
Abstract
Tumors of the central nervous system (CNS) comprise a broad and diverse collection of neoplasms within pediatric oncology. Yet when taken together pediatric brain and spine tumors represent the most common childhood cancer with an incidence of 5.57 per 100,000 annually and are a leading cause of cancer-related death in patients under 19 years of age (Ostrom et al. 2014; Siegel et al. 2015). Factors such as genetic predisposition, age, and sex play an increasingly significant role in understanding presentation, management, and etiology of childhood brain tumors. Although long-standing observations regarding general patterns of CNS tumors continue to be clinically useful, the introduction of molecular subtypes, such as in medulloblastoma and ependymoma, and the discovery of epigenetic regulators, such as in diffuse intrinsic pontine gliomas (DIPG) and other diffuse midline gliomas with H3K27M mutations, have repurposed epidemiological findings and reconceptualized CNS tumor classification (Louis et al. 2016). The elucidation of the molecular profile of pediatric CNS tumors has made it clear that epidemiology, viewed through a prism of genetics and epigenetics, can offer even greater insights into this incredibly challenging group of tumors. Epidemiology today considers not only environmental, parental, and birth factors that may increase the risk of pediatric CNS tumors, but also germline and molecular features that are causal or pathognomonic of tumor types and subtypes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

1.1 Introduction
Tumors of the central nervous system (CNS) comprise a broad and diverse collection of neoplasms within pediatric oncology. Yet when taken together pediatric brain and spine tumors represent the most common childhood cancer with an incidence of 5.57 per 100,000 annually and are a leading cause of cancer-related death in patients under 19 years of age (Ostrom et al. 2014; Siegel et al. 2015). Factors such as genetic predisposition, age, and sex play an increasingly significant role in understanding presentation, management, and etiology of childhood brain tumors. Although long-standing observations regarding general patterns of CNS tumors continue to be clinically useful, the introduction of molecular subtypes, such as in medulloblastoma and ependymoma, and the discovery of epigenetic regulators, such as in diffuse intrinsic pontine gliomas (DIPG) and other diffuse midline gliomas with H3K27M mutations, have repurposed epidemiological findings and reconceptualized CNS tumor classification (Louis et al. 2016). The elucidation of the molecular profile of pediatric CNS tumors has made it clear that epidemiology, viewed through a prism of genetics and epigenetics, can offer even greater insights into this incredibly challenging group of tumors. Epidemiology today considers not only environmental, parental, and birth factors that may increase the risk of pediatric CNS tumors, but also germline and molecular features that are causal or pathognomonic of tumor types and subtypes.
1.2 Astrocytomas and Other Gliomas
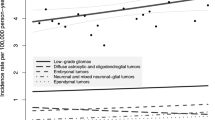
The gliomas are a heterogeneous group of tumors, comprised mostly of astrocytomas. Pediatric astrocytomas are divided into four grades by the World Health Organization (WHO), with pilocytic astrocytomas (WHO grade I) being the most common subtype of pediatric CNS tumor, comprising approximately 15% (Ostrom et al. 2014; Louis et al. 2007). The incidence of pilocytic astrocytomas in children in England and the USA is 0.75–0.97 per 100,000, and these tumors have an exceedingly low incidence of metastasis or malignant transformation (Ostrom et al. 2014; Stokland et al. 2010; Fisher et al. 2008; Arora et al. 2009). Although they may occur in any CNS location including the spine, they most commonly arise from the posterior fossa, optic pathway and hypothalamus, or brain stem (Fernandez et al. 2003; Gajjar et al. 1997; Hayostek et al. 1993; Khatib et al. 1994). Diffuse astrocytomas (WHO grade II), anaplastic astrocytomas (WHO grade III), and glioblastomas (WHO grade IV) have an incidence of 0.27, 0.08, and 0.15 per 100,000 children 0–14 years of age, respectively. Low-grade gliomas, which are comprised of WHO grade I and II astrocytomas as well as WHO grade I gangliogliomas, most commonly present with greater than 6 months of symptoms (Fisher et al. 2008). The incorporation of molecular characteristics in the 2016 WHO classification of tumors of the CNS will assist in a deeper epidemiological understanding by addressing distinct biologic entities, such as diffuse gliomas with IDH mutations and diffuse midline gliomas with H3K27M mutations (Louis et al. 2016).
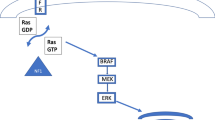
Children with pilocytic astrocytomas have excellent outcomes of >96% overall survival (OS) at 10 years, and patients with subtotal resections do not do significantly worse than patients with gross total resections (Ostrom et al. 2014; Gajjar et al. 1997). Posterior fossa tumors are common in children, with pilocytic astrocytomas being the second most common tumor arising in that location, behind only medulloblastoma; mean age of occurrence is 7.1 years (Smoots et al. 1998). Up to 60% of pilocytic astrocytomas are associated with a KIAA1549:BRAF fusion, which is associated with a better outcome (Becker et al. 2015; Jones et al. 2008). Optic pathway and hypothalamic astrocytomas are most often pilocytic astrocytomas, but other subtypes of low-grade gliomas also account for a small number of cases (Hoffman et al. 1993; Laithier et al. 2003). Optic pathway gliomas (OPGs) occur in approximately 15% of patients with neurofibromatosis type 1 (NF1), though they most often occur sporadically (Listernick et al. 1989). OPGs are reported to have a broad median age between 4.3 and 8.8 years, and those occurring in patients with NF1 present at a significantly earlier age than sporadic cases (Listernick et al. 1989; Nicolin et al. 2009; Singhal et al. 2002; Ahn et al. 2006; Janss et al. 1995; Khafaga et al. 2003; Jahraus and Tarbell 2006; Avery et al. 2011). The variation in age of presentation may be secondary to the presence of a cancer predisposition syndrome in NF1 patients, as well as the practice of asymptomatic surveillance imaging in that group, while 90% of sporadic cases present with new neurologic symptoms. Subependymal giant cell astrocytomas (SEGAs) are another WHO grade I astrocytoma subtype that develop almost exclusively in patients with tuberous sclerosis (TS), which occurs in 1 in 5600 live births (O’Callaghan et al. 1998). Five to twenty percent of patients with TS develop SEGAs, often in adolescence, but congenital cases have also been reported (Adriaensen et al. 2009; O’Callaghan et al. 2008; Hahn et al. 1991).
Several WHO grade II subtypes can be distinguished by histology and presentation. Pilomyxoid astrocytomas (WHO grade II) have a more aggressive course than pilocytic astrocytomas (WHO grade I), a greater propensity for growing in the hypothalamochiasmatic region, and often present earlier with a mean age of 3.3 years (Bhargava et al. 2013). Pleomorphic xanthoastrocytomas (WHO grade II) are typically located in the superficial temporal lobe; they classically present with seizures and have a median age at diagnosis of 20.5 years and an approximately 75% overall survival (Gallo et al. 2013; Perkins et al. 2012). These can rarely transform into a high-grade glioma.
Low-grade gliomas of the brain stem can be pilocytic astrocytomas or gangliogliomas, which typically occur dorsally and have the possibility of long-term cure. WHO grade II, III, and IV gliomas of the brain stem have dismal outcomes and together comprise diffuse intrinsic pontine glioma (DIPG). The 2016 WHO classification has adjusted that nomenclature in favor of diffuse midline gliomas, as diffuse gliomas of the pons, thalamus, and spinal cord may form a more biologically distinct category when H3K27M mutations are present (Louis et al. 2016; Shankar et al. 2016).
DIPGs arise most commonly in the ventral pons and comprise 10–15% of all pediatric CNS tumors and 80% of brain stem gliomas, affecting roughly 300 children in the USA each year (Ostrom et al. 2014; Ramos et al. 2013; Smith et al. 1998). Males and females are affected equally and the median age of presentation is 7 years (Lassiter et al. 1971; Lober et al. 2014; Veldhuijzen van Zanten et al. 2014). Presentation usually consists of a classic triad of ataxia, cranial nerve palsies, and pyramidal tract signs developing over 1 month, although atypical cases can present more slowly over several months (Fisher et al. 2000). It is now recognized that approximately 17% of patients undergo both local and distant neuraxis dissemination by 15 months, which is not far beyond the median overall survival of patients with DIPG, as only 10% of patients survive beyond 2 years and only 2–3% are considered long-term survivors (Gururangan et al. 2006; Hargrave et al. 2006; Jackson et al. 2013). Recently, 80% of DIPGs have been found to harbor mutations in K27M of histone 3.1 or 3.3, which are associated with mutations in ACVR1 and p53, respectively (Taylor et al. 2014; Wu et al. 2012).
High-grade gliomas (HGGs) occur much more frequently in adults, with an increasing incidence with age to a peak between the ages of 75 and 85 years (Ostrom et al. 2014). The outcomes of patients with high-grade gliomas appear to be inverse to patient age, as 5-year overall survivals for children less than three and those 3–14 years of age are 31–66% and 19%, respectively (Mathew et al. 2014). Glioblastoma has been reported in classic CNS tumor predisposition syndromes, such as neurofibromatosis, Li–Fraumeni, and Turcot syndromes, as well as in several genitourinary syndromes, such as Turner and Mayer–Rokitansky–Küster–Hauser syndrome, though the majority of cases are believed to be sporadic (Hanaei et al. 2015; Jeong and Yee 2014; Macy et al. 2012; Gonzalez and Prayson 2013).
1.3 Embryonal Tumors
Embryonal brain tumors are a diverse group of aggressive neoplasms, including medulloblastoma, primary neuroectodermal tumors (PNET), atypical rhabdoid/teratoid tumors (ATRT), and pineoblastoma, which share high mitotic activity and a predilection for dissemination throughout the neuraxis, and are all WHO grade IV (Louis et al. 2007). They account for 15% of CNS tumors in patients 0–14 years of age and 12% in those 0–19 years of age, with incidences of 0.78 and 0.64 per 100,000, respectively; these incidences have remained unchanged since at least 1990 (Ostrom et al. 2014; Johnston et al. 2014). Embryonal CNS tumors rarely occur outside of childhood with the median age at presentation being 7.3 years, and 44% of them being diagnosed between the ages of 4 and 9 years (Ostrom et al. 2014; Kool et al. 2012). Medulloblastomas, the most common malignant brain tumor in pediatrics, histologically appear as PNETs specifically arising in the posterior fossa (Northcott et al. 2011). A minority of medulloblastoma cases have been reported in patients with genetic predisposition syndromes such as Gorlin, Turcot B, Li–Fraumeni, ataxia telangiectasia, Nijmegen breakage, Rubenstein–Taybi, and Coffin–Siris syndromes (Distel et al. 2003; Hart et al. 1987; Larsen et al. 2014; Skomorowski et al. 2012; Taylor et al. 2001; Rogers et al. 1988). Overall, there is a male predominance of 1.5:1, with females reported to have superior outcomes, although again this is likely subgroup dependent, as there are fewer females in the higher risk Group 3 and 4, while more young females have sonic hedgehog (SHH) driven tumors (Louis et al. 2007; Northcott et al. 2011). Historically, patients clinically classified as average-risk had a 5-year OS of roughly 85%, while high-risk patients suffered poorer outcomes with near 70% OS and patients with large-cell anaplastic histology had particularly dismal outcomes (Kool et al. 2012; Gajjar et al. 2006; Packer et al. 2006; Tarbell et al. 2013; Ramaswamy et al. 2013). Overall, long-term survival in patients with medulloblastoma is achieved in only 66% of patients, with 10% suffering from secondary malignancies, 32% of which are secondary brain tumors (Ning et al. 2015).
Although particular subsets of medulloblastoma have long been suspected to behave differently, it is now commonly accepted that there are four distinct molecular subgroups: WNT, SHH, Group 3, and Group 4, which account for 11%, 28%, 27%, and 34% of cases, respectively (Kool et al. 2012; Northcott et al. 2011; Badiali et al. 1991). Prodromes may vary among the groups, ranging from only 2 weeks in patients with SHH tumors, to 4 weeks in patients with Group 3 tumors and 8 weeks in patients with WNT or Group 4 tumors (Ramaswamy et al. 2014). Furthermore, age of presentation varies as the incidence of SHH medulloblastomas is bimodal, peaking under 3 years and again over 15 years of age (Northcott et al. 2011). WNT and Group 4 both peak around age 11, but WNT tumors are essentially absent in infancy (Kool et al. 2012; Northcott et al. 2011). WNT tumors have no gender predominance, are the least frequent subgroup, and experience the best outcomes with greater than 90% overall survival (Ellison et al. 2011, 2011). Outcomes in patients with SHH tumors are inferior, though strongly age-dependent as the 10-year OS is 77% and 51% in infants and children, respectively (Kool et al. 2012; Ramaswamy et al. 2013). Despite presenting with metastatic disease in 17% of infants and 22% of children, SHH tumors most often recur locally (Kool et al. 2012; Ramaswamy et al. 2013). Group 3 and Group 4 occur nearly twice as often in males, accounting for the male predominance in medulloblastoma as a whole. Forty-seven percent of Group 3 medulloblastomas present with metastases and, while they do not have significantly worse prognoses than those without metastases, this subgroup overall suffers the poorest outcomes with long-term survival in less than 50% of patients (Kool et al. 2012; Northcott et al. 2011). Group 4 patients, on the other hand, have significantly different outcomes associated with the presence of metastases, ranging from nearly 40% OS (metastases present) to greater than 70% (metastases absent) (Kool et al. 2012). In patients that experience recurrence, the molecular subgroup remains consistent, and although outcomes are uniformly poor, Group 4 patients have the longest survival following recurrence (Ramaswamy et al. 2013).
Atypical teratoid/rhabdoid tumors (ATRTs) are embryonal CNS tumors with rhabdoid features that were initially described in the 1990s (Zuccoli et al. 1999). Since their initial description their incidence has increased, while the incidence of other PNETs has declined, more likely representative of a change in classification than a change in biological patterns of disease (Ostrom et al. 2014). The incidence of ATRT in childhood is approximately 0.1 per 100,000 with a peak between 1 and 2 years of age and no gender predisposition observed in the USA (Ostrom et al. 2014, b; Hilden et al. 2004; von Hoff et al. 2011; Woehrer et al. 2010). They account for 10% of CNS tumors in patients less than 1 year of age, but only 1.6% of all childhood brain tumors (Ostrom et al. 2014). The wide range of reported OS, between 28 and 48%, may be affected by delays in appropriate diagnosis, as one report noted a 5-year OS of only 15% in patients who were initially misdiagnosed (Ostrom et al. 2014; Hilden et al. 2004; von Hoff et al. 2011; Woehrer et al. 2010; Athale et al. 2009; Lafay-Cousin et al. 2012). Most reports conclude that metastatic disease at presentation is not prognostic, while descriptions of the prognostic impact of age differ (Ostrom et al. 2014; Hilden et al. 2004; von Hoff et al. 2011; Woehrer et al. 2010; Athale et al. 2009; Lafay-Cousin et al. 2012). The location of ATRTs, however, does appear to change with age, as patients under 1 year of age most commonly have infratentorial disease and the incidence of supratentorial disease increases with age (Ostrom et al. 2014). The characteristic loss of INI1 in these tumors is most commonly somatic, although germline mutations have been reported and can result in a rhabdoid tumor predisposition syndrome (RTPS) (Sredni and Tomita 2015; Taylor et al. 2000). The development of ATRTs has also been associated with low birth weight and twin pregnancies (Heck et al. 2013).
Pineoblastomas are malignant tumors of the pineal gland that, like other PNETs, are histologically similar to medulloblastomas, but display a distinct biology (Li et al. 2005). While some pineal tumors, such as germ cell tumors, occur more commonly in males, reports suggest pineoblastoma may be more common in females (Villa et al. 2012; Fauchon et al. 2000). Although patients with bilateral retinoblastomas may develop a pineoblastoma, “trilateral retinoblastoma” occurs in only 1% of patients with bilateral retinoblastoma and only in the setting of germline mutations (Ramasubramanian et al. 2013). While the majority of pineoblastoma cases appear sporadic, cases also have been reported as part of Turcot syndrome and with germline DICER1 mutations (Ikeda et al. 1998; Gadish et al. 2005; Sabbaghian et al. 2012).
1.4 Ependymoma
Virchow initially described ependymomas in the nineteenth century as CNS tumors originating from the walls of the ventricular system (Virchow 1863–67). Though ependymomas likely consist of several discrete subgroups that can be distinguished by location and molecular profile, most reports evaluate ependymomas as a whole or by grade, leaving their epidemiologic understanding incomplete. Ependymoma incidence in the USA is 0.3 and 0.29 per 100,000 children aged 0–14 years and 0–19 years, respectively, and has not increased since 1973; nearly one-third of cases occur in children under the age of 4 years (Ostrom et al. 2014; McGuire et al. 2009). Although 46% of ependymomas in adults are spinal, location varies according to age in children (Vera-Bolanos et al. 2015). The mean age for spinal, supratentorial, and infratentorial ependymomas are 12.2, 7.8, and 5 years, respectively (McGuire et al. 2009). The gender incidence may be affected by age and location, as the overall male-to-female ratio is 1.3:1, though males are more commonly affected by supratentorial ependymomas (1.4:1) and less commonly affected by spinal ependymoma (0.7:1) than females (McGuire et al. 2009; Dohrmann and Farwell 1976). Presentation with metastatic disease is rare in pediatric ependymomas but is more common in infants, although reports vary on whether supratentorial or infratentorial tumors are more likely to metastasize (Zacharoulis et al. 2008; Allen et al. 1998).
Currently, the treatment of ependymoma primarily varies according to age, grade, and location. In 2015, a new molecular classification was proposed though it has yet to be validated. It divides ependymomas into anatomical compartments: supratentorial (ST), posterior fossa (PF), and spinal (SP); tumors in each compartment are then divided into one of three subgroups: a subependymoma group and two other genetic or epigenetic subgroups (Pajtler et al. 2015). Supratentorial ependymomas are distinguished by either RELA fusions (ST-EPN-RELA), which occur at a median age of 8 years and result in frequent disease progressions, or YAP1 fusions (ST-EPN-YAP1), which occur at a median age of 1.4 years (Pajtler et al. 2015). Posterior fossa ependymomas are subdivided into those with a CpG methylator phenotype (PF-EPN-A), which account for 48% of all pediatric ependymomas and experience poor outcomes, and those that are not hypermethylated (PF-EPN-B), which often occur in older patients (EPN-PFB) (Pajtler et al. 2015; Parker et al. 2014; Witt et al. 2011).
Although histologic classification of WHO grade II or III in pediatric ependymoma may not offer prognostic significance, several WHO grade I subsets are clearly less aggressive neoplasms (Perilongo et al. 1997; Ross and Rubinstein 1989; Robertson et al. 1998). Subependymomas represent less than 1% of CNS tumors in children, are designated WHO grade I, and have essentially no metastatic potential (Scheinker 1945; Ragel et al. 2006). Myxopapillary ependymomas, also WHO grade I, have a median age of presentation of 36 years, yet are not uncommon in children with reports of patients as young as 6 years old being affected (Barton et al. 2010; Woesler et al. 1998). Despite their WHO grade I designation, the pediatric variant may be more aggressive than that seen in adults with a suggestion of dissemination in as many as 58% of patients (Fassett et al. 2005). Neurofibromatosis type II (NF2) is the most common hereditary predisposition for ependymoma, most often causing intramedullary spinal tumors of the cervical spine (Bianchi et al. 1994; Plotkin et al. 2011). Pediatric ependymomas have also been reported in Turcot B, MEN1, and Li–Fraumeni syndromes (Chan et al. 1999; Metzger et al. 1991).
1.5 Germ Cell Tumors
Germ cell tumors (GCTs) are a heterogeneous group of cancers with variable classification and nomenclature depending on the particular organ involvement. In the CNS, they are divided into germinomas, non-germinomatous germ cell tumors (NGGCT), and teratomas. The most common locations for GCTs are the suprasellar and pineal regions. GCTs account for 4% of pediatric CNS tumors with an incidence of 0.2 and 0.22 per 100,000 in children aged 0–14 and 0–19 years, respectively (Ostrom et al. 2014). Males account for 76% of all CNS GCTs, 58% of pituitary GCTs, and a remarkable 93% of pineal GCTs (Goodwin et al. 2009). In both sexes there is a small spike at birth and a much greater spike in adolescence with incidences peaking at roughly age 15. Race also influences incidence patterns, as in the USA nearly 20% of patients were Asian or Pacific Islander with an incidence of 0.26 per 100,000, double the 0.13 per 100,000 in white children 0–15 years of age (Goodwin et al. 2009). CNS GCTs also account for a greater percentage of pediatric CNS tumors in Japan, Korea, Taiwan, and China at 7.8%, 11.2%, 14%, and 7.9%, respectively (Cho et al. 2002; Mori and Kurisaka 1986; Wong et al. 2005; Zhou et al. 2008). Klinefelter syndrome is associated with the development of pediatric germ cell tumors including intracranial germinomas (Arens et al. 1988). Down syndrome and NF1 have also been reported in patients with intracranial germinomas (Hashimoto et al. 1995; Wong et al. 1995).
1.6 Family History
Despite the increasing awareness of CNS tumor genetic predispositions, further discussed within another chapter, there is still little evidence of the development of CNS tumors in the parents or siblings of affected children. The studies reporting increased pediatric CNS tumor incidence among siblings have been plagued by small numbers and an inability to exclude genetic predisposition syndromes; however, a larger Nordic cohort of patients showed no association among siblings outside of genetic predisposition syndromes (Draper et al. 1977; Farwell and Flannery 1984; Miller 1971; Winther et al. 2001). There have been several reports regarding the association of parental age with pediatric CNS tumors: two studies identified increased parental age as a risk factor, while one found only advanced maternal age to be a significant risk (Hemminki et al. 1999; Johnson et al. 2009; Yip et al. 2006). A review of Sweden’s Family-Cancer Database, consisting of over 13,000 CNS tumor diagnoses, found that oldest siblings were at increased risk for several childhood malignancies and this risk increased with the number of younger siblings (Altieri et al. 2006). The existence of three or more younger siblings resulted in a relative risk of 1.34, 2.3, 2.61, and 3.71 of astrocytoma, medulloblastoma, ependymoma, and meningioma, respectively (Altieri et al. 2006).
1.7 Birth History
As early as 1968, Kobayashi had published a report of the association between congenital anomalies and childhood cancer (Kobayashi et al. 1968). A review of 90,400 children found patients with congenital anomalies had a risk ratio of 5.8 (CI 3.7–9.1) of developing cancer in their first year of life (Agha et al. 2005). The risk was also increased for central nervous system and sympathetic nervous system tumors individually at a risk ratio of 2.5 (CI 1.8–3.4) and 2.2 (CI 1.4–3.4), respectively. A Bjørge et al. study of 5.2 million children and their families in Norway and Sweden also found patients with congenital anomalies had an increased cancer risk that extended into early adulthood (Bjorge et al. 2008). Furthermore, patients with CNS malformations were also at the highest risk of developing CNS malignancies, with a standardized incidence rate (SIR) of 58 (CI 41–80) and 8.3 (Louis et al. 2007; Stokland et al. 2010; Fisher et al. 2008; Arora et al. 2009; Fernandez et al. 2003; Gajjar et al. 1997; Hayostek et al. 1993; Khatib et al. 1994; Smoots et al. 1998; Becker et al. 2015; Jones et al. 2008; Hoffman et al. 1993) in Norway and Sweden, respectively. To assess potential cancer risk associated with congenital anomalies even outside of the setting of chromosomal defects, a review of the California Cancer Registry (CCR) found that between 1988 and 2004, children with congenital anomalies without chromosomal defects had a 1.8-fold increased risk of CNS cancer (Fisher et al. 2012). A further examination found a particularly increased risk in medulloblastoma (OR 1.7, CI 1.1–2.6), PNET (OR 3.64, CI 1.5–8.6), and germ cell tumors (OR 6.4, CI 2.1–19.6), as well as an increased risk in mothers with greater than two fetal losses after 20 weeks of gestation (OR 3.13, CI 1.3–7.4) (Partap et al. 2011).
Many large studies have evaluated the impact of birth weight on the risk of developing CNS tumors, with several suggesting an increased birth weight carries a greater relative risk, although the most common specific tumors types varied among studies (Bjorge et al. 2013; Harder et al. 2008; MacLean et al. 2010; Milne et al. 2008; Schmidt et al. 2010). In an examination matching each case (17,698) to 10 controls, Bjørge found an increased childhood cancer risk for higher birth weight infants, and also infants with larger head circumferences (Bjorge et al. 2013). Additionally, in an evaluation of Nordic children, Schmidt found a gestational age-adjusted birth weight of greater than 4.5 kg increased the risk of all CNS tumors (OR 1.27, CI 1.03–1.6), with the greatest increase among embryonal tumors (Schmidt et al. 2010). When 3733 CNS tumors from the CCR were matched to controls, Maclean et al. found an increased birth weight of 4 kg associated with an increased risk of CNS tumors, especially HGGs (MacLean et al. 2010). A meta-analysis of eight studies found that increased birth weight was associated with increased incidence of astrocytomas and medulloblastomas, but not ependymomas (Harder et al. 2008). Conversely, a study of over 600,000 live births in Western Australia between 1980 and 2004 found no association between birth size and the development of CNS tumors prior to age 14 (Milne et al. 2008).
1.8 Immune System
Although allergic conditions have been consistently reported as inversely associated with adult gliomas, reports in children have varied (Chen et al. 2011). In pediatrics, an initial report from the United Kingdom found that maternal asthma resulted in a decreased relative risk of their children developing a CNS tumor, particularly PNETs (Harding et al. 2008). Another study evaluating 272 matched case–control pairs in Canada found asthma associated inversely with the development of CNS tumors, especially ependymomas, while the relationship with eczema was not significant (Roncarolo and Infante-Rivard 2012). Furthermore, the use of asthma controller medications was found to be associated with an increased risk. However, a study of 352 pediatric brain tumors in Denmark, Norway, Sweden, and Switzerland found no association with asthma or eczema (Shu et al. 2014).
Studies evaluating the influence of prior infectious history on the development of pediatric CNS tumors have been conflicting. Harding et al. found infants without social interaction with other infants in the first year of life had an increased risk (OR 1.37, CI 1.08–1.75) of CNS tumors, especially PNET, compared to those who had such interaction (Harding et al. 2009). Attendance in day care also appeared to show a protective benefit, though not statistically significant. A Canadian study also found a reduced risk in patients with day care attendance, and, unlike Harding’s study, breastfeeding was found to be protective against the development of brain tumors (Shaw et al. 2006; Harding et al. 2007). Conversely, Anderson et al. found no association with day care attendance but that patients with more frequent sick days in the first 6 years of life had an increased incidence of gliomas and embryonal tumors (Andersen et al. 2013).
1.9 Environmental Exposure
Radiation therapy (RT), used decades ago to treat tinea capitis and more recently to treat childhood acute lymphoblastic leukemia (ALL), is known to cause secondary CNS tumors, especially meningiomas, p53 mutated glioblastomas, and PNETs (Kleinerman 2006; Ohgaki and Kleihues 2005). Fifty-three percent of secondary neoplasms in survivors of childhood ALL occur in the CNS and 89% of those are associated with prior cranial irradiation (Mody et al. 2008; Schmiegelow et al. 2013). The timing and outcome are dependent on pathology, as non-meningioma CNS tumors occur between 6.5 and 9.8 years and meningiomas occurred between 12.3 and 18.3 years after treatment, with OS of 18% and 96%, respectively (Schmiegelow et al. 2013). Prenatal diagnostic imaging has been evaluated as a potential cancer risk, but studies from the United Kingdom, Sweden, and Denmark did not describe a significant increase in pediatric CNS tumors in patients exposed to prenatal X-rays compared to controls (Mellemkjaer et al. 2006; Rajaraman et al. 2011; Stalberg et al. 2007). Diagnostic head X-rays also have not been associated with the development of CNS tumors (Khan et al. 2010). However, CT scans contribute to a slightly elevated risk of CNS tumors, with risk decreasing with increasing age at first CT scan exposure (Pearce et al. 2012; Mathews et al. 2013).
Magnetic fields, radio waves, and mobile phone use have not been found to be associated with an increase in pediatric brain tumors (Aydin et al. 2011; Elliott et al. 2010; Ha et al. 2007; Kheifets et al. 2010).
Although many different maternal medications have been evaluated, none have been found to consistently increase the risk of pediatric CNS tumors in offspring. A German study found an association between maternal prenatal antibiotic use and an increased risk of medulloblastoma (OR 2.07, CI 1.03–4.17) and astrocytoma (OR 2.26, CI 1.09–4.69) (Kaatsch et al. 2010). Although the odds ratio was similarly elevated in a Canadian study, the results were not statistically significant (OR 1.7, CI 0.8–3.6) (Shaw et al. 2006). A 2010 Swedish study evaluating potential associations with prenatal medications and the development of pediatric CNS tumors in children 0–14 years of age found no association with antibiotics, antifungals, antacids, analgesics, antiasthmatics, antiemetics, antihistamines, diuretics, folic acid, iron, laxatives, or vitamins, but did find an association with antihypertensives (OR 2.7, CI 1.1–6.5), particularly β-blockers (OR 5.3, CI 1.2–24.8) (Stalberg et al. 2010). An association between prenatal antihypertensive use and the development of pediatric CNS tumors, however, was not found in a German study evaluating pediatric CNS tumors diagnosed between 1992 and 1997 (Schuz et al. 2007). Amide or amine-containing medications can potentially be carcinogenic after conversion to N-nitroso compounds (NOCs) in the stomach, though three studies have all found little or no support for an association between maternal exposure and central nervous system tumors in subsequent children (Cardy et al. 2006; Carozza et al. 1995).
Prenatal vitamins, especially iron and folic acid, consistently have been shown to decrease the risk of pediatric CNS tumors (Bunin et al. 2005, 2006; Ortega-Garcia et al. 2010; Milne et al. 2012).
Although prenatal alcohol exposure can have a variety of toxic effects on the developing child, there is no clear increased risk of pediatric CNS tumors (Infante-Rivard and El-Zein 2007; Milne et al. 2013). The role of maternal tobacco smoking during pregnancy is unclear, as several reports have found no association (Filippini et al. 2002; Huncharek et al. 2002; Norman et al. 1996), while a review of the Swedish Birth Register of births between 1983 and 1997 found a hazard ratio of 1.24 (CI 1.01–1.53) (Brooks et al. 2004).
Pesticide exposure may have an association with pediatric CNS tumors. A review of 4723 patients from the North of England found no significant relationship between occupational exposure to pesticides and risk of any childhood cancer (Pearce et al. 2006). In contrast, a study from the USA found that paternal pesticide exposure was associated with an increased risk of his child developing an astrocytoma (OR 1.8, CI 1.1–31), but not PNET (Shim et al. 2009). A separate study investigating paternal hobbies did identify exposure to pesticides as increasing the risk of medulloblastoma and PNET (Rosso et al. 2008). An Australian study also found preconception exposure to pesticides increased the risk of pediatric CNS tumors (Greenop et al. 2013). The effect of residential pesticides may be contingent on particular predispositions as polymorphisms in PON1, a gene responsible for organophosphorous metabolism, may increase the risk of pediatric CNS tumors in exposed patients (Searles Nielsen et al. 2010).
An investigation of the risk of pediatric CNS tumors among children of parents working in a wide variety of occupations found no clear associations (Mazumdar et al. 2008). However, a separate analysis found that brain tumors were more common in children of mothers working in electronic component manufacturing (OR 13.78, CI 1.45–129) and garment and textile workers (IR 7.25, CI 1.42–37) (Ali et al. 2004). There also appears to be an increased incidence of CNS tumors among children whose parents are exposed to diesel fuel, but not other exhausts (Peters et al. 2013). Paternal polycyclic aromatic hydrocarbon exposure has also been linked to a subsequent increase in pediatric CNS tumors (OR 1.4, CI 1.1–1.7) (Cordier et al. 2004).
In conclusion, pediatric neuro-oncology is a rapidly evolving field in which molecular investigations are fueling a restructuring of tumor subgroups. Although pediatric CNS tumors have historically been distinguished by histopathology and location, driving mutations and epigenetic profiles are proving to not only be attractive therapeutic targets but also epicenters for new classifications. The challenge will be to integrate former classification systems with the latter, and, perhaps just as importantly, to frame our historical data according to the new groupings so that the decades of lessons learned in epidemiology can continue to be applied in the pursuit of improving outcomes for children with CNS tumors.
References
Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M (2009) Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 16(6):691–696
Agha MM, Williams JI, Marrett L, To T, Zipursky A, Dodds L (2005) Congenital abnormalities and childhood cancer. Cancer 103(9):1939–1948
Ahn Y, Cho BK, Kim SK, Chung YN, Lee CS, Kim IH et al (2006) Optic pathway glioma: outcome and prognostic factors in a surgical series. Childs Nerv Syst 22(9):1136–1142
Ali R, Yu CL, Wu MT, Ho CK, Pan BJ, Smith T et al (2004) A case-control study of parental occupation, leukemia, and brain tumors in an industrial city in Taiwan. J Occup Environ Med 46(9):985–992
Allen JC, Siffert J, Hukin J (1998) Clinical manifestations of childhood ependymoma: a multitude of syndromes. Pediatr Neurosurg 28(1):49–55
Altieri A, Castro F, Bermejo JL, Hemminki K (2006) Association between number of siblings and nervous system tumors suggests an infectious etiology. Neurology 67(11):1979–1983
Andersen TV, Schmidt LS, Poulsen AH, Feychting M, Roosli M, Tynes T et al (2013) Patterns of exposure to infectious diseases and social contacts in early life and risk of brain tumours in children and adolescents: an International Case-Control Study (CEFALO). Br J Cancer 108(11):2346–2353
Arens R, Marcus D, Engelberg S, Findler G, Goodman RM, Passwell JH (1988) Cerebral germinomas and Klinefelter syndrome. A review. Cancer 61(6):1228–1231
Arora RS, Alston RD, Eden TO, Estlin EJ, Moran A, Birch JM (2009) Age-incidence patterns of primary CNS tumors in children, adolescents, and adults in England. Neuro-Oncology 11(4):403–413
Athale UH, Duckworth J, Odame I, Barr R (2009) Childhood atypical teratoid rhabdoid tumor of the central nervous system: a meta-analysis of observational studies. J Pediatr Hematol Oncol 31(9):651–663
Avery RA, Fisher MJ, Liu GT (2011) Optic pathway gliomas. J Neuroophthalmol 31(3):269–278
Aydin D, Feychting M, Schuz J, Tynes T, Andersen TV, Schmidt LS et al (2011) Mobile phone use and brain tumors in children and adolescents: a multicenter case-control study. J Natl Cancer Inst 103(16):1264–1276
Badiali M, Pession A, Basso G, Andreini L, Rigobello L, Galassi E et al (1991) N-myc and c-myc oncogenes amplification in medulloblastomas. Evidence of particularly aggressive behavior of a tumor with c-myc amplification. Tumori 77(2):118–121
Barton VN, Donson AM, Kleinschmidt-DeMasters BK, Birks DK, Handler MH, Foreman NK (2010) Unique molecular characteristics of pediatric myxopapillary ependymoma. Brain Pathol 20(3):560–570
Becker AP, Scapulatempo-Neto C, Carloni AC, Paulino A, Sheren J, Aisner DL et al (2015) KIAA1549: BRAF gene fusion and FGFR1 hotspot mutations are prognostic factors in pilocytic astrocytomas. J Neuropathol Exp Neurol 74(7):743–754
Bhargava D, Sinha P, Chumas P, Al-Tamimi Y, Shivane A, Chakrabarty A et al (2013) Occurrence and distribution of pilomyxoid astrocytoma. Br J Neurosurg 27(4):413–418
Bianchi AB, Hara T, Ramesh V, Gao J, Klein-Szanto AJ, Morin F et al (1994) Mutations in transcript isoforms of the neurofibromatosis 2 gene in multiple human tumour types. Nat Genet 6(2):185–192
Bjorge T, Cnattingius S, Lie RT, Tretli S, Engeland A (2008) Cancer risk in children with birth defects and in their families: a population based cohort study of 5.2 million children from Norway and Sweden. Cancer Epidemiol Biomark Prev 17(3):500–506
Bjorge T, Sorensen HT, Grotmol T, Engeland A, Stephansson O, Gissler M et al (2013) Fetal growth and childhood cancer: a population-based study. Pediatrics 132(5):e1265–e1275
Brooks DR, Mucci LA, Hatch EE, Cnattingius S (2004) Maternal smoking during pregnancy and risk of brain tumors in the offspring. A prospective study of 1.4 million Swedish births. Cancer Causes Control 15(10):997–1005
Bunin GR, Gallagher PR, Rorke-Adams LB, Robison LL, Cnaan A (2006) Maternal supplement, micronutrient, and cured meat intake during pregnancy and risk of medulloblastoma during childhood: a children's oncology group study. Cancer Epidemiol Biomark Prev 15(9):1660–1667
Bunin GR, Kushi LH, Gallagher PR, Rorke-Adams LB, McBride ML, Cnaan A (2005) Maternal diet during pregnancy and its association with medulloblastoma in children: a children’s oncology group study (United States). Cancer Causes Control 16(7):877–891
Cardy AH, Little J, McKean-Cowdin R, Lijinsky W, Choi NW, Cordier S et al (2006) Maternal medication use and the risk of brain tumors in the offspring: the SEARCH international case-control study. Int J Cancer 118(5):1302–1308
Carozza SE, Olshan AF, Faustman EM, Gula MJ, Kolonel LN, Austin DF et al (1995) Maternal exposure to N-nitrosatable drugs as a risk factor for childhood brain tumours. Int J Epidemiol 24(2):308–312
Chan TL, Yuen ST, Chung LP, Ho JW, Kwan K, Fan YW et al (1999) Germline hMSH2 and differential somatic mutations in patients with Turcot’s syndrome. Genes Chromosomes Cancer 25(2):75–81
Chen C, Xu T, Chen J, Zhou J, Yan Y, Lu Y et al (2011) Allergy and risk of glioma: a meta-analysis. Eur J Neurol 18(3):387–395
Cho KT, Wang KC, Kim SK, Shin SH, Chi JG, Cho BK (2002) Pediatric brain tumors: statistics of SNUH, Korea (1959–2000). Childs Nerv Syst 18(1–2):30–37
Cordier S, Monfort C, Filippini G, Preston-Martin S, Lubin F, Mueller BA et al (2004) Parental exposure to polycyclic aromatic hydrocarbons and the risk of childhood brain tumors: the SEARCH International Childhood Brain Tumor Study. Am J Epidemiol 159(12):1109–1116
Distel L, Neubauer S, Varon R, Holter W, Grabenbauer G (2003) Fatal toxicity following radio- and chemotherapy of medulloblastoma in a child with unrecognized Nijmegen breakage syndrome. Med Pediatr Oncol 41(1):44–48
Dohrmann GJ, Farwell JR (1976) Ependymal neoplasms in children. Trans Am Neurol Assoc 101:125–129
Draper GJ, Heaf MM, Kinnier Wilson LM (1977) Occurrence of childhood cancers among sibs and estimation of familial risks. J Med Genet 14(2):81–90
Elliott P, Toledano MB, Bennett J, Beale L, de Hoogh K, Best N et al (2010) Mobile phone base stations and early childhood cancers: case-control study. BMJ 340:c3077
Ellison DW, Dalton J, Kocak M, Nicholson SL, Fraga C, Neale G et al (2011) Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathol 121(3):381–396
Ellison DW, Kocak M, Dalton J, Megahed H, Lusher ME, Ryan SL et al (2011) Definition of disease-risk stratification groups in childhood medulloblastoma using combined clinical, pathologic, and molecular variables. J Clin Oncol 29(11):1400–1407
Farwell J, Flannery JT (1984) Cancer in relatives of children with central-nervous-system neoplasms. N Engl J Med 311(12):749–753
Fassett DR, Pingree J, Kestle JR (2005) The high incidence of tumor dissemination in myxopapillary ependymoma in pediatric patients. Report of five cases and review of the literature. J Neurosurg 102(1 Suppl):59–64
Fauchon F, Jouvet A, Paquis P, Saint-Pierre G, Mottolese C, Ben Hassel M et al (2000) Parenchymal pineal tumors: a clinicopathological study of 76 cases. Int J Radiat Oncol Biol Phys 46(4):959–968
Fernandez C, Figarella-Branger D, Girard N, Bouvier-Labit C, Gouvernet J, Paz Paredes A et al (2003) Pilocytic astrocytomas in children: prognostic factors--a retrospective study of 80 cases. Neurosurgery 53(3):544–553. discussion 54–5
Filippini G, Maisonneuve P, McCredie M, Peris-Bonet R, Modan B, Preston-Martin S et al (2002) Relation of childhood brain tumors to exposure of parents and children to tobacco smoke: the SEARCH international case-control study. Surveillance of Environmental Aspects Related to Cancer in Humans. Int J Cancer 100(2):206–213
Fisher PG, Breiter SN, Carson BS, Wharam MD, Williams JA, Weingart JD et al (2000) A clinicopathologic reappraisal of brain stem tumor classification. Identification of pilocystic astrocytoma and fibrillary astrocytoma as distinct entities. Cancer 89(7):1569–1576
Fisher PG, Reynolds P, Von Behren J, Carmichael SL, Rasmussen SA, Shaw GM (2012) Cancer in children with nonchromosomal birth defects. J Pediatr 160(6):978–983
Fisher PG, Tihan T, Goldthwaite PT, Wharam MD, Carson BS, Weingart JD et al (2008) Outcome analysis of childhood low-grade astrocytomas. Pediatr Blood Cancer 51(2):245–250
Gadish T, Tulchinsky H, Deutsch AA, Rabau M (2005) Pinealoblastoma in a patient with familial adenomatous polyposis: variant of Turcot syndrome type 2? Report of a case and review of the literature. Dis Colon Rectum 48(12):2343–2346
Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE et al (2006) Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7(10):813–820
Gajjar A, Sanford RA, Heideman R, Jenkins JJ, Walter A, Li Y et al (1997) Low-grade astrocytoma: a decade of experience at St. Jude Children’s Research Hospital. J Clin Oncol 15(8):2792–2799
Gallo P, Cecchi PC, Locatelli F, Rizzo P, Ghimenton C, Gerosa M et al (2013) Pleomorphic xanthoastrocytoma: long-term results of surgical treatment and analysis of prognostic factors. Br J Neurosurg 27(6):759–764
Gonzalez EM, Prayson RA (2013) Glioblastoma arising in a patient with Mayer-Rokitansky-Kuster-Hauser syndrome. J Clin Neurosci 20(10):1464–1465
Goodwin TL, Sainani K, Fisher PG (2009) Incidence patterns of central nervous system germ cell tumors: a SEER Study. J Pediatr Hematol Oncol 31(8):541–544
Greenop KR, Peters S, Bailey HD, Fritschi L, Attia J, Scott RJ et al (2013) Exposure to pesticides and the risk of childhood brain tumors. Cancer Causes Control 24(7):1269–1278
Gururangan S, McLaughlin CA, Brashears J, Watral MA, Provenzale J, Coleman RE et al (2006) Incidence and patterns of neuraxis metastases in children with diffuse pontine glioma. J Neuro-Oncol 77(2):207–212
Ha M, Im H, Lee M, Kim HJ, Kim BC, Gimm YM et al (2007) Radio-frequency radiation exposure from AM radio transmitters and childhood leukemia and brain cancer. Am J Epidemiol 166(3):270–279
Hahn JS, Bejar R, Gladson CL (1991) Neonatal subependymal giant cell astrocytoma associated with tuberous sclerosis: MRI, CT, and ultrasound correlation. Neurology 41(1):124–128
Hanaei S, Habibi Z, Nejat F, Sayarifard F, Vasei M (2015) Pediatric glioblastoma multiforme in association with Turner's syndrome: a case report. Pediatr Neurosurg 50(1):38–41
Harder T, Plagemann A, Harder A (2008) Birth weight and subsequent risk of childhood primary brain tumors: a meta-analysis. Am J Epidemiol 168(4):366–373
Harding NJ, Birch JM, Hepworth SJ, McKinney PA (2008) Atopic dysfunction and risk of central nervous system tumours in children. Eur J Cancer 44(1):92–99
Harding NJ, Birch JM, Hepworth SJ, McKinney PA (2009) Infectious exposure in the first year of life and risk of central nervous system tumors in children: analysis of day care, social contact, and overcrowding. Cancer Causes Control 20(2):129–136
Harding NJ, Birch JM, Hepworth SJ, McKinney PA, Investigators U (2007) Breastfeeding and risk of childhood CNS tumours. Br J Cancer 96(5):815–817
Hargrave D, Bartels U, Bouffet E (2006) Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 7(3):241–248
Hart RM, Kimler BF, Evans RG, Park CH (1987) Radiotherapeutic management of medulloblastoma in a pediatric patient with ataxia telangiectasia. Int J Radiat Oncol Biol Phys 13(8):1237–1240
Hashimoto T, Sasagawa I, Ishigooka M, Kubota Y, Nakada T, Fujita T et al (1995) Down’s syndrome associated with intracranial germinoma and testicular embryonal carcinoma. Urol Int 55(2):120–122
Hayostek CJ, Shaw EG, Scheithauer B, O’Fallon JR, Weiland TL, Schomberg PJ et al (1993) Astrocytomas of the cerebellum. A comparative clinicopathologic study of pilocytic and diffuse astrocytomas. Cancer 72(3):856–869
Heck JE, Lombardi CA, Cockburn M, Meyers TJ, Wilhelm M, Ritz B (2013) Epidemiology of rhabdoid tumors of early childhood. Pediatr Blood Cancer 60(1):77–81
Hemminki K, Kyyronen P, Vaittinen P (1999) Parental age as a risk factor of childhood leukemia and brain cancer in offspring. Epidemiology 10(3):271–275
Hilden JM, Meerbaum S, Burger P, Finlay J, Janss A, Scheithauer BW et al (2004) Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol 22(14):2877–2884
Hoffman HJ, Soloniuk DS, Humphreys RP, Drake JM, Becker LE, De Lima BO et al (1993) Management and outcome of low-grade astrocytomas of the midline in children: a retrospective review. Neurosurgery 33(6):964–971
Huncharek M, Kupelnick B, Klassen H (2002) Maternal smoking during pregnancy and the risk of childhood brain tumors: a meta-analysis of 6566 subjects from twelve epidemiological studies. J Neuro-Oncol 57(1):51–57
Ikeda J, Sawamura Y, van Meir EG (1998) Pineoblastoma presenting in familial adenomatous polyposis (FAP): random association, FAP variant or Turcot syndrome? Br J Neurosurg 12(6):576–578
Infante-Rivard C, El-Zein M (2007) Parental alcohol consumption and childhood cancers: a review. J Toxicol Environ Health B Crit Rev 10(1–2):101–129
Jackson S, Patay Z, Howarth R, Pai Panandiker AS, Onar-Thomas A, Gajjar A et al (2013) Clinico-radiologic characteristics of long-term survivors of diffuse intrinsic pontine glioma. J Neuro-Oncol 114(3):339–344
Jahraus CD, Tarbell NJ (2006) Optic pathway gliomas. Pediatr Blood Cancer 46(5):586–596
Janss AJ, Grundy R, Cnaan A, Savino PJ, Packer RJ, Zackai EH et al (1995) Optic pathway and hypothalamic/chiasmatic gliomas in children younger than age 5 years with a 6-year follow-up. Cancer 75(4):1051–1059
Jeong TS, Yee GT (2014) Glioblastoma in a patient with neurofibromatosis type 1: a case report and review of the literature. Brain Tumor Res Treat 2(1):36–38
Johnson KJ, Carozza SE, Chow EJ, Fox EE, Horel S, McLaughlin CC et al (2009) Parental age and risk of childhood cancer: a pooled analysis. Epidemiology 20(4):475–483
Johnston DL, Keene D, Kostova M, Strother D, Lafay-Cousin L, Fryer C et al (2014) Incidence of medulloblastoma in Canadian children. J Neuro-Oncol 120(3):575–579
Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K et al (2008) Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68(21):8673–8677
Kaatsch P, Scheidemann-Wesp U, Schuz J (2010) Maternal use of antibiotics and cancer in the offspring: results of a case-control study in Germany. Cancer Causes Control 21(8):1335–1345
Khafaga Y, Hassounah M, Kandil A, Kanaan I, Allam A, El Husseiny G et al (2003) Optic gliomas: a retrospective analysis of 50 cases. Int J Radiat Oncol Biol Phys 56(3):807–812
Khan S, Evans AA, Rorke-Adams L, Orjuela MA, Shiminski-Maher T, Bunin GR (2010) Head injury, diagnostic X-rays, and risk of medulloblastoma and primitive neuroectodermal tumor: a Children’s Oncology Group study. Cancer Causes Control 21(7):1017–1023
Khatib ZA, Heideman RL, Kovnar EH, Langston JA, Sanford RA, Douglas EC et al (1994) Predominance of pilocytic histology in dorsally exophytic brain stem tumors. Pediatr Neurosurg 20(1):2–10
Kheifets L, Ahlbom A, Crespi CM, Feychting M, Johansen C, Monroe J et al (2010) A pooled analysis of extremely low-frequency magnetic fields and childhood brain tumors. Am J Epidemiol 172(7):752–761
Kleinerman RA (2006) Cancer risks following diagnostic and therapeutic radiation exposure in children. Pediatr Radiol 36(Suppl 2):121–125
Kobayashi N, Furukawa T, Takatsu T (1968) Congenital anomalies in children with malignancy. Paediatr Univ Tokyo 16:31–37
Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA et al (2012) Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropathol 123(4):473–484
Lafay-Cousin L, Hawkins C, Carret AS, Johnston D, Zelcer S, Wilson B et al (2012) Central nervous system atypical teratoid rhabdoid tumours: the Canadian paediatric brain tumour consortium experience. Eur J Cancer 48(3):353–359
Laithier V, Grill J, Le Deley MC, Ruchoux MM, Couanet D, Doz F et al (2003) Progression-free survival in children with optic pathway tumors: dependence on age and the quality of the response to chemotherapy--results of the first French prospective study for the French Society of Pediatric Oncology. J Clin Oncol 21(24):4572–4578
Larsen AK, Mikkelsen DB, Hertz JM, Bygum A (2014) Manifestations of Gorlin-Goltz syndrome. Dan Med J 61(5):A4829
Lassiter KR, Alexander E Jr, Davis CH Jr, Kelly DL Jr (1971) Surgical treatment of brain stem gliomas. J Neurosurg 34(6):719–725
Li MH, Bouffet E, Hawkins CE, Squire JA, Huang A (2005) Molecular genetics of supratentorial primitive neuroectodermal tumors and pineoblastoma. Neurosurg Focus 19(5):E3
Listernick R, Charrow J, Greenwald MJ, Esterly NB (1989) Optic gliomas in children with neurofibromatosis type 1. J Pediatr 114(5):788–792
Lober RM, Cho YJ, Tang Y, Barnes PD, Edwards MS, Vogel H et al (2014) Diffusion-weighted MRI derived apparent diffusion coefficient identifies prognostically distinct subgroups of pediatric diffuse intrinsic pontine glioma. J Neuro-Oncol 117(1):175–182
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A et al (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114(2):97–109
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK et al (2016) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820
MacLean J, Partap S, Reynolds P, Von Behren J, Fisher PG (2010) Birth weight and order as risk factors for childhood central nervous system tumors. J Pediatr 157(3):450–455
Macy ME, Birks DK, Barton VN, Chan MH, Donson AM, Kleinschmidt-Demasters BK et al (2012) Clinical and molecular characteristics of congenital glioblastoma. Neuro-Oncology 14(7):931–941
Mathew RK, O’Kane R, Parslow R, Stiller C, Kenny T, Picton S et al (2014) Comparison of survival between the UK and US after surgery for most common pediatric CNS tumors. Neuro-Oncology 16(8):1137–1145
Mathews JD, Forsythe AV, Brady Z, Butler MW, Goergen SK, Byrnes GB et al (2013) Cancer risk in 680,000 people exposed to computed tomography scans in childhood or adolescence: data linkage study of 11 million Australians. BMJ 346:f2360
Mazumdar M, Liu CY, Wang SF, Pan PC, Wu MT, Christiani DC et al (2008) No association between parental or subject occupation and brain tumor risk. Cancer Epidemiol Biomark Prev 17(7):1835–1837
McGuire CS, Sainani KL, Fisher PG (2009) Incidence patterns for ependymoma: a surveillance, epidemiology, and end results study. J Neurosurg 110(4):725–729
Mellemkjaer L, Hasle H, Gridley G, Johansen C, Kjaer SK, Frederiksen K et al (2006) Risk of cancer in children with the diagnosis immaturity at birth. Paediatr Perinat Epidemiol 20(3):231–237
Metzger AK, Sheffield VC, Duyk G, Daneshvar L, Edwards MS, Cogen PH (1991) Identification of a germ-line mutation in the p53 gene in a patient with an intracranial ependymoma. Proc Natl Acad Sci U S A 88(17):7825–7829
Miller RW (1971) Deaths from childhood leukemia and solid tumors among twins and other sibs in the United States, 1960–67. J Natl Cancer Inst 46(1):203–209
Milne E, Greenop KR, Bower C, Miller M, van Bockxmeer FM, Scott RJ et al (2012) Maternal use of folic acid and other supplements and risk of childhood brain tumors. Cancer Epidemiol Biomark Prev 21(11):1933–1941
Milne E, Greenop KR, Scott RJ, de Klerk NH, Bower C, Ashton LJ et al (2013) Parental alcohol consumption and risk of childhood acute lymphoblastic leukemia and brain tumors. Cancer Causes Control 24(2):391–402
Milne E, Laurvick CL, Blair E, de Klerk N, Charles AK, Bower C (2008) Fetal growth and the risk of childhood CNS tumors and lymphomas in Western Australia. Int J Cancer 123(2):436–443
Mody R, Li S, Dover DC, Sallan S, Leisenring W, Oeffinger KC et al (2008) Twenty-five-year follow-up among survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Blood 111(12):5515–5523
Mori K, Kurisaka M (1986) Brain tumors in childhood: statistical analysis of cases from the brain tumor registry of Japan. Childs Nerv Syst 2(5):233–237
Nicolin G, Parkin P, Mabbott D, Hargrave D, Bartels U, Tabori U et al (2009) Natural history and outcome of optic pathway gliomas in children. Pediatr Blood Cancer 53(7):1231–1237
Ning MS, Perkins SM, Dewees T, Shinohara ET (2015) Evidence of high mortality in long term survivors of childhood medulloblastoma. J Neuro-Oncol 122(2):321–327
Norman MA, Holly EA, Ahn DK, Preston-Martin S, Mueller BA, Bracci PM (1996) Prenatal exposure to tobacco smoke and childhood brain tumors: results from the United States West Coast childhood brain tumor study. Cancer Epidemiol Biomark Prev 5(2):127–133
Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29(11):1408–1414
O’Callaghan FJ, Martyn CN, Renowden S, Noakes M, Presdee D, Osborne JP (2008) Subependymal nodules, giant cell astrocytomas and the tuberous sclerosis complex: a population-based study. Arch Dis Child 93(9):751–754
O’Callaghan FJ, Shiell AW, Osborne JP, Martyn CN (1998) Prevalence of tuberous sclerosis estimated by capture-recapture analysis. Lancet 351(9114):1490
Ohgaki H, Kleihues P (2005) Epidemiology and etiology of gliomas. Acta Neuropathol 109(1):93–108
Ortega-Garcia JA, Ferris-Tortajada J, Claudio L, Soldin OP, Sanchez-Sauco MF, Fuster-Soler JL et al (2010) Case control study of periconceptional folic acid intake and nervous system tumors in children. Childs Nerv Syst 26(12):1727–1733
Ostrom QT, Chen Y, MdB P, Ondracek A, Farah P, Gittleman H et al (2014) The descriptive epidemiology of atypical teratoid/rhabdoid tumors in the United States, 2001–2010. Neuro-Oncology 16(10):1392–1399
Ostrom QT, Gittleman H, Liao P, Rouse C, Chen Y, Dowling J et al (2014) CBTRUS statistical report: primary brain and central nervous system tumors diagnosed in the United States in 2007–2011. Neuro-Oncology 16(Suppl 4):iv1–i63
Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL et al (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24(25):4202–4208
Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F et al (2015) Molecular classification of ependymal tumors across all CNS compartments, histopathological grades, and age groups. Cancer Cell 27(5):728–743
Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y et al (2014) C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 506(7489):451–455
Partap S, MacLean J, Von Behren J, Reynolds P, Fisher PG (2011) Birth anomalies and obstetric history as risks for childhood tumors of the central nervous system. Pediatrics 128(3):e652–e657
Pearce MS, Hammal DM, Dorak MT, McNally RJ, Parker L (2006) Paternal occupational exposure to pesticides or herbicides as risk factors for cancer in children and young adults: a case-control study from the North of England. Arch Environ Occup Health 61(3):138–144
Pearce MS, Salotti JA, Little MP, McHugh K, Lee C, Kim KP et al (2012) Radiation exposure from CT scans in childhood and subsequent risk of leukaemia and brain tumours: a retrospective cohort study. Lancet 380(9840):499–505
Perilongo G, Massimino M, Sotti G, Belfontali T, Masiero L, Rigobello L et al (1997) Analyses of prognostic factors in a retrospective review of 92 children with ependymoma: Italian Pediatric Neuro-Oncology Group. Med Pediatr Oncol 29(2):79–85
Perkins SM, Mitra N, Fei W, Shinohara ET (2012) Patterns of care and outcomes of patients with pleomorphic xanthoastrocytoma: a SEER analysis. J Neuro-Oncol 110(1):99–104
Peters S, Glass DC, Reid A, de Klerk N, Armstrong BK, Kellie S et al (2013) Parental occupational exposure to engine exhausts and childhood brain tumors. Int J Cancer 132(12):2975–2979
Plotkin SR, O’Donnell CC, Curry WT, Bove CM, MacCollin M, Nunes FP (2011) Spinal ependymomas in neurofibromatosis type 2: a retrospective analysis of 55 patients. J Neurosurg Spine 14(4):543–547
Ragel BT, Osborn AG, Whang K, Townsend JJ, Jensen RL, Couldwell WT (2006) Subependymomas: an analysis of clinical and imaging features. Neurosurgery 58(5):881–890. discussion 90
Rajaraman P, Simpson J, Neta G, Berrington de Gonzalez A, Ansell P, Linet MS et al (2011) Early life exposure to diagnostic radiation and ultrasound scans and risk of childhood cancer: case-control study. BMJ 342:d472
Ramasubramanian A, Kytasty C, Meadows AT, Shields JA, Leahey A, Shields CL (2013) Incidence of pineal gland cyst and pineoblastoma in children with retinoblastoma during the chemoreduction era. Am J Ophthalmol 156(4):825–829
Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ et al (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14(12):1200–1207
Ramaswamy V, Remke M, Shih D, Wang X, Northcott PA, Faria CC et al (2014) Duration of the pre-diagnostic interval in medulloblastoma is subgroup dependent. Pediatr Blood Cancer 61(7):1190–1194
Ramos A, Hilario A, Lagares A, Salvador E, Perez-Nunez A, Sepulveda J (2013) Brainstem gliomas. Semin Ultrasound CT MR 34(2):104–112
Robertson PL, Zeltzer PM, Boyett JM, Rorke LB, Allen JC, Geyer JR et al (1998) Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s Cancer Group. J Neurosurg 88(4):695–703
Rogers L, Pattisapu J, Smith RR, Parker P (1988) Medulloblastoma in association with the Coffin-Siris syndrome. Childs Nerv Syst 4(1):41–44
Roncarolo F, Infante-Rivard C (2012) Asthma and risk of brain cancer in children. Cancer Causes Control 23(4):617–623
Ross GW, Rubinstein LJ (1989) Lack of histopathological correlation of malignant ependymomas with postoperative survival. J Neurosurg 70(1):31–36
Rosso AL, Hovinga ME, Rorke-Adams LB, Spector LG, Bunin GR, Children’s Oncology G (2008) A case-control study of childhood brain tumors and fathers' hobbies: a Children’s Oncology Group study. Cancer Causes Control 19(10):1201–1207
Sabbaghian N, Hamel N, Srivastava A, Albrecht S, Priest JR, Foulkes WD (2012) Germline DICER1 mutation and associated loss of heterozygosity in a pineoblastoma. J Med Genet 49(7):417–419
Scheinker I (1945) Subependymoma: a newly recognized tumor of subependymal derivation. J Neurosurg 2:232–240
Schmidt LS, Schuz J, Lahteenmaki P, Trager C, Stokland T, Gustafson G et al (2010) Fetal growth, preterm birth, neonatal stress and risk for CNS tumors in children: a Nordic population- and register-based case-control study. Cancer Epidemiol Biomark Prev 19(4):1042–1052
Schmiegelow K, Levinsen MF, Attarbaschi A, Baruchel A, Devidas M, Escherich G et al (2013) Second malignant neoplasms after treatment of childhood acute lymphoblastic leukemia. J Clin Oncol 31(19):2469–2476
Schuz J, Weihkopf T, Kaatsch P (2007) Medication use during pregnancy and the risk of childhood cancer in the offspring. Eur J Pediatr 166(5):433–441
Searles Nielsen S, McKean-Cowdin R, Farin FM, Holly EA, Preston-Martin S, Mueller BA (2010) Childhood brain tumors, residential insecticide exposure, and pesticide metabolism genes. Environ Health Perspect 118(1):144–149
Shankar GM, Lelic N, Gill CM, Thorner AR, Van Hummelen P, Wisoff JH et al (2016) BRAF alteration status and the histone H3F3A gene K27M mutation segregate spinal cord astrocytoma histology. Acta Neuropathol 131(1):147–150
Shaw AK, Li P, Infante-Rivard C (2006) Early infection and risk of childhood brain tumors (Canada). Cancer Causes Control 17(10):1267–1274
Shim YK, Mlynarek SP, van Wijngaarden E (2009) Parental exposure to pesticides and childhood brain cancer: U.S. Atlantic coast childhood brain cancer study. Environ Health Perspect 117(6):1002–1006
Shu X, Prochazka M, Lannering B, Schuz J, Roosli M, Tynes T et al (2014) Atopic conditions and brain tumor risk in children and adolescents--an international case-control study (CEFALO). Ann Oncol 25(4):902–908
Siegel RL, Miller KD, Jemal A (2015) Cancer statistics, 2015. CA Cancer J Clin 65(1):5–29
Singhal S, Birch JM, Kerr B, Lashford L, Evans DG (2002) Neurofibromatosis type 1 and sporadic optic gliomas. Arch Dis Child 87(1):65–70
Skomorowski M, Taxier M, Wise W Jr (2012) Turcot syndrome type 2: medulloblastoma with multiple colorectal adenomas. Clin Gastroenterol Hepatol 10(10):A24
Smith MA, Freidlin B, Ries LA, Simon R (1998) Trends in reported incidence of primary malignant brain tumors in children in the United States. J Natl Cancer Inst 90(17):1269–1277
Smoots DW, Geyer JR, Lieberman DM, Berger MS (1998) Predicting disease progression in childhood cerebellar astrocytoma. Childs Nerv Syst 14(11):636–648
Sredni ST, Tomita T (2015) Rhabdoid tumor predisposition syndrome. Pediatr Dev Pathol 18(1):49–58
Stalberg K, Haglund B, Axelsson O, Cnattingius S, Pfeifer S, Kieler H (2007) Prenatal X-ray exposure and childhood brain tumours: a population-based case-control study on tumour subtypes. Br J Cancer 97(11):1583–1587
Stalberg K, Haglund B, Stromberg B, Kieler H (2010) Prenatal exposure to medicines and the risk of childhood brain tumor. Cancer Epidemiol 34(4):400–404
Stokland T, Liu JF, Ironside JW, Ellison DW, Taylor R, Robinson KJ et al (2010) A multivariate analysis of factors determining tumor progression in childhood low-grade glioma: a population-based cohort study (CCLG CNS9702). Neuro-Oncology 12(12):1257–1268
Tarbell NJ, Friedman H, Polkinghorn WR, Yock T, Zhou T, Chen Z et al (2013) High-risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol 31(23):2936–2941
Taylor MD, Gokgoz N, Andrulis IL, Mainprize TG, Drake JM, Rutka JT (2000) Familial posterior fossa brain tumors of infancy secondary to germline mutation of the hSNF5 gene. Am J Hum Genet 66(4):1403–1406
Taylor KR, Mackay A, Truffaux N, Butterfield YS, Morozova O, Philippe C et al (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46(5):457–461
Taylor MD, Mainprize TG, Rutka JT, Becker L, Bayani J, Drake JM (2001) Medulloblastoma in a child with Rubenstein-Taybi syndrome: case report and review of the literature. Pediatr Neurosurg 35(5):235–238
Veldhuijzen van Zanten SE, Jansen MH, Sanchez Aliaga E, van Vuurden DG, Vandertop WP, Kaspers GJ (2014) A twenty-year review of diagnosing and treating children with diffuse intrinsic pontine glioma in The Netherlands. Expert Rev Anticancer Ther:1–8
Vera-Bolanos E, Aldape K, Yuan Y, Wu J, Wani K, Necesito-Reyes MJ et al (2015) Clinical course and progression-free survival of adult intracranial and spinal ependymoma patients. Neuro-Oncology 17(3):440–447
Villa S, Miller RC, Krengli M, Abusaris H, Baumert BG, Servagi-Vernat S et al (2012) Primary pineal tumors: outcome and prognostic factors--a study from the Rare Cancer Network (RCN). Clin Transl Oncol 14(11):827–834
Virchow R (1863–67) Die krankhaften Geschwülste, vol 3. August Hirschwald, Berlin
von Hoff K, Hinkes B, Dannenmann-Stern E, von Bueren AO, Warmuth-Metz M, Soerensen N et al (2011) Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr Blood Cancer 57(6):978–985
Winther JF, Sankila R, Boice JD, Tulinius H, Bautz A, Barlow L et al (2001) Cancer in siblings of children with cancer in the Nordic countries: a population-based cohort study. Lancet 358(9283):711–717
Witt H, Mack SC, Ryzhova M, Bender S, Sill M, Isserlin R et al (2011) Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20(2):143–157
Woehrer A, Slavc I, Waldhoer T, Heinzl H, Zielonke N, Czech T et al (2010) Incidence of atypical teratoid/rhabdoid tumors in children: a population-based study by the Austrian brain tumor registry, 1996–2006. Cancer 116(24):5725–5732
Woesler B, Moskopp D, Kuchelmeister K, Schul C, Wassmann H (1998) Intracranial metastasis of a spinal myxopapillary ependymoma. A case report. Neurosurg Rev 21(1):62–65
Wong TT, Ho DM, Chang TK, Yang DD, Lee LS (1995) Familial neurofibromatosis 1 with germinoma involving the basal ganglion and thalamus. Childs Nerv Syst 11(8):456–458
Wong TT, Ho DM, Chang KP, Yen SH, Guo WY, Chang FC et al (2005) Primary pediatric brain tumors: statistics of Taipei VGH, Taiwan (1975–2004). Cancer 104(10):2156–2167
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J et al (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44(3):251–253
Yip BH, Pawitan Y, Czene K (2006) Parental age and risk of childhood cancers: a population-based cohort study from Sweden. Int J Epidemiol 35(6):1495–1503
Zacharoulis S, Ji L, Pollack IF, Duffner P, Geyer R, Grill J et al (2008) Metastatic ependymoma: a multi-institutional retrospective analysis of prognostic factors. Pediatr Blood Cancer 50(2):231–235
Zhou D, Zhang Y, Liu H, Luo S, Luo L, Dai K (2008) Epidemiology of nervous system tumors in children: a survey of 1,485 cases in Beijing Tiantan Hospital from 2001 to 2005. Pediatr Neurosurg 44(2):97–103
Zuccoli G, Izzi G, Bacchini E, Tondelli MT, Ferrozzi F, Bellomi M (1999) Central nervous system atypical teratoid/rhabdoid tumour of infancy. CT and mr findings. Clin Imaging 23(6):356–360
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Vitanza, N.A., Campen, C.J., Fisher, P.G. (2018). Epidemiology of Pediatric Central Nervous System Tumors. In: Gajjar, A., Reaman, G., Racadio, J., Smith, F. (eds) Brain Tumors in Children. Springer, Cham. https://doi.org/10.1007/978-3-319-43205-2_1
Download citation
DOI: https://doi.org/10.1007/978-3-319-43205-2_1
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-43203-8
Online ISBN: 978-3-319-43205-2
eBook Packages: MedicineMedicine (R0)