyte for every glutamine molecule transported out of the astrocyte and taken up by the neuron. Through coupling with glycolysis and the malate-aspartate shuttle, redox balance is also maintained [36].
The GGO cycle differs from the BCAA/BCKA and alanine/lactate shuttles in at least two related and significant ways. First, in the GGO cycle, the additional amino acid the neuron transfers to the astrocyte (neurotransmitter glutamate) plays a central, functional role in synaptic neurotransmission. In the BCAA/BCKA and alanine/lactate shuttles, the neuron transfers a neutral, non-polar amino acid to the astrocyte. Second, in the GGO cycle, the neuron transfers this additional amino acid (glutamate) via the direct mechanisms of glutamatergic neurotransmission (i.e., vesicular transport into the synaptic cleft and subsequent uptake into the astrocyte). Therefore no mechanism is needed to explain how synaptic activity is coupled to the shuttling of ammonia as is the case with the other proposed shuttles.
Evidence for GDH Acting in Opposite Directions in the Neuron and Astrocyte
A key requirement of all the proposed amino acid nitrogen shuttles is that GDH works in the forward direction in neurons and in the reverse direction in astrocytes. There is considerable evidence that GDH can support oxidative degradation of glutamate in astrocytes [62–69], and that this degradation is proportional to the concentration of extracellular glutamate [70]. In the direction of glutamate degradation, GDH produces ammonium ions and 2-OG and reduces NAD(P) to NAD(P)H. Paradoxically there is also considerable evidence for de novo synthesis of glutamate in astrocytes using GDH [8, 18, 20, 71–73]. The ability of the astrocytes to simultaneously support both glutamate synthesis and degradation and its implications for ammonia balance are discussed in “Amino Acid Shuttles for Maintaining Nitrogen Balance”.
Based on differences in concentrations of the reactant and product species in the neuron relative to the astrocyte it has been proposed that GDH in the neuron works in the forward direction in which GDH consumes ammonium ion and 2-OG and oxidizes the reduced form of nicotinamide adenine dinucleotide (NADH) to NAD+ [62, 74]. However, there is relatively little in vivo evidence of GDH being active in this direction in the neuron in vivo and studies which reported active glutamate synthesis [75, 76] have been questioned based on the absolute rate being much lower than anaplerotic glutamate synthesis arising through astrocytic GDH [20, 21, 29, 31].
The Purine Nucleotide Cycle as an Alternative to Ammonia Release by GDH
In addition to GDH there are other pathways of ammonia production. A pathway of particular relevance to the production of ammonia in the brain is the purine nucleotide cycle [77, 78], which is involved in the synthesis of adenosine mono phosphate (AMP) and a TCA cycle intermediate (fumarate). Free ammonia would then be released through the breakdown of AMP to inosine monophosphate (IMP). This cycle effectively regenerates AMP from its breakdown product, IMP, in a concerted two-step reaction (involving adenylosuccinate synthetase and adenylosuccinate lyase) converting aspartate to fumarate with hydrolysis of GTP to GDP and Pi. The net effect is the deamination of aspartate to release free ammonia. The enzymes of this pathway are very active in the brain, and more than sufficient to explain the rapid generation of ammonia during increased activity when energy turnover is high. Thus, ammonia production through this pathway must be viewed as a potential alternative to GDH as a source of glutamine amide nitrogen [41]. From the standpoint of the nitrogen shuttles described in this paper the purine nucleotide cycle could partially or completely replace the need for astrocytic GDH to act in the reverse direction if the amino nitrogen from neurotransmitter glutamate that enters the astrocyte through a GOG type of cycle was transferred by aspartate aminotransferase (AST) to aspartate and then aspartate and IMP were converted to fumarate and AMP by adenylosuccinate synthetase and adenylosuccinate lyase. Ammonia would then be released by the breakdown of AMP to IMP. Alternatively the nitrogen could initially be transferred to the astrocyte as a BCAA or alanine and be transferred to glutamate and then aspartate through the actions of BCAT, AAT, and AST. An alternate possibility is that the purine nucleotide cycle acts primarily in the neuron, as supported by the study of Knecht et al. [79] who found that AMP deaminase was localized to neurons and ependymal cells. If this is the case then the released ammonia would need to diffuse to the astrocytes in order to provide nitrogen for glutamine synthesis.
Glutamate Oxidation and Amino Acid Shuttles
In addition to the conversion of glutamate to glutamine in the glutamate/glutamine cycle astrocytes will degrade glutamate oxidatively in the TCA cycle [40, 63–65, 67–69]. Because neurons do not express pyruvate carboxylase in order to maintain neurotransmitter glutamate homeostasis a glutamine molecule must be synthesized by the astrocyte and returned to the neuron for every glutamate molecule oxidized. Based on in vivo studies in intact rats and humans the fraction of neurotransmitter glutamate that is oxidized and then resynthesized by anaplerosis is approximately 20 % of total glutamine synthesis in the astrocyte [18, 19, 21, 24–32].
A paradox in astrocyte glutamate oxidation is that if it depended completely on GDH the enzyme would have to work in both the forward and reverse direction as shown by the reaction equations below:
The first step in the complete oxidation of glutamate is deamination to 2-OG by GDH (Eq. 37). Further metabolism of 2-OG to malate occurs in the TCA cycle followed by successive actions of malic enzyme and pyruvate dehydrogenase complex (PDHC) producing acetyl-CoA (Eq. 38) which is oxidized to CO2 in the TCA cycle. This series of reactions is referred to as pyruvate recycling and has been demonstrated in glial cell cultures [68, 80]. Because neurons lack pyruvate carboxylase the loss of glutamate through oxidation in astrocytes must be replaced by anaplerosis to avoid deficits in neuronal glutamate/GABA neurotransmission. Astrocytes express mitochondrial pyruvate carboxylase (mPC) [2, 3] and cytosolic malic enzyme (cME) [81]. Because mPC is irreversible, this enzyme functions exclusively in the de novo synthesis of TCA cycle metabolites and their derivatives. However, cME has the potential to catalyze not only the carboxylation of pyruvate to form malate, but also the decarboxylation of malate to form pyruvate [82–84] and is believed to work primarily in the direction of pyruvate synthesis from malate in the astrocytes under normal conditions.
An alternate possibility is that astrocytic oxidation of glutamate is initiated by transamination as has been concluded by several groups in the field [85–88]. Hutson and coworkers formulated a version of the BCAA/BCKA shuttle which eliminates the need for bidirectional GDH through the use of mitochondrial BCAT to transaminate 2-OG to glutamate in the glutamine synthesis portion of this pathway [29, 37]. The reaction equations for this model describing the combined glutamate oxidation and glutamate resynthesis pathways in the astrocyte are given below and the model is shown schematically in Fig. 4.

Branched chain amino acids are transferred to the astrocytes from the neurons at a flux equal to the rate of glutamate oxidation (Vgluox) (Eq. 47) which is lower than the total rate of neurotransmitter glutamate release (Vcycle). Vcycle is defined here to include both neurotransmitter glutamate that is directly converted to glutamine as well as newly synthesized glutamine that is transferred to the neuron) (Eq. 49). As described above a fraction of the transferred glutamate is oxidized via the initial action of GDH (Eq. 50) followed by the TCA cycle and pyruvate recycling (Eq. 51). The need for having GDH work in the forward direction to produce glutamate is obviated by using mitochondrial BCATm transfer the amino group from a BCAA to 2-OG (Eq. 53) forming glutamate. The ammonia released from glutamate earlier by GDH (Eq. 50) is used by GS to synthesize glutamine (Eq. 54). From the standpoint of balancing nitrogen flows needed for glutamate oxidation the following relationship holds for the net fluxes through neuronal and astrocytic BCAT and GDH.
In contrast if the BCAA/BCKA shuttle is supporting the ammonia needs of the entire glutamate/glutamine cycle the flux balance equations would be:
Alternatively the alanine/lactate shuttle may be used to supply the needed ammonia to support glutamine resynthesis to replace oxidized neurotransmitter glutamate. The equivalent flux balance equations to Eqs. 55 and 56 would be given for the lactate/alanine shuttle by replacing VBCAT with VAAT.
Evidence for Amino Acid Shuttles Transferring Ammonia from Neurons to Astrocytes from Cell/Tissue Studies and Enzyme Localization Studies
Evidence for the BCAA/BCKA shuttle have primarily come from 15N and 14C labeling studies in cell cultures [37, 46, 47], and studies examining the compartmentation of the key enzymes in the shuttles [48]. Studies using 15N labeled leucine have shown that in cultured glial cells it can act as a significant nitrogen precursor for glutamine formation consistent with the net direction of BCAT towards formation of the ketoacid as proposed in the BCAA/BCKA shuttle [46, 47]. Conversely in synaptosomes the predominant direction of transamination via BCAT is towards leucine synthesis [47]. Adding leucine to cultured glial cells will increase their rate of glutamine release consistent with their having an important role in glutamine synthesis [37]. In addition the isotopic dilution of 15N labeled leucine in cultured glial cells and isolated retinas is much greater than that of U-13C-leucine which is consistent with a high rate of transamination by BCAT relative to dilution by leucine released by protein breakdown [37, 47, 89]. Consistent with cytosolic BCATc being used in the neuron for BCAA synthesis and shuttling, inhibition of cytosolic BCATc by gabapentin in ex vivo rat retinas led to reduced leucine transamination and a reduction in de novo synthesis of glutamine and glutamate from 14HCO3 − [29, 37, 89, 90].
Additional evidence for an active BCAA/BCKA shuttle has come from enzyme localization studies [48, 90]. The activity of the mitochondrial and cytosolic forms of the BCAT enzyme are many times higher than the activity of branched-chain-keto-acid-dehydrogenase which is the first step in the oxidation of BCKAs which is consistent with the shuttle activity being considerably higher than net breakdown of BCAAs for energy and total nitrogen balance [48, 49]. The localization of the mitochondrial isoform BCATm in the astrocytes and distribution of BCATc in the neurons is consistent with their role in shuttling nitrogen to the astrocytes for de novo glutamate synthesis [29, 37, 48, 49, 89, 91].
Similar to the BCATs the activity of alanine aminotransferase is high in the brain and labeling studies in cell culture have suggested that label can be transferred from neuronal alanine to glial glutamate and glutamine [53, 54]. However Bak et al. [32] reported no direct coupling between an alanine-lactate nitrogen shuttle and the glutamate-glutamine cycle in neuronal-astrocytic co-cultures, and that only the glutamate-glutamine cycle appears to be activity dependent.
Evaluation of the Rate of Amino Acid Shuttles from 13N and 15N Labeling Results in Rats In Situ
Labeled Ammonia Studies
Some insight into the potential rate of ammonia transfer by the amino acid cycles may be obtained from studies in which isotopically labeled ammonia (13N and 15N) or 15N labeled branched chain amino acid leucine was infused into rats and the rate of label incorporation into brain ammonia, glutamate, and glutamine pools was measured using magnetic resonance spectroscopy (MRS) or radioisotope methods. In the proposed BCAA/BCKA and alanine/lactate shuttles, neuronal GDH acting in the forward direction can incorporate ammonia produced from glutamine by glutaminase into 2-OG to produce glutamate. Therefore, if brain ammonia is isotopically labeled, then the initial flows of label into the N2 and N5 positions of astrocytic glutamine and neuronal glutamate are described by the following differential equations:
where *NH3 is the fractional enrichment of ammonia (either 13N or 15N) and [*N2-Glu] and [*N5-Gln] is the total labeled concentration of glutamate and glutamine respectively. For the BCAA/BCKA and alanine/lactate shuttles the rates of neuronal GDH and GS (VGDHN, VGS) must be equal to the rate of the glutamate/glutamine cycle (Eq. 56). Substituting Vcycle for VGS and VGDHN in Eqs. 57 and 58 yields an equal initial rate of labeling of the N2 and N5 positions of glutamate and glutamine (Eq. 59).
For the GGO cycle the total flux into the N2 and N5 pools would be ½Vcycle but the predicted initial rates of N5 and N2 labeling would be the same as described in Eq. 59.
Labeling of brain amino acids during acute infusion of 13 N and 15N ammonia has been measured [18, 20, 93, 94]. In the seminal study of Cooper and co workers [94] tracer amounts of 13N ammonia were infused into the carotid artery of awake rats and the labeling in the N2 positions of brain glutamate and glutamine and the N5 position of glutamine was measured at 20 min after the start of the infusion. The relative rate of 13N2 labeling to 13N5 labeling was approximately 1.5 % which is consistent with a very low rate of nitrogen incorporation by neuronal (or glial) GDH and amino acid shuttling. However an alternate explanation of the low ratio observed is that there is a large amount of dilution of ammonia label in the neuronal pool due to the high rate of PAG, especially at the early time point studied. Consistent with the possibility of a dilution is that the glutamate 13N2 concentration was 5 times lower than the glutamine 15N2 concentration (which should reflect the small astrocytic glutamate pool), although a lower rate of neuronal GDH is another alternative [20]. However arguing against dilution explaining the high N5/N2 ratio is that at the time point measured by Cooper and co-workers the amide glutamine nitrogen should have been highly labeled due to the high rate of the glutamate/glutamine cycle in awake rats [94]. In addition inhibition of GS by l-methionine-dl-sulfoximine (MSO) which would prevent trapping of labeled blood ammonia by the astrocytes did not lead to a significant increase in glutamate N2 labeling [94].
Due to sensitivity limitations studies of 15N labeling using 15N ammonia by in vivo 15N MRS have been performed under hyperammonemic conditions [18, 20, 30, 95, 96]. Under these conditions glial GDH may be induced to work in the forward direction due to elevated brain ammonia which should provide a maximum estimate of total brain GDH activity for trapping ammonia. Figure 5 shows the time courses of glutamine 15N5 and glutamate plus glutamine 15N2 during infusion of 15N ammonia as measured by in vivo 15N MRS [18]. The infusion raised plasma ammonia to approximately 0.39 mM which is considered to be hyperammonemic. During the initial 30 min of the infusion 15N2 labeling was not detectable relative to the initial 15N5 labeling which is proportional to Vcycle. After 30 min 15N2 labeling was detected and continued to increase at a constant rate. Using metabolic modeling it was calculated that the forward rate of GS was ~30 % of the rate of glutamine synthesis (the rest being due to the glutamate/glutamine cycle). More recently Cudalbu and colleagues [20] used 1H and 15N MRS to simultaneously measure 15N labeling and glutamine synthesis under similar conditions of acute hyperammonemia. As was found in the previous studies, the initial 15N5 labeling of glutamate was much higher than the combined 15N2 labeling of glutamate and glutamine. Using a two compartment metabolic model they concluded that the steady state N2 labeling rate corresponded to an anaplerosis rate that was 15 % of the rate of glutamine synthesis (0.3 μmol/g-min) with the remainder being due to the glutamate/glutamine cycle. The rate of anaplerosis matched the rate of glutamine accumulation during the infusion. In both of the 15N MRS studies the rate of the glutamate/glutamine cycle was consistent given the level of anesthesia with studies using 13C labeled substrates [18, 19, 21, 24–31, 97].

A representative set of time courses of [5-15N]glutamine (asterisk), [2-15N]glutamine/glutamate (multi symbol) and their summed concentrations (open circle) measured in vivo using 15N MRS. Reproduced with permission from Fig. 3 in Shen et al. [18]. The quantification of cerebral [5-15N] glutamine and [2-15N] glutamine/glutamate concentrations was based on in vitro analysis. The time course shows a several fold more rapid initial labeling of glutamine N5 than the combined positions of glutamate and glutamine N2, consistent with GS being at minimum on the order of 3–4 times more rapid than GDH under these conditions
The overall evidence from the in vivo labeled ammonia studies suggests that, even under conditions of presumably maximum stimulation (hyperammonemia) of ammonia fixation, the total forward activity of GDH is insufficient to support the ammonia needs of the glutamate/glutamine cycle, a conclusion also made by Cooper in a recent review [41]. The maximum rates of the BCAA/BCAT and alanine/lactate shuttles compatible with the data are on the order of 15–30 % of total glutamine synthesis. Because the GGO cycle requires half the rate of ammonia transfer from the neuron as the glutamate/glutamine cycle the measured activity of GDH would be sufficient to support 30–60 % of the ammonia shuttling needs of total glutamate trafficking.
The very low rate of N2 labeling relative to N5 during the early period of the isotopically labeled ammonia infusion [18, 93] may be due to the dilution of the labeled ammonia in the neuron by PAG-mediated hydrolysis of unlabeled glutamate. An alternate explanation is that astrocyte GDH activity is stimulated by hyperammonemia which is consistent with the finding of higher fractional N2 labeling of glutamine compared with glutamate [17, 19, 83] although stimulation of oxidation has also been reported for neuronal GDH [74]. However, intriguingly the upper value for the forward GDH reaction is very similar to what has been measured by 13C MRS for the fraction of anaplerotic glutamine production, consistent with the version of the BCKA/BCAA shuttle proposed by Hutson et al. [37, 90] discussed in “Amino Acid Shuttles for Maintaining Nitrogen Balance”.
Labeled Leucine Studies
An alternate in vivo strategy used to study the role of the BCAA/BCAT shuttle has involved infusion or oral feeding of 15N labeled leucine followed by the use of 15N MRS or mass spectroscopy to measure 15N labeled brain metabolites [98, 99]. Based on arterio-venous difference and transport studies leucine is the major branched chain amino acid taken up by the brain [90, 100–102]. In the study of Sakai and coworkers [98] [15N] leucine was fed to awake rats and brain and plasma 15N labeling was measured every hour over a 9 h period. The feeding regimen did not raise leucine levels enough to impact transport [101] so that the rates measured most likely reflect normal physiology. Total leucine uptake was determined from the accumulation of 15N into the major brain amino acid pools and was 0.02 μmol/g-min which is nearly equal to the maximum rate of leucine uptake in vivo as reported by Smith and coworkers [101]. This rate is also similar to rates reported for glutamine efflux from the brain under normo-ammonemic conditions supporting an important role for leucine in maintaining overall brain nitrogen balance [18, 25, 26]. It should be noted that although this rate is quite low over the 9 h study it was sufficient to account for ~50 % of the N2 labeling of glutamate and glutamine in the brain, and a similar fraction was found by Kanamori and coworkers using infused 15N-labeled leucine [99]. Originally the finding that branched chain amino acids can replace much of the brain amino acid nitrogen pool was interpreted as indicating a very low rate of cycling of nitrogen between neurons and astrocytes. However, total amino acid labeling from the blood reflects the exchange of nitrogen between the brain and blood pools and does not provide information about exchange between internal metabolic compartments such as between neurons and astrocytes, as has been previously pointed out by Hertz [103]. The low rate of 15N labeling from leucine is in agreement with findings from arterio-venous difference measurements that the total uptake of nitrogen from neutral amino acids (mainly leucine) by the brain from blood is nearly equal to the rate that nitrogen leaves the brain (primarily in the form of glutamine) [100, 102].
Figure 6 shows the time course of brain 15N leucine and [U-13C] leucine labeling relative to plasma from the study of Sakai and coworkers [91]. Minimal dilution of [U-13C] leucine was measured consistent with a low rate of leucine dilution due to protein turnover and leucine oxidation (15 %) relative to dilution by BCAT activity (85 %). The low dilution was consistent with previous studies in cell models [37, 46, 47] and the earlier study in vivo using [14C] leucine by Berl and Frigyesi [104]. Interestingly in the earlier 14C study a glutamine to glutamate labeling ratio of greater than 1 was found implying compartmentation of leucine metabolism primarily in the astroglia.

Time course of [15N] and [U-13C] leucine labeling in awake rats. Reproduced with permission from Sakai et al. [98]. The diet containing [15N] leucine or [U-13C] leucine was fed to the rat for 0.5–9 h after 12 h of fasting. [U-13C]- and [15N] enrichment of plasma leucine (open circles) and brain leucine (closed circles) were plotted. Insets show the time course of the isotopic enrichment ratio of brain to plasma leucine. a [U-13C] leucine b [15N] leucine. A 4–5 fold dilution of brain [15N] leucine is observed at all time points relative to plasma [15N] leucine consistent with in vivo BCAT activity being on the order of 4–5 times greater than the rate of leucine uptake into the brain (measured from total 15N incorporation into brain amino acids). The relatively small dilution of [U-13C] leucine in the brain indicates that the rate of leucine turnover due to protein breakdown is small compared with the transaminase activity
The rate of nitrogen exchange by BCAT (sum of neuronal and glial) was estimated from the dilution of brain leucine 15N labeling relative to plasma leucine 15N labeling using the following relationship (after adjustment for the low level of leucine turnover):
The rate VBCAT was found to be approximately 4–5 times higher than leucine transport with a flux of 0.1 μmol/g-min. As shown in Fig. 2 in the awake rat the rate of the glutamate/glutamine cycle is between 0.5 and 0.6 μmol/g-min so that the in vivo activity of BCAT is sufficient to provide the nitrogen needed to support the approximately 20 % of glutamine synthesis that would be required to replace oxidized glutamate through the BCAA/BCKA shuttle as proposed by Hutson et al. [37, 90].
Impact of Metabolic Heterogeneity
In the discussions in this paper it has been assumed that there is uniform metabolism within classes of neural cells, in particular astrocytes and glutamatergic neurons. However the interpretation of in vivo and in vitro data may be impacted by heterogeneity within cell neural classes as has been reported in cell cultures for mitochondria [105, 106] and mitochondrial pathways [107]. While the assessment of the potential quantitative impact of within cell class intracellular heterogeneity is beyond the scope of this paper, the rates of nitrogen metabolism reviewed here may be considered within class cell population averages to first order, and the mass balance requirements between neurons and astrocytes will not be affected. Future studies that compare different brain regions and cortical layers might provide fresh insight into the potential effects of metabolic heterogeneity.
Conclusions
The high in vivo flux of the glutamate/glutamine cycle puts a large demand on the return of ammonia released by neuronal PAG from the neurons to the astrocytes in order to maintain nitrogen balance. We have reviewed several amino acid shuttles that have been proposed for balancing the nitrogen flows between neurons and astrocytes in the glutamate/glutamine cycle. All of these shuttles depend on GDH working in opposite directions in the neurons and astrocytes, catalyzing reductive glutamate synthesis (forward reaction) in the neuron in order to capture the ammonia released by PAG, while catalyzing oxidative deamination of glutamate (reverse reaction) in the astrocytes to release ammonia for glutamine synthesis. Reanalysis of results from in vivo experiments using 13N and 15N labeled ammonia and 15N leucine in rats suggests that the maximum flux of the alanine/lactate or BCAA/BCAT shuttles between neurons and astrocytes are approximately 3–5 times lower than would be required to account for the ammonia transfer from neurons to astrocytes needed to support glutamine synthesis (amide nitrogen) to sustain the glutamate/glutamine cycle. However, in the rat brain both the total ammonia fixation rate by GDH and the total BCAT activity are sufficient to support a BCAA/BCKA shuttle, [37, 90], which would be sufficient to support the de novo synthesis of glutamine in the astrocyte to replace the ~20 % of neurotransmitter glutamate that is oxidized in the astrocyte. The same forward activity of neuronal GDH would support ~30 to 60 % of the ammonia needs of glutamine synthesis if the brain used alternate neurotransmitter cycles in which neurotransmitter glutamate and astrocyte synthesized TCA cycle intermediates act as a nitrogen shuttle [36]. A limitation of all in vivo studies in animals conducted to date is that none have directly shown transfer of nitrogen for glutamine amide synthesis, either as free ammonia or via an amino acid shuttle from the neurons to the astrocytes. In addition there is little direct evidence that neuronal GDH accounts for the majority of forward GDH activity in the brain. Future work will be needed, perhaps using methods for selectively labeling nitrogen in neurons, to conclusively establish the rate of amino acid nitrogen shuttles in vivo and their coupling to the glutamate/glutamine cycle.
References
Shephard GM (1994) The synaptic organization of the brain. Oxford University Press, Oxford
Yu AC, Drejer J, Hertz L, Schousboe A (1983) Pyruvate carboxylase activity in primary cultures of astrocytes and neurons. J Neurochem 41:1484–1487
Shank RP, Bennett GS, Freytag SO, Campbell GL (1985) Pyruvate carboxylase: an astrocyte-specific enzyme implicated in the replenishment of amino acid neurotransmitter pools. Brain Res 329:364–367
Van den Berg CJ, Garfinkel D (1971) A stimulation study of brain compartments. Metabolism of glutamate and related substances in mouse brain. Biochem J 123:211–218
Peng L, Hertz L, Huang R, Sonnewald U, Petersen SB, Westergaard N, Larsson O, Schousboe A (1993) Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev Neurosci 15:367–377
Schousboe A, Westergaard N, Sonnewald U, Petersen SB, Huang R, Peng L, Hertz L (1993) Glutamate and glutamine metabolism and compartmentation in astrocytes. Dev Neurosci 15:359–366
Schousboe A, Drejer J, Hertz L (1988) Uptake and release of glutamate and glutamine in neurons and astrocytes in primary cultures. In: Kvamme E (ed) Glutamine and glutamate in mammals. CRC Press, Boca Raton, FL, pp 21–38
Hertz L (2004) Intercellular metabolic compartmentation in the brain: past, present and future. Neurochem Int 45:285–296
Westergaard N, Sonnewald U, Unsgård G, Peng L, Hertz L, Schousboe A (1994) Uptake, release, and metabolism of citrate in neurons and astrocytes in primary cultures. J Neurochem 62:1727–1733
Peng LA, Schousboe A, Hertz L (1991) Utilization of alpha-ketoglutarate as a precursor for transmitter glutamate in cultured cerebellar granule cells. Neurochem Res 16:29–34
Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16:675–686
Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195:1356–1358
Patel AJ, Hunt A, Gordon RD, Balazs R (1982) The activities in different neural cell types of certain enzymes associated with the metabolic compartmentation glutamate. Brain Res 256:3–11
Aoki C, Kaneko T, Starr A, Pickel VM (1991) Identification of mitochondrial and non-mitochondrial glutaminase within select neurons and glia of rat forebrain by electron microscopic immunocytochemistry. J Neurosci Res 28:531–548
Würdig S, Kugler P (1991) Histochemistry of glutamate metabolizing enzymes in the rat cerebellar cortex. Neurosci Lett 130:165–168
Kvamme E, Svenneby G, Hertz L, Schousboe A (1982) Properties of phosphate activated glutaminase in astrocytes cultured from mouse brain. Neurochem Res 7:761–770
Hogstad S, Svenneby G, Torgner IA, Kvamme E, Hertz L, Schousboe A (1988) Glutaminase in neurons and astrocytes cultured from mouse brain: kinetic properties and effects of phosphate, glutamate, and ammonia. Neurochem Res 13:383–388
Shen J, Sibson NR, Cline G, Behar KL, Rothman DL, Shulman RG (1998) 15N-NMR spectroscopy studies of ammonia transport and glutamine synthesis in the hyperammonemic rat brain. Dev Neurosci 20:434–443
Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL (2002) Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci 22:1523–1531
Cudalbu C, Lanz B, Duarte JMN, Morgenthaler FD, Pilloud Y, Mlynárik V, Gruetter R (2012) Cerebral glutamine metabolism under hyperammonemia determined in vivo by localized (1)H and (15)N NMR spectroscopy. J Cereb Blood Flow Metab 32:696–708
Sibson NR, Mason GF, Shen J, Cline GW, Herskovits AZ, Wall JE, Behar KL, Rothman DL, Shulman RG (2001) In vivo (13)C NMR measurement of neurotransmitter glutamate cycling, anaplerosis and TCA cycle flux in rat brain during. J Neurochem 76:975–989
Gruetter R, Novotny EJ, Boulware SD, Mason GF, Rothman DL, Shulman GI, Prichard JW, Shulman RG (1994) Localized 13C NMR spectroscopy in the human brain of amino acid labeling from D-[1-13C]glucose. J Neurochem 63:1377–1385
Mason GF, Gruetter R, Rothman DL, Behar KL, Shulman RG, Novotny EJ (1995) Simultaneous determination of the rates of the TCA cycle, glucose utilization, alpha-ketoglutarate/glutamate exchange, and glutamine synthesis in human brain by NMR. J Cereb Blood Flow Metab 15:12–25
Mason GF, Petersen KF, de Graaf RA, Shulman GI, Rothman DL (2007) Measurements of the anaplerotic rate in the human cerebral cortex using 13C magnetic resonance spectroscopy and [1-13C] and [2-13C] glucose. J Neurochem 100:73–86
Sibson NR, Dhankhar A, Mason GF, Behar KL, Rothman DL, Shulman RG (1997) In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glutamine cycling. Proc Nat Acad Sci USA 94:2699–2704
Sibson NR, Dhankhar A, Mason GF, Rothman DL, Behar KL, Shulman RG (1998) Stoichiometric coupling of brain glucose metabolism and glutamatergic neuronal activity. Proc Nat Acad Sci USA 95:316–321
Gruetter R, Seaquist ER, Ugurbil K (2001) A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am J Physiol Endocrinol Metab 281:E100–E112
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, Petroff OA, Shulman GI, Shulman RG, Rothman DL (1999) Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Nat Acad Sci USA 96:8235–8240
Lieth E, LaNoue KF, Berkich DA, Xu B, Ratz M, Taylor C, Hutson SM (2001) Nitrogen shuttling between neurons and glial cells during glutamate synthesis. J Neurochem 76:1712–1723
Kanamori K, Ross BD (1993) 15 N n.m.r. measurement of the in vivo rate of glutamine synthesis and utilization at steady state in the brain of the hyperammonaemic rat. Biochem J 293(Pt 2):461–468
Oz G, Berkich DA, Henry P-G, Xu Y, LaNoue K, Hutson SM, Gruetter R (2004) Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J Neurosci 24:11273–11279
Patel AB, de Graaf RA, Mason GF, Kanamatsu T, Rothman DL, Shulman RG, Behar KL (2004) Glutamatergic neurotransmission and neuronal glucose oxidation are coupled during intense neuronal activation. J Cereb Blood Flow Metab 24:972–985
Rothman DL, De Feyter HM, de Graaf RA, Mason GF, Behar KL (2011) 13C MRS studies of neuroenergetics and neurotransmitter cycling in humans. NMR Biomed 24:943–957
Boumezbeur F, Mason GF, de Graaf RA, Behar KL, Cline GW, Shulman GI, Rothman DL, Petersen KF (2010) Altered brain mitochondrial metabolism in healthy aging as assessed by in vivo magnetic resonance spectroscopy. J Cereb Blood Flow Metab 30:211–221
Chhina N, Kuestermann E, Halliday J, Simpson LJ, Macdonald IA, Bachelard HS, Morris PG (2001) Measurement of human tricarboxylic acid cycle rates during visual activation by (13)C magnetic resonance spectroscopy. J Neurosci Res 66:737–746
Maciejewski PK, Rothman DL (2008) Proposed cycles for functional glutamate trafficking in synaptic neurotransmission. Neurochem Int 52:809–825
Hutson SM, Berkich D, Drown P, Xu B, Aschner M, LaNoue KF (1998) Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J Neurochem 71:863–874
Benjamin AM, Quastel JH (1975) Metabolism of amino acids and ammonia in rat brain cortex slices in vitro: a possible role of ammonia in brain function. J Neurochem 25:197–206
Marcaggi P, Coles JA (2001) Ammonium in nervous tissue: transport across cell membranes, fluxes from neurons to glial cells, and role in signalling. Prog Neurobiol 64:157–183
Sonnewald U, Westergaard N, Schousboe A (1997) Glutamate transport and metabolism in astrocytes. Glia 21:56–63
Cooper AJL (2012) The role of glutamine synthetase and glutamate dehydrogenase in cerebral ammonia homeostasis. Neurochem Res
Zwingmann C, Leibfritz D (2003) Regulation of glial metabolism studied by 13C-NMR. NMR Biomed 16:370–399
Chesler M, Kraig RP (1987) Intracellular pH of astrocytes increases rapidly with cortical stimulation. Am J Physiol 253:R666–R670
Chesler M, Kraig RP (1989) Intracellular pH transients of mammalian astrocytes. J Neurosci 9:2011–2019
Nagaraja TN, Brookes N (1998) Intracellular acidification induced by passive and active transport of ammonium ions in astrocytes. Am J Physiol 274:C883–C891
Yudkoff M (1997) Brain metabolism of branched-chain amino acids. Glia 21:92–98
Yudkoff M, Daikhin Y, Grunstein L, Nissim I, Stern J, Pleasure D (1996) Astrocyte leucine metabolism: significance of branched-chain amino acid transamination. J Neurochem 66:378–385
Cole JT, Sweatt AJ, Hutson SM (2012) Expression of mitochondrial branched-chain aminotransferase and α-keto-acid dehydrogenase in rat brain: implications for neurotransmitter metabolism. Front Neuroanat 6:18
Sweatt AJ, Wood M, Suryawan A, Wallin R, Willingham MC, Hutson SM (2004) Branched-chain amino acid catabolism: unique segregation of pathway enzymes in organ systems and peripheral nerves. Am J Physiol Endocrinol Metab 286:E64–E76
Hall TR, Wallin R, Reinhart GD, Hutson SM (1993) Branched chain aminotransferase isoenzymes. Purification and characterization of the rat brain isoenzyme. J Biol Chem 268:3092–3098
Fernstrom JD (2005) 4th Amino acid assessment workshop branched-chain amino acids and brain function, pp 1539–1546
Takanaga H, Mackenzie B, Peng J-B, Hediger MA (2005) Characterization of a branched-chain amino-acid transporter SBAT1 (SLC6A15) that is expressed in human brain. Biochem Biophys Res Commun 337:892–900
Peng L, Zhang X, Hertz L (1994) Alteration in oxidative metabolism of alanine in cerebellar granule cell cultures as a consequence of the development of the ability to utilize alanine as an amino group donor for synthesis of transmitter glutamate. Brain Res Dev Brain Res 79:128–131
Waagepetersen HS, Sonnewald U, Larsson OM, Schousboe A (2000) A possible role of alanine for ammonia transfer between astrocytes and glutamatergic neurons. J Neurochem 75:471–479
Pellerin L, Bergersen LH, Halestrap AP, Pierre K (2005) Cellular and subcellular distribution of monocarboxylate transporters in cultured brain cells and in the adult brain. J Neurosci Res 79:55–64
Pierre K, Pellerin L (2005) Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem 94:1–14
Shank RP, Campbell GL (1984) Glutamine, glutamate, and other possible regulators of alpha-ketoglutarate and malate uptake by synaptic terminals. J Neurochem 42:1162–1169
Shank RP, Campbell GL (1982) Glutamine and alpha-ketoglutarate uptake and metabolism by nerve terminal enriched material from mouse cerebellum. Neurochem Res 7:601–616
Westergaard N, Sonnewald U, Schousboe A (1994) Release of alpha-ketoglutarate, malate and succinate from cultured astrocytes: possible role in amino acid neurotransmitter homeostasis. Neurosci Lett 176:105–109
Schousboe A, Westergaard N, Waagepetersen HS, Larsson OM, Bakken IJ, Sonnewald U (1997) Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia 21:99–105
Inoue K, Fei Y-J, Zhuang L, Gopal E, Miyauchi S, Ganapathy V (2004) Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem J 378:949–957
Zaganas I, Waagepetersen HS, Georgopoulos P, Sonnewald U, Plaitakis A, Schousboe A (2001) Differential expression of glutamate dehydrogenase in cultured neurons and astrocytes from mouse cerebellum and cerebral cortex. J Neurosci Res 66:909–913
Yu AC, Schousboe A, Hertz L (1982) Metabolic fate of 14C-labeled glutamate in astrocytes in primary cultures. J Neurochem 39:954–960
Sonnewald U, Westergaard N, Petersen SB, Unsgård G, Schousboe A (1993) Metabolism of [U-13C]glutamate in astrocytes studied by 13C NMR spectroscopy: incorporation of more label into lactate than into glutamine demonstrates the importance of the tricarboxylic acid cycle. J Neurochem 61:1179–1182
McKenna MC, Tildon JT, Stevenson JH, Huang X (1996) New insights into the compartmentation of glutamate and glutamine in cultured rat brain astrocytes. Dev Neurosci 18:380–390
Gamberino WC, Berkich DA, Lynch CJ, Xu B, LaNoue KF (1997) Role of pyruvate carboxylase in facilitation of synthesis of glutamate and glutamine in cultured astrocytes. J Neurochem 69:2312–2325
Peng L, Swanson RA, Hertz L (2001) Effects of l-glutamate, d-aspartate, and monensin on glycolytic and oxidative glucose metabolism in mouse astrocyte cultures: further evidence that glutamate uptake is metabolically driven by oxidative metabolism. Neurochem Int 38:437–443
Waagepetersen HS, Qu H, Hertz L, Sonnewald U, Schousboe A (2002) Demonstration of pyruvate recycling in primary cultures of neocortical astrocytes but not in neurons. Neurochem Res 27:1431–1437
Hertz L, Hertz E (2003) Cataplerotic TCA cycle flux determined as glutamate-sustained oxygen consumption in primary cultures of astrocytes. Neurochem Int 43:355–361
McKenna MC, Sonnewald U, Huang X, Stevenson J, Zielke HR (1996) Exogenous glutamate concentration regulates the metabolic fate of glutamate in astrocytes. J Neurochem 66:386–393
Kaufman EE, Driscoll BF (1993) Evidence for cooperativity between neurons and astroglia in the regulation of CO2 fixation in vitro. Dev Neurosci 15:299–305
Hertz L, Peng L, Lai JC (1998) Functional studies in cultured astrocytes. Methods 16:293–310
Hertz L, Dringen R, Schousboe A, Robinson SR (1999) Astrocytes: glutamate producers for neurons. J Neurosci Res 57:417–428
Faff-Michalak L, Albrecht J (1993) Hyperammonemia and hepatic encephalopathy stimulate rat cerebral synaptic mitochondrial glutamate dehydrogenase activity specifically in the direction of glutamate oxidation. Brain Res 618:299–302
Hassel B, Brâthe A (2000) Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci 20:1342–1347
Hassel B (2001) Pyruvate carboxylation in neurons. J Neurosci Res 66:755–762
Schultz V, Lowenstein JM (1978) The purine nucleotide cycle. Studies of ammonia production and interconversions of adenine and hypoxanthine nucleotides and nucleosides by rat brain in situ. J Biol Chem 253:1938–1943
Schultz V, Lowenstein JM (1976) Purine nucleotide cycle. Evidence for the occurrence of the cycle in brain. J Biol Chem 251:485–492
Knecht K, Wiesmüller KH, Gnau V, Jung G, Meyermann R, Todd KG, Hamprecht B (2001) AMP deaminase in rat brain: localization in neurons and ependymal cells. J Neurosci Res 66:941–950
Sonnewald U, Westergaard N, Jones P, Taylor A, Bachelard HS, Schousboe A (1996) Metabolism of [U-13C5] glutamine in cultured astrocytes studied by NMR spectroscopy: first evidence of astrocytic pyruvate recycling. J Neurochem 67:2566–2572
Kurz GM, Wiesinger H, Hamprecht B (1993) Purification of cytosolic malic enzyme from bovine brain, generation of monoclonal antibodies, and immunocytochemical localization of the enzyme in glial cells of neural primary cultures. J Neurochem 60:1467–1474
Frenkel R (1972) Isolation and some properties of a cytosol and a mitochondrial malic enzyme from bovine brain. Arch Biochem Biophys 152:136–143
Bukato G, Kochan Z, Swierczyński J (1995) Different regulatory properties of the cytosolic and mitochondrial forms of malic enzyme isolated from human brain. Int J Biochem Cell Biol 27:1003–1008
Bukato G, Kochan Z, Swierczyński J (1995) Purification and properties of cytosolic and mitochondrial malic enzyme isolated from human brain. Int J Biochem Cell Biol 27:47–54
Rao VL, Murthy CR (1993) Transport and metabolism of glutamate by rat cerebellar mitochondria during ammonia toxicity. Molecular and chemical neuropathology/sponsored by the International Society for Neurochemistry and the World Federation of Neurology and research groups on neurochemistry and cerebrospinal fluid. 19: 297–312
Wysmyk-Cybula U, Faff-Michalak L, Albrecht J (1991) Effects of acute hepatic encephalopathy and in vitro treatment with ammonia on glutamate oxidation in bulk-isolated astrocytes and mitochondria of the rat brain. Acta Neurobiol Exp 51:165–169
Berkich DA, Ola MS, Cole J, Sweatt AJ, Hutson SM, LaNoue KF (2007) Mitochondrial transport proteins of the brain. J Neurosci Res 85:3367–3377
Balazs R, Haslam J (1965) Exchange transamination and the metabolism of glutamate in brain. Biochem J 94:131–141
LaNoue KF, Berkich DA, Conway M, Barber AJ, Hu LY, Taylor C, Hutson S (2001) Role of specific aminotransferases in de novo glutamate synthesis and redox shuttling in the retina. J Neurosci Res 66:914–922
Hutson SM, Lieth E, Lanoue KF (2001) Function of leucine in excitatory neurotransmitter metabolism in the central nervous. System 1(2):846–850
García-Espinosa MA, Wallin R, Hutson SM, Sweatt AJ (2007) Widespread neuronal expression of branched-chain aminotransferase in the CNS: implications for leucine/glutamate metabolism and for signaling by amino acids. J Neurochem 100:1458–1468
Bak LK, Sickmann HM, Schousboe A, Waagepetersen HS (2005) Activity of the lactate-alanine shuttle is independent of glutamate-glutamine cycle activity in cerebellar neuronal-astrocytic cultures. J Neurosci Res 79:88–96
Cooper AJ, Plum F (1987) Biochemistry and physiology of brain ammonia. Physiol Rev 67:440–519
Cooper AJ, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE (1979) The metabolic fate of 13 N-labeled ammonia in rat brain. J Biol Chem 254:4982–4992
Kanamori K, Ross BD (1995) Steady-state in vivo glutamate dehydrogenase activity in rat brain measured by 15 N NMR. J Biol Chem 270:24805–24809
Kanamori K, Parivar F, Ross BD (1993) A 15 N NMR study of in vivo cerebral glutamine synthesis in hyperammonemic rats. NMR Biomed 6:21–26
Patel AB, de Graaf RA, Mason GF, Rothman DL, Shulman RG, Behar KL (2005) The contribution of GABA to glutamate/glutamine cycling and energy metabolism in the rat cortex in vivo. Proc Nat Acad Sci USA 102:5588–5593
Sakai R, Cohen DM, Henry JF, Burrin DG, Reeds PJ (2004) Leucine-nitrogen metabolism in the brain of conscious rats: its role as a nitrogen carrier in glutamate synthesis in glial and neuronal metabolic compartments. J Neurochem 88:612–622
Kanamori K, Ross BD, Kondrat RW (1998) Rate of glutamate synthesis from leucine in rat brain measured in vivo by 15 N NMR. J Neurochem 70:1304–1315
Lying-Tunell U, Lindblad BS, Malmlund HO, Persson B (1981) Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol Scand 63:337–350
Smith QR, Momma S, Aoyagi M, Rapoport SI (1987) Kinetics of neutral amino acid transport across the blood-brain barrier. J Neurochem 49:1651–1658
Grill V, Bjorkman O, Gutniak M, Lindqvist M (1992) Brain uptake and release of amino acids in nondiabetic and insulin-dependent diabetic subjects: important role of glutamine release for nitrogen balance. Metab Clin Exp 41:28–32
Hertz L, Kala G (2007) Energy metabolism in brain cells: effects of elevated ammonia concentrations. Metab Brain Dis 22:199–218
Berl S, Frigyesi TL (1968) Metabolism of [14 C]leucine and [14 C]acetate in sensorimotor cortex, thalamus, caudate nucleus and cerebellum of the cat. J Neurochem 15:965–970
Waagepetersen HS, Hansen GH, Fenger K, Lindsay JG, Gibson G, Schousboe A (2006) Cellular mitochondrial heterogeneity in cultured astrocytes as demonstrated by immunogold labeling of alpha-ketoglutarate dehydrogenase. Glia 53:225–231
Sonnewald U, Hertz L, Schousboe A (1998) Mitochondrial heterogeneity in the brain at the cellular level. J Cereb Blood Flow Metab 18:231–237
Bakken IJ, White LR, Aasly J, Unsgård G, Sonnewald U (1997) Lactate formation from [U-13C]aspartate in cultured astrocytes: compartmentation of pyruvate metabolism. Neurosci Lett 237:117–120
Choi I-Y, Lei H, Gruetter R (2002) Effect of deep pentobarbital anesthesia on neurotransmitter metabolism in vivo: on the correlation of total glucose consumption with glutamatergic action. J Cereb Blood Flow Metab 22:1343–1351
de Graaf RA, Mason GF, Patel AB, Rothman DL, Behar KL (2004) Regional glucose metabolism and glutamatergic neurotransmission in rat brain in vivo. Proc Nat Acad Sci USA 101:12700–12705
van Eijsden P, Behar KL, Mason GF, Braun KPJ, de Graaf RA (2010) In vivo neurochemical profiling of rat brain by 1H-[13C] NMR spectroscopy: cerebral energetics and glutamatergic/GABAergic neurotransmission. J Neurochem 112:24–33
Wang J, Jiang L, Jiang Y, Ma X, Chowdhury GMI, Mason GF (2010) Regional metabolite levels and turnover in the awake rat brain under the influence of nicotine. J Neurochem 113:1447–1458
Yang J, Shen J (2005) In vivo evidence for reduced cortical glutamate-glutamine cycling in rats treated with the antidepressant/antipanic drug phenelzine. Neuroscience 135:927–937
Chowdhury GMI, Patel AB, Mason GF, Rothman DL, Behar KL (2007) Glutamatergic and GABAergic neurotransmitter cycling and energy metabolism in rat cerebral cortex during postnatal development. J Cereb Blood Flow Metab 27:1895–1907
Serres S, Raffard G, Franconi J-M, Merle M (2008) Close coupling between astrocytic and neuronal metabolisms to fulfill anaplerotic and energy needs in the rat brain. J Cereb Blood Flow Metab 28:712–724
Duarte JMN, Lanz B, Gruetter R (2011) Compartmentalized cerebral metabolism of [1,6-13C]Glucose determined by in vivo13C NMR Spectroscopy at 14.1 T. Frontiers in neuroenergetics. 3
Acknowledgments
The author’s would like to acknowledge the valuable suggestions by the reviewers and Gerald Dienel. In addition we acknowledge support from the National Institutes of Health 1R01AG034953-01A1 (DLR, HMDF) and R01MH095104 (KLB, DLR) and fellowship grant (#10A087) from AICR to HMDF. We also acknowledge Leif Hertz for his guidance, support, and insights over many years.
Author information
Authors and Affiliations
Corresponding author
Additional information
Special Issue: In Honor of Dr. Leif Hertz.
Rights and permissions
About this article
Cite this article
Rothman, D.L., De Feyter, H.M., Maciejewski, P.K. et al. Is there In Vivo Evidence for Amino Acid Shuttles Carrying Ammonia from Neurons to Astrocytes?. Neurochem Res 37, 2597–2612 (2012). https://doi.org/10.1007/s11064-012-0898-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-012-0898-7